

Abstract
Highly oxygenated organic molecules (HOM) are formed in the atmosphere via autoxidation involving peroxy radicals arising from volatile organic compounds (VOC). HOM condense on pre-existing particles and can be involved in new particle formation. HOM thus contribute to the formation of secondary organic aerosol (SOA), a significant and ubiquitous component of atmospheric aerosol known to affect the Earth’s radiation balance. HOM were discovered only very recently, but the interest in these compounds has grown rapidly. In this Review, we define HOM and describe the currently available techniques for their identification/quantification, followed by a summary of the current knowledge on their formation mechanisms and physicochemical properties. A main aim is to provide a common frame for the currently quite fragmented literature on HOM studies. Finally, we highlight the existing gaps in our understanding and suggest directions for future HOM research.
1. Introduction
Particles containing organic matter, termed organic aerosols (OA), are an important component of the atmosphere. They can affect climate either by interacting directly with solar radiation or by influencing cloud properties. Additionally, they can adversely affect human health. Organic aerosol is also responsible for a significant fraction of the total submicrometer aerosol mass.1 Despite extensive research on this topic, the sources of OA are still uncertain. OA is divided into primary (POA) and secondary (SOA) fractions. POA is directly emitted into the atmosphere, largely from combustion process, and SOA is formed in the atmosphere by oxidation of volatile gas-phase organic compounds emitted into the atmosphere either by biogenic or anthropogenic activity. The gas-phase compounds can be either abundant volatile organic compounds (VOC, for example, even methane, in certain conditions, is able to form SOA2) or less abundant but more potent intermediate volatile organic compounds (IVOC).3 It has been estimated that SOA is the biggest fraction of the total organic aerosol.3,4 However, the contribution of VOC to the formation of atmospheric SOA remains unclear, especially in the growth of newly formed particles from molecular clusters up to cloud condensation nuclei (CCN) size, where abundant organic vapors with very low volatility are required.
Recently, it has been discovered that some VOCs, such as monoterpenes (C10H16), are able to rapidly generate large amounts of low-volatility vapors5 through a gas-phase process called autoxidation.6 Through this oxidation mechanism, taking place under atmospheric conditions, it is possible to promptly form compounds such as C10H14O9 or C20H30O16. These vapors, which we will refer to as highly oxygenated organic molecules or HOM, are the topic of this Review. The first HOM observations were reported in laboratory studies of the ozonolysis of monoterpenes, and the laboratory mass spectra closely corresponded to observations from the boreal forest.5,7,8 Despite being a very recent discovery, the interest of HOM within the atmospheric science community has grown rapidly, spurring a wealth of follow-up studies. These range from detailed examination, both theoretical and experimental, of HOM formation mechanisms, to studies of the contribution of HOM to new-particle formation, and even estimates of the global importance of HOM for CCN concentrations. Great progress has been made in a short time, but at the same time the rapid developments have produced literature that is quite fragmented and potentially difficult to follow, as even the terminology has varied since the original studies. Furthermore, the chemistry itself is fascinating and of interest to a wide chemistry audience. For these reasons, we undertake this review in order to
-
1.
More carefully define the term “HOM”
-
2.
Summarize the current understanding on HOM formation mechanisms.
-
3.
Discuss findings on the atmospheric importance of HOM
-
4.
Highlight the existing gaps in our understanding and suggest where future research on HOM should be directed.
Section 2 of this Review clarifies the terminology of HOM. Section 3 discusses HOM detection and the relevant observation methods. Section 4 describes our current understanding of the formation pathways of HOM, and section 5 describes their properties and final fates in the atmosphere. Section 6 summarizes the identified atmospheric impacts of these compounds and where they have been observed. Finally, we conclude the Review with a perspective highlighting the need for future HOM studies with suggestions on where efforts should be directed to answer the most pressing open questions.
2. HOM Background
Both the term “HOM” as well as its definition have evolved over time, and it is extremely challenging for anyone new to the subject to follow the developments based on recent literature. This review attempts to summarize and clarify the current understanding. Detailed accounts on different aspects of HOM research are described in later sections, while the purpose of this section is to briefly clarify the key concepts and terminology. When discussing the term “HOM” in this section, it will be written in quotation marks. When referring to the HOM compound class, no quotation marks will be used, as usual.
2.1. Defining the Key Concepts
Without context, the term “highly oxygenated organic molecules” covers a very broad range of compounds. Therefore, “HOM” requires more distinct criteria to specify the group of compounds it has been used to describe. These criteria are discussed in section 2.1.2 below, but first the process of autoxidation is briefly described (section 2.1.1), as it is often an essential part of HOM formation.
2.1.1. Autoxidation Involving Peroxy Radicals
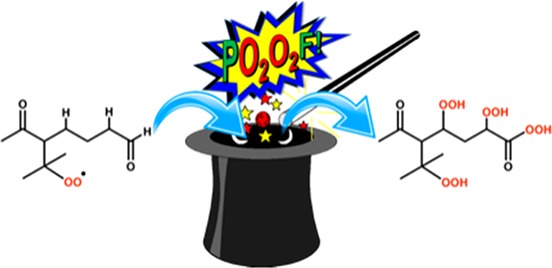
In the context of this review, we define autoxidation as the overall process in which a peroxy radical (RO2) first undergoes an intramolecular hydrogen-atom shift forming a hydroperoxide functionality and an alkyl radical, to which molecular oxygen then rapidly attaches to form a new, more oxidized, peroxy radical. The importance of this reaction sequence (see Figure 1 for an example) has been identified in a variety of different systems (see, e.g., Crounse et al., 2013, and references therein) but was long overlooked in the context of atmospheric chemistry.9−11 Only quite recent investigations have identified the relevance of autoxidation in atmospheric oxidation chemistry.5,6,9,12−17
Figure 1.

Autoxidation in OH-initiated oxidation of ketones. Following reaction with OH, the ketone C1 forms a peroxy radical (C2) which undergoes an H-shift isomerization to form a hydroperoxide with a radical center on the carbon atom from which the hydrogen atom was abstracted (C3). Subsequent rapid addition of O2 forms a new RO2 radical (C4), which undergoes another H-shift, in this case terminating the autoxidation process by loss of an OH radical, resulting in a dicarbonyl hydroperoxide. In this scheme, the steps between C2 and C4 define the autoxidation. Adapted with permission from ref (6). Copyright 2013 American Chemical Society.
Two other types of unimolecular mechanisms can also convert RO2 radicals into new, more oxidized RO2 radicals. First, in unsaturated RO2, such as those commonly formed in the oxidation of aromatics,18 ring closure reactions can take place, forming a new RO2 after O2 addition. Second, if bimolecular reactions transform the RO2 into an alkoxy radical (RO), it can undergo similar H-shifts as in Figure 1,19 ultimately forming new RO2. The first of these two reactions does not involve an H-shift, while in the second reaction the H-shift does not occur by a peroxy radical, and therefore neither is here defined as autoxidation involving peroxy radicals despite their similarity.
2.1.2. Highly Oxygenated Organic Molecules, HOM
The term “HOM” was initially defined through instrumental parameters for lack of a better description for the newly detected group of compounds. Recently our understanding of the composition and properties of HOM, as well as the selectivity of the instruments used to detect them, has increased considerably. The details of these insights are discussed in later sections, but in order to frame the rest of the review, we provide a set of criteria for classifying molecules as HOM. While we recommend that these criteria be used as guidelines in future publications for what constitutes HOM, each publication should explicitly define their usage of the term. We acknowledge that these criteria are not without potential problems, and these are discussed in conjunction with the specific criteria. Our aim is a definition which, as far as it is possible, agrees with the earlier usage of the term. At the same time, we do not in any way attempt to discourage the use of “highly oxygenated” for describing other types of molecules (e.g., sulfuric acid). However, it is our hope that the specific term “HOM” would be restricted to a certain group of compounds, as defined below.
Guidelines for classification of compounds as highly oxygenated organic molecules, HOM:
-
1.
HOM Are Formed via Autoxidation Involving Peroxy Radicals. Without this criterion, “HOM” would merely refer to any organic compound with high oxygen content. This would make it impossible to distinguish HOM from other compounds with very different formation processes and time scales and chemical characteristics. Compounds with high oxygen content have been observed in SOA long before HOM were identified, yet many of them are likely formed through other processes than autoxidation. For example, MBTCA (3-methyl-1,2,3-butanetricarboxylic acid, C8H12O6) and mannitol (C6H14O6) have been identified as markers for terpene SOA20 and airborne fungal spores,21 respectively, but neither is formed via autoxidation and therefore we recommend that they not be classified as HOM. Chemically, the key distinction between HOM and other compounds with similar elemental compositions which may be found in the atmosphere is that HOM contain at least one, and often multiple, hydroperoxide, peroxide, or peroxy acid groups. We recognize that most instruments are unable to determine the formation mechanisms or the exact functionalities of the detected molecules, and ultimately the classification as HOM will require supporting evidence. In laboratory studies, the rapid formation of molecules with six or more O atoms, under conditions where multiple OH reactions are unlikely, can be seen as one strong indication. If similar spectra are identified in the atmosphere, these can be argued to be HOM as well. The experimental and computational evidence that led to the conclusion that the observed “HOM” are formed by autoxidation are summarized in section 4.1.
-
2.
HOM Are Formed in the Gas Phase under Atmospherically Relevant Conditions. The importance of autoxidation (generally defined as oxidation by O2, implicitly implying that other oxidants are not needed, or are needed solely as initiators) has been known for a long time in many systems, such as liquid-phase22 and combustion23 chemistry. However, the term HOM has only been used to describe compounds formed under atmospheric conditions, and it is useful to keep the term to describe compounds of atmospheric relevance. The primary purpose of this criterion is to exclude chemistry at elevated temperatures, e.g., those found in combustion engines, far above anything in the atmosphere, and such processes are not reviewed here. In this review, we consider only gas-phase HOM formation, and the subsequent fate of the formed HOM in both gas and condensed phases. It is currently unknown whether or not autoxidation involving peroxy radical H-shifts and O2 addition is a competitive process also in the atmospheric condensed-phase; products of such processes are beyond the scope of our study.
-
3.
HOM Typically Contain Six or More Oxygen Atoms. As “highly oxygenated” is a matter of definition, and will generally depend on context, we propose this final criterion as a more explicit way to identify whether a compound could be classified as a HOM. Considering compounds like C20H30O10 (O:C = 0.5) and C2H4O2 (O:C = 1), only the former should be considered HOM, and therefore we include the absolute oxygen content as a criterion instead of O:C. We also acknowledge that a limit of six oxygen atoms is somewhat artificial, and in some cases also compounds with five oxygen atoms may be considered HOM. Similarly, pathways exist for molecules to reach six or more O atoms also without autoxidation, especially in cases where nitrate groups or dimers are formed.237,238 However, at six O atoms, a molecule already starts to be quite likely to have undergone RO2 isomerization. Another reason to include a limit on the oxygen content is that certain molecules, like the dicarbonyl hydroperoxide formed in Figure 1, can have undergone autoxidation and fulfils all other criteria, yet still only contain 3–4 oxygen atoms, and should thus not be considered HOM.
While the above constraints are closely aligned with what has been meant by the term HOM in most earlier studies, we recognize that several complications and limitations apply. Therefore, our recommendation is that future studies will utilize the guidelines provided above, while explicitly defining why compounds were classified as HOM, and specifically addressing if certain criteria are not met (or it cannot be verified whether they are met or not).
2.2. HOM in Relation to Other Classification Schemes
HOM have been equated with other, more commonly used, classification schemes in earlier work (see section 2.3). With improved understanding of HOM properties and formation mechanisms, the connection between HOM and other compound groups is now more complete. Here, we start by defining volatility classes (section 2.2.1), which provide a framework for section 2.2.2 where we attempt to schematically relate HOM to other commonly utilized classification schemes.
2.2.1. Volatility Classes
A very common naming scheme for various organic compounds is based on their volatility, as presented by Donahue et al. (2012).24 This scheme groups compounds into five classes based on their effective saturation concentration, C*, expressed in μg m–3. The classes are
-
1.
Extremely Low Volatility Organic Compounds (ELVOC), with C* < 3 × 10–5 μg m–3. These are molecules that will condense onto essentially any pre-existing cluster and may as well participate directly in new-particle formation involving only other ELVOC.
-
2.
Low Volatility Organic Compounds (LVOC), 3 × 10–5 < C* < 0.3 μg m–3. These are molecules that will condense onto any sufficiently large particle but may not condense onto the smallest particles due to high curvature (the Kelvin effect).
-
3.
Semivolatile Organic Compounds (SVOC), 0.3 < C* < 300 μg m–3. These are molecules that will exist in significant fractions in both the condensed and gas phases at equilibrium in the atmosphere.
-
4.
Intermediate Volatility Organic Compounds (IVOC), 300 < C* < 3 × 106 μg m–3. These are molecules with relatively low vapor pressure that nonetheless are almost exclusively in the gas phase in the atmosphere.
-
5.
Volatile Organic Compounds (VOC), C* > 3 × 106 μg m–3. These are molecules that are recognizably volatile under all circumstances and which, for the most part, dominate gas-phase oxidation chemistry in the troposphere.
The exact boundary between these classes depends on context, and so we have provided their qualitative definitions in addition to their quantitative definitions. For example, the limiting saturation vapor pressure between ELVOCs and LVOCs was revised downward by a factor of 10 in Tröstl et a., (2016) compared to Donahue et al. (2012) because the compounds on that margin do not develop a sufficient supersaturation to condense on the smallest particles under many conditions found in the atmosphere.25 The combined class (E)LVOCs (all pronounced “ell” VOCs) constitute molecules that condense to all but the smallest particles under almost all circumstances in the atmosphere. HOM primarily fall into this class, as seen in the following section.
2.2.2. Relations between the Concepts
With the definitions in the previous sections, we can map out the relation between HOM and other parameters in various ways. We provide different types of diagrams for visualizing the interrelations in this section but note that all of them should be read as indicative only. One reason is that our knowledge of HOM is not yet fully developed, and thus uncertainties in the relative and absolute positions of the different compound classes may yet shift slightly. Another reason is that we project compound classes onto parameter spaces where the classes are unlikely to have exact, well-defined boundaries. For example, higher oxygen content indicates a lower volatility, yet two molecules with identical elemental composition may have vastly different volatilities depending on their precise structure.26
By our definition, HOM are a subset of products formed by autoxidation (peroxy radical isomerization products, PRIP), as shown in the simple Euler diagram in Figure 2. The figure also includes the relevant volatility classes, showing that while PRIP may have various volatilities, HOM are primarily (E)LVOC, although some may be volatile enough to be classified as SVOC.
Figure 2.
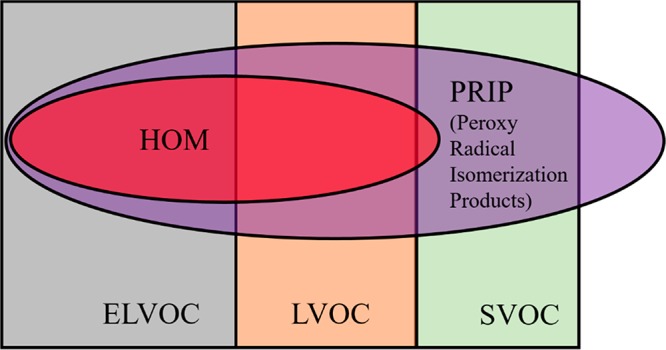
Euler diagram (not to scale) showing relation of HOM to products formed by autoxidation involving peroxy radicals (PRIP) and different volatility classes. The position of HOM in the diagram is estimated based on typical elemental formulas of HOM that have been published, and their volatility estimates by Kurtén et al., 2016.27
There are many commonly used parameter spaces in atmospheric chemistry, and we attempt to depict HOM and the volatility classes in some of these. First, if we position all molecules onto a grid defined by their carbon and oxygen numbers, we can easily plot where potential HOM can be found based on criterion 4 above (Figure 3). Whether a molecule in the red-shaded area is a HOM or not is determined by how it was formed. Figure 3 also includes the main volatility classes, although the regions of the C–O space that they cover is less well-defined, and thus these regions correspond to our current best estimates.
Figure 3.
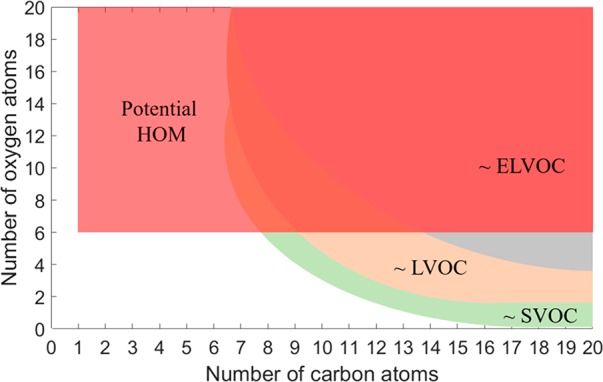
Relation between HOM and volatility classes in carbon–oxygen space. On the basis of the criterion of minimum of six oxygen atoms (see section 2.1.2), potential HOM are easily depicted in this space. However, the other criteria also need to be met in order to justify the assignment as HOM. The positions of the volatility classes can only be roughly estimated, as the precise functionalities will determine the volatility of the molecules. We also note that most of the top left part of the figure depicts purely hypothetical molecular compositions, but the red shaded area is not restricted for simplicity. As HOM often contain other heteroatoms than C and O, such restrictions would depend on what molecules can be considered plausible.
Another commonly used parameter space is that utilized in the 2D-VBS,24 plotting molecules as a function of average carbon oxidation state and volatility. This is the space where the volatility classes were defined, which makes them easy to position onto the graph (Figure 4). With estimates about average functionalities, we could also estimate the region where HOM would be observed in this parameter space. Again, the molecules in this red-shaded region will only count as HOM if they are PRIP.
Figure 4.
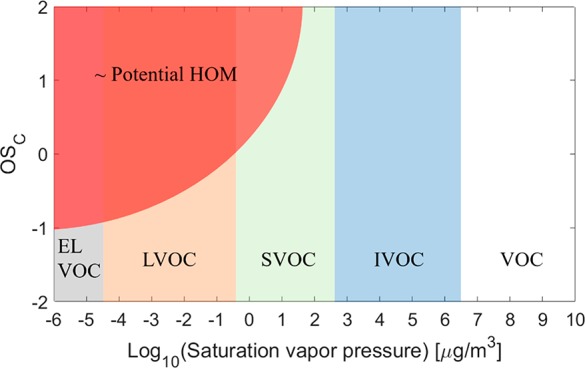
Relation between HOM and volatility classes in C*-OSC space. In this space, the volatility classes are well-defined, but the position of potential HOM will again depend on the exact functionality of the molecules. This C*-OSC space is discussed in more detail in section 5.1.
2.3. Historical Naming Conventions
The scientific progress on HOM has been very rapid in recent years, and unfortunately the naming conventions changed during the process. This means that earlier work will in many cases not have followed the guidelines outlined in section 2.1.2. In this section, we will shortly describe the previously used terminology in order to clarify the evolution of “HOM” and to provide a reference for how to interpret earlier work.
In the first publication where “HOM” was used,7 the molecules were described as “extremely oxidized pinene reaction products” and termed “highly oxidized multifunctional organic compounds”, also abbreviated “HOM”. A large number of later studies have used the term “HOM” as defined in this initial work. Once a follow-up study5 was able to quantify HOM and relate them to SOA formation, a more well-established name was utilized, and they were collectively referred to as ELVOC. This assumption of extremely low vapor pressure was motivated in part by utilizing existing structure–activity relationships (SAR) (e.g., (Donahue et al., 2011) for the observed elemental compositions).28 Ehn et al. (2014) also showed that the behavior of the observed molecules when in contact with chamber walls or aerosol particles was well-described when assuming nonvolatile molecules.5 However, HOM, according to the current understanding, contain a high number of (hydro)peroxide moieties, which is likely the reason for the SAR to under predict the vapor pressures of the compounds. Utilizing a quantum chemistry-based model to calculate vapor pressures for assumed HOM structures, a study27 suggested that a large fraction of α-pinene HOM were more likely to fall into the volatility class defined as LVOC and some even potentially to SVOC. In addition, laboratory measurements at the CLOUD experiment in CERN showed that a significant fraction of HOM involved in new-particle formation and growth only participated in the growth-phase once particles were larger than ∼2 nm.25 On the basis of these findings, the initial term “HOM” was found to be more suitable, although with the meaning “highly oxygenated molecules” to avoid any reference to the structure of the molecules. In this review, we incorporate the term “organic” explicitly into “highly-oxygenated organic molecules”, in order to make the definition more descriptive.
Regardless of which exact terminology was used to describe the highly oxygenated organic molecules in earlier studies, they have in practice been defined in most studies by the observational method by which they were discovered, as discussed below. To avoid needless assumptions on functionalities or vapor pressures, which the mass spectrometric detection methods rarely have been able to provide, we recommend that the terms “highly oxidized multifunctional” and ELVOC no longer be used to describe HOM. The term ELVOC should be used in accordance with its initial definition: molecules with C* < 3 × 10–5 μg m–3.
3. HOM Detection
HOM research would not have progressed even remotely to what it is today without the instrumental developments that led to their direct detection. The history of HOM research is thus closely intertwined with the history of instrumental advances, especially in the field of online chemical ionization mass spectrometry. Therefore, this section not only presents the techniques available for HOM detection but also includes (approximately in chronological order) the threshold moments that have led to our current understanding of HOM. In the following, we discuss the existing techniques for HOM detection in the gas (section 3.1) and particle (section 3.2) phases, and we outline the uncertainties and analytical challenges remaining for HOM detection, characterization, and quantification (section 3.3).
3.1. Gas Phase
3.1.1. Atmospheric Pressure Interface Time-Of-Flight Mass Spectrometer
The first observations of HOM were realized in the boreal forest in the form of N-containing ambient ions (e.g., C10H14NO10–, Figure 5A),29 but the elemental identification was only tentative at the time. The utilized instrument was the newly developed atmospheric pressure interface time-of-flight mass spectrometer (APi-TOF, Figure 6),30 manufactured by Tofwerk AG, Thun, Switzerland, and Aerodyne Research, Billerica, MA, USA. Because of the lack of an ion source, this instrument measures only naturally charged positive and negative ions in a mass-to-charge range up to several thousand Th, with an achievable detection limit <1 ion cm–3. The ions are guided in the APi from atmospheric pressure through three differentially pumped chambers containing two short segmented quadrupoles and an ion lens assembly, respectively, to the TOF. The ions are orthogonally extracted into the TOF onto a one- or three-reflection flight path, and their mass is determined based on their flight times. The one-reflection flight path (“V mode”) in the most commonly used TOF analyzer (the HTOF) has a resolution R (R = M/ΔM, M = mass, ΔM = full peak width at half-maximum of signal) of 3000–5000. Although used less frequently, the HTOF also has the potential for a three-reflection flight path (“W mode”) which can achieve nearly twice the resolution, at the expense of ion transmission. The specified mass accuracy is better than 4 ppm (0.0004%). Some APi-TOF models are equipped with different mass analyzers, either a compact TOF (CTOF) with R ≈ 1000 or a long TOF (LTOF) with achievable R > 10000.
Figure 5.

Mass spectra of naturally charged ions containing HOM measured by APi-TOF in the nighttime boreal forest in Hyytiälä (A), and in the Juelich Plant Atmosphere Chamber (JPAC) during experiments of α-pinene oxidation at low-OH conditions (B). HOM are predominantly detected as clusters with ambient NO3–. (C) HOM spectrum measured by nitrate CI-APi-TOF from α-pinene ozonolysis in the JPAC chamber, where the formation of HOM·NO3– clusters is augmented during the chemical ionization process. Adapted from with permission from ref (5). Copyright 2014 Springer Nature Limited. Adapted with permission from ref (7). Copyright 2012 Copernicus Publications. Adapted with permission from ref (29). Copyright 2010 Copernicus Publications.
Figure 6.

Schematic of APi-TOF and CI-APi-TOF or ToF-CIMS with different ion sources (a,b). Note that for the nitrate CI-APi-TOF, voltage settings may change depending on application, setup, and users; not all flow tube nitrate sources use the N2 flush as shown in inset in (a). Adapted with permission from ref (36). Copyright 2011 Copernicus Publications. Adapted with permission from ref (236). Copyright 2014 National Academy of Sciences.
The successful observation of HOM by the APi-TOF was a combination of several factors: high sensitivity to measure the low ion concentrations, high resolution to identify the elemental composition, and field deployability. Measurements of naturally charged atmospheric ions had already been conducted in the 1980s,31,32 but the low resolution of the employed quadrupole mass analyzers and their sharply decreasing transmission above a few hundred Th precluded identification of any sampled HOM.
Because the APi-TOF detects naturally charged ions, formed primarily from cosmic rays or radon decay in the atmosphere, the sensitivity of the instrument depends only on ion transmission (in addition to potential losses in sampling lines). For a well-tuned instrument, the transmission peaks around 1%.30,33,34
The conclusive elemental identification of the HOM observed in the boreal forest were made in a targeted chamber study looking at naturally charged ions during monoterpene oxidation by ozone (Figure 5B).7 Utilizing W mode (resolution ∼8000) and by varying conditions in the chamber, these experiments provided both the unambiguous elemental composition of the HOM ions as well as determination that the HOM were organic molecules clustered with NO3–. In other words, the C10H14NO10– was in fact C10H14O7·NO3–. This study was also the first to use the term “HOM”, although the abbreviation at that time was spelled out slightly differently (see section 2.3).
3.1.2. Chemical Ionization Mass Spectrometry
The main drawback of measuring naturally charged ions was the lack of quantification of the concentration of neutral HOM molecules. The realization that HOM were predominantly observed as clusters with NO3– meant that a chemical ionization (CI) source connected to the APi-TOF using nitrate as reagent ion35 should efficiently detect HOM. Since then, chemical ionization mass spectrometry has become the dominant tool for HOM measurement, with the overwhelming majority of HOM studies cited in this review being based on data from the nitrate CI-APi-TOF. In the following, we describe the nitrate CI-APi-TOF as well as other variants of CI mass spectrometers used for HOM detection with different reagent ions or different configurations of the ion source.
Unfortunately, the naming conventions of the chemical ionization mass spectrometers are not always consistent, even though most of them contain the same APi-TOF mass spectrometer (Figure 6). “CI-APi-TOF” commonly refers to an APi-TOF connected to an atmospheric pressure, Eisele-type chemical ionization inlet,35 as described in the next section. The term “ToF-CIMS”, on the other hand, has been used for instruments utilizing a low-pressure ion–molecule reaction (IMR) chamber coupled to an APi-TOF.36 However, as this distinction is not consistent across publications and research groups, we additionally provide the less common name in the section headings. Some publications may also simply use the term “CIMS” to describe either of these two instrument types. This is also the most common term in cases where the mass spectrometer is not an APi-TOF.
3.1.2.1. Nitrate CI-APi-TOF/Nitrate ToF-CIMS
The selectivity of nitrate ions toward highly functionalized organic compounds (featuring at least two suitably located hydrogen-bond donor functional groups such as hydroxy or hydroperoxy groups37) makes them ideal reagent ions for the observation of HOM and their precursor RO2. The development of the nitrate CI-APi-TOF (Figure 6) has thus greatly improved both HOM detection and quantification and facilitated the investigation of HOM formation pathways.5,38 The CI source added to the APi-TOF consists of a flow tube kept at ambient pressure based on the design of Eisele and Tanner et al.39 Nitrate ions are created from nitric acid (HNO3) in a sheath flow of clean air being exposed to either α radiation from a 10 MBq Am source,35 a corona discharge,40 or X-rays.41 In the ion reaction tube, sheath and sample air flow concentrically, and the nitrate ions are directed from sheath into sample flow via an electric field. HOM are primarily ionized via cluster formation with NO3– (as the earlier APi-TOF measurements revealed). From there, the ionized HOM are guided to the APi-TOF via its critical orifice.
HOM concentrations are established using the ratio of HOM-containing ions to reagent ions and a calibration factor C:42
For normal operating conditions, where ∑reagent ions ≫ ∑HOM-containing ions, the formula can be simplified to35
Because of the lack of HOM standards, C is most often determined with sulfuric acid,43,44 or in some cases using other molecules,5 with the assumption that HOM and the compounds used for calibration cluster with the reagent ion at similar, collision-limited, rates. Values for C commonly range between 109 and 2 × 1010 molecules cm–3. An additional correction factor can be applied for mass dependent ion transmission, if determined separately,33 or inlet losses.
3.1.2.2. CI-APi-TOF with Other Reagent Ions
There are indications that nitrate ionization may not be sensitive to all key HOM compounds (e.g., RO2 with a single H-bond donating group45). Therefore, other reagent ions such as acetate (C2H3O2–), lactate (C3H5O3–), and pyruvate (C3H3O3–) have also been used with the CI-APi-TOF.46,47 These reagent ions are created analogously to nitrate, starting from their conjugate acid. Like nitrate, these reagent ions are able to form stable clusters with HOM, or charged HOM are formed via proton transfer from the target molecule to the reagent ion. Acetate ionization seems to exhibit higher sensitivity toward HOM containing only one hydroperoxide moiety compared to nitrate ionization.45 At the same time, the generally increased sensitivity of acetate, lactate, and pyruvate to HOM and other organic compounds leads to more complex spectra comprising signals of other species as well, and these have thus far only been deployed under ultraclean laboratory conditions. Calibration factors C of the CI-APi-TOF with these reagent ions were calculated, assuming the ion–molecule reaction at the collision frequency and a negligible loss of formed clusters within the detection unit. Resulting HOM concentrations based on calculated C represent lower-limit values that are in line with the results applying calibration with sulfuric acid.45 Very recently, HOM detection in the positive mode with high sensitivity has been reported.48 Protonated n-propyl amine (C3H7NH3+) served as the reagent ion that forms stable clusters with HOM and less oxidized products as well. Calculated calibration factors have been used, identical to the approach with acetate, lactate, and pyruvate in the negative mode.
