

Abstract
Triple negative breast cancer (TNBC) is among the most aggressive breast cancer subtypes with poor prognosis. The purpose of this study is to better understand the molecular basis of TNBC as well as develop new therapeutic strategies. Our results demonstrate that HDAC9 is overexpressed in TNBC compared to non-TNBC cell lines and tissues. Furthermore, we show that HDAC9 overexpression is inversely proportional with miR-206 expression levels. Subsequent HDAC9 siRNA knockdown was then shown to restore miR-206 while also decreasing VEGF and MAPK3 levels. This study highlights HDAC9 as a mediator of invasion and angiogenesis in TNBC cells through VEGF and MAPK3 by modulating miR-206 expression and suggests that selective inhibition of HDAC9 may be an efficient route for TNBC therapy.
Keywords: HDAC9, triple negative breast cancer, microRNA, invasion, angiogenesis
1. Introduction
Two classes of enzymes impact the acetylation state of histone proteins — histone acetyltransferases (HATs) and histone deacetylases (HDACs) [1, 2]. The HDAC family of enzymes is involved in various biological processes including transcriptional control, growth arrest, and cell death, particularly in tumor development and proliferation [3–7]. A number of HDAC inhibitors have been characterized that inhibit tumor growth in vitro and in vivo at amounts that have little to no toxicity [8–15]. The results from these studies and their proposed mechanisms indicate that HDACs are excellent targets for cancer treatment. However, clinical benefits of selective versus broad HDAC inhibitors are unknown and the appropriateness of inhibitor may depend on tumor HDAC enzyme expression, enzyme selectivity of the inhibitor, and the desired effects. Although there are several published studies [16, 17] and numerous ongoing clinical trials involving HDAC inhibitors [18], very little is known about the roles of individual HDAC enzymes, and a formal assessment of HDAC enzyme expression is not routinely done. Emerging data suggests that the currently used HDAC inhibitors may differ significantly with regard to target selectivity. In addition, the expression of HDAC enzymes may vary considerably between normal and tumor tissues and likely between different phenotypes of tumor tissues. HDAC9 is thought to regulate gene expression through epigenetic modulation of the chromatin structure by catalyzing the deacetylation of histone proteins [19]. HDAC9 is also known to target non-histone proteins, such as forkhead box protein 3, ataxia telangiectasia group D-complementing protein (ATDC), and glioblastoma 1 protein, which are members of pathways implicated in carcinogenesis [20, 21]. More recently, aberrant HDAC9 expression has been observed in several types of cancers, including medulloblastoma [21], acute lymphoblastic leukemia [22], glioblastoma [23], osteosarcoma [24], and breast cancer [25]. HDAC9 has been shown to promote the growth of these tumors.
Triple negative breast cancer (TNBC) is one of the most clinically aggressive subtypes of breast cancer and is associated with overall poor prognosis [26–28]. TNBC lacks expression of the estrogen receptor (ER), progesterone receptor (PR) or human epidermal growth factor receptor 2 (her2/neu) [29, 30]; as a result, conventional therapies targeting each of these receptors proves to be unsuccessful in TNBC. Therefore, there is a major unmet need to better understand the molecular basis of this type of breast cancer as well as develop new therapeutic strategies. Emerging studies have highlighted microRNAs (miRNAs) as critical mediators of tumorigenesis, as their involvement have been well-established across several types of cancers, including breast, prostate, ovarian, and head and neck cancers [31–34]. Previous work from our group identified miR-206 as a critical tumor suppressor in TNBC and showed that downregulation of miR-206 lead to an upregulation of VEGF, MAPK3, and SOX9, crucial drivers of invasion and angiogenesis [35]. In this study, we demonstrate that overexpression of HDAC9 promotes the invasion and angiogenesis in TNBC by directly modulating miR-206 and provides a selective basis for TNBC treatment.
2. Materials and methods
2.1. Breast cancer cell lines and culture
The human breast cancer cell lines MDA-MB-231, MDA-MB-1739, HCC1395, MDA-MB-361, and SKBR3, were grown in RPMI1640 medium containing 10% FBS, 100 U/ml of penicillin sodium, and 100 µg/ml of streptomycin sulfate at 37 oC in a humidified atmosphere of 5% CO2. MCF-7, an ER-expressing breast cancer cell line, was cultured in DMEM medium containing 10% FBS plus 10 μg/ml of insulin. SKBR3 is a Her2/neu-expressing human breast cancer cell line, and MDA-MB-361 is positive for ER, PR, and Her2/neu. MDA-MB-231, MDA-MB-1739, and HCC1395 are all triple negative cell lines.
