

Abstract
Reactive oxygen species (ROS) induce different cellular stress responses but can also mediate cellular signaling. Augmented levels of ROS are associated with aging, cancer as well as various metabolic and neurological disorders. ROS can also affect the efficacy and adverse effects of drugs. Although proteins are key mediators of most ROS effects, direct studies of ROS-modulated-protein function in the cellular context are very challenging. Therefore the understanding of specific roles of different proteins in cellular ROS responses is still relatively rudimentary. In the present work we show that Mass Spectrometry-Cellular Thermal Shift Assay (MS-CETSA) can directly monitor ROS and redox modulations of protein structure at the proteome level. By altering ROS levels in cultured human hepatocellular carcinoma cell lysates and intact cells, we detected CETSA responses in many proteins known to be redox sensitive, and also revealed novel candidate ROS sensitive proteins. Studies in intact cells treated with hydrogen peroxide and sulfasalazine, a ROS modulating drug, identified not only proteins that are directly modified, but also proteins reporting on downstream cellular effects. Comprehensive changes are seen on rate-limiting proteins regulating key cellular processes, including known redox control systems, protein degradation, epigenetic control and protein translational processes. Interestingly, concerted shifts on ATP-binding proteins revealed redox-induced modulation of ATP levels, which likely control many cellular processes. Collectively, these studies establish CETSA as a novel method for cellular studies of redox modulations of proteins, which implicated in a wide range of processes and for the discovery of CETSA-based biomarkers reporting on the efficacy as well as adverse effects of drugs.
Keywords: Reactive oxygen species, Cellular thermal shift assay, Reactive cysteines, Quantitative proteomics, Glutathione, ATP levels, Sulfasalazine
Graphical abstract
Highlights
-
•
Redox-mediated protein modulations can be monitored by MS-CETSA.
-
•
CETSA signals reflect disulfide reorganization, covalent adduction, redox-sensitive metal level changes, and complex formation.
-
•
In-cell redox modulation by SSA, H2O2 or oligomycin affects cellular ATP levels.
-
•
In-cell CETSA identifies rate-limiting proteins controlling redox downstream effects.
1. Introduction
Reactive Oxygen Species (ROS) such as superoxide anion radicals (O2•−), hydrogen peroxide (H2O2) and hydroxyl radicals (HO•) can serve either as messengers in signal transduction, or in the induction of oxidative stress where excessive ROS can modify protein structure and result in direct damage on other biomolecules including DNA/RNA and lipids [1]. Oxidative stress has been implicated in various diseases including neurodegeneration, atherosclerosis, diabetes and cancer [2]. Malignant cancer cells are known to contain and tolerate higher ROS levels than normal cells, which is due to distorted cellular metabolism [3]. Based on this feature, there is a growing interest in manipulating ROS levels in anticancer therapies to selectively kill cancer cells without affecting normal cells [[4], [5], [6]]. Some drugs also induce ROS and redox stress as secondary effects, and for some cancer chemotherapies, such effects lead to immunogenic cell death which helps in activating the immune system against the tumour cells [7].
ROS effects are mediated by redox-sensitive proteins, which can undergo different reversible or irreversible chemical modifications upon oxidative stress including cysteine oxidation, carbonylation, methionine sulfoxidation, or via redox sensitive cofactors such as redox metals and in special cases, flavins (FAD, FMN) and nicotinamide cofactors (NAD, NADP) [[8], [9], [10]]. Among these, cysteine oxidation is the most well characterized modification, allowing certain ROS-sensitive proteins to function as signaling switches. Proteome-wide studies aimed at identifying proteins containing reactive cysteines have been carried out using different affinity enrichment probes such as oxidative isotope-coded affinity tag (OxICAT) [11], cysteine-reactive tandem mass tag (cysTMT) [12], and iTRAQ labelling of enriched cysteine-containing peptides (cysTRAQ) [13]. Such studies have yielded a valuable global overview of redox-sensitive proteins and reactive residues, but the synthetic nature of these chemical probes, and the fact that in some cases, experiments can only be performed in the lysate phase, can bias the repertoire of identified hits [14,15]. These methods are also less suited for interactive studies of redox modifications in complex cellular processes and systems. Given the importance of ROS modulated processes, and the fact that they are still hard to study in intact cells and tissues [16], there is a need for novel approaches to better understand ROS sensing and responding machineries of the cell in more detail in both normal and pathological situations.
In the present work, we implemented and validated the recently developed Cellular Thermal Shift Assay (CETSA) for studies of ROS-induced modulations of protein structure and activity in the cellular context. CETSA is a label-free and broadly applicable technique that was initially introduced to study the engagement of drug with target proteins in intact cells and tissues [17,18]. By combining the principles of CETSA and mass spectrometry (MS)-based quantitative proteomics, a more comprehensive assessment can be made regarding the stability effects on thousands of cellular proteins [19]. Recent developments further expanded the scope and application of MS-CETSA to reveal biochemical interactions of proteins with physiological ligands (e.g. other proteins, nucleic acids, metabolites and cofactors) [[20], [21], [22], [23]], demonstrating the potential of MS-CETSA to study various intracellular biological processes.
From previous CETSA studies [20,21,24], it has been shown that protein modifications such as phosphorylation can affect the thermal stability of the protein. Redox processes are known to affect the status of the thiol residues on proteins (i.e., formation and breaking of the disulfide bonds) in order to transmit biochemical information in the cell. These modifications on cysteine residues as well as other modifications such as glutathionylation and sulfenylation could potentially lead to changes in thermal stability of the proteins. Furthermore, these modifications could affect the ensemble of protein interaction states, which could also be reflected in MS-CETSA. Additionally, changes in redox-sensitive cofactors such as redox metals could also potentially affect association dynamics with the substrate protein, thereby yielding CETSA signals. To explore these hypotheses, we set out to pursue a CETSA-redoxome study.
In this study, we implemented a MS-CETSA workflow to analyse redox-sensitive proteins and applied it to hepatocellular carcinoma, HepG2 cell lysates and intact cells under different ROS conditions. We identified 240 glutathione- and/or H2O2-sensitive proteins that include many previously identified as well as novel redox-sensitive proteins. From in-cell studies of H2O2 and the redox-modulating drug sulfasalazine, we also identified many downstream affected proteins, which are potential mediators of ROS-induced processes. Notably, our first draft of the ROS-sensitive CETSA redoxome provides a repertoire of biomarkers for more detailed studies of redox processes in cells and tissues in different biological processes and in studies of drug action.
2. Material and methods
2.1. Cell lines and cell culture
Liver cancer HepG2 cell lines were obtained from ATCC. SNU398 cells (SNU398-SALL4) were obtained from Professor Daniel G. Tenen. Cells were cultured in DMEM (GE Healthcare) supplemented with 10% FBS (Biowest), 2 mM glutamine, 100 U/ml penicillin and 100 μg/ml streptomycin (ThermoFisher Scientific). Cells were maintained at 37 °C in a humidified atmosphere containing 5% CO2.
2.2. Oxidant or compound exposure
For lysate glutathione ITRR treatment, 250 μg HepG2 cell lysates (preparation described below) per reaction were incubated with ten different GSSG/GSH ratios (1:100, 1:20, 1:10, 1:5, 1:2, 1:1, 2:1, 5:1, 10:1, 100:1) at the final concentration equivalent to total 5 mM glutathione and 50 μl reaction volume for 1 min at room temperature. For lysate H2O2 ITDR treatment, same amount of protein lysates and reaction volume were incubated with ten different H2O2 concentrations (highest dosage 50 mM, a gradient formed with a dilution factor of 4) for 1 min. In-cell H2O2 ITDR experiment was conducted using 1 million HepG2 cells per reaction, where live cells suspended in 50 μl complete cell culture medium were incubated with a concentration gradient (same as the one used in lysate H2O2 ITDR treatment) of H2O2 solutions for 10 min in 5% CO2 incubator. Treated cells were thoroughly washed with PBS for CETSA sample preparation. In-cell oligomycin ITDR experiment was performed using 1 million SNU398 cells per reaction, where live cells were treated by ten dosages of oligomycin (highest dosage 100 μM, a gradient formed with a dilution factor of 5) in the presence of cell culture medium for 1 h in 5% CO2 incubator. In-cell Sulfasalazine ITDR experiment was conducted in a similar way (highest dosage 250 μM, a gradient formed with a dilution factor of 4) on adherent HepG2 cells in complete medium for 4 h in 5% CO2 incubator. In-cell Sulfasalazine ITTR treatment was done using 1 million HepG2 cells cultured in sufficient amount of complete medium over 24 h (0, 1, 2, 3, 4, 6, 8, 16, 20, 24 h). Compound addition was scheduled at different time points and all the cells were harvested at one single end point. Treated adherent cells were harvested by gentle scraping, washed in PBS and ready for downstream MS-CETSA sample preparation.
2.3. MS-CETSA sample preparation
Sample preparation for CETSA experiments was carried out as described previously [21,22]. For lysate experiments, PBS-washed HepG2 cells were resuspended in 1x kinase buffer containing 50 mM HEPES, pH 7.5, 5 mM beta-glycerophosphate, 0.1 mM sodium orthovanadate (Na3VO4), 10 mM MgCl2 (Sigma Aldrich), and 1x protease inhibitor (Nacalai), and lysed by multiple rounds of freeze-thaw procedure in combination with mechanical shearing. After centrifugation at 20,000 g for 20 min at 4 °C, clear cell lysates were collected for glutathione or H2O2 (Sigma Aldrich) treatment followed by heating at 37 °C and 52 °C (or 55 °C) for 3 min in a 96-well thermocycler. These samples were then centrifuged at 20,000 g for 20 min at 4 °C to remove aggregated proteins. For in-cell experiments, HepG2 or SNU398 cells treated by H2O2 or drugs (Sulfasalazine/oligomycin) (Sigma Aldrich) were then washed and resuspended in 50 μl PBS, and intact cells were heated at the indicated temperatures. After topping up with 50 μl of 2x kinase buffer, samples were lysed and subjected for centrifugation as aforementioned. After centrifugation, soluble protein supernatants were recovered for next step.
Protein concentrations of each sample were then determined by BCA assay (ThermoFisher Scientific) and same amount of soluble proteins (30 μg) were used for MS sample preparation. Protein denaturation and reduction was done with 10 mM TCEP (Sinopharm Chemical Reagent Co.) in the presence of 0.05% RapiGest (Waters) at 55 °C for 20 min and protein alkylation was done in the presence of 55 mM CAA (Sigma Aldrich) at room temperature for 30 min. Samples were then digested with 1.5 μg LysC (Wako) for 3 h and 1.5 μg Trypsin (Promega) for overnight at 37 °C. The Rapigest was acid-hydrolyzed with 1% TFA (Sigma Aldrich) at room temperature for 45 min. The peptide samples were then dried by a centrifugal vacuum evaporator (Eppendorf) and resolubilized in 100 mM TEAB (Sigma Aldrich) to 1 μg/μl 12 μg peptides from each ratio/dose/time-course treatment as a set were labelled with TMT10-plex reagents (ThermoFisher Scientific) for 1 h, quenched using 100 mM Tris (pH 7.4) and desalted with a C-18 Sep-Pak column (Waters) according to manufacturer's instructions. A liquid chromatography ÄKTA Micro system (GE Healthcare) coupled with a high pH reverse phase column (Agilent, Zorbax 300 Extend C-18 4.6 mm × 250 mm column) was used for peptide fractionation.
For the CETSA measurements of all ITRR, ITDR and ITTR samples, typically 52 °C was chosen as the heating temperature, and biological duplicates were acquired. In order to make sure the applied compounds would not lead to a global distortion of the protein samples, one measurement at 37 °C as protein level control was acquired in ITRR and ITDR experiments. In the ITTR experiment, 37 °C samples were prepared in biological duplicates in order to confidently measure the protein abundance changes over time.