3.1.2.3. Acetate and Iodide ToF-CIMS
The ToF-CIMS consists of the same mass spectrometer and atmospheric pressure interface as the CI-APi-TOF, but it uses a different CI source. While the nitrate CI-APi-TOF is highly selective toward HOM, the ToF-CIMS has mainly been used for the detection of a wide range of oxygenated volatile organic compounds (OVOC) and inorganic radicals (Lee et al. 2014, and references therein).49 The CI source of the CIMS consists of a reduced-pressure ion–molecule reaction (IMR) region36 kept at ∼100 mbar, with ports for up to two radioactive ion sources (Po-210, 10 mCi, NRD) oriented 90° apart and orthogonal to the sample flow (Figure 6). Empirically determined sensitivities of the CIMS with acetate and iodide are ∼20 counts s–1 ppt–1 for molecules with masses >200 Da,50,51 which in the case of iodide represents its sensitivity toward multifunctional organic molecules at the collision limit.52
With both acetate (proton abstraction)51 and iodide (cluster formation)50 as reagent ions, the detection of organic molecules with elemental formulas corresponding to HOM has been demonstrated in the field and laboratory.49,51,53 However, as structural information is not available, it is not possible to say whether the highly oxygenated organic compounds detected by the acetate and iodide CIMS are the same as those detected by the nitrate CIMS.
In addition, discrepancies between time series of compounds with the same molecular formula measured by a CI-APi-TOF and a ToF-CIMS deployed simultaneously exist,54 highlighting the need for further systematic comparisons of the different mass spectrometric techniques and reagent ions for HOM measurements in the gas phase.
3.1.2.4. Other Techniques
A proton transfer reaction-time-of-flight mass spectrometer using a new gas inlet and reaction chamber design (PTR3) was recently demonstrated to detect HOM, in good agreement with simultaneous measurements by CI-APi-TOFs.48,55,56 With the new inlet (using center-sampling and thus reducing wall losses significantly) and new reaction chamber (with a 30-fold longer reaction time and a 40-fold increase in pressure compared to standard PTRTOF), HOM present in the parts per quadrillion per volume (ppqv) range in the atmosphere can be measured.
Zhao et al. developed the so-called Cluster-CIMS, featuring a similar NO3– CI source as Figure 6a, but adapted to a quadrupole mass spectrometer, for gas-phase HOM detection.8,57
3.2. Particle Phase
The uncertain fate of HOM in the particle phase (as described in section 5) is mainly due to the very limited detection methods of particle-bound HOM. Methods are often indirect, or they present measurements of compounds with molecular formulas corresponding to HOM but without information on formation mechanisms. In some cases, these are also proposed to form through different chemical processes than the products discussed in this review. Of the limited number of studies reporting particle-phase HOM observations, most are based on offline particle samples and subsequent analysis of the extracted compounds by mass spectrometry. In the following, we will discuss both offline and online methods that have been used in studies reporting HOM in the condensed phase.
3.2.1. Offline Techniques
HOM are expected to contain hydroperoxide functionalities, as discussed in section 4, and several studies have shown that such compounds decompose in SOA on time scales of hours or less.58 In general, hydroperoxides are thermally unstable59 and can decompose in water under dark conditions.60 This reactivity, in addition to sensitivity toward different analytical steps (sampling, extraction, etc.) that are typically used to achieve molecular characterization via offline analyses, may severely limit the use of offline techniques to provide new insights on HOM molecular structures. Despite these limitations, the majority of reported HOM detections in the particle phase are based on offline techniques. Further, because a major role of HOM is in particle nucleation and growth below 3–10 nm diameter,25,61 even if HOM decompose into a more volatile molecule after an hour or so, the HOM may have effectively “done their job” in creating and nursing particles to a size where they can continue to grow.
Mutzel et al. identified around a dozen HOM,62 and a similar number of highly oxidized organosulfates (HOOS) believed to result from HOM acting as precursors in the particle phase from filter samples from both field and laboratory. Filters were derivatized with 2,4-dinitrophenylhydrazine (DNPH), and the samples analyzed by high-performance liquid chromatography electrospray ionization coupled to time-of-flight mass spectrometry (HPLC/ESI-ToFMS) and UPLC/ESI-Q-ToFMS. Derivatization with DNHP also yields structural information, and this study thus showed the presence of carbonyl groups in the detected compounds.
Molecules containing multiple peroxide functionalities (C8–10H12–18O4–9 monomers and C16–20H24–36O8–14 dimers) have been identified in α-pinene SOA.63 Those researchers analyzed filter samples with an electrospray ionization (ESI) drift-tube ion mobility spectrometer (DTIMS) interfaced to an APi-TOF. In this instrument, HOM molecules are cationized by the attachment of Na+ during ESI operated in the positive ion mode. Condensed-phase HOM were compared to molecules detected in the gas phase by iodide CIMS for evidence of their gas-to-particle conversion.
Krapf et al. used a long path absorption photometer (LOPAP) to measure condensed-phase OOH functional groups in α-pinene SOA formed in the laboratory collected on filters.58 They derived HOM yields from the resulting peroxide yields by assuming that HOM are composed of an average of about two OOH functional groups per molecule.
Tu et al. identified molecular formulas corresponding to HOM of nine compounds in biogenic SOA using a flow-tube reactor.64 They collected the particles on a Teflon coated, glass fiber filter, which then was extracted with an acetonitrile/water solution to prevent esterification of acid groups. The samples were analyzed by a Q extractive hybrid quadrupole-orbitrap mass spectrometer (Thermo Scientific, Waltham, MA, USA) coupled with a heated-electrospray ionization (HESI) probe.
Indirect information on the fate of HOM in the particle phase was derived from LC-MS and an aerosol flowing atmospheric-pressure Afterglow mass spectrometer (AeroFAPA-MS) measurements of particulate HOOS in central Europe.65 These measurements support the hypothesis of HOOS being formed by reactions of gas-phase HOM with particulate sulfate. Unstable HOM were speculated to be intermediates in the formation of high-molecular weight esters measured by liquid chromatography/electrospray ionization mass spectrometry (LC/ESI-MS).66
3.2.2. Online Techniques
There is only a small number of studies using online methods to detect HOM in the particle phase. Indirect indication of particulate HOM was presented by Zhang et al.,67 who used a particle-into-liquid sampler (PILS) integrated with UPLC/ESI-Q-ToFMS to demonstrate that hydroperoxy derivatives of pinonic acid are components present in the particle phase with molecular formulas corresponding to HOM.
A filter inlet for gases and aerosols (FIGAERO)51 was recently developed for the ToF-CIMS. The FIGAERO represents a quasi-online method of particle analysis, as it collects particles on a Teflon filter and subsequently desorbs them by a heated nitrogen flow. The desorbed molecules enter the IMR, where they are ionized and detected analogously to ToF-CIMS gas-phase sampling. The high sensitivity of the ToF-CIMS allows for relatively short sampling times even in remote areas (on the order of minutes to an hour). At the same time, thermal decomposition demonstrably influences the resulting mass spectra from FIGAERO measurements51,68,69 (and other desorption-based analysis methods), which may present certain limitations to HOM detection. However, laboratory and field measurements using the FIGAERO–CIMS with acetate and iodide as reagent ions51,53,70 demonstrate the potential of this technique for detection of organic molecules potentially attributable to HOM within the particle phase.
3.3. Uncertainties and Analytical Challenges of HOM Detection
In the previous paragraphs, we have outlined the various techniques available for HOM detection in both gas and particle phase. The typically low volatilities of HOM lead to low concentrations and a high propensity for losses during sampling, while their high oxygen content additionally makes them thermally labile. Although these challenges have been overcome by techniques such as those described above, further instrumental and analytical improvements could provide much new insight into HOM formation chemistry. Below, we summarize some of the current uncertainties and challenges related to HOM measurements:
-
1.
HOM detection: (a) Gas phase. Currently, subsecond processes are not measurable using available methods for HOM detection. This is in contrast to studies of low-temperature combustion processes where H-shifts and O2 additions have been successfully studied on millisecond time scales using pulsed laser initiation and direct photoionization by synchrotron-generated vacuum ultraviolet radiation. Whether such methods can be used for studies relevant for atmospheric processes remains to be seen. To our knowledge, such efforts have so far not been undertaken, but they may eventually provide an essential way forward by facilitating both faster measurements and more selective observations of radical intermediates. In general, fast reactions have generally utilized conditions far from atmospheric, e.g., low pressures and/or extremely high precursor concentrations, but these will most likely lead to very different reaction pathways. For example, at decreased pressures, the oxygen content will also be decreased and thus the (typically very fast) addition of O2 will become slower. (b) Particle phase. For both offline and online techniques, additional analytical steps are required for HOM detection in the particle phase compared to gas phase, e.g., filter extraction, sample derivatization, or thermal evaporation, processes that may lead to HOM decomposition.
-
2.
HOM quantification: Large uncertainties in HOM concentrations are due to a lack of authentic and/or appropriate surrogate standards that would be used to (1) characterize the losses throughout the analytical protocol, (2) obtain accurate quantification of the compounds of interest, and (3) to unambiguously elucidate molecular structures. However, the sensitivity toward HOM can be estimated based on other compounds, and kinetic limitations such as maximum collision rates as well as cluster strength measurements can be used to infer upper and lower bounds for the observed concentrations.52 Overall, the sensitivity of a CIMS to a specific molecule depends on the charging probability (governed mainly by thermodynamics) of the neutral molecule and the survival probability (governed mainly by kinetics) of the formed ion until detection. The type and amount of the reagent ion, as well as the strengths of the electric fields in the mass spectrometer, are examples of parameters that will influence this sensitivity,37,72 and good knowledge and control of these will facilitate more accurate quantification.
-
3.
HOM structure identification: The vast majority of studies on HOM use mass spectrometric techniques, where molecular structure information can only be inferred from molecular formulas, and in limited cases from the ion–molecule chemistry during the ionization process.71,72 This significantly limits our understanding of HOM physicochemical properties.
Despite these remaining uncertainties, we wish to emphasize that the identification of HOM in general is not in doubt. Following the initial observations of a new group of compounds with much higher oxygen content than predictions would have suggested, much effort was put on determining whether the observation could merely have been an artifact, e.g., through oxidation taking place inside the CI-APi-TOF. This option can be ruled out due to multiple supporting findings. First, similar HOM spectra have been acquired both using the CI-APi-TOF with active charging by NO3– and by just sampling naturally charged atmospheric ions into an APi-TOF.5,7 This shows that the HOM formation does not only happen in the CI inlet. Second, the residence time in the CI inlet is ∼200 ms, while the ambient ion lifetimes are on the order of a minute, making it unlikely that the clustering with NO3– itself would cause identical oxidation leading to the same HOM. Similarly, HOM formation have recently been observed also using very different (positive) reagent ions,48,56 and a very different (low-pressure) ionization inlet.56 Third, Berndt et al. (2016) directly probed the effect of decreased oxygen in the CI inlet and found no change in HOM concentrations, whereas the same change in the flow reactor used for VOC oxidation did cause a large decrease in HOM formation.46 Taken together, these observations clearly show that the HOM formation is not an instrumental artifact. Numerous other studies on HOM have shown remarkable consistency concerning the perturbations of HOM spectra as a function of the conditions under which they were formed, including different oxidants and the role of NOX.5,46,133,134
4. HOM Formation Mechanisms
In this section, we first (section 4.1) review the evidence that led to the hypothesis that HOM formation is driven by autoxidation involving sequential RO2 H-shifts and O2 additions. We then summarize the key features of this pseudo-unimolecular reaction sequence, focusing on reactions and reaction classes that are especially important for HOM formation and often possible (or at least competitive in atmospheric conditions) only for complex and oxidized RO2 or related species. General bimolecular RO2 reaction mechanisms will not be reviewed in detail, as most of these are well established in the literature on simpler RO2 (such as CH3OO, CH3CH2OO, etc.) and have recently been reviewed by Orlando and Tyndall (2012).73 Furthermore, experiments indicate that the overall reactivity of complex RO2 toward trace gases such as NO, NO2, and SO2 are mostly similar to those of simpler RO2.45 Bimolecular RO2 reactions will therefore be discussed in the context of their effect on HOM formation (section 4.2), highlighting possible differences between reactions of simple and complex RO2. Bimolecular reactions often terminate the autoxidation chain by generation of closed-shell association products, but they are also observed to support the oxidation progression by forming reactive radical intermediates (mainly alkoxy radicals) capable of continuing the sequential isomerization reactions.74 Finally, we will consider the external conditions (section 4.3), such as reactant concentrations and temperatures, that facilitate HOM formation in the laboratory and in the atmosphere and summarize the available information on HOM yields.
Much of the detailed molecular-level mechanistic understanding of HOM formation is based on computational chemistry, specifically on quantum chemical calculations combined with kinetic modeling. The methods of applied atmospheric computational chemistry have recently been reviewed75,76 and will not be discussed in detail here. For the RO2 H-shifts at the heart of HOM formation, comparison with the limited number of direct rate measurements6,12,77−79 (see also section 4.1.2) indicates that state-of-the-art calculations (including both coupled-cluster energy corrections, hydrogen atom tunneling corrections, and accounting for conformational complexity) are able to reproduce experimental H-shift rates to better than a factor of 10, possibly better than a factor of 5.12,79 While still far from quantitative, such calculations are thus able to qualitatively indicate whether a certain H-shift is atmospherically competitive or not and also reliably predict relative rates and trends between different compounds or functional groups. While, e.g., alkene + oxidant reactions have been studied extensively,75,76 the bimolecular reactions of complex polyfunctional RO2 relevant to HOM formation have so far received less computational attention. Reliable rate predictions for such reactions are further complicated by the presence of multiple radical centers, as well as low reaction barriers necessitating more elaborate kinetic modeling.80
4.1. Autoxidation Involving Peroxy Radicals As the Source of HOM
For the purpose of this review, HOM have already been defined (section 2.2) as products of autoxidation involving peroxy radicals. In this section, we briefly outline and review the evidence that lead to this conclusion (and the consequent definition). Most studies of HOM formation have initiated the oxidation processes by ozonolysis. The motivation for the first ozonolysis studies conducted by Ehn et al. (2012) in the Jülich Plant Atmosphere Chambers (JPAC)7 came from the observation of HOM products among natural ions observed in the nocturnal boreal atmosphere.29 As OH is not present in significant quantities at night, it was proposed and later confirmed that O3 reaction with biogenic monoterpenes is a source of this previously elusive highly oxidized gas-phase material. In subsequent studies,5,8,38,81,82 ozonolysis was confirmed to be an efficient pathway to the highly oxidized products. OH radicals have also been suggested to participate in HOM formation from alkenes, and the involvement of OH radicals in parallel with O3 has been inferred from ozonolysis experiments performed in presence and absence of OH scavengers,46,83 affecting the observed product distributions. For example, products that contain two hydrogen (H) atoms more than the parent hydrocarbon have been interpreted to originate from an oxidation pathway initiated by OH addition and terminated by a HO2 reaction (see section 4.2). Experiments using H2O2 as an OH source have recently confirmed that OH-oxidation can also produce HOM in the absence of O3, albeit possibly at somewhat lower yields.46 HOM formation has recently also been reported from NO3-initiated oxidation of α- and β-pinene.84 Presumably, chlorine (Cl) initiated oxidation will also produce HOM in suitable conditions, but to our knowledge there are as yet no reports on direct experiments of Cl - derived HOM.
4.1.1. Evidence for Autoxidation Involving RO2
The mass spectrometric observations of gas-phase HOM with more than six oxygen atoms presented a challenge for atmospheric chemistry modeling. Conventional understanding of atmospheric oxidation mechanisms, as implemented for example in current versions of the Master Chemical Mechanism, predicted that OH, O3, and NO3 initiated oxidation of hydrocarbons such as monoterpenes primarily leads to closed-shell products (first-generation products) with at most 5–6 oxygen atoms.
Highly oxidized products can be formed by sequential oxidation, for example, by another OH (or NO3) radical reacting with the first-generation products. This type of mechanism has been invoked to explain for example the formation of MBTCA (3-methyl-1,2,3-butanetricarboxylic acid) from the oxidation of α-pinene.20 Similarly, Molteni et al. (2018) found that a small fraction of HOM from OH oxidation of aromatics was likely formed through secondary OH reactions.85 Sequential O3 oxidation would require either multiple C=C bonds in the initial alkene (present in, e.g., limonene but absent in α-pinene), subsequent reaction steps forming C=C bonds, or reaction of ozonolysis-generated OH with the first-generation ozonolysis products. However, the rapid time scale (seconds to minutes) of HOM formation in many systems45,81,82,86 ruled out sequential oxidation as a significant source in these investigations. Furthermore, 18O3 labeling experiments5 demonstrate that the ozonolysis-generated monomer HOM contain precisely two oxygen atoms originating from ozone, with the third presumably having been lost in the well-established vinylhydroperoxide dissociation step.87 This observation ruled out participation of a second O3 molecule, as well as addition reactions of ozonolysis-generated OH. The majority of the oxygen atoms in the detected HOM species must thus originate from atmospheric O2, as no other oxygen-containing reactive molecule has sufficiently high concentrations to produce HOM on a time scale of seconds, even if collision-limit reactions between this “oxygen carrier” and the organic species undergoing oxidation are assumed. In loose analogy with definitions used in other fields of chemistry, such oxidation by atmospheric O2 (without other oxidants beyond the initiation reaction), has been termed “autoxidation”.6
Although the observational evidence strongly indicates that HOM production occurs in one generation of oxidation, this in no way precludes later-generation HOM formation in Earth’s atmosphere. As we shall show below, HOM formation typically represents a relatively minor pathway, with molar yields near 10% in many cases. Further, HOM formation is strongly enhanced by activating (oxidized) functional groups on a carbon backbone. Consequently, the 90% or so of the first-generation products that are not HOM are likely to undergo further oxidation in the atmosphere.88,89 The HOM yields from those subsequent oxidation steps may also be significant. However, there have been few experimental studies of later-generation oxidation and most laboratory experiments to date focus on first-generation oxidation products. At least one example does exist of later-generation oxidation: Schobesberger et al. studied OH oxidation of pinane diol, a representative first-generation oxidation product of a-pinene (formed via hydrolysis of the beta-hydroxy nitrate); they observed many products with an APi-TOF that would now be classified as HOM.90
HOM formation must thus involve multiple addition reactions of O2 to some intermediate species formed by the initial oxidation reaction. Spin conservation rules imply that such intermediates are radicals, as the addition of ground-state (triplet) O2 to closed-shell organic molecules, which almost invariably have singlet ground states, is typically very slow, as it is either spin-forbidden (in the case of ground-state singlet products) or endothermic (in the case of triplet or radical products). In the context of atmospheric C,H,O compounds, this limits the possible radical species to three main types: alkyl radicals (with the radical center on a carbon atom), alkoxy radicals (with the radical center on an O atom directly and solely bonded to a C atom), and peroxy radicals (with the radical center on a O atom bonded to another O atom). In addition, radical species may have resonance structures, which can usually be represented as combinations of two of these three radical types; for example, vinoxy radicals have both alkyl and alkoxy radical character. Reactions of O2 with peroxy radicals have not been measured (and are thus presumably very slow), while alkoxy radical + O2 reactions typically74 lead to closed-shell carbonyl compounds, e.g., via the channel RO + O2 → RC(O) + HO2. Alkyl radicals, on the other hand, are known to react almost instantaneously with O2, yielding peroxy radicals. Experimental rate coefficients for alkyl + O2 reactions are typically on the order of 10–12 molecules–1 cm3 s–1 at 298 K, and thus the lifetime of alkyl radicals is about 30 ns at the Earth’s surface.91 Vinoxy radicals are slightly less reactive with O2, but alkyl substitution has been found to increase also their reaction rates to around 10–12 molecules–1 cm3 s–1.92
HOM formation could thus be explained if a mechanism exits to convert peroxy radicals (RO2) back into more highly oxygenated alkyl radicals, thereby allowing the addition of further O2 molecules. At least two such RO2 reaction mechanisms are known: direct unimolecular hydrogen shifts (H-shifts) forming hydroperoxy-alkyl radicals, and indirect bimolecular reactions forming alkoxy radicals, which can then undergo rapid H-shifts to form hydroxyl-alkyl radicals. Alkoxy radical H-shifts can be fast, but they compete (see section 4.1.4) with other rapid pathways such as bond scissions and termination reactions with O2. It is therefore difficult to explain the formation of products with large numbers of oxygen atoms, and retaining the original number of carbon atoms, solely by alkoxy H-shifts. RO2 H-shifts are well-known reactions in combustion chemistry, and play a significant role in autoignition,93 and recently the products of these H-shift (denoted “QOOH”) have been directly observed.94,95 At atmospheric temperatures, RO2 H-shifts have usually been considered too slow to play a significant role. Also, for simple RO2 species, the H-shifts are usually thermodynamically unfavorable, i.e., the reverse rate is considerably faster than the forward rate. However, already a decade ago computational work on for example isoprene96,97 and α-pinene98 oxidation mechanisms demonstrated that some H-shifts could be rapid enough to be competitive with other RO2 reaction channels at certain atmospheric conditions.
Two different types of H/D isotope exchange experiments provide strong support for a HOM formation mechanism based on RO2 H-shifts. First, the HOM yields are strongly suppressed if the original hydrocarbon is deuterated,5,82 indicating that the rate-limiting reactions involve hydrogen or proton transfer reactions. These hydrogen transfer reactions are typically significantly faster for H (1H) compared to D (2H) nuclei due to a combination of quantum mechanical zero-point vibrational energy and tunneling effects.12,99 In the case of HOM originating from ozonolysis, part of this isotope effect is likely to originate from the 1,4 H-shift of the Criegee Intermediate, which is part of the formation pathway of the first-generation RO2. Second, heavy water (D2O) can be used to probe the number of “acidic” hydrogens (in practice, hydrogen atoms other than C–H, e.g., −OH and −OOH groups) in the HOM products, as these will rapidly be exchanged with deuterium, which can then be detected in the mass spectra. The ratio of acidic H atoms to O atoms in HOM compounds is comparably low, for example, for the C6H8O7 and C6H8O9 products derived from cyclohexene ozonolysis were found to have only 2 and 3 acidic H atoms, respectively.82 The majority of oxygen atoms can thus not be bound to, for example, OH groups, as this would lead to a much higher number of acidic H atoms. This observation rules out reaction pathways involving a large number of alkoxy (RO) H-shifts, as these would lead to products with multiple OH groups. In contrast, autoxidation involving peroxy radicals creates mainly hydroperoxy [−OOH] and peroxy acid [−(C=O)OOH] groups, with relatively low ratios of acidic H to O atoms. Compounds containing ester and ether groups would also satisfy the requirement of low ratios of acidic H to O atoms, although these are unlikely to form in the gas phase and would in any case tend to have different numbers of carbon atoms compared to the parent alkenes.
Experimental comparisons of different alkenes shed further light on the reaction mechanisms behind HOM formation. For example, 6-nonenal ozonolysis forms HOM similar to those from cyclohexene ozonolysis.5,82 This indicates that the excess energy from the initial ozonolysis reaction, which remains within a single species for cyclohexene, but is distributed over two products in the case of 6-nonenal, does not play a major role in HOM formation. This indirectly also suggests that HOM formation does not directly involve the high-energy Criegee Intermediates formed in ozonolysis, other than as precursors for the initial peroxy radical. The 18O3 labeling experiments further help rule out for example bimolecular reaction mechanisms based on Criegee Intermediates, as products of such reactions would, in the case of endocyclic alkene precursors, typically retain all three oxygen atoms from the O3 molecule. Additionally, HOM have also been observed from OH oxidation, implying that the Criegee Intermediates are not required.46,85,100,101
The dependence of HOM yields and composition on conditions (discussed in more detail in section 4.3) also support the hypothesis that HOM formation is primarily driven by peroxy radical H-shifts. HOM yields have been found to increase strongly with temperature (see sections 4.2 and 4.3), indicating that rate-limiting reactions have reasonably high activation energies (energy barriers). This in turn implies that such reactions are predominantly unimolecular, as bimolecular reactions with high barriers would have negligible yields at any atmospherically relevant temperature. Increasing concentrations of RO2 reaction partners such as NO or HO2 also generally suppresses HOM formation (although the detailed patterns are more complex, as discussed in section 4.2).
The final and most direct evidence for the key role of peroxy radical isomerization reactions in HOM formation comes from CIMS studies on systems undergoing autoxidation.5,38,45,81 In these mass spectra, elemental compositions corresponding to multiple generations of peroxy radicals are observed, separated by 32 mass units (corresponding to the addition of one O2). The assignment of mass peaks specific to peroxy radicals is corroborated by the presence of mass peaks corresponding to well-known termination products of uni- and bimolecular RO2 reactions: carbonyls, alcohols, and hydroperoxides. The observed dependence of the peaks assigned to peroxy radicals on the hydrocarbon loading also matches that expected for RO2 radicals: at low loadings where bimolecular reactions are negligible, the signals increase linearly. In contrast, at higher loadings, the radical signals increase roughly with the square root of the VOC oxidation rate, while the closed shell peaks increase approximately linearly.5,45,81 Laboratory experiments of OH initiated oxidation of 3-pentanone similarly show products from two steps of O2 addition followed by a unimolecular H-shift. The first step, which has a relatively slow H-shift ∼0.002 s–1, leads to a hydroperoxyketone. In the second step, the H-shift is accelerated to faster than 0.1 s–1 by the hydroperoxy group attached to the carbon from which H is abstracted (see section 4.1.2) and the product, a hydroperoxydiketone, has a significantly larger oxygen to carbon ratio compared to the parent 3-pentanone.6
4.1.2. General Trends for RO2 H-Shifts
The rates of the RO2 H-shifts driving HOM formation are strongly dependent on the precise chemical structure of the RO2. H-Shifts of simple monofunctional RO2 are too slow to play a role at room temperature, with typical rate coefficients at or below 10–3 s–1.6,102 In polyfunctional complex RO2, H-shift rates can be significantly enhanced by a combination of favorable transition state geometries and substituents or resonance structures activating the C–H bond (i.e., lowering the barrier to H atom transfer). When both of these preconditions are met, RO2 H-shift rates at 298 K can exceed 0.1 s–1 or sometimes even 1 s–1, as demonstrated experimentally for example for the methacrolein + OH,77 pentanone + OH,6 isoprene + OH,78 and hexane + OH + NO systems,12 and a/b-pinene + OH systems.ref239 See Table 1. for a list of experimentally measured RO2 H-shifts together with the available computational predictions. Note that none of the species listed in Table 1 are actually HOM by the definition used in this study (as they have less than six oxygen atoms), although many of them are potential HOM precursors.
Table 1. Measured and Calculated H-Shifts Rate Constants (Abstracted H Is Highlighted in Red).

For calculation details, see the individual references. All calculated rates in this table involve multiconformer transition state theory, with density functional theory geometries, coupled-cluster single-point energies, and tunneling corrections. The difference in the rate constants in the calculations on the isoprene + OH system originates in the density functional part of the calculation as the coupled cluster energy corrections were run on the butene backbone (i.e., without the methyl group present, in which case the 1OH and 4OH reactions are identical).
Systematic and predictive structure–activity relationships (SAR) for peroxyradical H-shifts have not yet been published. However, various general trends can be extracted from the studies published so far.6,12,79,82,102,103,242,243 Qualitative information on the relative likelihood of different RO2 H-shifts can also be inferred from structure–activity relationships published for the much more rapid RO H-shifts,104 as the general features strongly affecting the rates are similar. One of these features is the number of atoms in the cyclic H-shift transition state. The standard notation for the H-shifts discussed here is “1,n”, where n is the number of non-hydrogen atoms in the transition state ring, i.e., typically (n – 1) C atoms for RO and (n – 2) C atoms for RO2, with the ring in the cyclic transition state comprised of n + 1 atoms. As discussed in detail below, too small rings cause steric strain, while too large rings lead to entropic penalties. The chemical environment of the migrating H atom (e.g., substituents around a C–H group) can significantly affect the H-transfer barrier height, and thus change the reaction rate by several orders of magnitude, as illustrated in Figure 7. If the terminal oxygen atom of the RO2 group and the H atom being abstracted are separated by four atoms (1 O and 3 C), the H-shift is labeled a 1,5 H-shift, as illustrated in the inset in Figure 7. Substituents on the RO2 group itself generally have only minor effects on the C–H bond activity but may affect the transition state geometry by, for example, steric hindrance or hydrogen bonding.12,102,104 Substituents giving rise to resonance structures (for example, vinyl, acyloxy, or acylperoxy radicals) may also significantly affect reaction rates.
Figure 7.