2.2. Tissue samples and immunohistochemical staining
A formalin-fixed and paraffin-embedded breast cancer tissue array was obtained from US Biomax (Derwood, Maryland, USA). This is a breast cancer and matched metastatic carcinoma tissue array, including TNM and pathology grade, with ER, PR and Her-2 (neu) IHC results, 50 cases/100 cores. The sources and characteristics of archived breast tumor breast samples are summarized in Table 1. HDAC9 antibody (Abcam, Cat No. ab70954) was applied on slides at 1:500 and incubated for 1 hour at room temperature after deparaffinized, antigen-retrieved (DAKO, Cat No., S1699), and endogenous peroxidase block with 3% Hydrogen Peroxide (DAKO, Cat No., S2003,). Visualization and detection were established using DAKO EnVision+ Dual (mouse and rabbit) Link System-HRP (Cat No., K4061) with an incubation time of 30 minutes. The detailed staining procedure and semi-quantitative method of immunohistochemical staining of these tissue sections for HDAC9 are described in our previous paper [36] (DAKO, Cat No., K4061).
Table 1.
Characteristics and HDAC9 expression of tissue specimens of breast tumors.
| Total | T stage | Sex | Age | ||||
|---|---|---|---|---|---|---|---|
| T1&T2 | T3T4 | Male | Female | <50 | ≥50 | ||
| Triple Negative | 41 | 25 | 16 | 0 | 41 | 22 | 19 |
| Non-triple negative | 59 | 48 | 11 | 0 | 59 | 28 | 31 |
| Total | 100 | 73 | 27 | 0 | 100 | 50 | 50 |
Quantitative real-time RT-PCR
Regular and quantitative RT-PCR were performed following our previous protocol [37]. Primer sequences of miR-206, and U6 snRNA have been described in our previous report [35]. Primer sequences of HDAC9 are as follows: (GeneBank accession number BC152405), 5’- CAGCAACGAAAGACACTCCA-3’ and 5’- CAGAGGCAGTTTTTCGAAGG-3’. SYBR Green quantitative PCR reaction was carried out in a 15 μl reaction volume containing 2× PCR Master Mix (Applied Biosystems) per our previous reports [38, 39].
2.4. Construction of HDAC9 siRNAs and Transfection.
We designed and purchased the small interfering RNA (siRNA) duplexes against HDAC9 (Genbank accession no., BC152405) from ThermoFisher Scientific (Grand Island, NY, USA). The target sequence of HDAC9 siRNA is 5′-UAAAAUCUUCCUGCCCACCdTdT-3′. The nonspecific control siRNA duplexes were purchased from ThermoFisher Scientific with the same GC content as HDAC9 siRNAs. The siRNA or control oligonucleotides were transfected into TNBC MDA-MB-231 cells at a final concentration of 100 nM with Lipofectamine 2000 per the instructions.
2.5. Matrigel Invasion Assay
The invasion assay was performed by using a Matrigel invasion chamber from Corning Biocoat (Tewksbury, MA) as previously described [40]. 5×104 HDAC9 inhibitor-transfected or control oligonucleotide-transfected TNBC MDA-MB-231 cells were added into the top chambers. The Matrigel invasion chambers were then incubated for 20 hours in a humidified tissue culture incubator. Invading cells at the bottom was determined by counting the H&E-stained cells.
2.6. Matrigel plug assay and hemoglobin assay
For the in vivo angiogenesis assay (Matrigel plug assay), 2X105 MDA-MB-231 cells were mixed with 0.5 ml of growth factor-reduced Matrigel (Corning, Tewksbury, MA) and implanted subcutaneously into the flanks of nude mice. The following day, six mice in each group were treated with 100 µg/kg HDAC9 siRNAs or control oligonucleotides via daily subcutaneous injections between the two plugs on the back of the mice. For selective HDAC IIa inhibitor, TMP269, mice were administered with TMP269 by subcutaneous injections every other day at 15 mg/kg mice body weight between the two plugs on the back of the mice. The animals were sacrificed and the Matrigel plugs were excised 10 days after Matrigel injection. The excised plugs were homogenized and subjected to measure hemoglobin content with 100 µL of Drabkin’s solution (Sigma, St. Louis, MO) following manufactural instruction and our previous description [41].
2.7. Statistical analysis
Real-time RT-PCR reaction was run in triplicate for each sample and repeated at least 2 times, and the data were statistically analyzed with a Student T-test.
3. Results
3.1. HDAC9 is overexpressed in TNBC and is inversely correlated with miR-206
Expression levels of HDAC1–11 in three TNBC cell lines, MDA-MB-231, MDA-MB-1739, and HCC70 compared to those in three non-TNBC cell lines, MCF-7, MDA-MB-361, and SKBR3 were profiled with qRT-PCR analysis. Compared to non-TNBC cells, TNBC cells expressed much higher HDAC9 (Fig. 1A). Quantitative RT-PCR results reveal that expression levels of miR-206 are notably lower while HDAC9 levels are inversely higher in TNBC cell lines than those in non-TNBC cell lines (Fig. 1B). Furthermore, we analyzed the expression levels of HDAC9 proteins determined by immunohistochemical staining in breast cancer tissue samples. TNBC tissues express higher levels of HDAC9 compared to non-TNBC tissue samples (Fig. 1C). These results demonstrate that expression levels of HDAC9 are upregulated in TNBC tissues in comparison to non-TNBC tissue samples and are inversely correlated with the levels of miR-206.