2.4. LC-MS analysis
Peptide sample fractions were dried and resuspended in 1: 99 acetonitrile: water containing 0.5% (v/v) acetic acid and 0.06% TFA. 2 μg peptides per fraction were injected in Dionex UltiMate 3000 UPLC system coupled to a Q Exactive HF mass spectrometer (ThermoFisher Scientific). Separation was performed on a 50 cm × 75 μm EASY-Spray analytical column (ThermoFisher Scientific) with a pre-programmed gradient by mixing solvent A (0.1% formic acid in water) and solvent B (0.1% formic acid in acetonitrile) over 70 min. MS data was acquired using a top 12 data-dependent acquisition protocol at 60,000 resolution in MS1 and 45,000 resolution in MS2.
2.5. Protein identification and quantification
Raw MS data was analysed in Proteome Discoverer 2.1 software (ThermoFisher Scientific) for peptide and protein identification and quantification, using both Mascot 2.6.0 (Matrix Science) and Sequest HT (ThermoFisher Scientific). Obtained peptide sequences were mapped against a reviewed human proteome database downloaded from Uniprot containing 42105 sequence entries. Search parameters were set as the following: trypsin, missed cleavage sites ≤3, precursor mass tolerance 20 ppm, fragment mass tolerance 0.05Da. Dynamic modifications searched included: acetylation (protein N-terminus), oxidation (M), deamidation (NQ), and static modifications included: carbamidomethylation (C) and TMT10-plex (K and peptide N-terminus). False discovery rate (FDR) threshold was set to 1% at both PSM and peptide levels. Only the unique and razor peptides were used for protein assignment and abundance quantification. Isotopic correction of the reporter ions in each TMT channel was performed according to the product sheet. Only the master proteins in the protein group were used for downstream analysis.
2.6. Quantitative MS data analysis and visualization
Grouped protein abundances were analysed and visualized using in-house developed mineCETSA package as previously described [22]. Data extraction, clean-up and systematic scaling, curve fitting and plotting was performed accordingly. For each compound treatment experiment, an initial requirement was set that only proteins consistently quantified in all samples can be used for further hit selection.
In our ITRR, ITDR and ITTR 52 °C data, measured fold-changes for each protein across different ratios, dosages or time-courses would reflect protein thermal stability changes upon compound treatment. A strict hit selection criterion was applied to obtain target proteins possessing most robust thermal shifts. We require that target proteins should be quantified by at least three peptide spectrum matches (PSM) and their sigmoidal curve fitting (measured by R2 value) should be greater than 0.8. Minimum responsive level cut-off was set to median +2x MAD level, based on the median absolute deviation (MAD) calculated from all the readings from the first three ratio/dose/time-points in each treatment dataset. On top of this, we also require that fold-change levels of the last three ratio/dose/time-points should surpass a change of 30% compared to minimum responsive level. Importantly, the hit calling requires a thermal shift observed at 52 °C and a stable level at 37 °C for ITRR and ITDR datasets, namely proteins with shifts at 37 °C were excluded from hit selection, using the same criteria (PSM≥3, R2 > 0.8, MAD threshold = 2, fold-change>30%). In ITTR experiment, 37 °C measurement was used as protein level reference control to normalize 52 °C thermal shift measurement, in order to subtract any possible effects from protein abundance changes, i.e., at each time point, the real thermal shift is equal to: the measured thermal shift at 52 °C divided by the measured level change at 37 °C.
2.7. Expression and purification of recombinant proteins
Gene encoding human HAT protein (BC063003) was cloned into pSUMO-LIC vector and expressed in BL21 (DE3) Rosetta T1R E. coli (Novagen) in Terrific Broth media supplemented with kanamycin and chloramphenicol. Cells were cultured and induced with 0.5 mM isopropyl-beta-d-1-thiogalactopyranoside (IPTG) at 18 °C overnight, harvested and resuspended in lysis buffer (100 mM HEPES, 500 mM NaCl, 10 mM imidazole, 10% (v/v) glycerol at pH 8.0) supplemented with 1:1000 (v/v) EDTA-free protease inhibitor cocktail (Nacalai) and 250U/ml of Benzonase (Merck). After sonication, centrifugation and clarified by filtration, the protein extract was loaded onto Ni-NTA Superflow column (Qiagen), washed and eluted with 5 column volumes of elution buffer (20 mM HEPES, 500 mM NaCl, 500 mM imidazole, 10% (v/v) glycerol at pH 7.5). Eluate was then loaded onto a HiLoad 16/60 Superdex-200 column (GE Healthcare) and eluted with 1 column volumes of elution buffer (20 mM HEPES, 300 mM NaCl, 10% (v/v) glycerol at pH 7.5). Relevant protein fractions were pooled and concentrated using centrifugal force driven filter concentrators (VivaScience). Protein purity was assessed on SDS-PAGE and identity confirmed by mass spectrometry analysis. Protein concentration was determined by the absorbance at 280 nm using Nanodrop spectrophotometer (ThermoFisher Scientific).
2.8. In vitro reduction and oxidation
HepG2 cell extracts and purified HAT1 recombinant proteins, which were prepared as mentioned were treated by 1 mM GSSG alone or 1 mM GSSG in combination with 5 mM GSH for 10 min at room temperature. Free glutathione was removed by diluting the reaction with 10 vol designated buffers and filtering through Vivaspin 500 concentrator (Sartorius). Samples were then supplemented with NuPAGE LDS sample buffer (ThermoFisher Scientific) in the absence of reducing agent and without boiling; or in the presence of 100 mM DTT and boiled at 95 °C for 10 min, and used for western blotting analysis.
2.9. Western blot analysis
20 μg of total proteins from cell lysate or 200 ng of recombinant proteins were separated on NuPAGE Bis-Tris 4–12% Protein Gels (Invitrogen) and transferred to nitrocellulose membranes using iBlot system (Invitrogen). Membrane was blocked by 5% skimmed milk and incubated with primary antibodies for designated protein detection. The following antibodies were used in this study: anti-PRDX1 (#8499), anti-CBX3 (#2619) and anti-UBA2 (#8688) antibodies from Cell Signaling Technology; anti-AHS2 (A300-489A) from Bethyl Laboratories; anti-HAT1 (sc-390562) and anti-PRMT1 (sc-166963) from Santa Cruz Biotechnology. Membrane was then washed with PBS containing 0.1% Tween 20 (Sigma Aldrich) and incubated with corresponding secondary antibodies accordingly. Goat anti-mouse (#31430) or anti-rabbit (#31460) IgG (H+L) secondary antibodies were obtained from ThermoFisher Scientific. After thorough washing of membranes, chemiluminescence signals were visualized using Clarity ECL blotting substrates (Bio-Rad) and captured by ChemiDoc MP imaging system (Bio-Rad).
2.10. Protein interaction network generation and gene ontology analysis
Protein interaction network among CETSA hits of each treatment was retrieved by importing a list of Uniprot IDs into Cytoscape v.3.7.0 (https://cytoscape.org). In the embedded STRING interaction database (http://apps.cytoscape.org/apps/stringApp), a default confidence score cut-off at 0.4 was applied for each network retrieval. Each node represented one hit protein and edge width represented interaction score. Thermal shift profiles of each hit were mapped with a numeric table of corresponding thermal shift ratio and visualized as bar chart on top of the corresponding protein node. Node layouts of the networks were determined by yFiles Organic Layout plus manual adjustment for visual clarity.
Gene ontology functional enrichment was retrieved through STRING Enrichment function in Cytoscape using p-value cut-off at 0.05. For each hit list, the most representative and significantly enriched gene ontology biological process terms, cellular component terms and molecular function terms were plotted in bubble charts accordingly.
2.11. Protein complex analysis
Protein complex information within hits list was analysed by mapping the Uniprot ID of hits to the CORUM human protein complex database (http://mips.helmholtz-muenchen.de/corum/). The similarities of thermal shift profiles among each complex subunit protein were determined by Pearson correlation method.
2.12. Intracellular GSH/GSSG ratio determination
GSH and GSSG levels were measured using the GSH/GSSG-glo Glutathione Assay kit (Promega) according to manufacturer's instructions. In brief, HepG2 cells were cultured in 96-well white opaque plate at 5000 cells per well. Cells were treated with vehicle or compound to designated time-courses, washed and incubated with total or oxidized Glutathione Reagents and then with Luciferin Detection Reagent provided in kit. Luminescence reflecting total GSH equivalent or GSSG amount was measured using a luminometer. GSH/GSSG ratio was calculated directly from reading while the exact GSH and GSSG amount were calculated based on a GSH standard curve measured in parallel, and further normalized against total protein amount of each treatment determined by BCA assay (ThermoFisher Scientific).
2.13. Protein abundance profile analysis
Microarray data of Menadione-treated HepG2 cells was downloaded from NCBI (GEO series accession number GSE39291). Average RNA expression values from 0.5, 1, 2, 4, 8, and 24 h time points were normalized against average expression value of corresponding control at each time point and fold-changes over the first time point were calculated. Genes with |log2 fold-change|> 0.58 in at least one time point were considered with expression level changes. Similarly, protein abundance changes determined from our SSA ITTR 37 °C data from 0, 1, 2, 4, 8, 24 h time points were taken for fold-change calculation and proteins with |log2 fold-change|> 0.38 in at least one time point were considered with expression level changes. These two groups of genes/proteins with altered expression profile were used for further comparison and visualized in heat map.
2.14. InterPro domain analysis
Functional domain and family information of all the thermal shifted hits were retrieved from InterPro (https://www.ebi.ac.uk/interpro/). Protein hits were then clustered according to the contained domains and color-coded according to sample treatment. Domain and family clusters containing more than three proteins were kept for visualization and redundant domain terms were omitted.
2.15. Protein amino acid composition determination
A subset of 314 proteins (base), which exhibit minimal CETSA thermal shifts was randomly selected as a counterpart of 314 CETSA hits (hit) for analysis. For base subset selection, we require the proteins to be detected across all measurements, and to show minimal baseline thermal shifts determined by coefficient of variation (CV) < 10%. The canonical protein sequences in both subsets were downloaded from Uniprot and the composition percentages of each amino acid in each individual protein were calculated. Amino acid content percentage distribution in base and hit subsets were statistically evaluated using Mann–Whitney U test.
2.16. Cysteine pKa value and solvent accessibility evaluation
Between the aforementioned base and hit subset of proteins, the ones that possess functional domain details and available crystal structure information from Uniprot were used for cysteine sensitivity determination. Cpipe platform [25] which incorporated Surface Racer v5.0 [26] and PDB2PQR v1.7 [27] was applied to calculate cysteine pKa value and solvent accessibility. Briefly, PDB structures were input into Cpipe platform by specifying chain ID, and a number of cysteines' plots of exposure versus pKa were returned. Cysteines located to protein functional domains were recorded and a maximum of four cysteines were recorded for each protein. A potential sensitive cysteine was determined by the criteria: pKa ≤ 9.05, solvent accessibility>1.3 Å2 [28]. Predicted cysteines within protein crystal structures were visualized using PyMOL (https://pymol.org/2/).
