

Abstract
Ewing’s sarcoma is a rare and aggressive tumor which classified as peripheral primitive neuroectodermal tumor. It commonly arises in the long bones of the extremities and rarely in the head and neck region. Localization of the sinonasal tract is a rare occurrence thus the number of clinical studies published in the literature are limited. The diagnosis of this tumor requires a histopathological examination, immunohistochemistry and cytogenetic analysis. Ewing’s sarcomas are characterized by a CD99 positivity in immunohistochemistry stain and a t(11:22)(q24:q12) translocation in cytogenetic study. The treatment of choice is the multimodality treatment including surgery, radiotherapy and chemotherapy. This is a case report of sinonasal and orbital Ewing’s sarcoma in a 24-year-old male patient who presented with a history of right nasal obstruction, right eye pain and periorbital edema.
INTRODUCTION
Ewing’s sarcoma is a rare and aggressive tumor which classified as peripheral primitive neuroectodermal tumor [1]. It was first described by James Ewing, an American pathologist, in 1921 [2]. Ewing’s sarcoma commonly occurs in early childhood and adolescence, but rarely in adulthood [3]. Most cases arise in the long bones of the extremities [1, 2, 4]. Primary Ewing’s sarcoma of the head and neck is uncommon, hence it accounts for only 1–4% of all Ewing’s sarcomas [1, 4, 5]. Furthermore, sinonasal tract involvement is very rare and only few reported cases have been published in world literature [2, 4].
CASE REPORT
A 24-year-old male patient who presented with a history of right nasal obstruction, right eye pain, lower eyelid swelling and orbital swelling. Endoscopic examination revealed an obstructive mass occupying the right nasal cavity (Fig. 1). Cranial nerves were intact. The result of hematological and biochemical investigations were within normal limits.
Figure 1:
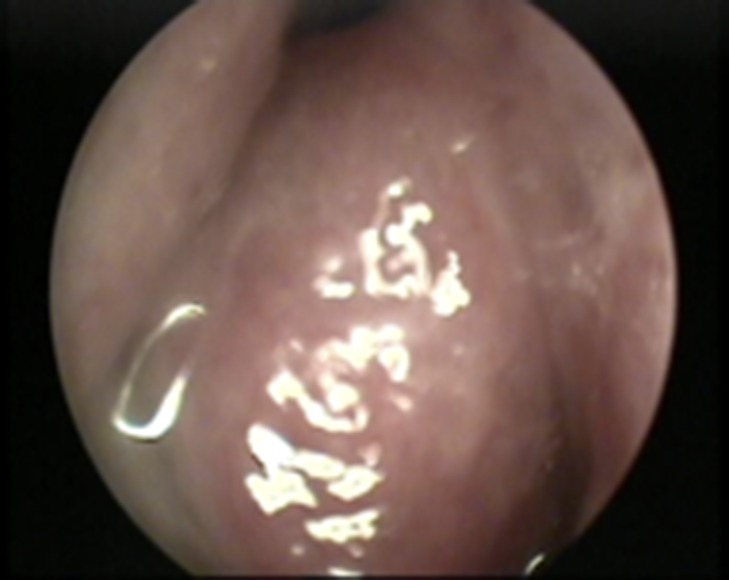
Nasal endoscopy image showing a soft tissue mass arising from the right lateral nasal wall.
On radiological evaluation, CT scan with contrast of the paranasal sinuses (PNS) revealed a mass involving the right ethmoid sinus with medial wall and orbital floor extension (Fig. 2). The subsequent magnetic resonance imaging (MRI) revealed an infiltrative soft tissue mass occupying the right ethmoid sinus, eroding inferio-medial orbital wall and extending to the extracoanal space (Fig. 3). Positron emission tomography (PET) scan demonstrated an ill-defined 4.5 × 4.2 cm2 mass lesion in the right nasal cavity and ethmoid sinus extending to the right medial orbital floor (Fig. 4). The scan did not reveal any associated lymphadenopathies.
