

Abstract

Determining atomic-level characteristics of molecules on two-dimensional surfaces is one of the fundamental challenges in chemistry. High-resolution nuclear magnetic resonance (NMR) could deliver rich structural information, but its application to two-dimensional materials has been prevented by intrinsically low sensitivity. Here we obtain high-resolution one- and two-dimensional 31P NMR spectra from as little as 160 picomoles of oligonucleotide functionalities deposited onto silicate glass and sapphire wafers. This is enabled by a factor >105 improvement in sensitivity compared to typical NMR approaches from combining dynamic nuclear polarization methods, multiple-echo acquisition, and optimized sample formulation. We demonstrate that, with this ultrahigh NMR sensitivity, 31P NMR can be used to observe DNA bound to miRNA, to sense conformational changes due to ion binding, and to follow photochemical degradation reactions.
Short abstract
Joint optimization of DNP formulation, target, and pulse sequence yields a factor >105 improvement in sensitivity of MAS NMR, allowing reactions of nucleic acids on flat supports to be monitored.
Introduction
The ability to detect and characterize molecules at the atomic scale, through the introduction of a range of physical methods, has transformed molecular and materials science over the past 50 years, leading to today’s structure-based understanding of chemistry. However, if the system under investigation is located at a surface with no periodicity, atomic-level characterization is still a major challenge. This is relevant to devices for advanced biosensors, solar energy conversion, targeted drug delivery systems, and electronic circuits, among others. Currently EXAFS, vibrational (IR and Raman) and electronic (UV–vis) spectroscopies, together with surface science techniques (XPS, auger, electron microscopy) and mass spectrometry (SIMS) are used to characterize surfaces, but it is challenging to obtain a complete picture of molecular structure that would enable a comprehensive understanding of function and operation, either because they provide average or bulk structural information or they are not applicable to amorphous supports.
Solid-state nuclear magnetic resonance (NMR) spectroscopy (in conjunction with other surface characterization techniques) would be the method of choice for characterizing surfaces. Several groups have developed NMR in this direction over the past few years with notable success in, for example, metal–organic frameworks (MOFs),1 cements,2 battery science,3 and catalysis.4,5 However, the detection limit of NMR is far too low to allow many modern materials to be examined. Even when using materials having high specific surface area (e.g., ∼1000 m2/g for mesoporous silica), the concentration of NMR active nuclei of interest often remains low (ca. 1 mmol of surface atoms/g), requiring many hours or even days to obtain solid-state NMR spectra with reasonable signal-to-noise ratio. Hyperpolarized xenon NMR has been used to probe lower surface areas indirectly, but magnetization transfer from xenon to obtain spectra of the surface, which has been demonstrated in some pioneering examples, is generally quite inefficient.6−9 As a result, low-surface-area materials are currently not amenable to high-resolution NMR studies, and with conventional NMR, macroscopically two-dimensional supports such as wafers, i.e., “flat” surfaces, are completely out of reach.
Here we demonstrate that, by combining recently introduced dynamic nuclear polarization (DNP) methods with multiple-echo (CPMG) acquisition and optimized sample formulations, it is possible to obtain solid-state NMR spectra of molecules immobilized on wafers with surface areas on the order of ca. 0.01 m2/g. We show how we can increase the sensitivity of NMR to the point that both one-dimensional (1D) and two-dimensional (2D) magic angle spinning (MAS) NMR spectra can be recorded from as little as 80 pmol of oligonucleotides such as DNA deposited onto supports with specific surface area estimated (see the SI, section I) to be less than 0.01 m2/g in 30 mg of sample: 2–3 orders of magnitude lower than for any previously reported analogues.10 We demonstrate that, with this ultrahigh NMR sensitivity, 31P NMR can be used to observe DNA bound to miRNA, to sense conformational changes due to ion binding, and to follow photochemical degradation reactions.
In recent years DNP, in which the spin polarization of unpaired electrons is transferred to surrounding atomic nuclei by saturation of the electron spin transitions using high-power microwave sources, has proven to be a useful means of sensitivity enhancement for solid-state NMR experiments on many materials.11−14 It is now routine to increase the nuclear magnetization by factors of 10–200 at magnetic field strengths near 10 T and temperatures around 100 K by adding an exogenous source of unpaired electrons to the sample of interest. This has been shown to be particularly useful to enhance the signals from surfaces by simple incipient wetness impregnation of the sample with a radical containing solution, which delivers 1H hyperpolarization to surface interfaces by spontaneous proton spin diffusion, followed by conventional cross-polarization (CP)15,16 transfer of enhanced proton magnetization to heteronuclei at the surface. This strategy has been dubbed DNP surface enhanced NMR spectroscopy (DNP-SENS).13,17
Polynucleotides composed of RNA and DNA are macromolecules which exhibit a rich diversity of structures to fulfill a wide variety of natural biological functions. X-ray diffraction and NMR spectroscopy have provided detailed structures of oligonucleotides that have been used to help understand their functions. Since their introduction over two decades ago,18,19 the development of arrays bearing biological molecules quickly transformed genomics research, and they remain an important tool in biology.20,21
In microarrays, where biological fragments such as oligonucleotides, small proteins, or antibodies22 are tethered to planar supports, the interactions that stabilize the native structure are likely to be affected by the presence of the surface. For oligonucleotides, this includes H-bonding motifs, groove geometries, and the interaction of the predominantly negatively charged phosphodiester backbone with cations. To refine and develop such systems, it would be important to determine structure-activity relationships in situ. The samples of interest here are thus wafers onto which various oligonucleotide species are deposited.
Approximately 30 mg of crushed glass wafers can be packed into a 3.2 mm rotor for NMR analysis, corresponding to, for example, roughly 300 pmol of a heptaribonucleotide for monolayer coverage (see the SI, section I). In comparison, 30 mg of a typical solid would typically correspond to about 300 μmol of bulk sample. This corresponds to a one-million-fold reduction in concentration for an RNA sample on the glass wafer. Since the sensitivity of an NMR experiment increases with the square root of the number of scans, if we assume that one needs 30 min to acquire a natural abundance 13C spectrum with conventional cross-polarization MAS with a good signal-to-noise ratio for a bulk organic solid, then one would require over 50 million years to obtain the same sensitivity from the molecules on the glass wafer. Equivalently, a 30 min experiment to detect 13C from 300 pmol of functionality per 30 mg would require scaling up to a 30 kg sample, all else being equal.