3. Results
3.1. Glutathione redox ratio-induced CETSA shifts
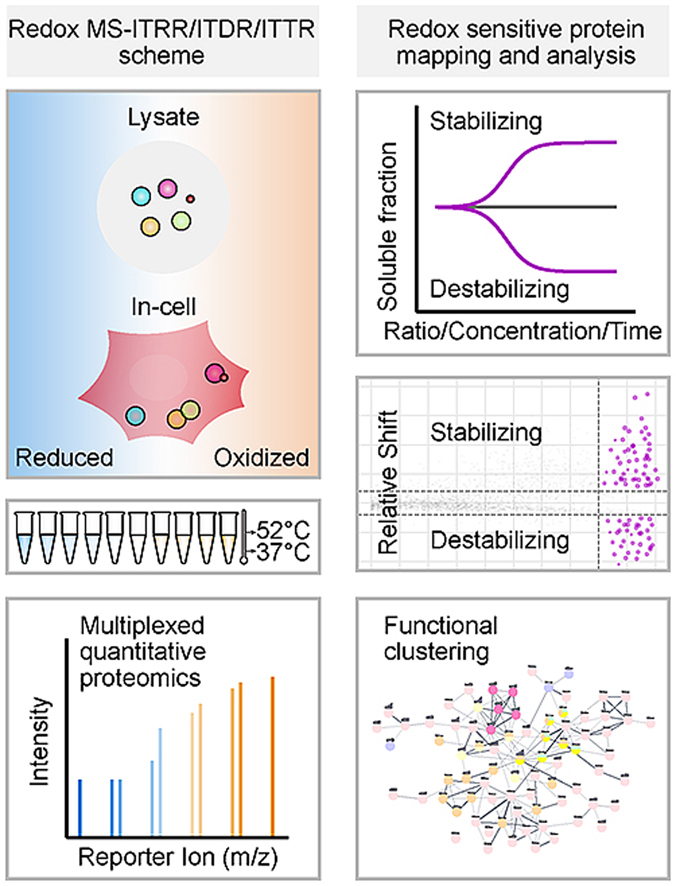
Cells have the ability to sense excessive oxidative stress and respond by mounting an antioxidant defence, counteracting the effects of increased ROS levels. The most abundant and principal intracellular antioxidant redox buffer is glutathione, which shuttles between reduced (GSH) and oxidized (GSSG) forms to balance cellular redox states. Generally, redox sensitive amino acids and cofactors of cytoplasmic proteins are maintained in a reduced state due to a high concentration of GSH (1–10 mM) and a high ratio (100–400:1) between GSH and GSSG [29]. Some proteins such as the thiol-disulfide oxidoreductases of the thioredoxin family, function by forming reversible disulfide bonds within the physiological GSH/GSSG ratio range, thereby transferring redox equivalents for chemical transformations. Given the physiological importance of the glutathione pools, and to shed light on the applicability of CETSA in revealing protein structure modulations induced by redox changes, we first conducted an isothermal MS-CETSA experiment [22] to investigate stability effects at the proteome level by varying the glutathione pool ratios. Different ratios (1:100–100:1) of GSH/GSSG redox pair were used at the final concentration of 5 mM GSH equivalent, to generate a redox gradient in HepG2 cell lysates. A short exposure time of 1 min was used to primarily capture proteins that directly interact with the glutathione species, while minimising downstream biochemical effects. Reactions were heated at 37 °C and 52 °C and samples were multiplexed using tandem mass tag (TMT) labelling (Fig. 1A) as previously described [22,30].
Fig. 1.

General workflow of redox ITRR/ITDR experiments and overview of protein stability changes induced by redox modulations. A, Schematic representation of workflow. HepG2 cell lysates or intact cells were incubated with different glutathione ratios or hydrogen peroxide concentrations and heated at 37 °C and 52 °C. Soluble fractions were harvested, digested and labelled with isobaric mass tags and quantified on an Orbitrap mass spectrometer. Representative mechanisms underlying ROS-induced CETSA shifts are depicted, including cysteine oxidation, disulfide formation or glutathionylation, intermolecular interaction, and cofactor level changes. Numbers of detected proteins and selected hits from each experiment are summarized in table. B, Heat map showing thermal shift profiles of target proteins from lysate glutathione (LG)-ITRR experiment. Each row represents one protein and each column is one ratio point of glutathione treatment. C, Scatter plot visualizing proteins with significant thermal stability changes in LG-ITRR experiment (orange and red dots). Proteins related to cell redox homeostasis are highlighted in red. The background non-responsive proteins are colored in gray.
From this isothermal ratio response (ITRR) analysis, 3999 proteins out of 8365 identified proteins passed the general CETSA quality control criteria (see methods) and were used in the subsequent analyses (Fig. 1A). By applying our criteria for hit selection (see methods), 112 proteins were identified with significant thermal stability changes at 52 °C in a ratio-dependent manner (Fig. 1B), while 37 °C protein level traces remained stable. Most of these proteins begin to show strong responses in the GSSG/GSH ratio region of 1:100–1:1, which includes the typical physiological glutathione ratio range [29].
As a confirmation that CETSA can indeed detect structural effects due to redox buffer modulations, a number of thiol-disulfide oxidoreductases were seen to shift across different glutathione ratios, including peroxiredoxins (PRDX), protein disulfide isomerases (PDI) and thioredoxin domain-containing proteins (TXNDC) (Fig. 1C). Thioredoxin, which is known to form intramolecular disulfides, however did not demonstrate thermal stability change, indicating that the detailed energetics of the redox forms for each protein determines whether or not there is a stability effect. 38 of 112 identified responders in the GSH/GSSG ITRR data have previously been shown in proteome-wide studies to carry reactive cysteine residues, typically sensitive to redox-induced modifications (Supplementary Fig. 1A, Supplementary Data 1). In Supplementary Fig. 1A, several functional clusters of these proteins are shown. One of the clusters includes the SUMO-activating enzymes (SAE) that are known to contain catalytic active-site thiols [31], for which both subunits (SAE1, UBA2) give similar shaped responses. Such correlated responses are consistent with previous demonstrations that subunits of protein complexes can shift together due to a co-aggregation phenomenon [21,32]. In this case only UBA2 is known to have a reactive cysteine, thus it might drive the thermal stabilization.
Several proteins involved in histone modification or chromatin remodelling also displayed CETSA responses. These include protein arginine N-methyltransferase 1 (PRMT1) and chromobox protein homolog 3 (CBX3), which are known to have reactive cysteine residues, as well as Set 1/Ash 2 histone methyltransferase complex subunit (ASH2) and histone acetyltransferase type B (HAT1) (Supplementary Fig. 1A-C, Supplementary Data 1) as potential novel redox-sensitive proteins. We were particularly interested in the HAT1 protein, which was not identified in previous proteome-wide studies as having redox active cysteines. Nevertheless, in the crystal structure of human HAT1 (PDB 2P0W), Cys 101 is modified by an acetamide group, suggesting that this residue is activated (Supplementary Fig. 1D). It further proves the advantage of MS-CETSA approach in capturing redox-sensitive proteins.
Histone H3 variant H3.1 (HIST1H3B) shows CETSA response in the GSH/GSSG ITRR data and is known from focused studies to be glutathionylated [33]. A modelling study suggests that Cys 110 is a modified residue which, when glutathionylated, induces a conformational change in helix α2 of H3.1, thereby compromising nucleosomal stability [33]. Interestingly, histone H1 variants that do not contain any cysteines remain unchanged in the CETSA dataset (Supplementary Fig. 1E), illustrating the specialized role of cysteines in the sensing of glutathione pool fluctuation for regulation of protein activity.
A subset of shifting proteins in the GSH/GSSG ITRR data that were not included in the previous proteomics studies of proteins with reactive cysteines, are extracellular or endoplasmic reticulum proteins carrying structural disulfides (e.g. SPOCK2, F2, CFI, CTGF, IGFBP1, AMBP) (Supplementary Fig. 1A). At the most reducing condition, these disulfides appear to have been reduced, leading to a destabilization of these proteins, as compared to the most oxidized condition.
A prominent shift was seen for STK24/MST3 (Supplementary Fig. 1A), a STE20-like kinase which is an effector of redox stress and an inducer of apoptosis [34]. Although the primary basis for the regulation of STK24 was shown to be at the transcriptional level, the stabilization seen in the CETSA data suggests a parallel direct regulatory mechanism induced by the GSH/GSSG ratios. STK24 is unusual in that its metal preferences in the kinase reaction include redox-sensitive metals (Mn2+, Co2+) as cofactors [35,36], where the GSH/GSSG ratios could potentially affect the concentration/coordination/redox states of these metals.
To further confirm two of the hits from the GSH/GSSG MS-ITRR dataset (Fig. 2A), we performed semi-quantitative western blotting (Fig. 2B). Due to the limited sensitivity of western blotting method, a heating challenge at 55 °C (as compared to 52 °C in the MS-ITRR) was used to get more distinct responses. Treated with a GSH/GSSG ratio gradient similar as in the MS-CETSA experiment, both PRDX1 and HAT1 showed consistent thermal shift responses (Fig. 2B). The induction of a shift by GSH/GSSG ratio changes was also confirmed for purified recombinant HAT1 protein (Fig. 2B lower panel). A matrix subjected to GSSG treatment in the absence or presence of GSH and resolved under reducing or non-reducing condition supported the notion of HAT1 oxidation inducing dimerization (Fig. 2C) as previously suggested [37].
Fig. 2.

Confirmation of protein sensitivity to changed glutathione redox ratios. A, MS-CETSA signals of PRDX1 and HAT1 proteins in LG-ITRR experiment. B, Western blotting-based CETSA validation of thermal stabilization of PRDX1 and HAT1 proteins in response to oxidation. C, SDS-PAGE analysis of PRDX1 and HAT1 proteins. HepG2 whole cell lysates (left and right panel) and HAT1 recombinant proteins (middle panel) were separated on SDS-PAGE under reducing (with DTT) or non-reducing (without DTT) conditions followed by western blotting detection. Arrows indicate protein monomers. The upper bands indicate dimer and oligomer formation of PRDX1 and HAT1 proteins upon GSSG addition, and inhibition in the presence of GSH.
3.2. Hydrogen peroxide induced CETSA shifts
To further assess redox effects on the human proteome with MS-CETSA, we studied hydrogen peroxide (H2O2), a diffusible mild oxidant, which is known to induce oxidation of protein cysteinyl thiols and co-factors. H2O2 is generated during redox stress where high levels of H2O2 can be deleterious [38]. H2O2 can also serve as a signaling messenger, mediating various physiological processes including cell proliferation, differentiation and migration [39,40].
To identify proteins with sensitivity to H2O2, we first performed an isothermal-dose response (ITDR) experiment in cell lysates with 1 min exposure to different doses of H2O2 (0–50 mM) followed by MS-CETSA data collection as described above. The short incubation time was used to minimise downstream effects of proteins directly affected by H2O2. 4585 proteins passed our overall quality criteria out of 8632 identified proteins in total from both heating temperatures (37 °C and 52 °C). 24 proteins passed our hit selection criteria having significant ITDR shifts at 52 °C (Fig. 3A upper panel) while at the same time showing stable levels at 37 °C. H2O2 reacts slowly with cysteine and mildly with Fe-S in vitro [41], and many redox-regulated proteins react with H2O2 much slower (∼1-500 M−1 S−1) than the most reactive proteins such as peroxiredoxins (∼107 M−1 S−1) [42]; this might explain the relatively small number of hits in this dataset.
Fig. 3.

Protein stability changes induced by hydrogen peroxide. A, Heat map showing thermal shift profiles of target proteins from lysate H2O2 (LH)-ITDR experiment (cluster labelled with purple bar) and in-cell H2O2 (IH)-ITDR experiment (cluster labelled with blue bar). Each row represents one protein and each column is one concentration point of H2O2 treatment. B, Scatter plot visualizing proteins with significant thermal stability changes in LH- and IH-ITDR experiment (upper and lower panel, respectively). In both, proteins related to cell redox homeostasis are highlighted in red. In the LH-ITDR data, iron binding proteins are highlighted in orange and proteins from ubiquitination system in green, the rest of the target proteins in purple. In the IH-ITDR data, common hits between IH and LH-ITDR data are colored in purple, the remaining targets in blue. The background non-responsive proteins are colored in gray. C, Gene ontology analysis for proteins with thermal stability changes in IH-ITDR experiment representing the top 20 highly enriched gene ontology terms including nucleotide binding and oxidation reduction process. Size of nodes indicates percentage of proteins in each term and color indicates significance of enrichment. D, STRING analysis for IH-ITDR hits with stability changes. Filled colors for each protein node encodes different functional categories and CETSA signal is visualized above each node. Edge between protein nodes represents interactions retrieved from the STRING database, whose width indicates interaction confidence. Clusters of interested functional protein groups are circled in the following colors - Red: SUMO-activating proteins; orange: NAD(P)H-binding proteins; purple: tRNA synthetases; green: helicases; blue: kinases. E, Destabilization of a group of ATP/GTP binding proteins from IH-ITDR experiment. F, Decreased ATP levels upon H2O2 addition in a dose-responsive manner. Statistical significance was calculated with one-way ANOVA, multiple comparisons of lowest-dose sample versus the rest, *p < 0.05.
