

Abstract
MbtH-like proteins (MLPs) are required for soluble expression and/or optimal activity of some adenylation (A) domains of nonribosomal peptide synthetases (NRPSs). Since A domains can interact with noncognate MLP partners, we investigated how the function of an A domain, TioK, involved in the biosynthesis of the bisintercalator thiocoraline, is altered by noncognate MLPs. Measuring TioK activity with 12 different MLPs from a variety of bacterial species using a radiometric assay suggested that the A domain substrate promiscuity could be altered by foreign MLPs. Kinetic studies and bioinformatics analysis expanded the complexity of MLP functions and interactions.
Keywords: Biosynthesis, Enzymology, Nonribosomal peptide synthetase, Protein-protein interaction, Thiocoraline
Graphical Abstract
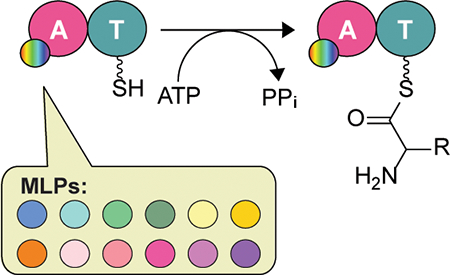
Eleven noncognate MbtH-like proteins (MLPs) from various nonribosomal peptide synthetase assembly-lines were shown to expand the substrate profile of the adenylation domain TioK from the thiocoraline biosynthetic gene cluster when compared to the cognate MLP TioT.
Introduction
Natural products are an important source of drugs and drug leads. In fact, from 1981 to 2014, natural products and their derivatives accounted for ~65% of all approved small molecule drugs worldwide.[1] Amongst these, natural products from the nonribosomal peptide (NRP) family were some of the most abundant. NRPs are biosynthesized by a family of mega-enzymes termed nonribosomal peptide synthetases (NRPSs).[2] NRPSs are composed of modules containing three fundamental domains: the adenylation (A), condensation (C), and thiolation (T) domains. Each module employs an amino acid or amino acid-derived molecule as a building block to construct the final peptidic structure. The core peptidic structure is derivatized by the inclusion of tailoring enzymes embedded in the modules of the NRPS assembly-line to produce a complex molecule. During NRP biosynthesis, a substrate building block is first selected by an A domain that adenylates it by using ATP to produce substrate-AMP. This activated substrate is then transferred onto a downstream partner T domain that is normally located on the same module as its partner A domain. The T domain then carries the substrate to a C domain, which catalyzes condensation with another building block tethered to a T domain from the adjacent upstream module. The assembled substrate then moves to another C domain on the next downstream module to condense with the next building block. This chain reaction is performed until it reaches the end of the assembly-line, whose product is usually released by a thioesterase (TE) domain.
It was recently discovered that mbtH-like genes are essential for the production of some NRPs, such as coelichelin and clorobiocin.[3] Two independent studies revealed that small, ~10-kDa, proteins encoded by mbtH-like genes are integral components of NRPSs, as these proteins stimulate the activity of A domains by binding in a 1:1 ratio.[4] The function of MbtH-like proteins (MLPs) has been further explored in various studies, which have also shown that they tightly interact with A domains and enhance their soluble expression,[5] activity,[6] and/or substrate specificity.[7] Mutation studies of MLPs have shown that the first two out of three conserved l-Trp residues and the first conserved l-Pro residue in MLP protein amino acid sequences are important for the activity of the MLP partner A domains (Figure S1).[4b, 5a] It was also shown that various MLPs can flexibly interact with a heterologously expressed A domain to change the kinetics of activation of its natural substrate.[6a, 8] Structural studies of MLPs[9] and MLP-A domain complexes[5g, 10] have provided insights as to how A domains interact with and are affected by MLPs. The MLP structures revealed that they contain three anti-parallel β-sheets followed by one or two α-helices.[9] The structures of MLP-A domain complexes confirmed that they interact with each other in a 1:1 ratio. In these complexes, three conserved l-Trp residues on MLPs located on the second and third β-sheets and on a loop after the first α-helix interact with an α-helix between the a6 and a7 conserved amino acid sequence motifs (Note: A domains contain 10 of these motifs, a1-a10)[11] of the partner A domain. MLPs do not directly communicate with the substrate to be activated since the binding region of the MLP is significantly distanced from the A domain active site.[10a] The A domain active site structure remains unchanged in the presence or absence of an MLP regardless of its origin.[10b] These studies suggested that MLPs work as stabilizers for A domains, but their exact role and effect on dictating the substrate to be activated by an A domain are still puzzling.
To investigate the potential effect of using noncognate MLPs on the substrate profile of an A domain, we selected to work with TioK from the biosynthetic gene cluster of the bisintercalator thiocoraline.[12] TioK is a minimal NRPS module consisting exclusively of an A and a T domain (Scheme 1). During thiocoraline biosynthesis, TioK serves as a platform for the β-hydroxylation of l-Trp catalyzed by a P450 enzyme, TioI, after activation by the TioK A domain and loading onto its T domain. β-Hydroxy-l-Trp is subsequently released by a type II TE, TioQ, for further modifications by TioF, TioG, TioH, TioL, and/or TioM.[5a, 13] This leads to the formation of 3-hydroxyquinaldic acid, a planar chromophore which is incorporated into the thiocoraline NRPS assembly-line by the A domain TioJ and a FabC carrier enzyme.[14] Like many NRPS A domains, TioK requires a MLP partner, TioT, for its soluble expression and activity.[5a] More recently TioT was also shown to be necessary for soluble expression or activity of other thiocoraline NRPS A domains, including TioN, TioS(A3), and TioS(A4).[5f, 5g, 15]
Scheme 1.

The function of TioK and abbreviated biosynthetic pathway for thiocoraline production.[5a, 12] The previously characterized enzymes (TioQ,[13b] TioJ/FabC,[14] TioN,[15] and TioS[5f, 5g]) are in black, whereas the yet to be biochemically investigated enzymes are in grey.
Herein, we investigated the effect of the MLP TioT and 11 noncognate MLPs on the promiscuity and activity of TioK by a well-established ATP-[32P]PPi exchange assay. To try to rationalize the result of the biochemical assays, the potential interactions between TioK and MLPs (including noncongate MLPs) were analyzed bioinformatically by using the solved crystal structures of A domain-MLP complexes.[5g, 10] This study provides a possible future application of using MLPs to enhance the promiscuity of A domains, which would be useful for combinatorial biosynthesis.
