

Abstract
Mendelian susceptibility to mycobacterial disease (MSMD) is caused by inborn errors of IFN-γ immunity. Since 1996, disease-causing mutations have been found in 11 genes, which, through allelic heterogeneity, underlie 21 different genetic disorders. We briefly review here progress in the study of molecular, cellular and clinical aspects of MSMD since the last comprehensive review published in 2014. Highlights include the discoveries of (i) a new genetic etiology, autosomal recessive SPPL2a deficiency, (ii) TYK2-deficient patients with a clinical phenotype of MSMD, (iii) an allelic form of partial recessive IFN-γR2 deficiency, and (iv) two forms of syndromic MSMD: RORγ/RORγT and JAK1 deficiencies. These recent findings illustrate how genetic and immunological studies of MSMD can shed a unique light onto the mechanisms of protective immunity to mycobacteria in humans.
Keywords: Mycobacterium, IFN-γ, primary immunodeficiency, next-generation sequencing
Graphical Abstract
BLURB FOR ETOC:
Mendelian susceptibility to mycobacterial disease (MSMD) is caused by inborn errors of IFN-? immunity. Since 1996, disease-causing mutations have been found in 11 genes, which, through allelic heterogeneity, underlie 21 different genetic disorders. We briefly review here progress in the study of molecular, cellular and clinical aspects of MSMD since the last comprehensive review published in 2014.
Introduction
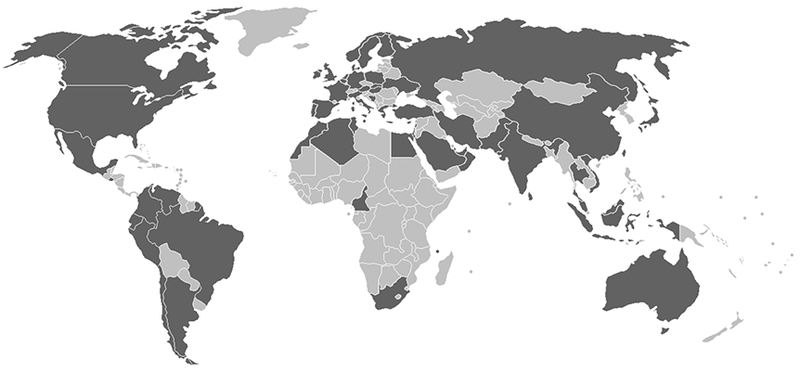
Mendelian susceptibility to mycobacterial disease (MSMD) is a rare inherited condition defined by selective susceptibility to weakly virulent mycobacteria, including Bacille Calmette-Guérin (BCG) vaccine substrains and various environmental mycobacteria (EM), in otherwise healthy patients without overt immunological abnormalities.1 Patients with MSMD may also suffer from bona fide tuberculosis, caused by Mycobacterium tuberculosis.2 A sizeable proportion of patients with MSMD also display invasive infections due to other intra-macrophagic microorganisms, such as Salmonella, or mucocutaneous infections caused by Candida species.1,3,4 Other infectious diseases have also been reported, albeit more rarely.1,4–7 Acquired and inherited immunodeficiencies conferring a predisposition to mycobacterial diseases in the context of other infections must first be excluded, before a diagnosis of MSMD can be reached.1,2,8 The most severe forms of MSMD lead to early-onset, disseminated, persistent, life-threatening mycobacterial disease, whereas the least severe forms can have a late onset, be relatively circumscribed, spontaneously improve with age, or even remain clinically silent due to incomplete penetrance.1 Clinical manifestations are, therefore, highly variable. Macrophage activation syndrome9–11 or vasculitis1,6,12–16 may occur in rare cases, probably as a consequence of uncontrolled infection. Since the discovery of its first genetic etiology in 1996, MSMD has been reported and a causal genetic lesion described in 501 individuals from 356 kindreds originating from 57 countries on five continents (Figure 1a). Over this period, the genetic dissection of MSMD in these patients has revealed this condition to be caused by inborn errors of IFN-γ immunity.1,3–7, 9–40,9,40–53 These findings confirm that IFN-γ, first described in 1965 as an antiviral IFN,54 is actually the macrophage-activating factor (MAF), as shown in 1983.55 Mutations of 11 different genes (IL12B, IL12RB1, ISG15, TYK2, IRF8, SPPL2A, CYBB, IFNGR1, IFNGR2, STAT1, NEMO) have been shown to cause MSMD (Table 1).1,17,39
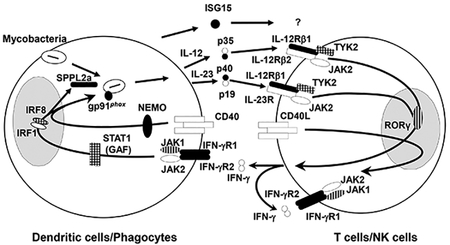
Figure 1 – Genetic spectrum of MSMD (a) Geographic distribution of patients with MSMD. (b) Cells involved in the production of and response to IFN-γ.