Of the 24 identified hit proteins, 9 have been shown in previous proteomics studies to have redox-sensitive cysteine residues (Supplementary Data 1). The two peroxiredoxins PRDX1 and PRDX5 are among the hits which responded early, consistent with previous reports [43]. Several iron-binding proteins are also part of the hit list including 1,2-dihydroxy-3-keto-5-methylthiopentene dioxygenase (ADI1), the redox-state dependent transcriptional coregulatory Pirin (PIR), as well as aconitase (ACO1), a rate-limiting enzyme in the tricarboxylic acid (TCA) cycle and a transcriptional regulator sensing cellular iron levels [44] (Fig. 3B upper panel). Previous studies have shown that H2O2 induces Fe release from the aconitase [4Fe-4S] cluster [45], which can explain the observed CETSA response. The formation of Fe2+-GSH complex has been implicated in chaperoning the labile cellular Fe2+ pool [46], and GSH depletion in the presence of H2O2 can therefore increase this iron pool [47]. As a consequence, the stability effects on iron binding proteins can therefore be mediated either by direct H2O2 oxidation modulating iron coordination/redox state, but also by H2O2-induced GSH depletion affecting cellular iron pools (Supplementary Fig. 2A).
Among the remaining hits in the H2O2 lysate experiment there are several proteins of the ubiquitination systems (USP24, USP5, SENP6, UBA3, NAE1) (Fig. 3B upper panel). USP24 and USP5 are ubiquitin erasers that cleave the isopeptide bond between ubiquitin and the target protein. Such a function is highly dependent on the catalytic cysteine, whose pKa value is lowered by surrounding His, Asp and Asn residues [48]. SENP6 belongs to SUMO proteases that contain catalytic cysteines. Although not detected in our dataset, another family member, SENP1, is known to be reversibly oxidized by H2O2, forming an intramolecular disulfide bond between Cys 603 (catalytic) and Cys 613 [49]. This suggests that a similar protective mechanism might be operative for SENP6. UBA3 and NAE1 form a complex to act as an E1 activating enzyme, and their profile similarities in our CETSA dataset (Supplementary Fig. 2B) support that they shift as a complex. A possible structural basis for the observed shift has been described by Walden et al. where NAE1's catalytic cysteine domain and UBA3's crossover loop undergo significant conformational changes upon NAE1-UBA3-NEDD8 complex formation during the E1 reaction cycle [50]. NEDD8 is unfortunately not detected in this dataset.
3.3. Hydrogen peroxide induced CETSA shifts in intact cells
CETSA has the unique capability to measure thermal shifts in intact cells and therefore detect in situ changes in protein interaction states, i.e. the extent to which a protein binds to physiological ligands, or structural effects on the protein induced by e.g. chemical modifications [23]. To study intracellular redox effects, we incubated HepG2 cells at different concentrations of H2O2 for 10 min, in order to detect early responses to redox changes. We collected MS-CETSA data as described above yielding 4163 proteins that fulfilled our overall quality control criteria, out of 8765 identified from two heating temperatures (37 °C and 52 °C). We observed the destabilization of many ribosomal proteins from this experiment (Supplementary Fig. 2C). In previous proteomics studies, elevated oxidation of ribosomal proteins by H2O2 show causal-dependent physiological effects on translational apparatus [51]. Cytosolic and mitochondrial ribosomal proteins displayed distinct thermal shift patterns suggesting differential sensitivity of the two translational machineries of the cell to H2O2. To facilitate the analysis of other proteins, these ribosomal proteins were omitted from the subsequent analyses of hits for the in-cell H2O2 data.
120 additional proteins passed our hit selection criteria having significant ITDR shifts at 52 °C, while showing stable levels at 37 °C (Fig. 3A lower panel). 7 of these were also seen in the lysate H2O2 data (PIR, OPLAH, ACO1, USP5, OTUB1, SENP6, CHORDC1). These include both proteins with redox sensitive cysteines and iron sensing/binding proteins. The two peroxiredoxins identified in the lysate H2O2 experiment (PRDX1 and PRDX5) exhibited tentative shifts but did not pass our selection criteria; instead PRDX3 and PRDX6 are selected as hits in the in-cell experiment (Fig. 3B lower panel). In total, 35 of the 120 in-cell H2O2 hits have in previous proteomics studies been shown to carry activated cysteine residues (Supplementary Data 1). Among them, the SUMO-activating enzyme subunits SAE1 and UBA2 also showed strong thermal stabilization in the mode of protein complex formation as aforementioned. The catalytic cysteines of UBA2 and another E2-conjugating enzyme UBC9 are known to form a disulfide bridge during oxidative stress thus inhibiting SUMO transfer to UBC9 [31]. Consistent with complex formation, or alternatively a coordinated reaction, we observed similar thermal response of UBC9 (Supplementary Fig. 2D). A pathway and gene ontology analysis and STRING analysis revealed the 120 hits to be distributed in a number of different pathways. Apart from redox process pathways (Fig. 3C), functional clusters involving nucleotide binding, which include a number of aminoacyl-tRNA synthetase and helicases (Fig. 3D) were found enriched.
The fact that we obtained more hits from in-cell H2O2 treatment than in the lysate experiment supports that many of these hits, in spite of the relatively short incubation time, could be due to indirect downstream effects of H2O2, induced by modifications of the primary target proteins. CETSA has recently been shown as an efficient means to monitor effects on proteins due to changes in the levels of physiological metabolites such as nucleotides [22]. In the in-cell H2O2 data, mitochondrial respiration Complex I subunits (NDUFS1, NUDFA2) displayed destabilization (Fig. 3D), potentially reflecting a distortion of Complex I upon H2O2 treatment. Both subunits have previously been shown to carry redox sensitive cysteines (Supplementary Data 1) and NDUFS1 is a core subunit of the peripheral arm of Complex I harbouring iron-sulfur clusters involved in electron transfer, suggesting potential mechanisms for the redox-induced CETSA shifts and regulation of Complex I activity.
We noticed that there was another responding subset encompassing ATP-binding proteins, in which most of them were destabilized (Fig. 3D and E). This is consistent with oxidative stress depleting intracellular ATP, likely as a consequence of inactivation of some key metabolic enzymes involved in ATP-generation [52]. We used a luciferase assay to measure ATP levels in the H2O2 treated samples, confirming ATP depletion in our HepG2 experiments (Fig. 3F). The destabilized ATP-binding proteins included several protein kinases (PANK4, STK4, CAMK2D, MAP4K5), helicases (DDX1, DDX6, DDX49, DHX38) and tRNA synthetases (DARS, SARS, TARS, AARS). In addition, several GTP-binding proteins (ARL2, SUCLG2, TUFM, ARF4, RAB13) were also destabilized (Fig. 3E), indicating a concomitant effect on GTP pools. As a reference experiment for the intracellular ATP effect, we analysed an unpublished ITDR MS-CETSA dataset available in our laboratory obtained from SNU398 liver cancer cells treated with oligomycin (0–100 μM), an inhibitor of mitochondrial F0F1-ATPase. Oligomycin is known to rapidly reduce ATP levels in the cell [53], and in this dataset we observed an ensemble of ATP binding proteins which were destabilized, including 5 proteins also being destabilized in the in-cell H2O2 datasets (PANK4, TUFM, KIF5B, OLA1, AARS) (Supplementary Fig. 2E). Several kinase family proteins (including DTYMK, PCK2, CMPK2, AKAP8 and GALK1) also showed similar shifts in the oligomycin and in-cell H2O2 experiments, although they didn't pass the strict hit selection criteria applied.
3.4. Time dependent CETSA shifts of sulfasalazine
With the above studies supporting that a multitude of redox effects on specific proteins can be monitored with CETSA, we proceeded with in-cell studies of sulfasalazine (SSA), a FDA-approved anti-rheumatic drug that is known to modulate ROS production [54]. Sepiapterin reductase (SPR) has been shown to be a direct target of SSA leading to the modulation of the biopterin pools with subsequent effects on redox processes e.g. leading to decreased H2O2 levels [55]. A recent study also supported the notion that SSA could block the xCT transporter, thereby reducing cysteine uptake, subsequently affecting de-novo glutathione synthesis decreasing the GSH/GSSG ratio [56].
In order to understand stability changes as well as protein expression changes in responding to SSA, an isothermal-time response (ITTR) experiment was performed (Fig. 4A). HepG2 cells were incubated with 250 μM SSA for varied time-courses (0–24 h), 4256 proteins passed our overall quality control criteria out of 9249 identified proteins in total from two heating temperatures (37 °C and 52 °C). Among the responding proteins, a large number of stabilized ribosomal proteins were observed (Supplementary Fig. 3A). A prominent thermal response starts from around 3–4 h for subunits of both the mitochondrial and cytosolic ribosomes, although the response is stronger for the latter.
Fig. 4.

Dose- and time-dependent thermal stability effects of Sulfasalazine. A, Schematic representation of the experimental workflow on Sulfasalazine (SSA). HepG2 intact cells were incubated for different time durations or with different SSA concentrations, washed and heated at 37 °C and 52 °C. Determination of soluble fractions were performed as described. Numbers of detected proteins and selected hits are summarized in table. B, Scatter plot visualizing proteins with significant thermal stability changes in the in-cell SSA-ITTR experiment. The known target SPR protein is highlighted in blue and a series of nucleotide-binding proteins are colored in black. Rest of the target proteins are highlighted in orange. The background non-responsive proteins are colored in gray. C, Stabilized MS-CETSA signals of SPR proteins from in-cell SSA-ITTR and SSA-ITDR experiments. D, Stabilization of a group of ATP/GTP binding proteins from SSA-ITTR experiment. E, Induced ATP levels upon SSA addition in a time-dependent manner. Statistical significance was calculated with one-way ANOVA, multiple comparisons of shortest time-point sample versus the rest, *p < 0.05. F, Gene ontology analysis for proteins with thermal stability changes in SSA-ITTR experiment demonstrating significantly enriched gene ontology terms (FDR<0.05) including nucleotide binding, spliceosome and glutamate-cysteine ligase complex. Size of nodes indicates percentage of proteins in each term and color indicates enrichment significance. G, Comparison of SSA-ITTR protein expression profiles to Menadione (Men) induced-RNA abundance changes from Deferme et al. [62]. H, Gene ontology analysis for SSA and Men overlapping hits showing enrichment of genes (FDR<0.05) involved in cell redox homeostasis and oxygen sensing. Size of nodes indicates percentage of proteins in each term and color indicates enrichment significance.
Excluding ribosomal proteins, 101 additional proteins passed our hit selection criteria, 21 of these were seen in the in-cell H2O2 ITDR experiment and 4 in the lysate glutathione ITRR experiment (Fig. 4B, Supplementary Fig. 3B). 18 additional proteins have previously been identified as having reactive cysteine residues in proteomics studies (Supplementary Data 1). SPR, the known direct target of SSA, was also identified as a strong and early responder to SSA treatment (Fig. 4C left panel). An ITDR experiment supports that SPR is engaged at low nanomolar concentrations of SSA after 4 h treatment (Fig. 4C right panel).
Interestingly, most of the proteins overlapping between the SSA ITTR and the in-cell ITDR H2O2 dataset shifted in opposing directions (Figs. 3D and 4D). In addition, these proteins are GTP- or ATP-binding proteins, or in protein complexes. Although some of these shifts could reflect direct redox modulation of reactive cysteine residues (e.g. ALDH18A1, known to have a redox active cysteine [57,58]), a likely explanation for most of these shifts is the modulation of purine triphosphate nucleotide levels in the SSA experiment in the opposite direction compared to the H2O2 experiment. Indeed, time-dependent measurement of ATP levels in the SSA experiment show a drastic increase in ATP in about 4 h which is the time point where the opposing SSA shifts start to build up (Fig. 4E). The fact that SSA could negatively regulate H2O2 formation [[59], [60], [61]] indicates that the opposing changes in ATP levels in the SSA and H2O2 dataset is related to a similar H2O2 regulated cellular mechanism in the two cases.