Results and Discussion
Choice of 12 MLPs
To assess the TioK activity with different MLPs, we selected 12 MLPs from a variety of bacterial species (Figure S1 and Table S1). These MLPs are well conserved (identity and similarity to TioT are 48–76% and 68–88%, respectively) and were chosen to not completely disrupt the TioK activity. CouY is found in the coumermycin A biosynthetic gene cluster.[16] Ecm8 is proposed to be the MLP partner of A domains found in the biosynthetic gene cluster of echinomycin, a bisintercalator analogue of thiocoraline.[17] KtzJ is the MLP involved in kutznerides biosynthesis. Kutznerides are circular NRPs, which have structures relatively similar to thiocoraline.[18] MbtH, from Mycobacterium tuberculosis, was the protein that inspired the name MbtH-like protein (MLP).[3a] MbtH is part of the mycobactin gene cluster. PA2412, from the pyoverdine gene cluster, is one of the most studied MLPs, since its structure has been solved as a monomeric form and a complex with an A domain, EntF.[9a, 10b] These five MLPs were selected based on the natural products that are produced by their gene clusters. The products all contain a tryptophan, phenylalanine, or another aromatic residue. We also selected six genes from other mycobacteria that had high similarity to MbtH. These included Mycobacterium smegmatis (MysmA.01649.b.A1 or MysmAbA), Mycobacterium avium (MyavA.01649.a.A1 and MyavA.01649.c.A1 or MyavAaA and MyavAcA), Mycobacterium paratuberculosis (MypaA.01649.a.A1 or MypaAaA), Mycobacterium marinum (MymaA.01649.c.A1 or MymaAcA), and Mycobacterium abscessus (MyabA.01649.a.A1 or MyabAaA).
Cloning and expression of TioK with 12 different MLPs
All MLP genes were cloned into the pACYCDuet-1 plasmid vector from the genomic DNA or from plasmids carrying the genes of interest, except for MyavAcA, which was cloned into the AVA0421 vector[19] (Table S2). The MLP genes, in their corresponding vectors, were transformed into E. coli BL21 (DE3) Δybdz::aac(3)iv, a strain lacking the endogenous MLP, containing the tiok gene in pET28a. After their expression, the excess of the various MLPs was removed by size exclusion chromatography in order to obtain precise concentrations of the TioK:MLP complexes. The 65.1-kDa TioK protein co-expressed and co-purified with the 11 different noncognate MLPs similarly to with the cognate TioT by NiII-NTA affinity chromatography (Figure S2).
Activity of TioK with 12 different MLPs: substrate profile
To assess if MLPs affect the TioK A domain versatility, we first investigated, by ATP-[32P]PPi exchange assay, the substrate specificity of TioK with 12 different MLPs and 14 amino acids (l-Trp, d-Trp, l-Phe, l-Tyr, l-Ser, l-Leu, l-Val, l-His, l-Asp, l-Ala, l-Cys, l-Thr, l-Ile, and l-Pro) (Figures 1 and 2). These amino acids were tested in our previous study reporting on the characterization of TioK co-expressed with TioT.[5a] Doing so, we demonstrated that the substrate promiscuity of TioK was increased by using noncognate MLPs. We observed that the activity for the natural substrate, l-Trp, was changed by 18–135% by using noncognate MLPs (Figure 2). The alteration of specificity was MLP-dependent.
Figure 1.

Bar graphs showing the % activity for the activation of various amino acids by TioK co-purified with the cognate MLP TioT (panel A) and 11 noncognate MLPs (panels B-L). The left Y axis for each graph represents the % activity for each amino acid, which is relative to that for the activation of l-Trp by TioK with the enzymes listed in the top left corner of each bar graph. The right Y axis for each graph represents the % exchange of [32P]PPi in the ATP-[32P]PPi exchange assays at the 2-h end point for each substrate activated by TioK with the enzymes listed in the top left corner of each bar graph. All experiments were performed in duplicate (n = 2) and are shown as individual data points.
Figure 2.

Bar graphs showing the ratio for the activation of various amino acids by TioK co-purified with 11 noncognate MLPs when compared to TioT. The activity for the TioK with the 11 noncognate MLPs is relative to that for the activation of each amino acids by TioK with TioT (not shown as the value is exactly 1 for this control). The amino acids selected were those that displayed enough activity to have statistically consistent values. The Y axis is in the logarithmic scale. The data are shown as individual points (n = 2).
For example, Ecm8, KtzJ, MbtH, and PA2412 increased the activity of TioK for almost all substrates tested, whereas Mycobacterium MLPs tended to increase the promiscuity towards l-Ser, l-Thr, l-Val, l-Leu, and l-His (Figure 1). The TioK activity profiles varied greatly depending on the MLP used (Figure 2). Amongst the substrates that displayed enough activity to be significantly measured, notable increases in activity (more than or equal to 10-fold; Figure 3) were observed for the activation of l-Ser by TioK with MyavAcA, MymaAcA, and MyabAaA, as well as l-Thr by TioK with MbtH. Notably, TioK with many Mycobacterium MLPs tended to display lower activity for amino acids with aromatic or non-polar side chains, but higher activity for those with hydroxyl- or thiol-containing side chains. Previous structural data showed that MLPs only interact with A domain surface residues, but not with those in its active site. However, the substrate profile data clearly indicated that interaction with foreign MLPs altered the A domain substrate specificity, which in fact supports the previous observation and the following idea that the surface structure/residues may be critical for determining the A domain substrate.[20] The interaction with different MLPs might affect the surface structure of TioK, which would cause a change in substrate specificity. Interestingly, a previous structural study of an A domain, EntF, with two different MLPs did not show any structural modification of this A domain.[10b] However, it is known that EntF purifies in a soluble and active form without an MLP. In contrast, TioK requires the presence of a MLP in order to be expressed and purified in a soluble and active form,[5a] which might explain why various noncognate MLPs affect the substrate profiles of this enzyme.
Figure 3.

