

Abstract
Biomaterial substrates can be engineered to present topographical signals to cells which, through interactions between the material and active components of the cell membrane, regulate key cellular processes and guide cell fate decisions. However, targeting mechanoresponsive elements that reside within the intracellular domain is a concept that has only recently emerged. Here, we show that mesoporous silicon nanoneedle arrays interact simultaneously with the cell membrane, cytoskeleton, and nucleus of primary human cells, generating distinct responses at each of these cellular compartments. Specifically, nanoneedles inhibit focal adhesion maturation at the membrane, reduce tension in the cytoskeleton, and lead to remodeling of the nuclear envelope at sites of impingement. The combined changes in actin cytoskeleton assembly, expression and segregation of the nuclear lamina, and localization of Yes-associated protein (YAP) correlate differently from what is canonically observed upon stimulation at the cell membrane, revealing that biophysical cues directed to the intracellular space can generate heretofore unobserved mechanosensory responses. These findings highlight the ability of nanoneedles to study and direct the phenotype of large cell populations simultaneously, through biophysical interactions with multiple mechanoresponsive components.
Keywords: nanoneedles, mechanotransduction, nuclear mechanics, cell−material interactions, super-resolution microscopy, porous silicon
Physical cues from the extracellular space are sensed at the cell membrane and initiate intracellular signaling cascades that ultimately influence cell fate and function.1−5 The rational design of materials that are employed as culture substrates enables investigation of how cells respond to physicochemical stimuli from the extracellular matrix (ECM). Indeed, cues such as substrate stiffness,3 micro/nanotopograhy,4 and spatial confinement6 can influence crucial cell functions, including regulation of gene expression programs, proliferation, and lineage specification.7 Due to the physical connection of the cell membrane to intracellular mechanoresponsive elements, forces generated at the cell–material interface can alter organelle structure and function, such as nuclear morphology, chromatin organization, and epigenetic status.5−12 However, most engineered materials interface exclusively with the cell membrane, and their effects on cell organelles—such as the cytoskeleton or the nucleus—are the consequence of stimuli originating from interactions at the membrane. Therefore, the influence of materials on the intracellular space can be considered indirect and is mediated by established mechanosensory signal transduction cascades. One central theme of these canonical pathways is the spatial regulation of the mechanoresponsive cofactors, Yes-associated protein (YAP) and transcriptional coactivator with a PDZ-binding motif (TAZ), which is mediated, in part, via the actomyosin contractile machinery.7 Several material systems have investigated how YAP/TAZ and cytoskeletal tension are influenced by changing physicochemical parameters,7,13−16 adding to literature that has provided exhaustive insight into how intracellular elements are affected by outside-in, canonical mechanosensing.17−23 In contrast, techniques such as micropipette aspiration,24 optical/magnetic tweezers,25 and atomic force microscopy26 have been used to directly probe individual organelles without relying upon material-derived cues, demonstrating that direct interaction with mechanosensitive organelles can induce changes in cell behaviors. However, their low throughput and complex setups limit their investigational and translational potential in more advanced tissue and in vivo models. The development of material systems to directly probe organelles within multiple cells simultaneously can enable the study of membrane-independent mechanosensing pathways within large and complex biological systems such as organotypic cultures and tissues, thus improving strategies for the modulation of cell behavior.
Arrays of high aspect ratio, vertically oriented nanostructures have recently garnered tremendous attention for their interactions with the intracellular component of cells in culture and tissues. These materials can deliver membrane-impermeant cargo to the cytosol,27−34 sense enzymatic activity,35,36 and stimulate/record electrical activity from within the cell.37,38 Importantly, interfacing these nanomaterials with cells does not noticeably alter their viability or metabolic activity, although it has a strong impact on mechanoresponsive elements within the cell. For example, cells on nanowires exhibit fewer adhesive structures2,39−42 and reduced cytoskeletal tension,2,15,17 alongside alterations to cellular8,29,43−50 and nuclear morphology.8,51 Although these observations have generated a wealth of understanding about the membrane-initiated response to nanowires, there remains an unmet need to understand the nature of the interactions between nanomaterials and the intracellular space, as well as how these events influence mechanosensory pathways.
To this end, we investigated the molecular and functional consequences of the interaction between porous silicon nanoneedles (nN) and specific mechanosensitive organelles in primary human cells and report canonical mechanosensing events alongside noncanonical responses of organelles to nanomaterial cues. We first show that interfacing porous silicon nN with cells prevents the formation and maturation of focal adhesions (FAs) at the cell–material interface, which leads to decreased cytoskeletal tension and reduced functional activity of mechanoresponsive transcriptional regulators. However, nN also induce a separate physical response in intracellular organelles: specifically, the actin cytoskeleton forms dense rings at sites of nN engagement, and the nuclear envelope undergoes type-specific remodeling of lamin A/C but not lamin B. Importantly, these processes are not dependent on intact actomyosin contractile machinery. Furthermore, nN induce a decoupling of YAP localization/activation and cell area, as well as physical segregation of lamin A at inward nuclear protrusions. The findings reported here reveal that porous silicon nN are a powerful tool to target intracellular organelles in multiple cells simultaneously and offer insight into the relationships between various mechanoresponsive cellular elements.
Results
Quantitative Morphometric Analysis
Human umbilical vein endothelial cells (HUVECs) and human mesenchymal stem cells (hMSCs) cultured on nN arrays for 6 h displayed extensive morphological alterations, as compared to the flat substrate controls (Figure 1A,B). Cells interacted directly with the nN (Figure 1A), which had a profound effect on the morphology of the entire cell population (Figure 1B). Importantly, most cells sunk into the sharp nN arrays and were not suspended on top of the structures (Figure S1). Using automated processing of immunofluorescence images, we performed quantitative morphometric analysis to extract and quantify the cellular features that were most heavily influenced by culture on nN substrates (Figures 1C and S2). Twenty-five features of cell morphology and actin textures were subsequently compared by linear discriminant analysis (LDA), which revealed that actin homogeneity, a measure of fiber size, reduced greatly on nN whereas protrusions extended farther radially from the nucleus (Figure 1D,E). Indeed, when compared to their respective flat controls, both cell types demonstrated a significant reduction in actin stress fiber density (Figure 1F), along with a greater number of high aspect ratio protrusions (Figure 1G,H). HUVECs also displayed an increased ratio of cortical-to-central actin on nN (Figure 1I), and the protrusions of hMSCs were aligned along the nN array, indicating that nN can guide protrusion formation (Figure 1J). Despite these significant morphological changes, culture on nN did not abolish cell proliferation, compromise the integrity of the nuclear envelope, or stimulate an apoptotic response, as indicated by positive staining for Ki67 (Figure S3), nuclear retention of heterogeneous nuclear ribonucleoprotein (hnRNP, Figure S4), and negative staining for caspase-3, respectively (Figure S5).
Figure 1.

Nanoneedle interaction with HUVECs and hMSCs reduces actin bundling and enhances actin-rich protrusions. (A) SEM images show direct interaction between cells and nN 6 h postseeding. Scale bars = 10 μm. (B) Wide-field immunofluorescence images of the actin cytoskeleton show drastic alterations to cell morphology on nN as compared to that on flat controls (green: phalloidin). HUVECs display a stellate morphology on nN, whereas hMSCs elongate along the nN array. Scale bars = 50 μm. (C) Workflow for extraction, quantification, and analysis of morphometric features using high-content imaging and automated cell segmentation algorithms. (D) Twenty-five features are compared by linear discriminant analysis (LDA) for the two cell types on the flat and nN substrates, and (E) most heavily influenced parameter measured is actin homogeneity. (F) Specific analysis of actin features reveals reduced stress fiber formation (actin bundling) on nN, compared to that on flat substrates for both cell types (box plots, minimum/maximum). (G,H) Image analysis quantification of actin features reveals longer protrusions with high aspect ratios on nN. (I) HUVECs exhibit increased levels of cortical versus central actin on nN (green: phalloidin). (J) hMSC actin cytoskeleton aligns to the nN array lattice. (I,J) Deconvolved maximum projection confocal immunofluorescence images (green: phalloidin). Scale bars = 50 μm. N ≥ 3 experimental replicates for all data; *p < 0.05, ***p < 0.001 between indicated groups.
Reduced Focal Adhesion Formation and Actomyosin Contractility
Such a significant morphological response to biophysical cues suggests a direct effect of nN on the mechanosensing cell machinery. Indeed, whereas both cell types formed dense FAs on flat substrates, smaller, more diffuse FAs appeared on nN (Figure 2A,B and Figure S6), and gene- and protein-level expression of vinculin was decreased (Figure 2C,D). The reduction in phosphorylated paxillin (pPax) on nN confirmed the limited maturation of FAs (Figure 2A). Cells on flat substrates demonstrated dense paxillin staining, as expected for a planar substrate upon which cells can readily form FAs and spread; however, cells on nN exhibited unordered staining patterns for both actin and paxillin (Figure S6A). Gene-level expression of multiple FA components, including focal adhesion kinase (FAK), paxillin (PAX), vinculin (VCL), and zyxin (ZYX), was significantly downregulated on nN (Figure 2C), and the expression of various integrins trended downward, with integrin β1 showing a significant reduction for both cell types on nN (Figure S7). Staining specific for phosphorylated myosin light chain (pMLC), which indicates active actomyosin contractility, was also significantly reduced for both cell types cultured on nN (Figure 2A,E), suggesting that hampered maturation of FAs on nN leads to attenuated intracellular tension.