Fig. 1.
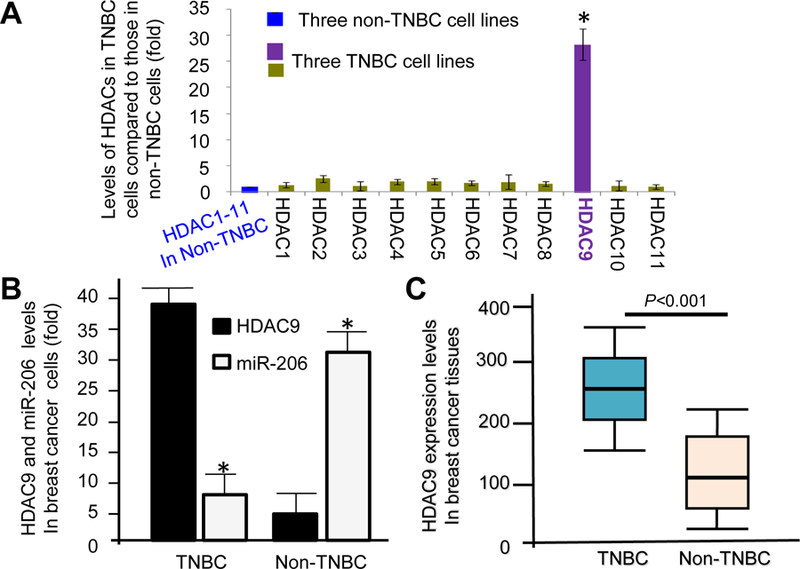
HDAC9 is overexpressed in TNBC cells and tissues. (A) Expression levels of HDAC1–9 mRNAs in three TNBC cell lines, MDA-MB-231, MDA-MB-1739, and HCC70 compared to those in three non-TNBC cell lines, MCF-7, MDA-MB-361, and SKBR3 determined by quantitative RT-PCR analysis. * P<0.001. (B) HDAC9 expression levels in triple negative breast cancer cell lines are inversely consistent with those of miR-206. * P<0.001. (C) A box and whisker plot diagram showing the comparison of HDAC9 levels in triple negative breast cancer tissues (n = 41), and non-TNBC breast cancer tissues (n =59) determined with HDAC9 IHC staining. Horizontal lines in the boxes represent the median HDAC9 value of each group (P<0.001). Top and bottom edges of the boxes indicate the score values of the 25th and the 75th percentile, respectively. Whiskers represent the highest and lowest values. The range is shown as a vertical line.
3.2. Inhibition of HDAC9 blocks the invasion of TNBC cells in vitro
To investigate whether selective HDAC9 inhibition blocks the invasion of TNBC cells, TNBC MDA-MB-231 cells were treated with TMP269, a selective class IIa HDAC inhibitor, or HDAC9 siRNA prior to Matrigel invasion assay. The invasive cells from treated groups were determined and compared to their controls by Matrigel invasion assay. Fig 2A shows representatives of invasive cell photographs from individual groups. Matrigel Invasion assay shows that the invasion of TNBC cells treated with selective class IIa HDAC inhibitor TMP269 was only 28% of that of the control (Fig. 2B). Similarly, HDAC9 siRNA treatment significantly blocked TNBC cell invasion. These results suggest that selective HDAC9 inhibition efficiently blocks the invasion of TNBC cells.
Fig. 2.
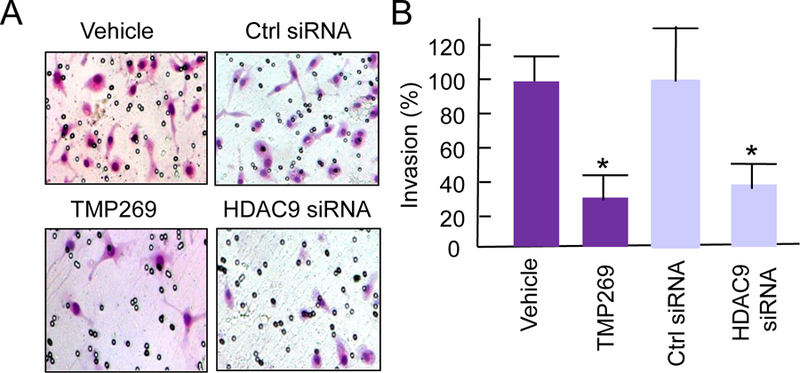
Selective HDAC9 inhibition blocks invasion of human TNBC MDA-MB-231 cells. (A) Representative photograph for invasion cells from HDAC9 siRNA-treated or TMP269-treated and their control groups. (B) Quantitative comparison of invasion cells of selective HDAC9 inhibition and their controls by Matrigel Invasion Assay. * P<0.01.