Representative sedimentation equilibrium data. Samples contained TioK-MbtH (1.24 μM, blue squares), TioK-MymaAcA (1.2 μM, pink circles), or TioK-MyavAcA (1.19 μM, green triangles) in 10 mM sodium phosphate, 200 mM NaCl pH 8.0. Samples were brought to sedimentation equilibrium at 12,000 rpm and 4 °C. The smooth curves are fits of Equation 1 to individual data sets. The residuals of these fits are shown in the upper panel.
Activity of TioK with 12 different MLPs: kinetics
To better understand the catalytic activity of TioK with the 12 different MLPs investigated, we calculated Michaelis-Menten kinetic parameters for two best substrates, l-Trp and l-Phe (Table 1). These kinetic data showed that Ecm8, KtzJ, and PA2412 are the three kinetically best noncognate MLPs among those tested. Interestingly, the substrate affinity (Km) was not significantly altered by noncognate MLPs, and some were even better when compared to the cognate TioK-TioT complex (0.8 to 1.5-fold for l-Trp and 1.0 to 1.1-fold for l-Phe). However, the maximum velocity (Vmax) was dramatically altered in the presence of some MLPs (0.08 to 1.3-fold activity for l-Trp and 0.08 to 1.4-fold activity for l-Phe), which was the major cause of decrease in overall kinetic activity of some noncognate MLPs (0.08 to 1.4-fold activity for l-Trp and 0.08 to 1.4-fold activity for l-Phe). To test if the different Vmax values result from dissociation of the noncognate MLPs from TioK after expression of the complex, purified Ecm8 was added into a TioK-MbtH complex reaction. If the variations in Vmax values were due to free TioK in solution after dissociation, the addition of Ecm8 would result in an increase in Vmax value compared to the reaction where Ecm8 was not added. No such increase was observed in the presence of Ecm8, indicating that TioK and noncognate MLPs are optimally bound even after expression. The effect of MLPs on kinetic parameters is opposite to that of a previous study about the activity of the A domain EntF with different MLPs, which displayed much greater dependence on Km than kcat.[8, 10b] However, as stated before, the activity of the EntF enzyme is not fully dependent on MLPs. These substrate profiles and kinetic data, when compared to previous studies, suggest that the exact role of MLPs cannot be explained by a single function, although it is obvious that MLPs are necessary for some A domains for their optimal activity.
Table 1.
Steady-state kinetic parameters for AMP derivatization by TioK with 12 different MLPs.
| Substrate | MLP | Km (mM(S)) | Vmax (μM(E)min−1) | Vmax/Km (μM(E)mM−1 min−1 mM(S)−1) |
|---|---|---|---|---|
| l-Trp | TioT (NHis)a | 0.10 ± 0.01 | 6.7 ± 0.1 | 67 ± 4 |
| CouY (NHis) | 0.13 ± 0.01 | 3.7 ± 0.1 | 29 ± 2 | |
| Ecm8 (NHis) | 0.09 ± 0.01 | 8.8 ± 0.1 | 93 ± 4 | |
| KtzJ (NHis) | 0.09 ± 0.01 | 5.7 ± 0.1 | 67 ± 2 | |
| MbtH (NHis) | 0.11 ± 0.01 | 2.9 ± 0.1 | 27 ± 1 | |
| PA2412 (NHis) | 0.15 ± 0.01 | 7.3 ± 0.1 | 50 ± 2 | |
| MysmA.01649.b.A1 (no tag) | 0.11 ± 0.01 | 1.2 ± 0.1 | 11 ± 1 | |
| MyavA.01649.a.A1 (no tag) | 0.09 ± 0.01 | 1.5 ± 0.1 | 16 ± 1 | |
| MyavA.01649.c.A1 (NHis) | 0.12 ± 0.01 | 1.6 ± 0.1 | 14 ± 1 | |
| MypaA.01649.a.A1 (no tag) | 0.09 ± 0.01 | 0.52 ± 0.01 | 6.0 ± 0.3 | |
| MymaA.01649.c.A1 (NHis) | 0.08 ± 0.01 | 1.4 ± 0.1 | 18 ± 1 | |
| MyabA.01649.a.A1 (NHis) | 0.13 ± 0.01 | 0.84 ± 0.01 | 6.5 ± 0.5 | |
| l-Phe | TioT (NHis)a | 1.1 ± 0.1 | 4.8 ± 0.1 | 4.5 ± 0.2 |
| CouY (NHis) | 1.1 ± 0.1 | 2.5 ± 0.1 | 2.4 ± 0.1 | |
| Ecm8 (NHis) | 1.1 ± 0.1 | 7.0 ± 0.1 | 6.3 ± 0.2 | |
| KtzJ (NHis) | 1.2 ± 0.1 | 4.8 ± 0.1 | 4.1 ± 0.2 | |
| MbtH (NHis) | 1.2 ± 0.1 | 2.4 ± 0.1 | 2.1 ± 0.1 | |
| PA2412 (NHis) | 1.1 ± 0.1 | 3.5 ± 0.1 | 3.3 ± 0.2 | |
| MysmA.01649.b.A1 (no tag) | 1.1 ± 0.1 | 0.85 ± 0.01 | 0.80 ± 0.03 | |
| MyavA.01649.a.A1 (no tag) | 1.1 ± 0.1 | 0.77 ± 0.04 | 0.70 ± 0.13 | |
| MyavA.01649.c.A1 (NHis) | 1.2 ± 0.1 | 1.2 ± 0.1 | 1.0 ± 0.1 | |
| MypaA.01649.a.A1 (no tag) | 1.2 ± 0.1 | 0.40 ± 0.01 | 0.34 ± 0.02 | |
| MymaA.01649.c.A1 (NHis) | 1.2 ± 0.1 | 0.62 ± 0.01 | 0.49 ± 0.03 | |
| MyabA.01649.a.A1 (NHis) | 1.11 ± 0.01 | 0.49 ± 0.01 | 0.45 ± 0.02 | |
| l-Trp | MbtH with additional Ecm8 | 0.11 ± 0.01 | 2.3 ± 0.1 | 21 ± 1 |
We previously published these data in ref [5a], but the kinetic parameters were newly measured to compare with the same purification methods as other protein complexes in this study. The variability +/− presented for kinetic parameters are the standard errors for the fitting of the curves. The units of concentration of substrate and enzyme are indicated as subscripts (S) and (E) on each unit, respectively. Note: Vmax (instead of kcat) is reported because only the relative concentrations of TioK-MLP complexes could be measured.