Proteins for which a mutation of the corresponding gene has been recognized to cause solely MSMD (IFN-γR1, IFN-γR2, SPPL2a, NEMO, gp91phox, IL-12p40, IL-12Rβ1, ISG15) are depicted in black, those responsible for syndromic MSMD (JAK1, RORγ) are depicted with vertical lines, those that can cause either MSMD or syndromic MSMD (IRF8, STAT1, TYK2) are depicted with crossed lines.
Table 1 –
Overview of diseases underlying MSMD
| Gene | Inheritance | Defect | Protein |
|---|---|---|---|
| IL12RB1 | AR | C | E− |
| AR | C | E+ | |
| IL12B | AR | C | E− |
| ISG15 | AR | C | E− |
| SPPL2A | AR | C | E− or E+ |
| IRF8 | AD | P | E+ |
| TYK2 | AR | C | E− |
| IFNGR1 | AR | C | E+ |
| AR | C | E− | |
| AD | P | E+++ | |
| AR | P | E+ | |
| IFNGR2 | AR | C | E+ |
| AR | C | E− | |
| AR | P | E+ of mutant protein | |
| AR | P | E+ of WT protein | |
| AD | P | E+ | |
| STAT1 | AD | P | E+P− |
| AD | P | E+B− | |
| AD | P | E+P−B− | |
| NEMO (IKBKG) | XR | P | E+ |
| CYBB | XR | P | E+ |
MSMD genetic etiologies may display autosomal recessive (AR), autosomal dominant (AD), or X-linked recessive (XR) inheritance. Defects may be complete (C) or partial (P). Expression (E) of the mutant protein may be abolished (E−), decreased or normal (E+), or increased (E+++). The mutant protein may be phosphorylated normally, unable to undergo phosphorylation (P−) or unable to bind DNA (B−).
The products of all these genes are involved in IFN-γ production (IL-12p40, IL-12Rβ1, TYK2, SPPL2a, ISG15), the response to IFN-γ (IFN-γR1, IFN-γR2, STAT1, gp91phox), or both (IRF8 and NEMO) (Figure 1b).1,17,39 MSMD can be inherited in an autosomal recessive (AR) (IL12RB1, IL12B, TYK2, IFNGR1, IFNGR2, ISG15, SPPL2A), autosomal dominant (AD) (IFNGR1, IFNGR2, STAT1, IRF8), or X-linked recessive (XR) manner (CYBB, NEMO).1,17,39 Allelic heterogeneity at the 11 loci underlies 21 different genetic forms of MSMD1,17,39, defined on the basis of (i) the functional deficiency being partial or complete, (ii) the protein being produced or not, and (iii) the mechanism underlying the dysfunction of expressed proteins (Table 1). The causal genetic lesions include single nucleotide variations, small deletions, duplications, insertions, or indels, and copy number variations (CNV; large deletions, insertions, or duplications).6, 42 Since the last comprehensive review on MSMD in 2014,1 three new genetic disorders have been reported, caused by mutations of TYK217 and SPPL2A,39 (two novel genetic etiologies) and IFNGR256 (a novel allelic form). Moreover, new mutations associated with the other 18 disorders have also been reported.5–7,10,12,18,22,25,30,32,34,36,42,43,45–49,57 We also discuss here two recently reported syndromic forms of MSMD: AR RORγ/RORγT and JAK1 deficiencies.58,59 Like three previously reported etiologies of syndromic MSMD, AR STAT1 and IRF8 deficiencies,1 and AD GATA2 deficiency,60 RORγ/RORγT and JAK1 deficiencies underlie mycobacterial diseases in the context of other infections.58,59 Unlike TYK2 deficiency, which also typically underlies mycobacterial disease and other infections,17 these five genetic etiologies of syndromic MSMD have not been diagnosed in patients with a pure form of MSMD limited to mycobacterial disease.