3.3. Knockout of HDAC9 inhibits the angiogenesis of TNBC tumors
To determine the effect of selective HDAC9 inhibition on angiogenesis in vivo, Matrigel plug assay was performed in nude mice. Briefly, a mixture of 2×105 TNBC MDA-MB-231 cells in 0.5 ml of growth factor reduced Matrigel was implanted at two subcutaneous sites. The mice in two groups were treated with 100 µg/kg HDAC9 siRNAs or control oligonucleotides via daily subcutaneous injections. For other two groups, mice received 15 mg/kg TMP269 or vehicle every other day by intraperitoneal injection between the two plugs on the back of the mice. Ten days after the Matrigel implant, the mice were sacrificed. The Matrigel plugs were excised, photographed, and processed to measure hemoglobin content by using Drabkin’s solution following manufacturer’ instructions. When MDA-MB-231 cells successfully promote neovasculature formation within the Matrigel plug, these neovasculatures allow tumor cells to proliferate much better than those without neovasculatures. Therefore, the control group with better angiogenesis in the Matrigel plug showed more red cells than the treated group (Fig. 3A). Fig. 3B summarizes the quantification of percentage of antiangiogenic efficacy based on hemoglobin content in 12 Matrigel plugs per group. In comparison to their controls, TMP269 treatment or HDAC9 siRNA transfection shows an obvious antiangiogenic effect with 76 % and 72% inhibition of angiogenesis, respectively. These results suggest that selective inhibition of HDAC9 effectively blocks TNBC angiogenesis.
Fig. 3.
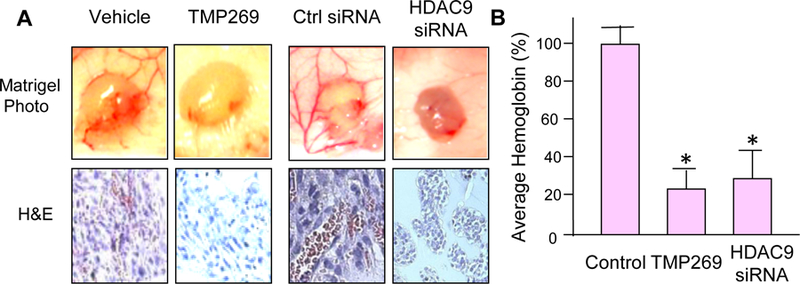
Efficacies of HDAC9 inhibition on Matrigel plug angiogenesis in vivo. (A) Microphotograph of Matrigel plug representatives from the control and the treated groups and representative pictures of H&E staining for Matrigel plug sections. (B) The column graph shows the comparison of average hemoglobin in plugs from the control and HDAC9-inhibited groups. * P<0.001.
3.4. HDAC9 regulates expression of VEGF and MAPK3 through miR-206 miRNA
To determine whether HDAC9 regulates miR-206 expression, HDAC9 was silenced with HDAC9 siRNA and then miR-206 expression levels were measured by quantitative RT-PCR analysis. The results show that selective HDAC9 inhibition significantly upregulated miR-206 expression (Fig. 4A). Furthermore, the results of a search for the predicted targets with TargetScan revealed that VEGF and MAPK3 are predicted targets of miR-206 (Fig. 4B). To confirm these results in vitro, TNBC MDA-MB-231 cells were treated with HDAC9 siRNA and then protein levels of VEGF and MAPK3 by Western blot analysis. The results demonstrated that selective inhibition of HDAC9 decreased the expression levels of VEGF and MAPK3 compared to their controls (Fig. 4C). These results demonstrated that selective HDAC9 inhibition increased the expression of VEGF and MAPK3 via upregulating miR-206 expression.
Fig. 4.
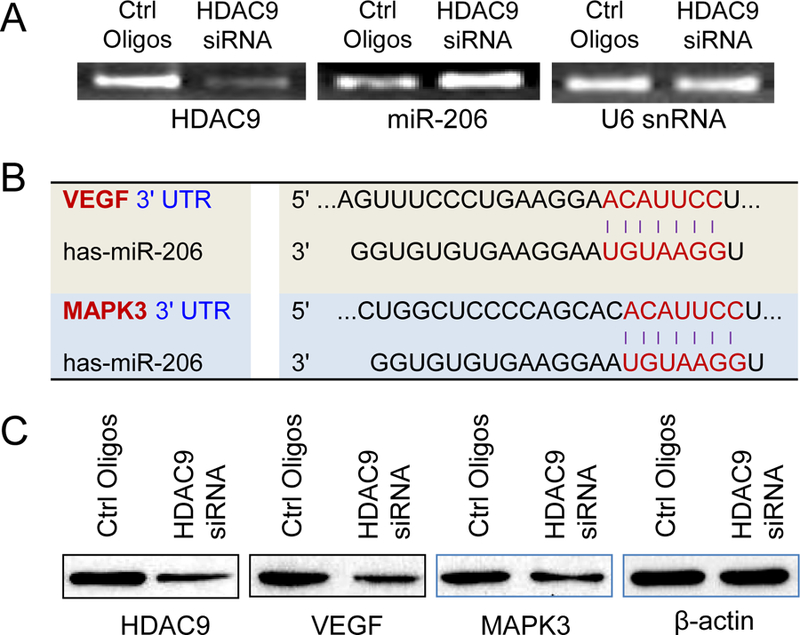
HDAC9 inhibition restores the miR-206 levels in TNBC cells and decreases VEGF and MAPK3 levels. (A) MiR-206 expression levels in TNBC MDA-MB-231 cells treated by HDAC9 siRNA. (B) Targeting sequences of miR-206 on 3’UTRs of VEGF and MAPK3. (C) HDAC9 inhibition decreased expression levels of VEGF and MAPK3 determined Western blot analysis.