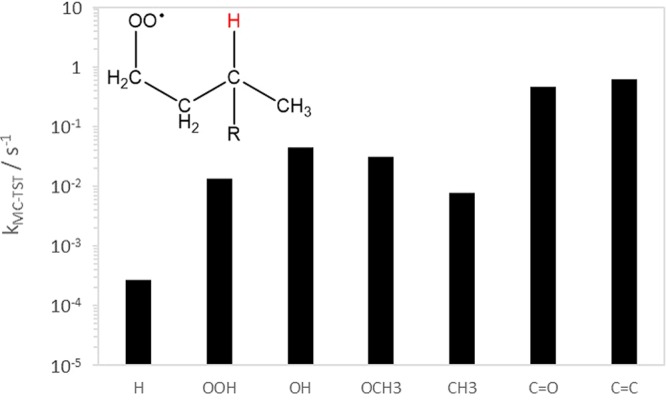
Effect of substitution (R and x-axis) on the calculated 1,5 H-shift rate constants.242
The first observed cases of atmospheric autoxidation all involved peroxy radicals with carbonyl, hydroxyl, and/or hydroperoxyl substituents. These are well-known to activate adjacent C–H bonds and thus promote both RO2 and RO H-shifts. For example, computational results indicate that the rate of H-abstraction at 298 K increases by several orders of magnitude when an OH or OOH substituent is added to the C atom from which the H is being abstracted (Figure 7).6,102 Mentel et al. (2015) observed HOM-formation from peroxy radicals generated by ozonolysis of several different endocyclic alkenes.38 The common characteristic of these RO2 is that they possess two aldehyde/ketone groups: one adjacent to the RO2 group, and one located further from it. Computational results on cyclohexene ozonolysis indicate that RO2 H-shifts from aldehyde groups can have rate constants on the order of 1 s–1.82 1,4 H-shifts from adjacent aldehyde carbons are expected to be somewhat slower because of steric strain in the transition state, though a 1,4 aldehydic H-shift rate constant of 0.5 s–1 has been measured for the methacrolein + OH system.77 Mentel et al. (2015) did not observe efficient HOM formation from the ozonolysis of Z-6-nonenol, even though the RO2 formed in this reaction could undergo a 1,7 H-shift from a primary alcohol carbon in addition to the 1,4 aldehydic H-shift.38 In contrast, the corresponding peroxy radical from Z-6-nonenal, with an aldehyde at the 1,7 position, was observed to form HOM. The reason for the lack of HOM formation in the Z-6-nonenol + O3 system can be explained by a combination of the 1,7 H-shift from the −CH2OH group being slower than that from the −CHO group, and by a unimolecular HO2 elimination reaction (see section 4.1.5). In addition to hydroxyl, hydroperoxyl, and carbonyl groups, computational studies indicate that ether groups such as −OCH3 also enhance the rate constant of RO2 H-shifts (Figure 7).95,102,105,106
Alkyl substitution also activates C–H bonds and thus promotes RO2 H-shifts (R = CH3 in Figure 7), a;though alkyl substitution alone is unlikely to lead to competitive H-shift rates in atmospheric conditions. In analogy with results on RO H-shifts,104 the effect of alkyl substitution is almost certainly additive to that of oxygenated functional groups, e.g., secondary alcohols have faster H-shifts than otherwise identical primary alcohols.6,102 It should be noted that the C3 and C4 rings found in some monoterpene derived peroxy radicals form special cases due to the ring strain associated with H-shifts from and across them. This ring strain is also manifested, e.g., as considerably higher C–H abstraction barriers in, e.g., cyclopropane compared to n-propane.107,108 Computational evidence indicates that H-shifts from such rings are likely to be slow, and that H-shifts across rings may also be sterically hindered compared to corresponding H-shifts without C3/C4 rings.109 In the α-pinene + O3 system, recent experiments involving selective deuteration indicate that 1,9 aldehydic H-shifts across the C4 ring (with a computed rate constant on the order of 0.0015 s–1)109 may nevertheless take place.63 Unfortunately, there are at the moment no direct and unambiguous measurements of room-temperature RO2 H-shift rates from or across C3 or C4 rings. Larger rings may sterically prevent some particular H-shifts (as shown for some RO2 species in the Δ3-carene and α-pinene + NO3 cases110), but, for example, the autoxidation of aromatic species very likely involves 1,4 H-shifts where both the RO2 group and the abstracted hydrogen are located on the same C6 ring.85 The −ONO2 substituent which arise in NO3 addition reactions as well in peroxy radicals formed from oxidation of RONO2 have been found to have minimal effect on the H-shift reactions of the H atoms attached the carbon on which the nitrate group is attached.
Resonance stabilization of the alkyl radical products of RO2 H-shifts also enhances the rate constant of H-shifts. This plays a significant role in the oxidation of dienes such as cycloheptadiene94 and isoprene.78 Similar to alkyl substitution, the effect of resonance stabilization is likely additive to that of other H-shift enhancing effects. In the isoprene + OH system, this additivity leads to experimental RO2 H-shift rates of up to 3.7 s–1 at 297 K, to our knowledge the fastest room-temperature RO2 H-shift (of a C–H hydrogen) reported for any atmospherically relevant system (see Table 1). The H-shifts where the hydrogen “scrambles” between two OO groups (OO–H–OO) are found theoretically to be even faster (see section 4.1.4).102,103
The number of atoms in the cyclic H-shift transition state affects RO2 H-shifts analogously to RO H-shifts. The most favorable geometries are in both cases usually found for 1,5-, 1,6-, and 1,7 H-shifts (6, 7, and 8 member cyclic transition states). For both RO and RO2 H-shifts, computational studies generally predict that 1,3 H-shifts are strongly hindered by steric strain, moderately hindered for 1,4 H-shifts, and even 1,5 H-shifts are sometimes computed to be slower than corresponding 1,6 H-shifts.6 For the case of RO H-shifts, computational predictions are in partial disagreement with the conclusion drawn from earlier experimental studies111 that only 1,5 H-shifts can take place, while, e.g., the SAR104 indicates that also 1,6 and 1,7 H-shifts can often be competitive. For the RO2 case, 1,4, 1,5, and 1,6 H-shifts have all been experimentally observed (see Table 1). In principle, it is well established that larger transition state rings suffer from entropic penalties, although the magnitude of this effect for RO2 H-shifts is highly uncertain, and also likely quite system-dependent.104 For example, hydrogen bonding effects may stabilize or destabilize transition states relative to reactants, and this effect may also strongly depend on the particular transition state ring.112,113 Disentangling pure ring-strain effects from, e.g., H-bonding effects, can therefore be difficult. For the cyclohexene + O3 system, calculations indicate that even 1,7 and 1,8 H-shifts can be quite rapid if the C–H bonds being attacked are active enough.82 Also, the steric hindrance for H-shifts across C3/C4 rings may partially be compensated for by a smaller entropic penalty for long-distance (e.g., 1,9) H-shifts, as the reactants cannot freely rotate over all the dihedral bonds separating the RO2 and the H atoms. In contrast, RO2 groups located on rings may suffer from additional steric constraints, for example, the RO2 formed in the NO3 oxidation of α-pinene and Δ3-carene were found to have very slow H-shifts due to steric effects.110
4.1.3. O2 Addition to Alkyl Radicals
Most mechanistic studies on atmospheric autoxidation have not explicitly investigated the addition of O2 to polyfunctional alkyl radicals but instead assumed that the reaction occurs irreversibly and at rates comparable to the oxygen addition reactions of smaller alkyl radicals. These are generally on the order of 10–12 molecules–1 cm3 s–1,91 leading (at O2 partial pressures on the order of 0.2 atm) to pseudo-unimolecular rates on the order of 107 s–1. Because this is many orders of magnitude larger than the rate of the H-shifts, or of bimolecular reactions with trace gases, the lack of interest in the specifics of the O2 addition reaction is understandable. However, there are some notable cases for which the exact kinetics of the O2 + alkyl reaction may be significant. First, O2 addition to many atmospherically relevant alkyl radicals can form multiple structural isomers or stereoisomers, which may have different reactivities. For example, in the OH or NO3 initiated oxidation of endocyclic monoterpenes, up to ten (α-pinene) first-generation peroxy radical isomers may be produced. These correspond to two different addition sites for OH or NO3 and four possible chiralities (two different R/S addition positions for each site with respect to the ring, and another two different R/S addition positions for the subsequent O2 addition). Each of these peroxy radicals may then differ in their reactivity, for example, due to varying steric constraints for the possible H-shifts.110 Thus, the exact branching ratio between the different O2 addition rates may be important for the subsequent oxidation or autoxidation mechanism, even if all the additions occur rapidly and irreversibly. Peroxy radical diastereoisomers differ in reactivity even for acyclic systems, as shown recently for the hexane + OH + NO system.12 Second, if the alkyl radical is resonance stabilized (as for example in aromatic systems85), the oxygen addition reaction may be reversible. This has recently been shown to be the case for the OH-initiated oxidation of isoprene,78,96 where the reversibility of the O2 addition leads to a very different distribution of peroxy radicals than what would be predicted based on forward rates alone. Third, in some cases, the alkyl radical may be able to undergo rapid unimolecular reactions, such as C–C bond scission reactions of both acyl radicals and cyclic alkyl radicals,13,110 carbonyl formation (via −OOH decomposition)113 or epoxide formation.114 These may also have rates on the order of 107 s–1, and estimating branching ratios would thus again require more accurate information on O2 addition rates.
4.1.4. Other Unimolecular Isomerization Reactions
RO radicals can be formed (in competition with other product channels) in bimolecular reactions of RO2 with NO, other RO2, and for complex RO2 also with HO2 (see section 4.2).115−118 Alkoxy radicals can undergo rapid H-shifts, with rate constants often faster than 103 s–1, leading to hydroxyl-alkyl and (after O2 addition) hydroxyl-peroxy radicals. This mechanism has been invoked to explain some of the HOM elemental compositions observed in measurements.38 The contribution of RO H-shifts (and other RO reactions) to HOM formation is likely to vary with the concentration of bimolecular coreactants. In clean conditions, RO formation is less likely, while higher NO, RO2. and possibly HO2 concentrations lead to greater RO formation. HOM formation in such conditions (to the extent that it happens) is thus more likely to involve RO reactions. Alkoxy H-shifts compete with both O2 reaction leading to closed-shell products and with C–C bond scission reactions, which lead to fragmentation or to ring opening in systems where the alkoxy carbon is located on a ring. On the basis of computational data, Kurtén et al. (2015)109 have suggested that ring-opening reactions may be necessary for autoxidation to proceed, e.g., in the O3 + α-pinene system, due to the C4 ring hindering most of the potential RO2 H-shifts. However, experimental evidence shows that HOM formation from O3 + α-pinene can occur also in conditions where bimolecular RO2 reactions are negligible.81
An important class of H-shift reactions neglected in the earliest autoxidation studies is the H atom “scrambling” between hydroperoxide and peroxy radical functional groups recently suggested by Jørgensen et al. (2016).102 These can be much faster than H-shifts from C–H groups, with computed and experimental rates often exceeding 103 s–1.102,103,ref240 While such H-shifts do not increase the overall oxygen content of the molecule, they can facilitate the autoxidation process in several ways. First, H atom “scrambling” between multiple COO groups (see Figure 8 for an example) may allow H-shifts that would otherwise be sterically hindered. Second, the equilibrium constants of “OOH···OO” H-shifts strongly favor peroxy acids over hydroperoxides.103 In other words, if a molecule has both C(O)OO and COO groups, the hydrogen atoms will predominantly be localized on the former and the radical centers on the latter. This acts to reduce the relative concentration acylperoxy radicals in systems undergoing autoxidation and also affects the probability of termination reactions as discussed in section 4.1.5
Figure 8.

Illustration of a scrambling OO···H···OO H-shift.
A related class of extremely rapid H-shifts from hydrogen atoms bound to oxygen has been suggested by Peeters and Nguyen (2012),119 who computationally predict that 1,6 H-shifts from enolic hydrogens (C=C–OH) may proceed at rates up to 106 s–1. While RO2 H-shifts from OH hydrogens are thermodynamically unfavorable, enolic H-shifts are significantly enhanced by resonance stabilization of the vinoxy radical formed in the reaction. However, systems containing enolic hydrogens are expected to be relatively rare in the atmosphere.
If the RO2 radical contains a C=C bond, cyclization steps forming cyclic peroxides (C–O–O–C) may be competitive with H-shifts.86,120 These reactions are known to play a role in the oxidation (including autoxidation) of aromatic compounds (as seen in the MCM aromatic mechanism).101 Cyclic peroxides likely help explain the very low numbers of acidic hydrogens observed in some H/D exchange experiments, such as the C6H7O10 product with only two acidic H observed in the ozonolysis of cyclohexadiene.86,120 In addition to aromatics and di- and trienes, endocyclization reactions may be important also for monoterpenes, where the C–C scission reactions of alkyl radicals lead to the formation of C=C bonds as part of the oxidation process. This has been suggested to be an important pathway, for example, in the OH-initiated oxidation of α-pinene,46,121 and cyclization may accordingly take place also in this system.
4.1.5. Unimolecular “Termination” and Fragmentation Channels
Many of the unimolecular fragmentation reactions involved in atmospheric autoxidation are already known from combustion chemistry.122,123 See Figure 9 for an illustration of the most important pathways. For example, H-abstraction at a carbon with an OOH group attached (Figure 9a) leads to prompt carbonyl formation and OH loss and thus termination of the autoxidation chain. This is generally believed to be the typical fate of the “QOOH” product of the first RO2 H-shift in combustion processes: the QOOH adds an O2 to form “OOQOOH”, which “back-isomerizes” by abstracting the H atom on the hydroperoxide carbon, leading to OH loss. For simple hydrocarbons with no other functional groups, this is likely to represent to dominant fate of OOQOOH, as no other hydrogen abstraction reaction is competitive. As described in the previous sections, HOM formation is made possible by the presence of additional functionalities (originating either from the parent hydrocarbon, or from the OH/O3/NO3 oxidation), which provide the OOQOOH with alternative, nonterminating H-abstraction pathways. Note that while unimolecular reactions leading to closed-shell organic products via, e.g., OH or HO2 loss are not termination reactions in a general sense, as the number of radical species remains unchanged, they are termination reactions from the point of view of HOM formation. The H atom scrambling discussed in section 4.1.4 tends to reduce the net rate of this OH loss channel by converting hydroperoxy groups into peroxy acid groups.
Figure 9.

Unimolecular fragmentation reactions of peroxy radicals. (a) OH loss following H-abstraction from a carbon with a −OOH group attached. (b) NO2 loss following H-abstraction from a carbon atom with a ONO2 group attached. (c) HO2 loss following H-abstraction from a carbon atom with an OH group attached (and subsequent O2 reaction). (d) CO loss following H-abstraction from an aldehydic carbon atom. (e) Epoxide formation.
Analogous to the OH loss pathway, H-abstraction from carbon atoms with nitrate groups attached (C–ONO2, Figure 9b) similarly leads to carbonyl formation and NO2 loss.98 Similarly, O2 addition to alkylhydroxy radicals leads to rapid HO2 loss and carbonyl formation (Figure 9c). Acyl radicals undergo C–CO scission reactions, forming carbon monoxide and a shorter-chain alkyl radical, in competition with O2 addition (Figure 9d). The relative rate of the two reactions likely varies between systems. For cyclohexene ozonolysis, modeling82 indicates that especially the first-generation acyl radical C–CO scission is very slow compared to the (assumed) rate of O2 addition. However, experiments indicate that both channels are competitive in atmospheric conditions, leading to a mixture of C5 and C6 products.45 HO2 loss leading to alkene formation has been suggested as a possible unimolecular termination reaction,82 but calculations indicate that this route is unlikely to be competitive in atmospheric conditions.124
Epoxide formation and associated OH loss from α-hydroperoxy alkyl radicals (Figure 9e) is known to play a role in OH-initiated oxidation of isoprene,125 although chemical activation (excess energy from previous reaction steps) has previously been thought to be necessary for the reaction to compete with O2 addition. However, recent experimental and computational results indicate that thermal epoxide formation (i.e., epoxide formation after collisional stabilization, without excess energy present) also may be competitive for sufficiently complex and oxidized α-hydroperoxy alkyl radicals, in which H-bonding functional groups are able to stabilize the transition state of the epoxy-forming reaction.114 The elemental composition of epoxide products is identical to that of the ketone products formed from OH loss of COOH groups, making it difficult to assess the relative importance of the two reaction routes solely based on mass.
4.2. Bimolecular RO2 Reactions
As discussed in section 4.1, HOM are formed by autoxidation steps in radical chain reactions mediated by RO2 and RO radical intermediates. Bimolecular reaction partners of peroxy radicals exert a significant influence on HOM formation. In general, gas-phase peroxy radicals are relatively unreactive free radicals and in atmospheric conditions react appreciably only with other RO2, NOx, and HO2 radicals.73,126,127 Each of these three main bimolecular RO2 reaction partners can in principle lead to either termination or propagation of the autoxidation chain. The propagation reactions all involve the formation of alkoxy radicals, which can then undergo H-shifts, O2 reaction or decomposition as discussed in section 4.1.4. The influence of these alkoxy H-shifts on HOM product distribution has been reported previously38,46 and was noted to shift the observed closed-shell products in O3-initiated autoxidation from odd oxygen compositions to even oxygen compositions, for example, from C6H8O7 to C6H8O8 in the case of cyclohexene.38
Most of the termination reactions, with the exception of ROOR dimer formation, are well-known from the extensive literature on small hydrocarbons in both atmospheric and combustion conditions.127 Propagation reactions with NO and RO2 are also well-known as important pathways in peroxy radical reactions.98,128 In contrast, much less is known about the RO2 + HO2 propagation channel, as it is probably important (or even possible) only for complex RO2 (as well as acyl RO2). In this section, we discuss the RO2 reaction channels, including both termination and propagation reactions, identified with each of the main reaction partners (other RO2, HO2, and NOx) and also briefly review proposed reactions with other coreactants. Special emphasis will be placed on acylperoxy radical reactions, as well as reaction mechanisms potentially forming HOM dimers.
To put the problem in context, kinetics measurements have shown that most reactions between RO2 and either HO2 or NO have similar rate constants, occurring at near 1 in 10 collisions (a rate constant near 10–11 cm3 molecules–1 s–1, with only a weak temperature dependence).111,126 In atmospheric chemistry, “high NO” or “low NO” thus in large part refers to the ratio NO:HO2. The HO2 concentration is often on the order of 10 pptv, and so for NO ≫ 10 pptv it is typically understood that the dominant RO2 loss process is reaction with NO. For peroxyacyl radicals, reaction with NO2 to form peroxyacyl nitrates is also important and often dominant. Further, because reactions of OH with oxygenated VOC (OVOC) often lead directly to HO2 formation (the simplest cases being reactions of OH with CO and CH2O), in general in the atmosphere HO2:RO2 > 1; however, laboratory experiments often isolate hydrocarbons without OVOC, and so it is not unusual for laboratory experiments to have HO2:RO2 < 1.
In contrast to the fairly uniform behavior of the reactions with HO2 and NO, the nature of the R group has a dramatic effect on the RO2 self-and cross reactions, as discussed below. Many of these reactions have rate constants slower than the rate constants with HO2 and NO (although with a strong temperature dependence), and so because the atmosphere also tends to have HO2:RO2 > 1, evidence for RO2 + R′O2 reactions in laboratory reactions is often regarded as evidence that laboratory conditions have strayed from atmospheric relevance. However, there are some R groups (notably peroxyacyl radicals) with very fast self-reaction rate constants, and in general if these self-and cross reactions have rate constants greater than 10–11 cm3 molecules s–1, it is possible for them to constitute a dominant loss process in the atmosphere. Further, relatively low HOM yields can still be important, and so even a minor role for RO2 + R′O2 reactions could be atmospherically significant (though in that case one would expect the overall branching to be extremely sensitive to photochemical conditions, i.e., HO2:NO:RO2, and even the specific composition mix of the RO2.
4.2.1. RO2 + R′O2
As reviewed, e.g., by Orlando and Tyndall,73 there are three main reaction channels for RO2 self- and cross-reactions:
| a |
| b |
| c |
Reaction channel (a) is a propagation channel, while (b) and (c) are termination reactions. If one or both of the RO2 reactants are acylperoxy radicals (RC(O)OO•), the reactions are typically exceptionally rapid.129−131
In the context of HOM, the formation of organic peroxide dimers in channel (c) is especially important, as it leads to products with more carbon atoms than the parent VOC. Highly oxidized gas-phase dimer compounds have been observed in multiple previous studies5,83,132,133 but without explicit information on their formation mechanism.
Detected dimer compositions include, for example, C10H14–16O9–12 for cyclopentene ozonolysis,38 C11–12H18–20O7–15 for cyclohexene ozonolysis,82 C18–20H28–32O10–18 for monoterpene ozonolysis,5 C29–30H46O10–20 for sesquiterpene ozonolysis,134 and C12H14Ox-C24H22Ox for aromatic OH oxidation.85 On the basis of the near-linear dependence of the concentrations of the dimer species (with composition C19–20H28–32O10–18 in α-pinene ozonolysis) on the reacted monoterpene concentration, Ehn et al.5 proposed reaction (c) as the formation mechanism for the dimers. Subsequent studies also found reaction (c) as the most likely pathway to observed gas-phase dimer formation. This was later supported by Berndt et al. (2018), who recently measured reaction (c) to be as fast as 10–10 cm3 molecule–1 s–1 for certain RO2 radicals.48 Mentel et al. (2015) showed for cyclopentene ozonolyis (in the presence of dark OH), that the most abundant C10-dimers can be explained assuming peroxides formed from the most abundant peroxy radicals.38
Reaction (c) is known to happen in liquid-phase hydrocarbon oxidation.135,136 It is often invoked in analogous gas-phase oxidation systems.98,127 However, there is little evidence that reaction (c) is at all possible for the extensively studied smaller hydrocarbons. Kan et al. (1980)137 and Niki et al. (1981)138 reported CH3OOCH3 as a minor product (<6%) from methylperoxy self-reaction.138 Niki et al. (1982)139 reported an upper limit for the C2H5OOC2H5 yield relative to the major product CH3CHO of 5% based on IR absorption experiments. Tyndall et al. (2001) discounted these earlier studies reporting experimental detection of organic peroxides by reaction (c) and stated: “There is no compelling evidence for peroxide formation in the self-reaction of CH3O2, or indeed in any reaction involving peroxy radicals”.140 In agreement with Tyndall, more recent experimental studies141 have found negligible yields for the dimerization channel. Theoretical studies have reported various mechanisms for the formation of CH3OOCH3 and/or CH3CH2OOCH2CH3 on both singlet and triplet potential energy surfaces.142−145 However, as pointed out by Dibble146 and by Vereecken and Francisco,75 the RO2 + RO2 reaction is a challenging system for computational chemistry methods, and none of the proposed potential energy surfaces can easily be reconciled with experimentally observed overall rates and/or yields. The recently proposed mechanism by Lee et al., invoking asymmetric cleavage of the tetroxide to form a weakly bound RO···O2···RO “cage”, followed by loss of O2, intersystem crossing, and alkoxy recombination to generate ROOR, does support the differences between secondary and tertiary peroxy radicals observed in liquid-phase experiments despite having rate-limiting barriers incompatible with the observed gas-phase reaction rates.143
Nonetheless, several observations point to reaction (c) as a source of gas-phase dimer compounds for highly oxidized, functionalized RO2. First, the experimentally observed dimer formation is severely affected by NO5, by RO2,35 by NO3,147 and by HO271 additions, all pointing to peroxy radicals as the important intermediates being intercepted. Second, the dimers observed in Ehn et al.5 were noted to have a dependence on reacted VOC, which would indicate their formation requiring second-order reactions of the type (c). Third, slower rise times of dimer signals compared to certain monomers have been observed in recent publications using atmospherically relevant VOC and oxidant concentrations148 indicating delayed dimer formation, occurring only after sufficient buildup of RO2 pool for mutual reactions to become significant. Fourth, in combined O3 and OH + cyclopentene experiments by Mentel et al. (2015), OH scavenging suppresses precisely the dimers (C10H16O9,11) that would form by recombination of the primary OH-derived cyclopentene peroxy radicals (C5H9O3) with the most abundant HOM peroxy radicals (C5H7O8,10).38 Fifth, of all other potential dimer-forming RO2 reaction partners, another RO2 seems the most likely candidate: RO2 addition to alkenes is generally associated with high barriers,149 R and RO radicals are too short-lived, and dimer formation is also observed in systems with low yields of stabilized Criegee Intermediates (sCI; for example, cyclohexene + O3)150 or indeed in systems lacking any Criegee Intermediates at all.46 Also, as discussed in section 4.1.1, ozonolysis experiments using 18O3 showed that HOM dimers of endocyclic monoterpenes retain precisely four of the original six oxygen atoms from the O3 addition reactions: sCI reaction products would be likely to contain five or six. While bimolecular reactions of sCIs have been proposed151 as a source of highly oxygenated dimer compounds, the experimental evidence thus indicates that they are unlikely to play a major role in HOM dimer formation.
Recently, Berndt et al. (2018) showed that multiple different RO2 radicals (originating from the ozonolysis of tetramethylethylene, combined with the OH-initiated oxidation of either 1,3,5-trimethylbenzene, 1-butene, isoprene, n-hexane, or methane) form dimer products with elemental masses corresponding to ROOR peroxides.48 The fastest dimer formation rate coefficients were comparable to those of the termination reactions with NO and HO2. For example, the observed rate coefficients of dimer formation from the self- and cross-reactions of RO2 radicals were in the range from 10–10 to 10–14 cm3 molecule–1 s–1, depending on the structure of the RO2 but appearing to be increasing with the size of the RO2 radical. This suggests that intermolecular interactions between the functional groups on the RO2 strongly affect the rate of dimer formation.48
Berndt et al. (2015) have also reported the influence of addition of another VOC to the oxidation mixture (mesitylene, 1,3,5-trimethylbenzene), used as an OH scavenger in the oxidation of cyclohexane.45 With introduction of this second VOC, two dimer products (C15H22O9 and C15H22O11) appeared in the nitrate CI-APi-TOF spectra, attributed to reaction (c) between cyclohexene (C6H10) and mesitylene (C9H12) oxidation products.
Some recent studies have suggested, in agreement with earlier work,152−154 that the ROOR dimer products formed in reaction (c) are predominantly diacylperoxides (RC(O)–OO–C(O)R).67,82,86 These are formed by rapid recombination reactions (c) of acylperoxy radicals, which in turn can be formed via rapid H-shifts from aldehydic C atoms (see section 4.1.2), followed by O2 addition. However, it should be noted that rapid “scrambling” H-shifts (see section 4.1.4) will also tend to decrease the concentrations of acylperoxy radicals in systems containing hydroperoxy groups. Also, the results of Berndt et al. (2018) demonstrate that acylperoxy radicals are not a necessary precondition for dimer formation.48
4.2.2. RO2 + NOx
NOx reactions with RO2 can either terminate or propagate the reaction chain:155
| e |
| f |
| g |
The termination reactions (e) and (g) are generally pressure-dependent. Reaction (e) is commonly the minor channel in RO2 + NO reaction, with the yield increasing as a function of the size of the hydrocarbon backbone, i.e., with degrees of freedom available to distribute the reaction energy.73,81 The RO2 + NO reaction was found by Yan et al. (2016) to be the dominant daytime source of HOM in Boreal forest conditions.133 Propagation reaction (f) is generally (at least for small RO2) the dominant pathway of peroxy radical reactions with NO. In addition to isomerization, fragmentation and reaction with O2, RO can react with NO and NO2 forming organic nitrites (RONO) and nitrates (RONO2), respectively, but are not expected to do so in atmospheric conditions due to their very short lifetimes.74
The NO2 reaction (g) leads to peroxynitrate formation. Whereas the organic nitrates formed in reaction (e) are fairly stable compounds, the peroxynitrates formed in reaction (g) from common primary, secondary, and tertiary RO2 are unstable and decompose back to RO2 and NO2, typically within a second time frame (Atkinson and Arey, 2003).128 However, for the special case of acyl peroxy (RC(O)O2) radicals, more stable adducts called peroxyacylnitrates (PANs, RC(O)OONO2) can be formed. On the basis of results for smaller PAN, these may decompose on a time scale ranging from minutes to weeks, depending mainly on the ambient temperature, and they are an important potential pathway for the long-range transport of NOx, and possibly also RC(O)O2. The formation of peroxyacylnitrates is usually also significantly faster than that of peroxynitrates.
4.2.3. RO2 + HO2
HO2 reactions are generally expected to terminate autoxidation by hydroperoxide (ROOH) formation shown in reaction channel (h):
| h |
However, for more complex (as well as acyl) RO2, other channels are possible. Recently, Praske et al. (2015)118 demonstrated that for RO2 formed in the OH oxidation of methyl vinyl ketone, reaction channel (h) only accounted for about 35% of the total RO2 + HO2 reactions, with two other channels also playing significant roles:
| i |
| j |
Reaction (i) represents a propagation reaction, with a yield of close to 50% in the methyl vinyl ketone + OH system.118,156 This reaction channel has also been suggested to have a high yield for acylperoxy radicals, where HO2 reactions have been observed to lead to radical chain branching.115,116 Reaction (j) represents an alternative termination pathway (from the point of view of the autoxidation chain), yielding slightly different products compared to reaction (h) and also affecting the radical balance by acting as a net source (compared to the immediate reactants) rather than a sink of HOx (OH + HO2). For acyl peroxy radicals, reaction (j) is impossible due to the lack of a hydrogen atom, but they can instead undergo a reaction forming carboxylic acid and O3:115,116
| k |
Reaction (k) is a termination reaction but regenerates an oxidant.
HO2 is omnipresent in hydrocarbon oxidation reactions.126 Reaction (h) has often been invoked71,85 to explain observations of products with more H atoms than in the parent hydrocarbon. For example, monoterpene oxidation products with 18 H atoms can be explained by OH addition to the parent monoterpene (C10H16), followed (possibly after one or more autoxidation steps) by termination via reaction (h).
Iyer et al. have directly tested the influence of HO2 on the autoxidation progression by introducing HO2 to the cyclohexene (C6H10) + O3 system.71 The HO2 addition was mainly observed to form hydroperoxides according to reaction (h), and this resulted in significantly more HOM compounds with 10 and 12 H atoms than observed in previous studies on cyclohexene autoxidation.38,45,82 The HO2 addition was also noted to suppress the dimer formation in reaction (c).