A new genetic etiology: AR SPPL2a deficiency
A new genetic etiology of MSMD has recently been described in three patients from two kindreds originating from Morocco and Turkey presenting BCG disease a few months after vaccination. Whole exome-sequencing (WES) and whole-genome linkage (WGL) analyses identified two different homozygous splice site mutations in SPPL2A, c.733+1G>A or c.1328–1G>A, in patients from these two kindreds.39 These splice site mutations disrupt the mRNA, creating aberrant transcripts without any leakiness, and, in overexpression systems, they result in a lack of protein production, or the production of a truncated protein. SPPL2A encodes signal peptide peptidase-like 2 A (SPPL2a), a protease with multiple substrates, including, in particular, the amino-terminal fragment of the HLA invariant chain (CD74)39, which is expressed by HLA-class II+ antigen-presenting cells. SPPL2a deficiency results in a deficit of conventional type 2 dendritic cells (cDC2), probably through accumulation of the toxic uncleaved CD74 amino-terminal fragment in these cells.39 This has been demonstrated for Sppl2a−/− mice, the equivalent DC phenotype of which is rescued by the genetic ablation of CD74.61 However, human SPPL2a deficiency does not significantly affect B-cell immunity,39 by contrast to what has been observed in mice with the corresponding defect.61–63 The immunological phenotype of SPPL2a-deficient patients is reminiscent of that of patients with AD IRF8 deficiency, who display a somewhat broader depletion of cDC.1 A binding site for IRF8 has been identified in the Sppl2a promoter in mouse macrophages, suggesting that the cDC2 deficit in AD-IRF8 deficiency may reflect an impairment of SPPL2A induction.39 Both AD IRF8 and AR SPPL2a deficiencies are also associated with a defect of IFN-γ production by mycobacterium-specific Th1* cells, a subset of CD4+ T cells secreting both IFN-γ and IL-17A/F.39 This suggests that cDC2 may be essential for the priming of Th1* cells through the presentation of mycobacterial antigen.39 SPPL2a deficiency thus causes MSMD through a quantitative defect of IL-12- and IL-23-producing cDC2s and through the impairment of IFN-γ production by mycobacterium-specific memory Th1* cells (Figure 1b and Table 1).39
Complete AR TYK2 deficiency and MSMD
Human TYK2 is a Janus kinase (JAK) involved in the pathways of response to IL-10, IL-12, IL-23 and IFN-α/β. The first patient with inherited complete AR TYK2 deficiency was reported in Japan in 2006.2,17 He suffered from typical signs of hyper-immunoglobulin E (IgE) syndrome (HIES): atopic dermatitis, high IgE levels, and recurrent cutaneous staphylococcal infections.2,17 He also had nontyphi Salmonella infections, lymphadenitis after BCG vaccination and a history of viral infections. In 2015, seven other patients from five unrelated families from Argentina, Iran, Morocco, and Turkey were reported, with homozygous nonsense or frameshift TYK2 mutations responsible for complete AR TYK2 deficiency.17 These patients displayed intracellular bacterial and/or viral infections, and none had the classic features of HIES.17 These seven patients had all been vaccinated with BCG. Four suffered from adverse reactions to BCG (either localized or regional (BCG-itis) or disseminated (BCG-osis)), another from abdominal tuberculosis, another from miliary tuberculosis, and only one had no history of mycobacterial disease.17 It is probable that their susceptibility to mycobacterial disease results from impaired (but not abolished) IL-12 and IL-23 responses, resulting in defective IFN-γ production by T cells and NK cells (Figure 1b and Table 1).17 Four of the seven patients also suffered from viral disease, consistent with their poor cellular responses to type I IFNs.17 However, one patient had a pure clinical phenotype of MSMD, and two had a phenotype of isolated tuberculosis.17 There is, thus, incomplete penetrance for both mycobacterial and viral infections in complete AR TYK2 deficiency. Intriguingly, a ninth patient was reported to suffer from HIES.57 Overall, among the nine patients with complete AR TYK2 deficiency, five had a history of BCG disease, and thus displayed phenotype of MSMD, due to poor cellular responses to IL-12 and IL-23.17,57 Two patients from one Japanese kindred with compound heterozygous frameshift and missense TYK2 mutations causing partial TYK2 deficiency were recently reported.64 Both displayed EBV-driven lymphoproliferative diseases but no MSMD.64
A new allelic form of AR partial IFN-γR2 deficiency