4. Discussion
It is now widely accepted that aberrant expression or activity of HDAC enzymes may lead to carcinogenesis and that specific HDAC enzymes are associated with particular malignancies; in turn, the inhibition of HDAC enzymes can result in therapeutic benefits in certain cancer types [42]. However, clinical benefits of selective versus broad HDAC inhibitors are unknown and the appropriateness of inhibitor may depend on tumor HDAC enzyme expression, enzyme selectivity of the inhibitor, and the desired effects. Although there are several published studies [16, 17] and numerous ongoing clinical trials involving HDAC inhibitors [18], very little is known about the roles of individual HDAC enzymes, and a formal assessment of HDAC enzyme expression is not routinely done. Emerging data suggests that the currently used HDAC inhibitors may differ significantly with regard to target selectivity. In addition, the expression of HDAC enzymes may vary considerably between normal and tumor tissues and likely between different phenotypes of tumor tissues. HDAC9 is thought to regulate gene expression through epigenetic modulation of the chromatin structure by catalyzing the deacetylation of histone proteins [19]. More recently, aberrant HDAC9 expression has been observed in several types of cancers, including medulloblastoma [21], acute lymphoblastic leukemia [22], glioblastoma [23], osteosarcoma [24], and breast cancer [25]. HDAC9 has been shown to promote the growth of these tumors. Our results demonstrate that HDAC9 is overexpressed in TNBC compared to non-TNBC cell lines and tissues. Furthermore, our data showed that selective inhibition of HDAC9 repressed the invasion and angiogenesis of TNBC cells. These findings reveal that HDAC9 promotes the invasion and angiogenesis of TNBC cells besides proliferation.
Several investigations have demonstrated that decreased miR-206 expression is involved in breast cancer proliferation [43, 44]. Our previous studies show that miR-206 is antagonistically involved in TNBC invasion and angiogenesis through modulating VEGF and MAPK3 [35]. Numerous investigations have demonstrated that VEGF actively promotes angiogenesis, metastasis, and chemoresistance [45–47]. MAPK overexpression has been shown to be associated with advanced stages and short survival in patients with some cancers [48, 49]. However, miR-206 has not been characterized as the targets of HDAC9 regulation. Our findings demonstrated that the selective inhibition of HDAC9 in siRNA-transfected TNBC MDA-MB-231 cells not only increased expression of miR-206, but also repressed the expression of VEGF and MAPK proteins and further inhibited TNBC cell invasion and angiogenesis. Here, we report HDAC9 for the first time as a mediator to modulate VEGF-mediated invasion and angiogenesis of TNBC tumor cells via modulating miR-206 expression. The results further confirmed that miR-206 actively regulates the expression of VEGF and MAPK3 in TNBC cells.
In conclusion, higher expression levels of HDAC9 are inversely correlated with miR-206 expression in TNBC cells. Furthermore, the selective inhibition of HDAC9 not only modulated the expression of miR-206, VEGF and MAPK3, but also particularly inhibited TNBC invasion and angiogenesis. Our findings suggested that HDAC9 overexpression in TNBC cells promotes the invasion and angiogenesis through VEGF and MAPK3 via regulating miR-206. These findings may be beneficial for better understanding TNBC regulation and designing personalized therapies for breast cancer patients.
Acknowledgements
This study was financially supported by a Research Grant from NIH NCI (R01CA165306).