Stoichiometric analysis of TioK-MLP complexes
In order to confirm that the TioK-MLP complexes studied are in the same ratio (e.g., 1:1/TioK:MLP) and the comparisons made above are valid, we performed sedimentation equilibrium experiments by analytical ultracentrifugation. Representative sedimentation equilibrium data sets are shown in Figure 3. The solid curves are fits of Equation 1 (In the Experimental Section) to these data. Small, symmetrical residuals (top panel) show that the three-species model embodied in Equation 1 is consistent with these data. This appears to be the most parsimonious model that accounts for all of the data. Tests of models with more species fit some data sets as well as Equation 1, but returned absorbance increments for the additional species that were below the threshold for accurate measurement. The results of analysis of the entire set of TioK-MLP complexes are summarized in Table 2. The dominant components in all samples were TioK-MLP complexes (mole fractions from A230 ratios >0.75). The weight-average molecular weights of these complexes fell into two narrow ranges; 7.7×104 < MC < 8.2×104, and 1.43×105 < MC < 1.52×105. The first corresponds well with molecular weights predicted for 1:1 TioK-MLP complexes; the second is compatible with values predicted for 2:2 stoichiometries.
Table 2.
Sedimentation equilibrium analysis of TioK-MLP complexes.
| Component MW | MW | Approximate mole fraction | MWobs | 95% CL MWobs | Multiple of MW(1:1) | ||||
|---|---|---|---|---|---|---|---|---|---|
| Samples | TioK | MLP | Predicted 1:1 | Free MLP | TioK-MLP complex | Aggregate | TioK-MLP complex | ||
| TioK-TioT | 65150 | 9678 | 74828 | 0.02 | 0.88 | 0.10 | 147932 | 15320 | 1.98 ± 0.07 |
| TioK-CouY | 65150 | 9692 | 74842 | 0.03 | 0.84 | 0.13 | 78810 | 13347 | 1.05 ± 0.18 |
| TioK-Ecm8 | 65150 | 9515 | 74665 | 0.01 | 0.76 | 0.23 | 144300 | 16227 | 1.93 ± 0.22 |
| TioK-KtzJ | 65150 | 9798 | 74948 | 0* | 0.92 | 0.08 | 143860 | 13755 | 1.92 ± 0.18 |
| TioK-MbtH | 65150 | 9695 | 74845 | 0.06 | 0.82 | 0.12 | 154299 | 5886 | 2.06 ± 0.08 |
| TioK-PA2412 | 65150 | 10056 | 75206 | 0* | 0.87 | 0.13 | 81762 | 6797 | 1.09 ± 0.09 |
| TioK-MysmAbA | 65150 | 8671 | 73821 | 0* | 0.84 | 0.16 | 78440 | 4879 | 1.06 ± 0.07 |
| TioK-MyavAaA | 65150 | 8885 | 74035 | 0.04 | 0.81 | 0.15 | 77110 | 5251 | 1.04 ± 0.07 |
| TioK-MyavAcA | 65150 | 11069 | 76219 | 0* | 0.93 | 0.07 | 149700 | 9980 | 1.96 ± 0.13 |
| TioK-MypaAaA | 65150 | 7897 | 73047 | 0.01 | 0.87 | 0.12 | 149440 | 12884 | 2.05 ± 0.18 |
| TioK-MymaAcA | 65150 | 10317 | 75467 | 0.01 | 0.92 | 0.07 | 146070 | 6346 | 1.94 ± 0.08 |
| TioK-MyabAaA | 65150 | 10368 | 75518 | 0* | 0.94 | 0.06 | 148240 | 9526 | 1.96 ± 0.13 |
Molecular weights (MW) and approximate mole fractions determined at sedimentation equilibrium. Mole fractions estimated from ratio of A230 values at the mid-point of each concentration gradient. Molecular weights represent averages of values obtained at 3 sample concentrations and 2 rotor speeds. 95% confidence limits (CL) were determined for each set of molecular weight values.
Below detection limit.
The 95% confidence limits (CL) on the molecular weights (MW) of TioK-TioT, TioK-CouY, TioK-Ecm8, TioK-KtzJ, and TioK-MypaAaA complexes are relatively large, and they do not rule out the possibility that some complexes might deviate from 1:1 (or 2:2) molar ratio. For these samples, the possible deviations are small, on the order of ± 1 MLP molecule. On the other hand, the molecular weights of the remaining 7 complexes are better specified and they rule out stoichiometries other than 1:1 or 2:2. It is intriguing that some complexes have 1:1 stoichiometry and some 2:2. Although we cannot currently account for this explanation, the fact that some 1:1 complexes do not dimerize while others do so to apparent completion indicates that TioK-MLP complexes within our protein set differ strongly in dimerization affinities.
Bioinformatics analysis of TioK with MLPs
In an attempt to rationalize the variations in TioK substrate profiles and kinetics observed, we analyzed the amino acid sequence alignment between the 12 tested MLPs (Figure S1). We concluded that a higher homology of the noncognate MLP to the cognate TioT does not necessarily result in similar activity of TioK. Although the two MLPs, Ecm8 and KtzJ, that gave the highest activity of TioK have the best identities (both 76%) and high sequence similarities (84% and 85%), TioK with PA2412 that has the least identity/similarity (48%/68%) showed the third highest activity (Table S1). A phylogenic tree analysis of MLPs also confirmed that PA2412 is the most distant relative of TioT when compared to other MLPs and that close relative does not necessarily lead to result similar to those from TioT, with the exception of the two closest relatives, Ecm8 and KtzJ (Figure S4). Based on sequence alignment (Figure S1), it seems that the amino acid at positions 50 and 59 (based on TioT numbering) of MLPs could play a role in dictating the activity of TioK. The MLPs that resulted in higher catalytic efficiency of TioK possess alanine and methionine at these positions, respectively, whereas those that resulted in lower catalytic efficiency of TioK have a polar amino acid and isoleucine residues at these positions, respectively.
