

Abstract
Understanding the function of human proteins is essential to decipher the molecular mechanisms of human diseases and phenotypes. Of the 17470 human protein coding genes in neXtProt 2018-01-17 database with unequivocal protein existence evidence (PE1), 1260 proteins do not have characterized functions. To reveal the function of poorly annotated human proteins, we developed a hybrid pipeline that creates protein structure prediction using I-TASSER and infers functional insights for the target protein from the functional templates recognized by COFACTOR. As a case study, the pipeline was applied to all 66 PE1 proteins with unknown or insufficiently specific function (uPE1) on human chromosome 17 as of neXtProt 2017-07-01. Benchmark testing on a control set of 100 well-characterized proteins randomly selected from the same chromosome shows high Gene Ontology (GO) term prediction accuracies of 0.69, 0.57, and 0.67 for molecular function (MF), biological process (BP) and cellular component (CC), respectively. Three pipelines of function annotations (homology detection, protein-protein interaction network inference, and structure template identification) have been exploited by COFACTOR. Detailed analyses show that structure template detection based on low-resolution protein structure prediction made the major contribution to enhancement of the sensitivity and precision of the annotation predictions, especially for cases that do not have sequence-level homologous templates. For the chromosome 17 uPE1 proteins, the I-TASSER/COFACTOR pipeline confidently assigned MF, BP and CC for 13, 33, and 49 proteins, respectively, with predicted functions ranging from sphingosine N-acyltransferase activity and sugar transmembrane transporter to cytoskeleton constitution. We highlight the 13 proteins with confident MF predictions; 11 of these are among the 33 proteins with confident BP predictions and 12 are among the 49 proteins with confident CC. This study demonstrates a novel computational approach to systematically annotate protein function in the human proteome and provides useful insights to guide experimental design and follow-up validation studies of these uncharacterized proteins.
Keywords: human proteome, chromosome 17, neXtProt protein existence levels, uPE1 proteins with unknown function, structure based function annotation, I-TASSER, COFACTOR
For TOC Only
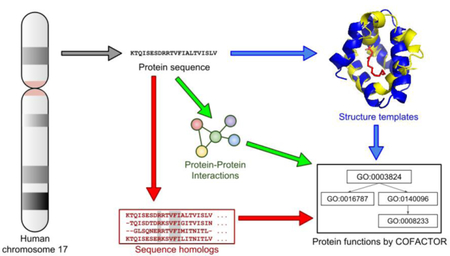
INTRODUCTION
As the direct carriers of biological functions in the human body, proteins participate in nearly all biological events, including catalysis of endogenous metabolites, regulation of most biological pathways, and formation of many subcellular structures. Understanding the function of human proteins has become an important prerequisite to uncover the secrets of human diseases and diverse phenotypes in modern biomedical studies. As a protein usually must be folded into specific tertiary structure in order to be functionally active, determining protein structure is an important avenue in protein function annotation.
Despite many years of community efforts in protein characterization, there is still a substantial number of proteins whose structure and biological functions are incomplete or unknown. Among all the 17470 confidently identified (PE1) human proteins in the neXtProt1 release 2018-01-17, there are 1260 uPE1 entries which do not have specific functional annotation (Supplementary Text S1). In the same neXtProt release, there are 6188 out of 17470 PE1 entries with experimental 3D structures but only 32 among the 1260 uPE1 proteins. The lack of structure and function annotations for many proteins in the human proteome limits our capability to understand their functional roles even in tissues with high expression. For example, of the 26 uPE1 proteins on chromosome 17 with immunohistochemistry data in Human Protein Atlas2 (retrieved on 2018-05-09), 24 have “high” expression in at least one tissue as detected by antibody studies. Similarly, 52 of the 66 uPE1 proteins on chromosome 17 (as of neXtProt 2017-08-01) have median RNA expression levels higher than 10 Transcripts per Million (TPM) in at least one tissue, as reported in GTEx3 version 7.
To alleviate the issue in protein structure and function annotations, we developed a hybrid pipeline which creates 3D structure prediction using I-TASSER4, with the functional insights deduced by COFACTOR5. Both I-TASSER and COFACTOR pipelines have been tested in community-wide blinded experiments, which demonstrate considerable reliability of structure modeling and functional annotations. For example, in CASP12, for 53 targets with template structures identified in PDB, I-TASSER generated correct folds with a TM-score >0.5 for 47 cases, where in 41 cases structures were driven closer to the native than the templates. For 39 free-modeling (FM) targets which do not have any similar fold in the PDB database, 11 were correctly folded by I-TASSER.6 In CASP9, the COFACTOR algorithm7 achieved a functional residue prediction precision of 72% and Matthews correlation coefficient 0.69 for the 31 function prediction targets, which were higher than those by all other methods in the experiment.7
The original version of COFACTOR8 was built on the transfer of function from structural templates detected by homologous and analogous structure alignments. That version of COFACTOR was used to suggest structure and function for dubious proteins in the human proteome (PE5).9 Recently, C. Zhang et al developed an extended version of COFACTOR with additional sequence and Protein-Protein Interaction (PPI) pipelines, which was tested in the most recent CAFA3 function annotation experiment.5, 10 According to the CAFA3 evaluation (https://www.synapse.org/#!Synapse:syn12299467) for GO term prediction in MF, BP, and CC aspects, COFACTOR achieved F1-scores (defined in Equation 1 below) 0.57, 0.60, and 0.61, respectively, which are 43%, 81%, and 17% higher in accuracy than the best baseline methods used by assessors. Additionally, we have used the I-TASSER/COFACTOR pipeline for proteome-wide structure and function modeling of E. coli proteins, and the predicted functions of three proteins have been validated by enzymatic assay and mutation experiments.11
In light of recent progress, we applied this pipeline to better annotate the human proteome as part of the HUPO Chromosome-centric Human Proteome Project (C-HPP).12 As a proof-of-principle study, we applied the I-TASSER/COFACTOR pipeline to all 66 uPE1 proteins from human chromosome 17 in neXtProt 2017-08-01 release to decipher the structure and function of these poorly annotated human proteins. The full prediction results as well as updated neXtProt annotations for these targets are available at https://zhanglab.ccmb.med.umich.edu/COFACTOR/chr17/.
MATERIALS AND METHODS
Protein structure and function prediction pipelines
Our computational workflow for structure-based function annotation of a given protein consists of two main components: structure modeling by I-TASSER and function annotation by COFACTOR (Figure 1). The pipeline is fully automated with the query sequence as the sole input.
Figure 1.
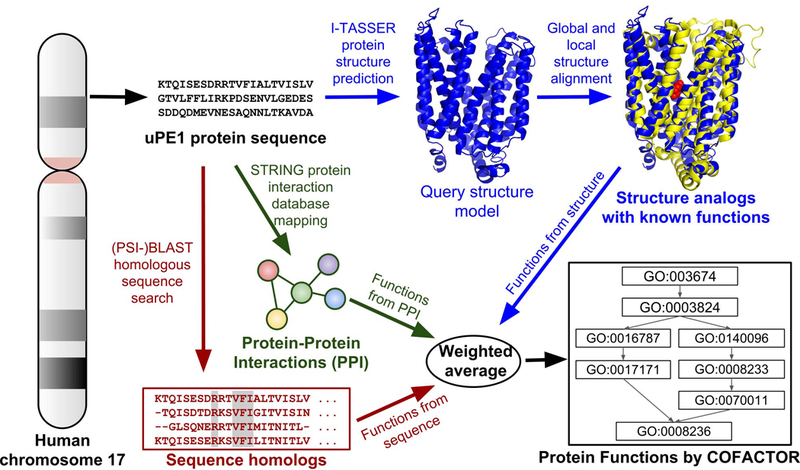
Flowchart of the hybrid I-TASSER/COFACTOR pipeline for protein structure and function prediction, applied to uPE1 proteins from human chromosome 17.
In the I-TASSER structure prediction stage, the query protein sequence is first threaded through a non-redundant PDB library (https://zhanglab.ccmb.med.umich.edu/library/) by LOMETS13, which is a locally-installed meta threading algorithm combining 10 different state-of-the-art threading programs14–22, to identify structure templates. Continuous fragments are excised from these template structures, which are subsequently assembled into full length structure by replica-exchange Monte Carlo (REMC) simulation implemented by I-TASSER. Tens of thousands of decoy conformations from the REMC simulation trajectory are then clustered by SPICKER23 by structure similarity. The centroid of the largest cluster, which corresponds to the conformation with lowest free energy, is selected to undergo structure refinement by FG-MD24 to obtain the final structure model. While I-TASSER typically reports up to five structure models, ranked in descending order of the size of cluster from which a model came, we use only the first I-TASSER model for subsequent function modeling. That is because the first model has the highest confidence score and on average is closer to native structure than the lower-ranked models.25
To obtain function annotation for the query structure model, the COFACTOR structure-based function prediction approach uses a modified TM-align26 structure alignment program to search the query structure against entries templates from the BioLiP27 structure-function database to identify structure templates with function annotations. The functions of structure templates are then transferred to query according to global structure similarity, active site local similarity and matching of sequence profiles between query and template, as measured by a combination of global and local structure alignments10. The combination of global and local structure similarity is critical to structure-based function annotation, as shown previously.10 If only global similarity is considered, the annotation result can be misled by fold promiscuity, where proteins sharing highly similar global topology can have very different functions.28 On the other hand, relying only on active site local structure similarity can also lead to false positive hits: ligand binding pockets with similar conformation can be associated with unrelated biochemical functions due to the very limited number of possible pocket structures.29 To further disentangle the structure promiscuity issue, the above structure-based function annotation is supplemented by the sequence-based approach, which extracts function annotations from BLAST and PSI-BLAST30 hits in the UniProt31 database search. Meanwhile, the protein-protein interaction (PPI) based approach infers function from UniProt sequences homologous to the query’s PPI partners, as defined by the STRING32 database. Each of the three structure, sequence, and PPI-based approaches provides a confidence score ranging from 0 to 1 for a given predicted GO term; the final consensus GO term prediction is a weighted average of the three approaches.
