

Abstract
Quantification of the multivalent interactions of influenza viruses binding at interfaces may provide ways to tackle key biological questions regarding influenza virulence and zoonoses. Yet, the deconvolution of the contributions of molecular and interfacial parameters, such as valency, interaction area, and receptor density, to the binding of whole viruses is hindered by difficulties in the direct determination of these parameters. We report here a chemical platform technology to study the binding of multivalent recombinant hemagglutinin (rHA) nanoparticles at artificial sialoglycan cell receptor-presenting interfaces in which all these parameters can be derived, thus allowing the desired full and quantitative binding analysis. SiO2 substrates were functionalized with supported lipid bilayers containing a targeted and tunable fraction of a biotinylated lipid, followed by the adsorption of streptavidin and biotinylated polyvalent 2,3- or 2,6-sialyl lactosamine (SLN). rHA nanoparticles were used as a virus mimic to provide a good prediction of the number of interactions involved in binding. Low nanomolar affinities and selectivities for binding at the 2,6-SLN platforms were observed for rHA particles from three different virus variants. When fitting the data to a multivalency model, the nanomolar overall affinity appears to be achieved by 6–9 HA–sugar molecular interaction pairs, which individually present a rapid association/dissociation behavior. This dynamic behavior may be an essential biological attribute in the functioning of the influenza virus.
Keywords: multivalency, influenza virus, interface, hemagglutinin, cell mimics, glycans
Influenza remains a threat to global health, causing millions of human infections and substantial mortality every year.1,2 Influenza A viruses are subtyped based on the antigenic properties of the two glycoproteins hemagglutinin (HA) and neuraminidase (NA).3 HA is responsible for binding of the virus to sialic acid (SA)-terminated carbohydrates present at cell membranes, and the resulting adsorption of the virus to a cell membrane embodies the onset of the infection. The initial virus attachment to the cell is regulated by multivalent interactions, where homotrimeric HA binds to SAs and multiple HA trimers are involved in the interaction with the carbohydrate-covered cell surface.4 The NA, instead, facilitates the release of the virus from the cell after reproduction by cleaving the SA residues present on the cell membrane.5
The overall affinity of the virus binding depends on the virus strain, expressed in the occurrence of different HA and NA subtypes, in combination with the specific form and density of SA presented at the membrane. Together, these factors determine the specificity of a virus for a particular host species. For example, avian influenza viruses bind preferentially to 2,3-sialyl-(N-acetyl-lactosamine) residues (2,3-SLN) while human influenza viruses show a preference for 2,6-sialyl-(N-acetyl-lactosamine) residues (2,6-SLN).6,7 Switching of a virus’ specificity to another host species occasionally occurs and may cause a pandemic when such a “zoonotic” virus further adapts to humans by improving its replication/transmission efficiencies.8,9 The latest example, the outbreak of the 2009 influenza pandemic,10 stresses the importance of a thorough understanding of the factors driving such events. Alteration of the binding specificity is essential at this first stage, and this involves, for instance, mutation at the HA binding site and/or reassortment of different HA and NA glycoproteins. However, subsequent adaptations appear to be required,8,9 including those that improve the functional balance between the HA and NA glycoproteins. To better understand these virus changes, it will be key to thoroughly understand the relationship between multivalent HA binding and virus infectivity at the molecular level.
A first step toward an improved molecular understanding is to provide a platform to study the binding selectivity of various virus strains for different SA residues and to quantify their interaction at these artificial cell surface mimics. Some examples of 2D sensor platforms bearing surface modifications for the study of the interaction of viruses with model cell receptors have been reported.8,11,12 These platforms, based on streptavidin-modified surfaces at which a fast and easy modification with biotinylated receptors or aptamers is achieved, allow an efficient detection of viruses. However, this type of surface modification does not resemble the structure and properties of the cell surface, such as, for example, the membrane fluidity. Moreover, the sugar density at cell membranes is known to affect the binding characteristics of the influenza virus strongly by influencing the valency and the multivalent effect of the overall interaction.13,14 Therefore, it is important to design platforms that allow a good control over the SA density at the surface.
Various methods have been developed to achieve control over surface densities of ligands or receptors, using self-assembled monolayers (SAMs) or (fluid) supported lipid bilayers (SLBs), to obtain static or laterally mobile layers, respectively.15−17 For example, the surface density of surface-exposed NTA(Ni) moieties was controlled using mixed SAMs of suitably modified thiols for His6-tag protein immobilization,18 the surface density of arginine–glycine–aspartic acid (RGD) peptide was varied using SAMs to investigate the effect on cell binding,19 and the surface density of biotin was controlled both using SAMs and SLBs to investigate the multivalent binding of streptavidin.20 However, so far, SLBs have not been used to quantify the interaction of influenza viruses.
A surface analytical technique such as biolayer interferometry (BLI) has proven to be suitable for the study of virus–platform interactions, and it allows the determination of the selectivity of different virus variants or mutants for specific SA residues at surfaces at which the SA density is varied.8 However, a molecular understanding of the avidity observed for the interactions between influenza viruses and SAs is required to better understand the mechanism of virus infection and the role of multivalent binding therein. Another important open question is whether the bound virus remains dynamic upon adsorption, which is in large part determined by the nature of the multivalent binding. Technical issues of the BLI technique, such as limited knowledge of the surface presentation of the sugars at the detection platforms, as well as biological ones, such as the inhomogeneity of whole virus samples regarding size distribution, contact area when adsorbing to a surface, and distribution of HA and NA proteins, prohibit a better understanding of the valency of the binding and the resulting multivalent effects.
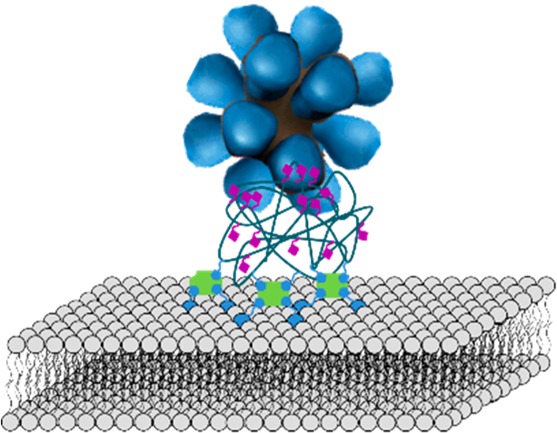
Here, we report a SA receptor-presenting SLB platform that functions as a mimic for a cell membrane and aims to provide control over the receptor density. Together with the use of recombinant protein clusters (“rosettes”) of HA (rHA) as virus models, this system enables control of the interaction area between the surface and the virus-like particle and thereby of the binding valency. The surface modification with SLBs provides a well-known cell membrane mimic that offers ease of preparation, controlled functionalization by incorporation of tunable fractions of functionalized lipids, and excellent nonfouling properties.21 By introducing tunable amounts of biotinylated lipids in the SLB, followed by the attachment of streptavidin (SAv), the surfaces have been functionalized with biotinylated human or avian receptors with control over their surface density. rHA rosettes bind selectively to the receptors, and their interactions have been quantified using quartz crystal microbalance with dissipation monitoring (QCM-D). The tunable sugar density at the SLB platform, together with the controlled valency of the rHA protein cluster in its interaction with this platform, enables the use of a multivalent binding model to quantify the multivalent interaction in terms of the individual affinity constant of a single HA-receptor site, the valency, and the receptor density-dependent, effective molarity.