Figure 2.

Nanoneedles inhibit focal adhesion formation and generation of intracellular tension. (A) Confocal maximum projection images 6 h postseeding. On flat substrates, dense vinculin staining is observed in stable focal adhesion (FA) complexes. Strong phosphorylated paxillin (pPax) and phosphorylated myosin light chain (pMLC) signal on flat substrates indicate FA maturation and active actomyosin contractile machinery, respectively. Cells on nN display diffuse vinculin staining and severely reduced pPax and pMLC signal. Scale bars: vinculin = 25 μm; pPax and pMLC = 50 μm. (B) Significant reduction in vinculin signal reveals reduced FA density on nN (box plots, minimum/maximum; N = 3). (C) qPCR indicates that culture on nN yields downregulation in gene expression for multiple FA components (focal adhesion kinase (FAK), paxillin (PAX), vinculin (VCL), and zyxin (ZYX); qPCR, N = 3, mean ± SD). (D) Western blot shows downregulation of vinculin protein expression on nN (HUVEC: N = 2, hMSC: N = 3, mean ± SD). (E) Quantification of pMLC signal intensity via image analysis confirms a significant reduction for both cell types cultured on nN, as compared to their respective controls (box plots, minimum/maximum, N ≥ 4); *p < 0.05, **p < 0.01, ***p < 0.001 between groups as indicated by the lines.
Modulation of YAP Localization and Function
Biophysical stimuli regulate the functionality of the transcription cofactors YAP and TAZ.7 When cytoskeletal tension can be generated, YAP and TAZ localize within the nucleus, activating their transcriptional program. However, when tension is interrupted, the cofactors are localized in the cytosol and their activity is reduced.7 The nuclear/cytosolic YAP ratio decreased on nN for both cell types (Figure 3A,B), and expression of the YAP target genes ankyrin repeat domain 1 (ANKRD1) and connective tissue growth factor (CTGF) was significantly decreased (Figure 3C), indicating reduced functional YAP activity in cells cultured on nN compared to those cultured on flat surfaces. On flat substrates, cell area correlated highly with YAP nuclear/cytosolic ratio (HUVEC: ρ = 0.592; hMSC: ρ = 0.465), but on nN, this correlation was notably lower (HUVEC: ρ = 0.184; hMSC: ρ = 0.263; Fisher’s R-to-Z transformation, HUVEC: p < 0.001; hMSC: p = 0.0183; Figure 3D). Taken together, these data demonstrate that nN induce cell spreading but uncouple changes in cell geometry from YAP activation.
Figure 3.

Nanoneedles reduce YAP activity and lessen the correlation between YAP activation and cell spreading. (A) Confocal microscopy shows nuclear YAP protein localization on flat substrates and cytosolic localization on nN (green: YAP). Scale bars = 50 μm. (B) Image analysis quantification of YAP localization shows significant reduction in the nuclear to cytoplasmic ratio of YAP on nN (minimum/maximum; N = 4). (C) qPCR analysis indicates reduced expression of the YAP target genes ankyrin repeat domain 1 (ANKRD1) and connective tissue growth factor (CTGF) (N = 4, mean ± SD). (D) Cell spread area and YAP nuclear localization correlate tightly on flat substrates, but correlation is weakened on nN (N = 3). (E) YAP localization following cell treatment with either the actin depolymerizing agent, LatB, or a small molecule to stimulate actin bundling, LPA. LatB treatment on flat substrates reduces nuclear YAP localization to levels comparable to untreated cells on nN. LatB treatment of cells on nN yields a small reduction in nuclear localization. LPA treatment on flat substrates did not affect YAP localization for HUVECs and marginally decreased this metric for hMSCs. On nN substrates, LPA had little effect on YAP localization. (minimum/maximum; N = 3); *p < 0.05, **p < 0.01, ***p < 0.001 between groups as indicated by the lines.
To determine if the loss of actin polymerization and/or actomyosin contractility underpinned decreased YAP nuclear translocation in cells plated on nN, we inhibited actin polymerization using latrunculin B (LatB) or upregulated actomyosin contractility by treating cells with lysophosphatidic acid (LPA).17 LatB treatment on nN further reduced the YAP ratio, although marginally (Figure 3E), whereas the YAP ratio of LatB-treated cells on flat control substrates was significantly reduced to levels comparable to those of untreated cells on nN. When cells were treated with LPA, the YAP ratio for cells on nN was not recovered back to levels observed for untreated or LPA-treated cells on flat control substrates, indicating that upregulation of signaling (biochemical) pathways that promote contractility and YAP activation on flat substrates is insufficient to promote YAP activity on nN. These data suggest that manipulation of actin polymerization and actomyosin contractility in cells on nN has only modest effects on YAP activity. Taken together, our data suggest that FA formation and/or turnover, and not changes in cell shape or actin organization, appears to be a principal driver of YAP activation.
Interaction with Mechanosensory Organelles
Interfacing of cells with nN also stimulated a physical response by the cytoskeleton and the nuclear envelope at sites of engagement (Figure 4). Analysis of morphometric parameters from whole populations on nN indicated actin homogeneity to be the most heavily influenced cell feature among those measured (Figure 1E). Indeed, dense actin rings formed at sites of nN interaction in both cell types (Figure 4A) at various heights along the nN, exhibiting a dynamic and short-lived nature (Supplementary Video 1). Strikingly, cells treated with LatB during the entire culture period still formed actin rings (Figure 4B), indicating that intact actomyosin contractility is not required for cytoskeletal structures to respond to nN. These data further support the idea that YAP nuclear translocation dynamics are not directly coupled to changes in actin polymerization.
Figure 4.

Nanoneedles interact with mechanoresponsive organelles. (A) Polymerized actin rings form at sites of nN interaction with both HUVECs and hMSCs, (HUVEC: structured illumination microscopy (SIM), single plane; hMSC: deconvolved confocal microscopy, single plane). Green: phalloidin, scale bars = 10 μm. Actin rings were located around the nN (deconvolved confocal z-stack; green: phalloidin, red: nN). Scale bar = 1 μm. (B) Confocal images demonstrate that actin rings still form even when cells are treated with the actin depolymerizing agent, LatB, for the entire 6 h culture period. (Green: phalloidin, single plane). Scale bars = 25 μm. (C) SIM of DAPI-stained nuclei and fluorescent nN shows physical displacement of the nucleus at nN sites (single plane; cyan: DAPI, red: nN). Scale bars = 5 μm. (D) SIM imaging shows lamin B distributing at the base of the nN, with lamin A localising throughout the needle length. (single plane; magenta: lamin A, yellow: lamin B). Scale bars = 5 μm. (E) Reslice images of the x−z plane from confocal z-stack images; lamin A signal increases around nN, whereas lamin B remains constant (red: nN, yellow: lamin B, magenta: lamin A). Scale bar = 2 μm. (F) Analysis of fold-change values of lamin A and B intensity along the lower nuclear envelope in resliced confocal images normalized to the signal measured at non-nN locations. (G) Normalized intensity values of lamin A and lamin B at the middle and top of nN. At sites of nuclear remodeling (i.e. lamin A/lamin B > 1) the A/B ratio increases exponentially along the nN axis (R2 = 0.808, n = 69 nN sites, n = 14 cells, N = 3). (H) qPCR analysis of nuclear lamina components shows increased LMNA but not LMNB expression on nN after 6 h in culture. (N = 4, mean ± SD). (I) Western blot and (J) analysis relative to GAPDH control reveal a decrease in protein-level lamin A after 6 h in culture. (K) Quantification of signal intensity for lamin A relative to lamin B images further confirm that a reduction in lamin A occurs following culture on nN substrates (N = 3 experiments); ***p < 0.001 between groups as indicated by the lines.
At the nucleus, nN physically displaced DNA at sites of engagement, as evidenced by areas where DAPI signal was absent (Figure 4C). In order to understand how the nN specifically interacted with the nuclear envelope, we analyzed the expression and localization of A- and B-type lamins, the intermediate filaments that provide structural integrity to the nuclear envelope.5 Three-dimensional structured illumination microscopy (3D SIM) showed intense lamin A signal at sites where nN impinged on the nucleus (Figure S9 and Supplementary Video 2). Of note, lamin remodeling was not observed at all nN sites (24% of hMSCs and 37% of HUVECs demonstrated remodeled nuclei), but where remodeling was evident, lamin A accumulated at the nN whereas lamin B remained equally distributed throughout the nuclear membrane (Figure 4D). Fold changes in lamin A and B signal along nuclear envelope as it wrapped around the nN were quantified using resliced x–z confocal images normalized to the nuclear membrane signal measured at non-nN locations (Figure 4E–G). At sites of remodeling, lamin A signal more than doubled on average and increased up to 4-fold relative to non-nN sites, whereas lamin B signal was mostly unchanged (Figure 4F), indicating that nN stimulate a dynamic response of lamin A, but not lamin B, that results in a segregation of the two nuclear envelope components. Furthermore, where remodeling occurred, the lamin A/B ratio increased exponentially toward the nN tip (Figure 4G). We further stained for the lamin A/C–C epitope, which is only accessible when the protein is not under tension,8 and found this to be located at the tips of the needles (Figure S10). This pattern of lamin A accumulation and relaxation suggests a local force application at the tip that remodels the nuclear membrane (Figure S11).