Assessment metrics for structure and function prediction
Following the standard practice of CAFA, the GO term prediction accuracy is mainly evaluated by maximum F1-score, i.e., the F-measure:
| (1) |
| (2) |
Here, pr(t) and re(t) are the prediction precision and recall, respectively, at confidence score cutoff ≥ t. Precision is defined as the number of correctly predicted GO terms tp(t) over the number of all predicted GO terms tp(t) + fp(t), while recall is defined as tp(t) divided by all GO terms annotated to query by neXtProt gold standard.
The structure modeling quality of I-TASSER is evaluated by TM-score33 between first I-TASSER model and native experimental structure. Ranging between 0 and 1, TM-score is a commonly used metric to assess structure similarity between two protein structures, with a TM-score>0.5 indicating the two conformations sharing the same topology:34
| (3) |
Here, L is the number of residues in a protein, Lali is the number of aligned residues, di is the distance between the ith aligned residue pair, and is a normalization factor that ensures TM-score is independent of protein size.
RESULTS AND DISCUSSION
Data Sets
The 66 uPE1 proteins from chromosome 17 were compiled from neXtProt release 2017-08-01. The detailed protocol for generating this list is specified in supplementary Text S2. While most of these uPE1 proteins do not have any GO term annotations for MF and BP, some of them have GO terms that are considered too generic by neXtProt to be qualified as “annotated” proteins, including protein binding, calcium binding, zinc binding, identical protein binding, and protein homooligomerization. As neXtProt does not consider GO CC terms when defining uPE1 proteins in the SPARQL query, some of these uPE1 proteins do have GO CC term annotations. For example, SYNGR2 (neXtProt ID: NX_O43760–1) is annotated as being located at “neuromuscular junction” (GO:0031594) and at “synaptic vesicle membrane” (GO:0030672) for CC based on its known role in modulating the localization of synaptophysin into synaptic-like microvesicles.35–36 Due to this known bias in how neXtProt treats GO CC terms for uPE1 proteins, we later discuss instances where our CC term prediction is different from existing neXtProt annotations.
The numbers of uPE1 proteins are “moving targets” due to new experimental evidence as well as evolving criteria reflected in excluded MF and BP terms. Thus neXtProt release 2017-08-01, which this study was based on, had 1218 uPE1 proteins proteome-wide and 66 uPE1 chromosome 17 proteins; neXtProt release 2018-01-17 has 1260 and 70, respectively (supplementary Text S1).
To establish the dependency of GO term prediction accuracy on confidence score of COFACTOR prediction, a benchmark set of 100 well-annotated proteins was randomly selected from the same chromosome according to the following criteria: (1) the protein has a protein neXtProt existence evidence level of PE1; and (2) it has experimental GO term annotation for all three aspects (MF, BP, CC) with “gold” evidence in neXtProt and with at least one of the seven high confidence evidence codes (EXP, IDA, IMP, IGI, IEP, TAS and IC) in UniProt, excluding non-specific GO terms such as protein binding mentioned above (Text S3). These seven UniProt-assigned evidence codes were used by CAFA for assessment of function predictions, and include five experimental evidence codes (EXP, IDA, IMP, IGI, and IEP) as well as two evidence codes assigned based on assertion of domain experts (TAS and IC). Our benchmark set includes a subset of 59 benchmark proteins with experimental structure information, on which I-TASSER achieves an average TM-score of 0.88 (Table S2).
Benchmark tests on structure and function prediction on well-annotated proteins
To evaluate the prediction accuracy of our approach, the hybrid I-TASSER/COFACTOR method was applied on the 100 well-annotated benchmark proteins. As control algorithms, we included three baseline GO term prediction methods, “BLAST”, “PSI-BLAST”, and “Naïve”, as implemented by CAFA experiments37–38. The “BLAST” and “PSI-BLAST” methods transfer function annotation by sequence identity of (PSI-)BLAST hits in UniProt, while “Naïve” predicts GO terms solely by the frequency of the GO term in the UniProt database regardless of input query. In addition to these three baseline methods, two representative state-of-the-art sequence-based function prediction methods, GoFDR39 and GOtcha40, are included. GoFDR was a top performing program in CAFA2 and transfers GO annotation from sequence homologs based on similarity of putative function discriminating residues. GOtcha infers function from BLAST hits using posterior probability calibrated for 37 representative organisms. To ensure that the benchmark performance on these well annotated proteins can be meaningfully extrapolated to uPE1 proteins, which usually lack experimentally characterized close homologs, we applied a stringent benchmark protocol of excluding any templates sharing >30% sequence identity with the query for both structure and function prediction. Since UniProt and neXtProt may have slightly different annotations for the same protein, GO term annotation with “GOLD” evidence was used as the gold standard for GO term prediction; we found no difference in conclusions if we use either UniProt or neXtProt annotation as gold standard (Table S1).
As shown in Figure 2, the sequence-based component in COFACTOR alone already outperforms all four control methods (BLAST, PSI-BLAST, Naïve, GOtcha and GoFDR) for all three aspects (MF, BP, and CC) of GO term prediction for the benchmark set of 100 PE1 proteins. Here it should be noted that, while COFACTOR and GoFDR use sequence homologs detected by BLAST and PSI-BLAST, both GoFDR and the sequence-based component in COFACTOR outperform the “BLAST” and “PSI-BLAST” control methods. This is because, while the “BLAST” and “PSI-BLAST” control methods report prediction confidence based only on the most significant sequence hit, both COFACTOR and GoFDR combine function annotations from multiple sequence homologs, which helps to enrich correct function annotations from multiple weakly homologous templates. Our sequence-based approach slightly outperforms GoFDR, probably because GoFDR heavily relies on comparison of functional discriminating residues, which are not easy to identify or align for non-homologous targets.
Figure 2.
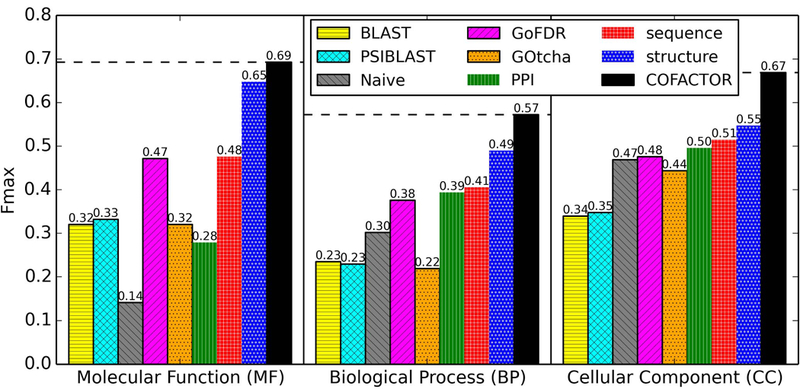
Fmax of different programs for predicting the three aspects of GO terms for the benchmark set of 100 PE1 proteins. “PPI”, “sequence”, and “structure” are the three component methods of “COFACTOR”. For each of the three GO term aspects, the horizontal dash line marks the Fmax of COFACTOR.
It should also be noted that, among the three components of COFACTOR, the structure-based pipeline provides the strongest contribution in function prediction. It has 36%, 21%, and 6% higher prediction accuracy than the sequence-based component and 132%, 24%, and 10% higher prediction accuracy than the PPI-based component in COFACTOR for prediction of the three GO term aspects MF, BP, and CC, respectively. These results underscore the importance of structure information for functional annotation of challenging protein targets with no or few characterized sequence homologs.
For all the three GO term aspects, the final consensus COFACTOR prediction consistently outperformed the most accurate component methods for each aspect, suggesting that each component method does have positive contribution towards final consensus prediction.