To further investigate this possibility, we used five available crystal structures of MLP-A domain complexes[5g, 10] to predict the possible amino acid residues involved in interactions between the 12 MLPs tested and TioK (Figure S1). The first of such structure was SlgN1 with an MLP tethered to the N-terminus of the A domain.[10a] The structures of the A domain involved in the biosynthesis of enterobactin, EntF, was solved with its natural MLP partner YbdZ and a noncognate MLP, PA2412.[10b] The structure of the A domain DhbF was also determined in complex with its MLP partner.[10c] Most recently, the structure of an interrupted A domain in the thiocoraline biosynthetic pathway, TioS(A4aM4A4b), was determined as a complex with TioT.[5g] The amino acid residues that are responsible for interactions by hydrogen bonds or salt bridges between these MLPs and their A domain partners are well conserved within the MLPs tested in this study, except for the residue corresponding to D34 of TioT (Figure S1). However, this residue of TioT does not interact with TioS(A4aM4A4b), and the side chain of this residue does not seem to affect the interaction that is in fact provided by the backbones of the peptides. Interestingly, in the five crystal structures, A50 of TioT that we predicted to possibly affect TioK activity, did not show any direct interactions with A domains. M59 displayed an interaction only in the EntF-YbdZ complex, but the interaction was provided by the backbone of the amino acids.
The amino acid sequence alignment between biochemically characterized A domains that require TioT for soluble expression and/or activity (TioK, TioS(A3a), and TioN) and structurally characterized A domains complexed with MLPs (TioS(A4a), EntF, DhbF, and SlgN1) suggested that the amino acid residues of A domains important for interactions with MLPs are also relatively well conserved (Figure S5). In addition to those A domains that required MLPs for their optimal functions, TioJ that is an A domain coded in the thiocoraline biosynthetic gene cluster, but does not need TioT for its solubility/function, was also included in the sequence alignment. This alignment showed that the amino acid sequence of the MLP interacting region was not conserved in TioJ (corresponding to R356-M376 of TioK) (Figure S5). In fact, there are two fully conserved A domain amino acid residues (corresponding to P357 and T360 of TioK) that interact with the 3rd of three highly conserved Trp residues in MLPs (corresponding to W26, W36, and W56 of TioT) although direct interactions of this type were not seen in the TioT-TioS(A4aM4A4b) complex. In contrast, another A domain residue (corresponding to L366 of TioK) known to interact with the first and/or second Trp residues is not conserved (Figure S5). This residue is also not conserved in A domains that share the same cognate MLP, TioT, in the thiocoraline biosynthetic gene cluster. Within these A domains, all non-conserved residues that interact with TioT use their backbone for interactions with the A domain, except for S362 of TioK. The side chain of TioK S362 would play an important role in the interactions whereas that of TioS(A4aM4A4b) E379 forms salt bridges with R45 of TioT. The side chain of TioK S362 would be able to form a hydrogen bond with R45 of TioT, however that of TioN A322 does not seem to establish any non-covalent interactions with the residue. This analysis showed that the MLP-A domain interactions are not fully conserved even though A domains share the same cognate MLP. In fact, TioN is the only A domain, which could be expressed in soluble form without co-expression with TioT (note that without TioT, the soluble TioN is not active), in biochemically characterized A domains in the thiocoraline biosynthetic gene cluster.[5a, 5f, 5g, 15] TioN is also unique among the biosynthetic pathways of homologous natural products. Therefore, TioN could be evolved separately from other NRPS enzymes in the thiocoraline biosynthetic gene cluster. Based on this simple bioinformatics analysis, it is clear that MLP-A domain interactions are much more complex than previously believed and a lot more structures of MLP-A domain complexes will be needed to help understand how the effect of noncognate MLPs on A domain substrate profiles arise.
Conclusions
In summary, we showed that noncognate MLPs could broaden the substrate promiscuity of TioK and alter its adenylating activity by affecting the enzyme’s turnover rate rather than the substrate affinity. The sedimentation equilibrium analyses by analytical ultracentrifugation showed that one TioK always had one MLP bound to it, either in monomeric (1:1) or dimeric (2:2) forms. By amino acid sequence analyses, we concluded that the amino acid residues involved in MLP-A domain interactions are fairly conserved, but not identical in A domains with a shared cognate MLP, and that sequence homology of MLPs does not directly correlate with their ability to alter A domain function, which is consistent with a previous study.[8] All these results indicated that MLP-A domain interactions are not simple and a lot remains to be understood. Based on the encouraging broadening of the substrate versatility of TioK by noncognate MLPs, future studies focused on engineering MLPs to enhance the promiscuity of A domains while retaining their activity will be warranted. Since inserting MLP genes into genomic DNA of microorganisms is relatively easy, this kind of MLPs could be useful tool in the future combinatorial biosynthesis.
Experimental Section
Bacterial strains, plasmids, materials, and instrumentation.
Restriction endonucleases, T4 DNA ligase, and Phusion DNA polymerase were purchased from New England Biolabs (Ipswich, MA, USA). DNA primers for PCR were purchased from Integrated DNA Technologies (Coralville, IA, USA) or Sigma-Aldrich (St Louis, MO, USA). The pACYCDuet-1 and pET28a vectors were purchased from Novagen (EMD Millipore, Billerica, MA, USA). Chemically competent E. coli TOP10 cells were purchased from Invitrogen (Carlsbad, CA, USA). The E. coli BL21 (DE3)ybdZ::aac(3)IV strain was generously provided by Prof. Michael G. Thomas (University of Wisconsin-Madison, USA). DNA sequencing was performed at the University of Michigan DNA sequencing core or at Eurofins Scientific (Louisville, KY, USA). All chemicals and reagents were purchased from Sigma-Aldrich and used without any further purification. [32P]PPi was bought from Perkin Elmer (Waltham, MA, USA).