Results and Discussion
Design and Characterization of the SLB Platform
To achieve control over the interaction area between the cell surface mimic and a virus particle, we have employed small rHA nanoparticles, also called rosettes, as a model for the influenza virus, while at the same time reducing complicating factors due to the heterogeneity of whole influenza viruses regarding their size, shape, and HA and NA fractions. Transmission electron microscopy (TEM) measurements showed that these rosettes are approximately 22 nm in size and consist of, on average, 10–12 recombinant HA0 (rHA) trimers embedded in a surfactant layer (see Supporting Information (SI), Figure S1).22,23 rHAs of different influenza A/H1N1 viruses, with affinities for different receptors, have been used here: influenza viruses A/California/07/2009 (Cal/09), A/New Caledonia/20/99 (NC/99), and A/Brisbane/59/07 (Bris/07) virus. All three viruses have been reported to bind preferentially to human sialic acid (2,6-SLN) residues.24−26
The SLB-based interaction platform was built up in a number of steps as schematically presented in Figure 1A. Unilamellar vesicles consisting of both 1,2-dioleoyl-sn-glycero-3-phosphocholine (DOPC) lipids and a targeted fraction of the lipid 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(biotinyl) (DOPE-biotin) were prepared by extrusion, using a polycarbonate membrane with 100 nm pore size, and their size was measured to be 78 ± 29 nm by dynamic light scattering (SI, Figure S2). Such vesicles are known to adhere to and rupture on oxidized glass substrates, resulting in SLBs that display biotin moieties at the SLB–water interface.20,27 SLBs consisting of zwitterionic DOPC lipids have been demonstrated to suppress nonspecific interactions effectively.21 This is of particular importance here, considering that the binding of viruses to the SLBs is based on multiple weak specific interactions, necessitating the suppression of nonspecific interactions.
Figure 1.

Schematic representation of the SLN-modified SLB platform and its interaction with rHA nanoparticles. (A) Step-by-step formation of the platform: the first step consists of the formation of the biotinylated SLB on a silicon oxide substrate presenting water at the interface, followed by adsorption of SAv and subsequently by the adsorption of biotinylated polyvalent SLN (biotin-PAAm-SLN) that can interact with the rosettes. (B) Molecular structure of the molecules used for the formation of the platform.
The density of biotin moieties displayed at the SLB can be conveniently controlled by mixing in the desired fraction, here varied from 0.1 to 5%, of the DOPE-biotin lipid during vesicle preparation. Subsequently, streptavidin (SAv) was bound to the surface by exploiting the strong biotin–SAv interaction. The surface-bound SAv presents additional free binding pockets, and these were used to bind a poly[N-(2-hydroxyethyl)acrylamide]-based (PAAm) polymer (biotin-PAAm-SLN, see Figure 1B), presenting both biotin and SLN moieties in a random fashion at the PAAm backbone. Polymers with an average of 22 SLN moieties and 5.5 biotins per polymer chain (thus with a ratio of 4 SLNs per biotin unit) were used. By controlling the biotin density in the SLB, the SAv density and ultimately the SLN density can be tuned. Such polymers are routinely used for the variation of SLN density in virus binding studies using BLI.28,29
QCM-D was used to monitor in situ the step-by-step formation of the SLB platform as well as the interaction of the rosettes with surface-bound SLNs (Figure 1A), thus allowing quantitative comparison of the experiments for different (2,6 and 2,3) SLN receptors and different rHA particles. An important consideration when using QCM-D for the (quantitative) analysis of biological entities at surfaces is the contribution of hydrated mass to the QCM-D output parameters, i.e., frequency (f) and dissipation (D). Because the associated water fraction is generally unknown, the relative surface coverages of bound molecules or particles can be determined from the QCM-D output (as the relative coverage will depend linearly on the frequency shift), but the absolute coverages cannot be determined exactly. Therefore, we primarily used the frequency shifts (Δf) to obtain relative coverages of rHA rosettes. Yet the relative contribution of the mass of hydrated water can still depend on the packing density of the adhered particles, especially at high packing densities, where hydration shells can significantly overlap, deviations from linearity may occur.30
In Figure 2A, an example of four parallel QCM-D measurements is presented where the DOPE-biotin fraction was varied between 0.1 and 5%. The first step, corresponding to the adsorption of the vesicles and their subsequent rupture, indicates the formation of high quality SLBs (i.e., Δf = −24 ± 1 Hz and ΔD < 0.5 × 10–6) on the SiO2-coated sensors.27 An intriguing property of SLBs is the lateral mobility of individual lipid constituents. The lateral mobility of the lipid bilayer, as well as of SAv bound to the biotin groups in a subsequent step, was confirmed using a fluorescently labeled lipid and fluorescently labeled SAv, respectively. DOPC-based SLBs with 1 mol % DOPE-biotin showed a lateral mobility of bound SAv similar to that of the native SLB, as was verified by fluorescence recovery after photobleaching (FRAP, see SI, Figure S3).
Figure 2.

Control over the SLN density at the SLB platform. (A) Example of four parallel QCM-D measurements showing SLB formation using DOPC vesicles with a molfraction x (0.1, 0.4, 1, and 5%) of DOPE-biotin followed by binding of SAv (0.5 μM) and subsequently of biotin-PAAm-2,6-SLN (4 μg/mL). Gray areas indicate the binding steps and white areas indicate buffer wash steps. All steps are under flow. (B) Correlation between QCM-D frequency shifts (Δf5, 5th overtone) of SAv binding as a function of the DOPE-biotin fraction. The red dashed line is a guide to the eye. (C) Correlation between QCM-D frequency shifts of the biotin-PAAm-2,6-SLN binding as a function of the frequency shift for SAv binding. The red dashed line is a linear fit to the data points.
Subsequently, all four substrates were washed with buffer and incubated with SAv. The frequency shift induced by adsorption of SAv as a function of the fraction of biotin in the SLB is reported in Figure 2B. The data shows a close-to-linear trend between the biotin density and the coverage of SAv up to 1% of DOPE-biotin, but the SAv coverage saturated at higher DOPE-biotin fractions. This observation suggests that above 1–2 mol % of DOPE-biotin, the surface reaches physical saturation with SAv, which is in agreement with previously reported dense packing of SAv at SLBs that were functionalized with 5% or 10% of biotin.31,32 The frequency shift of the subsequent binding of biotin-PAAm-2,6-SLN onto the SAv-modified substrates was found to be linearly related to the coverage of SAv that was reached in the preceding step (Figure 2C).
The results discussed above show that this platform is suited to tune the density of the sialic acid residues presented at the SLB platform. When we assume (i) that every SAv binds to two biotin moieties of DOPE-biotin present in the SLB,20 and (ii) that all biotin moieties of biotin-PAAm-SLN bind to and saturate the remaining available binding pockets of SAv, SLN densities values are estimated between 0.92 and 45.8 pmol/cm2 for 0.1 and 5% of biotin, respectively (see SI), here ignoring packing effects of SAv. Experimental average SLN densities ranged from 3.5 to 26 pmol/cm2, corresponding to 6.9 and 2.5 nm average spacing between SA residues, respectively. These values are based on an estimated 80% water content (based on experimental values ranging from 70% to 90% obtained for other large biomolecules)21 and the assumption that the Sauerbrey model is valid (a reasonable assumption considering the small increase in dissipation for the polymer binding step). The observed differences between the calculated and the experimental values for the SLN densities has reasonable explanations at both the lower and higher biotin densities: At a low fraction of DOPE-biotin in the SLB, and a concomitantly low coverage of SAv, the polymeric sugar probably binds to the surface with only two (of approximately 5.5) biotin moieties per polymer chain, allowing the attachment of a higher relative amount of polymer on the surface. At the other limit, the calculated value does not take into account the SAv saturation on the surface that occurs at a biotin percentage above 2% (Figure 2B), and thus not every biotin from DOPE-biotin can bind to SAv because of steric hindrance and, therefore, this omission leads to an overestimation of the polymer density on the surface. Yet, both the experimental and model values are average densities and do not take into account the probably inhomogeneous SLN distribution over the sensor surface which is related to the use of a polymer with a fixed degree of functionalization with biotin and SLN moieties. This issue is discussed in more detail below.
Rosette Binding on the SLB Platform
The specificity and selectivity of the interaction between the rHA rosettes and the SLN-displaying platform were investigated by studying the adsorption of the different nanoparticles onto SLBs with varying types and densities of SLN receptors. After verifying the formation of biotin-functionalized SLBs, the subsequent binding of SAv and of a biotinylated PAAm polymer, two different concentrations (0.14 and 0.56 nM) of Cal/07 rHA rosettes were flown over SLBs modified with biotin-PAAm (without SLN) or biotin-PAAm-2,6-SLN. Figure 3A shows the successful binding of rosettes to surfaces that were functionalized with 2,6-SLN, while no binding was observed in the absence of 2,6-SLN at the SLB. The higher dissipation signals relative to the obtained frequency shifts, in comparison with the results presented in Figure 2A, suggests that the rosettes interact with the SLB as intact particles. These results indicate that nonspecific interactions between the platform and the rosettes are negligible and that the binding of the rosettes to the SLN-functionalized platform is caused by specific, i.e., SLN-HA, interactions.