4.2.4. Other Bimolecular Reactions Affecting HOM Formation
Other bimolecular reactions consuming RO2 could also prevent HOM formation in the gas phase. Minor loss processes for peroxy radicals in the atmosphere include reactions with atmospheric oxidants, such as OH,157 Criegee Intermediates,75 and NO3 radicals.128 These reactions are potentially able to suppress and/or enhance HOM formation, but their involvement is currently highly uncertain at best. RO2 reactions with other common atmospheric constituents, such as SO2,158 etc., have also been inspected but found to be slow.
Criegee Intermediates (CI) are potentially important reaction partners in HOM formation because ozonolysis reactions of alkenes inevitably involve CI formation.28 Depending on the size of the parent hydrocarbon (and the pressure and temperature), some fraction of the CIs are collisionally stabilized and can then live long enough to react with multiple atmospheric constituents. Water, organic acids, and SO2 have been shown to be efficient atmospheric scavengers for stabilized CI.45,123,159−161 The role of CIs and/or sCIs in generating atmospheric oxidized material to participate in SOA formation has been a recurring theme during recent decades. However, according to current understanding,82 HOM formation from ozonolysis does not seem to significantly involve bimolecular CI reactions. Richters et al. (2016) added a large excess of acetic acid in a reaction mixture of β-caryophyllene to scavenge all sCI and found no effect on the highly oxygenated RO2 radical signals, suggesting that role of sCI in HOM formation was negligible.47 The CI are involved only as precursors for the first-generation RO2 via the well-known sequence of vinylhydroperoxide formation, dissociation, and O2 addition. As discussed in section 4.2.1, observational constraints point against a significant contribution of sCI reactions to HOM dimer formation.
4.3. Factors Affecting HOM Formation
4.3.1. Temperature
Temperature strongly affects unimolecular reaction rates. Qualitatively, unimolecular reaction rates increase with temperature, with the temperature sensitivity increasing exponentially with the height of the reaction barrier. For atmospherically relevant RO2 H-shifts with room-temperature rates around 0.1···1 s–1, a 20 K temperature increase/decrease typically leads to a factor of 5 increase/decrease in the reaction rate.12 Slower H-shifts are even more temperature sensitive. Temperature also affects the rates of bimolecular reactions, albeit much more weakly. Radical recombination reactions (such as dimer, RONOx, and ROOH formation discussed in section 4.2) often exhibit weakly negative temperature dependence.162,163 Currently, very little is known about the influence of temperature on detailed HOM formation. However, a strong temperature dependence is expected based on simple considerations. Provided that HOM formation derives from the internal H atom transfer autoxidation pathway described above, which necessarily has a relatively high activation energy, the autoxidation rate constant will drop substantially with decreasing temperature. In contrast, the radical–radical biomolecular termination reactions depend only weakly on temperature. Therefore, it is likely that HOM yields will have a strong positive temperature dependence. Frege et al. (2018) have reported HOM products at different temperatures, measured as natural ions in α-pinene ozonolysis initiated oxidation.164 As anticipated, they reported significantly less oxidized products and lower signal levels at the lowest temperatures, consistent with autoxidation formation pathway. Finally, Stolzenburg et al. (2018) have shown that this trend holds for neutral species and that at low temperatures such less oxidized HOM still contribute significantly to particle growth due to their reduced volatility.61
4.3.2. Concentrations of Bimolecular Reactants
The yield of HOM is ultimately controlled by the RO2 lifetime, which determines the branching ratio between unimolecular H-shifts and other RO2 fates (the branching ratio between autoxidation-propagating H-shifts and other unimolecular pathways, when present, is another significant controlling factor). Under most atmospheric and laboratory conditions, the RO2 lifetime is primarily determined by its reaction with NO (see section 4.2.2) and HO2 (see section 4.2.3). The importance of RO2 as a terminator increases with the VOC loading (specifically with the rate of VOC oxidation) and is usually much more important in laboratory settings, which often use higher VOC concentrations than found in the atmosphere. In laboratory studies, RO2 concentrations can easily be much higher than the HO2 concentration in the atmosphere. Assuming as an upper limit [RO2] = 1 × 1010 cm–3 and kRO2 = 1–100 × 10–12 cm3 s–1 (corresponding to the fastest known RO2 + RO2 reactions involving, e.g., acetyl peroxy radicals or large oxidized RO2),48,73 we see that RO2 lifetimes with respect to RO2 + RO2 reactions can be on the order of 1–100 s in high-VOC laboratory conditions. In typical experimental studies, the NO concentrations have in the past been relatively high, leading to relatively short RO2 lifetimes and limited autoxidation. More recently,77,165 experiments have appeared that have been conducted in chambers with rather pristine conditions, with [HO2] ∼ 25 ppt and [NO ] ∼ 50 ppt, giving atmospherically relevant RO2 lifetimes of up to 100 s and facilitating the investigation of H-shift reactions.
In ambient conditions, NO concentrations are typically high in polluted urban regions, with concentrations regularly exceeding tens of ppb (see Table 2 for representative concentrations). In more pristine environments, it drops to about 20 ppt. This leads to pseudo-first-order reaction rate constants of k′NO = k[NO] in the range 2 s–1 to 10–3 s–1 and hence RO2 lifetimes in the range of 0.5 to 1000 s (see Table 2). In comparison, HO2 concentrations in urban polluted areas are low (around 5 ppt) and increase to around 50 ppt in pristine regions. This leads to pseudo-first-order rate constants of k′HO2 = k[HO2] in the range of 10–3 to 10–2 s–1, and RO2 lifetimes of 1000 to 100 s (see Table 2). Thus, in pristine environments, the HO2 reaction will limit the RO2 lifetime, and in polluted areas the NO reaction will limit the RO2 lifetime. However, with the very fast acylperoxy self-reactions discussed above, in some circumstances RO2 self-reaction (and dimerization) may be the dominant bimolecular loss process even in the atmosphere. By combining these reactions, it can be seen that the RO2 lifetime will rarely exceed about 100 s. Hence for H-shift reactions to matter in the atmosphere, their rate constants need to exceed 10–2 s–1, and H-shifts slower than this can thus be ignored. Efficient methods to calculate (from quantum chemical methods) or estimate (from SARs) qualitative rate constants for H-shift reactions have proven extremely useful in assessing the involvement of H-shift reactions in atmospheric oxidation processes, especially in eliminating unlikely mechanisms. The accuracy of the calculated rates can be gauged from the results collected in Table 1. In urban environments, emission control measures are likely to decrease NO concentrations in the future and thus make H-shift based autoxidation more important. Together with increasing ambient temperatures due to global warming, the importance of autoxidation and HOM production is thus likely to increase in the future.
Table 2. Typical Atmospheric Concentration of RO2 Bimolecular Reactants HO2 and NO and the Associated RO2 Lifetimesa.
| urban | rural | forest | |
|---|---|---|---|
| [NO] | 10 ppb/0.5 s | 1 ppb/5 s | 20 ppt/200 s |
| [HO2] | 5 ppt/400 s | 20 ppt/100 s | 50 ppt/40 s |
We have used 8.5 × 10–12 cm3 molecule–1 s–1 for the RO2 + NO rate constant and 2 × 10–11 cm3 molecule–1 s–1 for the RO2 + HO2 rate constant.166 Typical NO and HO2 concentrations are taken from Ren et al. (2003), Holland et al. (2003), and Lelieveld et al. (2008).167−169 Note that the concentrations in any specific location may vary significantly from the typical values given here.
4.3.3. HOM Yields
Previously reported HOM yields have been collected in Table 3. To date, only ambient temperature and pressure HOM yields have been determined for a collection of endocyclic alkenes, a few straight chain model compounds, isoprene, and aromatics. All the determined HOM yields fall between 0.1% and 17%, with a mean of around 5%. As discussed above, the temperature has a strong influence on the oxidation progression through altering the rates of unimolecular isomerization reactions, thus influencing the HOM formation yields. However, as there are no direct temperature dependent yield studies, the influence of temperature on HOM formation can only be implied. Currently, only Frege et al. (2018) has reported temperature dependent data on HOM formation.164 They reported compositions of charged natural HOM ions measured by APi-ToF as a function of temperature. As expected due to sequential nature of the isomerizations, at lower temperature, less oxidized products were observed together with significantly lower signals (how this translates to yields is currently unclear).
Table 3. Determined Molar HOM Yields Using NO3– Ionization.
| compound | total yield (%) | due to O3 (%) | due to OH (%)a | ref |
|---|---|---|---|---|
| isoprene | 4b | 0.01 ± 0.005 | 0.03 ± 0.015 | Jokinen et al. 2015100 |
| Ng et al. 2008147 | ||||
| cyclopentene | 4.8 ± 0.2 | Berndt et al. 201545 | ||
| cyclohexene | 4 ± 2 | Ehn et al. 20145 | ||
| 4.5 ± 3.8 | Rissanen et al. 201482 | |||
| 6.0 ± 0.1 (7.3 ± 0.2)c | Berndt et al. 201545 | |||
| cycloheptene | 5.9 ± 0.1 | Berndt et al. 201545 | ||
| cyclooctene | 5.9 ± 0.1 | Berndt et al. 201545 | ||
| α-pinene | 7.0 ± 3.5 | Ehn et al. 20145 | ||
| 3.4 ± 1.0 | 3.4 ± 1.7 | 0.44 ± 0.22 | Jokinen et al. 2015100 | |
| 1.2 (+1.2/–0.72) | 1.2 (+1.2/–0.72) | Kirkby et al. 201683 | ||
| limonene | 17.0 ± 8.5 | 5.3 ± 2.7 | 0.93 ± 0.47 | Ehn et al. 20145 |
| Jokinen et al. 2015100 | ||||
| β-pinene | <0.1 | 0.12 ± 0.06 | 0.58 ± 0.29 | Ehn et al. 20145 |
| Jokinen et al. 2015100 | ||||
| myrcene | 0.47 ± 0.29 | 0.14 ± 0.07 | Jokinen et al. 2015100 | |
| β-caryophyllene | 1.7 ± 1.28 | Jokinen et al. 2016134 | ||
| 0.5 ± 0.3 | Richters et al. 2016170 | |||
| 1.8 ± 0.9c | Richters et al. 2016170 | |||
| α-cedrene | 0.6 ± 0.3 | Richters et al. 2016170 | ||
| 0.6 ± 0.3c | ||||
| α-humulene | 1.4 ± 0.7 | Richters et al. 2016170 | ||
| benzene | 0.2d | Molteni et al. 201885 | ||
| toluene | 0.1d | Molteni et al. 201885 | ||
| ethylbenzene | 0.2d | Molteni et al. 201885 | ||
| (o/m/p)-xylene | 1.0–1.7d | Molteni et al. 201885 | ||
| mesitylene | 0.6d | Molteni et al. 201885 | ||
| naphthalene | 1.8d | Molteni et al. 201885 | ||
| biphenyl | 2.5d | Molteni et al. 201885 | ||
Deduced by using an OH scavenger in an ozonolysis experiment.
Determined using CF3O– ionization.
Determined using acetate ionization.
Pure OH experiments. Note that almost all the experiments have been done at room temperature 20 ± 5 °C.
HOM yields are reported either for a certain oxidant (O3 or OH), in which the contribution of each pathway has been determined by applying an OH scavenger in an ozonolysis experiment, i.e., by subtracting the contribution of OH from the total product formation. The total yields have been obtained in ozonolysis studies without OH scavenging. Note that the incorporation of multiple O2 molecules into the relatively light hydrocarbon backbones makes the mass yield of HOM substantial in comparison to molar yields, and Ehn et al. translated a 6% molar yield to an approximate mass yield of 14% in the case of α-pinene ozonolysis.5
5. HOM Properties and Fates in the Atmosphere
5.1. Physical and Chemical Properties
The exact volatility of HOM is difficult to determine because most of the species have not been isolated. Vapor-pressure estimation thus relies on a combination of semiempirical and theoretical methods as well as experimental constraints. The semiempirical methods can be classified as composition–activity, group–contribution, and structure–activity methods. Composition activity means that only the molecular formula (i.e., CxHyOz) is known, and so typical or average functional groups and molecular structures must be assumed. The 2D-Volatility Basis Set (VBS) relies on composition–activity, relationships to estimate typical molecular composition when volatility and oxidation state is known.28 Group–contribution implies that the functional groups are known but the molecular structure is not (or it is unimportant). SIMPOL is a group–contribution method that has been tailored to compounds found in the atmosphere.171 Finally, SAR can include quite sophisticated treatments of intramolecular interactions such as internal hydrogen bonding to produce (presumably) more accurate estimates, provided that a sufficiently training set of similar compounds exists with well-known properties. The Dortmund family of methods rely on separately estimating normal boiling point and the enthalpy of vaporization to provide a general estimation of vapor pressure.172,173
A challenge with atmospheric organic aerosol in general and HOM in particular has been that the composition and structure of molecules comprising them have not been fully characterized. This was a major motivation for selecting the independent parameters of the 2D-VBS; it is possible to measure, e.g., O:C or OSC = 2O:C – H:C using aerosol mass spectrometers that do not isolate individual molecules.174,175 It is also possible to estimate volatility (C*) through a combination of differential loading (build up or dilution of aerosols176,177), heating (in chambers or thermal denuders178,179), isothermal evaporation method,180 and dynamical constraints based on simultaneous observation of gas- and condensed-phase behavior.25 Given known volatility and oxidation extent, it is then possible to estimate the probable chemical structure.
The original composition–activity relation presented with the 2D-VBS was based on the assumption that most condensed-phase molecules consisted of a mixture of carbonyl (=O) and alcohol (−OH) functional groups (with organic acids comprising of one of each) and then drew from SIMPOL as well as vapor-pressure literature to conclude that each carbonyl functional group depressed the volatility of an organic at 300 K by 1 order of magnitude and each alcohol functional group depressed the volatility by 2.4 orders of magnitude. However, the 2D-VBS has only two dimensions, and only one is composition, represented either by O:C or OSc. Further, composition constraints from ambient data often only provide O:C. Consequently, the original 2D-VBS parameters further include the assumption that =O and −OH exist in roughly equal proportions to give an overall reduction in volatility of 1.7 orders of magnitude per added oxygen.28 Additional constraints based on H:C can be incorporated into volatility estimations, but these also require assumptions about the distribution of functional groups and have typically been developed to constrain =O:–OH under the assumption that these functional groups dominate,181,182 where the 2D-VBS assumes that =O:–OH is 1:1.28 With strong evidence that HOM contain many −OOH and −O-O– functional groups, the additional information provided by H:C alone is minimal, without additional information on the functional group distribution. The full 2D-VBS implementation does include some nonlinear effects to describe nonideal interactions between nonpolar reduced compounds and polar oxidized compounds (interactions between C–C, O–O, and C–O) but those are not important for this discussion.28 Computational chemistry also suggests that the relatively flexible −OOH functional groups are prone to internal H-bonding, which can significantly increase volatility,27 and so the volatility estimates for compounds with multiple polar functional groups may be biased low.27,183 Dynamical models of particle growth rates based on similar volatility parametrizations have treated this uncertainty by shifting the overall distributions by one decade in either direction.61
That original VBS composition–activity relationship suggests that HOM with the composition C6H6O6 would have C* at 300 K = 10–3 μg m–3, just at the boundary between LVOC and ELVOC (as they were then defined28). This is why HOM were quickly labeled ELVOC.5 However, as described above, the chemistry responsible for HOM formation involves repeated H atom abstraction by peroxy radical moieties, producing exotic molecules with multiple hydroperoxide functional groups (−OOH). The extra O in the −OOH group is almost completely ineffective at reducing vapor pressure according to grou–contribution methods (both −OH and −OOH reduce volatility by about 2.5 orders of magnitude in SIMPOL),184 and it is possible that the added flexibility of the −OOH groups makes them more able to form internal hydrogen bonds, thus further increasing their volatility.110 Even though the multiple −OOH functional groups presumably render the HOM highly unstable chemically, their volatility is still important for condensation to very small particles, which grow an amount equal to their own diameter in a few minutes under typical ambient conditions (growth rates of 1–10 nm h–1).178
A revised set of 2D-VBS composition activity parameters can account for the increasing presence of −OOH groups with high O:C:
where nC0 = 25, bC = 0.475, bO = 0.2, and bCO = +0.9. nC and nO are the number of carbon and oxygen atoms in the compound. The upward curvature caused by the negative bCO term mimics a transition from organics dominated by −OH and =O at low O:C, consistent with the original 2D-VBS parameters and traditional VOC oxidation chemistry, to HOM dominated by −OOH at high O:C. The presence of organo-nitrates can also complicate vapor-pressure estimation. Their effect can be estimated by assuming that all nitrogens in an observed HOM (aside from any NO3– associated with the chemical ionization via nitrate clustering) are −ONO2 groups and the effect of each group is to lower C* by 2.5 decades.
Figure 10 shows our current best understanding of the HOM location within the 2D-VBS. The definite boundary is the nO = 6 contour, as indicated. The carbon isopleths are for nC = 9,10 and 18,19,20. This is because many of the HOM investigated so far are monomer and dimer products of monoterpene oxidation and so have these carbon numbers. Here it can be seen that the C10 monomer HOM still fall within the LVOC range but at the upper end instead of the lower end implied by the original 2D-VBS composition–activity relation. Only the C18–20 dimers, for the most part, fall in the ELVOC range.
Figure 10.

HOM saturation concentration (C°), average carbon oxidation state (OSC), and average O:C. Solid black lines extending from upper left to lower right are average carbon number (nC). Solid green curves bending from top to lower left are average oxygen number (nO). The broad volatility classes are indicated along the top, with corresponding colors in the figure; the number concentrations of compounds with O:C = 1 for the boundaries between these classes are indicated on the secondary x axis.
Although Figure 10 contains our current best knowledge of HOM volatilities, large differences (often several order of magnitude) still exist between state-of-the-art methods used for vapor pressure estimates of HOM.27 One of the main reasons for this is the ability of hydroperoxide and peroxy acid functionalities to form intramolecular hydrogen bonds, which causes an increase in the saturation vapor pressure which group contribution methods do not account for. The estimates given by the quantum chemical calculations also contain fairly large uncertainties, and in addition, the importance of possible heterogeneous reactions (reactive uptake) remains largely unknown. This remains one key obstacle for assessing the exact role of HOM in the formation of small clusters and particles.
5.2. Removal Mechanisms
5.2.1. Physical Removal
Because of their low or extremely low vapor pressure, HOM are expected to condense nearly irreversibly onto surfaces. We calculated lifetimes (τ = 1/CS; CS = condensation sink) of HOM through their loss rate to particles (i.e., condensation), considering different condensation sinks in order to cover the wide range of atmospheric aerosol concentrations encountered across the world. As shown in Figure 11, condensation is the major removal process currently recognized for the HOM, which is essentially linked to the presence of aerosol (i.e., CS).
Figure 11.
HOM lifetime estimated through their loss to particles (CS) and through their gas-phase oxidation in the presence of OH radicals. OH concentrations (i.e., 1.1 × 106 and 2 × 107 molecules cm–3) and CS (0.005 and 0.03 s–1) used for the estimations are based on previous measurements at Hyytiala186,187 and Beijing,188,189 respectively. A rate constant of 1 × 10–10 cm3 s–1 molecule–1 for the reaction (HOM + OH) was used, which provides an upper limit of the HOM lifetime vs oxidation processes.
5.2.2. Gas-Phase Reactions
No studies are available on the gas-phase oxidation of HOM. Therefore, only a modeling approach can be used to evaluate this potential removal process. Because no rate constants are available in the existing literature, a simple approximation is used to predict the atmospheric lifetime. The presence of multifunctional groups would likely enhance the reactivity with the atmospheric oxidants such as OH or NO3.155 Therefore, rate constants near the collision limit (i.e., 1 × 10–10 cm3 molecule–1 s–1) are likely. Ozone will only rarely participate in any potential gas-phase reactions of HOM due to the absence of double bonds in most HOM and the very slow reactivity of carbonyls with ozone.155 Finally, a wide range of OH radical concentrations (from 1 × 104 to 1 × 108 molecules cm–3) covers possible concentrations encountered across the world. As shown in Figure 11, removal of HOM through gas-phase oxidation processes is negligible in most of the atmosphere, especially considering that high concentration of oxidants is generally associated with SOA formation and thus large CS.
5.2.3. Condensed-Phase Reactions
While the HOM are recognized to condense nearly irreversibly, their fate within the aerosol phase is currently unknown. A large variety of accretion reactions such as oligomerization, hydration, or aldol condensation190 have been reported for other oxygenated species. In addition, it cannot be excluded that HOM containing peroxide and/or peroxy acid groups might act as oxidants and lead to the formation of accretion products through organic redox within the condensed phase. Recently, Mutzel and co-workers propose three fates of condensed HOM: they can (i) remain in the particle phase without a structural change, (ii) undergo fragmentation reactions leading to short-chain carbonyl compounds, and (iii) form larger SOA components.62 It is believed that these processes occur simultaneously, and many HOM will undergo further condensed-phase reactions and impact SOA composition and evolution. As the HOM contain a wide variety of functional groups, analogies with studies that have investigated the reactivity of oxygenated species such as peroxides (e.g., dimers), hydroperoxides, aldehydes, and/or ON can be attempted. However, it should be kept in mind that HOM reactivity may display important additive effects due to interactions between functional groups which are missing from the simpler analogous species and would complexify the situation.
Autoxidation reactions involving RO2 radicals lead to the formation of hydroperoxide groups, and so HOM are anticipated to contain a large number (up to 4–5) of −OOH groups. Hence, understanding the reactivity of such compounds within the aerosol phase is essential to evaluate the fate of HOM. However, a very limited number of studies on the stability and reactivity of the ROOH within the aerosol phase are available in the literature. Krapf et al. (2016) have recently reported that the half-lives of organic peroxides, including (hydro)peroxides, formed from the oxidation of biogenic compounds, are shorter than 1 h in the condensed phase under dark conditions.58 This is in contrast to an earlier work of Epstein et al. (2014) who estimated the lifetime of peroxides as long as 6 days under photolytic conditions.191 While the instability of such compounds is recognized, the quantitative understanding of the stability/reactivity of (hydro)peroxides in a complex matrix, such as SOA, remains challenging due to the lack of proper techniques to identify and monitor the (hydro)peroxides. Evidence of their reactivity has been obtained indirectly by either measuring the decomposition products or the formation of oxidized compounds from particle-phase processes involving the hydroperoxides. For example, Tong et al. (2016) have reported the formation of OH radicals from water extraction of SOA materials, which is proposed to be a result of the decomposition of hydroperoxides.60 Generation of OH radicals within the condensed or liquid phase can contribute to the formation of SOA from the formation of high molecular weight compounds such as oligomers.190 Other potentially important particle-phase processes involve peroxy hemiacetal formation from the reaction of hydroperoxides with aldehydes192 and the formation of organosulfates from the perhydrolysis.62,193,194
HOM monomers and dimers have been reported to also contain nitrate functional groups and are referred to in this review as organonitrates.62 Organonitrates (ON) are mainly formed from the reaction of nitric oxide (NO) with a RO2 radical or through NO3 radical oxidation. Laboratory195 and field studies49 have reported large concentrations of such compounds within the particle phase. Lee et al. (2016) reported that a large fraction of observed organonitrates are highly functionalized, containing from four to 11 oxygen atoms per molecule, and thus many of the organonitrates formed in forested areas could be classified as HOM.49 Using an observationally constrained diurnal zero-dimensional model, they estimated that the organonitrates had a short particle-phase lifetime (i.e., 2–4 h). Those ambient observations are consistent with recent laboratory studies who predicted that ONs are not stable within the particle phase, especially in acidic solution.196,197 Both studies revealed the uncharted role of acidity for the hydrolysis of HOM ONs.
In addition to particle-phase processes, HOM that do not condense near-irreversibly might also react at the gas–particle interface. By analogy to the subsequent reactivity of epoxides and lactones,198,199 certain functional groups undergo fast interfacial processes and lead to the formation of SOA. The presence of such functional groups cannot be ruled out, and certain HOM might undergo reactive uptake. For example, a recent study reported the large reactivity of highly oxidized oligomeric compounds formed from the ozonolysis of isoprene in the presence of acidic aerosol.200 Finally, under pristine environments, highly oxygenated RO2 might undergo further particle-phase processes and lead to the formation of a wide variety of products. For instance, Brüggemann et al. (2017) have recently proposed that such RO2 (e.g., C10H15O8) can react in the condensed phase and/or at the interface, yielding organosulfates (e.g., C10H16O10S).65
5.2.4. Photolysis
HOM contain peroxides, carbonyls, ON, and other photolabile functional groups that absorb light at actinic wavelengths, and it could be expected that HOM might also be degraded through photolysis processes in the condensed phase. Among the products present in SOA, organic peroxides are believed to be susceptible to photolysis,201,202 with lifetimes of about a few days. Additional laboratory studies have reported a pronounced decrease in the concentration of peroxides upon photolysis processes.203 Krapf et al. (2016) have, however, recently proposed that organic peroxides are not the main photolabile functional groups, as it is often proposed in the literature.58 Instead, they argue that carbonyl groups present in the HOM structures are responsible for the rapid photolysis (i.e., time scale of a few hours), explaining the photolytic degradation of α-pinene-derived SOA products such as HOM. This hypothesis is also consistent with recent reported SOA absorption cross-section data and other laboratory studies,204,205 signifying that carbonyls play a major role in the evolution of the SOA chemical composition through photolysis processes.206 Krapf et al. (2016) estimated that HOM monomers formed from the ozonolysis of α-pinene contain an average of two carbonyl functional groups and proposed that carbonyl species present in α-pinene-derived SOA have a photolysis rate of 0.75 × 10–4 s–1, suggesting that lifetimes of such species would be several hours under typical daytime conditions within the aerosol phase.58 Although more work is clearly needed to confirm such large reactivity of HOM upon irradiation, this study indicates that photolysis might be an important removal pathway of HOM within the particle phase.
6. HOM Atmospheric Observations and Impact
The majority of HOM studies have taken place in the laboratory, but several ambient observations have also been reported, as already discussed earlier. In this section, we summarize the atmospheric studies on HOM in both gas and aerosol phase (section 6.1), followed by discussion (section 6.2) on the impacts of HOM on aerosol formation in the atmosphere.
6.1. Ambient HOM Observation
6.1.1. Gas Phase
The first ambient observations of HOM were made at the SMEAR II boreal forest station in Finland29 using an APi-TOF to measure ambient ions, and subsequent studies have reported similar findings.7,132 Later studies of neutral HOM, often in monoterpene-influenced regions, using a CI-APi-TOF, have allowed the quantification of these compounds. Observations at rural background stations in Europe5,42,46,62,81,132,133,ref241 illustrate that the most abundant HOM species (including RO2, C10 monomers, C10 organonitrates, C20 dimers) can reach maximum concentrations of ∼107 molecules cm–3 with total HOM concentrations of 108 molecules cm–3, which is 1–2 orders of magnitude higher than typical sulfuric acid concentrations in Europe. The observed concentrations, and the presumably low volatility of HOM, led to the conclusions that HOM condensation may explain a large fraction of the growth of particles between 5 and 50 nm in diameter at SMEAR II.5
Several studies employing CI-APi-TOF have reported diurnal patterns of HOM.5,42,46,62,81,132,133,212 Generally, the dominating peaks in the CI-APi-TOF mass spectrum are attributed to C10 closed-shell monomers. While the total closed-shell C10 HOM monomer concentration shows a relatively weak diurnal pattern, the diurnal trends of many of the individual C10 HOM species are substantial. This is from the result of variation in both the formation and termination pathways of RO2 radicals, and potential impact from micro-meteorology.ref241 Organonitrate concentrations (main species C10H15O8N (m/Q 339 Th) and C10H15O9N (m/Q 355 Th)) reach their maximum concentrations during daytime, with substantially lower concentrations during nighttime, indicating that the main organonitrate source is via termination reactions between RO2 and NO. Lower concentrations of organonitrates are also observed during the night, likely originating from NO3 oxidation of monoterpenes.133 In contrast, the C18–C20 HOM dimers and most HOM RO2 concentrations reach their maximum concentrations during the night as NO decreases the lifetime of RO2 and prevents formation of HOM dimers during the daytime. Some specific RO2 (e.g., C10H17O9) reach maximum concentrations during daytime, illustrating that OH oxidation of monoterpenes also are an important source for HOM, which also was confirmed by laboratory experiments.46 Yan et al. (2016) found that HOM could be separated into nocturnal (ozone and NO3 radical oxidation) and daytime (ozone and OH oxidation, NO termination) groups using positive matrix factorization (PMF).133 The diurnal behavior of the total gas-phase HOM concentration shows two maxima, one around noon and another during late evening (Figure 12).133,207
Figure 12.

Diel profiles of six source categories of HOM measured at the SMEAR II station during the spring 2012.133 This figure demonstrates how formation mechanism and oxidation conditions affect the concentrations of HOM. These six categories were separated from ambient data using positive matrix factorization. The table on the right depicts the “fingerprint” molecules of each group. Adapted with permission from ref (133). Copyright 2016 Copernicus Publications.
Compounds with molecular formulas corresponding to HOM dimers observed using an iodide ToF-CIMS were studied in detail at the SMEAR II-station by Mohr et al. (2017).53 They found concentrations of ∼106–107 molecules cm–3 of dimeric monoterpene oxidation products (C16–20HyO6–9) in the gas phase and significant signals of corresponding ions in the condensed phase. Other higher m/Q HOM (C15), similar to those found from β-caryophyllene ozonolysis experiments, have also been identified at the SMEAR II station.134 Three different potential sesquiterpene HOM (C15H24O9, C15H22O9, and C15H24O7) were detected both in the naturally charged ions and neutral measurements. The neutral concentrations barely reached above detection limit, 4 × 104 molecules cm–3, with a maximum of ∼105 molecules cm–3.