A new form of partial IFN-γR2 deficiency was recently described in three patients from two kindreds from Turkey and India.56 The three patients developed BCG disease following vaccination. One of the patients died from M. chelonei infection at the age of five years.56 WES identified homozygous mutations in the first or second codon of IFNGR2 (c.1A>G and c.4delC).56 In an overexpression system, the two mutant proteins were produced, albeit in small amounts, and their function was impaired, as demonstrated by the cellular response to IFN-γ.56 Similar results were obtained for patients’ SV40-fibroblasts, Epstein-Barr transformed B lymphocytes (EBV-B cells), primary CD4+ T cells, and monocyte derived-macrophages (MDM) (homozygous for c.1A>G).56 The three patients had high plasma IFN-γ concentrations, as reported for patients with other forms of IFN-γR1 or IFN-γR2 deficiency.56 The impairment of the cellular response to IFN-γ was more severe than that in patients with previously reported forms of AR partial IFN-γR2 deficiency, but less severe than that in patients with AR complete IFN-γR2 deficiency.56 Interestingly, the two mutations led to a re-initiation of translation at proximal non-canonical codons located within the signal peptide, rather than at more distal AUG codons.56 The shorter signal peptide generated was sufficient for entry into and trafficking through the secretory pathway.56 These patients therefore had low levels of wild-type, full-length IFN-γR2 molecules on the surface of their cells. By contrast, the missense mutations underlying previously reported forms of partial AR IFN-γR2 deficiency result in the expression at the cell surface of abnormal and dysfunctional proteins. The mutations in the first or second codon result in the production of very low levels of normal IFN-γR2 proteins, i.e. a purely quantitative form of partial IFN-γR2 deficiency (Table 1).56
New mutations at known MSMD loci
Since 2014,1 34 new disease-causing mutations have been reported at six MSMD loci already discovered before this date, including IFNGR1,10,40,42,43,45,46,48,49 IFNGR2,5,10,47 IL12RB1,6,12,22,25,30 IL12B,7 STAT1,32–34 and NEMO.18,36–38 Interestingly, the two new hypomorphic NEMO mutations underlie mycobacterial diseases without anhidrotic ectodermal dysplasia (EDA).18,36–38 One of these mutations (c.1–16G>C) is located in the non-coding exon 1B of NEMO.36–38 Three patients harboring this mutation presented adulthood-onset disseminated mycobacterial disease.36 Another patient with the same phenotype carried the previously described65 c.1–16+1G>T NEMO mutation.36 A founder effect was described for the known p.W60* mutation, which is responsible for IL-12p40 deficiency in Saudi patients.19 By contrast, fewer CNVs have been reported, but those identified include an entire deletion of the IFNGR1 gene.42 Four large deletions and the first large duplication at the IL12RB1 locus were identified by targeted next-generation sequencing (NGS).6 Up to 7% of IL12RB1 mutations are CNVs, and the genetic structure of this locus leaves it prone to various Alu-mediated CNVs.6 There are probably many undetected CNVs at other MSMD loci.6 Finally, new mutations underlying complete AR IFN-γR2 deficiency5 and AD STAT1 deficiency32, 66 have been reported in two patients with a phenotype broader than the expected MSMD. Interestingly, both these patients actually suffered from two different primary immunodeficiencies (PIDS).5, 32, 66 The first also had IFN-αR1 deficiency,5 accounting for viral diseases, and the second had p40phox deficiency,32, 66 accounting for pyogenic bacterial diseases. This situation is reminiscent of that previously reported for patients with both ataxia-telangiectasia and IL-12Rβ1 deficiency.1 These findings highlight the importance of testing patients with a phenotype broader than expected, including the canonical MSMD phenotype, for other genetic diseases.
RORγ/RORγT and JAK1 deficiencies: new genetic etiologies of syndromic MSMD
Syndromic MSMD is typically defined as a combination of both mycobacterial disease and other infections associated with a more complex cellular phenotype. Known examples include AR STAT1 and TYK2 deficiencies, underlying mycobacterial and viral diseases,1,17 and AD GATA2 deficiency, underlying mycobacterial and viral diseases in the context of multiple myeloid and lymphoid abnormalities.1, 60 Two new disorders belonging to this group were recently discovered.58,59 A combination of WES and WGL identified homozygous RORC mutations in seven patients from three kindreds living in Chile, Israel, and Saudi Arabia, respectively.58 The patients had BCG-osis and chronic mucocutaneous candidiasis (CMC).58 The RORC gene can encode two protein isoforms that act as transcription factors: nuclear orphan receptor γ (RORγ), which is ubiquitously expressed, and RORγT, the expression of which is restricted to leukocytes. The mutations identified in the three kindreds were p.S38L, p.Q329*, p.Q441* for the RORγ isoform and p.S17L, p.Q308*, p.Q420* for the RORγT isoform. The mutant RORC alleles are loss-of-function.58 The patients displayed impaired lymphoid development with a small thymus, mild T lymphopenia, and small number of ILC3, MAIT cells and NKT cells.58 IL-17A/F secretion was impaired in T cells from the patients, accounting for CMC.58 IFN-γ secretion was normal in naïve or memory CD4+ T cells but strongly impaired in γδ T cells and Th1* cells, accounting for mycobacterial disease.58 Bi-allelic RORC mutations thus impair IL-17 and IFN-γ immunity, underlying candidiasis and mycobacteriosis, respectively.58 In addition, two homozygous variants (p.P733L, p.P832S) of the pseudokinase domain of JAK1 were recently identified by WES in a patient from Pakistan with atypical mycobacterial disease and a history of viral, fungal, and parasitic skin infections.59 This patient died from urothelial carcinoma at the age of 22 years.59 JAK1 is a tyrosine kinase involved in the intracellular signaling of many cytokines, including IFN-α/β and IFN-γ. Cellular responses to IFN-γ and IFN-α were impaired but not abolished by this mutant allele in an overexpression system (U4A cells), in primary fibroblasts, and in leukocytes from the patient.59 The p.P733L mutation was found to be hypomorphic and responsible for this cellular phenotype, whereas the p.P832S mutation was neutral. Impaired responses to IL-2, IL-4, IL-10 and IL-27 were also documented in leukocytes.59 AR partial JAK1 deficiency thus causes susceptibility to mycobacteria due to the impairment of IFN-γ signaling, and susceptibility to other infections due to defective responses to other cytokines, including IFN-α. This condition may also cause susceptibility to early-onset cancer.59