Figure 3.

Selectivity of the binding of HA rosettes at the SLB platform. (A) QCM-D results of the binding of A/California/07/2009 (Cal/09) rHA rosettes to SLBs modified with biotin-PAAm-2,6-SLN or biotin-PAAm (without SLN). (B) QCM-D results of the binding of A/Brisbane/59/07 (Bris/07) rHA rosettes to SLBs modified with biotin-PAAm-2,6-SLN or biotin-PAAm-2,3-SLN. Steps shown here were performed after (not shown): formation of SLB presenting biotin groups, subsequent binding of SAv, and of biotin-PAAm with/without SLN groups. DOPE-biotin densities were (A) 1% and (B) 0.4%. Gray areas indicate the binding steps and white areas indicate buffer wash steps. All steps were recorded under flow.
Various influenza viruses are known to bind selectively to specific glycan structures. Therefore, the selectivity of the binding of the Bris/07 rHA rosettes to both 2,6-SLN- and 2,3-SLN-functionalized SLBs was evaluated. A much higher (approximately 4-fold) QCM-D response was observed (see Figure 3B) for the 2,6-SLN-modified SLBs, indicating a preference for binding of the Bris/07 rHA virus particles to the human 2,6-sialic acid residues. This difference was not due to a different density of SA residues bound to the sensor surface because the coverages of biotin-PAAm-SLN at the biotin-SAv-modified SLB were comparable (<10% difference in Δf5). The rHA rosette binding was evaluated at two concentrations, showing an increased amount of binding at the higher concentration but with similar selectivity. Even more selective binding was observed in the case of binding of Cal/09 and NC/99 rHA clusters to 2,6-SLN-modified SLBs: no significant binding was observed for either of these clusters on 2,3-SLN surfaces, while significant binding was observed at 2,6-SLN surfaces (SI, Figure S4).
To determine the overall dissociation constant (Kd) of the interaction of Cal/09 rHA clusters with the 2,6-SLN-modified SLBs, solutions of Cal/09 rHA clusters at concentrations ranging from 0.14 to 4.2 nM were titrated at the surface and adsorptions were monitored with QCM-D. Biotin fractions in the SLB of 0.4 and 5 mol % were used, and Figure 4A reports the frequency shifts obtained in the titration performed with the SLB containing 0.4% of DOPE-biotin. Figure 4A shows clear binding steps at all concentrations, visible both in the frequency and dissipations signals, in agreement with the adsorption of soft particles. The adsorption steps reached a plateau in approximately 10 min, while flow of the respective solutions was maintained for 40 min, indicating that equilibrium was reached in each step. The reversibility of the particle binding is further indicated by the observed, though slow desorption upon switching the flow to buffer after the last particle solution. The data confirms qualitatively that the rosettes allow the assessment of their binding affinity by employing regular titrations performed under thermodynamic equilibrium.
Figure 4.

Affinity of Cal/09 rHA rosettes at the 2,6-SLN surface and effect of sugar density. (A) QCM-D titration of the Cal/09 rosettes at a 2,6-SLN-presenting SLB with 0.4 mol % of DOPE-biotin present in the SLB. Gray areas indicate the binding steps and white areas indicate buffer wash. All steps were performed under flow. Surface functionalization up to the rHA cluster binding was monitored as well but not shown here. (B) Binding curves from QCM-D titrations for Cal/09 rHA clusters at 2,6-SLN surfaces starting from 0.4% (red circles, left y-axis) and 5% (black squares, right y-axis) DOPE-biotin. Langmuir model fitting (solid lines) provided the binding constants shown.
Figure 4B shows the resulting binding data when plotting the plateau values of the frequency shift after each rosette binding step versus the concentration of the rosette. A Kd of 5.2 nM was found from fitting the 0.4 mol % DOPE-biotin data with a standard 1:1 (Langmuir) model. When the same type of titration was performed on a 2,6-SLN-modified SLB with a higher fraction (5 mol %) of DOPE-biotin, which resulted in a three times higher surface coverage of 2,6-SLN (based on Figure 2C), a very similar Kd of 9.4 nM was found (Figure 4B). In correspondence with this relatively strong binding, limited, but notable desorption was observed at the measurement time scale when washing the surface with buffer after the titration (Figure 4A).
The Langmuir fits of the dissociation constants (Figure 4B) require cofitting of the frequency shift plateau values that correspond to saturation of the surface with rHA nanoparticles. For the 0.4% and 5% platforms, these saturation frequencies, Δfmax, were 95 and 335 Hz, respectively. These values agree reasonably well with the relative differences in SLN receptor and SAv densities at these platforms as mentioned above. However, the plateau values estimated by these fits have a relatively large error because the titration data do not level off sufficiently to estimate more accurate values of the saturation levels. Limited stock concentrations of the rosettes prohibited us, however, from extending the titrations to higher concentrations.
QCM measurements of the binding of biotinylated lipid bilayer vesicles of 100 nm in diameter at SAv-modified SLBs showed a maximal binding frequency of about 150 Hz at dense vesicle coverage (see SI, Figure S5). This suggests that the here used smaller rHA rosettes probably bind in a close to dense fashion at the 5% platform and that the plateau frequency will most likely not be much larger than now estimated. Consequently, the estimated Kd values will not be much higher than the fitted values given above. Overall, this analysis indicates that the dissociation constants are definitely in the low nM regime.
Noteworthy, the Kd values for the interaction of the 0.4% and 5% platforms with the Cal/09 rosette are very similar. This seemingly contradicts published work performed on whole viruses which have shown strong dependencies of the binding affinity on the (polyvalent) sugar density.8,13,29 At the same time, however, it must be noted that these data may not be directly comparable, as these are often performed at only one virus concentration. As a consequence, full titrations (i.e., with different concentrations of virus to obtain different surface coverages), like done here for the rosettes, are normally not performed with whole viruses. For example, concentrations of 100 pM of whole influenza virus were used by Gamblin et al.13 to achieve either partial or full virus coverage of surfaces coated with the same biotin-PAAm-2,6-SLN polymer as used in this work and were used for monitoring relative differences between viruses without determining the binding constants.
Similar titrations were performed for rHA rosettes derived from NC/99 (at 0.4 mol % DOPE-biotin only) and Bris/07 (at 0.4 mol % and 5 mol % DOPE-biotin) (see SI, Figures S4 and S6), and the resulting overall dissociation constants are summarized in Table 1. Comparing the results for the three rosettes, very similar dissociation constants were found for the 2,6-SLN presenting surfaces, all in the low nM concentrations. Both the Cal/09 and Bris/07 rosettes showed (slightly) higher dissociation constants, i.e. weaker binding, at the 5% platforms in comparison to the 0.4% platforms. The differences in binding affinity may be due to the error involved with estimating the saturation frequency values, as indicated above, and possibly to small deviations from the linear frequency dependence at the densely packed 5% surface (see below), but it contrasts data on whole viruses that show generally stronger binding at higher receptor densities. Moreover, higher saturation values were obtained for Bris/07 clusters compared to the other clusters tested here. This may be due to a difference in size of the nanoparticles and, therefore, a different packing density on the surface, as well as a difference in the hydration of the rosettes. However, when the data were fitted by fixing the saturation values to 100 and 300 Hz at 0.4 mol % and 5 mol % DOPE-biotin, respectively, the Kd values obtained from the fitting did not decrease significantly: values of 1.9 and 6.4 nM were found for the lower and higher SLN densities, respectively, confirming the same low-nM Kd range already observed for the Cal/09 rosettes described above.
Table 1. Dissociation Constants for rHA Rosettes of Three Different Viruses at 2,6-SLN-Presenting Surfaces with 0.4 mol % or 5 mol % DOPE-Biotina.
| DOPE-biotin (mol %) | Kd/nM (Δfmax/Hz) Cal/09 | Kd/nM (Δfmax/Hz) Bris/07 | Kd/nM (Δfmax/Hz) NC/99 |
|---|---|---|---|
| 0.4 | 5.2 (95) | 3.2 (130) | 3.4 (71) |
| 5 | 9.4 (335) | 20 (739) |
In parentheses are given the saturation frequency shifts calculated from the Langmuir fits for each titration.