Increases in lamin A protein, such as those that occur during culture on matrices of high stiffness, can lead to upregulation of LMNA mRNA through the engagement of positive feedback loops.5 We thus set out to determine how displacement of lamin A protein from the nuclear envelope affects LMNA expression. The relative gene-level expression of lamin A (LMNA) increased for both cell types on nN, as compared to their respective flat control, whereas lamin B (LMNB) expression did not change (Figure 4H). Expression of the linker of nucleoskeleton and cytoskeleton (LINC) complex members, Nesprin-2 (SYNE2) and SUN2 (SUN2), was also unchanged (Figure S8). Western blot protein-level expression analysis showed an overall lamin A decrease on nN (Figure 4I,J), which was confirmed by quantitative image analysis of lamin A- and lamin B-stained cells (Figure 4K). Thus, displacement and destabilisation of lamin A by nN results in an upregulation of LMNA mRNA.
Recovery upon Nanoneedle Degradation
After 48 h in culture, nN were mostly degraded (Figure 5A) and cells recovered the characteristics observed on flat control substrates (Figure 5B). Specifically, actin stress fibers and vinculin-dense FAs were present at 48 h, and the ratio of nuclear/cytosolic YAP partially recovered (Figure 5C). The density of FAs on nN at 48 h, as indicated by vinculin staining, was similar to what was measured on flat control samples at 6 h (Figure 5D). Furthermore, 48 h post-nN interfacing, actin rings were not observed and lamin A remodeling of the nucleus was absent at sites of nN engagement (Figure 5B), indicating that direct nN interaction with organelles had been lost. The mechanoresponsive characteristics at 48 h therefore are similar to culture on a flat substrate with mature FA, engaged actomyosin machinery and restored nuclear envelope morphology (Figure 5E).
Figure 5.

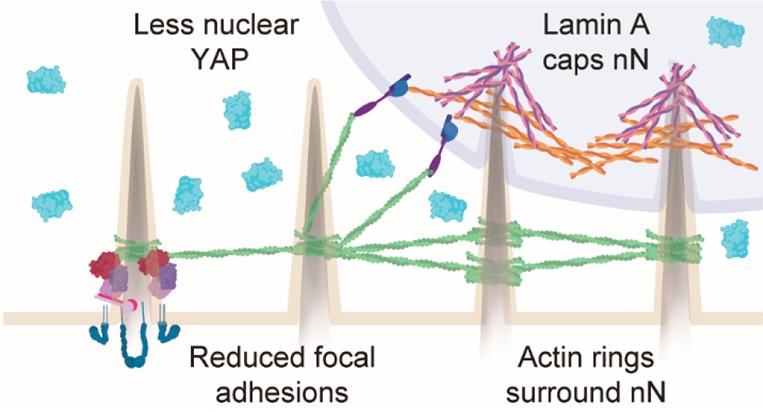
Nanoneedle degradation recovers mechanoresponsive cell behaviors. (A) SEM images show nN degradation after 48 h in culture. Scale bars = 1 μm, 2 μm inset. (B) Cell phenotype is restored on degraded nN as compared to flat control substrates at 6 h. Cells exhibit a spread actin cytoskeleton (green: phalloidin, scale bars = 50 μm), dense staining of vinculin-rich focal adhesions (red: vinculin, cyan: DAPI, scale bars = 25 μm), nuclear localization of YAP (green, scale bars = 50 μm), and an unimpinged nucleus (magenta: lamin A, cyan: DAPI, scale bars = 5 μm). (C) Image analysis shows a partial return of YAP localization to the nucleus and (D) increased focal adhesion (vinculin) density (box plots, minimum/maximum). (E) Schematic representation of the cell−nN interaction. Cells on flat substrates display firm focal adhesions, which allow for generation of intracellular tension, yielding YAP nuclear localization and subsequent transcriptional activity, and a uniform nuclear lamina composition. nN interfacing limits focal adhesion formation and maturation, directly stimulates actin ring formation, and results in segregation of lamin A and B at the nucleus. Furthermore, lamin A is downregulated at the protein level but upregulated at the gene level in response to interactions with nN.
Discussion
In the present study, we have demonstrated that nN interfacing simultaneously stimulates different mechanoresponsive organelles within primary human cells, inducing both canonical responses that arise from interactions at the membrane–material interface, as well as unreported mechanosensing events arising from interactions with the intracellular space. The degree of response to nN stimuli can differ across cell types, particularly at the cytoskeletal level (Figure 1G–J). Yet, the key features of this biophysical interaction, namely, the regulation of YAP and lamin A localization, FA interruption and actin accumulation, are preserved across cell types. Thus, despite the two cell types being inherently different (i.e., mesenchymal vs endothelial), our data suggest that conserved pathways drive the response to nN stimulation.
Uncoupling the mechanotransduction of stimuli that regulate cell contractility (i.e., mechanical cues) from those that regulate cell shape and spreading (i.e., geometric cues) using materials has historically been challenging as many engineered substrates modulate both simultaneously.12 Here, we show that nN greatly weaken the frequently observed correlation between cell area and YAP localization (Figure 3D), which has been established largely in cells cultured on flat substrates or micropillars.6 This finding indicates that geometric cues which increase cell spreading alone are insufficient to promote YAP activation on nN, suggesting that YAP activity might primarily be regulated by mechanical cues such as FA density on nN.
In addition to interactions with the membrane and actin network, nN also engaged with the nucleus and displaced the nuclear envelope, stimulating a dynamic remodeling process of A-type, but not B-type, lamin proteins (Figure 4). In structural models of the nuclear envelope, lamin A exhibits a viscous response to resist deformation whereas lamin B is an elastic component that preserves nuclear shape.5 Indeed, lamin A is required to prevent nuclear envelope rupture in the presence of external forces both in isolated nuclei25 and intact cells,24 and lamin A expression scales with tissue stiffness5 in order to protect the sensitive nuclear components from external pressures. In our system, lamin A, but not lamin B, responded to the mechanical stimulus by accumulating at nN sites, particularly showing a preferential accumulation at the nN tip, where the negative membrane curvature is strongest (Figure 4F). This agrees with the established protective role of A-type lamins in counteracting mechanical insults to the sensitive intranuclear cargo. Yet, lamin A protein levels decreased on nN, whereas gene expression increased (Figure 4). These data, together with the exposure of epitopes associated with relaxed lamin A (Figure S10), suggest that the nuclear envelope is under reduced tension at the sites of nN-nucleus engagement, leading to lamin A phosphorylation and degradation.52 Further, in contrast with previous reports showing that actomyosin contractility is necessary for nuclear envelope remodeling on nanopillars,8 we observe that lamin A remodeling still occurs in LatB-treated cells (Figure S12).
Lamin A and YAP localization tend to correlate in vitro when matrix rigidity is altered, until an overabundance of lamin A for very stiff substrates can prevent further nuclear YAP translocation.5 Instead, nN induce cytosolic YAP concomitantly with increased lamin A gene expression, providing insight into the complex relationship between the two factors (Figure S13). Material systems that simultaneously promote lamin A expression and cytosolic YAP localization have been absent in the literature, namely, because soft substrates that prevent nuclear accumulation of YAP also result in wrinkled nuclei with highly phosphorylated, inactive lamin A.7,52 By providing a platform for modulating multiple mechanoresponsive elements simultaneously, nN represent a useful tool for helping to deconstruct the YAP–lamin relationship. Adipogenic differentiation of stem cells on soft matrices is enhanced by low levels of lamin A and cytosolic YAP, whereas osteogenic differentiation on stiff matrices is heightened by high levels of lamin A and nuclear YAP.5,7 Due to their noncanonical regulation of lamin A and YAP, nN can expand the toolset to potentially direct cell fate and to improve our understanding of the role of biophysical cues in determining stem cell lineage.
The degradation of nN at 48 h in culture recovered the phenotype that was observed on flat substrates (Figure 5), highlighting the role of the physical nanofeatures in driving changes in mechanoresponsive organelles.
Conclusions
The data presented here highlight the ability of high aspect ratio nanostructures to mechanically stimulate remodeling of organelles in a reversible manner without loss of cell integrity. Further engineering of these nanostructures to modulate the cell’s response can be leveraged to induce isolated mechanosensory responses at the organelle level, to enable finer material-induced control over cell fate in tissue engineering.