Figure 2:
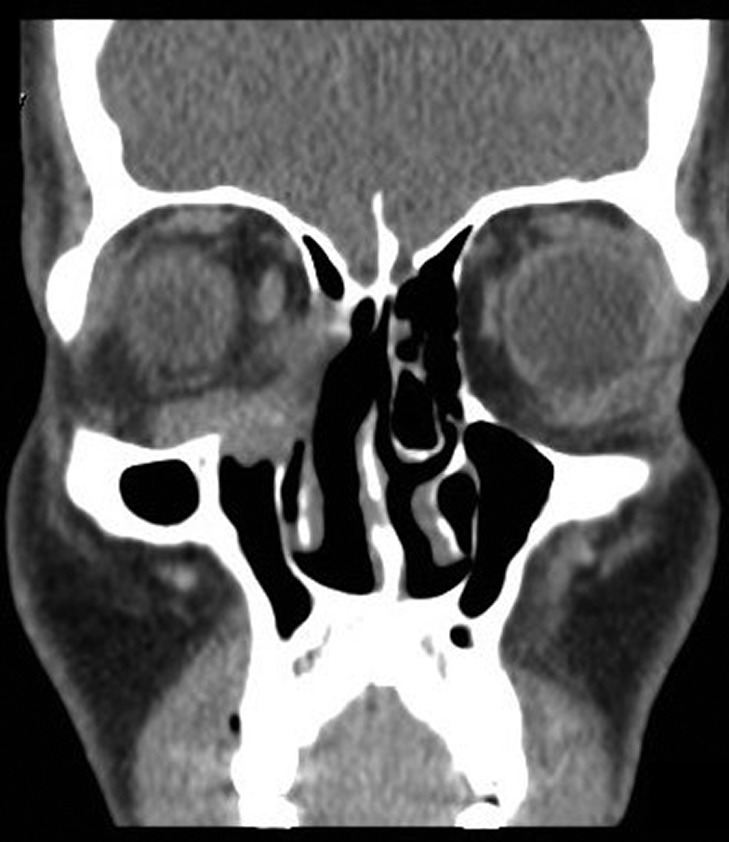
Contrast enhanced computed tomography scan. Coronal CT scan with contrast of PNS showing an enhancing soft tissue lesion in the right ethmoid, eroding the medial and inferior orbital walls.
Figure 3:
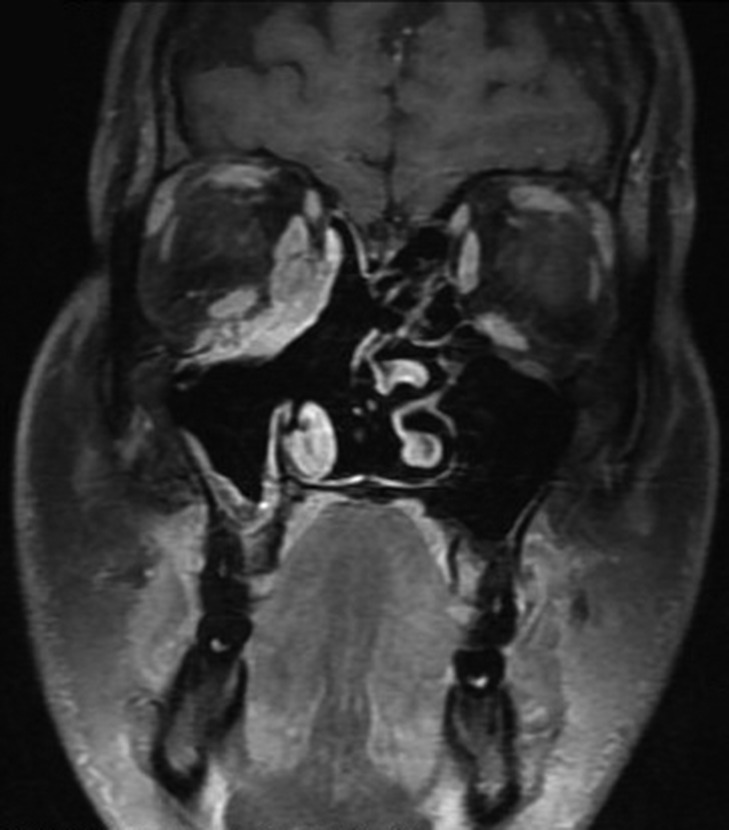
Magnetic resonance imaging (MRI) showing an enhancing soft tissue mass in the ethmoid sinus, eroding inferio-medial orbital wall.