Preparation of pCouY-pACYCDuet-1, pEcm8-pACYCDuet-1, pKtzJ-pACYCDuet-1, pMbtH-pACYCDuet-1, pTioT-pACYCDuet-1, pPA2412-pACYCDuet-1, pMysmA.01649.b.A1-pACYCDuet-1, pMyavA.01649.a.A1-pACYCDuet-1, pMyavA.01649.c.A1-AVA0421, pMypaA.01649.a.A1-pACYCDuet-1, pMymaA.01649.c.A1-pACYCDuet-1, pMyabA.01649.a.A1-pACYCDuet-1, and pTioK-pET28a overexpression constructs. The genes encoding CouY, Ecm8, KtzJ, and MbtH were PCR-amplified using the genomic DNA of Streptomyces rishiriensis, Streptomyces lasaliensis, Kutzneria sp. 744, and Mycobacterium tuberculosis H37Rv, respectively. The gene encoding PA2412 was amplified using a plasmid[9a] provided by Dr. Andrew M. Gulick (Hauptman Woodward Medical Research Institute Structural Biology, University at Buffalo, USA) as a template. The plasmids encoding MysmA.01649.b.A1, MyavA.01649.a.A1, MypaA.01649.a.A1, MymaA.01649.c.A1, and MyabA.01649.a.A1 were provided by Dr. Robin Stacy (Center for Infectious Disease Research, Seattle Structural Genomics Center for Infectious Disease (SSGCID), USA). The names for the Mycobacteria genes are SSGCID internal IDs and for which more information can be found at www.ssgcid.org. The internal SSGCID ID for MbtH is MytuD.01649.a. The primers used for the amplification of each gene are listed in Table S2. The amplified genes were inserted into the linearized pACYCDuet-1 vector via the corresponding EcoRI/HindIII, NcoI/HindIII, or BamHI/HindIII restriction sites, to give constructs that encode for NHis6-tagged (EcoRI/HindIII or BamHI/HindIII) or untagged (NcoI/HindIII) proteins. The preparation of pTioT-pACYCDuet-1 and pTioK-pET28a constructs was previously reported.[5a] After PCR amplification by using the primer set in Table S2, the gene encoding MyavA.01649.c.A1-AVA0421 was cloned into NruI/PmeI restriction site of the expression vector AVA0421 by ligation-independent cloning.[19] All clones were transformed into E. coli TOP10 competent cells. All expression clones were sequenced and showed perfect alignment with the reported sequences.
Co-overproduction and co-purification of TioK with 12 MbtH-like proteins.
The purified plasmid pTioK-pET28a was co-transformed with either pCouY-pACYCDuet-1, pEcm8-pACYCDuet-1, pKtzJ-pACYCDuet-1, pMbtH-pACYCDuet-1, pPA2412-pACYCDuet-1, pMysmA.01649.b.A1-pACYCDuet-1, pMyavA.01649.a.A1-pACYCDuet-1, pMyavA.01649.c.A1- AVA0421, pMypaA.01649.a.A1-pACYCDuet-1, pMymaA.01649.c.A1-pACYCDuet-1, pMyabA.01649.a.A1-pACYCDuet-1, or pTioT-pACYCDuet-1 into E. coli BL21 (DE3)ybdZ::aac(3)IV competent cells for protein co-expression and co-purification. Luria-Bertani (LB) medium (3 × 1 L) supplemented with kanamycin (50 μg/mL) and chloramphenicol (34 μg/mL) was inoculated with 10 mL of an overnight culture of each transformant and incubated at 28 °C. The cultures were grown to an OD600 of ~0.5, induced with 1 mM isopropyl-β-thiogalactopyranoside (IPTG), and shaken for an additional 16–18 h at 20 °C. Cells were harvested by centrifugation (5,000 rpm, 10 min, 4 °C) and resuspended in buffer A (25 mM Tris-HCl pH 8.0 adjusted at room temperature, 400 mM NaCl, and 10% glycerol). Cells were lysed (1 passage at 10,000–15,000 psi on an Avestin EmulsiFlex-C3 high-pressure homogenizer or by sonication (70% intensity, 2 s “on”, 10 s “off”, until a total time of 2 min was reached, repeated 3–4 times)), and the cell debris was removed by centrifugation (16,000 rpm, 45 min, 4 °C). Imidazole (final concentration of 2 mM) was added to the supernatant and then incubated with 3 mL of NiII-NTA agarose resin (Qiagen) at 4 °C for 2 h with gentle rocking. The resin was loaded onto a column and washed with 10 mL of buffer A containing 5 mM imidazole and then with 10 mL of buffer A containing 20 mM imidazole. The desired protein was eluted from the column with buffer A supplemented with a stepwise imidazole gradient (5 mL fractions of 20 mM (1x), 40 mM (1x), 60 mM (1x), 200 mM (2x), and 500 mM imidazole (2x)). Fractions containing the desired proteins [as determined by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE)] were combined and dialyzed at 4 °C against 2 L of buffer B (40 mM Tris-HCl pH 8.0 adjusted at room temperature, 200 mM NaCl, and 10% glycerol) overnight. Size exclusion chromatography was used to remove excess MLPs. All proteins were concentrated using Amicon Ultra PL-10. Protein concentrations were measured by the Bradford assay with bovine serum albumin (BSA) as the standard. Protein yields (in mg/L of culture) for TioK when co-expressed with MLPs were 1.2 (CouY), 0.7 (Ecm8), 5.7 (KtzJ), 1.4 (MbtH), 1.3 (PA2412), 1.2 (MysmA.01649.b.A1), 1.0 (MyavA.01649.a.A1), 0.9 (MyavA.01649.c.A1), 2.3 (MypaA.01649.a.A1), 3.2 (MymaA.01649.c.A1), 1.7 (MyabA.01649.a.A1), and 1.9 (TioT) (Figure S2). The excess of MLP from the size exclusion chromatograph was collected when possible. All proteins were flash frozen in liquid nitrogen and stored at −80 °C.
Substrate specificity and kinetic measurements for the A domain of TioK by ATP-[32P]PPi exchange assays.