SSA is also known to decrease the GSH/GSSG ratio as well as total GSH levels in cells (Supplementary Fig. 3C), the latter presumably due to the inhibition of the xCT transporter. This is consistent with the shift of PRMT1 in the SSA data (Supplementary Fig. 3D), and is also the top hit in the lysate glutathione experiment. However, the shift of the GSH/GSSG ratio in the SSA experiment is relatively small (i.e. 30%) for all time points except 24 h (Supplementary Fig. 3C), which might explain the small numbers of overlapping shifts with the GSH/GSSG ITRR data.
Gene ontology analysis was performed to elucidate biological processes enriched in the SSA MS-CETSA data where, in addition to processes involving typical nucleotide binding proteins (ARL2, SUCLG2, RHEB, TUFM, ARF4, SAR1A, ABCF1, PANK4, RAB13, ALDH18A1, DHX38, RTCA), poly(A) RNA binding and RNA splicing process were also enriched (Fig. 4F, Supplementary Fig. 3E). Enriched pathways also reflected the response of GCL complex (GCLC, GCLM) regulating cysteine and glutathione metabolic processes. Both subunits posted similar thermal stabilization, which could be due to GCLC binding to ATP, one of its co-substrates, but also due to direct modifications of known redox active cysteine residues (Supplementary Data 1).
Protein expression levels at 37 °C in the SSA MS-CETSA data revealed abundance changes of 830 proteins including 67 up-regulated ribosomal proteins. We conducted a comparison of our dataset with a previously published microarray RNA transcriptomics dataset obtained from HepG2 cells treated with menadione (Men), known to regulate ROS in a partly similar mechanism as SSA [[62], [63], [64], [65]]. Out of the 830 proteins with changed levels in the SSA data, 224 were found in both datasets, and 171 of these showed similar profiles (Fig. 4G). NRF2 is the key regulator of the transcriptional response to oxidative stress. Several well-characterized NRF2 targets were strongly induced, including GCLM, GSR, SLC7A11/xCT and SQSTM1 which boost the synthesis and regeneration of reducing moieties [66], as well as ASNS which responses to cell stress [67] (Supplementary Fig. 3F). This evidence reflected ROS induction, cellular antioxidant defence activation and compensation upon compound treatment.
Moreover, a number of the hypoxia-inducible factor HIF-α′s downstream targets (CA9, BNIP3, PDK1, PDK3, ATM, CTGF, TGFBR3, PLOD1, PLOD2, CDKN1A, CX3CL1) were found to be down-regulated in this dataset (Supplementary Fig. 3F), and relevant gene ontology terms including response to oxygen level and HIF-α signaling pathways were significantly enriched (Fig. 4H). These HIF-α target proteins are known to control cell proliferation, apoptosis, glycolysis, DNA damage response and other important biological processes, demonstrating diverse physiological outcomes upon SSA treatment. An inhibitory effect on HIF-α and downstream targets by SSA has previously been observed [[68], [69], [70]]; in addition, H2O2 is known to stabilize HIF-α through modulating the oxygen sensor prolyl hydroxylase (PHD) by oxidizing iron, one of its essential co-factors [[71], [72], [73]]. Interestingly, this oxygen sensor prolyl hydroxylase EGLN1/PHD2, which controls HIF-α hydroxylation and consequent degradation [[74], [75], [76], [77]], is stabilized in the SSA MS-CETSA data (Supplemenraty Fig. 3D). This might reflect an activation of EGLN1 by redox modulation leading to increased HIF-α degradation and concomitant downregulation of HIF-α target proteins. A mechanism for such regulation was recently proposed involving the inhibition of EGLN1/PHD2 activity by H2O2 induced dimerization [78]. If this is the explanation for the CETSA shift, this suggests that EGLN1/PHD2 dimerization leads to a destabilization of the protein, which is in contrast to HAT1 discussed above, where dimerization leads to stabilization.
3.5. Structural implications of CETSA responses of redox-sensitive proteins
Next, we conducted a closer analysis, using InterPro [79] of the entire collection of identified hits for the abovementioned redox CETSA experiments to assess clustering in specific structural and functional domains (Fig. 5A). In concert with GO analysis, there are 14 hits belonging to thioredoxin-like superfamily that includes thioredoxin domain-containing proteins, peroxiredoxins, protein disulfide isomerases and glutaredoxins. These hits contain at least one or two redox-sensitive cysteines enabling their oxidoreductase activity. Evidences have been shown that cysteine thiol oxidation of these enzymes results in significant conformational changes, which is reflected in the modulation of their stability. For instance, the peroxidatic and resolving cysteines in PRDX5 are more than 10 Å apart and are oriented in opposite directions, thus substantial structural rearrangements of both the loop-helix motif and the C-terminal arm are needed to form an intramolecular disulfide bond [80,81].
Fig. 5.

Structural determinants of cysteine-mediated redox sensing. A, Overrepresented InterPro domains in thermal shifted proteins. The domains appearing in at least three target proteins are included for visualization in the graph, and proteins are clustered based on the pattern of containing domains. B, Histogram (left) and violin plot (right) of the composition percentage of cysteines in Hit and Base proteins. Statistical significance was calculated with two sample t-tests. C, Boxplot of pKa value (left) and solvent accessibility area (right) of cysteines in Hit and Base protein groups. Statistical significance was calculated with two sample t-tests.
Outside the thioredoxin family, 15 hits were carrying diverse sub-classes of zinc finger motif inclusive of C2H2, RING and PHD types, in which multiple cysteine and histidine residues assemble in a tetrahedral geometry coordinating the Zn2+ ions [82]. Oxidative modification of cysteine thiols are known to cause Zn2+ release and protein structural alterations [83]. However, it is noteworthy that not every zinc finger protein showed significant thermal shifts upon oxidative stimuli in our dataset. As an example, ZFYVE9 and ZFYVE16 remained stable (Supplementary Fig. 4A), likely reflecting a distinct redox sensitivity of some FYVE zinc finger motifs (in these cases the signature sequences: N-terminal WXXD motif, R (R/K)HHCR patch and C-terminal RVC). Such selective sensitivity of zinc finger proteins has been reported previously where C2H2 zinc finger proteins were shown to be particularly sensitive to arsenic-induced oxidation [84].
12 additional hits contained protein kinase-like domains. All of these except one are from the glutathione ITRR (3 hits) or in-cell H2O2 ITDR dataset (8 hits). Although kinase hits in the H2O2 ITDR data could be due to changes in ATP concentration, the oligomycin data support that at least 5 of the H2O2 ITDR hits are not highly sensitive to ATP and might instead be due to structural redox modulations. We performed a prediction of redox-sensitive cysteines presented in these hits by evaluating their pKa values and residue surface accessibility using Surface Racer v5.0 and PDB2PQR v1.7, which are incorporated into the Cpipe platform [[25], [26], [27], [28], [25],42]. Interestingly, most of the predicted sensitive-cysteines are located nearby or within the catalytic core (Supplementary Fig. 4B) indicating that cysteine oxidation might affect the catalytic centre of these kinases; while some of them are located nearby regulatory loops. Another group of hits comprises GTPase/GTP-binding protein family members, including the Ras-related proteins (RAB32, RAB3B, RAB13), harbouring known redox sensitive-cysteine in a NKCD motif (Supplementary Fig. 4C) [85]. As mentioned above, some CETSA shifts in GTPases are likely due to fluctuations of the intracellular GTP concentrations. Additional structural domains that cluster in our data are shown in Fig. 5A.
To shed light on whether our hit selection enriched cysteine containing proteins, a calculation of amino acid composition was conducted for 314 proteins with major shifts (hit) and compared to a randomly selected counterpart of 314 proteins with minimal shifts (base). Overall cysteine composition in the hit protein group was significantly higher than that in the base group (Fig. 5B), while we did not observe significant difference on the composition of other amino acids (Supplementary Data 2). As mentioned earlier, there are two critical parameters that determine cysteine reactivity: exposure to solvent and protonation status, that is, thiols with low pKa values and higher residue surface accessibility are more reactive at physiological pH [28,42]. We used the Cpipe server to estimate pKa values and solvent accessibility for 70 hit proteins and 35 base proteins where structural information was available. Based on these theoretical estimates the hit proteins in our dataset exhibited significantly lower pKa value and larger solvent exposure compared to base proteins (Fig. 5C), suggesting that the hit proteins are enriched in potential functional or activated cysteine sites.
4. Discussion
CETSA has previously been used to effectively monitor the engagement of drugs with target proteins, as well as interactions of physiological ligands with proteins in lysates and in the cellular context [23]. In the present work, we have evaluated the potential of CETSA in detecting structural modulations of proteins due to typical redox modifications and applied this concept to study redox processes in intact cells. Using lysate experiments, we detected 132 proteins, which were sensitive to the changes of H2O2 concentrations and/or GSH/GSSG ratios. For H2O2 lysate experiment, it is likely that most, if not all, of the identified 24 protein shifts are due to direct H2O2-induced structural modulations, either as individual proteins or as part of protein complexes. These responders include thioredoxin and peroxiredoxin family members but also proteins from diverse families carrying iron sites or activated catalytic cysteine residues (Fig. 5A). In the GSH/GSSG experiment, nearly 45% of the responding proteins are known from previous studies to carry activated cysteine residues, structural disulfide bonds or metal binding sites which can be sensitive to GSH-adduction (Supplementary Data 1). It is likely that a significant number of the remaining hit proteins, previously not described to contain redox-sensitive cysteine residues, are either directly modified or in complexes which are sensitive to the GSH/GSSG ratio, but the extent of this remains unknown.
These novel redox-sensitive proteins likely include proteins carrying redox metals that do not have cysteine coordination, such as STK24, where free metal concentration and coordination/redox states can be modulated by GSH. Notably, this group of proteins have not been detected in the proteome-wide affinity enrichment studies discussed above. It should be noted that previous studies identified potential accessible and activated cysteine residues, and we identified redox sensitive cysteines, where redox modulations induce stability changes in the protein, it is not guaranteed that these cysteine residues are in functional sites. However, evolution often positions functional cysteine residues in pockets so they can be surrounded with hydrogen bonding residues modifying its pKa, as well as binding their substrates, in cases of enzymes. Redox modulations of pocket-localized cysteine residues could be expected to induce structural changes that yield stability changes, supporting that many of our CETSA hits might indicate functional modulations of these proteins. Alternatively, exposed cysteines could be involved in dimerization where the redox sensitive form is generated at a transient dimer interface [86].
In the GSH/GSSG ITRR analysis, as well as in the other datasets, we found a number of epigenetic regulators as well as chromatin remodelling proteins to be modified, consistent with emerging interconnection between cellular redox homeostasis and epigenetic regulations [87]. We also found frequent redox modulations of proteins in SUMOylation and ubiquitination systems, which are known to use catalytic cysteines to fulfil their function, and are therefore potential ROS-sensitive targets [88]. In our in-cell data, we can follow processes including the activation of different complexes in these pathways. Both epigenetic control and protein degradation are highly complex processes involving many different proteins and protein complexes, which are accessible for redox modulations in different cellular states and processes. Our data demonstrates that CETSA now provides a novel means to interactively assess which subsets of proteins are affected in these complicated processes in cells, and potentially tissues.
The in-cell data with SSA and H2O2 also display some of the direct redox interactions seen in the lysate experiment, including well-characterized proteins in cellular redox control, i.e. thioredoxin and peroxiredoxin families and iron-containing redox sensors. We also see a potential shift in EGLN1, which might explain the down regulation of hypoxia-induced genes by SSA. However, the majority of shifts in these experiments are presumably due to downstream effects of primary redox interactions. CETSA has recently been shown to be a distinct method in studying interactions with nucleotide binding proteins. Interestingly, we see clear concerted CETSA shifts of ensembles of ATP (and some GTP) binding proteins in the three in-cell datasets (SSA, H2O2 and oligomycin). Shifts in the H2O2 and oligomycin datasets are in the same direction and predominantly destabilizations, while shifts in the SSA data are predominantly stabilizations. This is consistent with the opposing changes of ATP concentrations in these datasets. In spite of the fact that ATP level changes are induced in different ways, and two different liver cell lines are used, there is still a set of proteins that give robust concerted ATP responses in all three datasets (e.g. PANK4, OLI1, PA2G4, KIF5B, GALK1, NARS). We propose that concerted shifts in this ensemble can now be used as a general indicator of ATP concentration changes in other liver-specific CETSA experiments. The exact mechanism leading to changes in ATP concentrations in the SSA and H2O2 experiments however remains uncertain. Previous work has proposed redox-induced inactivation of GAPDH as a possible mechanism [52], but our data also paves the way for alternative mechanism directly affecting mitochondrial respiration. The observed shifts in Complex I (NDUFS1, NDUFA2) in the H2O2 in-cell data might support a direct modulation of the iron-sulfur containing peripheral arm of Complex I by H2O2, where inhibition of electron transport would attenuate the generation of a proton gradient and thereby ATP production by mitochondrial ATP synthase.