As mentioned above, the relative contribution of hydrated water can depend on the packing density of the adhered entities, in this case the rHA rosettes. To evaluate whether possible hydration shell overlap influences the observed binding curves, we evaluated the dissipation signal as a function of the frequency shift for all titrations. SI, Figure S7, shows that these −Δf/ΔD plots were largely linear at 0.4 mol % DOPE-biotin density, whereas at 5 mol % DOPE-biotin density, the corresponding titrations showed nonlinear behavior with the ΔD leveling off at higher Δf values, which may be related to increasingly overlapping hydrations shells of the rHA rosettes. This indicates that the relative frequency shifts and the saturation frequencies are more reliable for the 0.4% platforms, and therefore we interpret the binding data obtained at the 5% platforms to be essentially very similar in affinity as observed for the 0.4% ones.
When we assume a plateau value to hold for a particular rosette (for example, of 130 Hz for Bris/07 at the 0.4% platform), Kd values for the 2,3-SLN platforms can be determined as well, leading to a Kd value of 26 nM of the Bris/07 rosette on the 0.4% platform. The 1 order of magnitude weaker binding to the 2,3-platform compared to the 2,6 is a clear signature of the difference in binding selectivity of the HA of this rosette.
As observed (Figure 4B, and Table 1, Δfmax values), the absolute amounts of binding are higher at the higher sugar densities. However, these results do not show the expected increased affinity for the higher sugar density. Instead, the affinities (Kd values) appear unaffected by the SLN surface density, suggesting that the sugar density sensed by the HA rosettes does not change with increasing densities of biotin-PAAm-SLN. Therefore, we here propose that this insensitivity of the rosette binding to the sugar density is caused by a fixed local SLN density due to restriction of the interaction area of a rosette to a single polymeric biotin-PAAm-SLN at the SLB platform.
Figure 5A shows a schematic top view of the surface and how a rosette particle interacts with it. Looking at the composition and structure of the SLN-modified polymer, the biotin-PAAm-SLN contains approximately 115 acrylamide monomer units with a stretched main chain of approximately 30 nm, of which, on average, 22 monomer units contain an SLN moiety and 5.5 a biotin group. One polymer chain can therefore bind 2–3 SAv proteins simultaneously. Indeed, adsorbed biotin-PAAm-SLN has been shown to cover an area with a diameter of 15 nm,33i.e., of 175 nm2, which can easily accommodate the area of the SAv molecules it binds to (approximately 25 nm2 per protein). From the SAv-bound biotin moieties located at the surface, small chain segments with a length of a few nm may stick out upward and sideward, exposing SLN units (and their linker chains) to provide additional flexibility.
Figure 5.

Interaction and contact area of an HA rosette binding to a single biotin-PAAm-SLN polymer at the SLB platform. (A) Schematic presentation of the constant local SLN density sensed by the HA rosettes for different biotin-PAAm-SLN coverages. (B) Representation of a single rosette interacting with three of its HA trimers with a single SLN-polymer at the SLB substrate.
The HA clusters have a diameter of 22 nm, assuming a diameter of about twice the length of one HA trimer.22 With 10–12 HA trimers, we can view such a particle as an icosahedron with an HA trimer at (almost) each apex, with an angle of 63° between neighboring trimers. This gives a contact area in which three trimers interact with the substrate, while the other trimers will be >5 nm away from the surface. At the protruding tips of the trimers, tip–tip distances of 15 nm can be estimated in this geometry, which is not far off from the 11 nm trimer–trimer distance in whole viruses.34,35 The somewhat larger tip–tip distance in the rosettes is a direct result of the much higher curvature of the smaller rHA rosettes compared to the whole virus.
Taken together, these considerations indicate an excellent match between the contact area of a rHA rosette and that of a single biotin-PAAm-SLN polymer molecule displayed at the sensor platform. This analysis supports the observed lack of density dependence in the binding behavior of the rHA rosettes, and it also explains the seemingly contradictory coverage-dependent virus binding observed in literature. Because the whole virus has a diameter of approximately 100 nm, it has a much larger contact area and valency with the substrate than an rHA nanoparticle. As a consequence, the virus can interact with multiple biotin-PAAm-2,6-SLN polymer molecules simultaneously, whereas the rosettes only bind to a single polymer molecule at a time, making the interaction of the whole virus sensitive to the polymeric SLN density whereas that of the rosette is not.13
The relatively small and well-defined contact area between an rHA rosette and the substrate allows for a detailed description of the multivalent interaction and the overall affinity resulting from the interaction. From an estimated interaction area involving three HA trimers, a valency of 6–9 can be estimated, depending on whether all three sites of a trimer can interact or not. For a tripodal arrangement of trimers, maximally two sites of each bonding trimer can be in direct contact with the substrate at any time. Yet, the third site of these trimers is at a distance of approximately 2.5 nm from the surface. From seeing the length and flexibility of the biotin-PAAm-SLN polymer and the linker connecting the SLN moieties to the polyacrylamide backbone, we estimate that this distance can be bridged easily by the SLN-modified polymer segments protruding from the surface.
The overall binding affinity, Kov (which is the inverse of Kd experimentally assessed above), of a multivalent ligand at a surface, following methodology developed earlier in our group,36 can be described as follows (eq 1).
| 1 |
Here, Ki is the intrinsic affinity constant of a single interaction pair, here between an HA monomeric binding site and an SLN moiety, EM is the effective molarity, which is a measure for the probability of intramolecular bond formation applicable to additional interaction pairs formed upon formation of the first intermolecular interaction, and n is the valency of the multivalent interaction. Here, we ignore statistical prefactors and differences in probabilities of intramolecular bond formation resulting from the tripodal trimeric arrangement, which would formally need a nested multivalent approach. Yet, because of the flexibility of the biotin-PAAm-SLN polymer, and the similar distances between binding sites with an HA trimer (5 nm) and between sites from neighboring trimers (7–8 nm), we here assume one value for EM to hold for all intramolecular binding steps of the rHA particle to the surface. Furthermore, eq 1 only holds when the multivalent enhancement factor, KiEM, which is a measure of how much Kov is enhanced when an additional binding site is added to the multivalent interaction, is substantially larger than 1.37
From the titrations with the rHA rosettes at the 0.4% platforms (see Table 1) and the nanomolar Kd values found here, Kov values can be calculated to be approximately 2–3 × 108 M–1. Values for the monovalent interaction affinity (Ki) of SLN with HA have been reported in the literature for different influenza variants, and we here assume a value of 1000 M–1 (a Kd of 1 mM).13 When assuming all sites of three HA trimers to be involved (n = 9), KiEM can be calculated to be approximately 5, leading to an EM value of approximately 5 mM. For two sites per HA trimer (n = 6), KiEM can be calculated to be around 12, leading to an EM value of approximately 12 mM. It should be noted that this analysis of the KiEM and EM values is rather insensitive to changes in n and Kov. This is a direct result of the exponential relationship shown in eq 1. When rewritten as eq 2, it becomes clear that KiEM is only logarithmically dependent on the ratio Kov/Ki and inversely on the number of intramolecular bonds, n – 1.
| 2 |
Therefore, limited accuracy in the determination of Kov can be tolerated, as even variations of an order of magnitude have limited influence on KiEM. Likewise, the range of n values assumed here has only limited effect on KiEM, as already shown above. Errors in Ki have a similarly low effect on KiEM but have of course a direct influence on EM.
EM values on the order of 10 mM are not unreasonable for such surface densities, and slightly higher values (on the order of 100 mM) have been obtained for cyclodextrin host–guest surface assemblies that have a higher surface receptor density.38 Besides the difference in binding site density, the lower EM value obtained for the rHA rosettes compared to other systems might be attributed to the rigidity of the rosettes interacting with the receptor surface. It should be noted that assuming a lower valency leads to a higher EM value needed to explain the overall affinity, as explained above.