We optimized the ensemble of factors leading to best overall NMR sensitivity. As has been discussed extensively,23 the overall sensitivity factor, F, with respect to a reference experiment can be expressed as a product of several factors, which here include
| 1 |
with Σ† being the overall sensitivity factor due to the DNP process.24
| 2 |
The different terms are discussed in the following. We first maximize Σ† by maximizing εDNP, the DNP enhancement factor, by using the most recently introduced state-of-the-art polarization sources. These are organic dinitroxide radicals designed to have the correct spacing and relative orientation of the two nitroxide groups, as well as to have long electron spin relaxation times at 100 K.25,26 Using the biradical TEKPol,27 surface DNP enhancements of between 50 and 200 have been obtained using 1,1,2,2-tetrachloroethane (TCE) as an impregnating solvent. For glass plates we estimate εDNP ∼ 150 with TEKPol in TCE. θsurface is the fraction of observable nuclei on the support, due to both depolarization28,29 and direct paramagnetic quenching effects. For the solvent resonances in a bulk solution of TEKPol in TCE, θ has been measured to be 0.35,26 which we adopt as our estimate of θsurface. dformulation takes into account the fact that a radical containing solution must be added to the sample to achieve DNP which may lead to some dilution as compared to a dry powder. In the case of DNP-SENS a minimal volume of solvent only impregnates the surface, such that dformulation ∼ 1. The ratio S100K/S298K accounts for the relative Boltzmann polarization as well as, e.g., an improvement in the probe quality factors. The Boltzmann factor would be ∼2.8, and we have previously measured an overall factor of 3.7.24 The typical polarization build-up time for TEKPol in TCE is around 2.5 s, which defines the recycle time in the DNP experiment. This can be compared to a typical room temperature T1 value around 5 s. This leads to a value for (T1/TDNP)1/2 of 1.4. As a result of the above considerations a reasonable estimate of Σ† is 280.
We now turn our attention to optimizing the other
parameters of
the NMR experiment contained in F. The most promising
target for the detection of NMR signals from the nucleic acids is
the highly sensitive 31P nuclei of the phosphodiester groups
that are 100% abundant. Compared to our benchmark of a single carbon-13
nucleus at natural abundance, the detection of, e.g., the 6 nearly
equivalent backbone 31P atoms of a heptaribonucleotide
fragment yields a factor 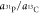 of 550, and a factor
of 550, and a factor 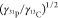 of 1.3. Finally, the
term Iexp0/Iref refers to the possibility to optimize
the type of experiment used
for acquisition to obtain more signal, I0, from each acquisition. Here the disordered nature of the surface
causes a slight distribution of 31P resonance frequencies
which exceeds the homogeneous (intrinsic) line width. In such cases,
Carr–Purcell–Meiboom–Gill (CPMG) echo acquisition
schemes30 can yield factors exceeding 10
in the best cases.31−33 Due to transverse relaxation induced by the presence
of the nitroxide centers, in most DNP experiments the gain in sensitivity
is usually limited to a factor of 3.24
of 1.3. Finally, the
term Iexp0/Iref refers to the possibility to optimize
the type of experiment used
for acquisition to obtain more signal, I0, from each acquisition. Here the disordered nature of the surface
causes a slight distribution of 31P resonance frequencies
which exceeds the homogeneous (intrinsic) line width. In such cases,
Carr–Purcell–Meiboom–Gill (CPMG) echo acquisition
schemes30 can yield factors exceeding 10
in the best cases.31−33 Due to transverse relaxation induced by the presence
of the nitroxide centers, in most DNP experiments the gain in sensitivity
is usually limited to a factor of 3.24
Taken together, if we combine efficient magic angle spinning DNP at 100 K, with the most efficient polarization sources, careful sample formulation by impregnation, detection of abundant spins, and CPMG acquisition, then we estimate we can achieve F over half a million for the DNA sample on the glass plate. Hence, high-resolution 31P MAS spectra from these samples should be possible within a few hours of acquisition.
Results and Discussion
Observation of Signals from Nucleic Acids on Flat Supports
Figure 1 shows 1D DNP-SENS CP-CPMG MAS 31P NMR spectra of heptameric oligocytidine strands (dS(C7), C = cytidine) deposited on three different materials: sapphire and borosilicate glass wafers for optical microscopy, and a fused silica tube used for EPR. The phosphodiester (P(OR)2O2–) groups of DNA strands are replaced by phosphorothioester (P(OR)2OS–) functional groups, providing a unique 31P chemical shift signature near 55 ppm. This signal is clearly observed and comes through most strongly for the sapphire system, for which there are 300 pmol of DNA on the surface and where the enhancement by DNP was the highest.
Figure 1.

Echo-reconstructed 1D DNP-SENS CP-CPMG 31P spectra from the given amounts of dS(C7) strands deposited on various supports. The total acquisition time for each experiment ranged between 20 and 28 h. The table indicates the DNP enhancement and maximum amount of phosphorothioester functionalized DNA that was analyzed. The specific surface area of the samples is <0.01 m2/g.
It is already known that different materials lead to higher degrees of polarization in the wetting phase, all else being equal, and here the sapphire leads to enhancements which exceed 500 in the solvent, and a correspondingly strong surface signal, in accordance with previously reported experiments.34
Each system also exhibits signals which are centered around −5 ppm, and which should not originate from the nucleic acids. As discussed in the SI, section VI, control experiments on the frozen 12 mM TEKPol/TCE impregnating solution and on impregnated microcrystals of l-histidine hydrochloride monohydrate do not generate such signals, whereas control experiments on sapphire and borosilicate glass wafers that were not subjected to the oligonucleotide immobilization procedure do. This implies that this signal originates from phosphate-like species embedded in the bulk material, revealed through the very high detection sensitivity obtained here. The fused silica system also exhibits a relatively strong signal around −145 ppm. This is a rather unusual 31P chemical shift and most likely corresponds to an unidentified source of trace hexafluorophosphate anion. We note that the very low levels of detection obtained here can also lead to other unexpected signals from accidental inclusion of trace impurities. Figures S4 and S5 show the example of a peak at 28 ppm which can be identified as an impurity in the impregnating solvent.
Detection Limits and miRNA Binding
Figure 2 shows fully optimized 1D DNP-SENS CP-CPMG MAS 31P spectra from 200 pmol of a hybrid RNA/DNA duplex r(A10)–dS(T10) (A = adenosine, T = thymidine) deposited on sapphire coverslips. In panel A, we see that it is possible to observe the well-resolved signal from the phosphorothioester groups in the DNA components with S/N greater than 20 in less than 1 h. This corresponds to the detection of a signal originating from no more than 1.8 nmol of phosphorothioester functional groups. The signal at −1 ppm, corresponding to the ordinary phosphodiester groups of the complementary RNA strand, exhibits a S/N ratio approaching 60. This signal is not fully resolved from the impurity signals but is nonetheless clearly distinguishable here as it is narrower than the overlapping signals (see Figure S6).
Figure 2.