Figure 4:
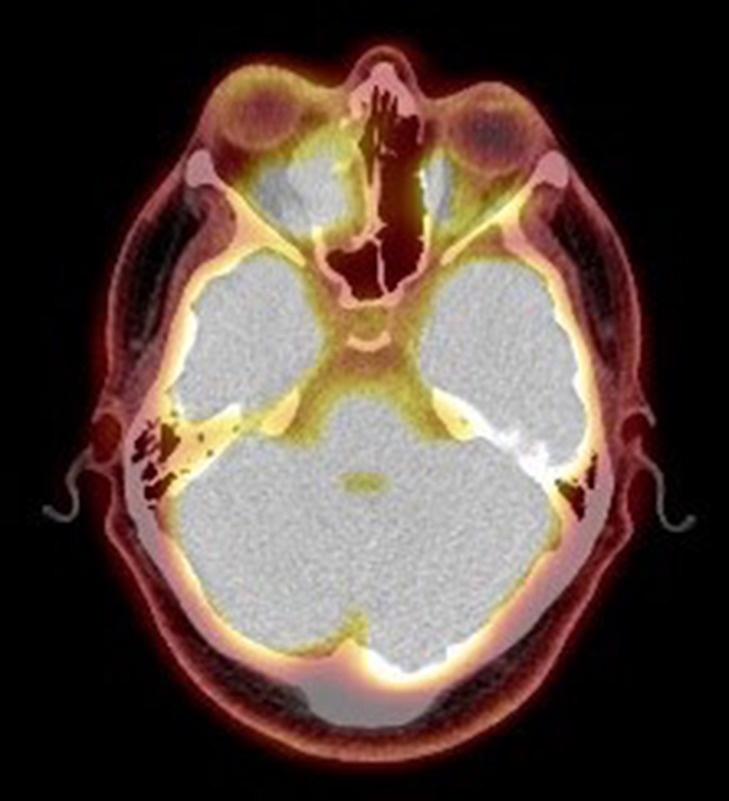
Positron emission tomography scan. PET scan showing an ill-defined 4.5 × 4.2 cm2 mass lesion in the right nasal cavity, maxillary and ethmoid sinuses extending to the right medial orbital floor.
Histological analysis of a biopsy from the mass revealed a small rounded blue cell tumor suggestive of Ewing’s sarcoma/primitive neuroectodermal tumor. Immunohistochemistry showed the neoplastic cells are positive for CD99, S-100 protein and vimintin. However, neuron specific enolase (NSE), smooth muscle actin (SMA), desmin, myogenin, CD45, synaptophysin and pan-cytokeratin were all negative. KI-67 index was 30–40%.
Subsequently, molecular study using fluorescence in situ hybridization (FISH) had shown rearrangement of EWSR1 gene in 100% of the analyzes nuclei that confirm the diagnosis of Ewing’s sarcoma. The patient underwent endoscopic excision of the tumor followed by chemotherapy and radiotherapy treatment. MRI 6-months post treatment showed complete resolution of the disease (Fig. 5). The patient remains symptom-free during 2 years follow-up and maintains a very good quality of life.