Apparently and noteworthy for this system, KiEM is well above 1, confirming the validity of eq 1, but is at the same time only moderately high, on the order of 10. The moderate nature of the value of KiEM indicates that an increase of the valency of the system has only a moderate effect on the overall affinity. In other words, the here observed difference of approximately 5 orders of magnitude in affinity between the monovalent (Ki) and multivalent interaction (Kov) is reached with 6–9 molecular interaction pairs (of which 1 is regarded as intermolecular, and 5–8 as intramolecular), so less than 1 order of magnitude per added site. Such avidities are not uncommon for biological systems like influenza inhibitors39 but are in contrast to much stronger multivalent effects observed for synthetic systems where KiEM values of >1000 have been observed.38,40
What is the biological relevance of the multivalent enhancement factor KiEM? Apart from the molecular understanding of what a binding site contributes to the overall affinity increase in a multivalent system, it gives insight into the dynamics of a system. The ratio of lifetimes of the bound and unbound states of an interaction pair in an intramolecular system is given by KiEM:1, and therefore the corresponding relative bound/unbound fractions by KiEM/(KiEM + 1) and 1/(KiEM + 1), respectively. For moderate values of KiEM, say ranging from 0.1 to 10, these bound and unbound lifetimes are of the same order of magnitude, indicating that each interaction pair is dynamically equilibrating between its bound and unbound states, the frequency of which is dictated by the intrinsic dissociation rate constant, kd,i. For considerably stronger multivalent systems with KiEM ≫10, for example, >1000 as observed before for cyclodextrin surfaces,38,40 the bound lifetime is orders of magnitude higher than the unbound one, and consequently the bound/unbound dynamics is reduced. This kinetic trapping at high KiEM has been observed in an earlier study,39 where only a divalent guest showed measurable surface diffusion along a cyclodextrin-coated surface, while mobility of a trivalent guest was not observed due to too strong binding.
The binding energy landscape of a multivalent particle at a surface is therefore described by both a thermodynamic parameter (the difference between the overall and monovalent affinities) as well as a kinetic parameter (the average fraction of bound sites). Figure 6 shows this energy landscape graphically, by plotting the bound fraction, KiEM/(KiEM + 1), and the avidity parameter, log Kov – log Ki, as a function of the multivalency parameters, i.e., the valency, n, and the multivalent enhancement factor, KiEM. When KiEM < 0.1, the system behaves basically as a monovalent system: Kov ≈ Ki and the bound fraction of the interaction sites (and for each site individually) is below 10%. At high KiEM, >10, Kov scales as given by eq 1, and thus log Kov, is linearly dependent on n and on log KiEM. That means that systems with high valencies reach very high Kov values. Taken together with a bound fraction that approaches 1, indicating that all sites are practically all of the time in the bound state, such systems get kinetically trapped: neither spontaneous desorption, which would require dissociation of all binding sites, nor interfacial mobility, which is based on partial site dissociation, are possible under these circumstances. In between these extremes, we call these systems “weakly multivalent”: for moderate values of KiEM, ranging from 0.1 to 10 (as indicated green in Figure 6), the system is multivalent and dynamic at the same time. Additional binding sites do contribute to the overall affinity, but with less than 1 order of magnitude, and the fractions of bound and unbound sites are comparable as well as their lifetimes. Therefore, such systems can exhibit dynamic behavior, especially in processes like interfacial mobility in which only partial site dissociation is needed. We coin this part of the multivalent binding energy landscape to be called the “sweet spot”.
Figure 6.
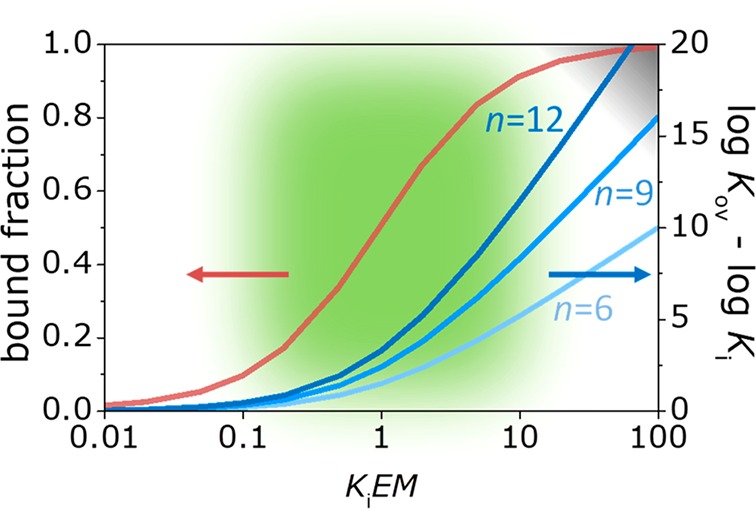
Energy landscape of multivalent interactions. Average bound fraction (left y axis) of all interaction pairs at any given time, given by KiEM/(KiEM + 1), and avidity parameter log Kov – log Ki (right y axis), here for three cases with n = 6, 9, and 12, as a function of KiEM. The dark-gray area (right upper corner) indicates the kinetic trap: slowing dynamics of the system when the number of potentially interacting sites, n, increases at high KiEM; the bound fraction approaches 1 and log Kov scales with log KiEM and with n. The green area indicates the “sweet spot”: the multivalent enhancement factor is not too low (KiEM < 0.1, bound fraction <0.1), where an increased valency does not lead to enhanced multivalent binding, nor too high (KiEM > 10, bound fraction >0.9), where the system becomes kinetically trapped.
As a result of the above analysis, we believe that the biological origin of the here observed weakly multivalent behavior of HA rosettes is inherent to the function of the adhesion process of influenza in real life: a virus may bind to a cell surface or to the mucus layer, but the interaction remains dynamic until a site is found at the cell surface where endocytosis is induced. This notion also provides insight in why the intrinsic binding affinity of an HA site of influenza is always of the same order of magnitude: mutations that would take the virus–cell surface interaction outside the sweet spot would either render the virus nonbinding or running into a kinetic trap upon interaction, which are both detrimental for virus proliferation.
How realistic is the rosette–SLB interaction for mimicking the interaction of whole viruses at cell surfaces and for understanding the interaction at a quantitative level? This question has aspects that affect the platform and those that deal with the rosette as a virus-like particle. While the cell surface with its glycocalyx is a tremendously complex system,41 we here look only to the density of the displayed glycans. On the cell level, the quantitative determination of the glycan density is a recent and important technical development, and densities of 107 sialic acids per cell have been reported.42 However, knowledge on local areal density, type of glycan, and determinations on relevant cell types will have to be performed to provide a more quantitative comparison. At a more technical level, the BLI method employs SAv-coated surfaces which have a similar density43 as the SLB platform developed here. Because the BLI method has been used on whole viruses,8,13 we are confident that our platform can exhibit the right glycan densities to study the binding of whole viruses with similar quantitative rigor as the rosettes described here.
When comparing smaller virus-like particles, such as the rHA clusters employed here, with whole viruses, some considerations need to be discussed. As stated above, a virus is considerably larger than a rosette, and some differences are expected for these systems for their binding behavior on platforms like the one reported here. First of all, as larger particles experience slower diffusion toward the surface and whole viruses are normally applied at lower particle concentrations, reaching thermodynamic equilibrium for whole viruses binding to the interaction platform might be slower, and therefore verification of equilibrium will be necessary when designing full titration experiments to acquire thermodynamic binding constants. Moreover, several examples have been reported in the literature where binding of whole viruses is observed on platforms using a fixed concentration of 100 pM.8,13 Therefore, relevant values of Kov of the interaction of these viruses are expected to be in the range of, approximately, 1010–1013 M–1, which are significantly higher than the ones measured for the rHA clusters reported here. Obviously, higher values of Kov are expected, as the contact area of a whole virus with the platform (or host cell) surface is expected to be much larger than the one of a rosette, and consequently many more HA trimers are involved in the overall interaction. When we assume that, for example, only 5% of the outer surface area of a virus binds to the platform surface and that the trimer–trimer distance in a virus is 11 nm,34,35 an average of approximately 12 HA trimers, i.e., 36 HA–sugar molecular interaction pairs, is involved in the interaction with the platform surface, which will increase the avidity accordingly. When we assume at the same time that the HA densities at a whole virus and a rosette are similar, and we note that EM in the KiEM factor is primarily governed by this density, the individual HA–sugar interaction pairs will remain dynamic, which will cause the overall binding behavior of the virus to be dynamic as well. Future work on studying the overall binding behavior of whole viruses and their dynamics will have to be performed to verify this behavior experimentally.
