

Abstract
Whereas drugs used for maintenance/escalation therapy do not maintain their beneficial effect after cessation of therapy, some new highly effective therapies can show prolonged treatment effects after a short treatment course. Such therapies have been named pulsed immune reconstitution therapies or pulsed immunosuppressive therapies, and typical representatives are alemtuzumab and cladribine. Autologous haematopoietic stem cell transplantation could be considered as the strongest immune reconstitution therapy. Both alemtuzumab and cladribine induce depletion of lymphocytes, and a common mechanism of action is preferential depletion of class-switched and unswitched memory B-cells. Whereas CD-19+ B-lymphocytes repopulate within 6 months, CD4+ T-cells repopulate at a slower rate, taking 1–2 years to reach the lower level of normal. In general, the depletion of lymphocytes is more profound and the repletion of T-cells is slower after alemtuzumab than after cladribine treatment. Both drugs have a strong effect on relapses and magnetic resonance imaging (MRI) activity, and reduce disability worsening. The therapeutic effect is maintained beyond the period of active treatment in a large proportion of patients, which is best documented for alemtuzumab. Adverse effects include reactivation of latent infections such as tuberculosis and risk of herpes zoster. The main disadvantage in alemtuzumab-treated patients is the risk of secondary immune-mediated disorders. Pulsed immune reconstitution therapy is an option as initial therapy in relapsing-remitting multiple sclerosis patients with high disease activity and in patients on treatment with another disease-modifying therapy with significant relapse and/or MRI activity.
Keywords: alemtuzumab, autologous haematopoietic stem cell transplantation, cladribine, multiple sclerosis, MS, multiple sclerosis treatment, pulsed immune reconstitution therapy
Today the traditional treatment algorithm for relapsing-remitting multiple sclerosis (RRMS) is to initiate treatment with a moderately effective injectable or oral disease-modifying therapy (DMT) and to escalate treatment to a highly effective DMT in patients with a suboptimal treatment response.1 A smaller fraction of highly active patients has been treated with a highly effective treatment as the initial therapy, typically fingolimod or natalizumab.2
Whereas the effect of fingolimod and natalizumab is not maintained after cessation of therapy, which in some patients may even result in rebound disease activity,3 some new highly effective therapies can show prolonged treatment effects in a proportion of patients after a short course of treatment. Such therapies have been named pulsed immunosuppressive therapies or pulsed immune reconstitution therapies and are thought to induce long-term qualitative beneficial changes in the adaptive immune system.4
The strongest immune reconstitution therapy is probably intense immunosuppression with stem cell support or autologous haematopoietic stem cell transplantation (HSCT), which, however, differs from the before-mentioned therapies by being administered only as a single treatment course.5 HSCT is, therefore, only briefly mentioned in the present article.
A proposal for categorization of DMTs divides treatments into drugs for maintenance/escalation therapy and into pulsed immune reconstitution therapies. The principles of maintenance/escalation therapy and pulse immune reconstitution therapy is shown in Figure 1.
Figure 1.
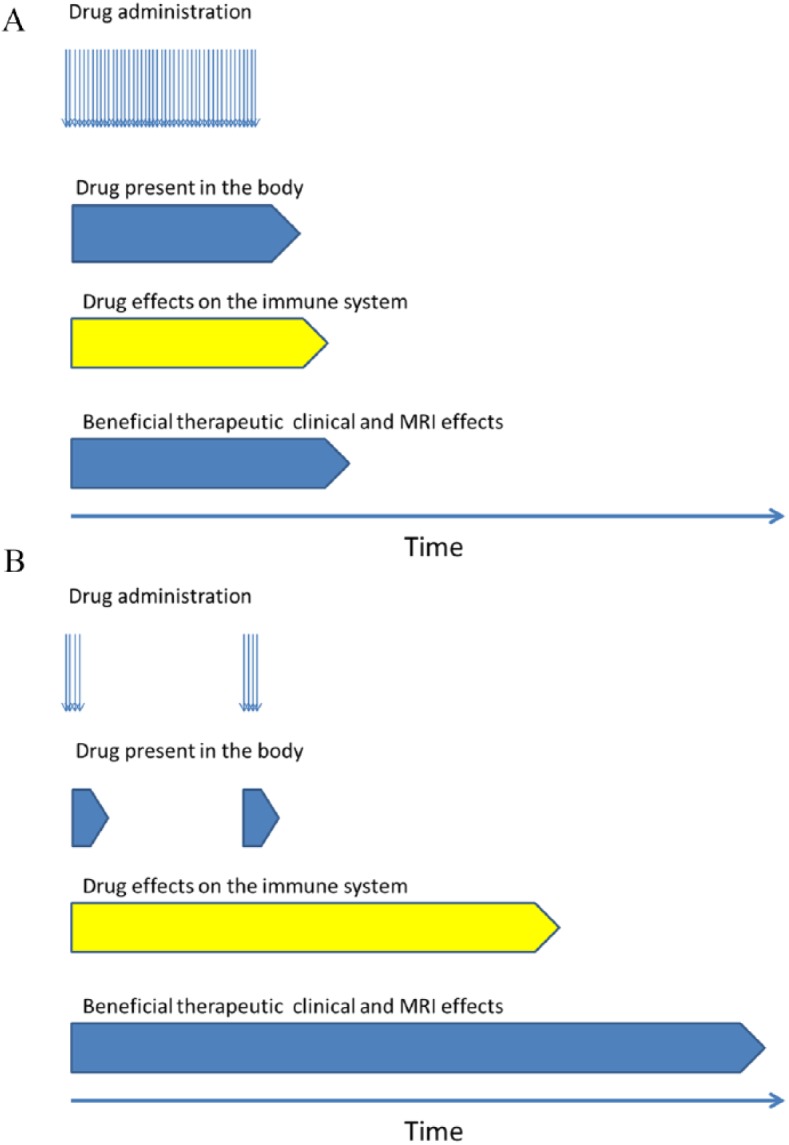
Principles of maintenance/escalation therapy (A) pulsed immune reconstitution therapy (B)
MRI, magnetic resonance imaging.