Figure 5:
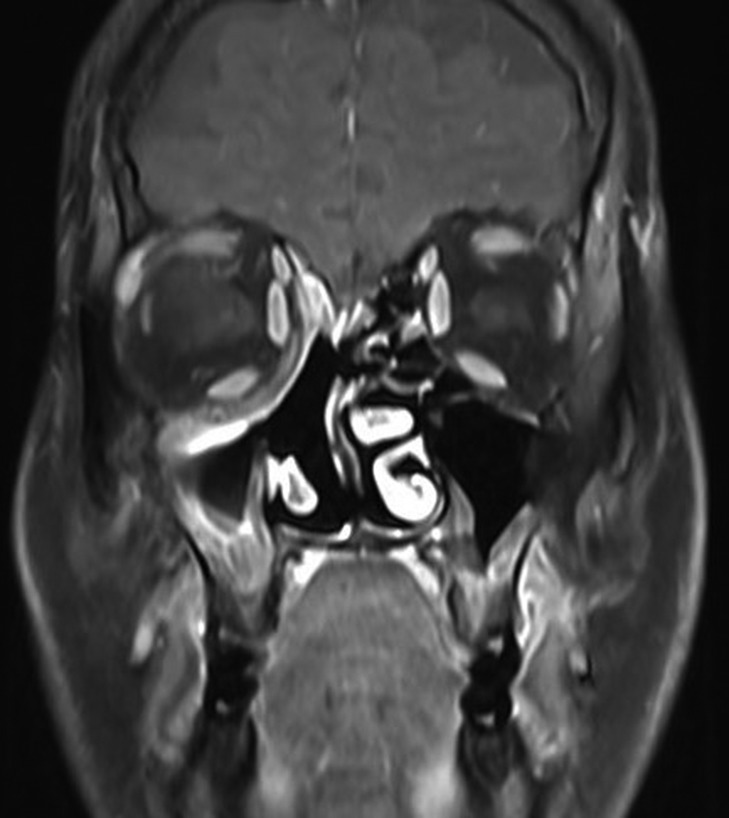
Magnetic resonance imaging after treatment. MRI post treatment showing total resolution of the tumor.
DISCUSSION
Ewing’s sarcoma is an aggressive tumor that commonly arises in the long bones or pelvis and less frequently in the soft tissue of the trunk and extremities [1, 2]. It is a rare disease comprising 4–6% of all primary bone tumors [1, 5, 6]. Ewing’s sarcoma of the head and neck is uncommon, accounting for 1–4% of all cases [1, 4, 5]. Moreover, Ewing’s sarcomas in the nasal cavity and PNS are extremely rare [1, 5]. Pediatrics and adolescents are more susceptible to the disease [3]. About 80% of patients are younger than 20 years of age with the highest incidence in the second decade of life [2, 3, 6]. Ewing’s sarcoma is more common in white populations, and have a slight male predominance [4, 5].
Clinically, majority of the patients are presented with enlarging mass, nasal obstruction, rhinorrhea and epistaxis [4, 7]. Other symptoms and signs are due to mass effect of the tumor [4, 7]. In particular, patients with periorbital extension may present with proptosis, periorbital edema and decrease visual acuity [7]. Approximately 15–30% of the patients are presented with metastasis at time of diagnosis [7]. The most common sites of distant metastasis are the lungs and bones [7]. Microscopically, Ewing’s sarcomas are composed of uniform small round cells with round nuclei containing fine chromatin, scanty clear or eosinophilic cytoplasm and PAS‑positive intracytoplasmic glycogen granules [2, 7, 8].
Diagnosing sinonasal Ewing’s sarcoma is extremely challenging as several small round neoplasms must be excluded [1, 2, 9]. The differential diagnosis is widely broad including rhabdomyosarcoma, lymphoma, undifferentiated carcinoma, melanoma and olfactory neuroblastoma [2, 6]. The diagnosis of Ewing’s sarcoma can be established after a careful evaluation using radiological study, histopathological examination, immunohistochemistry, and cytogenetic analysis [2, 9]. The immunohistochemical profile usually shows CD99 and Fli-1 positivity [2, 8]. In 2011, Hafezi reviewed 14 cases of sinonasal Ewing’s sarcoma [6]. All cases were positive for CD99 in the immunohistochemistry staining [6]. In the present case, immunohistochemistry revealed positive staining for CD99, S-100 protein and vimintin. A definitive diagnosis of Ewing’s sarcoma can be achieved through molecular analysis that detect EWSR1/FLI-1 fusion [1]. This fusion causes a specific t(11:22)(q24:q12) chromosomal translocation [1].