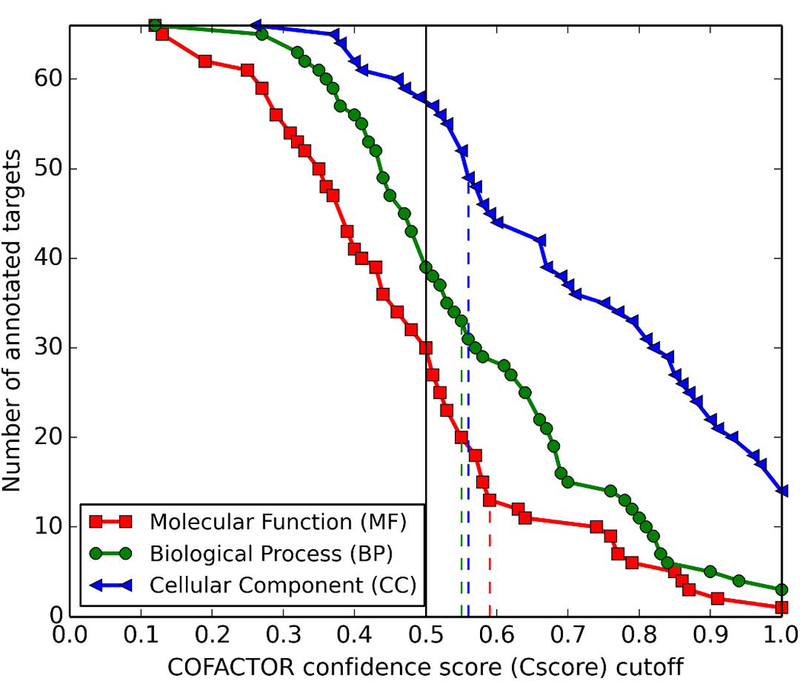
To determine reasonable GO term prediction confidence (Cscore) cutoffs in the I-TASSER/COFACTOR pipeline, we show in Figure 3 the relation between Cscore and prediction accuracy (F-measure). The highest F-measures for MF, BP, and CC are achieved when we choose Cscore cutoffs > 0.59, > 0.55, and > 0.56, respectively.
Figure 3.
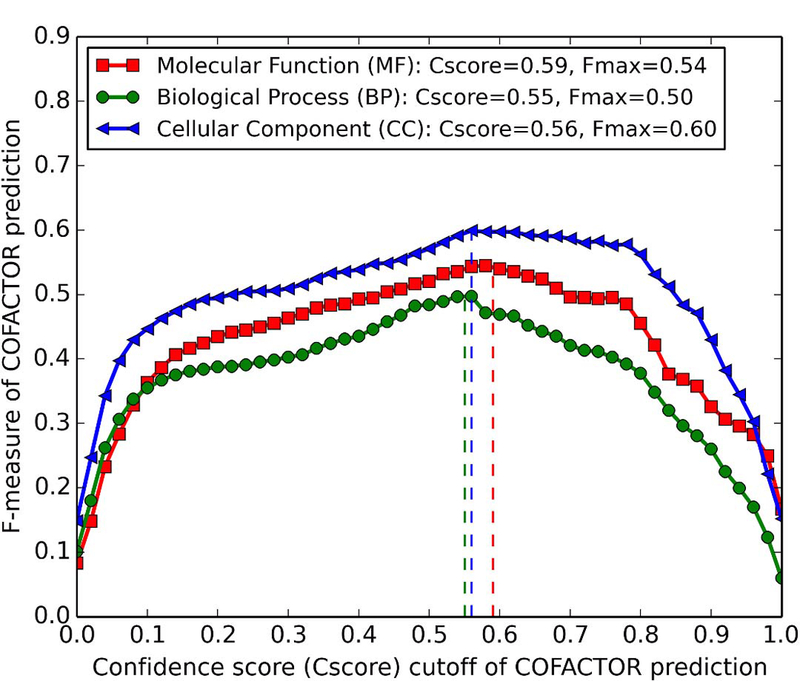
F-measures of COFACTOR prediction versus confidence score cutoffs for the three aspects of GO terms. From left to right, the three vertical dashed lines indicate Cscores 0.55 (green), 0.56 (blue), and 0.59 (red) which are Cscore cutoffs corresponding to the highest F-measure for BP, CC, and MF, respectively.
Since the input of COFACTOR function prediction pipeline is the I-TASSER structure model, we check the dependency of function prediction accuracy on the I-TASSER structure model for the subset of 59 benchmark proteins with experimental structure information (Table S2). Interestingly, I-TASSER structure model quality (in terms of TM-score) is only moderately correlated to GO term prediction accuracy by structure-based pipeline in COFACTOR: the Pearson correlation coefficients between TM-score and F-measure for MF, BP, and CC are 0.44, 0.40, and 0.43, respectively. The correlations between TM-score and F-measure of final consensus COFACTOR function prediction are 0.29, 0.25, and 0.16 for MF, BP, and CC, respectively. Such weak dependency of our function prediction accuracy on I-TASSER structure quality can be partially attributed to the two sequence and PPI-based component methods, which compensate the structure-based pipeline when the I-TASSER model quality is low. For example, the I-TASSER model of the ZNHIT3 protein (neXtProt ID: NX_Q15649–1) has a relatively low TM-score of 0.47 to its native structure (PDB entry 5l85 chain A), which is one of the reasons for the low F-measures of structure-based function prediction (0.35, 0.00, and 0.18, respectively, for MF, BP and CC). Yet, after combining with the sequence and PPI-based methods, the final COFACTOR prediction has much higher F-measures of 0.46, 0.52, and 0.60 for the three GO term aspects. These data suggest that, while accurate structure modeling is certainly desirable for I-TASSER/COFACTOR pipeline, our function annotation approach is not severely biased by low structure modeling quality for targets that are challenging for structure modeling.
As a specific example of the I-TASSER and COFACTOR modeling, we show in Figure 4 the TP53 protein (neXtProt ID: NX_P04637–1) from chromosome 17. As the most extensively studied tumor-suppressor protein and the guardian of the genome41, TP53 is the transcription factor that regulates the expression of multiple downstream cell cycle-related proteins in response to DNA damage. Accordingly, a list of the most confident COFACTOR predictions for TP53 includes “damaged DNA binding” (GO:0003684, Cscore 0.97), “p53 binding” (GO:0002039, Cscore 0.97), and “transcription factor activity, sequence-specific DNA binding” (GO:0003700, Cscore 0.92) for MF; “regulation of cell cycle” (GO:0051726, Cscore 1.00) for BP, and nuclear chromatin (“GO:0000790”, Cscore 0.90) for CC, which are all highly consistent with what we know about TP53. It should be noted that such high confidence prediction resulted from consensus of multiple weakly homologous function templates, as any template sharing >30% sequence identity to query was excluded. Meanwhile, while the native full-length structure of TP53 is unavailable, its DNA binding domain was experimentally determined, which has a striking structure similarity of TM-score 0.96 to its respective portion in the I-TASSER model, despite the model being predicted without any homologous template. The top COFACTOR hit for structure-based function annotation is the CEP-1 (PDB entry 4qo1 chain B, Figure 4 right, TM-score 0.49 to TP53), a transcript factor from C. elegans that is also involved in pathways for DNA-damage response and cell cycle regulation.
Figure 4.
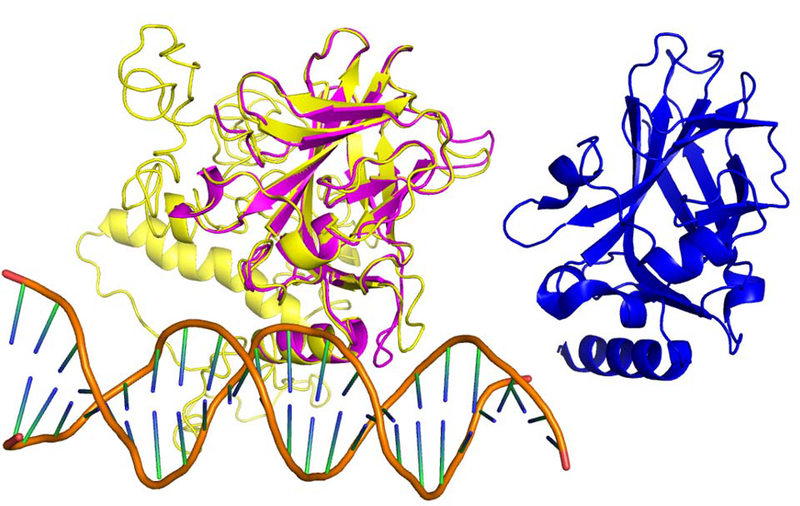
I-TASSER model of full length TP53 (yellow), which has a high TM-score 0.96 to its native structure for the DNA binding domain (PDB entry 1tup chain B, pink). The double stranded DNA associated with 1tup is shown in the lower left cartoon. The top COFACTOR structure template (PDB entry 1t4w chain A) with a similar beta sandwich topology is shown in blue on the right.
Summary of Predicted Structure and Functions of the 66 uPE1 Proteins
For the 66 chromosome 17 uPE1 proteins, the same I-TASSER/COFACTOR pipeline is used, except that homologous templates are not excluded, because we want to obtain the best possible structure and function modeling results for these real prediction targets. Among the first ranked I-TASSER model of these uPE1 proteins, models of 12 proteins are predicted to have correct fold (estimated TM-score >0.5), while 13 are predicted to have roughly correct fold (estimated TM-score >0.4 and ≤0.5).
For prediction of GO terms for these uPE1 proteins, using Cscores >0.59, >0.55, and >0.56 established by Figure 3 as thresholds for reliable COFACTOR prediction for MF, BP, and CC, respectively, we obtained confident predictions for 13, 33, and 49 proteins for the respective GO term aspects (Figure 5). If these stringent Cscore cutoffs are slightly relaxed such that we also consider predicted GO terms with Cscore > 0.5, the number of uPE1 proteins with predicted GO terms will be increased to 30, 39, and 58 for MF, BP, and CC, respectively, as listed (shaded) by Table S3, which summarizes all predicted functions for all 66 uPE1 proteins.