To determine the substrate specificity of the A domain of TioK, ATP-[32P]PPi exchange assays were performed at room temperature in 100 μL reactions containing Tris-HCl (75 mM, pH 7.5 adjusted at room temperature), MgCl2 (10 mM), TCEP (5 mM, pH 7.0 adjusted at room temperature), ATP (5 mM), the amino acid substrate (5 mM), and Na4P2O7 (1 mM, spiked with approximately 400,000 cpm of [32P]PPi per reaction). The reaction was initiated with the addition of TioK (co-purified with MLPs) (0.16 mg/mL). After 2 h incubation, the reaction was quenched with 500 μL of suspension solution (1.6% (w/v) activated charcoal, 4.5% (w/v) Na4P2O7, and 3.5% (v/v) perchloric acid in H2O). The charcoal was pelleted by centrifugation (13,000 rpm, 7 min, room temperature) and washed twice with 500 μL of wash solution (4.5% (w/v) Na4P2O7 and 3.5% (v/v) perchloric acid in H2O). The pellet was then resuspended in 500 μL of H2O followed by resuspension in 5 mL of scintillation cocktail, whose radioactivity was measured by a liquid scintillation counter (Figures 1 and 2). In order to determine the kinetic parameters for TioK substrates, reactions were carried out at room temperature with varying concentrations of the amino acid substrate (0.05, 0.1, 0.25, 0.5, 1, 1.75, 2.5, 5, 10, and up to 15 mM until the substrate inhibits the enzyme function) (Table 1). The reactions were initiated by the addition of an amino acid substrate to a mixture of Tris-HCl (75 mM, pH 7.5 adjusted at room temperature), MgCl2 (10 mM), TCEP (5 mM, pH 7.0 adjusted at room temperature), ATP (5 mM), Na4P2O7 (1 mM, spiked with approximately 400,000 cpm of [32P]PPi per reaction), and TioK (co-purified with MLPs) (0.16 mg/mL). To test the possibility of existence of free TioK, Ecm8 (0.03 mg/mL; note: Ecm8 has a 6.5-fold lower molar mass than TioK) was added into the TioK-MbtH reaction solution. The reactions were quenched after 15 min for TioK co-expressed with TioT, CouY, Ecm8, KtzJ, MbtH, or PA2412, or 20 min for TioK co-expressed with MysmA.01649.b.A1, MyavA.01649.a.A1, MyavA.01649.c.A1, MypaA.01649.a.A1, MymaA.01649.c.A1, or MyabA.01649.a.A1, and all experiments were performed at least in duplicate for each substrate concentration. Michaelis-Menten kinetic parameters were calculated in SigmaPlot and are presented in Table 1.
Sedimentation equilibrium analyses.
Protein samples were dialyzed at 4 °C against 10 mM sodium phosphate and 200 mM NaCl at pH 8.0. Analytical ultracentrifugation was performed at 20 °C in a Beckman XL-A centrifuge using an AN60Ti rotor, with scanning at 230 nm. Three sample concentrations (nominally 0.4 μM, 0.8 μM, and 1.2 μM) were analyzed at two rotor speeds (12,000 and 16,000 rpm). Equilibration was considered complete when scans taken 6 h apart were indistinguishable. Samples typically contained a trace of free MLP protein, TioK-MLP complex, and a small amount of high molecular weight protein aggregate. Data from these systems were analyzed with equation 1.
| (Eq. 1) |
In this equation, A(r) is the absorbance at radial position r, and αMLP, αC, and αAgg are absorbances of MLP protein, the TioK-MLP complex, and protein aggregate, respectively, at the reference position, r0. The symbol ε represents a baseline offset that accounts for radial position-independent differences in the absorbances of different cell assemblies.[21] The reduced molecular weights of MLP, TioK-MLP complex, and protein aggregate are given by σMLP = MMLP(1 - MLP ρ)ω2/(2RT), σC = MC(1 - C ρ)ω2/(2RT), and σAgg = MAgg (1 - Agg ρ)ω2/(2RT). The molecular weights of MLP protein, TioK-MLP complex, and protein aggregate are MMLP, MC, and MAgg, respectively; ρ is the solvent density, ω is the rotor angular velocity, R is the gas constant, and T is the temperature (Kelvin). The partial specific volumes () of MLP and TioK-MLP complexes as well as the density of sample buffer were calculated using the program SEDNTERP (Figure 3 and Table 2).[22]
Phylogenetic tree creation for MLPs.
MLPs were retrieved from UniProt. The protein sequences were aligned by using ClustalW.[23] The phylogenetic tree was created by neighbour joining[24] by using MEGA7 (Figure S4).[25] No bootstrap test was performed to generate the tree. The evolutionary distances (shown by the branches) were computed using the Poisson correction method[26] and are in the units of the number of amino acid substitutions per site. The UniProt IDs for the MLPs studied were A0QF81 (MyavAaA), A0QHN9 (MyavAcA), Q3L893 (MysmAbA), B2HHJ4 (MymaAcA), Q9F8V3 (CouY), P9WIP4 (MbtH), Q73XY9 (MypaAaA), A8CF84 (KtzJ), Q333U6 (TioT), Q0X0B7 (Ecm8), B1MAR0 (MyabAaA), and Q9I169 (PA2412).
Supplementary Material
Acknowledgements
This work was supported by a NSF CAREER Award MCB-1149427 (to S.G.-T.), the Department of Pharmaceutical Sciences (to S.G.-T.) and the Department of Molecular and Cellular Biochemistry at the Univesity of Kentucky (to M.G.F.), and in part with Federal funds awarded to the Seattle Structural Genomics Center for Infectious Disease (SSGCID) from the National Institute of Allergy and Infectious Diseases, National Institute of Health (NIH), Department of Health and Human Services, under contract No. HHSN272201200025C (to S.G.-T. and G.W.B). S.M. is a recipient of a 2018 long-term visit fellowship from the Yamada Science Foundation, Japan. We thank Dr. Robin Stacy for collaborating on this project. We thank Dr. Wenjing Chen for preliminary cloning and expression of TioK with some MLPs.
Footnotes
Supporting information for this article is given via a link at the end of the document.