Concluding remarks
Over the last years, the genetic dissection of patients with isolated MSMD or syndromic MSMD has shed new light on the molecular and cellular bases of human immunity to mycobacteria (Figure 1b). An enigma not addressed here concerns the occurrence of mycobacterial disease in some patients with gain-of-function STAT1 mutations.67 This is paradoxical, as STAT1 deficiency underlies mycobacterial disease by decreasing cellular responses to IFN-γ.1 One would have predicted patients with enhanced cellular responses to IFN-γ not to be expected to be prone to mycobacterial disease, and even perhaps being protected from them. This mystery remains to be solved, but the last four years have clearly revealed four new molecular players in MSMD players at the molecular level: SPPL2a, TYK2, JAK1 and RORγ/RORγT. They have also revealed two new MSMD players at the cellular level, IFN-γ-producing Th1* and γδ T cells.58 They have also collectively confirmed the importance of previously identified molecules, encoded by known MSMD loci. Finally, they have added weight to the previous suggestion that IL-12- and IL-23-producing cDC2s are essential for protective immunity to mycobacteria.39 Admittedly, the contribution of these cells to this condition will be proven only if genetic defects exclusively preventing their development or function are discovered. The study of MSMD is far from complete. MSMD is the most studied of a handful of “Mendelian infections” that provided early support for a genetic theory of infectious diseases, paving the way for the study of monogenic but non-Mendelian infections.68,69 Only about half the MSMD patients in our laboratory cohort have a known single-gene inborn error of IFN-γ immunity. A molecular diagnosis of MSMD makes it possible to offer families genetic counseling and paves the way for treatment options based on a better understanding of the pathogenesis of mycobacterial disease. For example, IFN-γ therapy should be considered as the natural treatment, in conjunction with antibiotics, in all patients who do not have a complete lack of cellular responses to this cytokine. Next-generation sequencing (NGS) techniques, including both WES and whole-genome sequencing (WGS) in particular, are now accelerating genetic dissection in the remaining MSMD patients without a genetic diagnosis. This work will lead to the discovery of new genetic etiologies39,70 and will improve the screening of mutations not easily detectable by Sanger sequencing, such as CNV.6 NGS is a powerful tool that will help to decipher the genetic and clinical heterogeneity of MSMD,70 paving the way for similar studies on tuberculosis.2,71
Disclosures and acknowledgement
We thank both branches of the Laboratory of Human Genetics of Infectious Diseases for helpful discussions. The Laboratory of Human Genetics of Infectious Diseases is supported by the National Institute of Allergy and Infectious Diseases grant number 5R37AI095983, the Rockefeller University, the St. Giles Foundation, National Institute of Health and Medical Research (INSERM), Paris Descartes University, the Integrative Biology of Emerging Infectious Diseases Laboratory of Excellence (ANR-10-LABX-62-IBEID) and the French National Research Agency under the “Investments for the future” program (grant number ANR-10-IAHU-01), ANR-GENMSMD (ANR-16-CE17–0005–01 for JB) and SRC2017 (for JB). XFK was supported by the Jerome Lejeune Foundation, the Stony Wold-Herbert Fund, the Choh-Hao Li Memorial Fund Scholar Award and the Shanghai Educational Development Foundation. RMB was funded by a European Molecular Biology Organization (EMBO) Long-Term fellowship. NRA was supported by Conacyt No 277639 and Stony Wold Herbert Fund. COQ is supported by ANR-HGDIFD (ANR-14-CE15–006–01).
Footnotes
The authors have no conflict of interest to declare.
References