Figure 5.
Number of uPE1 proteins with GO term prediction at different Cscore thresholds. The solid black vertical line marks the Cscore=0.5, while the red, green, and blue dashed vertical lines indicate Cscore cutoffs 0.59, 0.55, and 0.56 for MF, BP, and CC, respectively. Here, GO terms associated with more than 20% of proteins in the UniProt database are excluded, because these GO terms, such as “protein binding”, are too general to provide meaningful insight into their specific function.
As a concise entry to Table S3, we list the top 13 uPE1 proteins with highest Cscores for MF GO terms in Table 1.
Table 1.
A concise table for 13 uPE1 proteins with high confidence predicted functions for MF. For each of the three aspects, MF, BP and CC, the GO term with the highest confidence and the GO term with Cscore >0.5 that can provide specific biological insight are listed, with the Cscore enclosed by parentheses. The four entries discussed as case studies in the following sections are indicated with asterisks. The entries are in descending order according to MF Cscore.
| NeXtProt ID (Gene Name) | Molecular Function (MF) | Biological Process (BP) | Cellular Component (CC) | |
|---|---|---|---|---|
| 1* | NX_Q8TBR7-2 (FAM57A) | GO:0016740 (1.00) transferase activity GO:0050291 (0.99) sphingosine N-acyltransferase activity |
GO:0032502 (0.69) developmental process GO:0007420 (0.54) brain development |
GO:0005887 (1.00) integral component of plasma membrane GO:0005886 (1.00) plasma membrane |
| 2 | NX_Q12767–1 (TMEM94) | GO:0022892 (0.91) substrate-specific transporter activity GO:0046873 (0.57) metal ion transmembrane transporter activity |
GO:0065008 (0.80) regulation of biological quality GO:0030001 (0.56) metal ion transport |
GO:0005654 (1.00) nucleoplasm |
| 3 | NX_Q5BKU9-1 (OXLD1) | GO:0016491 (0.87) oxidoreductase activity GO:0004128 (0.73) cytochrome-b5 reductase activity, acting on NAD(P)H |
GO:0015701 (0.90) bicarbonate transport GO:0008652 (0.53) cellular amino acid biosynthetic process |
GO:0005739 (0.90) Mitochondrion GO:0005737 (0.66) cytoplasm |
| 4* | NX_A6NGC4-1 (TLCD2) | GO:0016740 (0.86) transferase activity GO:0050291 (0.76) sphingosine N-acyltransferase activity |
GO:0006643 (0.76) membrane lipid metabolic process GO:0006672 (0.73) ceramide metabolic process |
GO:0016021 (1.00) integral component of membrane GO:0005783 (0.75) endoplasmic reticulum |
| 5* | NX_O43934–1 (MFSD11) | GO:0005215 (0.85) transporter activity GO:0005351 (0.66) sugar:proton symporter activity |
GO:0006810 (0.82) transport GO:0008643 (0.68) carbohydrate transport |
GO:0016021 (1.00) integral component of membrane GO:0005887 (0.77) integral component of plasma membrane |
| 6 | NX_Q9P298–1 (HIGD1B) | GO:0016740 (0.79) transferase activity GO:0061630 (0.71) ubiquitin protein ligase activity |
GO:0043234 (0.88) protein complex GO:0005634 (0.71) nucleus |
|
| 7 | NX_Q2TAL5-1 (SMTNL2) | GO:0008092 (0.77) cytoskeletal protein binding | GO:0016043 (0.70) cellular component organization GO:0048856 (0.59) anatomical structure development |
GO:0005737 (0.66) Cytoplasm GO:0044430 (0.50) cytoskeletal part |
| 8 | NX_Q9BQS6-1 (HSPB9) | GO:0042802 (0.76) identical protein binding GO:0051082 (0.52) unfolded protein binding |
GO:0050896 (0.82) response to stimulus GO:0042981 (0.51) regulation of apoptotic process |
GO:0005634 (0.97) Nucleus GO:0005737 (0.96) cytoplasm |
| 9 | NX_Q96LD4–1 (TRIM47) | GO:0004842 (0.76) ubiquitin-protein transferase activity | GO:0031323 (0.54) regulation of cellular metabolic process GO:0019538 (0.54) protein metabolic process |
GO:0005737 (0.57) cytoplasm |
| 10 | NX_Q8N7B9-1 (EFCAB3) | GO:0043169 (0.74) cation binding | GO:0019538 (0.58) protein metabolic process | GO:0016020 (0.82) Membrane GO:0005737 (0.68) cytoplasm |
| 11* | NX_Q6AI12–1 (ANKRD40) | GO:0008092 (0.62) cytoskeletal protein binding GO:0030507 (0.57) spectrin binding |
GO:0060255 (0.62) regulation of macromolecule metabolic process GO:0016043 (0.60) cellular component organization |
GO:0005737 (0.77) Cytoplasm GO:0043234 (0.51) protein complex |
| 12 | NX_Q6UX52-1 (C17orf99) | GO:0004872 (0.63) receptor Activity GO:0019199 (0.50) transmembrane receptor protein kinase activity |
GO:0032502 (0.68) developmental process GO:0030030 (0.54) cell projection organization |
GO:0031224 (1.00) intrinsic component of membrane GO:0005887 (0.63) integral component of plasma membrane |
| 13 | NX_Q3MHD2-1 (LSM12) | GO:0003723 (0.59) RNA binding | GO:0090304 (0.79) nucleic acid metabolic process GO:0016070 (0.73) RNA metabolic process |
GO:0005576 (0.55) extracellular region |
It can also be observed that the number of confidently annotated proteins is smaller for MF compared to BP and CC. This is partially due to the fact that, while most of these 66 uPE1 proteins lack close sequence homologs, the majority (56 of 66) have known or inferred PPI information, which COFACTOR can take advantage of in BP and CC prediction. For example, the uPE1 protein C17orf82 (neXtProt ID: NX_Q86X59–1) does not have any strong sequence or structure template hit, but interacts with proteins known to be involved in developmental processes or cellular component organization (https://string-db.org/network/9606.ENSP00000335229). Using the homologs of these PPI partners, COFACTOR deduces that the target protein is involved in “cellular component organization” (GO:0016043, Cscore=0.55) and “developmental process” (GO:0032502, Cscore=0.52). While PPI is informative of BP and CC, it is not as useful for MF prediction, because proteins that physically interact with each other do not necessarily share the same molecular function (MF), even though they generally are involved in the same pathway (BP) at the same subcellular location (CC). This phenomenon is revealed in Figure 2 (green bars), where the prediction accuracy of PPI for MF GO terms is 39% and 79% lower than that in BP and CC prediction, respectively.
Among the uPE1 proteins with relatively confidently predicted functions (Figure 6), 7 are associated with cytoskeleton (GO:0008092 “cytoskeletal protein binding” for MF and GO:0044430 “cytoskeletal part” for CC), while another 7 are putative transmembrane transporters (GO:0022857 “transmembrane transporter activity” for MF). Other notable predicted biological functions shared by multiple uPE1 proteins include nucleic acid binding (GO:0003676 “nucleic acid binding” for MF and GO:0090304 “nucleic acid metabolic process” for BP), ubiquitin-dependent protein degradation (GO:0004842 “ubiquitin-protein transferase activity” for MF and GO:0006511 “ubiquitin-dependent protein catabolic process” for BP), and N-acylsphingosine synthesis (GO:0050291 “sphingosine N-acyltransferase activity” for MF). Here we include both GO terms predicted with the stringent Cscore cutoffs 0.59, 0.55, and 0.56 for MF, BP, and CC, respectively (Figure 6, gray), and the GO terms predicted with the relaxed Cscore cutoffs 0.50 for all three aspects (Figure 6, white). There is no major difference in the source of prediction (structure, sequence, or PPI), the distribution of prevalent GO terms (Table S3) or the Fmax that resulted from the two sets of Cscore cutoffs (Figure 3).
Figure 6.
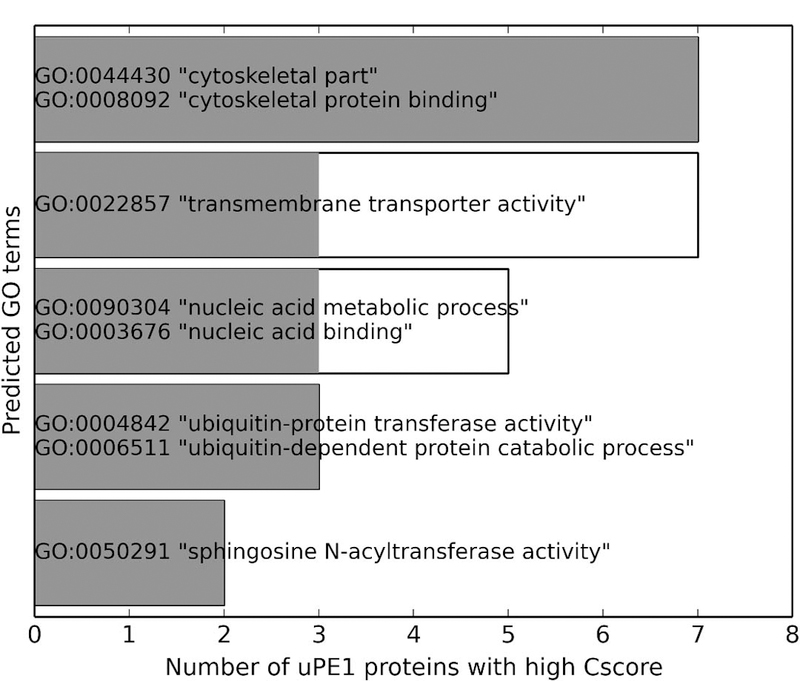
Notable GO terms predicted with high Cscore for multiple uPE1 proteins. White bars show the number of proteins predicted with Cscore > 0.5 for given GO terms, while the gray bars show the number of proteins predicted with Cscore > 0.59, 0.55, and 0.56 for MF, BP, and CC, respectively.