Echo-reconstructed 1D DNP-SENS CP-CPMG 31P spectra from monolayers of oligonucleotide duplexes deposited on sapphire coverslip. (A) Maximum 200 pmol of the oligonucleotide duplex r(A10)–dS(T10) after 0.94 h of acquisition, giving a S/N ratio of 21 for the well-resolved phosphorothioester (“PS”) signal at 55 ppm. (B) Maximum 80 pmol of DNA probe d(ACAATCAGCTAAT*C*GACACTGCCTA), with phosphorothioester groups between the positions labeled with *, after addition of 160 pmol of the nearly complementary miRNA hsa-miR34a-5p, r(UGGCAGUGUCUUAGCUGGUUGU). The maximum amount of phosphorothioester functional groups that were analyzed is given above the corresponding signals. A slight shift of the probe PS signal in panel B relative to the duplex signal in panel A may be the result of miRNA induced bulging at the PS groups. A schematic of this possible bulged structure is given in the inset. * indicate spinning sidebands which occur in different positions due to different spinning rates for the two experiments. Complete acquisition parameters and signal processing procedures are given in the SI.
In Figure 2B, we show signals arising from a microarray-like setup in which a DNA monolayer, with sequence ACAATCAGCTAAT*G*ACACTGCCTA (C = cytidine, G = guanosine), containing two PS linkages as indicated, is first deposited onto the sapphire. Subsequently, a 2-fold excess of a microRNA (miRNA) hsa-miR34a-5p (UGGCAGUGUCUUAGCUGGUUGU, U = uradine), a prominent tumor suppressor,35 is added to the surface. Mature miRNAs are short natural RNAs which, in contrast to small interfering RNAs (siRNAs), are characterized by a few mismatched base pairs in their duplex regions, a concept which is also reflected by the probe design in this study. In this system, a maximum of 160 pmol of the phosphorothioester groups is present, corresponding to 80 pmol of the DNA. Nonetheless, we clearly observe signals from these groups in less than 48 h of acquisition. This is, to our knowledge, the lowest level of detection for MAS NMR to date. The PS resonance of the DNA probe in Figure 2B is slightly shielded relative to the duplex PS signal in Figure 2A by about 1–2 ppm, indicating a slight difference in the average chemical environments. In response to the miRNA, the DNA is expected to adopt a bulged structure in which the phosphorothioester linkages would undergo conformational changes due to the geometric constraints imposed by base pairing over the rest of the duplex.36 The observed shift may arise from this conformational change. This is consistent with Figure S7, where the measured PS shift of the DNA probe on its own is closer to that of the duplex system.
Two-Dimensional NMR from Nucleic Acids on Flat Supports
The 31P nuclear shielding anisotropy is also a powerful fingerprint of structural changes37 and has been previously used to investigate surface binding interactions of the mononucleotide dAMP adsorbed onto high-surface-area mesoporous alumina.38 It can be accurately measured in two-dimensional NMR experiments which correlate spinning sidebands in one dimension to the isotropic spectrum in the other. The sensitivity of the optimized 31P DNP-SENS CPMG experiments is such that two-dimensional experiments can now be realized even for such small sample quantities. Echo train acquisition can be appended to the 2D-PASS39 experiment to maintain a level of sensitivity enhancement similar to that of CPMG.40Figure 3 shows 31P 2D correlation spectra obtained from dS(C7) strands deposited on a sapphire support in the presence of Na+. The 1D sideband-free spectrum consists of a distribution of isotropic chemical shifts from +8 to −15 ppm, corresponding to phosphorus impurities embedded in the support, as well as a somewhat narrower signal at 55 ppm, corresponding to the phosphorothioester groups. Vertical cross-sections of the spectrum give spinning sideband profiles at the given isotropic shift. We see that the shielding anisotropy of the embedded phosphorus is smaller than that of the phosphorothioester groups, as indicated by its smaller vertical span. The sideband profile of the 55 ppm cross-section was analyzed to determine the nuclear shielding anisotropy of the dS(C7) phosphorothioester groups, assuming the profile can be modeled using a single tensor, to obtain a shielding anisotropy of ζσ = 110 ppm and an asymmetry parameter of ησ = 0.4. These values are similar to those previously reported of ordinary phosphodiester groups in nucleic acid crystals,41 indicating that in our case sulfurization has very little effect on the principal values of the anisotropy, despite its strong isotropic deshielding influence.
Figure 3.

Echo-reconstructed 2D DNP-SENS CP PASS-PIETA 31P NMR spectra from 200 pmol of dS(C7) strands deposited on sapphire. The sum projection at the top of the (sheared) 2D-PASS spectrum presents the isotropic spectrum. The vertical cross-sections give the sideband profile corresponding to a given isotropic frequency. The profiles at 55 ppm fit well to a single-site model of the nuclear shielding anisotropy, with parameters ζσ = (107.0 ± 5.4) ppm, ησ = 0.48 ± 0.08, as shown in the SI, section VII. The MAS rate was 3125 Hz. Complete acquisition parameters and signal processing procedures are given in the SI.
Monitoring Reactions of Nucleic Acids on Flat Supports
Figure 4 shows how the 31P NMR spectra of the flat surface systems can be used to monitor the response of oligonucleotide monolayers due to exposure of the slides to different reactive conditions. The first case is shown in Figure 4A, illustrating that the phosphodiester chemical shift is a probe of different cationic environments. Exposing plates with short RNA strands (sequence: UGCAUAU) to Mg2+ produces a clear shift and broadening of the 31P signal maximum as compared to Na+. An interpretation consistent with this observation is that the phosphodiester resonance is shifting to higher frequency in response to magnesium coordination while the signal from phosphorus impurities embedded in the support surface is unaffected. A change in the 31P isotropic shift of up to +10 ppm has been predicted when portions of the RNA backbone undergo conformational changes,42 which can be engendered, for example, through bidentate coordination of the magnesium ion.43 This behavior stands in contrast to that observed for the dS(C7) strands described in Figure 3, where no shift is observed upon exposure to Mg2+ (Figure S8). This suggests that the phosphorothioester groups on the surface interact more weakly with Mg2+ than the phosphodiester groups, in line with reports of reduced phosphorothioester affinity for Mg2+.41
Figure 4.