Methods
Fabrication of Nanoneedles
Nanoneedles were fabricated according to our established protocol28,35 on 100 mm diameter p-type doped Si wafers with 0.01 Ω·cm resistivity. A 1200 Å film of low stress silicon nitride was deposited by low-pressure chemical vapor deposition (Scottish Microelectronics Centre, UK). With an MA6 mask aligner (Suss Microtech, Germany), a pattern consisting of 0.6 μm dots with 2 μm pitch was transferred into a layer of NR9-250P photoresist (Futurrex, USA) spin-coated on the substrate. The pattern was transferred into the low stress nitride film with a 2 min 30 s reactive ion etching in CF4 gas in an Oxford NGP80 (20 sccm, 200 W, 100 mTorr, Oxford Instruments, UK). The native oxide layer was stripped by soaking for 2 min in 10% v/v HF solution. The substrate was rapidly transferred in a 10% v/v HF solution of 0.02 M AgNO3 and incubated for 2 min for electroless deposition of Ag nanoparticles. The substrate was transferred to a 10% v/v HF solution containing 0.12 M H2O2 to undergo metal-assisted chemical etching for 8 min 30 s, forming porous pillar structures. The substrate was washed repeatedly in water and dried under N2 stream. Reactive ion etching in SF6 gas for 2 min 30 s (20 sccm, 100 mTorr, 250 W, Oxford NGP80) formed the final conical nN structures. The wafer was diced into 8 × 8 mm dies for subsequent use (DISCO Technologies, Japan). The typical nN had 3–4 μm length, a base diameter of 600 nm, and an apical diameter below 100 nm.
Preparation of Substrates
Samples were prepared as previously described. Substrate surfaces were activated using an oxygen plasma cleaner (10 min, Plasma Prep 5, Gala Instrumente, Germany) and then functionalized with 3-aminotriethoxysilane (APTES, Sigma-Aldrich, A3648) by liquid-phase conjugation in an ethanoic solution of 2% v/v APTES for 2 h. Following repeat washes in absolute ethanol (Sigma-Aldrich 32221), the nN arrays were dried under nitrogen. To generate fluorescent substrates, 0.0005% w/v 5 carboxytetramethylrhodamine N-succinimidyl ester (TAMRA, Sigma-Aldrich 53048) in phosphate-buffered saline (PBS) or 0.05 mg/mL fluorescein isothiocyanate isomer I (Sigma-Aldrich F7250) in PBS was conjugated to the APTES amine group with 2 h incubation followed by repeated washes with PBS and water. Nonfluorescent samples were sterilized under UV light for at least 20 min prior to cell experiments.
Cell Culture
Human umbilical vein endothelial cells (HUVECs, Lonza) were expanded and seeded in endothelial growth medium-2 (EGM-2, Lonza) according to the manufacturer’s instructions. hMSCs were used between passages 4 and 6, and HUVECs were used between passages 5 and 10. For 6 h experiments, hMSCs were seeded at a density of 20 000 viable cells/cm2 and HUVECs were seeded at a density of 30 000 viable cells/cm2, as determined by Trypan Blue exclusion. For 48 h experiments, hMSCs and HUVECs were seeded at different densities on flat and nN substrates to avoid confluent overgrowth. For hMSCs, cells were seeded at 2500 and 8375 cells/cm2 for flat and nN substrates, respectively. For HUVECs, cells were seeded at 3000 and 12 500 cells/cm2 for flat and nN substrates, respectively.
Human mesenchymal stem cells (hMSCs, Lonza Ltd., Basel, Switzerland) were expanded in serum-free, chemically defined medium (MSCGM-CD) with supplements (TheraPEAK, Lonza), as per the manufacturer’s instructions. When ∼80% confluent, hMSCs were detached with 0.05% v/v trypsin-EDTA, reseeded at a density of 100–500 cell/cm2, and cultured for 7–14 days before reaching confluence. For interfacing with nN or flat substrates, hMSCs were seeded in minimum essential medium alpha (αMEM, Gibco ThermoFisher Scientific, Paisley, United Kingdom) with 10% v/v MSC-qualified fetal bovine serum (FBS, Gibco) and 1% v/v penicillin/streptomycin (P/S, Gibco).
Latrunculin B, Lysophosphatidic Acid, and Staurosporine Treatment
For treatment with Latrunculin B (LatB) or lysophosphaticid acid (LPA), cells were cultured for 5 h on flat or nN substrates, and then the medium was changed to include either dimethyl sulfoxide control (DMSO, Sigma-Aldrich, 1:10000), LatB (Sigma-Aldrich, 1:10000 in DMSO, 100 nM final concentration), or LPA (Santa Cruz Biotechnology, 1:500, 10 μm final concentration). Cells were then cultured for 1 h in the treated condition before end point experiments.
For treatment with staurosporine, cells were cultured for the entire 6 h time course in either DMSO (1:10000) or staurosporine (Abcam 120056, in 1:10000 DMSO, final concentration 1 μM). After 6 h, cells were treated with fluorescent wheat germ agglutinin (WGA-555, ThermoFisher W32464, 1:200) and CellEvent caspase-3/7 green detection reagent (ThermoFisher C10423, 4 μM) in PBS with 5% v/v FBS for 30 min. Cells were then fixed with 3.7% w/v PFA, washed twice with PBS, and fluorescent images were captured to detect caspase activity.
Immunocytochemistry and Imaging
Cells were fixed in 3.7% w/v paraformaldehyde (PFA, Sigma-Aldrich) in PBS for 15 min at room temperature, then washed twice with PBS. For treatment with the cytoskeletal stabilization buffer (CSK, vinculin images), cells were incubated with CSK (10 mM PIPES, 50 mM NaCl, 3 mM MgCl2, 300 mM sucrose, 0.5% v/v Triton-X 100) for 1 min at 4 °C prior to fixation, following an established protocol.53 Cells were then permeabilized with 0.25% v/v Triton X (Sigma-Aldich) for 10 min and blocked with 5% v/v donkey serum for 1–2 h. Primary antibodies were diluted in fresh 0.1% w/v bovine serum albumin (BSA, Sigma-Aldrich) in PBS and added to the cells overnight at 4 °C. Samples were then washed three times with PBS for 5 min before being incubated with secondary antibodies (1:500) in 0.1% w/v BSA for 60–90 min at room temperature; cells were then washed three more times with PBS for 5 min. Where applicable, samples were incubated with AlexaFluor-conjugated phalloidin (1:100–1:200 in 0.1% w/v BSA) for 1 h. All samples were counterstained with DAPI (1:1000, 1 μg/mL final concentration) for 5 min and stored upside down in Vectashield (H-1000 Vector Laboratories, Peterborough, United Kingdom) in glass-bottom chamber slides for imaging (Nunc, ThermoFisher Scientific). Antibody information is listed in Supplementary Table 1. Confocal imaging was performed on a Leica SP5 microscope (Leica Microsystems, Wetzler, Germany), and z-stacks were collected with a 63× 1.4 NA oil-immersion objective lens at 700 nm step size and with a pixel size of 240 nm. Wide-field imaging was performed with an Axio Observer automated microscope (Carl Zeiss Meditec, Jena, Germany) with a 20× 0.8 NA dry objective and a pixel size of 240 nm imaged at 16 bits per pixel (Hamamatsu Flash4 sCMOS). 3D structured illumination microscopy (3D SIM) imaging was performed at room temperature with an Elyra PS.1 (Carl Zeiss). A 63× 1.4 NA oil-immersion objective lens was used, with three orientation angles of the excitation grid and five phases acquired for each image with a 110 nm z-step and a pixel size of 32 nm imaged at 16 bits per pixel on an Andor Zyla. SIM processing was performed with the SIM module of the Zen software package (Carl Zeiss), then TIF stacks of processed SIM data were exported. The SIM data sets were then turned into projection images using ImageJ software.
Live Cell Imaging
HUVECs were seeded in 35 mm plates for next day 70% confluency (100 k/cm2). A ratio of 3:1 FuGENE HD transfection reagent (Promega, E2311)/DNA (Lifeact plasmid) with 1 μg of DNA was made up in Opti-MEM I reduced serum medium, GlutaMAX (Life Technologies, 51985-026), and 100 μL was added to the cells in EBM-2 basal medium (Lonza CC-3156) with 2% FBS. After 6 h, the medium was replaced with EGM. Forty-eight hours after transfection, cells were trypsinized and seeded on the nN in 24-well plates at a seeding density determined by the transfection efficiency. Following 2 h of incubation, to allow adherence of cell to the nN, the nN were inverted and placed down in an 8-well chamber slide containing EGM media and imaged on a wide-field Ti-E Eclipse microscope (Nikon-Minato, Japan) with a 20× 0.8 NA dry objective. The cells were imaged every 15 min for 2 h and 45 min with z-stacks of 5 μm range and 500 nm spacing.
Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
Cells on nN or flat substrates were incubated with Trizol reagent (Life Technologies), mixed with chloroform (5:1 Trizol/chloroform), and separated by centrifugation (12 000g, 15 min, 4 °C). The RNA contained within the aqueous phase was then isolated with RNeasy columns (Qiagen), according to the manufacturer’s instructions. cDNA was synthesized using a reverse transcription kit (Applied Biosystems, Life Technologies, product #4368814), and qRT-PCR was performed with a SYBR Green master mix (Applied Biosystems, Life Technologies, product #1179401K) with 2–5 ng of cDNA and 250–500 nM each of forward and reverse primers, using either a StepOne Plus or QuantStudio6 machine (Applied Biosystems). The qRT-PCR protocol was slightly different for the two machines. For the StepOne Plus, the protocol included the following: 95 °C for 20 s followed by 40 cycles of denaturation at 95 °C for 3 s and annealing at a temperature between 55 and 60 °C for 30 s. For the QuantStudio6, the protocol included the following: 95 °C for 20 s followed by 40 cycles of denaturation at 95 °C for 1 s and annealing at a temperature between 55 and 60 °C for 20 s. On both machines, a melt curve was subsequently performed in all reactions to ensure that a single amplicon was generated for each target gene. Cycles-to-threshold (Ct) values were automatically obtained using the ThermoFisher Scientific Cloud Software for qPCR file processing (https://www.thermofisher.com/uk/en/home/cloud.html). These values were subsequently exported to an Excel file and manually processed to generate fold change expression values. The expression of each gene of interest was normalized to the geometric mean54 of the expression of at least two housekeeping genes (PPIA, RPL13A, and/or HPRT1), generating the ΔC(t) value, and expression of 2–ΔΔC(t) relative to the flat control for each cell type, and N ≥ 3 experimental replicates are reported. Statistical analysis information is listed in the relevant section below. Custom primers were purchased from Invitrogen and tested for specificity prior to use. Sequences are listed in Supplementary Table 2.
Extraction of Cell Lysates and Western Blotting
Medium from cells on nN or flat substrates was gently aspirated, and cells were rinsed two times with ice cold PBS. Cell lysate from 8 chips were extracted in 300 μL of cell lysate buffer (4 M urea, 150 mM NaCl, PhosSTOP (Roche), and complete EDTA-free protease inhibitor cocktail (Roche)) by scraping on ice. Lysates were sonicated using an immersion probe for 10 s pulse at 200 W. Insoluble protein was removed by centrifuging at 15 000g for 10 min at 4 °C. Protein was quantified using Qubit protein assay (Q33211, Thermo) and Qubit fluorometric quantification instrument (Thermo). Protein samples were prepared with 4× sample buffer containing β-mercaptoethanol in a ratio of 3:1 and heated at 80 °C for 5 min. SDS-PAGE electrophoresis was conducted using TGS running buffer (Bio-Rad) for 45 min at 100 V. Gel transfer was conducted using Transblot-turbo (Biorad). Blots were probed with primary mouse anti-human vinculin (Abcam ab18058, 1:1000) and secondary (Li-Cor IR 680, 1:1000) antibodies in iBind fluorescent solution (SLF1019, Thermo) using the respective iBind Flex western device. Blots were analyzed for intensity of fluorescent band using a Li-Cor Odyssey imaging system.
Scanning Electron Microscopy
Cells on nN or flat substrates were fixed in 2.5% v/v glutaraldehyde solution (Sigma) for 1 h in PBS at room temperature and then washed three times in PBS. PBS buffer was substituted with 0.1 M sodium cacodylate buffer (Electron Microscopy Sciences, USA), and cells were washed twice for 5 min. Cells were postfixed in 1% v/v osmium tetroxide for 1 h in 0.1 M sodium cacodylate buffer and subsequently washed with distilled water two times for 5 min. Samples were dehydrated in a series of ethanol dilutions (20, 30, 50, 70, 80, 90% v/v ethanol in water), treated with 100% ethanol four times for 5 min, after which they were treated with hexamethyldisilazane for 5 min and air-dried. Samples were mounted and sputtered with 10 nm of chromium (Q150, Quorum) and imaged using Sigma300 (Zeiss) scanning electron microscope with a working distance of 10 mm and an accelerating voltage of 5 keV.
Quantifying Actin Stress Fibers
Actin stress fibers were quantified from confocal images. To highlight stress fibers, the actin channel was filtered by performing convolution with a 6 by 10 kernel with the central two columns containing a positive value and outer four columns containing a negative value. Convolution was performed with orientations of the kernel at 3° intervals between 0 and 180°, and the maximum value for each pixel over 60 orientations was then selected. Other kernel shapes and orientation regimes were tested; however, this choice emphasized stress fibers most effectively, as judged by visual inspection (Figure S2).
Thresholding was then performed on these images, and the subsequent binary mask appeared to match stress fibers well. Regions below 100 pixels in size were removed as these were largely noise.
Quantifying Focal Adhesion Density
To determine the focal adhesion density, the number of focal adhesion regions in the image, divided by the image area covered by actin (determined by a similar threshold operation to blurred actin image) was calculated. Significance was determined by pairwise t test between 10 analyzed fields of view taken over two experimental repeats.
Quantitative Cell Morphology Analysis
Quantification of cell and protrusions morphology was performed on tiled wide-field microscopy images using the MATLAB image analysis toolbox. Background correction was performed by negating the image with itself, following very large Gaussian blur. Marker controlled watershed segmentation was then performed on DAPI channel to identify nuclei. Nuclei touching the border, or below a threshold intensity, were filtered. To identify the cell cytoplasm, thresholding was performed on the actin channel to identify the image region containing cells. Watershed on the actin channel resulted in extensive mis-segmentation, thus utilizing nuclei as markers, and the actin containing image region as the boundary, marker-controlled watershed segmentation was performed on the YAP channel. This resulted in effective detection of cell boundaries with the substrate and with other cells. Cells with mean actin intensity similar to background were filtered out, as were those touching the border.
Feature Extraction
Features describing cell and protrusion morphology were extracted from cell and nuclei segments. For intensity and texture properties, the original images were used, without background correction; however, images were log transformed, and subsequently the 10th percentile intensity was deducted to align background intensity to zero, thus reducing technical intensity variations between replicates. Protrusion regions are defined by applying a large erosion and dilation operation to the whole cell segment. This generated a highly rounded core, region of cytoplasm not in this core are defined as protrusions. A list of the features extracted for linear discriminant analysis is given below.
Morphology (except Actin Channel Intensity)
Cell area
Cell major axis length: length of the equivalent ellipse based upon second order moments
Cell minor axis length: width of the equivalent ellipse
Cell eccentricity: eccentricity of the equivalent ellipse
Cell extent: proportion of pixels within the bounding box
Cell solidity: proportion of pixels in the convex
Cell perimeter: perimeter length of cell segment
Cell roundness: defined as
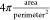
Cell channel intensity: mean actin intensity: not included for LDA
Number of protrusions per cell
Mean protrusion area per cell
Max protrusion area per cell
Total protrusion area per cell
Mean length of protrusions per cell (major axis length of equivalent ellipse)
Max length of protrusion per cell
Total length of protrusions per cell
Mean width of protrusion per cell (minor axis length of equivalent ellipse)
Max width of protrusion per cell
Total width of protrusion per cell
Mean extent of protrusions per cell, defined as length from nuclei center of mass to furthest point in protrusion region minus length from nuclei center of mass to nearest point in protrusion region.
Max extent of protrusion per cell
Total extent of protrusion per cell
Actin Texture Calculated from Gray Level Co-occurrence Matrix (GLCM)
Contrast: variance between neighboring pixels
Correlation: correlation between neighboring pixels
Energy: angular second moment of the image
Homogeneity: a measure of how sharp are gradients within the image
Additional Features
Nuclear centroid X position
Nuclear centroid Y position
Nuclear orientation angle of equivalent ellipse
Cell orientation angle of equivalent ellipse
Mean intensity of central actin (following erosion of cytoplasmic segment mask)
Mean intensity of cortical actin (mask of cytoplasmic ring region that is eroded to form central region)
Protrusion Features
Index of cell protrusion. This links protrusions to the unique cell ID in the corresponding single cell data file.
Protrusion area
Length of protrusion (major axis length of equivalent ellipse)
Width of protrusion (minor axis length of equivalent ellipse)
Protrusion orientation angle of equivalent ellipse
Extent, defined as length from nuclei center of mass to furthest point in protrusion region minus length from nuclei center of mass to nearest point in protrusion region
Linear Discriminant Analysis
LDA between flat substrate and nN was performed on cells pooled over 3 experimental repeats for HUVEC and hMSC cells, with two technical repeats for two experiments, and one technical replicate for one experiment. Each cell type was analyzed independently, where cells were labeled as “Flat” or “nN” and the features described above were extracted for all cells. R2 values for HUVEC and hMSC cells were 0.286 and 0.343, respectively; p < 0.001 in both cases. Also, LDA applied to data following random permutation of class labels led to insignificant separation. These R2 values correspond to 75 and 78% correct substrate classification of HUVEC and hMSC cells, respectively. The box plot in Figure 1 shows a random sample of 900 cells from each group. This analysis was conducted with the Scikit-Learn module for Python.55
YAP Localization Analysis
YAP intensities and localization were calculated following cell and nuclei segmentation as described above. Three features on YAP localization were recorded:
Nuclear YAP intensity: taken as the median of the nuclear YAP intensities
Cytoplasmic YAP intensity: the cytoplasmic segments was eroded by a fixed width (10px), such that pixels bordering the substrate and other cell were not included, subsequently the median YAP intensity from this region was taken.
Log nuclear to cytoplasmic YAP ratio
YAP nuclear to cytoplasmic localization changes both in wild-type and following drug and DMSO treatment were recorded from a pooled random sample of 180 cells taken over two experimental repeats, with two technical repeats per experiment. Significance of differences was calculated using a pair wise t test between single cell populations.