Conclusion
We have reported the development of a platform that mimics the multivalent interaction of influenza A viruses at host cell membranes. The use of biotinylated supported lipid bilayers (SLBs) provides control over the type and density of sialoglycan receptors on the surface. Selectivity for human sialic acid residues was established as expected for the here used recombinant hemagglutinin (rHA) rosettes. Low nanomolar affinities for the rHA rosettes binding to the SA-presenting surfaces were obtained from full titration curves. Because of the small size of the rosette and its limited number of HA trimers compared to the whole virus, the interaction area and the valency in binding to a surface can be estimated relatively well. This allowed us to assess the extent of multivalent binding, and quite low multivalent enhancement factors, KiEM, of about 5–10 were found, indicating that each additional binding site contributes with less than 1 order of magnitude to the overall binding affinity of the rHA particle. Because of the similar HA site density present at whole influenza A viruses, we assume that these have similarly low multivalent enhancement factors as the rosettes studied here.
By evaluating the relationship between the overall binding affinity Kov, the individual Ki, the valency n, and the effective molarity EM, a binding energy landscape has been sketched in which a sweet spot is evident for weakly multivalent systems, that is, for systems in which the individual binding sites equilibrate between bound and unbound with similar frequencies, rendering the multivalent system dynamic in nature. Future work will be focused on scaling the interaction area and valency to the values found in whole viruses. In addition, the methodology and analysis developed here may be applied to screen for future antiviral drugs or antibodies, which have the potential to block influenza virus binding.39,44 Finally, the quantitative assessment of weak multivalency may also be applicable to study different virus–host cell interactions, or other biological systems in which multivalent interactions drive cellular responses, such as the mono- or low-valency ligand interactions triggering the B-cell antigen receptor.45−47
Materials and Methods
Materials
Chemicals were purchased from Sigma-Aldrich and Acros Organics. Commercial lipids were obtained from Avanti Polar Lipids. Streptavidin labeled with Alexa Fluor 488 (SAv488) was obtained from ThermoFisher. HEPES buffer contained 0.01 M HEPES and 0.15 M sodium chloride was made using Milli-Q water (MQ, Millipore, 18.2 mΩ) and adjusted to pH 7.4 at 25 °C using sodium hydroxide. Biotin-PAA-SLN was obtained from Lectinity and used as received. rHA protein clusters were obtained from Protein Sciences Corporation. The concentration of the stock solutions of the protein clusters used in this work ranged from 100 nM to 246 nM.
QCM-D Measurements
QCM-D measurements were performed using a Qsense analyzer (Biolin Scientific). Measurements were performed at 22 °C and operated with four parallel flow chambers, using two Ismatec peristaltic pumps with a flow rate of 100 μL/min. Throughout this work, the fifth overtone was used for the normalized frequency (Δf5) and dissipation (ΔD5). SiO2-coated sensors (QSX303, Biolin Scientific) were used. During every rHA protein cluster addition, solutions were recycled.
Large Unilamellar Vesicles (LUV) and Supported Lipid Bilayer (SLB) Formation
1,2-Dioleoyl-sn-glycero-3-phosphocholine (DOPC) and 1,2-dioleoyl-sn-glycero-3-phosphoethanolamine-N-(cap biotinyl), sodium salt (biotin-PE), were stored in chloroform at −20 °C. The headgroup modified lipid–dye conjugate, Texas Red-1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine (TR, ThermoFisher scientific) was stored in methanol at −20 °C. Dissolved lipids were mixed in desired molar ratios before use and dried under a flow of nitrogen in a glass vial in order to create a film of lipid material at the glass wall. This film was further dried under vacuum for at least 1 h and subsequently hydrated by vortexing with Milli-Q water to form multilamellar vesicles at 1 mg/mL. The lipid suspension was extruded 11 times through a polycarbonate membrane (Whatman) with 100 nm pore size, resulting in large unilamellar vesicles (LUVs) that were stored in the refrigerator and used within 2 weeks. For SLB fabrication, vesicles were diluted to a concentration of 0.1 mg/mL in HEPES directly before use. SLB formation was achieved by flowing this solution on a cleaned and activated surface. For flat QCM-D sensors or glass bottom well plates, cleaning was performed using a 2 wt % sodium dodecyl sulfate (SDS) solution and thorough rising with Milli-Q. Activation was performed with 30 min UV/ozone treatment (for QCM-D sensors) using a Bioforce chamber (Nanosciences) or overnight incubation in 2% Hellmanex and again thorough Milli-Q rinsing (for well plates). The quality of SLBs was monitored by fluorescence recovery after photobleaching (FRAP) or in situ by QCM-D (where high quality SLB defined as Δf = −24 ± 1 Hz and ΔD < 0.5 × 10–6). After SLB formation, care was taken to keep the surface submerged in buffer and without bubbles.
Fluorescence Recovery after Photobleaching (FRAP)
A DOPC SLB was doped with 0.2 mol % of TR and 1% of biotin-PE. Subsequently, the surface was incubated with 0.2 μM SAv488 for 1 h and washed carefully with buffer for at least 15 times. Using a confocal microscope, a spot of 10 μm diameter was bleached, and subsequently, the fluorescence intensity in this bleached region was monitored. The intensity was normalized and corrected for acquisition bleaching by using the fluorescence intensity in a location not too close to the bleached spot. The FRAP protocol consisted of 11 imaging loops (1 s interval) before bleaching, 10 loops bleaching with no delay in between loops, and 300 loops of recovery (1 s interval). For confocal microscopy, a Nikon confocal (A1) microscope was used equipped with a 488 nm laser and a 525/50 nm emission filter and with a 561 nm laser with a 595/50 nm emission filter. In microscopy images displayed in this work, contrast and brightness were adapted for using ImageJ.
Transmission Electron Microscopy (TEM)
The original solution of rHA was diluted 1:2 with PBS. Then 5 μL of the suspension was pipetted onto a hydrophilized (by 60 s glow discharging at 8 W in a BALTEC MED 020 device (Leica Microsystems, Wetzlar, Germany)) Formvar-supported carbon-covered microscopical copper grid (400 mesh). After 30 s, a piece of filter paper was used to remove excess fluid. Subsequently, 5 μL of a contrast-enhancing heavy metal staining solution (1% phosphotungstic acid, pH 7.4) was applied and blotted again after 45 s. After air-drying, a standard holder was used to transfer the sample into a Talos L120C microscope (Thermo Fisher Scientific Inc., Waltham, Massachusetts, USA) equipped with a LaB6-cathode operated at an acceleration voltage of 120 kV. Micrographs were recorded with a 4k × 4k Ceta 16 M camera at a nominal magnification of 57000×.
Acknowledgments
We thank Dr. Kai Ludwig, Freie Universität Berlin, core facility “BioSupraMol,” for the TEM measurements, and Manon Cox and Indresh Srivastava from Protein Sciences corporation for providing the rHA clusters. J.H. acknowledges the Institute of Advanced Study, Durham University, UK, for hosting him as a Fellow and for fruitful discussions. This work was financially supported by the Marie Curie Innovative Training Network MULTI-APP (no. 642793) and the Volkswagen Foundation. M.V. and P.J. thank The Netherlands Organization for Scientific Research, NWO (Vidi Grant no. 723.012.106) for funding.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsnano.8b09410.