Prominent shifts of both mitochondrial and cytosolic ribosomal proteins are also seen in the in-cell datasets, which likely reflect specific activity modulations of protein translation. This is consistent with previous studies supporting redox control of translation. However, we cannot distinguish whether the CETSA shifts are due to direct redox modulations of ribosomal proteins, or by downstream effects from other interactions, such as changes in ATP and GTP concentrations, which are also known to control translational activity [89,90].
In summary, the present work confirms that structural changes due to redox modifications can be detected for many proteins using CETSA. The work also provides a first draft of proteins that report on such changes, i.e., the CETSA redoxome. Redox modulations of proteins are notoriously difficult to study directly in intact cells, but CETSA now provides a new avenue for direct and interactive studies of redox process in different cellular systems. Some of the shifts elucidated in this work concerns activity modulation of key redox control proteins, and these shifts can now be directly applied as biomarkers in studies of ROS or other redox effects. As both direct interactions and downstream effects are seen in the in-cell data, further work will be needed to dissect the origin of these effects on many individual proteins. However, we envisage that with accumulating CETSA data on redox studies, the association between redox modifications and downstream effects on many responding proteins will emerge, similar to some of the proteins in the present study. As MS-CETSA is applicable to studies of a wide range of samples including tissues, CETSA now also offers a novel way to provide detailed insights into operative aspects of ROS and other redox processes in specific diseases, as well as during therapy.
Author contributions
Conceptualization, W.S. and P.N.; Methodology, P.N., W.S., L.Y.D., Y.T.L., R.M.S., and N.P.; Investigation, W.S., H.Y., F.L., and J.L.T.; Formal Analysis, W.S., L.Y.D., and P.N.; Validation, W.S., B.P., T.Z. and M.W.C.; Resources, P.N.; Writing-Original Draft, W.S. and P.N.; Writing-Review & Editing, W.S., L.Y.D., N.P., and P.N.; Supervision, N.P., D.G.T., and P.N.
Author declaration
P.N. is the inventor of patents related to the CETSA method and the co-founder of Pelago Bioscience AB. All the other authors declare no competing interests.
Acknowledgements
We thank Drs. Chris Soon Heng Tan, Kelly Hew, Loo Chien Wang, Ying Yu Liang and Lars Abrahmsen for discussions, and the Protein Production Platform, Nanyang Technological University for recombinant protein preparation. P.N. acknowledges a startup grant from Nanyang Technological University and grants from the Swedish Research Council, the Swedish Cancer Society, Radiumhemmets funds and the Knut and Alice Wallenberg foundation. This research is also supported partly by the Singapore Ministry of Health’s National Medical Research Council, under its MOHIAFCAT2/004/2015 to P.N. and R.M.S, A*STAR BMRC YIG2015 grant to R.M.S.
This research is also supported by the Singapore Ministry of Health's National Medical Research Council under its Singapore Translational Research (STaR) Investigator Award (to D.G.T.), and by the National Research Foundation Singapore and the Singapore Ministry of Education under its Research Centres of Excellence initiative.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2019.101168.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
Confirmation of protein stability changes induced by a series of glutathione redox ratios. Related to Fig. 1, Fig. 2. A, STRING analysis for LG-ITRR hits with stability changes. CETSA signal is visualized as a bar plot above each node. Edge between protein nodes represents protein-protein interactions retrieved from the STRING database, whose width indicates interaction confidence. Protein node with purple label stands for known redox-sensitive proteins from publications. Clusters of functional protein groups that are discussed in text are circled in colors as follows- Red: SUMO-activating proteins; purple: epigenetic regulators; blue: extracellular or endoplasmic reticulum proteins carrying structural disulfides. B, MS-CETSA signals of epigenetic regulating proteins PRMT1, ASH2L and CBX3 proteins from LG-ITRR experiment. C, Western blot validation of thermal stabilization of PRMT1, ASH2L and CBX3 proteins. D, Structure of human HAT1 (PDB accession 2P0W) (gray) showing the position of predicted redox-sensitive cysteines C27 (yellow spheres), C101 (blue spheres) and C168 (orange spheres). E, Distinct thermal stability changes of histone variants in the LG-ITRR experiment. Yellow and green curves (left) represent the ITRR response of cysteine-bearing histone H3 variant while gray and brown curves for the ITRR response of non-cysteine containing histone H1 variants.
CETSA shifts related to protein complex formation and metabolite concentration changes upon H2O2 exposure. Related to Fig. 3. A, Decreased GSH/GSSG ratios and GSH amounts upon H2O2 treatment in HepG2 cells. Statistical significance was calculated with two sample t-tests, *p < 0.05. B, Similar ITDR-CETSA responses for subunits of the E1 activating enzyme UBA3-NAE1 functional complex. C, Abundance (37 °C) and stability (52 °C) profiles of all the detected mitochondrial and cytosolic ribosomal proteins passed QC from IH-ITDR experiment. Names of the presented ribosomal protein subunits are listed in table. D, Similar ITDR-CETSA responses among subunits of the E1 activating enzymes and E2-conjugating enzyme UBA2-SAE1-UBC9 transient functional complex, together with a schematic of the working mechanism, modified from Bossis et al. [31]. E, STRING analysis for oligomycin-ITDR hits with stability changes. SNU398 cells were incubated with different oligomycin concentrations, and heated at 37 °C and 52 °C. Filled colors for each protein node encode different functional categories and 52 °C CETSA signal is visualized above each node. Edge between protein nodes represents interactions retrieved from the STRING database, whose width indicates interaction confidence. Protein node with blue label stands for common hits also identified in IH-ITDR experiment. ATP-binding proteins demonstrated significant and consistent destabilization.
Overview and functional clustering of proteins with abundance and stability changes in SSA-ITTR experiment. Related to Fig. 4. A, Abundance (37 °C) and stability (52 °C) profiles of all the detected mitochondrial and cytosolic ribosomal proteins passed QC from SSA-ITTR experiment. Names of the presented ribosomal protein subunits are listed in table. B, Venn diagram showing determined hits from four treatments (LG-ITRR, LH-ITDR, IH-ITDR, SSA-ITTR experiments) in this study. All the 316 proteins (314 proteins without distinguishing 2 protein isoforms) are presented in the graph. C, GSH/GSSG ratios and GSH amounts decrease upon SSA treatment in a time-dependent manner. Statistical significance was calculated with one-way ANOVA, multiple comparison of shortest time-point sample versus the rest, *p < 0.05. D, Confirmation of thermal stabilization of PRMT1, one common hit between LG-ITRR and SSA-ITTR experiments. Left, western blotting validating thermal stabilization of PRMT1 proteins. Middle, stabilized MS-CETSA signals of PRMT1 from SSA-ITTR experiment. Right, stabilized MS-CETSA signals of EGLN1 from SSA-ITTR experiment. E, STRING analysis for SSA-ITTR hits with thermal stability changes at 52 °C. Filled color for each protein node encodes different functional category and CETSA signal is visualized above each node. Edges between protein nodes represent interactions retrieved from the STRING database, whose width indicates interaction confidence. Protein node with blue label or green label stands for common hits also identified in IH-ITDR or LG-ITRR experiments, respectively. F, STRING analysis for SAA-ITTR target proteins with abundance changes. Filled colors for each protein node encode different functional categories and protein level profiles at 37 °C is visualized above each node. Edge between protein nodes represents interactions retrieved from the STRING database, whose width indicates interaction confidence. Several NRF2 targets are clustered in the purple circle.
Structural determinants of cysteine-mediated redox sensing. Related to Fig. 5. A, Distinct thermal stability responses of zinc finger protein subsets. FYVE domain-containing zinc finger proteins displayed minimal CETSA shifts in our datasets. B, Superimposed structure of several kinases and predicted redox-sensitive cysteines. Structures of kinase domains for CDK16 (PDB accession 3MTL), SRC (4MXO), STK4 (3COM), CAMK2D (6AYW), SRPK2 (5MYV), STK24 (3CKW) and GSK3B (1I09) are superimposed, and the positions of predicted redox-sensitive cysteines are shown in spheres. C, Cysteine containing-NKCD sequence motif in RAB proteins.