Abbreviations:
- HDAC9 HDAC
histone deacetylase
- TNBC
Triple negative breast cancer
- miRNA
microRNA
- VEGF
Vascular endothelial growth factor
- MAPK
Mitogen-activated protein kinase
- siRNA
small interfering RNA
- RT-PCR
reverse transcription polymerase chain reaction
References
- [1].Gregory PD, Wagner K, Horz W, Histone acetylation and chromatin remodeling, Exp Cell Res, 265 (2001) 195–202. [DOI] [PubMed] [Google Scholar]
- [2].Deckert J, Struhl K, Histone acetylation at promoters is differentially affected by specific activators and repressors, Mol Cell Biol, 21 (2001) 2726–2735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Marks PA, Richon VM, Kelly WK, Chiao JH, Miller T, Histone deacetylase inhibitors: development as cancer therapy, Novartis Foundation symposium, 259 (2004) 269–281; discussion 281–268. [PubMed] [Google Scholar]
- [4].Kelly WK, O’Connor OA, Marks PA, Histone deacetylase inhibitors: from target to clinical trials, Expert Opin Investig Drugs, 11 (2002) 1695–1713. [DOI] [PubMed] [Google Scholar]
- [5].Arts J, de Schepper S, Van Emelen K, Histone deacetylase inhibitors: from chromatin remodeling to experimental cancer therapeutics, Current medicinal chemistry, 10 (2003) 2343–2350. [DOI] [PubMed] [Google Scholar]
- [6].Sengupta N, Seto E, Regulation of histone deacetylase activities, J Cell Biochem, 93 (2004) 57–67. [DOI] [PubMed] [Google Scholar]
- [7].Dokmanovic M, Marks PA, Prospects: histone deacetylase inhibitors, J Cell Biochem, 96 (2005) 293–304. [DOI] [PubMed] [Google Scholar]
- [8].Phiel CJ, Zhang F, Huang EY, Guenther MG, Lazar MA, Klein PS, Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen, The Journal of biological chemistry, 276 (2001) 36734–36741. [DOI] [PubMed] [Google Scholar]
- [9].Saito A, Yamashita T, Mariko Y, Nosaka Y, Tsuchiya K, Ando T, Suzuki T, Tsuruo T, Nakanishi O, A synthetic inhibitor of histone deacetylase, MS-27–275, with marked in vivo antitumor activity against human tumors, Proceedings of the National Academy of Sciences of the United States of America, 96 (1999) 4592–4597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Nakajima H, Kim YB, Terano H, Yoshida M, Horinouchi S, FR901228, a potent antitumor antibiotic, is a novel histone deacetylase inhibitor, Exp Cell Res, 241 (1998) 126–133. [DOI] [PubMed] [Google Scholar]
- [11].Richon VM, Emiliani S, Verdin E, Webb Y, Breslow R, Rifkind RA, Marks PA, A class of hybrid polar inducers of transformed cell differentiation inhibits histone deacetylases, Proceedings of the National Academy of Sciences of the United States of America, 95 (1998) 3003–3007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Kim YB, Lee KH, Sugita K, Yoshida M, Horinouchi S, Oxamflatin is a novel antitumor compound that inhibits mammalian histone deacetylase, Oncogene, 18 (1999) 2461–2470. [DOI] [PubMed] [Google Scholar]
- [13].Butler LM, Agus DB, Scher HI, Higgins B, Rose A, Cordon-Cardo C, Thaler HT, Rifkind RA, Marks PA, Richon VM, Suberoylanilide hydroxamic acid, an inhibitor of histone deacetylase, suppresses the growth of prostate cancer cells in vitro and in vivo, Cancer research, 60 (2000) 5165–5170. [PubMed] [Google Scholar]
- [14].Butler LM, Webb Y, Agus DB, Higgins B, Tolentino TR, Kutko MC, LaQuaglia MP, Drobnjak M, Cordon-Cardo C, Scher HI, Breslow R, Richon VM, Rifkind RA, Marks PA, Inhibition of transformed cell growth and induction of cellular differentiation by pyroxamide, an inhibitor of histone deacetylase, Clin Cancer Res, 7 (2001) 962–970. [PubMed] [Google Scholar]
- [15].Meinke PT, Colletti SL, Doss G, Myers RW, Gurnett AM, Dulski PM, Darkin-Rattray SJ, Allocco JJ, Galuska S, Schmatz DM, Wyvratt MJ, Fisher MH, Synthesis of apicidin-derived quinolone derivatives: parasite-selective histone deacetylase inhibitors and antiproliferative agents, Journal of medicinal chemistry, 43 (2000) 4919–4922. [DOI] [PubMed] [Google Scholar]