- 1.Bustamante J, Boisson-Dupuis S, Abel L, et al. Mendelian susceptibility to mycobacterial disease: genetic, immunological, and clinical features of inborn errors of IFN-gamma immunity. Semin Immunol 2014;26:454–470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Boisson-Dupuis S, Bustamante J, El-Baghdadi J, et al. Inherited and acquired immunodeficiencies underlying tuberculosis in childhood. Immunol Rev 2015;264:103–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Glanzmann B, Moller M, Moncada-Velez M, et al. Autosomal Dominant IFN-gammaR1 Deficiency Presenting with both Atypical Mycobacteriosis and Tuberculosis in a BCG-Vaccinated South African Patient. J Clin Immunol 2018;38:460–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hatipoglu N, Guvenc BH, Deswarte C, et al. Inherited IL-12Rbeta1 Deficiency in a Child With BCG Adenitis and Oral Candidiasis: A Case Report. Pediatrics. 2017;140:e20161668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hoyos-Bachiloglu R, Chou J, Sodroski CN, et al. A digenic human immunodeficiency characterized by IFNAR1 and IFNGR2 mutations. J Clin Invest 2017;127:4415–4420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rosain J, Oleaga-Quintas C, Deswarte C, et al. A Variety of Alu-Mediated Copy Number Variations Can Underlie IL-12Rbeta1 Deficiency. J Clin Immunol 2018;38:617–627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Parvaneh N, Barlogis V, Alborzi A, et al. Visceral leishmaniasis in two patients with IL-12p40 and IL-12Rbeta1 deficiencies. Pediatr Blood Cancer. 2017;64:e26362. [DOI] [PubMed] [Google Scholar]
- 8.Esteve-Sole A, Sologuren I, Martinez-Saavedra MT, et al. Laboratory evaluation of the IFN-gamma circuit for the molecular diagnosis of Mendelian susceptibility to mycobacterial disease. Crit Rev Clin Lab Sci 2018;55:184–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Staines-Boone AT, Deswarte C, Venegas Montoya E, et al. Multifocal Recurrent Osteomyelitis and Hemophagocytic Lymphohistiocytosis in a Boy with Partial Dominant IFN-gammaR1 Deficiency: Case Report and Review of the Literature. Front Pediatr 2017;5:75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tesi B, Sieni E, Neves C, et al. Hemophagocytic lymphohistiocytosis in 2 patients with underlying IFN-gamma receptor deficiency. J Allergy Clin Immunol 2015;135:1638–1641. [DOI] [PubMed] [Google Scholar]
- 11.Muriel-Vizcaino R, Yamazaki-Nakashimada M, Lopez-Herrera G, et al. Hemophagocytic Lymphohistiocytosis as a Complication in Patients with MSMD. J Clin Immunol 2016;36:420–422. [DOI] [PubMed] [Google Scholar]
- 12.Louvain de Souza T, de Souza Campos Fernandes RC, Azevedo da Silva J, et al. Microbial Disease Spectrum Linked to a Novel IL-12Rbeta1 N-Terminal Signal Peptide Stop-Gain Homozygous Mutation with Paradoxical Receptor Cell-Surface Expression. Front Microbiol. 2017;8:616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Nabhani S, Ginzel S, Miskin H, et al. Deregulation of Fas ligand expression as a novel cause of autoimmune lymphoproliferative syndrome-like disease. Haematologica. 2015;100:1189–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Tan C, Cagdas-Ayvaz D, Metin A, et al. Clinical and genetic features of IL12Rb1 deficiency: Single center experience of 18 patients. Turk J Pediatr 2016;58:356–361. [DOI] [PubMed] [Google Scholar]
- 15.Mandola AB, Broides A, Nahum A. Salmonella typhi infection causing prolonged chronic illness and cutaneous leucocytoclastic vasculitis in a patient with IL-12 receptor β1 deficiency. LymphoSign Journal 2017;4:25–29. [Google Scholar]
- 16.Khamassi I, Ben Ali M, Ben Mustapha I, et al. Salmonella enteriditis inducing cutaneous leucocytoclasic vasculitis: An unusual complication in a patient with an interleukine- 12 receptor beta-1 deficiency. Tunis Med 2015;93:328–329. [PubMed] [Google Scholar]
- 17.Kreins AY, Ciancanelli MJ, Okada S, et al. Human TYK2 deficiency: Mycobacterial and viral infections without hyper-IgE syndrome. J Exp Med 2015;212:1641–1662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Karaca N, Aksu G, Ulusoy E, et al. Disseminated BCG Infectious Disease and Hyperferritinemia in a Patient With a Novel NEMO Mutation. J Investig Allergol Clin Immunol 2016;26:268–271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alodayani AN, Al-Otaibi AM, Deswarte C, et al. Mendelian Susceptibility to Mycobacterial Disease Caused by a Novel Founder IL12B Mutation in Saudi Arabia. J Clin Immunol 2018;38:278–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alaki EM, Aljobair F, Alaklobi F, et al. Chronic Disseminated Salmonellosis in a Patient With Interleukin-12p40 Deficiency. Pediatr Infect Dis J 2018;37:90–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Zahid MF, Ali SA, Jehan F, et al. Recurrent Salmonellosis in a Child with Complete IL-12Rbeta1 Deficiency. J immunodefic Disord 2014;3:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dhalla F, Fox H, Davenport EE, et al. Chronic mucocutaneous candidiasis: characterization of a family with STAT-1 gain-of-function and development of an ex-vivo assay for Th17 deficiency of diagnostic utility. Clin Exp Immunol 2016;184:216–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Gokturk B, Reisli I, Caliskan U, et al. Infectious diseases, autoimmunity and midline defect in a patient with a novel bi-allelic mutation in IL12RB1 gene. Turk J Pediatr 2016;58:331–336. [DOI] [PubMed] [Google Scholar]
- 24.Lovell JP, Foruraghi L, Freeman AF, et al. Persistent nodal histoplasmosis in nuclear factor kappa B essential modulator deficiency: Report of a case and review of infection in primary immunodeficiencies. J Allergy Clin Immunol 2016;138:903–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Sarrafzadeh SA, Mahloojirad M, Nourizadeh M, et al. Mendelian Susceptibility to Mycobacterial Disease due to IL-12Rbeta1 Deficiency in Three Iranian Children. Iran J Public Health 2016;45:249–254. [PMC free article] [PubMed] [Google Scholar]