TEM, DLS, and FRAP measurements; QCM-D binding profiles for titrations of rHA clusters and Langmuir binding data; QCM-D binding profile of biotinylated lipid bilayer vesicles of 100 nm; ΔD5/−Δf5plot for rHA cluster titrations; calculations details of quantification of receptors on a surface (PDF)
Author Contributions
D.D.I. and M.L.V. contributed equally. The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Molinari N.-A. M.; Ortega-Sanchez I. R.; Messonnier M. L.; Thompson W. W.; Wortley P. M.; Weintraub E.; Bridges C. B. The Annual Impact of Seasonal Influenza in the US: Measuring Disease Burden and Costs. Vaccine 2007, 25, 5086–5096. 10.1016/j.vaccine.2007.03.046. [DOI] [PubMed] [Google Scholar]
- Iuliano A. D.; Roguski K. M.; Chang H. H.; Muscatello D. J.; Palekar R.; Tempia S.; Cohen C.; Gran J. M.; Schanzer D.; Cowling B. J.; Wu P.; Kyncl J.; Ang L. W.; Park M.; Redlberger-Fritz M.; Yu H.; Espenhain L.; Krishnan A.; Emukule G.; van Asten L.; et al. Estimate of Global Seasonal Influenza-Associated Mortality: A Modelling Study. Lancet 2018, 391, 1285–1300. 10.1016/S0140-6736(17)33293-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Medina R. A.; García-Sastre A. Influenza A Viruses: New Research Developments. Nat. Rev. Microbiol. 2011, 9, 590–603. 10.1038/nrmicro2613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mammen M.; Choi S.-K.; Whitesides G. M. Polyvalent Interactions in Biological Systems: Implications for Design and Use of Multivalent Ligands and Inhibitors. Angew. Chem., Int. Ed. 1998, 37, 2754–2794. . [DOI] [PubMed] [Google Scholar]
- Wagner R.; Wolff T.; Herwig A.; Pleschka S.; Klenk H.-D. Interdependence of Hemagglutinin Glycosylation and Neuraminidase as Regulators of Influenza Virus Growth: A Study by Reverse Genetics. J. Virol. 2000, 74, 6316–6323. 10.1128/JVI.74.14.6316-6323.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connor R. J.; Kawaoka Y.; Webster R. G.; Paulson J. C. Receptor Specificity in Human, Avian, and Equine H2 and H3 Influenza Virus Isolates. Virology 1994, 205, 17–23. 10.1006/viro.1994.1615. [DOI] [PubMed] [Google Scholar]
- Matrosovich M.; Tuzikov A.; Bovin N.; Gambaryan A.; Klimov A.; Castrucci M. R.; Donatelli I.; Kawaoka Y. Early Alterations of the Receptor-Binding Properties of H1, H2, and H3 Avian Influenza Virus Hemagglutinins after Their Introduction into Mammals. J. Virol. 2000, 74, 8502–8512. 10.1128/JVI.74.18.8502-8512.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vachieri S. G.; Xiong X.; Collins P. J.; Walker P. A.; Martin S. R.; Haire L. F.; Zhang Y.; McCauley J. W.; Gamblin S. J.; Skehel J. J. Receptor Binding by H10 Influenza Viruses. Nature 2014, 511, 475–477. 10.1038/nature13443. [DOI] [PubMed] [Google Scholar]
- Herfst S.; Imai M.; Kawaoka Y.; Fouchier R. A. M.. Avian Influenza Virus Transmission to Mammals. In Influenza Pathogenesis and Control; Springer International Publishing, 2014; Vol. 1, pp 137–155. [DOI] [PubMed] [Google Scholar]
- Garten R. J.; Davis C. T.; Russell C. A.; Shu B.; Lindstrom S.; Balish A.; Sessions W. M.; Xu X.; Skepner E.; Deyde V.; Okomo-Adhiambo M.; Gubareva L.; Barnes J.; Smith C. B.; Emery S. L.; Hillman M. J.; Rivailler P.; Smagala J.; de Graaf M.; Burke D. F.; et al. Antigenic and Genetic Characteristics of Swine-Origin 2009 A(H1N1) Influenza Viruses Circulating in Humans. Science 2009, 325 (325), 197–201. 10.1126/science.1176225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai H.; Wang R.; Hargis B.; Lu H.; Li Y. A SPR Aptasensor for Detection of Avian Influenza Virus H5N1. Sensors 2012, 12, 12506–12518. 10.3390/s120912506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carvalho S. B.; Moleirinho M. G.; Wheatley D.; Welsh J.; Gantier R.; Alves P. M.; Peixoto C.; Carrondo M. J. T. Universal Label-Free In-Process Quantification of Influenza Virus-Like Particles. Biotechnol. J. 2017, 12, 1700031. 10.1002/biot.201700031. [DOI] [PubMed] [Google Scholar]
- Xiong X.; Coombs P. J.; Martin S. R.; Liu J.; Xiao H.; McCauley J. W.; Locher K.; Walker P. A.; Collins P. J.; Kawaoka Y.; Skehel J. J.; Gamblin S. J. Receptor Binding by a Ferret-Transmissible H5 Avian Influenza Virus. Nature 2013, 497, 392–396. 10.1038/nature12144. [DOI] [PubMed] [Google Scholar]
- Jung H.; Robison A. D.; Cremer P. S. Multivalent Ligand-Receptor Binding on Supported Lipid Bilayers. J. Struct. Biol. 2009, 168, 90–94. 10.1016/j.jsb.2009.05.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gooding J. J.; Parker S. G.; Lu Y.; Gaus K. Molecularly Engineered Surfaces for Cell Biology: From Static to Dynamic Surfaces. Langmuir 2014, 30, 3290–3302. 10.1021/la4037919. [DOI] [PubMed] [Google Scholar]
- Satav T.; Huskens J.; Jonkheijm P. Effects of Variations in Ligand Density on Cell Signaling. Small 2015, 11, 5184–5199. 10.1002/smll.201500747. [DOI] [PubMed] [Google Scholar]
- Koçer G.; Jonkheijm P. Guiding hMSC Adhesion and Differentiation on Supported Lipid Bilayers. Adv. Healthcare Mater. 2017, 6, 1600862. 10.1002/adhm.201600862. [DOI] [PubMed] [Google Scholar]
- Wasserberg D.; Cabanas-Danés J.; Prangsma J.; O’Mahony S.; Cazade P.-A.; Tromp E.; Blum C.; Thompson D.; Huskens J.; Subramaniam V.; Jonkheijm P. Controlling Protein Surface Orientation by Strategic Placement of Oligo-Histidine Tags. ACS Nano 2017, 11, 9068–9083. 10.1021/acsnano.7b03717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houseman B. T.; Mrksich M. The Microenvironment of Immobilized Arg-Gly-Asp Peptides Is an Important Determinant of Cell Adhesion. Biomaterials 2001, 22, 943–955. 10.1016/S0142-9612(00)00259-3. [DOI] [PubMed] [Google Scholar]
- Dubacheva G. V.; Araya-Callis C.; Geert Volbeda A.; Fairhead M.; Codée J.; Howarth M.; Richter R. P. Controlling Multivalent Binding through Surface Chemistry: Model Study on Streptavidin. J. Am. Chem. Soc. 2017, 139, 4157–4167. 10.1021/jacs.7b00540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Weerd J.; Karperien M.; Jonkheijm P. Supported Lipid Bilayers for the Generation of Dynamic Cell-Material Interfaces. Adv. Healthcare Mater. 2015, 4, 2743–2779. 10.1002/adhm.201500398. [DOI] [PubMed] [Google Scholar]
- Rhodes D. G.; Holtz K.; Robinson P.; Wang K.; McPherson C. E.; Cox M. M.; Srivastava I. K. Improved Stability of Recombinant Hemagglutinin Using a Formulation Containing Sodium Thioglycolate. Vaccine 2015, 33, 6011–6016. 10.1016/j.vaccine.2015.09.029. [DOI] [PubMed] [Google Scholar]
- Böttcher C.; Ludwig K.; Herrmann A.; van Heel M.; Stark H. Structure of Influenza Haemagglutinin at Neutral and at Fusogenic pH by Electron Cryo-Microscopy. FEBS Lett. 1999, 463, 255–259. 10.1016/S0014-5793(99)01475-1. [DOI] [PubMed] [Google Scholar]