- [16].Chiu HW, Yeh YL, Wang YC, Huang WJ, Chen YA, Chiou YS, Ho SY, Lin P, Wang YJ, Suberoylanilide hydroxamic acid, an inhibitor of histone deacetylase, enhances radiosensitivity and suppresses lung metastasis in breast cancer in vitro and in vivo, PLoS One, 8 (2013) e76340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Tate CR, Rhodes LV, Segar HC, Driver JL, Pounder FN, Burow ME, Collins-Burow BM, Targeting triple-negative breast cancer cells with the histone deacetylase inhibitor panobinostat, Breast Cancer Res, 14 (2012) R79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Millward M, Price T, Townsend A, Sweeney C, Spencer A, Sukumaran S, Longenecker A, Lee L, Lay A, Sharma G, Gemmill RM, Drabkin HA, Lloyd GK, Neuteboom ST, McConkey DJ, Palladino MA, Spear MA, Phase 1 clinical trial of the novel proteasome inhibitor marizomib with the histone deacetylase inhibitor vorinostat in patients with melanoma, pancreatic and lung cancer based on in vitro assessments of the combination, Investigational new drugs, 30 (2012) 2303–2317. [DOI] [PubMed] [Google Scholar]
- [19].Clocchiatti A, Florean C, Brancolini C, Class IIa HDACs: from important roles in differentiation to possible implications in tumourigenesis, Journal of cellular and molecular medicine, 15 (2011) 1833–1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Yuan J, Adamski R, Chen J, Focus on histone variant H2AX: to be or not to be, FEBS Lett, 584 (2010) 3717–3724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Milde T, Oehme I, Korshunov A, Kopp-Schneider A, Remke M, Northcott P, Deubzer HE, Lodrini M, Taylor MD, von Deimling A, Pfister S, Witt O, HDAC5 and HDAC9 in medulloblastoma: novel markers for risk stratification and role in tumor cell growth, Clin Cancer Res, 16 (2010) 3240–3252. [DOI] [PubMed] [Google Scholar]
- [22].Moreno DA, Scrideli CA, Cortez MA, de Paula Queiroz R, Valera ET, da Silva Silveira V, Yunes JA, Brandalise SR, Tone LG, Differential expression of HDAC3, HDAC7 and HDAC9 is associated with prognosis and survival in childhood acute lymphoblastic leukaemia, Br J Haematol, 150 (2010) 665–673. [DOI] [PubMed] [Google Scholar]
- [23].Yang R, Wu Y, Wang M, Sun Z, Zou J, Zhang Y, Cui H, HDAC9 promotes glioblastoma growth via TAZ-mediated EGFR pathway activation, Oncotarget, 6 (2015) 7644–7656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Zhao YX, Wang YS, Cai QQ, Wang JQ, Yao WT, Up-regulation of HDAC9 promotes cell proliferation through suppressing p53 transcription in osteosarcoma, International journal of clinical and experimental medicine, 8 (2015) 11818–11823. [PMC free article] [PubMed] [Google Scholar]
- [25].Lapierre M, Linares A, Dalvai M, Duraffourd C, Bonnet S, Boulahtouf A, Rodriguez C, Jalaguier S, Assou S, Orsetti B, Balaguer P, Maudelonde T, Blache P, Bystricky K, Boulle N, Cavailles V, Histone deacetylase 9 regulates breast cancer cell proliferation and the response to histone deacetylase inhibitors, Oncotarget, (2016). [DOI] [PMC free article] [PubMed]
- [26].Bauer KR, Brown M, Cress RD, Parise CA, Caggiano V, Descriptive analysis of estrogen receptor (ER)-negative, progesterone receptor (PR)-negative, and HER2-negative invasive breast cancer, the so-called triple-negative phenotype: a population-based study from the California cancer Registry, Cancer, 109 (2007) 1721–1728. [DOI] [PubMed] [Google Scholar]
- [27].Schneider BP, Winer EP, Foulkes WD, Garber J, Perou CM, Richardson A, Sledge GW, Carey LA, Triple-negative breast cancer: risk factors to potential targets, Clin Cancer Res, 14 (2008) 8010–8018. [DOI] [PubMed] [Google Scholar]
- [28].Dent R, Trudeau M, Pritchard KI, Hanna WM, Kahn HK, Sawka CA, Lickley LA, Rawlinson E, Sun P, Narod SA, Triple-negative breast cancer: clinical features and patterns of recurrence, Clin Cancer Res, 13 (2007) 4429–4434. [DOI] [PubMed] [Google Scholar]
- [29].Rakha EA, Ellis IO, Triple-negative/basal-like breast cancer: review, Pathology, 41 (2009) 40–47. [DOI] [PubMed] [Google Scholar]
- [30].Irvin WJ Jr., Carey LA, What is triple-negative breast cancer?, Eur J Cancer, 44 (2008) 2799–2805. [DOI] [PubMed] [Google Scholar]