Beside forested rural background locations, observations of HOM have also been made in an industrial area in Finland,208 high altitude stations,2,209 and using a ToF-CIMS in a (semi)polluted location in Brent, AL, USA.49,210,211 One study utilized an iodide ToF-CIMS in an aircraft to obtain HOM measurements over the southeastern USA.50
Observations from the USA concentrate on isoprene derived HOM during the Southern Oxidant and Aerosol Study (SOAS) during the summer of 2013. Brophy and Farmer (2015) reported observations mainly of carboxylic acids but also a possible product of autoxidation involving biogenic peroxy radicals (C9H14O5) and six oxidation products of isoprene with elemental composition of C5H10On (n=3–6). The most detailed investigation of isoprene derived products of autoxidation involving peroxy radicals from the SOAS campaign was published by Krechmer et al. (2015). They observed 14 different product isomers generated by autoxidation involving isoprene-derived peroxy radicals (C5H10O5–6 and C5H12O5–6) from a chamber experiment and from ambient air using nitrate ion chemical ionization. Several of the isomers showed a strong diurnal patterns following solar radiation, with concentrations varying between <0.1 and 0.5 pptv (Figure 13).211
Figure 13.

Diurnal cycles of ambient measurements during the SOAS field study from a 10-day period beginning 22 June 2013: (a) key LVOC identified in the laboratory, and (b) OH and ISOPOOH. Also shown in (a) is the LVOC production rate (k[ISOPOOH][OH]) multiplied by a yield of ∼1%. All of the diurnal cycles peak around 13:00 CDT when solar radiation and OH are at peak levels. (c) ISOPOOH SOA estimated with factor analysis of ambient AMS data and ambient IEPOX. Adapted with permission from ref (211). Copyright 2015 American Chemical Society.
Two publications show evidence of HOM in the free troposphere during some specific sunny days.209,212 Negative naturally charged ions and neutral HOM were observed at the Jungfraujoch, Switzerland, site of the International Foundation High Altitude Research Stations. Three most abundant peaks observed in both ionic and neutral forms were associated with C8H10O11NO3–, C9H12O11NO3–, and C10H14O11NO3–.212 Even though the chemical formulas of these HOM differ from those of HOM generated by monoterpene oxidation, both span a similar mass range and O:C. The first concentrations published from a high-altitude station are <3 × 106 molecules cm–3 for neutral HOM. A complete list of observed ambient ions and neutral HOM measured at Jungfraujoch station are listed in the Bianchi et al. (2016) Supporting Information.212 Frege et al. (2017) also reported HOM measured over a longer period from the same station. They observed naturally charged ions both in positive and negative ion mode and found oxygenated organic compounds both during the day and night. Positive ions were mostly nitrogen containing, and it was ambiguous whether the spectra contained HOM or amines. However, this may be the first ambient observation of positively charged HOM in the ambient air.209
Table 4 lists studies reporting ambient observations of HOM in the gas-phase or in SOA, and which measurement technique that was employed.
Table 4. List of Ambient Observations of HOM in the Gas-Phase and in SOA.
| location | techniquea | reagent ion: | gas | SOA | neutral | ion | ref |
|---|---|---|---|---|---|---|---|
| SMEAR II, Finland | CI-APi-TOF | NO3– | X | X | Kulmala et al. 2013213 | ||
| Zhao et al. 20138 | |||||||
| Ehn et al. 20145 | |||||||
| Yan et al. 2014133 | |||||||
| Jokinen et al. 201481 | |||||||
| Berndt et al. 201646 | |||||||
| Jokinen et al. 2016134 | |||||||
| Jokinen et al. 2017207 | |||||||
| Bianchi et al. 2017132 | |||||||
| APi-TOF (−) | X | X | Ehn et al. 201029 | ||||
| Ehn et al. 20127 | |||||||
| Lehtipalo et al. 2011214 | |||||||
| Jokinen et al. 2016134 | |||||||
| Bianchi et al. 2017132 | |||||||
| FIGAERO | I– | X | X | X | Mohr et al. 201753 | ||
| Porvoo, Finland | CI-APi-TOF | NO3– | X | X | Sarnela et al. 2015208 | ||
| Michelstadt–Vielbrunn/Odenwald, Germany | CI-APi-TOF | NO3– | X | X | Kürten et al. 201642 | ||
| Melpitz, Germany | HPLC-ESI-TOFMS | X | X | Mutzel et al. 201562 | |||
| CI-APi-TOF | NO3– | X | X | Jokinen et al. 201481 | |||
| CI-APi-TOF | NO3– | X | X | ||||
| Jungfraujoch, Switzerland | CI-APi-TOF | NO3– | X | Bianchi et al. 2016212 | |||
| APi-TOF (−) | X | X | Bianchi et al. 2016212 | ||||
| APi-TOF (+) | Frege et al. 2017209 | ||||||
| Brent, Alabama, USA | CIMS | NO3– | X | X | Krechmer et al. 2015211 | ||
| I– | X | X | Brophy and Farmer 2015210 | ||||
| FIGAERO | I– | X | X | X | Lee et al. 201649 | ||
| flights over GA, TN, AR, MO | CIMS | I– | X | X | Lee et al. 201450 | ||
We report the name of the technique that was mentioned in the publication.
6.1.2. Particle Phase
The fate of the HOM after condensing onto the particulate phase is not well characterized. While ambient observations of HOM in the gas phase have been reported, studies characterizing their presence in the condensed phase remain sparse due to the analytical challenges involved in sampling and analyzing such compounds (section 3). While many observations of highly functionalized compounds in SOA sampled in field studies have been reported over the past decade, many of these compounds are not proposed to form via the chemistry discussed in this review (i.e., autoxidation) and are not referred to as HOM.
The presence of HOM in the particle phase was first suggested by Mutzel et al. (2015) with measurements done in central Europe.62 Using off-line analysis, the authors identified seven compounds attributed to HOM. Because of the lack of standards, however, the authors were not able to quantify these products. Using a similar analytical technique, Brüggeman et al. (2017) reported the presence of HOM in a similar environment.65 Off-line methods suffer from low temporal resolution, hindering the characterization of the fundamental physicochemical processes involving HOM. Taking advantage of the FIGAERO–CIMS, a few studies have been able to monitor the evolution of molecules with elemental composition that can be classified as HOM in aerosol sampled in the USA49,68 and Finland.53 Moreover, the FIGAERO–CIMS was able to confirm the observed temperature dependence of HOM participating in NPF during chamber experiments.61 Overall, no published studies have provided a complete description or quantification of the HOM in aerosols sampled in both ambient and laboratory environments.
6.2. Atmospheric Impact
6.2.1. SOA Formation
Very few studies have specifically investigated the contribution of HOM to atmospheric SOA formation, and in particular their contribution to the growth of new particles. In measurements conducted in the boreal forest, estimated HOM concentrations were found to be in the range needed to explain growth of 5–50 nm particles.5 However, this assumes that the measured HOM condense irreversibly, that the measured HOM concentrations are representative of the entire planetary boundary layer (PBL), and that no particle-phase reactions occur (infinite HOM lifetime). Jokinen et al. (2015) implemented experimental results of HOM yields from BVOCs into a global model framework and estimated the impact of HOM on atmospheric cloud condensation nuclei (CCN) formation.100 In that study, HOM were treated as effectively nonvolatile and contributed together with sulfuric acid to the growth of new particles, starting at 1 nm in diameter. The simulations indicated that the SOA formation from HOM increased the number of cloud condensation nuclei (CCN), especially at high water vapor supersaturations. They also showed that HOM primarily contribute to the number of CCN in clean remote regions with low levels of anthropogenic primary particle emissions and where new particle formation has a substantial contribution to the number of CCN.100
A few studies have used atmospheric chemistry transport models in combination with atmospheric aerosol observations to evaluate the contribution of HOM (ELVOC) from monoterpenes to the OA mass and initial particle growth. In the western Mediterranean region, HOM (ELVOC) has been estimated to contribute to 10% and 15% of the PM1 OA mass during the summers 2012 and 2013, respectively.215 At the SORPES station in Nanjing, China, simulations find that HOM (ELVOC) from monoterpenes can explain ∼30% of the observed particle growth below 10 nm in diameter when the air masses are originating over forest regions in South China.216
Recently, Öström et al. (2017) published the first study where a detailed autoxidation mechanism involving peroxy radicals and leading to HOM formation from monoterpenes was implemented and used in the process-based aerosol-chemistry transport model ADCHEM.185 The HOM formation mechanism, which was coupled to the Master Chemical Mechanism (MCMv3.3.1)166,217,218 was based on previous experiment and theoretical results on HOM formation with absolute HOM yields similar to those reported by Ehn et al. (2014). According to Öström et al. (2017), the growth of new particles from 1.5 nm into the 50 nm in diameter size range is dominated by condensation of HOM formed from monoterpenes oxidized by ozone and OH.185 However, the modeled particle growth is relatively sensitive to the method used to estimate the HOM pure-liquid saturation vapor pressures (p0). Even using the functional group contribution method SIMPOL,184 which generally predicts orders-of-magnitude lower HOM p0 than state-of-the-art quantum chemical calculations,27 most HOM that were simulated to contribute to the SOA formation, are not classified as ELVOC (Figure 14).
Figure 14.

Results from Öström et al. (2017) illustrating the volatility distribution of the modeled HOM and other organic molecules formed from BVOC oxidation at the subarctic forest field station Pallas in Finland.185 The organic molecules in the particle phase (p) have been divided into HOM monomers without nitrate functional groups (HOM monomers), HOM dimers, organonitrates (ON), and other organic molecules from MCM that are LVOC and SVOC. The C* of the HOM species were estimated with SIMPOL.184 The volatility distribution represents the average results from simulations of 10 individual new particle formation and growth events covering in total 17 days at the Pallas field station. The organic molecule volatility distribution was generated by distributing the modeled gas- and particle-phase mass concentrations of each organic molecule species (in total more than 700 species) into discrete logarithmically spaced volatility bins according to their individual C* at 298 K. However, the actual fractions of the organic compounds found in the gas and particle phase do not represent their expected equilibrium gas–particle partitioning at 298 K but instead how the organic molecules actually were distributed between the gas and particle phase in the aerosol dynamics model at the ambient conditions at Pallas (e.g., temperature in the range of 265–295 K). Adapted with permission from ref (185). Copyright 2017 Copernicus Publications..
Although ground- (or near-ground-) based observations of HOM in the gas phase exist (see section 6.1.1), it is not straightforward to quantify the sources and sinks of HOM in the atmosphere. The observed HOM concentrations at the ground or above the canopy depend on at least the following variables: (i) their formation rate, which depends on the concentration of BVOCs, NO, HO2, O3, and OH, (ii) the rate of vertical mixing in the PBL, (iii) the volatility of the HOM (and thus their condensation loss from the gas phase), and (iv) the dry deposition rates of HOM. The later are likely substantial because of the low volatility, presumably high reactivity, and water solubility of HOM. By assuming that all HOM are nonvolatile, the particle growth rates and condensation sink of HOM can be estimated at the location of the measurement site, but this does not necessarily represent the average particle growth rates in the entire planetary boundary layer. Second, if many HOM are LVOC (or even SVOC) and not ELVOC, their lifetime in the gas phase may be longer and their formation rates and contribution to particle growth will be overestimated. Thus, to constrain the contribution of HOM to the growth of particles in the atmosphere, methods are needed that combine both observations and models that can provide detailed information about the gas-phase chemistry, HOM volatility (i.e., p0 and reactivity in the condensed phase), aerosol dynamics, dry deposition losses, and the planetary boundary layer mixing. To our knowledge, there exist no published studies that provide a complete closure of the modeled and observed HOM(g), the particle composition, and growth rates.
6.2.2. New Particle Formation
For several years, it has been speculated that organic compounds could contribute to the early growth of atmospheric new particles. Kulmala et al., (1998) predicted that very low-volatile organic vapors, with saturation vapor concentrations less than 105 molecules per cubic centimeter (C* < 4 × 10–5 μg m–3), must have a dominant role in the early growth of new particles over the boreal forest.219 Since then, many studies have addressed the influence of organic compounds on the formation and growth of new particles.220−224 However, before the discovery of HOM, the properties and formation pathways of these molecules were not known.225
The recent experiments conducted in CERN at the CLOUD chamber proved that HOM are able to cluster efficiently with sulfuric acid and form new particles.90,226 These experiments have also shown that new particle formation rates derived from these clustering processes may explain the nucleation observed in the ambient atmosphere, specifically over the boreal forest.90,226 Sulfuric acid–HOM clusters have been observed in the boreal forest.132 However, until very recently, it was unclear if HOM can form clusters without sulfuric acid at saturation ratios typical of ambient conditions. Recent laboratory experiments performed in the CLOUD chamber at CERN show that HOM alone can form new particles at atmospheric conditions.83 The authors speculated that new particle formation and growth by HOM could have been responsible for a large fraction of the CCN during the preindustrial era.227,228 However, the importance of pure HOM nucleation in the current-day atmosphere is still unclear, especially if other nucleation agents are present.
Quantum chemical calculations indicate that strong intramolecular hydrogen bonds in HOM containing multiple hydroperoxide- and peroxyacid groups weakens the intermolecular interactions between HOM molecules, and also HOM and sulfuric acid.27,229,230 This implies that most HOM may be more volatile than indicated by state-of-the-art functional group contribution methods27 and less prone to form new clusters. Indeed, both experimental and theoretical studies also indicate that C10 monomer HOM, as shown in Figure 10, are too volatile to participate in nucleation (or even growth below 2 nm).25 Further, positively charged clusters during α-pinene ozonolysis experiments in the CLOUD chamber contain only even multiples of 10 carbons, starting with C20 dimers, followed by C40, C60, etc.164 It is only at very low temperatures (−25 °C) that C30 clusters appear in the APi-TOF spectra.164
Thus, it appears that only the C20 HOM dimers from α-pinene oxidation can be involved in purely biogenic nucleation. Even in that case, the ELVOC HOM dimers do not appear to nucleate at the collision frequency (i.e., it is not kinetic nucleation). Chuang et al. (2017) estimated that dimers with C* ≤ 10–8 μg m–3 nucleate with a probability of only 10–4 at 300 K. However, because the dimers span a wide range of volatility and thus saturation ratios, a more nuanced view is likely that the collisional efficiency for nucleation ranges from unity (kinetic nucleation) for dimers below some threshold volatility and then taper off as volatility increases (and thus cluster evaporation lifetimes decrease).231 This is also consistent with the strong observed ion enhancement in pure biogenic nucleation by almost a factor of 100 for [HOM] ≤ 107 cm–3 reported in Kirby et al.83 This can only occur if the corresponding neutral clusters are (on average) relatively inefficient at new particle formation.
Ambient observations also suggest that HOM with low vapor pressure (ELVOC) are involved in NPF. The first indication that HOM contribution to atmospheric nucleation was found by Kulmala et al. (2013) who detected positive correlation with certain HOM and the 1.5–2 nm cluster concentration during active aerosol formation.213 The first direct ambient evidence that HOM could nucleate by themselves (or at negligible sulfuric acid concentration) was found in the free troposphere at the Jungfraujoch station.212
Despite the large amount of VOC that are present/emitted in the boreal forest,232 pure HOM nucleation has never been observed during the daytime. This can be explained by the ubiquity of sulfuric acid at relatively high concentrations, which favors formation of HOM–H2SO4 clusters132 and excludes pure organic nucleation.90,226 However, during the night when the surface HOM dimer concentration are highest,133 a large number of clusters formed almost exclusively by HOM is frequently observed.132,214,233 The clusters are followed by formation of nocturnal particles/ions that generally do not grow to sizes larger than a few nm in diameter (at times reaching ∼6 nm).214,233,234 Additionally, during some of those nights, up to 40 carbon atom HOM clusters (four α-pinene oxidized units) were identified in the ambient air.132 This may indicate that pure organic nucleation is occurring but that the overall pool of LVOC HOM monomers is insufficient to drive subsequent growth of the particles.
Generally, it can be concluded that HOM have an important role during continental planetary boundary layer NPF events, during the cluster formation (nucleation) and/or the early particle growth (sub-2–10 nm diameter size range).
7. Summary and Perspectives
This review summarizes the current state of knowledge on highly oxygenated organic molecules (HOM), a class of organic compounds formed by gas-phase autoxidation involving peroxy radicals in the atmosphere. Their initial discovery was made possible through improved detection techniques over the past decade, and together with the indispensable support from quantum chemical calculations, mechanistic insights into their formation pathways have been achieved. In parallel, deeper understanding of the significant impacts of HOM on the formation of secondary organic aerosol and new particles have been realized. However, despite the general understanding that has been attained, key details of many important aspects of HOM formation and properties remain elusive. In a recent perspective, Ehn et al. (2017) listed several challenges for the chemical kinetics community pertaining to HOM.235 Below, we expand on these and describe some of the most urgent needs for continued fast progress within the exciting field of HOM research.
While autoxidation is known to be critical in HOM formation, there is a lack of direct measurements of individual H-shift rates, not to mention the temperature dependence of these (which is likely to be highly variable over the range of typical temperatures in the atmosphere). Such measurements would greatly facilitate computational validation, but they are challenging due to the short time scales involved in many of the isomerization steps. More generally, HOM quantification is, as discussed in section 3, still very uncertain, relying on sensitivity estimates from either proxy calibrants such as H2SO4, or kinetic limitations such as maximum collision rates. Simpler and more robust calibration techniques would be of great value. Equally important as the quantification is the structure elucidation of HOM. However, current mass spectrometric techniques only yield information about the HOM molecular formulas, which severely hampers the development of detailed formation mechanisms for many atmospherically relevant systems. Thus, there is a need for new analytical techniques and innovative experimental setups to allow fast- and detailed characterization of HOM.
Certain systems, such as the ozonolysis of α-pinene, have been studied extensively, yet an unambiguous mechanistic understanding of HOM formation is still lacking. At the same time, many other relevant precursor molecules have also been probed, and a large fraction of these have shown considerable HOM yields with one or more atmospheric oxidants. To our knowledge, no targeted laboratory studies have been carried out with other oxidants than ozone and OH radicals, although NO3 radicals have been shown to be important in the night-time formation of HOM in the boreal forest. HOM formation has been observed from isoprene, monoterpenes, sesquiterpenes, and aromatics, and one can expect that other compound groups also may be found to have surprisingly high HOM yields. We therefore encourage future work to determine HOM yields using different oxidants, different VOC, and in different combinations of these.
As with many aspects of atmospheric chemistry, HOM are also greatly influenced by NOx. The role of NO and NO2 in HOM formation needs further investigation from a mechanistic point of view. NO radicals to large extent control the fate of peroxy radicals: at high concentrations, NO + RO2 reactions may outcompete autoxidation propagation reactions, while at low concentrations, NO can change the HOM product distribution by decreasing HOM dimer formation from RO2 + RO2 reactions. HOM dimer formation, which itself is a process that has eluded a conclusive theoretical explanation (see section 4.2.1), has been linked to formation of new particles, and therefore a detailed description of this branching ratio is critical from a climatic perspective.
For understanding the ultimate atmospheric impacts of HOM, not only their concentrations need to be known but also their physicochemical properties such as volatility and reactivity. For example, their fate in the condensed phase remains unresolved, as are their interactions with other gas-phase species associated with new-particle formation, i.e., sulfuric acid and bases such as ammonia and amines.
Finally, as mentioned in section 6, a more complete network of atmospheric HOM observations are needed in order to understand the sources and importance of HOM in different environments. For example, the Amazon should be a very large source of HOM formed from isoprene and monoterpenes, yet no studies including HOM observations from this region exist. Ideally, HOM measurements should be carried out at different sites as well as at different altitudes in order to understand the vertical distribution. This would help resolve the impact of low temperatures on HOM formation and HOM clustering and contribution to new particle formation. We now know that HOM can contribute to formation of new particles without the presence of sulfuric acid, but it is important to better quantify where and when these mechanisms are (or were) important for atmospheric aerosol formation. This will help us to better understand preindustrial aerosol formation before the presence of anthropogenic SO2 emissions contributed to atmospheric new particle formation.
Acknowledgments
We thank Ugo Molteni, Liine Heikkinen, Rasmus V. Otkjær, and Kristian H. Møller for useful discussion. We thank the CSC Centre for Scientific Computing in Finland and the Danish Center for Scientific Computing from the Copenhagen University. We also thank the Center for Exploitation of Solar Energy founded by the University of Copenhagen, the Swedish Strategic Research Program MERGE, and the European Regional Development Fund (project MOBTT42). This research has received funding from the Swiss National Science Foundation (P2EZP2_168787), the Academy of Finland (grant no. 1315203, 266388 and 299574), the U.S. National Science Foundation (grant nos. AGS 1801897, ACS 1508526), the U.S. Department of Energy’s Office of Science (grant no. DE-SC0018221), and the European Research Council starting grant COALA (grant no. 638703).
Biographies
Federico Bianchi, born in Bergamo, Italy, in 1984, graduated in chemistry from University of Milan. After working for four years as a Marie Curie student at the CLOUD project in CERN and at the Paul Scherrer Institute, he received his Ph.D. in atmospheric chemistry from the Eidgenössische Technische Hochschule (ETH) Zürich (2014). In 2015, he took his postdoc at the University of Helsinki, and in 2016 he was awarded with the Postdoc.Mobility fellowships given by the Swiss National Science Foundation. In 2017, he received the Arne Richter Award for Outstanding Early Career Scientists given by the European Geosciences Union. After being awarded with an Academy of Finland Postdoctoral Researchers, he was appointed as Assistant Professor on atmosphere and cryosphere interactions at the University of Helsinki. His research interests are the formation of new particles in several environments, from pristine free troposphere to polluted megacities. Currently, his group is focusing on understanding preindustrial atmosphere and the influence of biogenic highly oxygenated organic molecules on aerosol formation.
Theo Kurtén, born in Helsinki, Finland, in 1980, graduated in physical chemistry from the University of Helsinki in 2005 and received his Ph.D. in physics from the same institution in 2007. His supervisors were Professors Hanna Vehkamäki and Markku Kulmala. After postdocs in Clermont-Ferrand (France) and Copenhagen (Denmark), he returned to Helsinki as a University Lecturer in Chemistry in 2012 and received an Academy of Finland Research Fellowship in 2013. He is a computational chemist studying reactive sulfur, nitrogen, and carbon compounds in the atmosphere, with a focus on the formation, degradation, and clustering of condensing vapors. His group specializes in discovering and modeling complex reaction mechanisms, predicting thermodynamical properties of atmospheric oxidation products, and modelling the chemical ionisation of both reactive intermediates and products.
Matthieu Riva received his diploma in environmental and analytical chemistries from the University of Bordeaux in 2013. After doing his postdoctoral research in atmospheric chemistry at the University of North Carolina at Chapel Hill in the Department of Environmental Sciences and Engineering, USA, and at the University of Helsinki, Finland, he then joined the Centre National de la Recherche Scientifique (CNRS) as senior researcher in 2018 at the Institute of Research on Catalysis and Environment at Lyon, France. His research aims at a better understanding and characterization of heterogeneous processes in the troposphere. He has a wide experience with the detailed chemical characterization of gas- and particle-phase chemistries using online and offline mass spectrometric techniques.
Claudia Mohr received her Ph.D. in environmental sciences in 2011 from ETH Zurich/Paul Scherrer Institute, Switzerland. After postdoctoral research positions at the University of Washington, Seattle, Washington, USA, and at the Karlsruhe Institute of Technology, Germany, she joined the faculty of the Department of Environmental Science and Analytical Chemistry (ACES) at Stockholm University, Sweden, as an Assistant Professor in 2017. In the same year, she was selected as Wallenberg Academy Fellow. The characterization of the chemical composition of organic aerosol particles and organic trace gases by means of advanced mass spectrometric techniques, and the investigation of their sources, formation, and transformation processes, and fate in the atmosphere in the field and laboratory, lie at the heart of her research.
Matti P. Rissanen is an Assistant Professor of experimental aerosol science at the Tampere University (TU), Finland, since January 2019. He completed his undergraduate studies in chemistry at the University of Helsinki with honors, where he later also received his Ph.D. in physical chemistry with the highest possible grade, Laudatur. Since then, he has been a Finnish Academy funded Post-Doctoral Researcher at the Division of Atmospheric Sciences at the Physics Department, also at the University of Helsinki, with research focus on gas-phase reactions creating low volatile material that serves as a substrate for atmospheric new particle formation. Rissanen has also remained active with his previous post in the gas kinetics laboratory and is still frequently publishing papers on chemical kinetics mostly relevant to low temperature combustion and atmospheric chemistry.
Pontus Roldin, born in Sweden, in 1983, graduated in environmental engineering at Lund University in 2007 and received his Ph.D. in physics at Lund University in 2013. He spent three years as postdoctoral researcher in the Atmosphere Modelling Group led by Dr Michael Boy at the University of Helsinki (2014–2016). Since 2017, he is working as a senior scientist in the aerosol research group at the division of Nuclear Physics at Lund University. His main interests are aerosol dynamics, gas-phase chemistry, and secondary aerosol formation.
Torsten Berndt received his Ph.D. in physical chemistry from the Technical University of Leuna–Merseburg under the supervision of Professor Klaus Scherzer (1992). He is a research chemist at the Leibniz Institute for Tropospheric Research in Leipzig. His work is focused on laboratory studies in the field of gas-phase oxidation of organic compounds, including kinetics and chemical mechanisms. Of particular interest are the reactions of Criegee intermediates and RO2 radicals.
John D. Crounse received his B.S. and Ph.D. in chemistry from Andrews University and the California Institute of Technology (Caltech), respectively. He is currently a senior staff scientist at Caltech. His research interests include the atmospheric photooxidation mechanisms of organic compounds and how these transformations impact the composition of the atmosphere.
Paul O. Wennberg is the R. Stanton Avery Professor of Atmospheric Chemistry and Environmental Science and Engineering at the California Institute of Technology, where he also directs The Linde Center for Global Environmental Science. He received a B.A. and Ph.D. in chemistry from Oberlin College and Harvard University, respectively. His research interests include atmospheric photochemistry and global carbon cycle science.
Thomas F. Mentel, born in Giessen, Germany, in 1957, graduated in chemistry (1984) and received his Ph.D. in physical chemistry (1988) from Philipps University Marburg under the supervision of Professor W. A. P. Luck. He was a postdoc at the Department of Chemistry, Princeton University, with Professor G. Scoles (1989–1991). Since 1991, he is affiliated with Institute of Tropospheric Chemistry (now IEK-8) at the Research Center Jülich, where he is leading the Heterogeneous Reactions Group (since 2001) and the Aerosol Section (since 2006). Since 2015, he is appointed as Adjunct Professor for Aerosol Chemistry at the Department of Chemistry and Molecular Biology, Göteborg University. His current research interests are mechanisms of VOC oxidation and secondary organic aerosol formation, microphysics and chemistry of aerosols, and mass spectrometric techniques in atmospheric applications.
Jürgen Wildt, born 1950 in Fischenich, Germany, graduated in physics (1977) and received his Ph.D. in physical chemistry (1979) from Rheinische Friedrich Wilhelms University Bonn under the supervision of Professor Dr. E. H. Fink. He was postdoc at the Bergische Universität Wuppertal (1979–1990) and made his postdoctoral lecture qualification in 1989. From 1990 to 2016, he was affiliated at Research Centre Jülich (1990–2001, Institute for Polluted Atmosphere, and 2001–2016, Institute Phytosphere). With his group, he developed and built the Jülich Plant Atmosphere Chamber to study the mechanisms of trace gas exchange between plants and atmosphere. Since 2016, he is retired and working as a consultant at IEK-8, Research Center Jülich. His current research interest is the conjunction between radical chemistry and particle formation.
Heikki Junninen, Senior Research Fellow, Ph.D., has a long history in atmospheric sciences. He graduated in environmental chemistry from the University of Kuopio (2001) and received his Ph.D. in physics at the University of Helsinki (2014). He has expertise on aerosol and mass spectrometric instrumentation, software development, and statistical modelling. He is leading the development of software package for mass spectrometry data analysis and data management software for environmental stations. He was awarded by Estonian Research Council with a Top Scientist Grant on “Chemical composition and interactions in atmosphere: from gases to aerosols and climate change” (2017), now acting as a Senior Research Fellow and a group leader at the University of Tartu, Institute of Physics. He has educated three Master and two Bachelor students and is currently supervising three Doctoral students and one Masters student. He has published 133 peer reviewed papers (Nature 6, Science 4), h-factor 43, and has 6534 citations. He is highly cited scientist (Clarivate Analytics, 2018) and owns six patents and three nonpatented invention reports.
Tuija Jokinen, born in Nokia, Finland, in 1982, graduated in analytical chemistry from University of Helsinki in 2008 and after two sabbatical years started to work on her Ph.D. at the Division of Atmospheric Sciences under the supervision of Associate Professor Mikko Sipilä and Professor Markku Kulmala. After participating several intensive field campaigns during her Ph.D. studies, she visited the Chemistry Department at TROPOS for 6 months as a visiting researcher under the supervision of Dr. Torsten Berndt to conduct laboratory experiments. She received her Ph.D. in physics from the University of Helsinki in 2015 and received the prestigious University of Helsinki dissertation award for her doctoral dissertation. She immediately started her Postdoc in the United States of America at the University of California in the ultrafine aesorol group led by Professor James N. Smith. After a year, she returned to University of Helsinki and started working as a research coordinator for ACTRIS. Her current tasks concentrate in establishing calibration centers for sub 10 nm particles and aerosol precursor molecules and clusters. She is still a passionate mass spectrometer experimentalist, and her research interests are the interactions of biosphere and atmosphere in the polar regions of the planet.