The prognosis depends on the site of the primary tumor, the presence of distant metastasis at presentation, and the age of the patient [2, 3]. Researchers have found that patients younger than 15 years old and patients with axial and sinonasal tract diseases have a better prognosis [2, 3]. Furthermore, a good prognosis can be expected if the disease has not metastasized [2–4]. Disseminated disease at time of presentation is associated with 22% 5-year survival compared to 55% in patients without metastases [2–4]. However, the survival rate in patients without metastatic disease has improved to 86% [6, 10]. This increase in survival rate could be explained by the recent advances in the treatment [6, 10].
The treatment of choice in Ewings sarcoma is the multimodality treatment that includes surgery, radiotherapy and chemotherapy [2]. In our case, based on the histological diagnosis, immunohistochemistry and cytogenetics confirmation, the patient was treated with surgery followed by chemotherapy and radiotherapy. The patient remains symptom-free and maintains a very good quality of life after 2 years follow-up.
CONCLUSION
Ewing’s sarcoma originating from the sinonasal tract is very rare. Although the diagnosis of the disease is challenging, it is feasible using histopathological examination, immunohistochemical study and cytogenetic analysis. Treatment includes a multidisciplinary approach with surgery followed by chemotherapy and radiotherapy.
CONFLICT OF INTEREST STATEMENT
The authors declare that they have no conflict of interest. Informed consent was obtained from all individual participants included in the study.
FUNDING
No funding obtained.
REFERENCES
- 1. Liang J. Sinonasal Ewing sarcoma: a case report and literature review. Perm J 2018; 10.7812/tpp/17-086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yeshvanth S, Ninan K, Bhandary S, Lakshinarayana KP, Shetty J, Makannavar J. Rare case of extraskeletal ewings sarcoma of the sinonasal tract. J Cancer Res Ther 2012;8:142–4. 10.4103/0973-1482.95197. [DOI] [PubMed] [Google Scholar]
- 3. Howarth K, Khodaei I, Karkanevatos A, Clarke R. A sinonasal primary Ewing’s sarcoma. Int J Pediatr Otorhinolaryngol 2004;68:221–4. 10.1016/j.ijporl.2003.09.011. [DOI] [PubMed] [Google Scholar]
- 4. Souheil J, Skander K, Sawssen D, Sana M, Delia Y, Khalil M, et al. Ewing sarcomas of the sino-nasal tract and maxillary bone. Egyptian J Ear Nose Throat Allied Sci 2016;17:147–53. 10.1016/j.ejenta.2016.08.001. [DOI] [Google Scholar]
- 5. Balamuth NJ, Womer RB. Ewing’s sarcoma. Lancet Oncol 2010;11:184–92. 10.1016/s1470-2045(09)70286-4. [DOI] [PubMed] [Google Scholar]
- 6. Hafezi S, Seethala RR, Stelow EB, Mills SE, Leong IT, MacDuff E, et al. Ewing’s family of tumors of the sinonasal tract and maxillary bone. Head Neck Pathology 2010;5:8–16. 10.1007/s12105-010-0227-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Vaccani J, Forte V, De Jong A, Taylor G. Ewing’s sarcoma of the head and neck in children. Int J Pediatr Otorhinolaryngol 1999;48:209–16. 10.1016/s0165-5876(99)00030-0. [DOI] [PubMed] [Google Scholar]
- 8. Simons SA, Bridge JA, Leon ME. Sinonasal small round blue cell tumors: an approach to diagnosis. Semin Diagn Pathol 2016;33:91–103. 10.1053/j.semdp.2015.09.010. [DOI] [PubMed] [Google Scholar]
- 9. Suzuki T, Yasumatsu R, Nakashima T, Arita S, Yamamoto H, Nakagawa T. Primary Ewing’s sarcoma of the sinonasal tract: a case report. Case Rep Oncol 2017;10:91–7. 10.1159/000455040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. La TH, Meyers PA, Wexler LH, Alektiar KM, Healey JH, Laquaglia MP, et al. Radiation therapy for Ewing’s sarcoma: results from Memorial Sloan-Kettering in the modern era. Int J Radiat Oncol Biol Phys 2006;64:544–50. 10.1016/j.ijrobp.2005.07.299. [DOI] [PubMed] [Google Scholar]