- [31].Sung H, Jeon S, Lee KM, Han S, Song M, Choi JY, Park SK, Yoo KY, Noh DY, Ahn SH, Kang D, Common genetic polymorphisms of microRNA biogenesis pathway genes and breast cancer survival, BMC cancer, 12 (2012) 195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Liu J, Liu J, Wei M, He Y, Liao B, Liao G, Li H, Huang J, Genetic variants in the microRNA machinery gene GEMIN4 are associated with risk of prostate cancer: a case-control study of the Chinese Han population, DNA and cell biology, 31 (2012) 1296–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Liang D, Meyer L, Chang DW, Lin J, Pu X, Ye Y, Gu J, Wu X, Lu K, Genetic variants in MicroRNA biosynthesis pathways and binding sites modify ovarian cancer risk, survival, and treatment response, Cancer research, 70 (2010) 9765–9776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Zhang X, Yang H, Lee JJ, Kim E, Lippman SM, Khuri FR, Spitz MR, Lotan R, Hong WK, Wu X, MicroRNA-related genetic variations as predictors for risk of second primary tumor and/or recurrence in patients with early-stage head and neck cancer, Carcinogenesis, 31 (2010) 2118–2123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Liang Z, Bian X, Shim H, Downregulation of microRNA-206 promotes invasion and angiogenesis of triple negative breast cancer, Biochem Biophys Res Commun, 477 (2016) 461–466. [DOI] [PubMed] [Google Scholar]
- [36].Shim H, Lau SK, Devi S, Yoon Y, Cho HT, Liang Z, Lower expression of CXCR4 in lymph node metastases than in primary breast cancers: potential regulation by ligand-dependent degradation and HIF-1alpha, Biochem Biophys Res Commun, 346 (2006) 252–258. [DOI] [PubMed] [Google Scholar]
- [37].Liang Z, Brooks J, Willard M, Liang K, Yoon Y, Kang S, Shim H, CXCR4/CXCL12 axis promotes VEGF-mediated tumor angiogenesis through Akt signaling pathway, Biochem Biophys Res Commun, 359 (2007) 716–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Livak KJ, Schmittgen TD, Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method, Methods (San Diego, Calif, 25 (2001) 402–408. [DOI] [PubMed] [Google Scholar]
- [39].Liang Z, Wu H, Xia J, Li Y, Zhang Y, Huang K, Wagar N, Yoon Y, Cho HT, Scala S, Shim H, Involvement of miR-326 in chemotherapy resistance of breast cancer through modulating expression of multidrug resistance-associated protein 1, Biochemical pharmacology, 79 (2010) 817–824. [DOI] [PubMed] [Google Scholar]
- [40].Liang Z, Yoon Y, Votaw J, Goodman M, William L, Shim H, Silencing of CXCR4 blocks breast cancer metastasis, Cancer research, 65 (2005) 967–971. [PMC free article] [PubMed] [Google Scholar]
- [41].Zhu A, Zhan W, Liang Z, Yoon Y, Yang H, Grossniklaus HE, Xu J, Rojas M, Lockwood M, Snyder JP, Liotta DC, Shim H, Dipyrimidine amines: a novel class of chemokine receptor type 4 antagonists with high specificity, Journal of medicinal chemistry, 53 (2010) 8556–8568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Bolden JE, Peart MJ, Johnstone RW, Anticancer activities of histone deacetylase inhibitors, Nat Rev Drug Discov, 5 (2006) 769–784. [DOI] [PubMed] [Google Scholar]
- [43].Elliman SJ, Howley BV, Mehta DS, Fearnhead HO, Kemp DM, Barkley LR, Selective repression of the oncogene cyclin D1 by the tumor suppressor miR-206 in cancers, Oncogenesis, 3 (2014) e113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Georgantas RW 3rd, Streicher K, Luo X, Greenlees L, Zhu W, Liu Z, Brohawn P, Morehouse C, Higgs BW, Richman L, Jallal B, Yao Y, Ranade K, MicroRNA-206 induces G1 arrest in melanoma by inhibition of CDK4 and Cyclin D, Pigment cell & melanoma research, 27 (2014) 275–286. [DOI] [PubMed] [Google Scholar]
- [45].Schuurbiers OC, Kaanders JH, van der Heijden HF, Dekhuijzen RP, Oyen WJ, Bussink J, The PI3-K/AKT-pathway and radiation resistance mechanisms in non-small cell lung cancer, J Thorac Oncol, 4 (2009) 761–767. [DOI] [PubMed] [Google Scholar]
- [46].Bussink J, van der Kogel AJ, Kaanders JH, Activation of the PI3-K/AKT pathway and implications for radioresistance mechanisms in head and neck cancer, Lancet Oncol, 9 (2008) 288–296. [DOI] [PubMed] [Google Scholar]
- [47].Jameel JK, Rao VS, Cawkwell L, Drew PJ, Radioresistance in carcinoma of the breast, Breast (Edinburgh, Scotland), 13 (2004) 452–460. [DOI] [PubMed] [Google Scholar]
- [48].Mukherjee R, McGuinness DH, McCall P, Underwood MA, Seywright M, Orange C, Edwards J, Upregulation of MAPK pathway is associated with survival in castrate-resistant prostate cancer, British journal of cancer, 104 (2011) 1920–1928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Wang C, Cigliano A, Delogu S, Armbruster J, Dombrowski F, Evert M, Chen X, Calvisi DF, Functional crosstalk between AKT/mTOR and Ras/MAPK pathways in hepatocarcinogenesis: implications for the treatment of human liver cancer, Cell Cycle, 12 (2013) 1999–2010. [DOI] [PMC free article] [PubMed] [Google Scholar]