- Ward B. J.; Landry N.; Trépanier S.; Mercier G.; Dargis M.; Couture M.; D’Aoust M.-A.; Vézina L.-P. Human Antibody Response to N-Glycans Present on Plant-made Influenza Virus-Like Particle (VLP) Vaccines. Vaccine 2014, 32, 6098–6106. 10.1016/j.vaccine.2014.08.079. [DOI] [PubMed] [Google Scholar]
- McBrayer A.; Camp J. V.; Tapp R.; Yamshchikov V.; Grimes S.; Noah D. L.; Jonsson C. B.; Bruder C. E. Course of Seasonal Influenza A/Brisbane/59/07 H1N1 Infection in the Ferret. Virol. J. 2010, 7, 149. 10.1186/1743-422X-7-149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daum L. T.; Canas L. C.; Smith C. B.; Klimov A.; Huff W.; Barnes W.; Lohman K. L. Genetic and Antigenic Analysis of the first A/New Caledonia/20/99-like H1N1 Influenza Isolates Reported in the Americas. Emerging Infect. Dis. 2002, 8, 408–412. 10.3201/eid0804.010311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lind T. K.; Cárdenas M. Understanding the Formation of Supported Lipid Bilayers via Vesicle Fusion—A Case that Exemplifies the Need for the Complementary Method Approach. Biointerphases 2016, 11, 020801 10.1116/1.4944830. [DOI] [PubMed] [Google Scholar]
- Xiong X.; Martin S. R.; Haire L. F.; Wharton S. A.; Daniels R. S.; Bennett M. S.; McCauley J. W.; Collins P. J.; Walker P. A.; Skehel J. J.; Gamblin S. J. Receptor Binding by an H7N9 Influenza Virus from Humans. Nature 2013, 499, 496–499. 10.1038/nature12372. [DOI] [PubMed] [Google Scholar]
- Collins P. J.; Vachieri S. G.; Haire L. F.; Ogrodowicz R. W.; Martin S. R.; Walker P. A.; Xiong X.; Gamblin S. J.; Skehel J. J. Recent Evolution of Equine Influenza and the Origin of Canine Influenza. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 11175–11180. 10.1073/pnas.1406606111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reviakine I.; Johannsmann D.; Richter R. P. Hearing What You Cannot See and Visualizing What You Hear: Interpreting Quartz Crystal Microbalance Data from Solvated Interfaces. Anal. Chem. 2011, 83, 8838–8848. 10.1021/ac201778h. [DOI] [PubMed] [Google Scholar]
- Reviakine I.; Brisson A. Streptavidin 2D Crystals on Supported Phospholipid Bilayers: Toward Constructing Anchored Phospholipid Bilayers. Langmuir 2001, 17, 8293–8299. 10.1021/la010626i. [DOI] [Google Scholar]
- Larsson C.; Rodahl M.; Höök F. Characterization of DNA Immobilization and Subsequent Hybridization on a 2D Arrangement of Streptavidin on a Biotin-modified Lipid Bilayer Supported on SiO2. Anal. Chem. 2003, 75, 5080–5087. 10.1021/ac034269n. [DOI] [PubMed] [Google Scholar]
- Bovin N. V. Polyacrylamide-based Glycoconjugates as Tools in Glycobiology. Glycoconjugate J. 1998, 15, 431–446. 10.1023/A:1006963717646. [DOI] [PubMed] [Google Scholar]
- Wasilewski S.; Calder L. J.; Grant T.; Rosenthal P. B. Distribution of Surface Glycoproteins on Influenza A Virus Determined by Electron Cryotomography. Vaccine 2012, 30, 7368–7373. 10.1016/j.vaccine.2012.09.082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris A.; Cardone G.; Winkler D. C.; Heymann J. B.; Brecher M.; White J. M.; Steven A. C. Influenza Virus Pleiomorphy Characterized by Cryoelectron Tomography. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 19123–19127. 10.1073/pnas.0607614103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huskens J.; Mulder A.; Auletta T.; Nijhuis C. A.; Ludden M. J. W.; Reinhoudt D. N. A Model for Describing the Thermodynamics of Multivalent Host–Guest Interactions at Interfaces. J. Am. Chem. Soc. 2004, 126, 6784–6797. 10.1021/ja049085k. [DOI] [PubMed] [Google Scholar]
- Hunter C. A.; Anderson H. L. What is Cooperativity?. Angew. Chem., Int. Ed. 2009, 48, 7488–7499. 10.1002/anie.200902490. [DOI] [PubMed] [Google Scholar]
- Mulder A.; Huskens J.; Reinhoudt D. N. Multivalency in Supramolecular Chemistry and Nanofabrication. Org. Biomol. Chem. 2004, 2, 3409–3424. 10.1039/b413971b. [DOI] [PubMed] [Google Scholar]
- Bhatia S.; Lauster D.; Bardua M.; Ludwig K.; Angioletti-Uberti S.; Popp N.; Hoffmann U.; Paulus F.; Budt M.; Stadtmuller M.; Wolff T.; Hamann A.; Bottcher C.; Herrmann A.; Haag R. Linear Polysialoside Outperforms Dendritic Analogs for Inhibition of Influenza Virus Infection In Vitro and In Vivo. Biomaterials 2017, 138, 22–34. 10.1016/j.biomaterials.2017.05.028. [DOI] [PubMed] [Google Scholar]
- Perl A.; Gomez-Casado A.; Thompson D.; Dam H. H.; Jonkheijm P.; Reinhoudt D. N.; Huskens J. Gradient-Driven Motion of Multivalent Ligand Molecules along a Surface Functionalized with Multiple Receptors. Nat. Chem. 2011, 3, 317–322. 10.1038/nchem.1005. [DOI] [PubMed] [Google Scholar]
- Ohtsubo K.; Marth J. D. Glycosylation in Cellular Mechanisms of Health and Disease. Cell 2006, 126, 855–867. 10.1016/j.cell.2006.08.019. [DOI] [PubMed] [Google Scholar]
- Chen Y.; Ding L.; Liu T.; Ju H. Arrayed Profiling of Multiple Glycans on Whole Living Cell Surfaces. Anal. Chem. 2013, 85, 11153–11158. 10.1021/ac403150n. [DOI] [PubMed] [Google Scholar]
- Guo H.; Rabouw H.; Slomp A.; Dai M.; van der Vegt F.; van Lent J. W. M.; McBride R.; Paulson J. C.; de Groot R. J.; van Kuppeveld F. J. M.; de Vries E.; de Haan C. A. M. Kinetic Analysis of the Influenza A Virus HA/NA Balance Reveals Contribution of NA to Virus-Receptor Binding and NA-Dependent Rolling on Receptor-containing Surfaces. PLoS Pathog. 2018, 14, e1007233 10.1371/journal.ppat.1007233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bangaru S.; Zhang H.; Gilchuk I. M.; Voss T. G.; Irving R. P.; Gilchuk P.; Matta P.; Zhu X.; Lang S.; Nieusma T.; Richt J. A.; Albrecht R. A.; Vanderven H. A.; Bombardi R.; Kent S. J.; Ward A. B.; Wilson I. A.; Crowe J. E. A Multifunctional Human Monoclonal Neutralizing Antibody that Targets a Unique Conserved Epitope on Influenza HA. Nat. Commun. 2018, 9, 2669. 10.1038/s41467-018-04704-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim Y.-M.; Pan J. Y.-J.; Korbel G. A.; Peperzak V.; Boes M.; Ploegh H. L. Monovalent Ligation of the B Cell Receptor Induces Receptor Activation but Fails to Promote Antigen Presentation. Proc. Natl. Acad. Sci. U. S. A. 2006, 103, 3327–3332. 10.1073/pnas.0511315103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avalos A. M.; Bilate A. M.; Witte M. D.; Tai A. K.; He J.; Frushicheva M. P.; Thill P. D.; Meyer-Wentrup F.; Theile C. S.; Chakraborty A. K.; Zhuang X.; Ploegh H. L. Monovalent Engagement of the BCR Activates Ovalbumin-Specific Transnuclear B Cells. J. Exp. Med. 2014, 211, 365–379. 10.1084/jem.20131603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Villar R. F.; Patel J.; Weaver G. C.; Kanekiyo M.; Wheatley A. K.; Yassine H. M.; Costello C. E.; Chandler K. B.; McTamney P. M.; Nabel G. J.; McDermott A. B.; Mascola J. R.; Carr S. A.; Lingwood D. Reconstituted B Cell Receptor Signaling Reveals Carbohydrate-Dependent Mode of Activation. Sci. Rep. 2016, 6, 36298. 10.1038/srep36298. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.