References
- [1].Newman DJ, Cragg GM, J. Nat. Prod 2016, 79, 629–661. [DOI] [PubMed] [Google Scholar]
- [2].a) Walsh CT, Nat. Prod. Rep 2016, 33, 127–135; [DOI] [PubMed] [Google Scholar]; b) Weissman KJ, Nat. Chem. Biol 2015, 11, 660–670. [DOI] [PubMed] [Google Scholar]
- [3].a) Quadri LE, Sello J, Keating TA, Weinreb PH, Walsh CT, Chem. Biol 1998, 5, 631–645; [DOI] [PubMed] [Google Scholar]; b) Lautru S, Oves-Costales D, Pernodet JL, Challis GL, Microbiology 2007, 153, 1405–1412; [DOI] [PubMed] [Google Scholar]; c) Wolpert M, Gust B, Kammerer B, Heide L, Microbiology 2007, 153, 1413–1423; [DOI] [PubMed] [Google Scholar]; d) Ochsner UA, Wilderman PJ, Vasil AI, Vasil ML, Mol. Microbiol 2002, 45, 1277–1287. [DOI] [PubMed] [Google Scholar]
- [4].a) Felnagle EA, Barkei JJ, Park H, Podevels AM, McMahon MD, Drott DW, Thomas MG, Biochemistry 2010, 49, 8815–8817; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhang W, Heemstra JR Jr., Walsh CT, Imker HJ, Biochemistry 2010, 49, 9946–9947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].a) Zolova OE, Garneau-Tsodikova S, MedChemComm 2012, 3, 950–955; [Google Scholar]; b) Heemstra JR Jr., Walsh CT, Sattely ES, J. Am. Chem. Soc 2009, 131, 15317–15329; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Zolova OE, Garneau-Tsodikova S, Antibiot J. 2014, 67, 59–64; [DOI] [PubMed] [Google Scholar]; d) Imker HJ, Krahn D, Clerc J, Kaiser M, Walsh CT, Chem. Biol 2010, 17, 1077–1083; [DOI] [PMC free article] [PubMed] [Google Scholar]; e) McMahon MD, Rush JS, Thomas MG, Bacteriol J. 2012, 194, 2809–2818; [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Mori S, Garzan A, Tsodikov OV, Garneau-Tsodikova S, Biochemistry 2017, 56, 6087–6097; [DOI] [PubMed] [Google Scholar]; g) Mori S, Pang AH, Lundy TA, Garzan A, Tsodikov OV, Garneau-Tsodikova S, Nat. Chem. Biol 2018, 14, 428–430. [DOI] [PubMed] [Google Scholar]
- [6].a) Boll B, Taubitz T, Heide L, J. Biol. Chem 2011, 286, 36281–36290; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Zhang C, Kong L, Liu Q, Lei X, Zhu T, Yin J, Lin B, Deng Z, You D, PLoS One 2013, 8, e56772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Davidsen JM, Bartley DM, Townsend CA, J. Am. Chem. Soc 2013, 135, 1749–1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Schomer RA, Thomas MG, Biochemistry 2017, 56, 5380–5390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].a) Drake EJ, Cao J, Qu J, Shah MB, Straubinger RM, Gulick AM, J. Biol. Chem 2007, 282, 20425–20434; [DOI] [PubMed] [Google Scholar]; b) Buchko GW, Kim CY, Terwilliger TC, Myler PJ, Tuberculosis (Edinb) 2010, 90, 245–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].a) Herbst DA, Boll B, Zocher G, Stehle T, Heide L, J. Biol. Chem 2013, 288, 1991–2003; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Miller BR, Drake EJ, Shi C, Aldrich CC, Gulick AM, J. Biol. Chem 2016, 291, 22559–22571; [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Tarry MJ, Haque AS, Bui KH, Schmeing TM, Structure 2017, 25, 783–793. e784. [DOI] [PubMed] [Google Scholar]
- [11].Gulick AM, ACS Chem. Biol 2009, 4, 811–827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Lombó F, Velasco A, Castro A, de la Calle F, Brana AF, Sanchez-Puelles JM, Mendez C, Salas JA, ChemBioChem 2006, 7, 366–376. [DOI] [PubMed] [Google Scholar]
- [13].a) Sheoran A, King A, Velasco A, Pero JM, Garneau-Tsodikova S, Mol. Biosyst 2008, 4, 622–628; [DOI] [PubMed] [Google Scholar]; b) Mady AS, Zolova OE, Millan MA, Villamizar G, de la Calle F, Lombó F, Garneau-Tsodikova S, Mol. Biosyst 2011, 7, 1999–2011. [DOI] [PubMed] [Google Scholar]
- [14].Mori S, Shrestha SK, Fernandez J, Millan M. Alvarez San, Garzan A, Al-Mestarihi AH, Lombó F, Garneau-Tsodikova S, Biochemistry 2017, 56, 4457–4467. [DOI] [PubMed] [Google Scholar]
- [15].Al-Mestarihi AH, Villamizar G, Fernandez J, Zolova OE, Lombó F, Garneau-Tsodikova S, J. Am. Chem. Soc 2014, 136, 17350–17354. [DOI] [PubMed] [Google Scholar]
- [16].Wang ZX, Li SM, Heide L, Antimicrob. Agents Chemother 2000, 44, 3040–3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Watanabe K, Hotta K, Praseuth AP, Koketsu K, Migita A, Boddy CN, Wang CC, Oguri H, Oikawa H, Nat. Chem. Biol 2006, 2, 423–428. [DOI] [PubMed] [Google Scholar]
- [18].Fujimori DG, Hrvatin S, Neumann CS, Strieker M, Marahiel MA, Walsh CT, Proc. Natl. Acad. Sci., U. S. A 2007, 104, 16498–16503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Choi R, Kelley A, Leibly D, Hewitt SN, Napuli A, Van Voorhis W, Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun 2011, 67, 998–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Evans BS, Chen Y, Metcalf WW, Zhao H, Kelleher NL, Chem. Biol. 2011, 18, 601–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].a) Lebowitz J, Lewis MS, Schuck P, Protein Sci. 2002, 11, 2067–2079; [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Balbo A, Schuck P, (Eds.: Golemics E, Adams PD), Cold Spring Harbor Press, New York, 2005. [Google Scholar]
- [22].Laue TM, Shah BD, Ridgeway TM, Pelletier SL, in Analytical ultracentrifugation in biochemistry and polymer science. (Eds.: Harding SE, Rowe AJ, Horton JC), The Royal Society of Chemistry, Cambridge, England, 1992, pp. 90–125. [Google Scholar]
- [23].Larkin MA, Blackshields G, Brown NP, Chenna R, McGettigan PA, McWilliam H, Valentin F, Wallace IM, Wilm A, Lopez R, Thompson JD, Gibson TJ, Higgins DG, Bioinformatics 2007, 23, 2947–2948. [DOI] [PubMed] [Google Scholar]
- [24].Saitou N, Nei M, Mol. Biol. Evol 1987, 4, 406–425. [DOI] [PubMed] [Google Scholar]
- [25].Kumar S, Stecher G, Tamura K, Mol. Biol. Evol 2016, 33, 1870–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Zuckerkandl E, Pauling L, in Evolving genes and proteins, Academic Press, New York, 1965, pp. 97–166. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.