References
- 1.Cross C.E. Oxygen radicals and human disease. Ann. Intern. Med. 1987;107(4):526–545. doi: 10.7326/0003-4819-107-4-526. [DOI] [PubMed] [Google Scholar]
- 2.Berlett B.S., Stadtman E.R. Protein oxidation in aging, disease, and oxidative stress. J. Biol. Chem. 1997;272(33):20313–20316. doi: 10.1074/jbc.272.33.20313. [DOI] [PubMed] [Google Scholar]
- 3.Cairns R.A., Harris I.S., Mak T.W. Regulation of cancer cell metabolism. Nat. Rev. Cancer. 2011;11(2):85–95. doi: 10.1038/nrc2981. [DOI] [PubMed] [Google Scholar]
- 4.Trachootham D., Alexandre J., Huang P. Targeting cancer cells by ROS-mediated mechanisms: a radical therapeutic approach? Nat. Rev. Drug Discov. 2009;8(7):579–591. doi: 10.1038/nrd2803. [DOI] [PubMed] [Google Scholar]
- 5.Gorrini C., Harris I.S., Mak T.W. Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 2013;12(12):931–947. doi: 10.1038/nrd4002. [DOI] [PubMed] [Google Scholar]
- 6.Raza M.H. ROS-modulated therapeutic approaches in cancer treatment. J. Cancer Res. Clin. Oncol. 2017;143(9):1789–1809. doi: 10.1007/s00432-017-2464-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Krysko D.V. Immunogenic cell death and DAMPs in cancer therapy. Nat. Rev. Cancer. 2012;12(12):860–875. doi: 10.1038/nrc3380. [DOI] [PubMed] [Google Scholar]
- 8.Dalle-Donne I. Protein carbonylation in human diseases. Trends Mol. Med. 2003;9(4):169–176. doi: 10.1016/s1471-4914(03)00031-5. [DOI] [PubMed] [Google Scholar]
- 9.Valko M. Redox- and non-redox-metal-induced formation of free radicals and their role in human disease. Arch. Toxicol. 2016;90(1):1–37. doi: 10.1007/s00204-015-1579-5. [DOI] [PubMed] [Google Scholar]
- 10.Green J., Paget M.S. Bacterial redox sensors. Nat. Rev. Microbiol. 2004;2(12):954–966. doi: 10.1038/nrmicro1022. [DOI] [PubMed] [Google Scholar]
- 11.Leichert L.I. Quantifying changes in the thiol redox proteome upon oxidative stress in vivo. Proc. Natl. Acad. Sci. U. S. A. 2008;105(24):8197–8202. doi: 10.1073/pnas.0707723105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Murray C.I. Identification and quantification of S-nitrosylation by cysteine reactive tandem mass tag switch assay. Mol. Cell. Proteomics. 2012;11(2) doi: 10.1074/mcp.M111.013441. M111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tambor V. CysTRAQ - a combination of iTRAQ and enrichment of cysteinyl peptides for uncovering and quantifying hidden proteomes. J. Proteom. 2012;75(3):857–867. doi: 10.1016/j.jprot.2011.09.027. [DOI] [PubMed] [Google Scholar]
- 14.Yang J., Carroll K.S., Liebler D.C. The expanding landscape of the thiol redox proteome. Mol. Cell. Proteomics. 2016;15(1):1–11. doi: 10.1074/mcp.O115.056051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Lennicke C. Redox proteomics: methods for the identification and enrichment of redox-modified proteins and their applications. Proteomics. 2016;16(2):197–213. doi: 10.1002/pmic.201500268. [DOI] [PubMed] [Google Scholar]
- 16.Chio I.I.C., Tuveson D.A. ROS in cancer: the burning question. Trends Mol. Med. 2017;23(5):411–429. doi: 10.1016/j.molmed.2017.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martinez Molina D. Monitoring drug target engagement in cells and tissues using the cellular thermal shift assay. Science. 2013;341(6141):84–87. doi: 10.1126/science.1233606. [DOI] [PubMed] [Google Scholar]
- 18.Martinez Molina D., Nordlund P. The cellular thermal shift assay: a novel biophysical assay for in situ drug target engagement and mechanistic biomarker studies. Annu. Rev. Pharmacol. Toxicol. 2016;56:141–161. doi: 10.1146/annurev-pharmtox-010715-103715. [DOI] [PubMed] [Google Scholar]
- 19.Savitski M.M. Tracking cancer drugs in living cells by thermal profiling of the proteome. Science. 2014;346(6205) doi: 10.1126/science.1255784. [DOI] [PubMed] [Google Scholar]
- 20.Becher I. Pervasive protein thermal stability variation during the cell cycle. Cell. 2018;173(6):1495–1507. doi: 10.1016/j.cell.2018.03.053. e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dai L. Modulation of protein-interaction states through the cell cycle. Cell. 2018;173(6):1481–1494. doi: 10.1016/j.cell.2018.03.065. e13. [DOI] [PubMed] [Google Scholar]
- 22.Lim Y.T. An efficient proteome-wide strategy for discovery and characterization of cellular nucleotide-protein interactions. PLoS One. 2018;13(12) doi: 10.1371/journal.pone.0208273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dai L. Horizontal cell biology: monitoring global changes of protein interaction states with the proteome-wide cellular thermal shift assay (CETSA) Annu. Rev. Biochem. 2019 doi: 10.1146/annurev-biochem-062917-012837. In press. [DOI] [PubMed] [Google Scholar]
- 24.Drake W.R. New use for CETSA: monitoring innate immune receptor stability via post-translational modification by OGT. J. Bioenerg. Biomembr. 2018;50(3):231–240. doi: 10.1007/s10863-018-9754-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Soylu I., Marino S.M. Cpipe: a comprehensive computational platform for sequence and structure-based analyses of Cysteine residues. Bioinformatics. 2017;33(15):2395–2396. doi: 10.1093/bioinformatics/btx181. [DOI] [PubMed] [Google Scholar]
- 26.Tsodikov O.V., Record M.T., Jr., Sergeev Y.V. Novel computer program for fast exact calculation of accessible and molecular surface areas and average surface curvature. J. Comput. Chem. 2002;23(6):600–609. doi: 10.1002/jcc.10061. [DOI] [PubMed] [Google Scholar]
- 27.Dolinsky T.J. PDB2PQR: an automated pipeline for the setup of Poisson-Boltzmann electrostatics calculations. Nucleic Acids Res. 2004;32:W665–W667. doi: 10.1093/nar/gkh381. (Web Server issue) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sanchez R. Prediction of reversibly oxidized protein cysteine thiols using protein structure properties. Protein Sci. 2008;17(3):473–481. doi: 10.1110/ps.073252408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gilbert H.F. Thiol/disulfide exchange equilibria and disulfide bond stability. Methods Enzymol. 1995;251:8–28. doi: 10.1016/0076-6879(95)51107-5. [DOI] [PubMed] [Google Scholar]
- 30.Franken H. Thermal proteome profiling for unbiased identification of direct and indirect drug targets using multiplexed quantitative mass spectrometry. Nat. Protoc. 2015;10(10):1567–1593. doi: 10.1038/nprot.2015.101. [DOI] [PubMed] [Google Scholar]
- 31.Bossis G., Melchior F. Regulation of SUMOylation by reversible oxidation of SUMO conjugating enzymes. Mol. Cell. 2006;21(3):349–357. doi: 10.1016/j.molcel.2005.12.019. [DOI] [PubMed] [Google Scholar]
- 32.Tan C.S.H. Thermal proximity coaggregation for system-wide profiling of protein complex dynamics in cells. Science. 2018;359(6380):1170–1177. doi: 10.1126/science.aan0346. [DOI] [PubMed] [Google Scholar]
- 33.Garcia-Gimenez J.L. Histone h3 glutathionylation in proliferating mammalian cells destabilizes nucleosomal structure. Antioxid. Redox. Signal. 2013;19(12):1305–1320. doi: 10.1089/ars.2012.5021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen C.B. Mammalian sterile 20-like kinase 3 (MST3) mediates oxidative-stress-induced cell death by modulating JNK activation. Biosci. Rep. 2009;29(6):405–415. doi: 10.1042/BSR20090096. [DOI] [PubMed] [Google Scholar]
- 35.Schinkmann K., Blenis J. Cloning and characterization of a human STE20-like protein kinase with unusual cofactor requirements. J. Biol. Chem. 1997;272(45):28695–28703. doi: 10.1074/jbc.272.45.28695. [DOI] [PubMed] [Google Scholar]
- 36.Lu T.J. Zinc ion acts as a cofactor for serine/threonine kinase MST3 and has a distinct role in autophosphorylation of MST3. J. Inorg. Biochem. 2005;99(6):1306–1313. doi: 10.1016/j.jinorgbio.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 37.Cumming R.C. Protein disulfide bond formation in the cytoplasm during oxidative stress. J. Biol. Chem. 2004;279(21):21749–21758. doi: 10.1074/jbc.M312267200. [DOI] [PubMed] [Google Scholar]
- 38.D'Autreaux B., Toledano M.B. ROS as signalling molecules: mechanisms that generate specificity in ROS homeostasis. Nat. Rev. Mol. Cell Biol. 2007;8(10):813–824. doi: 10.1038/nrm2256. [DOI] [PubMed] [Google Scholar]
- 39.Sundaresan M. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science. 1995;270(5234):296–299. doi: 10.1126/science.270.5234.296. [DOI] [PubMed] [Google Scholar]
- 40.Rhee S.G. Hydrogen peroxide: a key messenger that modulates protein phosphorylation through cysteine oxidation. Sci. STKE. 2000;2000(53) doi: 10.1126/stke.2000.53.pe1. p. pe1. [DOI] [PubMed] [Google Scholar]
- 41.Winterbourn C.C., Metodiewa D. Reactivity of biologically important thiol compounds with superoxide and hydrogen peroxide. Free Radic. Biol. Med. 1999;27(3–4):322–328. doi: 10.1016/s0891-5849(99)00051-9. [DOI] [PubMed] [Google Scholar]
- 42.Winterbourn C.C., Hampton M.B. Thiol chemistry and specificity in redox signaling. Free Radic. Biol. Med. 2008;45(5):549–561. doi: 10.1016/j.freeradbiomed.2008.05.004. [DOI] [PubMed] [Google Scholar]
- 43.Rhee S.G. Cell signaling. H2O2, a necessary evil for cell signaling. Science. 2006;312(5782):1882–1883. doi: 10.1126/science.1130481. [DOI] [PubMed] [Google Scholar]
- 44.Chen X.J. Aconitase couples metabolic regulation to mitochondrial DNA maintenance. Science. 2005;307(5710):714–717. doi: 10.1126/science.1106391. [DOI] [PubMed] [Google Scholar]
- 45.Bulteau A.L., Ikeda-Saito M., Szweda L.I. Redox-dependent modulation of aconitase activity in intact mitochondria. Biochemistry. 2003;42(50):14846–14855. doi: 10.1021/bi0353979. [DOI] [PubMed] [Google Scholar]
- 46.Hider R.C., Kong X.L. Glutathione: a key component of the cytoplasmic labile iron pool. Biometals. 2011;24(6):1179–1187. doi: 10.1007/s10534-011-9476-8. [DOI] [PubMed] [Google Scholar]
- 47.Kaur D. Glutathione depletion in immortalized midbrain-derived dopaminergic neurons results in increases in the labile iron pool: implications for Parkinson's disease. Free Radic. Biol. Med. 2009;46(5):593–598. doi: 10.1016/j.freeradbiomed.2008.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Komander D. Mechanism, specificity and structure of the deubiquitinases. Subcell. Biochem. 2010;54:69–87. doi: 10.1007/978-1-4419-6676-6_6. [DOI] [PubMed] [Google Scholar]
- 49.Xu Z. Molecular basis of the redox regulation of SUMO proteases: a protective mechanism of intermolecular disulfide linkage against irreversible sulfhydryl oxidation. FASEB J. 2008;22(1):127–137. doi: 10.1096/fj.06-7871com. [DOI] [PubMed] [Google Scholar]
- 50.Walden H. The structure of the APPBP1-UBA3-NEDD8-ATP complex reveals the basis for selective ubiquitin-like protein activation by an E1. Mol. Cell. 2003;12(6):1427–1437. doi: 10.1016/s1097-2765(03)00452-0. [DOI] [PubMed] [Google Scholar]
- 51.Topf U. Quantitative proteomics identifies redox switches for global translation modulation by mitochondrially produced reactive oxygen species. Nat. Commun. 2018;9(1):324. doi: 10.1038/s41467-017-02694-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Reichmann D., Voth W., Jakob U. Maintaining a healthy proteome during oxidative stress. Mol. Cell. 2018;69(2):203–213. doi: 10.1016/j.molcel.2017.12.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Eguchi Y., Shimizu S., Tsujimoto Y. Intracellular ATP levels determine cell death fate by apoptosis or necrosis. Cancer Res. 1997;57(10):1835–1840. [PubMed] [Google Scholar]
- 54.Mottonen T. Comparison of combination therapy with single-drug therapy in early rheumatoid arthritis: a randomised trial. FIN-RACo trial group. Lancet. 1999;353(9164):1568–1573. doi: 10.1016/s0140-6736(98)08513-4. [DOI] [PubMed] [Google Scholar]
- 55.Chidley C. A yeast-based screen reveals that sulfasalazine inhibits tetrahydrobiopterin biosynthesis. Nat. Chem. Biol. 2011;7(6):375–383. doi: 10.1038/nchembio.557. [DOI] [PubMed] [Google Scholar]
- 56.Gout P.W. Sulfasalazine, a potent suppressor of lymphoma growth by inhibition of the x(c)- cystine transporter: a new action for an old drug. Leukemia. 2001;15(10):1633–1640. doi: 10.1038/sj.leu.2402238. [DOI] [PubMed] [Google Scholar]