Reactive response of oligonucleotide monolayers brought about subjecting the surfaces to different conditions, as reported by changes in their echo-reconstructed 1D DNP-SENS CP-CPMG 31P spectra. (A) Reaction from 300 pmol of short RNA strands (sequence: UGCAUAU) deposited on crushed borosilicate glass plates. The initial spectrum (cyan) of the sodium coordinated strands was acquired in 44.7 h. Plates were then subjected to soaking treatments in 1 M MgCl2 solution. The plates that were exposed to the magnesium soaks give the NMR spectrum shown in dark blue. (B) Reaction from 300 pmol of oligonucleotides r(xUGCAUGU), tagged at a terminal phosphorothioester group with a photolabile dinitrobenzhydryl tag (“x”) due to irradiation of the plates with ultraviolet light. In the absence of UV irradiation, a typical PS shift of 54 ppm is recorded. From a disk exposed to 60 min of irradiation at 365 nm (30 min per side), the PS signal disappears and a significant amount of new signal around 20 ppm, corresponding to the 31P chemical shift of bis(phosphoryl)disulfide (PSSP) groups, appears. Both samples have the same phosphodiester concentration; therefore, signal intensities are normalized to the intensity of PO signal using the spinning sidebands near 88 ppm. This is more accurate than using the stronger PO signal at −1 ppm for normalization since, at MAS rates above 10 kHz, this spinning sideband is relatively free of contributions by impurity signals due to their smaller shielding anisotropy (vide supra). Complete acquisition parameters and signal processing procedures are given in the SI.
Finally, Figure 4B shows the 31P NMR spectrum of a monolayer of an oligonucleotide possessing a single terminal phosphorothioester group tagged with a dinitrobenzhydryl (DNB) group.44 We clearly observe the peak at around 54 ppm that is the signature of the phosphorothioester. We compare this to the spectrum of an otherwise identical slide, but where the sample is exposed to ultraviolet light for 1 h. We see that the exposure to UV light leads to clear changes in the 31P NMR spectra. The PS signal at 54 ppm disappears completely, and we observe significant new spectral intensity in the region around 20 ppm. The most likely rationale for these changes is cleavage of DNB and dimerization of the oligonucleotides. Under the influence of the UV light, the DNB tag is designed to cleave, allowing an opportunity for the phosphorothioester group to react. The resulting bis(phosphoryl)disulfide functional group has been characterized by solution NMR,45,46 with characteristic 31P chemical shifts between 15 and 25 ppm, in line with the new signals that appear in Figure 4B. In Figure S9 we show a comparison of 31P NMR spectra including a disk exposed to 2 min of UV irradiation, where the intensity of the two peaks is intermediate, consistent with the proposed surface-based degradation process operating on the time scale of minutes. In Figure S10, we show LC-MS data which provides evidence that oligonucleotide dimerization happens in aqueous solution when DNB tagged oligonucleotides are exposed to UV. It is worth noting that to measure the 31P chemical shifts of these oligonucleotide dimers by solution NMR would require 1–3 orders of magnitude more sample, requiring multiple weeks of oligonucleotide synthesis.
Conclusion
We have shown that state-of-the-art hyperpolarization methods for surface enhanced solid-state NMR spectroscopy can provide atomic-level information pertaining to the chemical environments of biologically relevant functionalities on two-dimensional surfaces. Specifically, here high-resolution 31P NMR signals from oligonucleotide species deposited onto the surface of sapphire wafers are acquired in less than 1 h. The outstanding sensitivity of the approach used here allows us to record one- and two-dimensional NMR experiments to observe DNA bound to miRNA, to sense conformational changes due to ion binding, and to follow photochemical degradation reactions.
With the introduction here of a multifaceted, sensitivity-optimized approach incorporating DNP-SENS and yielding surface-based NMR signals for systems with specific surface areas on the order of 0.01 m2/g, and as the achievable sensitivity by DNP continues to increase,47,48 solid-state NMR is now in position to address structural questions of direct relevance to microarray function and quality control, including three-dimensional structures49 and the surface/fragment interactions which are crucial to solving the performance and reproducibility problems of many protein-based microarrays.50,51
Experimental Section
Oligonucleotide Synthesis
Chemicals were purchased from Sigma-Aldrich (Steinheim, Germany), Fluorochem (Hadfield, United Kingdom), and TCI (Eschborn, Germany). Phosphoramidites were obtained from Thermo Fisher Scientific (Waltham, MA). Oligonucleotides were synthesized on a MM12 synthesizer from Bio Automation (BioAutomation Corp., Irving, TX) using 5 mg 500 Å UnyLinker CPG (ChemGenes, Wilmington, MA). For RNA synthesis, standard 2′-TBDMS phosphoramidites were used with a coupling time of 2 × 90 s. For DNA synthesis, coupling time was 2 × 60 s. Phosphoramidites were prepared as 0.08 M solutions in dry acetonitrile (ACN); the activator 5-(benzylthio)-1H-tetrazole (Biosolve BV, Valkenswaard, Netherlands) was prepared as a 0.24 M solution in dry ACN. Oxidizer was prepared as a 0.02 M I2 solution in THF/pyridine/H2O (70:20:10, % v/v); sulfurization to give a diastereoisomeric mixture of phosphorothioester oligonucleotides was carried out using a 0.1 M solution of 3-((N,N-dimethylaminomethylidene)amino)-3H-1,2,4-dithiazole-5-thione (DDTT; Glen Research, Sterling, VA) in dry pyridine/ACN (9:1). Capping reagent A was THF/lutidine/acetic anhydride (8:1:1), and capping reagent B was 16% N-methylimidazole/THF. Detritylations were performed using 3% dichloroacetic acid in dichloromethane. A 5′-phosphorothioate cap was introduced via a dinitrobenzhydryl-phosphporamidite (DNB) as described previously in combination with a sulfurization step.44 The 5′-DNB photocage was removed after sapphire deposition through irradiation at 365 nm for 30 min/side. All oligonucleotide sequences are given in 5′–3′ direction.
Oligonucleotide Purification
Oligonucleotides were cleaved from the solid support, and the protecting groups on the exocyclic amino groups and the backbone were removed using a 1:1 mixture of 40% aqueous methylamine and 25% aqueous ammonia (AMA) for 1 h at 65 °C. The 2′-O-tert-butyldimethylsilyl groups (TBDMS) were removed using a fresh mixture of N-methyl-2-pyrrolidone (60 μL), triethylamine (30 μL), and triethylamine trihydrofluoride (40 μL) at 70 °C for 2 h. Desilylation was quenched with trimethylethoxysilane (200 μL), and then diisopropyl ether (200 μL) was added to precipitate the oligonucleotide. The precipitate was dissolved in H2O and purified DMT-on on an Agilent 1200 series HPLC fitted with a Waters XBridge oligonucleotide BEH C18 column, 10 × 50 mm, 2.5 μm at 65 °C. Fractions were pooled, dried in a SpeedVac instrument, and treated for 15 min with 40% acetic acid at room temperature. After drying in a SpeedVac instrument, oligonucleotides were dissolved in H2O and subjected to a second purification on RP-HPLC. Buffer A was 0.1 M triethylammonium acetate, pH 8.0. Buffer B was methanol. The gradient for DMT-on purification is 20–60% buffer B in 5 min, flow rate 5 mL/min, and that for DMT-off purification is 5–25% buffer B in 8 min, flow rate 5 mL/min.