Case Studies of Predicted Function of uPE1 Proteins
For this section, we selected four uPE1 proteins whose specific biological functions are predicted with a high MF Cscore by COFACTOR (Table 1) plus one uPE1 protein predicted with a high CC Cscore (Table S1) for manual interpretation of their likely structure and function, as well as the origin of the function assertion by our pipeline.
MFSD11 (neXtProt ID: NX_O43934–1) is a hard function prediction target with neither experimentally solved structure nor any functionally characterized sequence homolog sharing >30% sequence identity. The I-TASSER structure model of this target shows a multi-pass transmembrane helical protein topology with high confidence: the TM-score of the model, as estimated by statistical significance of threading template hits and convergence of folding simulation,25 is as high as 0.86. The structure model superposes well to a proton:xylose symporter (PDB entry 4gby chain A, Figure 7), from which COFACTOR asserted that the MF for the target protein of interest is “sugar transmembrane transporter activity” (GO:0051119, Cscore=0.74). This function prediction is consistent with a previous study42, which suggested that MFSD11 may be a membrane protein that transports soluble molecules and is involved in energy regulation.
Figure 7.
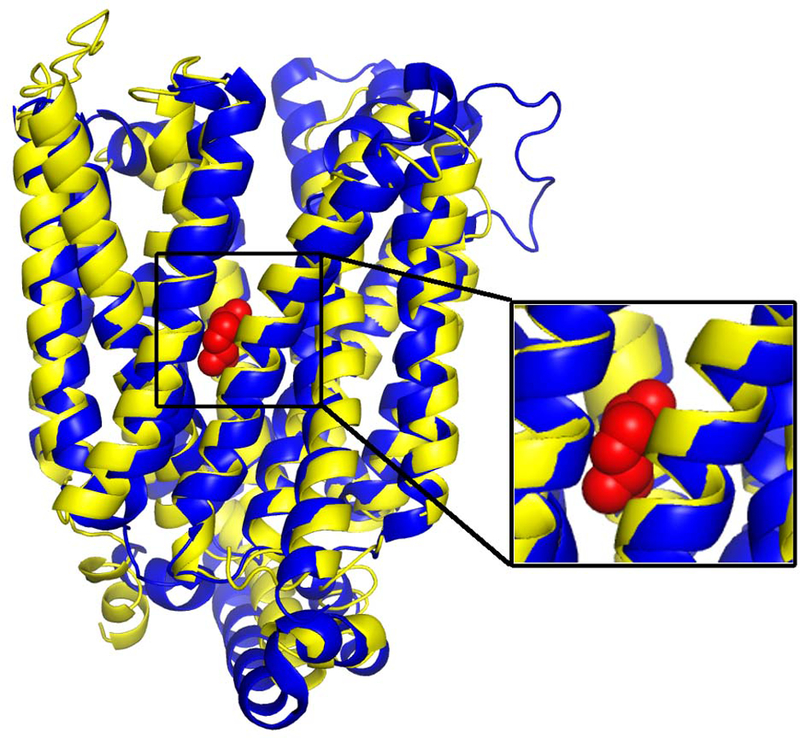
I-TASSER model of MFSD11 (yellow) superposed to the E. coli proton:xylose symporter (PDB entry 4gby chain A, blue) with TM-score=0.86. The xylose ligand from 4gbyA is shown in red spheres in the inset.
FAM57A and TLCD2 (neXtProt ID: NX_Q8TBR7–2 and NX_A6NGC4–1, respectively) are two protein coding genes located at p13.3 region on chromosome 17, separated from each other by 0.96 million base pairs. COFACTOR considers both proteins as sphingosine N-acyltransferases (GO:0050291, Cscore=0.99 for FAM57A and Cscore=0.76 for TLCD2) in terms of MF. These proteins have sequence identity of only 0.24; the lack of confident predictions for the binding sites makes it infeasible to assess the active site similarity for these proteins. Sphingosine is an important phospholipid constituent of the cell membrane, and is consistent with both proteins’ I-TASSER structure models, which adopt a fold typical of membrane-associated proteins (Figure 8). Moreover, FAM57A is homologous to FAM57B (neXtProt ID: NX_Q71RH2–1) with sequence identity 0.46. FAM57B is already annotated as sphingosine N-acyltransferases, which further confirms the function assertion.
Figure 8.
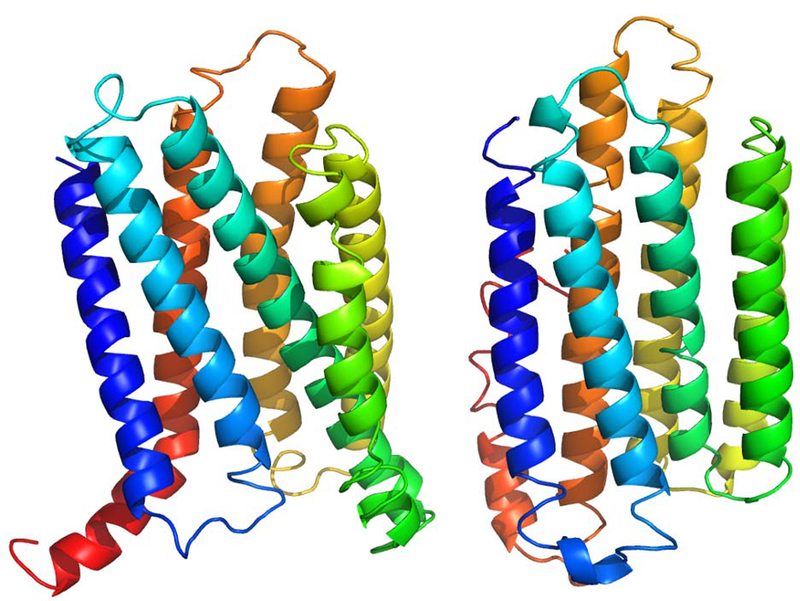
I-TASSER models of FAM57A (left) and TLCD2 (right). Both proteins are colored in spectrum with blue to red marking N- to C-termini.
ANKRD40 (neXtProt ID: NX_Q6AI12–1) is another hard function prediction target without functionally characterized close sequence homologs. I-TASSER predicts the target as an ankyrin repeat (Figure 9) with an estimated TM-score of 0.51. Based on the known role of ankyrin repeat-containing proteins in cytoskeleton anchoring, COFACTOR predicts the molecular function of ANKRD40 as “cytoskeletal protein binding” (GO:0008092, Cscore=0.62), “spectrin binding” (GO:0030507, Cscore=0.57), and “cytoskeletal adaptor activity” (GO:0008093, 0.57).
Figure 9.
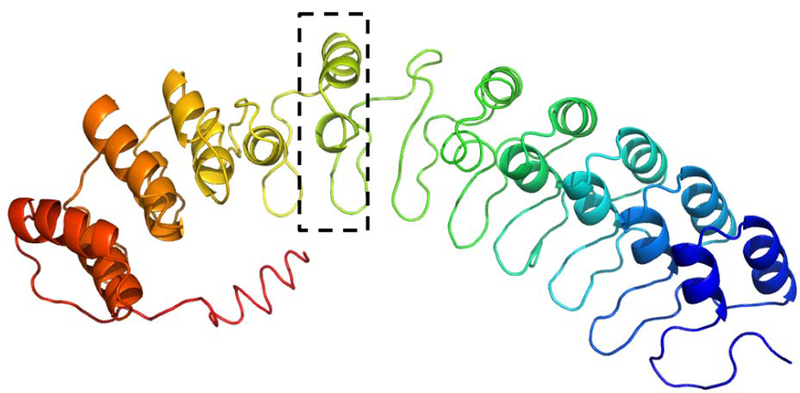
I-TASSER structure of ANKRD40 with nine consecutive ankyrin repeat units, each consisting of two helices linked by a loop. One ankyrin repeat unit is indicated in dashed rectangle.
Another interesting protein, based on CC prediction, is CCDC57 (neXtProt ID: NX_Q2TAC2–1), a large protein with 916 residues. While neither the sequence-based nor the PPI-based pipeline gives much hint to the function, the structure-based pipeline found that 17 of all 19 structure templates identified by the I-TASSER model belong to “phosphatidylinositol 3-kinase complex” (GO:0005942, Cscore=0.89) for CC (Figure 10). This is consistent with COFACTOR’s molecular function annotation “phosphatidylinositol 3-kinase activity” (GO:0035004, Cscore=0.31) and biological process annotation “inositol lipid-mediated signaling” (GO:0048017, Cscore=0.41), even though both function predictions have relatively low to moderate Cscores. Phosphatidylinositol triphosphate (PI3P) is a phospholipid found in membranes that helps to recruit a range of proteins, many of which are involved in protein trafficking; we conclude that CCDC57 has a related function.