- 57.Fu L. Systematic and quantitative assessment of hydrogen peroxide reactivity with cysteines across human proteomes. Mol. Cell. Proteomics. 2017;16(10):1815–1828. doi: 10.1074/mcp.RA117.000108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Yang J. Site-specific mapping and quantification of protein S-sulphenylation in cells. Nat. Commun. 2014;5:4776. doi: 10.1038/ncomms5776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Rust C. Sulfasalazine reduces bile acid induced apoptosis in human hepatoma cells and perfused rat livers. Gut. 2006;55(5):719–727. doi: 10.1136/gut.2005.077461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Couto D. Scavenging of reactive oxygen and nitrogen species by the prodrug sulfasalazine and its metabolites 5-aminosalicylic acid and sulfapyridine. Redox Rep. 2010;15(6):259–267. doi: 10.1179/135100010X12826446921707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Yang S. Sulfa drugs inhibit sepiapterin reduction and chemical redox cycling by sepiapterin reductase. J. Pharmacol. Exp. Ther. 2015;352(3):529–540. doi: 10.1124/jpet.114.221572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Deferme L. Time series analysis of oxidative stress response patterns in HepG2: a toxicogenomics approach. Toxicology. 2013;306:24–34. doi: 10.1016/j.tox.2013.02.001. [DOI] [PubMed] [Google Scholar]
- 63.Lacher S.E. Identification of a functional antioxidant response element at the HIF1A locus. Redox Biol. 2018;19:401–411. doi: 10.1016/j.redox.2018.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Ivanova D. Vitamin K: redox-modulation, prevention of mitochondrial dysfunction and anticancer effect. Redox Biol. 2018;16:352–358. doi: 10.1016/j.redox.2018.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Na Y.R. Menadione and ethacrynic acid inhibit the hypoxia-inducible factor (HIF) pathway by disrupting HIF-1alpha interaction with p300. Biochem. Biophys. Res. Commun. 2013;434(4):879–884. doi: 10.1016/j.bbrc.2013.04.044. [DOI] [PubMed] [Google Scholar]
- 66.Ma Q. Role of nrf2 in oxidative stress and toxicity. Annu. Rev. Pharmacol. Toxicol. 2013;53:401–426. doi: 10.1146/annurev-pharmtox-011112-140320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Gwinn D.M. Oncogenic KRAS regulates amino acid homeostasis and asparagine biosynthesis via ATF4 and alters sensitivity to L-asparaginase. Cancer Cell. 2018;33(1):91–107 e6. doi: 10.1016/j.ccell.2017.12.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhou J., Schmid T., Brune B. Tumor necrosis factor-alpha causes accumulation of a ubiquitinated form of hypoxia inducible factor-1alpha through a nuclear factor-kappaB-dependent pathway. Mol. Biol. Cell. 2003;14(6):2216–2225. doi: 10.1091/mbc.E02-09-0598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Patel J. Regulation of hypoxia inducible factors (HIF) in hypoxia and normoxia during placental development. Placenta. 2010;31(11):951–957. doi: 10.1016/j.placenta.2010.08.008. [DOI] [PubMed] [Google Scholar]
- 70.Lu H. Chemotherapy triggers HIF-1-dependent glutathione synthesis and copper chelation that induces the breast cancer stem cell phenotype. Proc. Natl. Acad. Sci. U. S. A. 2015;112(33):E4600–E4609. doi: 10.1073/pnas.1513433112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hellfritsch J. Knockout of mitochondrial thioredoxin reductase stabilizes prolyl hydroxylase 2 and inhibits tumor growth and tumor-derived angiogenesis. Antioxid. Redox. Signal. 2015;22(11):938–950. doi: 10.1089/ars.2014.5889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Gerald D. JunD reduces tumor angiogenesis by protecting cells from oxidative stress. Cell. 2004;118(6):781–794. doi: 10.1016/j.cell.2004.08.025. [DOI] [PubMed] [Google Scholar]
- 73.Pan Y. Multiple factors affecting cellular redox status and energy metabolism modulate hypoxia-inducible factor prolyl hydroxylase activity in vivo and in vitro. Mol. Cell Biol. 2007;27(3):912–925. doi: 10.1128/MCB.01223-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lando D. Oxygen-dependent regulation of hypoxia-inducible factors by prolyl and asparaginyl hydroxylation. Eur. J. Biochem. 2003;270(5):781–790. doi: 10.1046/j.1432-1033.2003.03445.x. [DOI] [PubMed] [Google Scholar]
- 75.Jaakkola P. Targeting of HIF-alpha to the von Hippel-Lindau ubiquitylation complex by O2-regulated prolyl hydroxylation. Science. 2001;292(5516):468–472. doi: 10.1126/science.1059796. [DOI] [PubMed] [Google Scholar]
- 76.Kaelin W.G., Jr., Ratcliffe P.J. Oxygen sensing by metazoans: the central role of the HIF hydroxylase pathway. Mol. Cell. 2008;30(4):393–402. doi: 10.1016/j.molcel.2008.04.009. [DOI] [PubMed] [Google Scholar]
- 77.Keith B., Johnson R.S., Simon M.C. HIF1alpha and HIF2alpha: sibling rivalry in hypoxic tumour growth and progression. Nat. Rev. Cancer. 2011;12(1):9–22. doi: 10.1038/nrc3183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Lee G. Oxidative dimerization of PHD2 is responsible for its inactivation and contributes to metabolic reprogramming via HIF-1alpha activation. Sci. Rep. 2016;6:18928. doi: 10.1038/srep18928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Finn R.D. InterPro in 2017-beyond protein family and domain annotations. Nucleic Acids Res. 2017;45(D1):D190–D199. doi: 10.1093/nar/gkw1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wood Z.A., Poole L.B., Karplus P.A. Peroxiredoxin evolution and the regulation of hydrogen peroxide signaling. Science. 2003;300(5619):650–653. doi: 10.1126/science.1080405. [DOI] [PubMed] [Google Scholar]
- 81.Barford D. The role of cysteine residues as redox-sensitive regulatory switches. Curr. Opin. Struct. Biol. 2004;14(6):679–686. doi: 10.1016/j.sbi.2004.09.012. [DOI] [PubMed] [Google Scholar]
- 82.Chasapis C.T. Zinc and human health: an update. Arch. Toxicol. 2012;86(4):521–534. doi: 10.1007/s00204-011-0775-1. [DOI] [PubMed] [Google Scholar]
- 83.Kroncke K.D., Klotz L.O. Zinc fingers as biologic redox switches? Antioxid. Redox. Signal. 2009;11(5):1015–1027. doi: 10.1089/ARS.2008.2269. [DOI] [PubMed] [Google Scholar]
- 84.Zhou X. Selective sensitization of zinc finger protein oxidation by reactive oxygen species through arsenic binding. J. Biol. Chem. 2015;290(30):18361–18369. doi: 10.1074/jbc.M115.663906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Heo J. Redox control of GTPases: from molecular mechanisms to functional significance in health and disease. Antioxid. Redox. Signal. 2011;14(4):689–724. doi: 10.1089/ars.2009.2984. [DOI] [PubMed] [Google Scholar]
- 86.Cremers C.M., Jakob U. Oxidant sensing by reversible disulfide bond formation. J. Biol. Chem. 2013;288(37):26489–26496. doi: 10.1074/jbc.R113.462929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Garcia-Gimenez J.L. Role of glutathione in the regulation of epigenetic mechanisms in disease. Free Radic. Biol. Med. 2017;112:36–48. doi: 10.1016/j.freeradbiomed.2017.07.008. [DOI] [PubMed] [Google Scholar]
- 88.Stankovic-Valentin N., Melchior F. Control of SUMO and ubiquitin by ROS: signaling and disease implications. Mol. Aspect. Med. 2018;63:3–17. doi: 10.1016/j.mam.2018.07.002. [DOI] [PubMed] [Google Scholar]
- 89.Gaal T. Transcription regulation by initiating NTP concentration: rRNA synthesis in bacteria. Science. 1997;278(5346):2092–2097. doi: 10.1126/science.278.5346.2092. [DOI] [PubMed] [Google Scholar]
- 90.Schneider D.A., Gaal T., Gourse R.L. NTP-sensing by rRNA promoters in Escherichia coli is direct. Proc. Natl. Acad. Sci. U. S. A. 2002;99(13):8602–8607. doi: 10.1073/pnas.132285199. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Confirmation of protein stability changes induced by a series of glutathione redox ratios. Related to Fig. 1, Fig. 2. A, STRING analysis for LG-ITRR hits with stability changes. CETSA signal is visualized as a bar plot above each node. Edge between protein nodes represents protein-protein interactions retrieved from the STRING database, whose width indicates interaction confidence. Protein node with purple label stands for known redox-sensitive proteins from publications. Clusters of functional protein groups that are discussed in text are circled in colors as follows- Red: SUMO-activating proteins; purple: epigenetic regulators; blue: extracellular or endoplasmic reticulum proteins carrying structural disulfides. B, MS-CETSA signals of epigenetic regulating proteins PRMT1, ASH2L and CBX3 proteins from LG-ITRR experiment. C, Western blot validation of thermal stabilization of PRMT1, ASH2L and CBX3 proteins. D, Structure of human HAT1 (PDB accession 2P0W) (gray) showing the position of predicted redox-sensitive cysteines C27 (yellow spheres), C101 (blue spheres) and C168 (orange spheres). E, Distinct thermal stability changes of histone variants in the LG-ITRR experiment. Yellow and green curves (left) represent the ITRR response of cysteine-bearing histone H3 variant while gray and brown curves for the ITRR response of non-cysteine containing histone H1 variants.
CETSA shifts related to protein complex formation and metabolite concentration changes upon H2O2 exposure. Related to Fig. 3. A, Decreased GSH/GSSG ratios and GSH amounts upon H2O2 treatment in HepG2 cells. Statistical significance was calculated with two sample t-tests, *p < 0.05. B, Similar ITDR-CETSA responses for subunits of the E1 activating enzyme UBA3-NAE1 functional complex. C, Abundance (37 °C) and stability (52 °C) profiles of all the detected mitochondrial and cytosolic ribosomal proteins passed QC from IH-ITDR experiment. Names of the presented ribosomal protein subunits are listed in table. D, Similar ITDR-CETSA responses among subunits of the E1 activating enzymes and E2-conjugating enzyme UBA2-SAE1-UBC9 transient functional complex, together with a schematic of the working mechanism, modified from Bossis et al. [31]. E, STRING analysis for oligomycin-ITDR hits with stability changes. SNU398 cells were incubated with different oligomycin concentrations, and heated at 37 °C and 52 °C. Filled colors for each protein node encode different functional categories and 52 °C CETSA signal is visualized above each node. Edge between protein nodes represents interactions retrieved from the STRING database, whose width indicates interaction confidence. Protein node with blue label stands for common hits also identified in IH-ITDR experiment. ATP-binding proteins demonstrated significant and consistent destabilization.
Overview and functional clustering of proteins with abundance and stability changes in SSA-ITTR experiment. Related to Fig. 4. A, Abundance (37 °C) and stability (52 °C) profiles of all the detected mitochondrial and cytosolic ribosomal proteins passed QC from SSA-ITTR experiment. Names of the presented ribosomal protein subunits are listed in table. B, Venn diagram showing determined hits from four treatments (LG-ITRR, LH-ITDR, IH-ITDR, SSA-ITTR experiments) in this study. All the 316 proteins (314 proteins without distinguishing 2 protein isoforms) are presented in the graph. C, GSH/GSSG ratios and GSH amounts decrease upon SSA treatment in a time-dependent manner. Statistical significance was calculated with one-way ANOVA, multiple comparison of shortest time-point sample versus the rest, *p < 0.05. D, Confirmation of thermal stabilization of PRMT1, one common hit between LG-ITRR and SSA-ITTR experiments. Left, western blotting validating thermal stabilization of PRMT1 proteins. Middle, stabilized MS-CETSA signals of PRMT1 from SSA-ITTR experiment. Right, stabilized MS-CETSA signals of EGLN1 from SSA-ITTR experiment. E, STRING analysis for SSA-ITTR hits with thermal stability changes at 52 °C. Filled color for each protein node encodes different functional category and CETSA signal is visualized above each node. Edges between protein nodes represent interactions retrieved from the STRING database, whose width indicates interaction confidence. Protein node with blue label or green label stands for common hits also identified in IH-ITDR or LG-ITRR experiments, respectively. F, STRING analysis for SAA-ITTR target proteins with abundance changes. Filled colors for each protein node encode different functional categories and protein level profiles at 37 °C is visualized above each node. Edge between protein nodes represents interactions retrieved from the STRING database, whose width indicates interaction confidence. Several NRF2 targets are clustered in the purple circle.
Structural determinants of cysteine-mediated redox sensing. Related to Fig. 5. A, Distinct thermal stability responses of zinc finger protein subsets. FYVE domain-containing zinc finger proteins displayed minimal CETSA shifts in our datasets. B, Superimposed structure of several kinases and predicted redox-sensitive cysteines. Structures of kinase domains for CDK16 (PDB accession 3MTL), SRC (4MXO), STK4 (3COM), CAMK2D (6AYW), SRPK2 (5MYV), STK24 (3CKW) and GSK3B (1I09) are superimposed, and the positions of predicted redox-sensitive cysteines are shown in spheres. C, Cysteine containing-NKCD sequence motif in RAB proteins.