After purification, oligonucleotides were precipitated as their respective Na+, K+, or Mg2+ salt by adding 3 M sodium acetate, pH 5.5 (or potassium acetate or magnesium chloride) to a final concentration of 0.3 M and 3 volumes of ethanol. Supernatant was discarded; the pellet was washed with 75% ethanol and dissolved in RNase-free H2O.
Oligonucleotide Fixation
Two methods were used to fixate oligonucleotides onto the coverslips.
For the Borosilicate Glass Systems Described in Figure 4A
Standard Nr. 1. ThermoScientific borosilicate coverslips, 0.13–0.16 mm in thickness, were immersed in Piranha solution (∼2:1 H2SO4/H2O2) overnight at room temperature, washed with ultrapure water and methanol, and stored overnight in a desiccator with CaCl2 under house vacuum (∼20 Torr). Azide functionalization of the glass surface, using 0.5 g of cleaned glass plates, was achieved using the procedure of Godula et al.52 with details provided in the SI, section II. A mixed sequence heptaribonucleotide was immobilized to the surface of the glass plates using a 2′-O-propargyl group at its 3′-terminus. The surface functionalization was conducted using a similar protocol to that described previously53,54 for the reaction of propargyl-modified RNAs on controlled pore glass (CPG). Quantitative conversion can be expected under these conditions. Specifically, 0.9 mL of a 1:1 H2O/MeOH mixture, TBTA ligand (tris(benzyltriazoylmethyl)amine, 100 equiv, 35 μmol in 200 μL of dimethylformamide (DMF)), sodium ascorbate (100 equiv, 35 μmol in 100 μL of H2O), CuSO4·5H2O (10 equiv, 3.5 μmol in 100 μL of H2O), and the alkyne-modified RNA with sequence 5′-UGCAUAU (1 equiv, 350 nmol in 100 μL of H2O) were successively added to 160 mg of azide-modified glass plates in an Eppendorf tube. The mixture was shaken vigorously for 16 h at 45 °C on an Eppendorf shaker. The solution was removed, and the residual glass plates were washed sequentially with DMF (1 mL), 0.1 N EDTA (in water, pH = 8, 1 mL, two times), DMF (1 mL, two times), and acetonitrile (1 mL, three times), and dried in vacuo overnight to afford the (sodium coordinated) RNA-coated glass plates.
For All Sapphire Supports and the Glassy Supports Described in Figure 1
Sapphire coverslips (WST-102; diameter 10 mm, thickness 0.20 mm; U.Q.G. Optics Limited, Cambridge, England) were coated on each side with the amount of oligonucleotide corresponding to 0.9 nmol of each phosphate species (PS or PO) out of 80 μL aqueous solutions. For example, to coat a sapphire disk with 200 pmol of the hybrid duplex described in Figure 2A, 80 μL of a 1.25 μM solution was deposited on each side of the disk and air-dried overnight. Each of the individual decamer strands features 9 phosphodiester or phosphorothioester linkages; therefore 200 pmol of the hybrid duplex corresponds to 1.8 nmol of each phosphate species.
For the Fused Silica Support in Figure 1
The coating was achieved by lyophilization of the oligonucleotide solution in an EPR tube (ARMAR AG, Döttingen, Switzerland).
DNP-SENS
Following deposition of the nucleotides, and any subsequent treatment, coverslips were crushed in an alumina mortar. The polarizing agent, TEKPol or TEKPol2 dissolved in 1,1,2,2-tetrachloroethane, was added by incipient wetness impregnation, and the resulting wet material was packed into a 3.2 mm o.d. sapphire rotor. Care was taken to avoid any residual trace impurities on the packing tools or the mortar or in the solvent. Details pertaining to specific sample formulations are given in the SI, section III. Once packed, the sapphire rotor was plugged with a silicone or polyfluoroethylene insert and capped with a zirconia drive tip. The DNP-SENS NMR experiments were carried out at a nominal field strength of 9.4 T (1H Larmor frequency 400.21 MHz) using either a Bruker AV I or AV III HD spectrometer and 263 GHz DNP gyrotron as a continuous source of monochromatic microwaves.55 After initially inserting the sample into the stator at ∼100 K and allowing the TCE to freeze, the sample was ejected back into the sample hold chamber at room temperature where it was allowed to thaw.34 For each sample that was exposed to air, between 4 and 10 thawing cycles were performed prior to each NMR experiment to remove dissolved oxygen from the TCE and improve the efficiency of DNP. MAS rates between 10 and 15 kHz were used for the DNP enhanced CPMG experiments. An MAS rate of 3125 Hz was used for the DNP enhanced PASS-PIETA experiments. A more extensive table of NMR parameters used for each sample is given in the SI, section IV.
NMR Signal Processing
All signal processing was performed with RMN.56 A matched filter was applied to the echo train during whole echo reconstruction to further improve its S/N ratio. For CPMG the contribution of the initial FID was also included, as in an ordinary CP experiment. A table of apodization parameters and experimental sensitivities of the final processed data sets are given in the SI, section V. Neither direct apodization nor zero filling was applied when extracting site profiles for sideband analysis to avoid inducing an artificial correlation between neighboring points along the isotropic dimension. All spectra are referenced internally to the chemical shift of H3PO4(l) via the DNP enhanced 1H signal maximum of the TCE, which was set to 6.2 ppm. Shearing and scaling transformations were used to remove the zeroth-rank (isotropic) contribution from the chemical shift anisotropy,40 converting the indirect dimension into a sideband order dimension.
Spinning Sideband Analysis
To characterize chemical shift anisotropy tensors, sideband profiles from the PASS experiments were analyzed using a home-built simulator coded in C. The fitting algorithm implemented a Markov chain biased walk to explore the (δZZ, δYY) parameter space near an initial guess to determine probability distributions for the NMR parameters. Additional details about the algorithm and a summary of the sideband analysis, including best fit histograms for the sideband profiles and the sideband correlation spectrum for the r(GAAGAGAAGC)·dS(GCTTCTCTTC) duplex in Figure 3, are provided in the Supporting Information, section VI.
LC-MS
The integrities of purified oligonucleotides were confirmed by LC-MS analysis on an Agilent 1200/6130 system fitted with a Waters Acquity UPLC OST C-18 column (2.1 × 50 mm, 1.7 μm) at 65 °C, with a gradient of 5–35% buffer B in 14 min with a flow rate of 0.3 mL/min. Gradient for the comparison of the 5′-PS-DNB capped RNA r(xUGCAUGU) before and after UV irradiation shown in Figure S10 was 2–50% buffer B in 10 min. Buffer A was aqueous hexafluoroisopropanol (0.4 M) containing triethylamine (15 mM). Buffer B was methanol.