Figure 10.
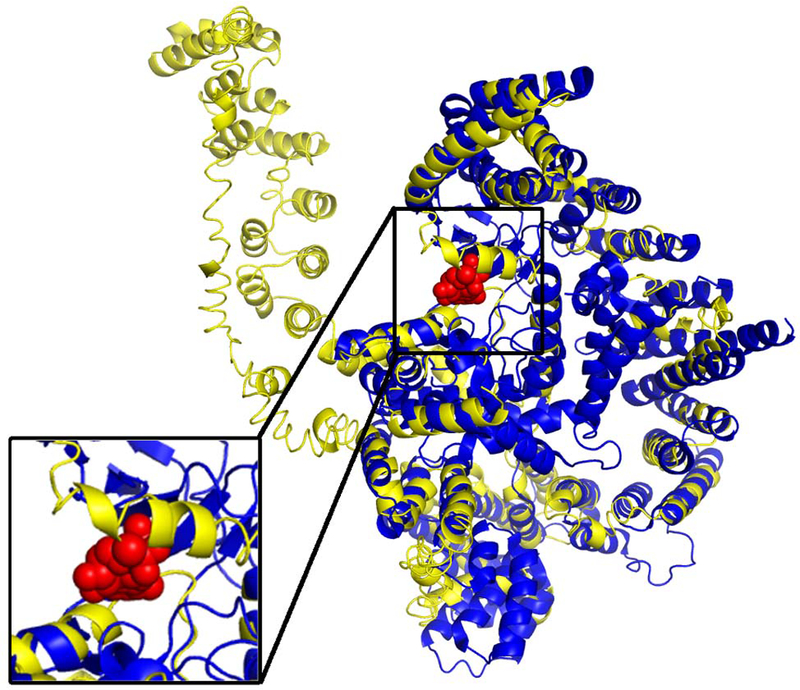
I-TASSER model of CCDC57 (yellow) superposed to PDB entry 4jsp chain A (blue), one of the many structure templates associated with phosphoinositide 3-kinase complex. The ligand bound to the 4jsp structure is phosphothiophosphoric acid-adenylate ester (red spheres), which is a small molecule analog of ATP, one of the substrates of phosphoinositide 3-kinases.
Comparing COFACTOR Prediction with Very Recent Function Annotations
The list of 66 uPE1 proteins was originally curated based on the lack of function annotations in neXtProt release 2017-08-01. Two previously unannotated proteins have new characterized functions. When we were drafting this manuscript, neXtProt release 2018-01-17 became available, with a finding that EVI2B (neXtProt ID: NX_P34910-1) regulates hematopoietic stem cell division and granulocyte differentiation.43 COFACTOR failed to predict the highly specific BP function of this protein, only suggesting it is an “integral component of plasma membrane” (GO:0005887, Cscore=1.00) for which UniProt gave the same CC term. In contrast, a recently published report characterized TRIM47 (neXtProt ID: NX_Q96LD4-1) as an E3 ubiquitin ligase;44 the corresponding function annotation has not yet been updated in neXtProt 2018-01-17. I-TASSER/COFACTOR predicted the GO MF for TRIM47 as “ubiquitin-protein transferase activity” (GO:0004842, Cscore=0.76).
Function Predictions that are Inconsistent with Database Annotations
For the uPE1 proteins investigated in this study, there are two cases where the I-TASSER/COFACTOR prediction is conflicting with existing annotations especially for subcellular localization (GO CC terms).
The first protein, TMEM94 (neXtProt ID: NX_Q12767-1), is annotated as “integral component of membrane” (GO:0016021) for CC in both neXtProt and UniProt with 10 predicted transmembrane helices based on automated annotation with IEA (Inferred from Electronic Annotation) evidence code by UniProt (https://www.uniprot.org/keywords/KW-0812) without experimental validation. Consistent with that database annotation, COFACTOR assigns “substrate-specific transporter activity” (GO:0022892, Cscore=0.91) for MF and “metal ion transport” (GO:0030001, Cscore=0.56) for BP, both of which are associated with transmembrane transport.
We present TMEM94 as an example for inconsistency of CC prediction and neXtProt annotation. The CC result of COFACTOR for this protein is “nucleoplasm” (GO:0005654, Cscore=1.00). This COFACTOR annotation, which has no counterpart in neXtProt, is generated by our sequence-based pipeline, whose function library contains the UniProt GO term of TMEM94 from year 2017 (line 382 of https://www.uniprot.org/uniprot/Q12767.txt?version=119). This UniProt annotation, labeled by UniProt with evidence “IDA:HPA” (inferred from direct assay, as reported by Human Protein Atlas database), originated from immunofluorescence experiments conducted in three human cell lines reported in the Human Protein Atlas (https://www.proteinatlas.org/ENSG00000177728-TMEM94/cell). Interestingly, while UniProt up to version 2017_02 contained the “nucleoplasm” annotation, this annotation is recently dropped by UniProt (https://www.uniprot.org/uniprot/Q12767?version=119&version=120&diff=true) even though the Human Protein Atlas experiments have not been invalidated. Since we do not exclude sequence homologs when predicting uPE1 functions, the COFACTOR sequence-based pipeline ends up hitting the TMEM94 protein itself as the “template” for its CC prediction. These differences in database annotations require further experimental efforts to determine the true or at least primary cellular component/localization of this protein.
Another example is C17orf99 (neXtProt ID: NX_Q6UX52-1), a putative human cytokine. The mouse ortholog of C17orf99 was recently established as a new 27 kDa cytokine called Interleukin 40 (IL-40), which is secreted by activated B cells.45 Since the UniProt annotation was updated during the peer review process of this manuscript, neither the COFACTOR function library nor the current neXtProt database (version 2018-01-17) includes this annotation. In our PSI-BLAST search for C17orf99 against human proteome (https://www.uniprot.org/proteomes/UP000005640, protein list last modified May 26, 2018), none of the top hits is cytokine, whereas the most significant hits within the human proteome are FCRL2 (neXtProt ID: NX_Q96LA5) and FCRL5 (neXtProt ID: NX_Q96RD9); both are transmembrane receptors involved in B cell development, which resulted in our pipeline’s predicted CC term of C17orf99 is “intrinsic component of membrane” (GO:0031224, Cscore=1.00). Nevertheless, the UniProt CC designation as “extracellular region” (GO:0005576) due to the predicted N-terminal signal peptide (https://www.nextprot.org/entry/NX_Q6UX52/sequence) and reported cytokine function may be preferable.
These contradictions in function annotations underscore the difficulty in CC prediction, which is a common challenge among many function prediction programs. In fact, it was observed in the CAFA2 experiment that almost none of the state-of-the-art programs could outperform the “Naïve” baseline in terms of CC prediction.38 In the future, we will address the challenges in CC prediction by incorporation of amino acid composition and local sequence signatures such as predicted transmembrane regions and signal peptides into the COFACTOR function annotation algorithm.
CONCLUSIONS
As a pilot study on prediction of functions for uncharacterized human proteins, we have carried out a comprehensive survey of PE1 proteins on chromosome 17 using the composite I-TASSER and COFACTOR structure and function annotation pipeline, which has been extensively tested in the community-wide CASP and CAFA experiments.6–7, 10 The prediction accuracy of the pipeline was examined on 100 randomly-selected well-characterized proteins from this chromosome, and achieved a high F-measures of 0.69, 0.57, and 0.67 for MF, BP, and CC aspects of GO term predictions, respectively. The structure-based function prediction component of this pipeline is the main contributor of prediction accuracy for the non-homologous protein targets. Applying the pipeline on all of the 66 poorly- or non-characterized uPE1 proteins coded by genes on chromosome 17, we are able to infer the specific biological function with high confidence for 13, 33, and 49 uPE1 proteins for MF, BP, and CC aspects, respectively. The majority of these function inferences could not be achieved using traditional sequence-based function annotation approaches. We give extensive details for the 13 highest-rated predictions for Molecular Functions, plus structural findings for 5 case studies.
As a proof-of-concept, we started with the set of 66 uPE1 proteins on human chromosome 17 only. The pipeline can be readily extended to all 1260 uPE1 proteins from the entire human proteome, as well as 677 additional unannotated human proteins in neXtProt categories PE2, PE3, and PE4 (https://www.nextprot.org/proteins/search?mode=advanced&queryId=NXQ_00022). The work along this line is in progress.
We hope our modeling results will stimulate the interest of molecular and cell biologists and assist them to design appropriate experiments that could validate the computational predictions and, more importantly, elucidate the structure and biological function of these proteins in human tissues and cells.
Supplementary Material
ACKNOWLEDGMENT
We are grateful to Dr. Lydie Lane of neXtProt for help in understanding the Gene Ontology terms that do not qualify for sufficiently specific function annotations in neXtProt and for critical review of the manuscript. We thank Dr. Peter L. Freddolino for insightful discussions. We also thank the reviewers for stimulating comments. This work used the Extreme Science and Engineering Discovery Environment (XSEDE)46, which is supported by National Science Foundation (ACI-1548562).