Markku Kulmala, Academy Professor, directs the Institute for Atmospheric and Earth System Research (INAR) and has served as Professor at the University of Helsinki since 1996. Kulmala also acts as coordinator for the Centre of Excellence, appointed by the Academy of Finland the first time in 2002 and the Digital Belt and Road Program International Center of Excellence at the University of Helsinki. Previously, he has directed the Nordic Center of Excellence, appointed by Nordforsk (CRAICC), which is the largest joint Nordic research and innovation initiative to date, aiming to strengthen research and innovation regarding climate change issues in the Nordic and high-latitude regions. He has received several international awards such as the Smoluchovski Award (1997), the International Aerosol Fellow Award (2004), the Wilhelm Bjerkenes medals (2007), Fuchs Memorial Award (2010), Litke Gold Medal of the Russian Geographical Society (2015), the honorary title of Academician of Science (Finland and China), Wihuri International Prize 2017, Distinguished Visiting Fellow (IIASA), and World Academy of Sciences fellow.
Douglas R. Worsnop is Vice President of Aerodyne Research and FiDiPro Professor in the Institute of Atmospheric Research (INAR, Physics) at the University of Helsinki. His research focuses on chemical kinetics and aerosol chemistry, specializing in laboratory and field studies of the interactions of gases and aerosol particles using applied mass spectrometry. He received a B.A. in Chemistry at Hope College (1974) and a Ph.D. in Chemistry at Harvard University (1982), applying molecular beam reactive scattering under Professor Dudley Herschbach. As a Humboldt Fellow in Physics at the University of Freiburg (1982–1985), he synthesized solvated electron clusters in molecular beams in the group of Professor Helmut Haberland. A Fellow of AAAS, AGU, and AAAR, he is winner of the Benjamin Liu Prize for Aerosol Instrumentation (AAAR), the Yoram Kaufmann Award for Unselfish Cooperation in Research (AGU), the Fuchs Memorial Award (IARA) for Outstanding Research in Aerosol Science, and the American Chemical Society (ACS) Award for Creativity in Environmental Science.
Joel A. Thornton was born in St. Johnsbury, VT, graduated from Dartmouth College (Chemistry) in 1996, and received his Ph.D. (chemistry) from the University of California, Berkeley, working with Professor Ronald Cohen. After a two-year postdoctoral fellowship at the University of Toronto with Professor Jonathan P. D. Abbatt (Chemistry), he joined the faculty in the Department of Atmospheric Sciences at the University of Washington in 2004. His current research interests include the transformations of reactive organic molecules and oxides of nitrogen and their influence on the air quality and climate impacts of atmospheric aerosol particles.
Neil Donahue was born in Pittsburgh Pennsylvania, USA, in 1963 and received an AB in Physics from Brown University in 1985 as well as a Ph.D. in Meteorology from MIT in 1991. He spent the rest on the 1990s as a researcher in the group of James G. Anderson at Harvard studying gas-phase radical-molecule kinetics before returning to Pittsburgh to take a faculty position at Carnegie Mellon University, where he is now the Thomas Lord Professor of Chemistry in the Departments of Chemistry, Chemical Engineering, and Engineering and Public Policy. He is a fellow of the American Geophysical Union and has received the Pittsburgh and Esselen Awards from the American Chemical Society. His current research covers a wide range of topics associated with the oxidation chemistry of atmospheric organic compounds, focusing especially on processes that lead to the formation and growth of organic particulate matter.
Henrik G. Kjaergaard is Professor of Physical Chemistry in the Department of Chemistry at the University of Copenhagen. He received his Ph.D. from Odense University, was a postdoctoral fellow at University of Guelph, and before moving to Copenhagen, was a Professor at the University of Otago. His research interests include of hydrogen bonding in molecular complexes and atmospheric gas-phase reaction kinetics.
Mikael Ehn, born in Vaasa, Finland, in 1980, graduated in Physics from the University of Helsinki in 2005 and received his Ph.D. in aerosol physics from the same institution in 2010 under the supervision of Professor Markku Kulmala. After two years as a postdoc at the Research Center Jülich, Germany, with Dr. Thomas Mentel, he was appointed University Lecturer in physics in 2013 and Associate Professor (tenure track) in 2016, at the University of Helsinki. In 2014, he received a European Research Council Starting Grant and in 2018 an Academy of Finland Research Fellowship. His current research focuses on processes leading from volatile organic emissions to low-volatile vapors and secondary organic aerosol, primarily utilizing various mass spectrometric techniques.
The authors declare no competing financial interest.
References
- Jimenez J. L.; Canagaratna M. R.; Donahue N. M.; Prevot A. S. H.; Zhang Q.; Kroll J. H.; DeCarlo P. F.; Allan J. D.; Coe H.; et al. Evolution of organic aerosols in the atmosphere. Science 2009, 326, 1525–1529. 10.1126/science.1180353. [DOI] [PubMed] [Google Scholar]
- Bianchi F.; Barmet P.; Stirnweis L.; El Haddad I.; Platt S. M.; Saurer M.; Lotscher C.; Siegwolf R.; Bigi A.; et al. Contribution of methane to aerosol carbon mass. Atmos. Environ. 2016, 141, 41–47. 10.1016/j.atmosenv.2016.06.036. [DOI] [Google Scholar]
- Donahue N. M.; Robinson A. L.; Pandis S. N. Atmospheric organic particulate matter: From smoke to secondary organic aerosol. Atmos. Environ. 2009, 43, 94–106. 10.1016/j.atmosenv.2008.09.055. [DOI] [Google Scholar]
- Hallquist M.; Wenger J. C.; Baltensperger U.; Rudich Y.; Simpson D.; Claeys M.; Dommen J.; Donahue N. M.; George C.; et al. The formation, properties and impact of secondary organic aerosol: current and emerging issues. Atmos. Chem. Phys. 2009, 9, 5155–5236. 10.5194/acp-9-5155-2009. [DOI] [Google Scholar]
- Ehn M.; Thornton J. A.; Kleist E.; Sipilä M.; Junninen H.; Pullinen I.; Springer M.; Rubach F.; Tillmann R.; et al. A large source of low-volatility secondary organic aerosol. Nature 2014, 506, 476–480. 10.1038/nature13032. [DOI] [PubMed] [Google Scholar]
- Crounse J. D.; Nielsen L. B.; Jorgensen S.; Kjaergaard H. G.; Wennberg P. O. Autoxidation of organic compounds in the atmosphere. J. Phys. Chem. Lett. 2013, 4, 3513–3520. 10.1021/jz4019207. [DOI] [Google Scholar]
- Ehn M.; Kleist E.; Junninen H.; Petäjä T.; Lönn G.; Schobesberger S.; Dal Maso M.; Trimborn A.; Kulmala M.; et al. Gas phase formation of extremely oxidized pinene reaction products in chamber and ambient air. Atmos. Chem. Phys. 2012, 12, 5113–5127. 10.5194/acp-12-5113-2012. [DOI] [Google Scholar]
- Zhao J.; Ortega J.; Chen M.; McMurry P. H.; Smith J. N. Dependence of particle nucleation and growth on high-molecular-weight gas-phase products during ozonolysis of α-pinene. Atmos. Chem. Phys. 2013, 13, 7631–7644. 10.5194/acp-13-7631-2013. [DOI] [Google Scholar]
- Perrin O.; Heiss A.; Doumenc F. d. r.; Sahetchian K. Homogeneous and heterogeneous reactions of the n-C5H11O, n-C5H10OH and OOC5H10OH radicals in oxygen. Analytical steady state solution by use of the Laplace transform. J. Chem. Soc., Faraday Trans. 1998, 94, 2323–2335. 10.1039/a803340d. [DOI] [Google Scholar]
- Jorand F.; Heiss A.; Sahetchian K.; Kerhoas L.; Einhorn J. Identification of an unexpected peroxide formed by successive isomerization reactions of the n-butoxy radical in oxygen. J. Chem. Soc., Faraday Trans. 1996, 92, 4167–4171. 10.1039/ft9969204167. [DOI] [Google Scholar]
- Blin-Simiand N.; Jorand F.; Keller K.; Fiderer M.; Sahetchian K. Ketohydroperoxides and ignition delay in internal combustion engines. Combust. Flame 1998, 112, 278–282. 10.1016/S0010-2180(97)81778-6. [DOI] [Google Scholar]
- Praske E.; Otkjær R. V.; Crounse J. D.; Hethcox J. C.; Stoltz B. M.; Kjaergaard H. G.; Wennberg P. O. Atmospheric autoxidation is increasingly important in urban and suburban North America. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 64. 10.1073/pnas.1715540115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vereecken L.; Muller J. F.; Peeters J. Low-volatility poly-oxygenates in the OH-initiated atmospheric oxidation of [alpha]-pinene: impact of non-traditional peroxyl radical chemistry. Phys. Chem. Chem. Phys. 2007, 9, 5241–5248. 10.1039/b708023a. [DOI] [PubMed] [Google Scholar]
- Blin-Simiand N.; Jorand F.; Sahetchian K.; Brun M.; Kerhoas L.; Malosse C.; Einhorn J. Hydroperoxides with zero, one, two or more carbonyl groups formed during the oxidation of n-dodecane. Combust. Flame 2001, 126, 1524–1532. 10.1016/S0010-2180(01)00264-4. [DOI] [Google Scholar]
- Heiss A.; Sahetchian K. Isomerization reactions of the n-C4H9O and n-OOC4H8OH radicals in oxygen. Int. J. Chem. Kinet. 1996, 28, 531–544. 10.1002/(SICI)1097-4601(1996)28:7<531::AID-KIN5>3.0.CO;2-W. [DOI] [Google Scholar]
- Jorand F.; Heiss A.; Perrin O.; Sahetchian K.; Kerhoas L.; Einhorn J. Isomeric hexyl-ketohydroperoxides formed by reactions of hexoxy and hexylperoxy radicals in oxygen. Int. J. Chem. Kinet. 2003, 35, 354–366. 10.1002/kin.10136. [DOI] [Google Scholar]
- Perrin O.; Heiss A.; Sahetchian K.; Kerhoas L.; Einhorn J. Determination of the isomerization rate constant HOCH2CH2CH2CH(OO center dot)CH3 ->(HOCHCH2CH2CH)-H-center dot(OOH)CH3. Importance of intramolecular hydroperoxy isomerization in tropospheric chemistry. Int. J. Chem. Kinet. 1998, 30, 875–887. 10.1002/(SICI)1097-4601(1998)30:12<875::AID-KIN2>3.0.CO;2-8. [DOI] [Google Scholar]
- Birdsall A. W.; Andreoni J. F.; Elrod M. J. Investigation of the Role of Bicyclic Peroxy Radicals in the Oxidation Mechanism of Toluene. J. Phys. Chem. A 2010, 114, 10655–10663. 10.1021/jp105467e. [DOI] [PubMed] [Google Scholar]
- Atkinson R. Atmospheric reactions of alkoxy and beta-hydroxyalkoxy radicals. Int. J. Chem. Kinet. 1997, 29, 99–111. 10.1002/(SICI)1097-4601(1997)29:2<99::AID-KIN3>3.0.CO;2-F. [DOI] [Google Scholar]
- Muller L.; Reinnig M. C.; Naumann K. H.; Saathoff H.; Mentel T. F.; Donahue N. M.; Hoffmann T. Formation of 3-methyl-1,2,3-butanetricarboxylic acid via gas phase oxidation of pinonic acid - a mass spectrometric study of SOA aging. Atmos. Chem. Phys. 2012, 12, 1483–1496. 10.5194/acp-12-1483-2012. [DOI] [Google Scholar]
- Claeys M.; Kourtchev I.; Pashynska V.; Vas G.; Vermeylen R.; Wang W.; Cafmeyer J.; Chi X.; Artaxo P.; et al. Polar organic marker compounds in atmospheric aerosols during the LBA-SMOCC 2002 biomass burning experiment in Rondônia, Brazil: sources and source processes, time series, diel variations and size distributions. Atmos. Chem. Phys. 2010, 10, 9319–9331. 10.5194/acp-10-9319-2010. [DOI] [Google Scholar]
- Ingold K. U. Inhibition of the autoxidation of organic substances in the liquid phase. Chem. Rev. 1961, 61, 563–589. 10.1021/cr60214a002. [DOI] [Google Scholar]
- Cox R. A.; Cole J. A. Chemical aspects of the autoignition of hydrocarbon-air mixtures. Combust. Flame 1985, 60, 109–123. 10.1016/0010-2180(85)90001-X. [DOI] [Google Scholar]
- Donahue N. M.; Kroll J. H.; Pandis S. N.; Robinson A. L. A two-dimensional volatility basis set - Part 2: Diagnostics of organic-aerosol evolution. Atmos. Chem. Phys. 2012, 12, 615–634. 10.5194/acp-12-615-2012. [DOI] [Google Scholar]
- Trostl J.; Chuang W. K.; Gordon H.; Heinritzi M.; Yan C.; Molteni U.; Ahlm L.; Frege C.; Bianchi F.; et al. The role of low-volatility organic compounds in initial particle growth in the atmosphere. Nature 2016, 533, 527–531. 10.1038/nature18271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kroll J. H.; Seinfeld J. H. Chemistry of secondary organic aerosol: Formation and evolution of low-volatility organics in the atmosphere. Atmos. Environ. 2008, 42, 3593–3624. 10.1016/j.atmosenv.2008.01.003. [DOI] [Google Scholar]
- Kurtén T.; Tiusanen K.; Roldin P.; Rissanen M.; Luy J.-N.; Boy M.; Ehn M.; Donahue N. α-Pinene autoxidation products may not have extremely low saturation vapor pressures despite high O:C ratios. J. Phys. Chem. A 2016, 120, 2569–2582. 10.1021/acs.jpca.6b02196. [DOI] [PubMed] [Google Scholar]
- Donahue N. M.; Epstein S. A.; Pandis S. N.; Robinson A. L. A two-dimensional volatility basis set: 1. Organic-aerosol mixing thermodynamics. Atmos. Chem. Phys. 2011, 11, 3303–3318. 10.5194/acp-11-3303-2011. [DOI] [Google Scholar]
- Ehn M.; Junninen H.; Petaja T.; Kurten T.; Kerminen V. M.; Schobesberger S.; Manninen H. E.; Ortega I. K.; Vehkamaki H.; et al. Composition and temporal behavior of ambient ions in the boreal forest. Atmos. Chem. Phys. 2010, 10, 8513–8530. 10.5194/acp-10-8513-2010. [DOI] [Google Scholar]
- Junninen H.; Ehn M.; Petäjä T.; Luosujärvi L.; Kotiaho T.; Kostiainen R.; Rohner U.; Gonin M.; Fuhrer K.; et al. A high-resolution mass spectrometer to measure atmospheric ion composition. Atmos. Meas. Tech. 2010, 3, 1039–1053. 10.5194/amt-3-1039-2010. [DOI] [Google Scholar]
- Eisele F. L. Natural and anthropogenic negative-ions in the troposphere. J. Geophys. Res. 1989, 94, 2183–2196. 10.1029/JD094iD02p02183. [DOI] [Google Scholar]
- Eisele F. L. Natural and transmission-line produced positive-ions. J. Geophys. Res. 1989, 94, 6309–6318. 10.1029/JD094iD05p06309. [DOI] [Google Scholar]
- Heinritzi M.; Simon M.; Steiner G.; Wagner A. C.; Kürten A.; Hansel A.; Curtius J. Characterization of the mass-dependent transmission efficiency of a CIMS. Atmos. Meas. Tech. 2016, 9, 1449–1460. 10.5194/amt-9-1449-2016. [DOI] [Google Scholar]
- Ehn M.; Junninen H.; Schobesberger S.; Manninen H. E.; Franchin A.; Sipila M.; Petaja T.; Kerminen V. M.; Tammet H.; et al. An instrumental comparison of mobility and mass measurements of atmospheric amall ions. Aerosol Sci. Technol. 2011, 45, 522–532. 10.1080/02786826.2010.547890. [DOI] [Google Scholar]
- Jokinen T.; Sipilä M.; Junninen H.; Ehn M.; Lönn G.; Hakala J.; Petäjä T.; Mauldin R. L. Iii; Kulmala M.; Worsnop D. R. Atmospheric sulphuric acid and neutral cluster measurements using CI-Api-TOF. Atmos. Chem. Phys. 2012, 12, 4117–4125. 10.5194/acp-12-4117-2012. [DOI] [Google Scholar]
- Bertram T. H.; Kimmel J. R.; Crisp T. A.; Ryder O. S.; Yatavelli R. L. N.; Thornton J. A.; Cubison M. J.; Gonin M.; Worsnop D. R. A field-deployable, chemical ionization time-of-flight mass spectrometer. Atmos. Meas. Tech. 2011, 4, 1471–1479. 10.5194/amt-4-1471-2011. [DOI] [Google Scholar]
- Hyttinen N.; Kupiainen-Määttä O.; Rissanen M. P.; Muuronen M.; Ehn M.; Kurtén T. Modeling the charging of highly oxidized cyclohexene ozonolysis products using nitrate-based chemical ionization. J. Phys. Chem. A 2015, 119, 6339–6345. 10.1021/acs.jpca.5b01818. [DOI] [PubMed] [Google Scholar]
- Mentel T. F.; Springer M.; Ehn M.; Kleist E.; Pullinen I.; Kurtén T.; Rissanen M.; Wahner A.; Wildt J. Formation of highly oxidized multifunctional compounds: autoxidation of peroxy radicals formed in the ozonolysis of alkenes – deduced from structure–product relationships. Atmos. Chem. Phys. 2015, 15, 6745–6765. 10.5194/acp-15-6745-2015. [DOI] [Google Scholar]
- Eisele F. L.; Tanner D. J. Measurement of the gas phase concentration of H2SO4 and methane sulfonic acid and estimates of H2SO4 production and loss in the atmosphere. J. Geophys. Res. Atmos. 1993, 98, 9001–9010. 10.1029/93JD00031. [DOI] [Google Scholar]
- Kurten A.; Rondo L.; Ehrhart S.; Curtius J. Performance of a corona ion source for measurement of sulfuric acid by chemical ionization mass spectrometry. Atmos. Meas. Tech. 2011, 4, 437–443. 10.5194/amt-4-437-2011. [DOI] [Google Scholar]
- Massoli P.; Stark H.; Canagaratna M. R.; Krechmer J. E.; Xu L.; Ng N. L.; Mauldin R. L.; Yan C.; Kimmel J.; et al. Ambient measurements of highly oxidized gas-phase molecules during the southern oxidant and aerosol study (SOAS) 2013. ACS Earth Space Chem. 2018, 2, 653–672. 10.1021/acsearthspacechem.8b00028. [DOI] [Google Scholar]
- Kürten A.; Bergen A.; Heinritzi M.; Leiminger M.; Lorenz V.; Piel F.; Simon M.; Sitals R.; Wagner A. C.; Curtius J. Observation of new particle formation and measurement of sulfuric acid, ammonia, amines and highly oxidized organic molecules at a rural site in central Germany. Atmos. Chem. Phys. 2016, 16, 12793–12813. 10.5194/acp-16-12793-2016. [DOI] [Google Scholar]
- Kürten A.; Rondo L.; Ehrhart S.; Curtius J. Performance of a corona ion source for measurement of sulfuric acid by chemical ionization mass spectrometry. Atmos. Meas. Tech. 2011, 4, 437–443. 10.5194/amt-4-437-2011. [DOI] [Google Scholar]
- Kürten A.; Rondo L.; Ehrhart S.; Curtius J. Calibration of a chemical ionization mass spectrometer for the measurement of gaseous sulfuric acid. J. Phys. Chem. A 2012, 116, 6375–6386. 10.1021/jp212123n. [DOI] [PubMed] [Google Scholar]
- Berndt T.; Richters S.; Kaethner R.; Voigtländer J.; Stratmann F.; Sipilä M.; Kulmala M.; Herrmann H. Gas-phase ozonolysis of cycloalkenes: formation of highly oxidized ro2 radicals and their reactions with NO, NO2, SO2, and other RO2 radicals. J. Phys. Chem. A 2015, 119, 10336–10348. 10.1021/acs.jpca.5b07295. [DOI] [PubMed] [Google Scholar]
- Berndt T.; Richters S.; Jokinen T.; Hyttinen N.; Kurten T.; Otkjaer R. V.; Kjaergaard H. G.; Stratmann F.; Herrmann H.; Sipilä M.; Kulmala M.; Ehn M. Hydroxyl radical-induced formation of highly oxidized organic compounds. Nat. Commun. 2016, 7, 13677. 10.1038/ncomms13677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richters S.; Herrmann H.; Berndt T. Different pathways of the formation of highly oxidized multifunctional organic compounds (HOMs) from the gas-phase ozonolysis of β-caryophyllene. Atmos. Chem. Phys. 2016, 16, 9831–9845. 10.5194/acp-16-9831-2016. [DOI] [Google Scholar]
- Berndt T.; Scholz W.; Mentler B.; Fischer L.; Herrmann H.; Kulmala M.; Hansel A. accretion product formation from self- and cross-reactions of RO2 radicals in the atmosphere. Angew. Chem., Int. Ed. 2018, 57, 3820–3824. 10.1002/anie.201710989. [DOI] [PubMed] [Google Scholar]
- Lee B. H.; Mohr C.; Lopez-Hilfiker F. D.; Lutz A.; Hallquist M.; Lee L.; Romer P.; Cohen R. C.; Iyer S.; et al. Highly functionalized organic nitrates in the southeast United States: Contribution to secondary organic aerosol and reactive nitrogen budgets. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 1516–1521. 10.1073/pnas.1508108113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee B. H.; Lopez-Hilfiker F. D.; Mohr C.; Kurtén T.; Worsnop D. R.; Thornton J. A. An Iodide-adduct High-Resolution Time-of-Flight Chemical-Ionization mass spectrometer: application to atmospheric inorganic and organic compounds. Environ. Sci. Technol. 2014, 48, 6309–6317. 10.1021/es500362a. [DOI] [PubMed] [Google Scholar]
- Lopez-Hilfiker F. D.; Mohr C.; Ehn M.; Rubach F.; Kleist E.; Wildt J.; Mentel T. F.; Carrasquillo A. J.; Daumit K. E.; et al. Phase partitioning and volatility of secondary organic aerosol components formed from α-pinene ozonolysis and OH oxidation: the importance of accretion products and other low volatility compounds. Atmos. Chem. Phys. 2015, 15, 7765–7776. 10.5194/acp-15-7765-2015. [DOI] [Google Scholar]
- Lopez-Hilfiker F. D.; Iyer S.; Mohr C.; Lee B. H.; D’Ambro E. L.; Kurtén T.; Thornton J. A. Constraining the sensitivity of iodide adduct chemical ionization mass spectrometry to multifunctional organic molecules using the collision limit and thermodynamic stability of iodide ion adducts. Atmos. Meas. Tech. 2016, 9, 1505–1512. 10.5194/amt-9-1505-2016. [DOI] [Google Scholar]
- Mohr C.; Lopez-Hilfiker F. D.; Yli-Juuti T.; Heitto A.; Lutz A.; Hallquist M.; D’Ambro E. L.; Rissanen M. P.; Hao L.; Schobesberger S.; Kulmala M.; Mauldin R. L.; Makkonen L.; Sipilä M.; Petäjä T.; Thornton J. A. Ambient observations of dimers from terpene oxidation in the gas phase: Implications for new particle formation and growth. Geophys. Res. Lett. 2017, 44, 2958. 10.1002/2017GL072718. [DOI] [Google Scholar]
- Isaacman-VanWertz G.; Massoli P.; O’Brien R. E.; Nowak J. B.; Canagaratna M. R.; Jayne J. T.; Worsnop D. R.; Su L.; Knopf D. A.; et al. Using advanced mass spectrometry techniques to fully characterize atmospheric organic carbon: current capabilities and remaining gaps. Faraday Discuss. 2017, 200, 579–598. 10.1039/C7FD00021A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernhammer A. K.; Fischer L.; Mentler B.; Heinritzi M.; Simon M.; Hansel A. Production of highly oxygenated organic molecules (HOMs) from trace contaminants during isoprene oxidation. Atmos. Meas. Tech. 2018, 11, 4763–4773. 10.5194/amt-11-4763-2018. [DOI] [Google Scholar]
- Breitenlechner M.; Fischer L.; Hainer M.; Heinritzi M.; Curtius J.; Hansel A. PTR3: An instrument for studying the lifecycle of reactive organic carbon in the Atmosphere. Anal. Chem. 2017, 89, 5824–5831. 10.1021/acs.analchem.6b05110. [DOI] [PubMed] [Google Scholar]
- Zhao J.; Eisele F. L.; Titcombe M.; Kuang C. G.; McMurry P. H. Chemical ionization mass spectrometric measurements of atmospheric neutral clusters using the cluster-CIMS. J. Geophys. Res. 2010, 115, JD012606. 10.1029/2009JD012606. [DOI] [Google Scholar]
- Krapf M.; El Haddad I.; Bruns E. A.; Molteni U.; Daellenbach K. R.; Prévôt A. S. H.; Baltensperger U.; Dommen J. Labile peroxides in secondary organic aerosol. Chem 2016, 1, 603–616. 10.1016/j.chempr.2016.09.007. [DOI] [Google Scholar]
- Bach R. D.; Ayala P. Y.; Schlegel H. B. A reassessment of the bond dissociation energies of peroxides. An ab initio study. J. Am. Chem. Soc. 1996, 118, 12758–12765. 10.1021/ja961838i. [DOI] [Google Scholar]
- Tong H.; Arangio A. M.; Lakey P. S. J.; Berkemeier T.; Liu F.; Kampf C. J.; Brune W. H.; Pöschl U.; Shiraiwa M. Hydroxyl radicals from secondary organic aerosol decomposition in water. Atmos. Chem. Phys. 2016, 16, 1761–1771. 10.5194/acp-16-1761-2016. [DOI] [Google Scholar]
- Stolzenburg D.; Fischer L.; Vogel A. L.; Heinritzi M.; Schervish M.; Simon M.; Wagner A. C.; Dada L.; Ahonen L. R.; et al. Rapid growth of organic aerosol nanoparticles over a wide tropospheric temperature range. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 9122–9127. 10.1073/pnas.1807604115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mutzel A.; Poulain L.; Berndt T.; Iinuma Y.; Rodigast M.; Böge O.; Richters S.; Spindler G.; Sipilä M.; et al. Highly oxidized multifunctional organic compounds observed in tropospheric particles: a field and laboratory study. Environ. Sci. Technol. 2015, 49, 7754–7761. 10.1021/acs.est.5b00885. [DOI] [PubMed] [Google Scholar]
- Zhang X.; Lambe A. T.; Upshur M. A.; Brooks W. A.; Gray Bé A.; Thomson R. J.; Geiger F. M.; Surratt J. D.; Zhang Z.; et al. Highly oxygenated multifunctional compounds in α-pinene secondary organic aerosol. Environ. Sci. Technol. 2017, 51, 5932–5940. 10.1021/acs.est.6b06588. [DOI] [PubMed] [Google Scholar]
- Tu P.; Hall W. A.; Johnston M. V. Characterization of highly oxidized molecules in fresh and aged biogenic secondary organic aerosol. Anal. Chem. 2016, 88, 4495–4501. 10.1021/acs.analchem.6b00378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brüggemann M.; Poulain L.; Held A.; Stelzer T.; Zuth C.; Richters S.; Mutzel A.; van Pinxteren D.; Iinuma Y.; et al. Real-time detection of highly oxidized organosulfates and BSOA marker compounds during the F-BEACh 2014 field study. Atmos. Chem. Phys. 2017, 17, 1453–1469. 10.5194/acp-17-1453-2017. [DOI] [Google Scholar]
- Kahnt A.; Vermeylen R.; Iinuma Y.; Safi Shalamzari M.; Maenhaut W.; Claeys M. High-molecular-weight esters in α-pinene ozonolysis secondary organic aerosol: Structural characterization and mechanistic proposal for their formation from highly oxygenated molecules. Atmos. Chem. Phys. 2018, 18, 8453–8467. 10.5194/acp-18-8453-2018. [DOI] [Google Scholar]
- Zhang X.; McVay R. C.; Huang D. D.; Dalleska N. F.; Aumont B.; Flagan R. C.; Seinfeld J. H. Formation and evolution of molecular products in α-pinene secondary organic aerosol. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 14168–14173. 10.1073/pnas.1517742112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Hilfiker F. D.; Mohr C.; D’Ambro E. L.; Lutz A.; Riedel T. P.; Gaston C. J.; Iyer S.; Zhang Z.; Gold A.; et al. Molecular composition and volatility of organic aerosol in the southeastern U.S.: Implications for IEPOX derived SOA. Environ. Sci. Technol. 2016, 50, 2200–2209. 10.1021/acs.est.5b04769. [DOI] [PubMed] [Google Scholar]