Focal Adhesion Quantification
To identify focal adhesions, a similar approach to actin stress fiber identification was employed. Confocal images of cells treated with CSK and then stained for vinculin were used for image analysis. Vinculin channel images were filtered by performing convolution with a 5 by 6 kernel with the central two columns containing a positive value and outer two columns containing a negative value. Convolution was performed with orientations of the kernel at 10° intervals between 0 and 90°, the maximum value for each pixel over the 9 orientations was then selected. Following thresholding similar to actin bundle quantification (Figure S2), regions below 10 pixels in size were removed as these were largely noise.
Again, to determine the focal adhesion density, the number of focal adhesion regions in the image divided by the image area covered by actin (determined by a similar threshold operation) was calculated. Significance was determined by pairwise t test between 10 analyzed fields of view taken over two experimental repeats.
Quantification of Lamin Signal
In Figure 4E–G, single cells were isolated from cropped confocal z-stack images for analysis using ImageJ software (National Institutes of Health, Bethesda, MD, USA). A line was drawn in the x–y plane along a row of nN interacting with the nuclear envelope, and a reslice image was created (reslice shown in Figure 4I). Reslice line width was 25 pixels for Figure 4F and 20 pixels for Figure 4G. From the reslice images, one of two quantifications was performed. For data in Figure 4F, a line of 20 pixel width was created in the reslice image and hand-drawn along the base of the nuclear envelope and up the entire z-height of each individual nN. Along this line, the signal for lamin A and lamin B channels were recorded (test line). In another area of the nuclear envelope where nN were not interacting (i.e., top nuclear envelope or space between nN interfacing), an additional line of 20 pixel width was drawn, and the lamin A and lamin B signal values over this line were averaged as a normalization factor for that specific reslice image (normalization line). Then, all values recorded from the test line were normalized to the respective average calculated from the normalization line, providing a fold change measurement for each lamin channel within that particular reslice image.
To obtain the data in Figure 4G, reslice images were prepared in the same way, and two additional lines were prepared for measurement of signal within the reslice image. Two identical lines of 20 pixel width were drawn in the y-direction (that is, perpendicular to the nN height). One line was placed in the middle of the nN z-height (middle line) and an identical line was z-shifted to the top of the nN z-height (top line), and signals for nN, lamin A, and lamin B channels were recorded. Similar to above, a normalization line was also recorded from an area within the reslice along a 20 pixel width line where nN were not interacting. The troughs within the nN signal were detected from the inflection points around local maxima of the nN signal from the middle line. Lamin A and/or lamin B signals were then integrated from trough-to-trough distances for the middle and top lines. This analysis was performed because measurement of lamin A or B signal at specific points along the nN width in the original reslice image did not reveal the accumulation of the protein signal around the nN in the y-direction. Integrated values were subsequently normalized to the respective average values calculated from the normalization line, and the ratio of integrated lamin A/B at the nN middle and top is reported. In Figure S11, the integrated values for lamin A or B at the middle or top were normalized and reported.
Statistical Analysis
Statistics on biochemical data were performed with GraphPad Prism software (La Jolla, CA, USA). When comparing two groups, a Student’s unpaired t test was performed. When comparing more than two groups, a one-way analysis of variance (ANOVA) was performed, followed by pairwise comparisons via Bonferroni’s multiple comparison test. For the comparison of lamin A/B ratios in Figure 4, a nonlinear exponential curve was fit to the data set, and the best-fit values are reported. For image analysis, MatLab (Mathworks, Cambridge, UK) was used to analyze data and prepare charts.
For qPCR analysis, three to four experimental replicates were performed, with at least two biological replicates within each experiment. For each experimental replicate, the expression of the gene of interest (GOI) was normalized to the geometric mean of at least two housekeeping genes (HKGs) following previous methods,56 which generated the ΔCt value. For each cell type, the average was calculated for the normalized GOI expression on flat samples, and expression of all groups was then normalized to these values. This resulted in an expression value equal to 1 for HUVEC or hMSC flat samples but carried a nonzero standard deviation that reflected the intraexperimental heterogeneity of biological replicates. In order to propagate this error, the relative standard deviation (RSD) was calculated for all groups of interest, where
where N is the number of experimental replicates (N = 3–4) and SD_expRep represents the standard deviation of the normalized expression for each group within each experimental replicate. This approach allowed for the variability within each experiment to be propagated. Of note, for lamin A and lamin B gene expression in Figure 4, the expression of all groups was normalized to HUVEC flat so that cell type-specific differences in lamin signal could be observed. For Figure S13A–D, ΔCt values for each GOI were compared as a ratio and were not normalized to the expression of the flat control cells (i.e., ANKRD1/LMNA describes ΔCt_ANKRD1/ΔCt_LMNA).
For all box plots, the 25th and 75th quartiles are represented, the line is the median, and the whiskers extend to the minimum and maximum data points, unless the point is a statistical outlier (and is therefore shown in red). For all experiments, p < 0.05 was considered significant.
Data Availability
Raw data are available upon request from rdm-enquiries@imperial.ac.uk.
Acknowledgments
C.S.H. and A.E.G.C. are associated with the Institute for Chemical Biology at Imperial College London, which is supported by funding from the Engineering and Physical Sciences Research Council (EPSRC, EP/F500416/1). S.W.C. was supported by the Whitaker International Program, Institute of International Education, USA, and the European Commission (H2020-MSCA-IF-2014 655698). S.C. was funded through the STRATiGRAD program at Imperial College London. S.G. was funded by a PhD studentship in Biomedicine and Bioengineering in Osteoarthritis, Imperial College London. M.J.P.C. was funded by the Fundação para a Ciência e Tecnologia (FCT), Portugal (Grant PD/BD/105775/2014). L.dO.M was supported by the European Commission FP7 (“SAVVY project”, number 310445). C.B. is funded by the Stand Up to Cancer campaign for Cancer Research UK, and Cancer Research UK Programme Foundation Award (C37275/1A20146). C.C. acknowledges funding from the Royal Society (NF110513) and the European Commission (PIIF-GA-2011-302638 ERC StG 759577). M.B., C.C., and M.M.S. acknowledge support from the Rosetrees Trust, the Stoneygate Trust, the Bloom Foundation, and the Robert Luff Foundation. M.M.S. acknowledges support from the ERC Seventh Framework Programme Consolidator grant “Naturale CG” (616417), the Wellcome Trust Senior Investigator Award (098411/Z/12/Z) and the grant “Bio-functionalized Nanomaterials for Ultra-sensitive Biosensing” (EP/K020641/1) funded by EPSRC. M.M.S. acknowledges support from the ERC Seventh Framework Programme Consolidator grant “Naturale CG” (616417), the Wellcome Trust Senior Investigator Award (098411/Z/12/Z), the grant “Bio-functionalized Nanomaterials for Ultra-sensitive Biosensing” (EP/K020641/1) funded by EPSRC, and the Research Council of Norway through its Centres of Excellence scheme (262613). Imaging was performed at the Facility for Imaging by Light Microscopy (FILM) at Imperial College London, which is part-supported by funding from the Biotechnology and Biological Sciences Research Council (BBSRC, BB/L015129/1). The authors would like to acknowledge K. Pchelintseva and M. Chee for their help with Western blotting. The authors would like to acknowledge K. Pchelintseva and M. Chee for their help with Western blotting, and A. Nogiwa-Valdez for support with data management.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acsnano.8b06998.
Author Present Address
■ Department of Computational Biology, University of Lausanne, Lausanne, Switzerland, and SIB Swiss Institute of Bioinformatics
Author Present Address
○ LBCMCP, Centre de Biologie Intégrative (CBI), Université de Toulouse, CNRS/UPS, 31062 Toulouse Cedex, France
Author Present Address
⬡ Centre for Craniofacial and Regenerative Biology, King’s College London, London, UK.
Author Contributions
& Joint first co-authors.