LA-ICPMS
Depth profiling of the supports (without oligonucleotide immobilization) for elemental phosphorus was carried out using a GeoLas C Elan DRC6100plus instrument. For robust quantitative analysis, an external calibration (SRM NIST 610) was carried out using Si/Al as internal standard and assuming a pure SiO2/Al2O3 matrix composition of the samples. Three random points at one slice were ablated fully using a 120 μm ablation crater at 10 Hz repetition rate to obtain a depth profile. The results of the analysis are as follows: borosilicate glass, 20.9 ± 2.1 mg/kg; fused silica, 26 ± 5 mg/kg; sapphire, 8 ± 5 mg/kg; uniform profiles.
Safety Declaration
The hazards encountered over the course of this work were routine and well handled by standard operating procedures.
Acknowledgments
Lyophilization of the fused silica system was carried out with the help of Kurt Hauenstein. Dr. David Gajan provided support during the early stages of DNP experiments. Prof. P. Tordo, Dr. O. Ouari and Dr. G. Casano (Aix-Marseille Université) are thanked for supplying TEKPol. Financial support comes from ERC Advanced Grant 320860, Swiss National Science Foundation Grant 205321_169612, and the NCCR RNA and Disease.
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acscentsci.8b00916.
Sample calculations for oligonucleotide coverage; text and tables elaborating upon experimental protocols and data processing; and further results, discussion, and figures in support of the results of the main text, including control experiments (PDF)
Author Contributions
The manuscript was prepared through the contribution of all authors.
The authors declare no competing financial interest.
Supplementary Material
References
- Kong X.; Deng H.; Yan F.; Kim J.; Swisher J. A.; Smit B.; Yaghi O. M.; Reimer J. A. Mapping of functional groups in metal-organic frameworks. Science 2013, 341 (6148), 882–885. 10.1126/science.1238339. [DOI] [PubMed] [Google Scholar]
- Smith B. J.; Rawal A.; Funkhouser G. P.; Roberts L. R.; Gupta V.; Israelachvili J. N.; Chmelka B. F. Origins of saccharide-dependent hydration at aluminate, silicate, and aluminosilicate surfaces. Proc. Natl. Acad. Sci. U. S. A. 2011, 108 (22), 8949–8954. 10.1073/pnas.1104526108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T.; Leskes M.; Yu W.; Moore A. J.; Zhou L.; Bayley P. M.; Kim G.; Grey C. P. Cycling Li-O2 batteries via LiOH formation and decomposition. Science 2015, 350 (6260), 530–533. 10.1126/science.aac7730. [DOI] [PubMed] [Google Scholar]
- Blanc F.; Berthoud R.; Copéret C.; Lesage A.; Emsley L.; Singh R.; Kreickmann T.; Schrock R. R. Direct observation of reaction intermediates for a well defined heterogeneous alkene metathesis catalyst. Proc. Natl. Acad. Sci. U. S. A. 2008, 105 (34), 12123–12127. 10.1073/pnas.0802147105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang J.; Jorda J. L.; Yu J.; Baumes L. A.; Mugnaioli E.; Diaz-Cabanas M. J.; Kolb U.; Corma A. Synthesis and structure determination of the hierarchical meso-microporous zeolite ITQ-43. Science 2011, 333 (6046), 1131–1134. 10.1126/science.1208652. [DOI] [PubMed] [Google Scholar]
- Raftery D.; Long H.; Meersmann T.; Grandinetti P. J.; Reven L.; Pines A. High-field NMR of adsorbed xenon polarized by laser pumping. Phys. Rev. Lett. 1991, 66 (5), 584. 10.1103/PhysRevLett.66.584. [DOI] [PubMed] [Google Scholar]
- Haake M.; Pines A.; Reimer J. A.; Seydoux R. Surface-enhanced NMR using continuous-flow laser-polarized xenon. J. Am. Chem. Soc. 1997, 119 (48), 11711–11712. 10.1021/ja9713587. [DOI] [Google Scholar]
- Raftery D.; MacNamara E.; Fisher G.; Rice C. V.; Smith J. Optical pumping and magic angle spinning: Sensitivity and resolution enhancement for surface NMR obtained with laser polarized xenon. J. Am. Chem. Soc. 1997, 119 (37), 8746–8747. 10.1021/ja972035d. [DOI] [Google Scholar]
- Jänsch H. J.; Gerhard P.; Koch M. 129Xe on Ir(111): NMR study of xenon on a metal single crystal surface. Proc. Natl. Acad. Sci. U. S. A. 2004, 101 (38), 13715–13719. 10.1073/pnas.0405426101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sangodkar R. P.; Smith B. J.; Gajan D.; Rossini A. J.; Roberts L. R.; Funkhouser G. P.; Lesage A.; Emsley L.; Chmelka B. F. Influences of dilute organic adsorbates on the hydration of low-surface-area silicates. J. Am. Chem. Soc. 2015, 137 (25), 8096–8112. 10.1021/jacs.5b00622. [DOI] [PubMed] [Google Scholar]
- Becerra L. R.; Gerfen G. J.; Temkin R. J.; Singel D. J.; Griffin R. G. Dynamic nuclear polarization with a cyclotron resonance maser at 5 T. Phys. Rev. Lett. 1993, 71 (21), 3561–3564. 10.1103/PhysRevLett.71.3561. [DOI] [PubMed] [Google Scholar]
- Hall D. A.; Maus D. C.; Gerfen G. J.; Inati S. J.; Becerra L. R.; Dahlquist F. W.; Griffin R. G. Polarization-enhanced NMR spectroscopy of biomolecules in frozen solution. Science 1997, 276 (5314), 930–932. 10.1126/science.276.5314.930. [DOI] [PubMed] [Google Scholar]
- Rossini A. J.; Zagdoun A.; Lelli M.; Lesage A.; Copéret C.; Emsley L. Dynamic nuclear polarization surface enhanced NMR spectroscopy. Acc. Chem. Res. 2013, 46 (9), 1942–1951. 10.1021/ar300322x. [DOI] [PubMed] [Google Scholar]
- Kobayashi T.; Perras F. A.; Slowing I. I.; Sadow A. D.; Pruski M. Dynamic nuclear polarization solid-state NMR in heterogeneous catalysis research. ACS Catal. 2015, 5 (12), 7055–7062. 10.1021/acscatal.5b02039. [DOI] [Google Scholar]