- Stark H.; Yatavelli R. L. N.; Thompson S. L.; Kang H.; Krechmer J. E.; Kimmel J. R.; Palm B. B.; Hu W. W.; Hayes P. L.; et al. Impact of thermal decomposition on thermal desorption instruments: advantage of thermogram analysis for quantifying volatility distributions of organic species. Environ. Sci. Technol. 2017, 51, 8491–8500. 10.1021/acs.est.7b00160. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Yee L. D.; Lee B. H.; Curtis M. P.; Worton D. R.; Isaacman-VanWertz G.; Offenberg J. H.; Lewandowski M.; Kleindienst T. E.; et al. Monoterpenes are the largest source of summertime organic aerosol in the southeastern United States. Proc. Natl. Acad. Sci. U. S. A. 2018, 115, 2038–2043. 10.1073/pnas.1717513115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer S.; He X.; Hyttinen N.; Kurtén T.; Rissanen M. P. Computational and experimental investigation of the detection of HO2 radical and the products of its reaction with cyclohexene ozonolysis derived RO2 radicals by an iodide-based chemical ionization mass spectrometer. J. Phys. Chem. A 2017, 121, 6778–6789. 10.1021/acs.jpca.7b01588. [DOI] [PubMed] [Google Scholar]
- Hyttinen N.; Otkjær R. V.; Iyer S.; Kjaergaard H. G.; Rissanen M. P.; Wennberg P. O.; Kurtén T. Computational comparison of different reagent ions in the chemical ionization of oxidized multifunctional compounds. J. Phys. Chem. A 2018, 122, 269–279. 10.1021/acs.jpca.7b10015. [DOI] [PubMed] [Google Scholar]
- Orlando J. J.; Tyndall G. S. Laboratory studies of organic peroxy radical chemistry: an overview with emphasis on recent issues of atmospheric significance. Chem. Soc. Rev. 2012, 41, 6294–6317. 10.1039/c2cs35166h. [DOI] [PubMed] [Google Scholar]
- Orlando J. J.; Tyndall G. S.; Wallington T. J. The atmospheric chemistry of alkoxy radicals. Chem. Rev. 2003, 103, 4657–4689. 10.1021/cr020527p. [DOI] [PubMed] [Google Scholar]
- Vereecken L.; Francisco J. S. Theoretical studies of atmospheric reaction mechanisms in the troposphere. Chem. Soc. Rev. 2012, 41, 6259–6293. 10.1039/c2cs35070j. [DOI] [PubMed] [Google Scholar]
- Vereecken L.; Glowacki D. R.; Pilling M. J. Theoretical chemical kinetics in tropospheric chemistry: methodologies and applications. Chem. Rev. 2015, 115, 4063–4114. 10.1021/cr500488p. [DOI] [PubMed] [Google Scholar]
- Crounse J. D.; Knap H. C.; Ørnsø K. B.; Jørgensen S.; Paulot F.; Kjaergaard H. G.; Wennberg P. O. Atmospheric fate of methacrolein. 1. Peroxy radical isomerization following addition of OH and O2. J. Phys. Chem. A 2012, 116, 5756–5762. 10.1021/jp211560u. [DOI] [PubMed] [Google Scholar]
- Teng A. P.; Crounse J. D.; Wennberg P. O. Isoprene peroxy radical dynamics. J. Am. Chem. Soc. 2017, 139, 5367–5377. 10.1021/jacs.6b12838. [DOI] [PubMed] [Google Scholar]
- Møller K. H.; Otkjær R. V.; Hyttinen N.; Kurtén T.; Kjaergaard H. G. Cost-effective implementation of multiconformer transition state theory for peroxy radical hydrogen shift reactions. J. Phys. Chem. A 2016, 120, 10072–10087. 10.1021/acs.jpca.6b09370. [DOI] [PubMed] [Google Scholar]
- Bao J. L.; Truhlar D. G. Variational transition state theory: theoretical framework and recent developments. Chem. Soc. Rev. 2017, 46, 7548–7596. 10.1039/C7CS00602K. [DOI] [PubMed] [Google Scholar]
- Jokinen T.; Sipilä M.; Richters S.; Kerminen V. M.; Paasonen P.; Stratmann F.; Worsnop D.; Kulmala M.; Ehn M.; et al. Rapid autoxidation forms highly oxidized ro2 radicals in the atmosphere. Angew. Chem., Int. Ed. 2014, 53, 14596–14600. 10.1002/anie.201408566. [DOI] [PubMed] [Google Scholar]
- Rissanen M. P.; Kurten T.; Sipila M.; Thornton J. A.; Kangasluoma J.; Sarnela N.; Junninen H.; Jorgensen S.; Schallhart S.; et al. The formation of highly oxidized multifunctional products in the ozonolysis of cyclohexene. J. Am. Chem. Soc. 2014, 136, 15596–15606. 10.1021/ja507146s. [DOI] [PubMed] [Google Scholar]
- Kirkby J.; Duplissy J.; Sengupta K.; Frege C.; Gordon H.; Williamson C.; Heinritzi M.; Simon M.; Yan C.; et al. Ion-induced nucleation of pure biogenic particles. Nature 2016, 533, 521–524. 10.1038/nature17953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nah T.; Sanchez J.; Boyd C. M.; Ng N. L. photochemical aging of α-pinene and β-pinene secondary organic aerosol formed from nitrate radical oxidation. Environ. Sci. Technol. 2016, 50, 222–231. 10.1021/acs.est.5b04594. [DOI] [PubMed] [Google Scholar]
- Molteni U.; Bianchi F.; Klein F.; El Haddad I.; Frege C.; Rossi M. J.; Dommen J.; Baltensperger U. Formation of highly oxygenated organic molecules from aromatic compounds. Atmos. Chem. Phys. 2018, 18, 1909–1921. 10.5194/acp-18-1909-2018. [DOI] [Google Scholar]
- Rissanen M. P.; Kurtén T.; Sipilä M.; Thornton J. A.; Kausiala O.; Garmash O.; Kjaergaard H. G.; Petäjä T.; Worsnop D. R.; et al. Effects of chemical complexity on the autoxidation mechanisms of endocyclic alkene ozonolysis products: from methylcyclohexenes toward understanding α-pinene. J. Phys. Chem. A 2015, 119, 4633–4650. 10.1021/jp510966g. [DOI] [PubMed] [Google Scholar]
- Gutbrod R.; Schindler R. N.; Kraka E.; Cremer D. Formation of OH radicals in the gas phase ozonolysis of alkenes, the unexpected role of carbonyl oxides. Chem. Phys. Lett. 1996, 252, 221–229. 10.1016/0009-2614(96)00126-1. [DOI] [Google Scholar]
- Donahue N. M.; Chuang W.; Epstein S. A.; Kroll J. H.; Worsnop D. R.; Robinson A. L.; Adams P. J.; Pandis S. N. Why do organic aerosols exist? Understanding aerosol lifetimes using the two-dimensional volatility basis set. Environ. Chem. 2013, 10, 151–157. 10.1071/EN13022. [DOI] [Google Scholar]
- Donahue N. M.; Henry K. M.; Mentel T. F.; Kiendler-Scharr A.; Spindler C.; Bohn B.; Brauers T.; Dorn H. P.; Fuchs H.; et al. Aging of biogenic secondary organic aerosol via gas-phase OH radical reactions. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 13503–13508. 10.1073/pnas.1115186109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schobesberger S.; Junninen H.; Bianchi F.; Lonn G.; Ehn M.; Lehtipalo K.; Dommen J.; Ehrhart S.; Ortega I. K.; et al. Molecular understanding of atmospheric particle formation from sulfuric acid and large oxidized organic molecules. Proc. Natl. Acad. Sci. U. S. A. 2013, 110, 17223–17228. 10.1073/pnas.1306973110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park J.; Jongsma C. G.; Zhang R. Y.; North S. W. OH/OD initiated oxidation of isoprene in the presence of O-2 and NO. J. Phys. Chem. A 2004, 108, 10688–10697. 10.1021/jp040421t. [DOI] [Google Scholar]
- Oguchi T.; Miyoshi A.; Koshi M.; Matsui H.; Washida N. Kinetic study on reactions of 1-and 2-methylvinoxy radicals with O-2. J. Phys. Chem. A 2001, 105, 378–382. 10.1021/jp001826q. [DOI] [Google Scholar]
- Zador J.; Taatjes C. A.; Fernandes R. X. Kinetics of elementary reactions in low-temperature autoignition chemistry. Prog. Energy Combust. Sci. 2011, 37, 371–421. 10.1016/j.pecs.2010.06.006. [DOI] [Google Scholar]
- Savee J. D.; Papajak E.; Rotavera B.; Huang H. F.; Eskola A. J.; Welz O.; Sheps L.; Taatjes C. A.; Zador J.; Osborn D. L. Direct observation and kinetics of a hydroperoxyalkyl radical (QOOH). Science 2015, 347, 643–646. 10.1126/science.aaa1495. [DOI] [PubMed] [Google Scholar]
- Wang S. N.; Wang L. M. The atmospheric oxidation of dimethyl, diethyl, and diisopropyl ethers. The role of the intramolecular hydrogen shift in peroxy radicals. Phys. Chem. Chem. Phys. 2016, 18, 7707–7714. 10.1039/C5CP07199B. [DOI] [PubMed] [Google Scholar]
- Peeters J.; Muller J. F.; Stavrakou T.; Nguyen V. S. Hydroxyl radical recycling in isoprene oxidation driven by hydrogen bonding and hydrogen tunneling: the upgraded LIM1 mechanism. J. Phys. Chem. A 2014, 118, 8625–8643. 10.1021/jp5033146. [DOI] [PubMed] [Google Scholar]
- Peeters J.; Nguyen T. L.; Vereecken L. HOx radical regeneration in the oxidation of isoprene. Phys. Chem. Chem. Phys. 2009, 11, 5935–5939. 10.1039/b908511d. [DOI] [PubMed] [Google Scholar]
- Vereecken L.; Peeters J. Decomposition of substituted alkoxy radicals-part I: a generalized structure-activity relationship for reaction barrier heights. Phys. Chem. Chem. Phys. 2009, 11, 9062–9074. 10.1039/b909712k. [DOI] [PubMed] [Google Scholar]
- Sage A. M.; Donahue N. M. Deconstructing experimental rate constant measurements: Obtaining intrinsic reaction parameters, kinetic isotope effects, and tunneling coefficients from kinetic data for OH plus methane, ethane and cyclohexane. J. Photochem. Photobiol., A 2005, 176, 238–249. 10.1016/j.jphotochem.2005.09.011. [DOI] [Google Scholar]
- Jokinen T.; Berndt T.; Makkonen R.; Kerminen V.-M.; Junninen H.; Paasonen P.; Stratmann F.; Herrmann H.; Guenther A. B.; et al. Production of extremely low volatile organic compounds from biogenic emissions: Measured yields and atmospheric implications. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 7123–7128. 10.1073/pnas.1423977112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S.; Wu R.; Berndt T.; Ehn M.; Wang L. Formation of highly oxidized radicals and multifunctional products from the atmospheric oxidation of alkylbenzenes. Environ. Sci. Technol. 2017, 51, 8442–8449. 10.1021/acs.est.7b02374. [DOI] [PubMed] [Google Scholar]
- Jorgensen S.; Knap H. C.; Otkjaer R. V.; Jensen A. M.; Kjeldsen M. L. H.; Wennberg P. O.; Kjaergaard H. G. Rapid hydrogen shift scrambling in hydroperoxy-substituted organic peroxy radicals. J. Phys. Chem. A 2016, 120, 266–275. 10.1021/acs.jpca.5b06768. [DOI] [PubMed] [Google Scholar]
- Knap H. C.; Jørgensen S. Rapid Hydrogen Shift Reactions in Acyl Peroxy Radicals. J. Phys. Chem. A 2017, 121, 1470–1479. 10.1021/acs.jpca.6b12787. [DOI] [PubMed] [Google Scholar]
- Vereecken L.; Peeters J. A structure-activity relationship for the rate coefficient of H-migration in substituted alkoxy radicals. Phys. Chem. Chem. Phys. 2010, 12, 12608–12620. 10.1039/c0cp00387e. [DOI] [PubMed] [Google Scholar]
- Orlando J. J. The atmospheric oxidation of diethyl ether: chemistry of the C2H5–O–CH(Ȯ)CH3 radical between 218 and 335 K. Phys. Chem. Chem. Phys. 2007, 9, 4189–4199. 10.1039/b706819k. [DOI] [PubMed] [Google Scholar]
- Collins E. M.; Sidebottom H. W.; Wenger J. C.; Calvé S. L.; Mellouki A.; LeBras G.; Villenave E.; Wirtz K. The influence of reaction conditions on the photooxidation of diisopropyl ether. J. Photochem. Photobiol., A 2005, 176, 86–97. 10.1016/j.jphotochem.2005.09.004. [DOI] [Google Scholar]
- Tian Z.; Fattahi A.; Lis L.; Kass S. R. Cycloalkane and cycloalkene C–H bond dissociation energies. J. Am. Chem. Soc. 2006, 128, 17087–17092. 10.1021/ja065348u. [DOI] [PubMed] [Google Scholar]
- Clarke J. S.; Kroll J. H.; Donahue N. M.; Anderson J. G. Testing frontier orbital control: Kinetics of OH with ethane, propane, and cyclopropane from 180 to 360K. J. Phys. Chem. A 1998, 102, 9847–9857. 10.1021/jp982922i. [DOI] [Google Scholar]
- Kurtén T.; Rissanen M. P.; Mackeprang K.; Thornton J. A.; Hyttinen N.; Jørgensen S.; Ehn M.; Kjaergaard H. G. Computational study of hydrogen shifts and ring-opening mechanisms in α-pinene ozonolysis products. J. Phys. Chem. A 2015, 119, 11366–11375. 10.1021/acs.jpca.5b08948. [DOI] [PubMed] [Google Scholar]
- Kurtén T.; Møller K. H.; Nguyen T. B.; Schwantes R. H.; Misztal P. K.; Su L.; Wennberg P. O.; Fry J. L.; Kjaergaard H. G. Alkoxy radical bond scissions explain the anomalously low secondary organic aerosol and organonitrate yields from α-pinene + NO3. J. Phys. Chem. Lett. 2017, 8, 2826–2834. 10.1021/acs.jpclett.7b01038. [DOI] [PubMed] [Google Scholar]
- Atkinson R. Gas-phase tropospheric chemistry of organic compounds: a review. Atmos. Environ. 2007, 41, 200–240. 10.1016/j.atmosenv.2007.10.068. [DOI] [Google Scholar]
- Lizardo-Huerta J. C.; Sirjean B.; Bounaceur R.; Fournet R. Intramolecular effects on the kinetics of unimolecular reactions of β-HOROȮ and HOQ̇OOH radicals. Phys. Chem. Chem. Phys. 2016, 18, 12231–12251. 10.1039/C6CP00111D. [DOI] [PubMed] [Google Scholar]
- Vereecken L.; Peeters J. Theoretical investigation of the role of intramolecular hydrogen bonding in β-hydroxyethoxy and β-hydroxyethylperoxy radicals in the tropospheric oxidation of ethene. J. Phys. Chem. A 1999, 103, 1768–1775. 10.1021/jp9826930. [DOI] [Google Scholar]
- D’Ambro E. L.; Møller K. H.; Lopez-Hilfiker F. D.; Schobesberger S.; Liu J.; Shilling J. E.; Lee B. H.; Kjaergaard H. G.; Thornton J. A. Isomerization of second-generation isoprene peroxy radicals: epoxide formation and implications for secondary organic aerosol yields. Environ. Sci. Technol. 2017, 51, 4978–4987. 10.1021/acs.est.7b00460. [DOI] [PubMed] [Google Scholar]
- Groß C. B. M.; Dillon T. J.; Schuster G.; Lelieveld J.; Crowley J. N. direct kinetic study of oh and O3 formation in the reaction of CH3C(O)O2 with HO2. J. Phys. Chem. A 2014, 118, 974–985. 10.1021/jp412380z. [DOI] [PubMed] [Google Scholar]
- Hasson A. S.; Kuwata K. T.; Arroyo M. C.; Petersen E. B. Theoretical studies of the reaction of hydroperoxy radicals (HO2) with ethyl peroxy (CH3CH2O2), acetyl peroxy (CH3C(O)O2), and acetonyl peroxy (CH3C(O)CH2O2) radicals. J. Photochem. Photobiol., A 2005, 176, 218–230. 10.1016/j.jphotochem.2005.08.012. [DOI] [Google Scholar]
- Le Crane J.-P.; Rayez M.-T.; Rayez J.-C.; Villenave E. A reinvestigation of the kinetics and the mechanism of the CH3C(O)O2 + HO2 reaction using both experimental and theoretical approaches. Phys. Chem. Chem. Phys. 2006, 8, 2163–2171. 10.1039/B518321A. [DOI] [PubMed] [Google Scholar]
- Praske E.; Crounse J. D.; Bates K. H.; Kurtén T.; Kjaergaard H. G.; Wennberg P. O. Atmospheric fate of methyl vinyl ketone: peroxy radical reactions with NO and HO2. J. Phys. Chem. A 2015, 119, 4562–4572. 10.1021/jp5107058. [DOI] [PubMed] [Google Scholar]
- Peeters J.; Nguyen T. L. Unusually fast 1,6-H shifts of enolic Hydrogens in peroxy radicals: formation of the first-generation C2 and C3 carbonyls in the oxidation of Isoprene. J. Phys. Chem. A 2012, 116, 6134–6141. 10.1021/jp211447q. [DOI] [PubMed] [Google Scholar]
- Richters S.; Pfeifle M.; Olzmann M.; Berndt T. endo-Cyclization of unsaturated RO2 radicals from the gas-phase ozonolysis of cyclohexadienes. Chem. Commun. 2017, 53, 4132–4135. 10.1039/C7CC01350G. [DOI] [PubMed] [Google Scholar]
- Peeters J.; Vereecken L.; Fantechi G. The detailed mechanism of the OH-initiated atmospheric oxidation of alpha-pinene: a theoretical study. Phys. Chem. Chem. Phys. 2001, 3, 5489–5504. 10.1039/b106555f. [DOI] [Google Scholar]
- Miller J. A.; Pilling M. J.; Troe J. Unravelling combustion mechanisms through a quantitative understanding of elementary reactions. Proc. Combust. Inst. 2005, 30, 43–88. 10.1016/j.proci.2004.08.281. [DOI] [Google Scholar]
- Taatjes C. A. Uncovering the Fundamental Chemistry of Alkyl + O2 Reactions via Measurements of Product Formation. J. Phys. Chem. A 2006, 110, 4299–4312. 10.1021/jp056997f. [DOI] [PubMed] [Google Scholar]
- Hyttinen N.; Knap H. C.; Rissanen M. P.; Jørgensen S.; Kjaergaard H. G.; Kurtén T. Unimolecular HO2 loss from peroxy radicals formed in autoxidation is unlikely under atmospheric conditions. J. Phys. Chem. A 2016, 120, 3588–3595. 10.1021/acs.jpca.6b02281. [DOI] [PubMed] [Google Scholar]
- Paulot F.; Crounse J. D.; Kjaergaard H. G.; Kurten A.; St. Clair J. M.; Seinfeld J. H.; Wennberg P. O. unexpected epoxide formation in the gas-phase photooxidation of isoprene. Science 2009, 325, 730–733. 10.1126/science.1172910. [DOI] [PubMed] [Google Scholar]
- Atkinson R. Kinetics and mechanisms of the gas-phase reactions of the hydroxyl radical with organic-compounds under atmospheric conditions. Chem. Rev. 1986, 86, 69–201. 10.1021/cr00071a004. [DOI] [Google Scholar]
- Lightfoot P. D.; Cox R. A.; Crowley J. N.; Destriau M.; Hayman G. D.; Jenkin M. E.; Moortgat G. K.; Zabel F. Organic peroxy radicals: Kinetics, spectroscopy and tropospheric chemistry. Atmos. Environ., Part A 1992, 26, 1805–1961. 10.1016/0960-1686(92)90423-I. [DOI] [Google Scholar]
- Atkinson R.; Arey J. Atmospheric degradation of volatile organic compounds. Chem. Rev. 2003, 103, 4605–4638. 10.1021/cr0206420. [DOI] [PubMed] [Google Scholar]
- Moortgat G.; Veyret B.; Lesclaux R. Absorption-spectrum and kinetics of reactions of the acetylperoxy radical. J. Phys. Chem. 1989, 93, 2362–2368. 10.1021/j100343a032. [DOI] [Google Scholar]
- Shallcross D. E.; Raventos-Duran M. T.; Bardwell M. W.; Bacak A.; Solman Z.; Percival C. J. A semi-empirical correlation for the rate coefficients for cross- and self-reactions of peroxy radicals in the gas-phase. Atmos. Environ. 2005, 39, 763–771. 10.1016/j.atmosenv.2004.09.072. [DOI] [Google Scholar]
- Villenave E.; Lesclaux R. Kinetics of the cross reactions of CH3O2 and C2H5O2 radicals with selected peroxy radicals. J. Phys. Chem. 1996, 100, 14372–14382. 10.1021/jp960765m. [DOI] [Google Scholar]
- Bianchi F.; Garmash O.; He X.; Yan C.; Iyer S.; Rosendahl I.; Xu Z.; Rissanen M. P.; Riva M.; et al. The role of highly oxygenated molecules (HOMs) in determining the composition of ambient ions in the boreal forest. Atmos. Chem. Phys. 2017, 17, 13819–13831. 10.5194/acp-17-13819-2017. [DOI] [Google Scholar]
- Yan C.; Nie W.; Äijälä M.; Rissanen M. P.; Canagaratna M. R.; Massoli P.; Junninen H.; Jokinen T.; Sarnela N.; et al. Source characterization of highly oxidized multifunctional compounds in a boreal forest environment using positive matrix factorization. Atmos. Chem. Phys. 2016, 16, 12715–12731. 10.5194/acp-16-12715-2016. [DOI] [Google Scholar]
- Jokinen T.; Kausiala O.; Garmash O.; Perakyla O.; Junninen H.; Schobesberger S.; Yan C.; Sipila M.; Rissanen M. P. Production of highly oxidized organic compounds from ozonolysis of beta-caryophyllene: laboratory and field measurements. Boreal Env. Res. 2016, 21, 262–273. [Google Scholar]
- Bartlett P. D.; Traylor T. G. Oxygen-18 tracer studies of alkylperoxy radicals 0.1. Cumylperoxy radical and chain termination in autoxidation of cumene. J. Am. Chem. Soc. 1963, 85, 2407. 10.1021/ja00899a014. [DOI] [Google Scholar]
- Bennett J. E.; Brown D. M.; Mile B. The equilibrium between tertiary alkylperoxy-radicals and tetroxide molecules. J. Chem. Soc. D 1969, 504–505. 10.1039/c29690000504. [DOI] [Google Scholar]
- Kan C. S.; Calvert J. G.; Shaw J. H. Reactive channels of the CH3O2-CH3O2 reaction. J. Phys. Chem. 1980, 84, 3411–3417. 10.1021/j100462a020. [DOI] [Google Scholar]
- Niki H.; Maker P. D.; Savage C. M.; Breitenbach L. P. A ft-ir study of a transitory product in the gas-phase ozone-ethylene reaction. J. Phys. Chem. 1981, 85, 1024–1027. 10.1021/j150608a020. [DOI] [Google Scholar]
- Niki H.; Maker P. D.; Savage C. M.; Breitenbach L. P. Fourier-transform infrared studies of the self-reaction of C2H5O2 radicals. J. Phys. Chem. 1982, 86, 3825–3829. 10.1021/j100216a023. [DOI] [Google Scholar]
- Tyndall G. S.; Cox R. A.; Granier C.; Lesclaux R.; Moortgat G. K.; Pilling M. J.; Ravishankara A. R.; Wallington T. J. Atmospheric chemistry of small organic peroxy radicals. J. Geophys. Res. Atmos. 2001, 106, 12157–12182. 10.1029/2000JD900746. [DOI] [Google Scholar]
- Noell A. C.; Alconcel L. S.; Robichaud D. J.; Okumura M.; Sander S. P. near-infrared kinetic spectroscopy of the HO2 and C2H5O2 self-reactions and cross reactions. J. Phys. Chem. A 2010, 114, 6983–6995. 10.1021/jp912129j. [DOI] [PubMed] [Google Scholar]
- Ghigo G.; Maranzana A.; Tonachini G. Combustion and atmospheric oxidation of hydrocarbons: Theoretical study of the methyl peroxyl self-reaction. J. Chem. Phys. 2003, 118, 10575–10583. 10.1063/1.1574316. [DOI] [Google Scholar]
- Lee R.; Gryn’ova G.; Ingold K. U.; Coote M. L. Why are sec-alkylperoxyl bimolecular self-reactions orders of magnitude faster than the analogous reactions of tert-alkylperoxyls? The unanticipated role of CH hydrogen bond donation. Phys. Chem. Chem. Phys. 2016, 18, 23673–23679. 10.1039/C6CP04670C. [DOI] [PubMed] [Google Scholar]
- Liang Y. N.; Li J.; Wang Q. D.; Wang F.; Li X. Y. Computational study of the reaction mechanism of the methylperoxy self-reaction. J. Phys. Chem. A 2011, 115, 13534–13541. 10.1021/jp2048508. [DOI] [PubMed] [Google Scholar]
- Zhang P.; Wang W. L.; Zhang T. L.; Chen L.; Du Y. M.; Li C. Y.; Lu J. Theoretical study on the mechanism and kinetics for the self-reaction of C2H5O2 radicals. J. Phys. Chem. A 2012, 116, 4610–4620. 10.1021/jp301308u. [DOI] [PubMed] [Google Scholar]
- Dibble T. S. Failures and limitations of quantum chemistry for two key problems in the atmospheric chemistry of peroxy radicals. Atmos. Environ. 2008, 42, 5837–5848. 10.1016/j.atmosenv.2007.11.005. [DOI] [Google Scholar]
- Ng N. L.; Kwan A. J.; Surratt J. D.; Chan A. W. H.; Chhabra P. S.; Sorooshian A.; Pye H. O. T.; Crounse J. D.; Wennberg P. O.; et al. Secondary organic aerosol (SOA) formation from reaction of isoprene with nitrate radicals (NO3). Atmos. Chem. Phys. 2008, 8, 4117–4140. 10.5194/acp-8-4117-2008. [DOI] [Google Scholar]
- Sarnela N.; Jokinen T.; Duplissy J.; Yan C.; Nieminen T.; Ehn M.; Schobesberger S.; Heinritzi M.; Ehrhart S.; et al. Measurement-model comparison of stabilized Criegee intermediate and highly oxygenated molecule production in the CLOUD chamber. Atmos. Chem. Phys. 2018, 18, 2363–2380. 10.5194/acp-18-2363-2018. [DOI] [Google Scholar]
- Stark M. S. Addition of peroxyl radicals to alkenes and the reaction of oxygen with alkyl radicals. J. Am. Chem. Soc. 2000, 122, 4162–4170. 10.1021/ja993760m. [DOI] [Google Scholar]
- Donahue N. M.; Drozd G. T.; Epstein S. A.; Presto A. A.; Kroll J. H. Adventures in ozoneland: down the rabbit-hole. Phys. Chem. Chem. Phys. 2011, 13, 10848–10857. 10.1039/c0cp02564j. [DOI] [PubMed] [Google Scholar]
- Kristensen K.; Cui T.; Zhang H.; Gold A.; Glasius M.; Surratt J. D. Dimers in α-pinene secondary organic aerosol: effect of hydroxyl radical, ozone, relative humidity and aerosol acidity. Atmos. Chem. Phys. 2014, 14, 4201–4218. 10.5194/acp-14-4201-2014. [DOI] [Google Scholar]
- McDowell C. A.; Sharples L. K. The photochemical oxidation of aldehydes in the gaseous phase.1. The kinetics of the photochemical oxidation of acetaldehyde. Can. J. Chem. 1958, 36, 251–257. 10.1139/v58-034. [DOI] [Google Scholar]
- McDowell C. A.; Sifniades S. Oxygen-18 tracer evidence for the termination mechanism in the photochemical oxidation of acetaldehyde. Can. J. Chem. 1963, 41, 300–307. 10.1139/v63-046. [DOI] [Google Scholar]
- Ziemann P. J. evidence for low-volatility diacyl peroxides as a nucleating agent and major component of aerosol formed from reactions of O3 with cyclohexene and homologous compounds. J. Phys. Chem. A 2002, 106, 4390–4402. 10.1021/jp012925m. [DOI] [Google Scholar]
- Atkinson R. Atmospheric chemistry of VOCs and NOx. Atmos. Environ. 2000, 34, 2063–2101. 10.1016/S1352-2310(99)00460-4. [DOI] [Google Scholar]
- Schwantes R. H.; Teng A. P.; Nguyen T. B.; Coggon M. M.; Crounse J. D.; St. Clair J. M.; Zhang X.; Schilling K. A.; Seinfeld J. H.; Wennberg P. O. Isoprene NO3 oxidation products from the RO2 + HO2 pathway. J. Phys. Chem. A 2015, 119, 10158–10171. 10.1021/acs.jpca.5b06355. [DOI] [PubMed] [Google Scholar]
- Muller J. F.; Liu Z.; Nguyen V. S.; Stavrakou T.; Harvey J. N.; Peeters J. The reaction of methyl peroxy and hydroxyl radicals as a major source of atmospheric methanol. Nat. Commun. 2016, 7, 13213. 10.1038/ncomms13213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurtén T.; Lane J. R.; Jørgensen S.; Kjaergaard H. G. A computational study of the oxidation of SO2 to SO3 by gas-phase organic oxidants. J. Phys. Chem. A 2011, 115, 8669–8681. 10.1021/jp203907d. [DOI] [PubMed] [Google Scholar]
- Huang H. L.; Chao W.; Lin J. J. M. Kinetics of a Criegee intermediate that would survive high humidity and may oxidize atmospheric SO2. Proc. Natl. Acad. Sci. U. S. A. 2015, 112, 10857–10862. 10.1073/pnas.1513149112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sipila M.; Jokinen T.; Berndt T.; Richters S.; Makkonen R.; Donahue N. M.; Mauldin R. L.; Kurten T.; Paasonen P.; et al. Reactivity of stabilized Criegee intermediates (sCIs) from isoprene and monoterpene ozonolysis toward SO2 and organic acids. Atmos. Chem. Phys. 2014, 14, 12143–12153. 10.5194/acp-14-12143-2014. [DOI] [Google Scholar]