- 26.Alinejad Dizaj M, Mortaz E, Mahdaviani SA, et al. Susceptibility to mycobacterial disease due to mutations in IL-12Rbeta1 in three Iranian patients. Immunogenetics. 2018;70:373–379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Arias AA, Perez-Velez CM, Orrego JC, et al. Severe Enteropathy and Hypogammaglobulinemia Complicating Refractory Mycobacterium tuberculosis Complex Disseminated Disease in a Child with IL-12Rbeta1 Deficiency. J Clin Immunol 2017;37:732–738. [DOI] [PubMed] [Google Scholar]
- 28.Sogkas G, Atschekzei F, Schacht V, et al. First Association of Interleukin 12 Receptor Beta 1 Deficiency with Sjogren’s Syndrome. Front Immunol 2017;8:885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Stray-Pedersen A, Sorte HS, Samarakoon P, et al. Primary immunodeficiency diseases: Genomic approaches delineate heterogeneous Mendelian disorders. J Allergy Clin Immunol 2017;139:232–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kadayifci EK, Karaaslan A, Atici S, et al. IL12Rbeta1 defect presenting with massive intra-abdominal lymphadenopathy due to Mycobacterium intracellulare infection. Asian Pac J Allergy Immunol 2017;35:161–165. [DOI] [PubMed] [Google Scholar]
- 31.Ling G, Ling E, Broides A, et al. IL-12 receptor 1beta deficiency with features of autoimmunity and photosensitivity. Autoimmunity. 2016;49:143–146. [DOI] [PubMed] [Google Scholar]
- 32.Kagawa R, Fujiki R, Tsumura M, et al. Alanine-scanning mutagenesis of human signal transducer and activator of transcription 1 to estimate loss- or gain-of-function variants. J Allergy Clin Immunol 2017;140:232–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Boudjemaa S, Dainese L, Heritier S, et al. Disseminated Bacillus Calmette-Guerin Osteomyelitis in Twin Sisters Related to STAT1 Gene Deficiency. Pediatr Dev Pathol 2017;20:255–261. [DOI] [PubMed] [Google Scholar]
- 34.Ueki M, Yamada M, Ito K, et al. A heterozygous dominant-negative mutation in the coiled-coil domain of STAT1 is the cause of autosomal-dominant Mendelian susceptibility to mycobacterial diseases. Clin Immunol 2017;174:24–31. [DOI] [PubMed] [Google Scholar]
- 35.Esteve-Sole A, Sánchez-Dávila SP, Deyà-Martínez A, et al. Severe BCG-osis Misdiagnosed as Multidrug-Resistant Tuberculosis in an IL-12Rβ1-Deficient Peruvian Girl. Journal of Clinical Immunology. 2018;38:712–716. [DOI] [PubMed] [Google Scholar]
- 36.Hsu AP, Zerbe CS, Foruraghi L, et al. IKBKG (NEMO) 5’ untranslated splice mutations lead to severe, chronic disseminated mycobacterial infections. Clin Infect Dis 2018;67:456–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Huppmann AR, Leiding JW, Hsu AP, et al. Pathologic Findings in NEMO Deficiency: A Surgical and Autopsy Survey. Pediatr Dev Pathol 2015;18:387–400. [DOI] [PubMed] [Google Scholar]
- 38.Braue J, Murugesan V, Holland S, et al. NF-kappaB Essential Modulator Deficiency Leading to Disseminated Cutaneous Atypical Mycobacteria. Mediterr J Hematol Infect Dis 2015;7:e2015010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kong XF, Martinez-Barricarte R, Kennedy J, et al. Disruption of an antimycobacterial circuit between dendritic and helper T cells in human SPPL2a deficiency. Nat Immunol 2018;19:973–985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Holzer U, Reinhardt K, Lang P, et al. Influence of a mutation in IFN-gamma receptor 2 (IFNGR2) in human cells on the generation of Th17 cells in memory T cells. Hum Immunol 2013;74:693–700. [DOI] [PubMed] [Google Scholar]
- 41.Olbrich P, Martinez-Saavedra MT, Perez-Hurtado JM, et al. Diagnostic and therapeutic challenges in a child with complete interferon-gamma receptor 1 deficiency. Pediatr Blood Cancer. 2015;62:2036–2039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.de Vor IC, van der Meulen PM, Bekker V, et al. Deletion of the entire interferon-gamma receptor 1 gene causing complete deficiency in three related patients. J Clin Immunol 2016;36:195–203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gutierrez MJ, Kalra N, Horwitz A, et al. Novel Mutation of Interferon-gamma Receptor 1 Gene Presenting as Early Life Mycobacterial Bronchial Disease. J Investig Med High Impact Case Rep 2016;4:2324709616675463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Olbrich P, Falcon-Neyra L, Molinos-Quintana A, et al. First Documented Case of Influenza A (H3N2 Subtype) Infection in a Patient With Complete Interferon Gamma Receptor 1 Deficiency: A Call for Systemic Vaccination Strategies. Pediatr Infect Dis J 2016;35:712–713. [DOI] [PubMed] [Google Scholar]
- 45.Martinez-Morales MC, Deswarte C, Castaneda-Casimiro J, et al. [Disseminated infection by M. tuberculosis complex in patient with IFN-gamma receptor 1 complete deficiency]. Rev Alerg Mex 2017;64:499–504. [DOI] [PubMed] [Google Scholar]
- 46.Khanolkar A, Kirschmann DA, Caparelli EA, et al. CD4 T cell-restricted IL-2 signaling defect in a patient with a novel IFNGR1 deficiency. J Allergy Clin Immunol 2018;141:435–439 e437. [DOI] [PubMed] [Google Scholar]
- 47.Kamoun C, Morsheimer M, Sullivan KE, et al. Successful unrelated cord blood transplant for complete IFN-gamma receptor 2 deficiency. J Allergy Clin Immunol 2016;138:1489–1491. [DOI] [PubMed] [Google Scholar]