- Pines A.; Gibby M. G.; Waugh J. S. Proton-enhanced NMR of dilute spins in solids. J. Chem. Phys. 1973, 59 (2), 569–590. 10.1063/1.1680061. [DOI] [Google Scholar]
- Schaefer J.; Stejskal E. O. Carbon-13 nuclear magnetic resonance of polymers spinning at the magic angle. J. Am. Chem. Soc. 1976, 98 (4), 1031–1032. 10.1021/ja00420a036. [DOI] [Google Scholar]
- Lesage A.; Lelli M.; Gajan D.; Caporini M. A.; Vitzthum V.; Miéville P.; Alauzun J.; Roussey A.; Thieuleux C.; Mehdi A.; Bodenhausen G.; Coperet C.; Emsley L. Surface enhanced NMR spectroscopy by dynamic nuclear polarization. J. Am. Chem. Soc. 2010, 132 (44), 15459–15461. 10.1021/ja104771z. [DOI] [PubMed] [Google Scholar]
- Pease A. C.; Solas D.; Sullivan E. J.; Cronin M. T.; Holmes C. P.; Fodor S. P. Light-generated oligonucleotide arrays for rapid DNA sequence analysis. Proc. Natl. Acad. Sci. U. S. A. 1994, 91 (11), 5022–5026. 10.1073/pnas.91.11.5022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schena M.; Shalon D.; Davis R. W.; Brown P. O. Quantitative monitoring of gene expression patterns with a complementary DNA Microarray. Science 1995, 270 (5235), 467–470. 10.1126/science.270.5235.467. [DOI] [PubMed] [Google Scholar]
- Brown P. O.; Botstein D. Exploring the new world of the genome with DNA microarrays. Nat. Genet. 1999, 21 (1s), 33–37. 10.1038/4462. [DOI] [PubMed] [Google Scholar]
- Malone J. H.; Oliver B. Microarrays, deep sequencing and the true measure of the transcriptome. BMC Biol. 2011, 9, 34. 10.1186/1741-7007-9-34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- MacBeath G.; Schreiber S. L. Printing proteins as microarrays for high-throughput function determination. Science 2000, 289 (5485), 1760–1763. [DOI] [PubMed] [Google Scholar]
- Ernst R. R.; Bodenhausen G.; Wokaun A.. Principles of nuclear magnetic resonance in one and two dimensions; Clarendon Press: Oxford, 1987. [Google Scholar]
- Rossini A. J.; Zagdoun A.; Lelli M.; Gajan D.; Rascón F.; Rosay M.; Maas W. E.; Copéret C.; Lesage A.; Emsley L. One hundred fold overall sensitivity enhancements for Silicon-29 NMR spectroscopy of surfaces by dynamic nuclear polarization with CPMG acquisition. Chem. Sci. 2012, 3 (1), 108–115. 10.1039/C1SC00550B. [DOI] [Google Scholar]
- Ysacco C.; Rizzato E.; Virolleaud M.-A.; Karoui H.; Rockenbauer A.; Moigne F. L.; Siri D.; Ouari O.; Griffin R. G.; Tordo P. Properties of dinitroxides for use in dynamic nuclear polarization (DNP). Phys. Chem. Chem. Phys. 2010, 12 (22), 5841–5845. 10.1039/c002591g. [DOI] [PubMed] [Google Scholar]
- Kubicki D. J.; Casano G.; Schwarzwaelder M.; Abel S.; Sauvee C.; Ganesan K.; Yulikov M.; Rossini A. J.; Jeschke G.; Coperet C.; Lesage A.; Tordo P.; Ouari O.; Emsley L. Rational design of dinitroxide biradicals for efficient cross-effect dynamic nuclear polarization. Chem. Sci. 2016, 7 (1), 550–558. 10.1039/C5SC02921J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zagdoun A.; Casano G.; Ouari O.; Schwarzwälder M.; Rossini A. J.; Aussenac F.; Yulikov M.; Jeschke G.; Copéret C.; Lesage A.; Tordo P.; Emsley L. Large molecular weight nitroxide biradicals providing efficient dynamic nuclear polarization at temperatures up to 200 K. J. Am. Chem. Soc. 2013, 135 (34), 12790–12797. 10.1021/ja405813t. [DOI] [PubMed] [Google Scholar]
- Thurber K. R.; Tycko R. Perturbation of nuclear spin polarizations in solid state NMR of nitroxide-doped samples by magic-angle spinning without microwaves. J. Chem. Phys. 2014, 140 (18), 184201. 10.1063/1.4874341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mentink-Vigier F.; Paul S.; Lee D.; Feintuch A.; Hediger S.; Vega S.; Paëpe G. D. Nuclear depolarization and absolute sensitivity in magic-angle spinning cross effect dynamic nuclear polarization. Phys. Chem. Chem. Phys. 2015, 17 (34), 21824–21836. 10.1039/C5CP03457D. [DOI] [PubMed] [Google Scholar]
- Meiboom S.; Gill D. Modified spin-echo method for measuring nuclear relaxation times. Rev. Sci. Instrum. 1958, 29 (8), 688–691. 10.1063/1.1716296. [DOI] [Google Scholar]
- Larsen F. H.; Jakobsen H. J.; Ellis P. D.; Nielsen N. C. Sensitivity-enhanced quadrupolar-echo NMR of half-integer quadrupolar nuclei. Magnitudes and relative orientation of chemical shielding and quadrupolar coupling tensors. J. Phys. Chem. A 1997, 101 (46), 8597–8606. 10.1021/jp971547b. [DOI] [Google Scholar]
- Wiench J. W.; Lin V. S. Y.; Pruski M. 29Si NMR in solid state with CPMG acquisition under MAS. J. Magn. Reson. 2008, 193 (2), 233–242. 10.1016/j.jmr.2008.05.007. [DOI] [PubMed] [Google Scholar]
- Chaudhari S. R.; Berruyer P.; Gajan D.; Reiter C.; Engelke F.; Silverio D. L.; Coperet C.; Lelli M.; Lesage A.; Emsley L. Dynamic nuclear polarization at 40 kHz magic angle spinning. Phys. Chem. Chem. Phys. 2016, 18 (15), 10616–10622. 10.1039/C6CP00839A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubicki D. J.; Rossini A. J.; Purea A.; Zagdoun A.; Ouari O.; Tordo P.; Engelke F.; Lesage A.; Emsley L. Amplifying dynamic nuclear polarization of frozen solutions by incorporating dielectric particles. J. Am. Chem. Soc. 2014, 136 (44), 15711–15718. 10.1021/ja5088453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misso G.; Di Martino M. T.; De Rosa G.; Farooqi A. A.; Lombardi A.; Campani V.; Zarone M. R.; Gullà A.; Tagliaferri P.; Tassone P.; Caraglia M. Mir-34: A new weapon against cancer?. Mol. Ther.--Nucleic Acids 2014, 3, e195 10.1038/mtna.2014.47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hüsken D.; Goodall G.; Blommers M. J. J.; Jahnke W.; Hall J.; Häner R.; Moser H. E. Creating RNA bulges: Cleavage of RNA in RNA/DNA duplexes by metal ion catalysis. Biochemistry 1996, 35 (51), 16591–16600. 10.1021/bi961700c. [DOI] [PubMed] [Google Scholar]