- Welz O.; Eskola A. J.; Sheps L.; Rotavera B.; Savee J. D.; Scheer A. M.; Osborn D. L.; Lowe D.; Booth A. M.; Xiao P.; Khan M. A. H.; Percival C. J.; Shallcross D. E.; Taatjes C. A. Rate Coefficients of C1 and C2 criegee intermediate reactions with formic and acetic acid near the collision limit: direct kinetics measurements and atmospheric implications. Angew. Chem., Int. Ed. 2014, 53, 4547–4550. 10.1002/anie.201400964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkinson R.; Baulch D. L.; Cox R. A.; Crowley J. N.; Hampson R. F.; Hynes R. G.; Jenkin M. E.; Rossi M. J.; Troe J.; Wallington T. J. Evaluated kinetic and photochemical data for atmospheric chemistry: Volume IV – gas phase reactions of organic halogen species. Atmos. Chem. Phys. 2008, 8, 4141–4496. 10.5194/acp-8-4141-2008. [DOI] [Google Scholar]
- Ziemann P. J.; Atkinson R. Kinetics, products, and mechanisms of secondary organic aerosol formation. Chem. Soc. Rev. 2012, 41, 6582. 10.1039/c2cs35122f. [DOI] [PubMed] [Google Scholar]
- Frege C.; Ortega I. K.; Rissanen M. P.; Praplan A. P.; Steiner G.; Heinritzi M.; Ahonen L.; Amorim A.; Bernhammer A. K.; et al. Influence of temperature on the molecular composition of ions and charged clusters during pure biogenic nucleation. Atmos. Chem. Phys. 2018, 18, 65–79. 10.5194/acp-18-65-2018. [DOI] [Google Scholar]
- Crounse J. D.; Paulot F.; Kjaergaard H. G.; Wennberg P. O. Peroxy radical isomerization in the oxidation of isoprene. Phys. Chem. Chem. Phys. 2011, 13, 13607–13613. 10.1039/c1cp21330j. [DOI] [PubMed] [Google Scholar]
- Saunders S. M.; Jenkin M. E.; Derwent R. G.; Pilling M. J. Protocol for the development of the Master Chemical Mechanism, MCM v3 (Part A): tropospheric degradation of non-aromatic volatile organic compounds. Atmos. Chem. Phys. 2003, 3, 161–180. 10.5194/acp-3-161-2003. [DOI] [Google Scholar]
- Holland F.; Hofzumahaus A.; Schafer R.; Kraus A.; Patz H. W. Measurements of OH and HO2 radical concentrations and photolysis frequencies during BERLIOZ. J. Geophys. Res. Atmos. 2003, 108, PHO2-1–PHO2-23. 10.1029/2001JD001393. [DOI] [Google Scholar]
- Lelieveld J.; Butler T. M.; Crowley J. N.; Dillon T. J.; Fischer H.; Ganzeveld L.; Harder H.; Lawrence M. G.; Martinez M.; et al. Atmospheric oxidation capacity sustained by a tropical forest. Nature 2008, 452, 737–740. 10.1038/nature06870. [DOI] [PubMed] [Google Scholar]
- Ren X. R.; Harder H.; Martinez M.; Lesher R. L.; Oliger A.; Shirley T.; Adams J.; Simpas J. B.; Brune W. H. HOx concentrations and OH reactivity observations in New York City during PMTACS-NY2001. Atmos. Environ. 2003, 37, 3627–3637. 10.1016/S1352-2310(03)00460-6. [DOI] [Google Scholar]
- Richters S.; Herrmann H.; Berndt T. Highly oxidized RO2 radicals and consecutive products from the ozonolysis of three sesquiterpenes. Environ. Sci. Technol. 2016, 50, 2354–2362. 10.1021/acs.est.5b05321. [DOI] [PubMed] [Google Scholar]
- Pankow J. F.; Barsanti K. C. The carbon number-polarity grid: A means to manage the complexity of the mix of organic compounds when modeling atmospheric organic particulate matter. Atmos. Environ. 2009, 43, 2829–2835. 10.1016/j.atmosenv.2008.12.050. [DOI] [Google Scholar]
- Nannoolal Y.; Rarey J.; Ramjugernath D. Estimation of pure component properties: Part 3. Estimation of the vapor pressure of non-electrolyte organic compounds via group contributions and group interactions. Fluid Phase Equilib. 2008, 269, 117–133. 10.1016/j.fluid.2008.04.020. [DOI] [Google Scholar]
- Nannoolal Y.; Rarey J.; Ramjugernath D.; Cordes W. Estimation of pure component properties: Part 1. Estimation of the normal boiling point of non-electrolyte organic compounds via group contributions and group interactions. Fluid Phase Equilib. 2004, 226, 45–63. 10.1016/j.fluid.2004.09.001. [DOI] [Google Scholar]
- DeCarlo P. F.; Dunlea E. J.; Kimmel J. R.; Aiken A. C.; Sueper D.; Crounse J.; Wennberg P. O.; Emmons L.; Shinozuka Y.; et al. Fast airborne aerosol size and chemistry measurements above Mexico City and Central Mexico during the MILAGRO campaign. Atmos. Chem. Phys. 2008, 8, 4027–4048. 10.5194/acp-8-4027-2008. [DOI] [Google Scholar]
- Kroll J. H.; Donahue N. M.; Jimenez J. L.; Kessler S. H.; Canagaratna M. R.; Wilson K. R.; Altieri K. E.; Mazzoleni L. R.; Wozniak A. S.; et al. Carbon oxidation state as a metric for describing the chemistry of atmospheric organic aerosol. Nat. Chem. 2011, 3, 133–139. 10.1038/nchem.948. [DOI] [PubMed] [Google Scholar]
- Grieshop A. P.; Logue J. M.; Donahue N. M.; Robinson A. L. Laboratory investigation of photochemical oxidation of organic aerosol from wood fires 1: measurement and simulation of organic aerosol evolution. Atmos. Chem. Phys. 2009, 9, 1263–1277. 10.5194/acp-9-1263-2009. [DOI] [Google Scholar]
- Presto A. A.; Donahue N. M. Investigation of α-Pinene + Ozone Secondary Organic Aerosol Formation at Low Total Aerosol Mass. Environ. Sci. Technol. 2006, 40, 3536–3543. 10.1021/es052203z. [DOI] [PubMed] [Google Scholar]
- Riipinen I.; Pierce J. R.; Donahue N. M.; Pandis S. N. Equilibration time scales of organic aerosol inside thermodenuders: Evaporation kinetics versus thermodynamics. Atmos. Environ. 2010, 44, 597–607. 10.1016/j.atmosenv.2009.11.022. [DOI] [Google Scholar]
- Stanier C. O.; Pathak R. K.; Pandis S. N. measurements of the volatility of aerosols from α-pinene ozonolysis. Environ. Sci. Technol. 2007, 41, 2756–2763. 10.1021/es0519280. [DOI] [PubMed] [Google Scholar]
- Vaden T. D.; Imre D.; Beranek J.; Shrivastava M.; Zelenyuk A. Evaporation kinetics and phase of laboratory and ambient secondary organic aerosol. Proc. Natl. Acad. Sci. U. S. A. 2011, 108, 2190–2195. 10.1073/pnas.1013391108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chhabra P. S.; Lambe A. T.; Canagaratna M. R.; Stark H.; Jayne J. T.; Onasch T. B.; Davidovits P.; Kimmel J. R.; Worsnop D. R. Application of high-resolution time-of-flight chemical ionization mass spectrometry measurements to estimate volatility distributions of α-pinene and naphthalene oxidation products. Atmos. Meas. Tech. 2015, 8, 1–18. 10.5194/amt-8-1-2015. [DOI] [Google Scholar]
- Daumit K. E.; Kessler S. H.; Kroll J. H. Average chemical properties and potential formation pathways of highly oxidized organic aerosol. Faraday Discuss. 2013, 165, 181–202. 10.1039/c3fd00045a. [DOI] [PubMed] [Google Scholar]
- Bilde M.; Barsanti K.; Booth M.; Cappa C. D.; Donahue N. M.; Emanuelsson E. U.; McFiggans G.; Krieger U. K.; Marcolli C.; et al. Saturation Vapor Pressures and Transition Enthalpies of Low-Volatility Organic Molecules of Atmospheric Relevance: From Dicarboxylic Acids to Complex Mixtures. Chem. Rev. 2015, 115, 4115–4156. 10.1021/cr5005502. [DOI] [PubMed] [Google Scholar]
- Pankow J. F.; Asher W. E. SIMPOL.1: a simple group contribution method for predicting vapor pressures and enthalpies of vaporization of multifunctional organic compounds. Atmos. Chem. Phys. 2008, 8, 2773–2796. 10.5194/acp-8-2773-2008. [DOI] [Google Scholar]
- Öström E.; Putian Z.; Schurgers G.; Mishurov M.; Kivekäs N.; Lihavainen H.; Ehn M.; Rissanen M. P.; Kurtén T.; et al. Modeling the role of highly oxidized multifunctional organic molecules for the growth of new particles over the boreal forest region. Atmos. Chem. Phys. 2017, 17, 8887–8901. 10.5194/acp-17-8887-2017. [DOI] [Google Scholar]
- Dal Maso M.; Kulmala M.; Lehtinen K. E. J.; Mäkelä J. M.; Aalto P.; O’Dowd C. D. Condensation and coagulation sinks and formation of nucleation mode particles in coastal and boreal forest boundary layers. J. Geophys. Res. Atmos. 2002, 107, PAR2-1–PAR2-10. 10.1029/2001JD001053. [DOI] [Google Scholar]
- Petäjä T.; Mauldin R. L. III; Kosciuch E.; McGrath J.; Nieminen T.; Paasonen P.; Boy M.; Adamov A.; Kotiaho T.; Kulmala M. Sulfuric acid and OH concentrations in a boreal forest site. Atmos. Chem. Phys. 2009, 9, 7435–7448. 10.5194/acp-9-7435-2009. [DOI] [Google Scholar]
- Tan Z.; Rohrer F.; Lu K.; Ma X.; Bohn B.; Broch S.; Dong H.; Fuchs H.; Gkatzelis G. I.; et al. Wintertime photochemistry in Beijing: observations of ROx radical concentrations in the North China Plain during the BEST-ONE campaign. Atmos. Chem. Phys. 2018, 18, 12391–12411. 10.5194/acp-18-12391-2018. [DOI] [Google Scholar]
- Wu Z.; Hu M.; Liu S.; Wehner B.; Bauer S.; Ma ßling A.; Wiedensohler A.; Petäjä T.; Dal Maso M.; Kulmala M. New particle formation in Beijing, China: Statistical analysis of a 1-year data set. J. Geophys. Res. Atmos. 2007, 112, JD007406. 10.1029/2006JD007406. [DOI] [Google Scholar]
- Herrmann H.; Schaefer T.; Tilgner A.; Styler S. A.; Weller C.; Teich M.; Otto T. Tropospheric aqueous-phase chemistry: kinetics, mechanisms, and its coupling to a changing gas phase. Chem. Rev. 2015, 115, 4259–4334. 10.1021/cr500447k. [DOI] [PubMed] [Google Scholar]
- Epstein S. A.; Blair S. L.; Nizkorodov S. A. Direct photolysis of α-pinene ozonolysis secondary organic aerosol: effect on particle mass and peroxide content. Environ. Sci. Technol. 2014, 48, 11251–11258. 10.1021/es502350u. [DOI] [PubMed] [Google Scholar]
- Tobias H. J.; Ziemann P. J. thermal desorption mass spectrometric analysis of organic aerosol formed from reactions of 1-tetradecene and O3 in the presence of alcohols and carboxylic acids. Environ. Sci. Technol. 2000, 34, 2105–2115. 10.1021/es9907156. [DOI] [Google Scholar]
- Riva M.; Budisulistiorini S. H.; Zhang Z.; Gold A.; Surratt J. D. Chemical characterization of secondary organic aerosol constituents from isoprene ozonolysis in the presence of acidic aerosol. Atmos. Environ. 2016, 130, 5–13. 10.1016/j.atmosenv.2015.06.027. [DOI] [Google Scholar]
- Riva M.; Da Silva Barbosa T.; Lin Y.-H.; Stone E. A.; Gold A.; Surratt J. D. Chemical characterization of organosulfates in secondary organic aerosol derived from the photooxidation of alkanes. Atmos. Chem. Phys. 2016, 16, 11001–11018. 10.5194/acp-16-11001-2016. [DOI] [Google Scholar]
- Faxon C.; Hammes J.; Le Breton M.; Pathak R. K.; Hallquist M. Characterization of organic nitrate constituents of secondary organic aerosol (SOA) from nitrate-radical-initiated oxidation of limonene using high-resolution chemical ionization mass spectrometry. Atmos. Chem. Phys. 2018, 18, 5467–5481. 10.5194/acp-18-5467-2018. [DOI] [Google Scholar]
- Rindelaub J. D.; Borca C. H.; Hostetler M. A.; Slade J. H.; Lipton M. A.; Slipchenko L. V.; Shepson P. B. The acid-catalyzed hydrolysis of an α-pinene-derived organic nitrate: kinetics, products, reaction mechanisms, and atmospheric impact. Atmos. Chem. Phys. 2016, 16, 15425–15432. 10.5194/acp-16-15425-2016. [DOI] [Google Scholar]
- Rindelaub J. D.; McAvey K. M.; Shepson P. B. The photochemical production of organic nitrates from α-pinene and loss via acid-dependent particle phase hydrolysis. Atmos. Environ. 2015, 100, 193–201. 10.1016/j.atmosenv.2014.11.010. [DOI] [Google Scholar]
- Gaston C. J.; Riedel T. P.; Zhang Z.; Gold A.; Surratt J. D.; Thornton J. A. reactive uptake of an isoprene-derived epoxydiol to submicron aerosol particles. Environ. Sci. Technol. 2014, 48, 11178–11186. 10.1021/es5034266. [DOI] [PubMed] [Google Scholar]
- Surratt J. D.; Chan A. W. H.; Eddingsaas N. C.; Chan M.; Loza C. L.; Kwan A. J.; Hersey S. P.; Flagan R. C.; Wennberg P. O.; Seinfeld J. H. Reactive intermediates revealed in secondary organic aerosol formation from isoprene. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 6640–6645. 10.1073/pnas.0911114107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riva M.; Budisulistiorini S. H.; Zhang Z.; Gold A.; Thornton J. A.; Turpin B. J.; Surratt J. D. Multiphase reactivity of gaseous hydroperoxide oligomers produced from isoprene ozonolysis in the presence of acidified aerosols. Atmos. Environ. 2017, 152, 314–322. 10.1016/j.atmosenv.2016.12.040. [DOI] [Google Scholar]
- Badali K. M.; Zhou S.; Aljawhary D.; Antiñolo M.; Chen W. J.; Lok A.; Mungall E.; Wong J. P. S.; Zhao R.; Abbatt J. P. D. Formation of hydroxyl radicals from photolysis of secondary organic aerosol material. Atmos. Chem. Phys. 2015, 15, 7831–7840. 10.5194/acp-15-7831-2015. [DOI] [Google Scholar]
- Henry K. M.; Donahue N. M. photochemical aging of α-pinene secondary organic aerosol: effects of OH radical sources and photolysis. J. Phys. Chem. A 2012, 116, 5932–5940. 10.1021/jp210288s. [DOI] [PubMed] [Google Scholar]
- Surratt J. D.; Murphy S. M.; Kroll J. H.; Ng N. L.; Hildebrandt L.; Sorooshian A.; Szmigielski R.; Vermeylen R.; Maenhaut W.; et al. chemical composition of secondary organic aerosol formed from the photooxidation of isoprene. J. Phys. Chem. A 2006, 110, 9665–9690. 10.1021/jp061734m. [DOI] [PubMed] [Google Scholar]
- Mang S. A.; Henricksen D. K.; Bateman A. P.; Andersen M. P. S.; Blake D. R.; Nizkorodov S. A. contribution of carbonyl photochemistry to aging of atmospheric secondary organic aerosol. J. Phys. Chem. A 2008, 112, 8337–8344. 10.1021/jp804376c. [DOI] [PubMed] [Google Scholar]
- Pan X.; Underwood J. S.; Xing J. H.; Mang S. A.; Nizkorodov S. A. Photodegradation of secondary organic aerosol generated from limonene oxidation by ozone studied with chemical ionization mass spectrometry. Atmos. Chem. Phys. 2009, 9, 3851–3865. 10.5194/acp-9-3851-2009. [DOI] [Google Scholar]
- George C.; Ammann M.; D’Anna B.; Donaldson D. J.; Nizkorodov S. A. Heterogeneous photochemistry in the atmosphere. Chem. Rev. 2015, 115, 4218–4258. 10.1021/cr500648z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jokinen T.; Kontkanen J.; Lehtipalo K.; Manninen H. E.; Aalto J.; Porcar-Castell A.; Garmash O.; Nieminen T.; Ehn M.; Kangasluoma J.; Junninen H.; Levula J.; Duplissy J.; Ahonen L. R.; Rantala P.; Heikkinen L.; Yan C.; Sipilä M.; Worsnop D. R.; Bäck J.; Petäjä T.; Kerminen V.-M.; Kulmala M. Solar eclipse demonstrating the importance of photochemistry in new particle formation. Sci. Rep. 2017, 7, 45707. 10.1038/srep45707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarnela N.; Jokinen T.; Nieminen T.; Lehtipalo K.; Junninen H.; Kangasluoma J.; Hakala J.; Taipale R.; Schobesberger S.; et al. Sulphuric acid and aerosol particle production in the vicinity of an oil refinery. Atmos. Environ. 2015, 119, 156–166. 10.1016/j.atmosenv.2015.08.033. [DOI] [Google Scholar]
- Frege C.; Bianchi F.; Molteni U.; Trostl J.; Junninen H.; Henne S.; Sipila M.; Herrmann E.; Rossi M. J.; et al. Chemical characterization of atmospheric ions at the high altitude research station Jungfraujoch (Switzerland). Atmos. Chem. Phys. 2017, 17, 2613–2629. 10.5194/acp-17-2613-2017. [DOI] [Google Scholar]
- Brophy P.; Farmer D. K. A switchable reagent ion high resolution time-of-flight chemical ionization mass spectrometer for real-time measurement of gas phase oxidized species: characterization from the 2013 southern oxidant and aerosol study. Atmos. Meas. Tech. 2015, 8, 2945–2959. 10.5194/amt-8-2945-2015. [DOI] [Google Scholar]
- Krechmer J. E.; Coggon M. M.; Massoli P.; Nguyen T. B.; Crounse J. D.; Hu W. W.; Day D. A.; Tyndall G. S.; Henze D. K.; et al. formation of low volatility organic compounds and secondary organic aerosol from isoprene hydroxyhydroperoxide low-NO oxidation. Environ. Sci. Technol. 2015, 49, 10330–10339. 10.1021/acs.est.5b02031. [DOI] [PubMed] [Google Scholar]
- Bianchi F.; Trostl J.; Junninen H.; Frege C.; Henne S.; Hoyle C. R.; Molteni U.; Herrmann E.; Adamov A.; et al. New particle formation in the free troposphere: A question of chemistry and timing. Science 2016, 352, 1109–1112. 10.1126/science.aad5456. [DOI] [PubMed] [Google Scholar]
- Kulmala M.; Kontkanen J.; Junninen H.; Lehtipalo K.; Manninen H. E.; Nieminen T.; Petaja T.; Sipila M.; Schobesberger S.; et al. Direct observations of atmospheric aerosol nucleation. Science 2013, 339, 943–946. 10.1126/science.1227385. [DOI] [PubMed] [Google Scholar]
- Lehtipalo K.; Sipila M.; Junninen H.; Ehn M.; Berndt T.; Kajos M. K.; Worsnop D. R.; Petaja T.; Kulmala M. Observations of nano-CN in the nocturnal boreal forest. Aerosol Sci. Technol. 2011, 45, 499–509. 10.1080/02786826.2010.547537. [DOI] [Google Scholar]
- Chrit M.; Sartelet K.; Sciare J.; Pey J.; Marchand N.; Couvidat F.; Sellegri K.; Beekmann M. Modelling organic aerosol concentrations and properties during ChArMEx summer campaigns of 2012 and 2013 in the western Mediterranean region. Atmos. Chem. Phys. 2017, 17, 12509–12531. 10.5194/acp-17-12509-2017. [DOI] [Google Scholar]
- Huang X.; Zhou L.; Ding A.; Qi X.; Nie W.; Wang M.; Chi X.; Petäjä T.; Kerminen V. M.; et al. Comprehensive modelling study on observed new particle formation at the SORPES station in Nanjing, China. Atmos. Chem. Phys. 2016, 16, 2477–2492. 10.5194/acp-16-2477-2016. [DOI] [Google Scholar]
- Jenkin M. E.; Saunders S. M.; Pilling M. J. The tropospheric degradation of volatile organic compounds: A protocol for mechanism development. Atmos. Environ. 1997, 31, 81–104. 10.1016/S1352-2310(96)00105-7. [DOI] [Google Scholar]
- Jenkin M. E.; Young J. C.; Rickard A. R. The MCM v3.3.1 degradation scheme for isoprene. Atmos. Chem. Phys. 2015, 15, 11433–11459. 10.5194/acp-15-11433-2015. [DOI] [Google Scholar]
- Kulmala M.; Toivonen A.; Makela J. M.; Laaksonen A. Analysis of the growth of nucleation mode particles observed in Boreal forest. Tellus, Ser. B 1998, 50, 449–462. 10.3402/tellusb.v50i5.16229. [DOI] [Google Scholar]
- Laaksonen A.; Kulmala M.; O’Dowd C. D.; Joutsensaari J.; Vaattovaara P.; Mikkonen S.; Lehtinen K. E. J.; Sogacheva L.; Dal Maso M.; et al. The role of VOC oxidation products in continental new particle formation. Atmos. Chem. Phys. 2008, 8, 2657–2665. 10.5194/acp-8-2657-2008. [DOI] [Google Scholar]
- Metzger A.; Verheggen B.; Dommen J.; Duplissy J.; Prevot A. S. H.; Weingartner E.; Riipinen I.; Kulmala M.; Spracklen D. V.; et al. Evidence for the role of organics in aerosol particle formation under atmospheric conditions. Proc. Natl. Acad. Sci. U. S. A. 2010, 107, 6646–6651. 10.1073/pnas.0911330107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Dowd C. D.; Aalto P.; Hmeri K.; Kulmala M.; Hoffmann T. Aerosol formation - Atmospheric particles from organic vapours. Nature 2002, 416, 497–498. 10.1038/416497a. [DOI] [PubMed] [Google Scholar]
- Zhang R. Y.; Suh I.; Zhao J.; Zhang D.; Fortner E. C.; Tie X. X.; Molina L. T.; Molina M. J. Atmospheric new particle formation enhanced by organic acids. Science 2004, 304, 1487–1490. 10.1126/science.1095139. [DOI] [PubMed] [Google Scholar]
- Zhang R. Y.; Wang L.; Khalizov A. F.; Zhao J.; Zheng J.; McGraw R. L.; Molina L. T. Formation of nanoparticles of blue haze enhanced by anthropogenic pollution. Proc. Natl. Acad. Sci. U. S. A. 2009, 106, 17650–17654. 10.1073/pnas.0910125106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riipinen I.; Yli-Juuti T.; Pierce J. R.; Petaja T.; Worsnop D. R.; Kulmala M.; Donahue N. M. The contribution of organics to atmospheric nanoparticle growth. Nat. Geosci. 2012, 5, 453–458. 10.1038/ngeo1499. [DOI] [Google Scholar]
- Riccobono F.; Schobesberger S.; Scott C. E.; Dommen J.; Ortega I. K.; Rondo L.; Almeida J.; Amorim A.; Bianchi F.; et al. Oxidation products of biogenic emissions contribute to nucleation of atmospheric particles. Science 2014, 344, 717–721. 10.1126/science.1243527. [DOI] [PubMed] [Google Scholar]
- Gordon H.; Kirkby J.; Baltensperger U.; Bianchi F.; Breitenlechner M.; Curtius J.; Dias A.; Dommen J.; Donahue N. M.; et al. Causes and importance of new particle formation in the present-day and preindustrial atmospheres. J. Geophys. Res. Atmos. 2017, 122, 8739–8760. 10.1002/2017JD026844. [DOI] [Google Scholar]
- Gordon H.; Sengupta K.; Rap A.; Duplissy J.; Frege C.; Williamson C.; Heinritzi M.; Simon M.; Yan C.; et al. Reduced anthropogenic aerosol radiative forcing caused by biogenic new particle formation. Proc. Natl. Acad. Sci. U. S. A. 2016, 113, 12053–12058. 10.1073/pnas.1602360113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elm J.; Myllys N.; Hyttinen N.; Kurtén T. Computational study of the clustering of a cyclohexene autoxidation product C6H8O7 with itself and sulfuric acid. J. Phys. Chem. A 2015, 119, 8414–8421. 10.1021/acs.jpca.5b04040. [DOI] [PubMed] [Google Scholar]
- Elm J.; Myllys N.; Luy J.-N.; Kurtén T.; Vehkamäki H. The effect of water and bases on the clustering of a cyclohexene autoxidation product C6H8O7 with sulfuric acid. J. Phys. Chem. A 2016, 120, 2240–2249. 10.1021/acs.jpca.6b00677. [DOI] [PubMed] [Google Scholar]
- Chuang W. K.; Donahue N. M. Dynamic consideration of smog chamber experiments. Atmos. Chem. Phys. 2017, 17, 10019–10036. 10.5194/acp-17-10019-2017. [DOI] [Google Scholar]
- Guenther A.; Hewitt C. N.; Erickson D.; Fall R.; Geron C.; Graedel T.; Harley P.; Klinger L.; Lerdau M.; et al. A global-model of natural volatile organic-compound emissions. J. Geophys. Res. 1995, 100, 8873–8892. 10.1029/94JD02950. [DOI] [Google Scholar]
- Rose C.; Zha Q. Z.; Dada L.; Yan C.; Lehtipalo K.; Junninen H.; Mazon S. B.; Jokinen T.; Sarnela N.; Sipilä M.; Petäjä T.; Kerminen V.-M.; Bianchi F.; Kulmala M. Observations of biogenic ion-induced cluster formation in the atmosphere. Sci. Adv. 2018, 4, eaar5218. 10.1126/sciadv.aar5218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazon S. B.; Kontkanen J.; Manninen H. E.; Nieminen T.; Kerminen V. M.; Kulmala M. A long-term comparison of nighttime cluster events and daytime ion formation in a boreal forest. Boreal Env. Res. 2016, 21, 242–261. [Google Scholar]
- Ehn M.; Berndt T.; Wildt J.; Mentel T. Highly oxygenated molecules from atmospheric autoxidation of hydrocarbons: a prominent challenge for chemical kinetics studies. Int. J. Chem. Kinet. 2017, 49, 821–831. 10.1002/kin.21130. [DOI] [Google Scholar]
- Kürten A.; Jokinen T.; Simon M.; Sipilä M.; Sarnela N.; Junninen H.; Adamov A.; Almeida J.; Amorim A.; et al. Neutral molecular cluster formation of sulfuric acid–dimethylamine observed in real time under atmospheric conditions. Proc. Natl. Acad. Sci. U. S. A. 2014, 111, 15019–15024. 10.1073/pnas.1404853111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ziemann P. J. Evidence for low-volatility diacyl peroxides as a nucleating agent and major component of aerosol formed from reactions of O3 with cyclohexene and homologous compounds. J. Phys. Chem. A 2002, 106, 4390–4402. 10.1021/jp012925m. [DOI] [Google Scholar]
- Claflin M. S.; Ziemann P. J. Identification and quantitation of aerosol products of the reaction of β-pinene with NO3 radicals and implications for gas- and particle-phase reaction mechanisms. J. Phys. Chem. A 2018, 122, 3640–3652. 10.1021/acs.jpca.8b00692. [DOI] [PubMed] [Google Scholar]
- Xu L.; Moller K. H.; Crounse J. D.; Otkjaer R. V.; Kjaergaard H. G.; Wennberg P. O. Unimolecular reactions of peroxy radicals formed in the oxidation of a-pinene and b-pinene by hydroxyl radicals. J. Phys. Chem. A 2019, 10.1021/acs.jpca.8b11726. [DOI] [PubMed] [Google Scholar]
- Praske E.; Otkjaer R. V.; Crounse J. D.; Hethcox J. C.; Stoltz B. M.; Kjaergaard H. G.; Wennberg P. O. Intermolecular hydrogen shift chemistry of hydroperoxy-substituted peroxy radicals. J. Phys. Chem. A 2019, 123, 590–600. 10.1021/acs.jpca.8b09745. [DOI] [PubMed] [Google Scholar]
- Zha Q.; Yan C.; Junninen H.; Riva M.; Sarnela N.; Aalto J.; Quéléver L.; Schallhart S.; Dada L.; Heikkinen L.; Peräkylä O.; Zou J.; Rose C.; Wang Y.; Mammarella I.; Katul G.; Vesala T.; Worsnop D. R.; Kulmala M.; Petäjä T.; Bianchi F.; Ehn M. Vertical characterization of highly oxygenated molecules (HOMs) below and above a boreal forest canopy. Atmos. Chem. Phys. 2018, 18, 17437–17450. 10.5194/acp-18-17437-2018. [DOI] [Google Scholar]
- Otkjær R. V.; Jakobsen H. H.; Tram C. M.; Kjaergaard H. G. Calculated hydrogen shift rate constants in substituted alkyl peroxy radicals. J. Phys. Chem. A 2018, 122, 8665–8673. 10.1021/acs.jpca.8b06223. [DOI] [PubMed] [Google Scholar]
- Møller K. H.; Bates K.; Kjaergaard H. G. The importance of peroxy radical hydrogen shift reactions in atmospheric isoprene oxidation. J. Phys. Chem. A 2019, 123, 920–932. 10.1021/acs.jpca.8b10432. [DOI] [PubMed] [Google Scholar]