Author Contributions
C.S.H. and S.W.C. designed and performed all experiments, data analysis, and prepared the manuscript; C.C. and M.B. fabricated the nanoneedles; S.C. and L.O.M. performed image and data analysis and prepared the manuscript; M.J.P.C., S.G., and M.B. performed experiments and prepared the manuscript; C.C. and M.M.S. designed the research plan; A.E.G.C., C.B., C.C., and M.M.S. prepared the manuscript; M.M.S. supervised the project. C.S.H. and S.W.C. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Crowder S. W.; Leonardo V.; Whittaker T.; Papathanasiou P.; Stevens M. M. Material Cues as Potent Regulators of Epigenetics and Stem Cell Function. Cell Stem Cell 2016, 18, 39–52. 10.1016/j.stem.2015.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalby M. J.; Biggs M. J. P.; Gadegaard N.; Kalna G.; Wilkinson C. D. W.; Curtis A. S. G. Nanotopographical Stimulation of Mechanotransduction and Changes in Interphase Centromere Positioning. J. Cell. Biochem. 2007, 100, 326–338. 10.1002/jcb.21058. [DOI] [PubMed] [Google Scholar]
- Engler A. J.; Sen S.; Sweeney H. L.; Discher D. E. Matrix Elasticity Directs Stem Cell Lineage Specification. Cell 2006, 126, 677–689. 10.1016/j.cell.2006.06.044. [DOI] [PubMed] [Google Scholar]
- McMurray R. J.; Gadegaard N.; Tsimbouri P. M.; Burgess K. V.; McNamara L. E.; Tare R.; Murawski K.; Kingham E.; Oreffo R. O. C.; Dalby M. J. Nanoscale Surfaces for the Long-Term Maintenance of Mesenchymal Stem Cell Phenotype and Multipotency. Nat. Mater. 2011, 10, 637–644. 10.1038/nmat3058. [DOI] [PubMed] [Google Scholar]
- Swift J.; Ivanovska I. L.; Buxboim A.; Harada T.; Dingal P. C. D. P.; Pinter J.; Pajerowski J. D.; Spinler K. R.; Shin J. W.; Tewari M.; Rehfeldt F.; Speicher D. W.; Discher D. E. Nuclear Lamin-A Scales with Tissue Stiffness and Enhances Matrix-Directed Differentiation. Science 2013, 341, 1240104. 10.1126/science.1240104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBeath R.; Pirone D. M.; Nelson C. M.; Bhadriraju K.; Chen C. S. Cell Shape, Cytoskeletal Tension, and RhoA Regulate Stem Cell Lineage Commitment. Dev. Cell 2004, 6, 483–495. 10.1016/S1534-5807(04)00075-9. [DOI] [PubMed] [Google Scholar]
- Dupont S.; Morsut L.; Aragona M.; Enzo E.; Giulitti S.; Cordenonsi M.; Zanconato F.; Le Digabel J.; Forcato M.; Bicciato S.; Elvassore N.; Piccolo S. Role of YAP/TAZ in Mechanotransduction. Nature 2011, 474, 179–184. 10.1038/nature10137. [DOI] [PubMed] [Google Scholar]
- Hanson L.; Zhao W.; Lou H. Y.; Lin Z. C.; Lee S. W.; Chowdary P.; Cui Y.; Cui B. Vertical Nanopillars for in Situ Probing of Nuclear Mechanics in Adherent Cells. Nat. Nanotechnol. 2015, 10, 554–562. 10.1038/nnano.2015.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ihalainen T. O.; Aires L.; Herzog F. A.; Schwartlander R.; Moeller J.; Vogel V. Differential Basal-to-Apical Accessibility of Lamin A/C Epitopes in the Nuclear Lamina Regulated by Changes in Cytoskeletal Tension. Nat. Mater. 2015, 14, 1252–1261. 10.1038/nmat4389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D. H.; Wirtz D. Cytoskeletal Tension Induces the Polarized Architecture of the Nucleus. Biomaterials 2015, 48, 161–172. 10.1016/j.biomaterials.2015.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovett D. B.; Shekhar N.; Nickerson J. A.; Roux K. J.; Lele T. P. Modulation of Nuclear Shape by Substrate Rigidity. Cell. Mol. Bioeng. 2013, 6, 230–238. 10.1007/s12195-013-0270-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makhija E.; Jokhun D. S.; Shivashankar G. V. Nuclear Deformability and Telomere Dynamics are Regulated by Cell Geometric Constraints. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, E32–E40. 10.1073/pnas.1513189113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dupont S. Role of YAP/TAZ in Cell-Matrix Adhesion-Mediated Signalling and Mechanotransduction. Exp. Cell Res. 2016, 343, 42–53. 10.1016/j.yexcr.2015.10.034. [DOI] [PubMed] [Google Scholar]
- Elosegui-Artola A.; Andreu I.; Beedle A. E. M.; Lezamiz A.; Uroz M.; Kosmalska A. J.; Oria R.; Kechagia J. Z.; Rico-Lastres P.; Le Roux A. L.; Shanahan C. M.; Trepat X.; Navajas D.; Garcia-Manyes S.; Roca-Cusachs P. Force Triggers YAP Nuclear Entry by Regulating Transport Across Nuclear Pores. Cell 2017, 171, 1397–1410.e14. 10.1016/j.cell.2017.10.008. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Cooper L. F.; Zhang X.; Zhang Y.; Deng F.; Song J.; Yang S. Titanium Nanotubes Induce Osteogenic Differentiation through the FAK/RhoA/YAP Cascade. RSC Adv. 2016, 6, 44062–44069. 10.1039/C6RA04002K. [DOI] [Google Scholar]
- Nardone G.; Oliver-De La Cruz J.; Vrbsky J.; Martini C.; Pribyl J.; Skládal P.; Pešl M.; Caluori G.; Pagliari S.; Martino F.; Maceckova Z.; Hajduch M.; Sanz-Garcia A.; Pugno N. M.; Stokin G. B.; Forte G. YAP Regulates Cell Mechanics by Controlling Focal Adhesion Assembly. Nat. Commun. 2017, 8, 15321. 10.1038/ncomms15321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ankam S.; Lim C. K.; Yim E. K. F. Actomyosin Contractility Plays a Role in MAP2 Expression during Nanotopography-Directed Neuronal Differentiation of Human Embryonic Stem Cells. Biomaterials 2015, 47, 20–28. 10.1016/j.biomaterials.2015.01.003. [DOI] [PubMed] [Google Scholar]
- Cosgrove B. D.; Mui K. L.; Driscoll T. P.; Caliari S. R.; Mehta K. D.; Assoian R. K.; Burdick J. A.; Mauck R. L. N-Cadherin Adhesive Interactions Modulate Matrix Mechanosensing and Fate Commitment of Mesenchymal Stem Cells. Nat. Mater. 2016, 15, 1297–1306. 10.1038/nmat4725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mosqueira D.; Pagliari S.; Uto K.; Ebara M.; Romanazzo S.; Escobedo-Lucea C.; Nakanishi J.; Taniguchi A.; Franzese O.; Di Nardo P.; Goumans M. J.; Traversa E.; Pinto-Do-Ó P.; Aoyagi T.; Forte G. Hippo Pathway Effectors Control Cardiac Progenitor Cell Fate by Acting as Dynamic Sensors of Substrate Mechanics and Nanostructure. ACS Nano 2014, 8, 2033–2047. 10.1021/nn4058984. [DOI] [PubMed] [Google Scholar]
- Musah S.; Morin S. A.; Wrighton P. J.; Zwick D. B.; Jin S.; Kiessling L. L. Glycosaminoglycan-Binding Hydrogels Enable Mechanical Control of Human Pluripotent Stem Cell Self-Renewal. ACS Nano 2012, 6, 10168–10177. 10.1021/nn3039148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song L.; Wang K.; Li Y.; Yang Y. Nanotopography Promoted Neuronal Differentiation of Human Induced Pluripotent Stem Cells. Colloids Surf., B 2016, 148, 49–58. 10.1016/j.colsurfb.2016.08.041. [DOI] [PubMed] [Google Scholar]
- Teo B. K. K.; Ankam S.; Chan L. Y.; Yim E. K. F. Nanotopography/Mechanical Induction of Stem-Cell Differentiation. Methods Cell Biol. 2010, 98, 241–294. 10.1016/S0091-679X(10)98011-4. [DOI] [PubMed] [Google Scholar]
- Yim E. K. F.; Pang S. W.; Leong K. W. Synthetic Nanostructures Inducing Differentiation of Human Mesenchymal Stem Cells into Neuronal Lineage. Exp. Cell Res. 2007, 313, 1820–1829. 10.1016/j.yexcr.2007.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pajerowski J. D.; Dahl K. N.; Zhong F. L.; Sammak P. J.; Discher D. E. Physical Plasticity of the Nucleus in Stem Cell Differentiation. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 15619–15624. 10.1073/pnas.0702576104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guilluy C.; Osborne L. D.; Van Landeghem L.; Sharek L.; Superfine R.; Garcia-Mata R.; Burridge K. Isolated Nuclei Adapt to Force and Reveal a Mechanotransduction Pathway in the Nucleus. Nat. Cell Biol. 2014, 16, 376–381. 10.1038/ncb2927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu H.; Wen J.; Xiao Y.; Liu J.; Hopyan S.; Radisic M.; Simmons C. A.; Sun Y. In Situ Mechanical Characterization of the Cell Nucleus by Atomic Force Microscopy. ACS Nano 2014, 8, 3821–3828. 10.1021/nn500553z. [DOI] [PubMed] [Google Scholar]