Funding Sources
This work was supported in part by the National Institutes of Health (GM083107 and GM116960 to Y.Z.; P30ES017885 and U24CA210967 to G.S.O.) and the National Science Foundation (DBI1564756 to Y.Z.).
ABBREVIATIONS
- PE1
Protein Evidence at protein level.
- uPE1
unknown function with PE1 evidence.
- GO
Gene Ontology.
- MF
Molecular Function.
- BP
Biological Process.
- CC
Cellular Component.
- CAFA
Critical Assessment of Function Annotation.
- CASP
Critical Assessment of protein Structure Prediction.
- PDB
Protein Data Bank.
Footnotes
Notes
The authors declare no competing financial interest. G.S.O., a co-author of this paper, is a guest editor of the Journal of Proteome Research. This does not alter the authors’ compliance to Journal of Proteome Research editorial policies or peer-review processes.
REFERENCES
- 1.Gaudet P; Michel PA; Zahn-Zabal M; Britan A; Cusin I; Domagalski M; Duek PD; Gateau A; Gleizes A; Hinard V; de Laval VR; Lin JJ; Nikitin F; Schaeffer M; Teixeira D; Lane L; Bairoch A, The neXtProt knowledgebase on human proteins: 2017 update. Nucleic Acids Res 2017, 45 (D1), D177–D182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Uhlen M; Fagerberg L; Hallstrom BM; Lindskog C; Oksvold P; Mardinoglu A; Sivertsson A; Kampf C; Sjostedt E; Asplund A; Olsson I; Edlund K; Lundberg E; Navani S; Szigyarto CA; Odeberg J; Djureinovic D; Takanen JO; Hober S; Alm T; Edqvist PH; Berling H; Tegel H; Mulder J; Rockberg J; Nilsson P; Schwenk JM; Hamsten M; von Feilitzen K; Forsberg M; Persson L; Johansson F; Zwahlen M; von Heijne G; Nielsen J; Ponten F, Tissue-based map of the human proteome. Science 2015, 347 (6220). [DOI] [PubMed] [Google Scholar]
- 3.Carithers LJ; Ardlie K; Barcus M; Branton PA; Britton A; Buia SA; Compton CC; DeLuca DS; Peter-Demchok J; Gelfand ET; Guan P; Korzeniewski GE; Lockhart NC; Rabiner CA; Rao AK; Robinson KL; Roche NV; Sawyer SJ; Segre AV; Shive CE; Smith AM; Sobin LH; Undale AH; Valentino KM; Vaught J; Young TR; Moore HM; Consortium G, A Novel Approach to High-Quality Postmortem Tissue Procurement: The GTEx Project. Biopreserv Biobank 2015, 13 (5), 311–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang JY; Yan RX; Roy A; Xu D; Poisson J; Zhang Y, The I-TASSER Suite: protein structure and function prediction. Nature Methods 2015, 12 (1), 7–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhang CX; Freddolino PL; Zhang Y, COFACTOR: improved protein function prediction by combining structure, sequence and protein-protein interaction information. Nucleic Acids Res 2017, 45 (W1), W291–W299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Zhang CX; Mortuza SM; He BJ; Wang YT; Zhang Y, Template-based and free modeling of I-TASSER and QUARK pipelines using predicted contact maps in CASP12. Proteins 2018, 86, 136–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schmidt T; Haas J; Cassarino TG; Schwede T, Assessment of ligand-binding residue predictions in CASP9. Proteins 2011, 79, 126–136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Roy A; Yang JY; Zhang Y, COFACTOR: an accurate comparative algorithm for structure-based protein function annotation. Nucleic Acids Res 2012, 40 (W1), W471–W477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dong QW; Menon R; Omenn GS; Zhang Y, Structural Bioinformatics Inspection of neXtProt PE5 Proteins in the Human Proteome. J Proteome Res 2015, 14 (9), 3750–3761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zhang C; Zheng W; Freddolino PL; Zhang Y, MetaGO: Predicting Gene Ontology of non-homologous proteins through low-resolution protein structure prediction and protein–protein network mapping. Journal of molecular biology 2018, 430 (15), 2256–2265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang C; Rahimpour M; Freddolino PL; Zhang Y, Proteome-wide Structure-Based Function Prediction Reveals Roles of Proteins Responsible for E. coli Fitness. In US HUPO 14th Annual Conference, Minneapolis, MN, USA, 2018. [Google Scholar]
- 12.Menon R; Panwar B; Eksi R; Kleer C; Guan YF; Omenn GS, Computational Inferences of the Functions of Alternative/Noncanonical Splice Isoforms Specific to HER2+/ER-/PR-Breast Cancers, a Chromosome 17 C-HPP Study. J Proteome Res 2015, 14 (9), 3519–3529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu ST; Zhang Y, LOMETS: A local meta-threading-server for protein structure prediction. Nucleic Acids Res 2007, 35 (10), 3375–3382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wu ST; Zhang Y, MUSTER: Improving protein sequence profile-profile alignments by using multiple sources of structure information. Proteins 2008, 72 (2), 547–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Soding J, Protein homology detection by HMM-HMM comparison. Bioinformatics 2005, 21 (7), 951–960. [DOI] [PubMed] [Google Scholar]
- 16.Xu Y; Xu D, Protein threading using PROSPECT: Design and evaluation. Proteins-Structure Function and Genetics 2000, 40 (3), 343–354. [PubMed] [Google Scholar]
- 17.Yan RX; Xu D; Yang JY; Walker S; Zhang Y, A comparative assessment and analysis of 20 representative sequence alignment methods for protein structure prediction. Scientific Reports 2013, 3, 2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Jaroszewski L; Rychlewski L; Li ZW; Li WZ; Godzik A, FFAS03: a server for profile-profile sequence alignments. Nucleic Acids Res 2005, 33, W284–W288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Madera M, Profile Comparer: a program for scoring and aligning profile hidden Markov models. Bioinformatics 2008, 24 (22), 2630–2631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lobley A; Sadowski MI; Jones DT, pGenTHREADER and pDomTHREADER: new methods for improved protein fold recognition and superfamily discrimination. Bioinformatics 2009, 25 (14), 1761–1767. [DOI] [PubMed] [Google Scholar]
- 21.Xu D; Jaroszewski L; Li ZW; Godzik A, FFAS-3D: improving fold recognition by including optimized structural features and template re-ranking. Bioinformatics 2014, 30 (5), 660–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou HY; Zhou YQ, Single-body residue-level knowledge-based energy score combined with sequence-profile and secondary structure information for fold recognition. Proteins 2004, 55 (4), 1005–1013. [DOI] [PubMed] [Google Scholar]
- 23.Zhang Y; Skolnick J, SPICKER: A clustering approach to identify near-native protein folds. Journal of Computational Chemistry 2004, 25 (6), 865–871. [DOI] [PubMed] [Google Scholar]
- 24.Zhang J; Liang Y; Zhang Y, Atomic-Level Protein Structure Refinement Using Fragment-Guided Molecular Dynamics Conformation Sampling. Structure 2011, 19 (12), 1784–1795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang Y, I-TASSER server for protein 3D structure prediction. Bmc Bioinformatics 2008, 9 (1), 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhang Y; Skolnick J, TM-align: a protein structure alignment algorithm based on the TM-score. Nucleic Acids Res 2005, 33 (7), 2302–2309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yang JY; Roy A; Zhang Y, BioLiP: a semi-manually curated database for biologically relevant ligand-protein interactions. Nucleic Acids Res 2013, 41 (D1), D1096–D1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nagano N; Orengo CA; Thornton JM, One fold with many functions: The evolutionary relationships between TIM barrel families based on their sequences, structures and functions. Journal of Molecular Biology 2002, 321 (5), 741–765. [DOI] [PubMed] [Google Scholar]