- Copéret C.; Liao W.-C.; Gordon C. P.; Ong T.-C. Active sites in supported single-site catalysts: An NMR perspective. J. Am. Chem. Soc. 2017, 139 (31), 10588–10596. 10.1021/jacs.6b12981. [DOI] [PubMed] [Google Scholar]
- Fry R. A.; Kwon K. D.; Komarneni S.; Kubicki J. D.; Mueller K. T. Solid-state NMR and computational chemistry study of mononucleotides adsorbed to alumina. Langmuir 2006, 22 (22), 9281–9286. 10.1021/la061561s. [DOI] [PubMed] [Google Scholar]
- Antzutkin O. N.; Shekar S. C.; Levitt M. H. Two-dimensional sideband separation in magic-angle-spinning NMR. J. Magn. Reson., Ser. A 1995, 115 (1), 7–19. 10.1006/jmra.1995.1142. [DOI] [Google Scholar]
- Walder B. J.; Dey K. K.; Kaseman D. C.; Baltisberger J. H.; Grandinetti P. J. Sideband separation experiments in NMR with phase incremented echo train acquisition. J. Chem. Phys. 2013, 138 (17), 174203. 10.1063/1.4803142. [DOI] [PubMed] [Google Scholar]
- Pecoraro V. L.; Hermes J. D.; Cleland W. W. Stability constants of magnesium and cadmium complexes of adenine nucleotides and thionucleotides and rate constants for formation and dissociation of magnesium-ATP and magnesium-ADP. Biochemistry 1984, 23 (22), 5262–5271. 10.1021/bi00317a026. [DOI] [PubMed] [Google Scholar]
- Benda L.; Sochorová Vokáčová Z.; Straka M.; Sychrovský V. Correlating the 31P NMR chemical shielding tensor and the 2JP,C spin--spin coupling constants with torsion angles ζ and α in the backbone of nucleic acids. J. Phys. Chem. B 2012, 116 (12), 3823–3833. 10.1021/jp2099043. [DOI] [PubMed] [Google Scholar]
- Petrov A. S.; Bowman J. C.; Harvey S. C.; Williams L. D. Bidentate RNA--magnesium clamps: On the origin of the special role of magnesium in RNA folding. RNA 2011, 17 (2), 291–297. 10.1261/rna.2390311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradère U.; Halloy F.; Hall J. Chemical synthesis of long RNAs with terminal 5′-phosphate groups. Chem. - Eur. J. 2017, 23 (22), 5210–5213. 10.1002/chem.201700514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krawczyk E.; Skowrońska A. A new synthesis of bis-phosphoryl disulfides [(RO)2P(O)S]2. Phosphorus Sulfur Relat. Elem. 1980, 9 (2), 189–192. 10.1080/03086648008078238. [DOI] [Google Scholar]
- Krawczyk E.; Skowrońska A.; Michalski J. Reaction of three-coordinate phosphorus compounds with organophosphorus pseudohalogens 3. Phosphonium and phosphorane intermediates in the desulfurization and deoxygenation of bis(phosphinoyl) disulfides. Influence of Lewis acids on the reaction chemoselectivity. J. Chem. Soc., Perkin Trans. 1 1994, 0 (1), 89–99. 10.1039/P19940000089. [DOI] [Google Scholar]
- Lelli M.; Chaudhari S. R.; Gajan D.; Casano G.; Rossini A. J.; Ouari O.; Tordo P.; Lesage A.; Emsley L. Solid-state dynamic nuclear polarization at 9.4 and 18.8 T from 100 K to room temperature. J. Am. Chem. Soc. 2015, 137 (46), 14558–14561. 10.1021/jacs.5b08423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhari S. R.; Wisser D.; Pinon A. C.; Berruyer P.; Gajan D.; Tordo P.; Ouari O.; Reiter C.; Engelke F.; Copéret C.; Lelli M.; Lesage A.; Emsley L. Dynamic nuclear polarization efficiency increased by very fast magic angle spinning. J. Am. Chem. Soc. 2017, 139 (31), 10609–10612. 10.1021/jacs.7b05194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berruyer P.; Lelli M.; Conley M. P.; Silverio D. L.; Widdifield C. M.; Siddiqi G.; Gajan D.; Lesage A.; Copéret C.; Emsley L. Three-dimensional structure determination of surface sites. J. Am. Chem. Soc. 2017, 139 (2), 849–855. 10.1021/jacs.6b10894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cretich M.; Damin F.; Chiari M. Protein microarray technology: how far off is routine diagnostics?. Analyst 2014, 139 (3), 528–542. 10.1039/C3AN01619F. [DOI] [PubMed] [Google Scholar]
- Grawe R. W.; Knotts IV T. A. The effects of tether placement on antibody stability on surfaces. J. Chem. Phys. 2017, 146 (21), 215102. 10.1063/1.4983705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Godula K.; Rabuka D.; Nam K. T.; Bertozzi C. R. Synthesis and microcontact printing of dual end-functionalized mucin-like glycopolymers for microarray applications. Angew. Chem., Int. Ed. 2009, 48 (27), 4973–4976. 10.1002/anie.200805756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pradère U.; Brunschweiger A.; Gebert L. F. R.; Lucic M.; Roos M.; Hall J. Chemical synthesis of mono- and bis-labeled pre-microRNAs. Angew. Chem., Int. Ed. 2013, 52 (46), 12028–12032. 10.1002/anie.201304986. [DOI] [PubMed] [Google Scholar]
- Pradère U.; Hall J. Site-specific difunctionalization of structured RNAs yields probes for microRNA maturation. Bioconjugate Chem. 2016, 27 (3), 681–687. 10.1021/acs.bioconjchem.5b00661. [DOI] [PubMed] [Google Scholar]
- Rosay M.; Tometich L.; Pawsey S.; Bader R.; Schauwecker R.; Blank M.; Borchard P. M.; Cauffman S. R.; Felch K. L.; Weber R. T.; Temkin R. J.; Griffin R. G.; Maas W. E. Solid-state dynamic nuclear polarization at 263 GHz: spectrometer design and experimental results. Phys. Chem. Chem. Phys. 2010, 12 (22), 5850–5860. 10.1039/c003685b. [DOI] [PMC free article] [PubMed] [Google Scholar]
- PhySy Ltd, RMN, Version 1.8; PhySy Ltd.: Grandview Heights, Ohio; www.physyapps.com.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.