- Chiappini C.; Martinez J. O.; De Rosa E.; Almeida C. S.; Tasciotti E.; Stevens M. M. Biodegradable Nanoneedles for Localized Delivery of Nanoparticles in Vivo: Exploring the Biointerface. ACS Nano 2015, 9, 5500–5509. 10.1021/acsnano.5b01490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiappini C.; De Rosa E.; Martinez J. O.; Liu X.; Steele J.; Stevens M. M.; Tasciotti E. Biodegradable Silicon Nanoneedles Delivering Nucleic Acids Intracellularly Induce Localized in Vivo Neovascularization. Nat. Mater. 2015, 14, 532–539. 10.1038/nmat4249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mumm F.; Beckwith K. M.; Bonde S.; Martinez K. L.; Sikorski P. A Transparent Nanowire-Based Cell Impalement Device Suitable for Detailed Cell-Nanowire Interaction Studies. Small 2013, 9, 263–272. 10.1002/smll.201201314. [DOI] [PubMed] [Google Scholar]
- Peer E.; Artzy-Schnirman A.; Gepstein L.; Sivan U. Hollow Nanoneedle Array and Its Utilization for Repeated Administration of Biomolecules to the Same Cells. ACS Nano 2012, 6, 4940–4946. 10.1021/nn300443h. [DOI] [PubMed] [Google Scholar]
- Shalek A. K.; Robinson J. T.; Karp E. S.; Lee J. S.; Ahn D.-R.; Yoon M.-H.; Sutton A.; Jorgolli M.; Gertner R. S.; Gujral T. S.; MacBeath G.; Yang E. G.; Park H. Vertical Silicon Nanowires as a Universal Platform for Delivering Biomolecules into Living Cells. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 1870–1875. 10.1073/pnas.0909350107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vandersarl J. J.; Xu A. M.; Melosh N. A. Nanostraws for Direct Fluidic Intracellular Access. Nano Lett. 2012, 12, 3881–3886. 10.1021/nl204051v. [DOI] [PubMed] [Google Scholar]
- Xie X.; Xu A. M.; Leal-Ortiz S.; Cao Y.; Garner C. C.; Melosh N. A. Nanostraw-Electroporation System for Highly Efficient Intracellular Delivery and Transfection. ACS Nano 2013, 7, 4351–4358. 10.1021/nn400874a. [DOI] [PubMed] [Google Scholar]
- Xu A. M.; Aalipour A.; Leal-Ortiz S.; Mekhdjian A. H.; Xie X.; Dunn A. R.; Garner C. C.; Melosh N. A. Quantification of Nanowire Penetration into Living Cells. Nat. Commun. 2014, 5, 3613. 10.1038/ncomms4613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiappini C.; Liu X.; Fakhoury J. R.; Ferrari M. Biodegradable Porous Silicon Barcode Nanowires with Defined Geometry. Adv. Funct. Mater. 2010, 20, 2231–2239. 10.1002/adfm.201000360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemyj S. V. Busses Made by “L’vovskij Avtobusnyj Zavod” Company, Which Have Been Equipped with Engines Made by Yaroslavl’ Engine Works. Avtomobil’naya Promyshlennost 2002, 13, 8–9. [Google Scholar]
- Robinson J. T.; Jorgolli M.; Shalek A. K.; Yoon M. H.; Gertner R. S.; Park H. Vertical Nanowire Electrode Arrays as a Scalable Platform for Intracellular Interfacing to Neuronal Circuits. Nat. Nanotechnol. 2012, 7, 180–184. 10.1038/nnano.2011.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie C.; Lin Z.; Hanson L.; Cui Y.; Cui B. Intracellular Recording of Action Potentials by Nanopillar Electroporation. Nat. Nanotechnol. 2012, 7, 185–190. 10.1038/nnano.2012.8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park Y. S.; Yoon S. Y.; Park J. S.; Lee J. S. Deflection Induced Cellular Focal Adhesion and Anisotropic Growth on Vertically Aligned Silicon Nanowires with Differing Elasticity. NPG Asia Mater. 2016, 8, e249–e249. 10.1038/am.2016.5. [DOI] [Google Scholar]
- Qi S.; Yi C.; Ji S.; Fong C. C.; Yang M. Cell Adhesion and Spreading Behavior on Vertically Aligned Silicon Nanowire Arrays. ACS Appl. Mater. Interfaces 2009, 1, 30–34. 10.1021/am800027d. [DOI] [PubMed] [Google Scholar]
- Bonde S.; Berthing T.; Madsen M. H.; Andersen T. K.; Buch-Månson N.; Guo L.; Li X.; Badique F.; Anselme K.; Nygård J.; Martinez K. L. Tuning InAs Nanowire Density for HEK293 Cell Viability, Adhesion, and Morphology: Perspectives for Nanowire-Based Biosensors. ACS Appl. Mater. Interfaces 2013, 5, 10510–10519. 10.1021/am402070k. [DOI] [PubMed] [Google Scholar]
- Berthing T.; Bonde S.; Rostgaard K. R.; Madsen M. H.; Sorensen C. B.; Nygård J.; Martinez K. L. Cell Membrane Conformation at Vertical Nanowire Array Interface Revealed by Fluorescence Imaging. Nanotechnology 2012, 23, 415102. 10.1088/0957-4484/23/41/415102. [DOI] [PubMed] [Google Scholar]
- Bucaro M. A.; Vasquez Y.; Hatton B. D.; Aizenberg J. Fine-Tuning the Degree of Stem Cell Polarization and Alignment on Ordered Arrays of High-Aspect-Ratio Nanopillars. ACS Nano 2012, 6, 6222–6230. 10.1021/nn301654e. [DOI] [PubMed] [Google Scholar]
- Kim S. Y.; Yang E. G. Collective Behaviors of Mammalian Cells on Amine-Coated Silicon Nanowires. Nanotechnology 2013, 24, 455704. 10.1088/0957-4484/24/45/455704. [DOI] [PubMed] [Google Scholar]
- Persson H.; Købler C.; Mølhave K.; Samuelson L.; Tegenfeldt J. O.; Oredsson S.; Prinz C. N. Fibroblasts Cultured on Nanowires Exhibit Low Motility, Impaired Cell Division, and DNA Damage. Small 2013, 9, 4006–4016. 10.1002/smll.201300644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beckwith K. S.; Cooil S. P.; Wells J. W.; Sikorski P. Tunable High Aspect Ratio Polymer Nanostructures for Cell Interfaces. Nanoscale 2015, 7, 8438–8450. 10.1039/C5NR00674K. [DOI] [PubMed] [Google Scholar]
- Cha K. J.; Hong J. M.; Cho D. W.; Kim D. S. Enhanced Osteogenic Fate and Function of MC3T3-E1 Cells on Nanoengineered Polystyrene Surfaces with Nanopillar and Nanopore Arrays. Biofabrication 2013, 5, 025007 10.1088/1758-5082/5/2/025007. [DOI] [PubMed] [Google Scholar]
- Jahed Z.; Zareian R.; Chau Y. Y.; Seo B. B.; West M.; Tsui T. Y.; Wen W.; Mofrad M. R. K. Differential Collective- and Single-Cell Behaviors on Silicon Micropillar Arrays. ACS Appl. Mater. Interfaces 2016, 8, 23604–23613. 10.1021/acsami.6b08668. [DOI] [PubMed] [Google Scholar]
- Kim D. J.; Lee G.; Kim G. S.; Lee S. K. Statistical Analysis of Immuno-Functionalized Tumor-Cell Behaviors on Nanopatterned Substrates. Nanoscale Res. Lett. 2012, 7, 637. 10.1186/1556-276X-7-637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J.; Kang B. S.; Hicks B.; Chancellor T. F.; Chu B. H.; Wang H. T.; Keselowsky B. G.; Ren F.; Lele T. P. The Control of Cell Adhesion and Viability by Zinc Oxide Nanorods. Biomaterials 2008, 29, 3743–3749. 10.1016/j.biomaterials.2008.05.029. [DOI] [PubMed] [Google Scholar]
- Hanson L.; Lin Z. C.; Xie C.; Cui Y.; Cui B. Characterization of the Cell-Nanopillar Interface by Transmission Electron Microscopy. Nano Lett. 2012, 12, 5815–5820. 10.1021/nl303163y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buxboim A.; Swift J.; Irianto J.; Spinler K. R.; Dingal P. C. D. P.; Athirasala A.; Kao Y. R. C.; Cho S.; Harada T.; Shin J. W.; Discher D. E. Matrix Elasticity Regulates Lamin-A,C Phosphorylation and Turnover with Feedback to Actomyosin. Curr. Biol. 2014, 24, 1909–1917. 10.1016/j.cub.2014.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamashita H.; Ichikawa T.; Matsuyama D.; Kimura Y.; Ueda K.; Craig S. W.; Harada I.; Kioka N. The Role of the Interaction of the Vinculin Proline-Rich Linker Region with Vinexin in Sensing the Stiffness of the Extracellular Matrix. J. Cell Sci. 2014, 127, 1875–1886. 10.1242/jcs.133645. [DOI] [PubMed] [Google Scholar]
- Vandesompele J.; De Preter K.; Pattyn F.; Poppe B.; Van Roy N.; De Paepe A.; Speleman F. Accurate Normalization of Real-Time Quantitative RT-PCR Data by Geometric Averaging of Multiple Internal Control Genes. Genome Biol. 2002, 3, research0034.1. 10.1186/gb-2002-3-7-research0034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínková N.; Nová P.; Sablina O. V.; Graphodatsky A. S.; Zima J. Karyotypic Relationships of the Tatra Vole (Microtus Tatricus). Folia Zool. 2004, 53, 279–284. [Google Scholar]
- Malysheva G. V. Predicting the Endurance of Adhesive Joints. Polym. Sci., Ser. D 2014, 7, 145–147. 10.1134/S1995421214020105. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Raw data are available upon request from rdm-enquiries@imperial.ac.uk.