- 29.Skolnick J; Gao M, Interplay of physics and evolution in the likely origin of protein biochemical function. P Natl Acad Sci USA 2013, 110 (23), 9344–9349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Altschul SF; Madden TL; Schaffer AA; Zhang JH; Zhang Z; Miller W; Lipman DJ, Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res 1997, 25 (17), 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bateman A; Martin MJ; O’Donovan C; Magrane M; Alpi E; Antunes R; Bely B; Bingley M; Bonilla C; Britto R; Bursteinas B; Bye-A-Jee H; Cowley A; Da Silva A; De Giorgi M; Dogan T; Fazzini F; Castro LG; Figueira L; Garmiri P; Georghiou G; Gonzalez D; Hatton-Ellis E; Li WZ; Liu WD; Lopez R; Luo J; Lussi Y; MacDougall A; Nightingale A; Palka B; Pichler K; Poggioli D; Pundir S; Pureza L; Qi GY; Rosanoff S; Saidi R; Sawford T; Shypitsyna A; Speretta E; Turner E; Tyagi N; Volynkin V; Wardell T; Warner K; Watkins X; Zaru R; Zellner H; Xenarios I; Bougueleret L; Bridge A; Poux S; Redaschi N; Aimo L; Argoud-Puy G; Auchincloss A; Axelsen K; Bansal P; Baratin D; Blatter MC; Boeckmann B; Bolleman J; Boutet E; Breuza L; Casal-Casas C; de Castro E; Coudert E; Cuche B; Doche M; Dornevil D; Duvaud S; Estreicher A; Famiglietti L; Feuermann M; Gasteiger E; Gehant S; Gerritsen V; Gos A; Gruaz-Gumowski N; Hinz U; Hulo C; Jungo F; Keller G; Lara V; Lemercier P; Lieberherr D; Lombardot T; Martin X; Masson P; Morgat A; Neto T; Nouspikel N; Paesano S; Pedruzzi I; Pilbout S; Pozzato M; Pruess M; Rivoire C; Roechert B; Schneider M; Sigrist C; Sonesson K; Staehli S; Stutz A; Sundaram S; Tognolli M; Verbregue L; Veuthey AL; Wu CH; Arighi CN; Arminski L; Chen CM; Chen YX; Garavelli JS; Huang HZ; Laiho K; McGarvey P; Natale DA; Ross K; Vinayaka CR; Wang QH; Wang YQ; Yeh LS; Zhang J; Consortium U, UniProt: the universal protein knowledgebase. Nucleic Acids Res 2017, 45 (D1), D158–D169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Szklarczyk D; Franceschini A; Wyder S; Forslund K; Heller D; Huerta-Cepas J; Simonovic M; Roth A; Santos A; Tsafou KP; Kuhn M; Bork P; Jensen LJ; von Mering C, STRING v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res 2015, 43 (D1), D447–D452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang Y; Skolnick J, Scoring function for automated assessment of protein structure template quality. Proteins 2004, 57 (4), 702–710. [DOI] [PubMed] [Google Scholar]
- 34.Xu JR; Zhang Y, How significant is a protein structure similarity with TM-score=0.5? Bioinformatics 2010, 26 (7), 889–895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Belfort GM; Kandror KV, Cellugyrin and synaptogyrin facilitate targeting of synaptophysin to a ubiquitous synaptic vesicle-sized compartment in PC12 cells. Journal of Biological Chemistry 2003, 278 (48), 47971–47978. [DOI] [PubMed] [Google Scholar]
- 36.Belfort GM; Bakirtzi K; Kandror KV, Cellugyrin induces biogenesis of synaptic-like microvesicles in PC12 cells. Journal of Biological Chemistry 2005, 280 (8), 7262–7272. [DOI] [PubMed] [Google Scholar]
- 37.Radivojac P; Clark WT; Oron TR; Schnoes AM; Wittkop T; Sokolov A; Graim K; Funk C; Verspoor K; Ben-Hur A; Pandey G; Yunes JM; Talwalkar AS; Repo S; Souza ML; Piovesan D; Casadio R; Wang Z; Cheng JL; Fang H; Goughl J; Koskinen P; Toronen P; Nokso-Koivisto J; Holm L; Cozzetto D; Buchan DWA; Bryson K; Jones DT; Limaye B; Inamdar H; Datta A; Manjari SK; Joshi R; Chitale M; Kihara D; Lisewski AM; Erdin S; Venner E; Lichtarge O; Rentzsch R; Yang HX; Romero AE; Bhat P; Paccanaro A; Hamp T; Kassner R; Seemayer S; Vicedo E; Schaefer C; Achten D; Auer F; Boehm A; Braun T; Hecht M; Heron M; Honigschmid P; Hopf TA; Kaufmann S; Kiening M; Krompass D; Landerer C; Mahlich Y; Roos M; Bjorne J; Salakoski T; Wong A; Shatkay H; Gatzmann F; Sommer I; Wass MN; Sternberg MJE; Skunca N; Supek F; Bosnjak M; Panov P; Dzeroski S; Smuc T; Kourmpetis YAI; van Dijk ADJ; ter Braak CJF; Zhou YP; Gong QT; Dong XR; Tian WD; Falda M; Fontana P; Lavezzo E; Di Camillo B; Toppo S; Lan L; Djuric N; Guo YH; Vucetic S; Bairoch A; Linial M; Babbitt PC; Brenner SE; Orengo C; Rost B; Mooney SD; Friedberg I, A large-scale evaluation of computational protein function prediction. Nature Methods 2013, 10 (3), 221–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jiang YX; Oron TR; Clark WT; Bankapur AR; D’Andrea D; Lepore R; Funk CS; Kahanda I; Verspoor KM; Ben-Hur A; Koo DCE; Penfold-Brown D; Shasha D; Youngs N; Bonneau R; Lin A; Sahraeian SME; Martelli PL; Profiti G; Casadio R; Cao RZ; Zhong Z; Cheng JL; Altenhoff A; Skunca N; Dessimoz C; Dogan T; Hakala K; Kaewphan S; Mehryary F; Salakoski T; Ginter F; Fang H; Smithers B; Oates M; Gough J; Toronen P; Koskinen P; Holm L; Chen CT; Hsu WL; Bryson K; Cozzetto D; Minneci F; Jones DT; Chapman S; Dukka BKC; Khan IK; Kihara D; Ofer D; Rappoport N; Stern A; Cibrian-Uhalte E; Denny P; Foulger RE; Hieta R; Legge D; Lovering RC; Magrane M; Melidoni AN; Mutowo-Meullenet P; Pichler K; Shypitsyna A; Li B; Zakeri P; ElShal S; Tranchevent LC; Das S; Dawson NL; Lee D; Lees JG; Sillitoe I; Bhat P; Nepusz T; Romero AE; Sasidharan R; Yang HX; Paccanaro A; Gillis J; Sedeno-Cortes AE; Pavlidis P; Feng S; Cejuela JM; Goldberg T; Hamp T; Richter L; Salamov A; Gabaldon T; Marcet-Houben M; Supek F; Gong QT; Ning W; Zhou YP; Tian WD; Falda M; Fontana P; Lavezzo E; Toppo S; Ferrari C; Giollo M; Piovesan D; Tosatto SCE; del Pozo A; Fernandez JM; Maietta P; Valencia A; Tress ML; Benso A; Di Carlo S; Politano G; Savino A; Rehman HU; Re M; Mesiti M; Valentini G; Bargsten JW; van Dijk ADJ; Gemovic B; Glisic S; Perovic V; Veljkovic V; Veljkovic N; Almeida-e-Silva DC; Vencio RZN; Sharan M; Vogel J; Kansakar L; Zhang S; Vucetic S; Wang Z; Sternberg MJE; Wass MN; Huntley RP; Martin MJ; O’Donovan C; Robinson PN; Moreau Y; Tramontano A; Babbitt PC; Brenner SE; Linial M; Orengo CA; Rost B; Greene CS; Mooney SD; Friedberg I; Radivojac P, An expanded evaluation of protein function prediction methods shows an improvement in accuracy. Genome Biol 2016, 17 (1), 184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gong QT; Ning W; Tian WD, GoFDR: A sequence alignment based method for predicting protein functions. Methods 2016, 93, 3–14. [DOI] [PubMed] [Google Scholar]
- 40.Martin DMA; Berriman M; Barton GJ, GOtcha: a new method for prediction of protein function assessed by the annotation of seven genomes. Bmc Bioinformatics 2004, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Strachan T; Read AP, An overview of mutation, polymorphism, and DNA repair. Human molecular genetics 1999, 2, 316. [Google Scholar]
- 42.Perland E; Lekholm E; Eriksson MM; Bagchi S; Arapi V; Fredriksson R, The Putative SLC Transporters Mfsd5 and Mfsd11 Are Abundantly Expressed in the Mouse Brain and Have a Potential Role in Energy Homeostasis. Plos One 2016, 11 (6), e0156912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Zjablovskaja P; Kardosova M; Danek P; Angelisova P; Benoukraf T; Wurm AA; Kalina T; Sian S; Balastik M; Delwel R; Brdicka T; Tenen DG; Behre G; Fiore F; Malissen B; Horejsi V; Alberich-Jorda M, EVI2B is a C/EBP alpha target gene required for granulocytic differentiation and functionality of hematopoietic progenitors. Cell Death Differ 2017, 24 (4), 705–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ji YX; Huang Z; Yang X; Wang XZ; Zhao LP; Wang PX; Zhang XJ; Alves-Bezerra M; Cai L; Zhang P; Lu YX; Bai L; Gao MM; Zhao H; Tian S; Wang Y; Huang ZX; Zhu XY; Zhang Y; Gong J; She ZG; Li F; Cohen DE; Li HL, The deubiquitinating enzyme cylindromatosis mitigates nonalcoholic steatohepatitis. Nat Med 2018, 24 (2), 213.-+. [DOI] [PubMed] [Google Scholar]
- 45.Catalan-Dibene J; Vazquez MI; Luu VP; Nuccio SP; Karimzadeh A; Kastenschmidt JM; Villalta SA; Ushach I; Pone EJ; Casali P; Raffatellu M; Burkhardt AM; Hernandez-Ruiz M; Heller G; Hevezi PA; Zlotnik A, Identification of IL-40, a Novel B Cell-Associated Cytokine. J Immunol 2017, 199 (9), 3326–3335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Towns J; Cockerill T; Dahan M; Foster I; Gaither K; Grimshaw A; Hazlewood V; Lathrop S; Lifka D; Peterson GD; Roskies R; Scott JR; Wilkins-Diehr N, XSEDE: Accelerating Scientific Discovery. Computing in Science & Engineering 2014, 16 (5), 62–74. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.