- 48.van de Vosse E, van Dissel JT. IFN-gammaR1 defects: Mutation update and description of the IFNGR1 variation database. Hum Mutat 2017;38:1286–1296. [DOI] [PubMed] [Google Scholar]
- 49.Cakan M, Keskindemirci G, Aydogmus C, et al. Coexistence of early onset sarcoidosis and partial interferon-gamma receptor 1 deficiency. Turk J Pediatr 2016;58:545–549. [DOI] [PubMed] [Google Scholar]
- 50.Sharma VK, Pai G, Deswarte C, et al. Disseminated Mycobacterium avium complex infection in a child with partial dominant interferon gamma receptor 1 deficiency in India. J Clin Immunol 2015;35:459–462. [DOI] [PubMed] [Google Scholar]
- 51.Antonietti J, Retornaz K, Bernasconi A, et al. [Disseminated BCG disease revealing a partial deficiency in receptor 1 interferon gamma]. Arch Pediatr 2015;22:971–973. [DOI] [PubMed] [Google Scholar]
- 52.Quispel WT, Stegehuis-Kamp JA, Santos SJ, et al. Intact IFN-gammaR1 expression and function distinguishes Langerhans cell histiocytosis from mendelian susceptibility to mycobacterial disease. J Clin Immunol 2014;34:84–93. [DOI] [PubMed] [Google Scholar]
- 53.Holland SM, Pierce VM, Shailam R, et al. Case 28–2017. A 13-Month-Old Girl with Pneumonia and a 33-Year-Old Woman with Hip Pain. N Engl J Med 2017;377:1077–1091. [DOI] [PubMed] [Google Scholar]
- 54.Wheelock EF. Interferon-Like Virus-Inhibitor Induced in Human Leukocytes by Phytohemagglutinin. Science. 1965;149:310–311. [PubMed] [Google Scholar]
- 55.Nathan CF, Murray HW, Wiebe ME, et al. Identification of interferon-gamma as the lymphokine that activates human macrophage oxidative metabolism and antimicrobial activity. J Exp Med 1983;158:670–689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Oleaga-Quintas C, Deswarte C, Metin A, et al. A novel form of partial recessive IFN-γR2 deficiency caused by mutations of the initiation and second codon Human Molecular Genetics. 2018;in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fuchs S, Kaiser-Labusch P, Bank J, et al. Tyrosine kinase 2 is not limiting human antiviral type III interferon responses. Eur J Immunol 2016;46:2639–2649. [DOI] [PubMed] [Google Scholar]
- 58.Okada S, Markle JG, Deenick EK, et al. IMMUNODEFICIENCIES. Impairment of immunity to Candida and Mycobacterium in humans with bi-allelic RORC mutations. Science. 2015;349:606–613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Eletto D, Burns SO, Angulo I, et al. Biallelic JAK1 mutations in immunodeficient patient with mycobacterial infection. Nat Commun 2016;7:13992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Donadieu J, Lamant M, Fieschi C, et al. Natural history of GATA2 deficiency in a survey of 79 French and Belgian patients. Haematologica. 2018;103:1278–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Beisner DR, Langerak P, Parker AE, et al. The intramembrane protease Sppl2a is required for B cell and DC development and survival via cleavage of the invariant chain. J Exp Med 2013;210:23–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bergmann H, Yabas M, Short A, et al. B cell survival, surface BCR and BAFFR expression, CD74 metabolism, and CD8- dendritic cells require the intramembrane endopeptidase SPPL2A. J Exp Med 2013;210:31–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Schneppenheim J, Dressel R, Huttl S, et al. The intramembrane protease SPPL2a promotes B cell development and controls endosomal traffic by cleavage of the invariant chain. J Exp Med 2013;210:41–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Nemoto M, Hattori H, Maeda N, et al. Compound heterozygous TYK2 mutations underlie primary immunodeficiency with T-cell lymphopenia. Sci Rep 2018;8:6956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Mooster JL, Cancrini C, Simonetti A, et al. Immune deficiency caused by impaired expression of nuclear factor-kappaB essential modifier (NEMO) because of a mutation in the 5’ untranslated region of the NEMO gene. J Allergy Clin Immunol 2010;126:127–132 e127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.van de Geer A, Nieto-Patlan A, Kuhns DB, et al. Inherited p40phox deficiency differs from classic chronic granulomatous disease. J Clin Invest 2018;128:3957–3975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Toubiana J, Okada S, Hiller J, et al. Heterozygous STAT1 gain-of-function mutations underlie an unexpectedly broad clinical phenotype. Blood. 2016;127:3154–3164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Casanova JL. Severe infectious diseases of childhood as monogenic inborn errors of immunity. Proc Natl Acad Sci U S A 2015;112:E7128–7137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Casanova JL. Human genetic basis of interindividual variability in the course of infection. Proc Natl Acad Sci U S A 2015;112:E7118–7127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Meyts I, Bosch B, Bolze A, et al. Exome and genome sequencing for inborn errors of immunity. J Allergy Clin Immunol 2016;138:957–969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Abel L, Fellay J, Haas DW, et al. Genetics of human susceptibility to active and latent tuberculosis: present knowledge and future perspectives. Lancet Infect Dis 2018;18:e64–e75. [DOI] [PMC free article] [PubMed] [Google Scholar]