Immunomodulatory drugs and drugs for chronic immunosuppression need to be administered continuously to maintain their therapeutic efficacy, while pulsed immune reconstitution therapies are administered in short courses and in many patients the therapeutic efficacy is maintained well beyond the active treatment period.2 Some drugs may be difficult to place according to the classification – for example, ocrelizumab, which might be either called a chronic immunosuppressive or pulsed immune reconstitution therapy. Ocrelizumab is administered as pulsed therapy every 6 months, but it has not been fully elucidated whether the therapeutic efficacy is maintained over a period beyond the next planned administration.6
Mitoxantrone represents drugs causing nonselective immunosuppression and has been used as induction therapy.7–9 The difference between an induction therapy and a pulsed immune reconstitution therapy is that induction therapy involves administration of a highly effective drug (e.g. mitoxantrone) after which the treatment is deescalated to a moderately effective therapy (e.g. glatiramer acetate),9 whereas pulsed immune reconstitution therapy (e.g. alemtuzumab) is administration of a highly active drug followed by long-term drug-free observation until new disease activity appears. Mitoxantrone could be used as both induction or pulsed immune reconstitution therapy, but there are no data supporting the use as pulsed immune reconstitution therapy. We decided not to include mitoxantrone in the present article because the safety record includes frequent severe adverse events, and, therefore, mitoxantrone is not recommended for routine use in patients with RRMS.10
In the present article we review the mechanism of action, efficacy and safety of the pulsed immune reconstitution therapies alemtuzumab, cladribine and, in brief, HSCT.
Alemtuzumab
Mechanism of action
Alemtuzumab is a humanized monoclonal antibody directed against the CD52 molecule, which is present on the cell surface of lymphocytes but also at lower levels on monocytes, macrophages, eosinophils and NK cells. Bone marrow-derived haematopoietic precursor cells lack CD52 on the surface, which allows lymphocyte reconstitution following treatment with alemtuzumab.11,12
Alemtuzumab causes a long-lasting depletion of lymphocytes, especially T-cells, mediated by three different mechanisms: antibody-dependent cell-mediated cytotoxicity, complement-dependent cytotoxicity and apoptosis.13 Only minor effects are seen in cells of the innate immune system. Neutropenia occurs in approximately 10% of patients after each treatment course; it is usually mild but can be severe in rare cases.14
Alemtuzumab preferentially depletes class-switched and unswitched memory B-cells.15 CD-19+ B-lymphocytes repopulate within approximately 3–6 months and show an increase to 124–165% of baseline levels at 12 months. CD8+ cytotoxic T-cells will reach the lower normal level after 9–12 months, while CD4+ T-cells repopulate at a slower rate, taking 1–2 years to reach the lower level of normal; and prolonged CD4+ T-cell depletion may last many years in some patients.16,17 Depletion is especially long-lasting for the subset of pro-inflammatory CD20 positive T-cells.18 Monocytes reach baseline levels after 3 months.19,20
The faster B-cell than T-cell recovery might set the stage for unregulated B-cell expansion and antibody production in response to self-antigens, and after repletion the peripheral blood is dominated by immature B-cells coupled to increased serum levels of BAFF (B-cell activating factor), which have previously been associated with B-cell-related autoimmune disorders.19,21
The overshoot in repletion of B-cells has also in MS been associated with development of secondary autoimmune disorders, although no direct relationship between B-cell repopulation kinetics and autoimmunity has been reported. After alemtuzumab treatment, T-cells are mainly memory T-cells originating from the pool of cells that escaped deletion and from cell populations in the bone marrow, lymphoid structures and thymus.19,20 A high rate of cells proliferating from the pool of T-cells that escaped deletion is associated with a higher risk of development of secondary autoimmune disorders, while a higher proportion of newly generated T-cells tend to imply a lower risk of autoimmunity, probably because of the higher clonal diversity of the resulting T-cell pool.22 The repletion after alemtuzumab includes expansion of regulatory T-cells (Tregs) with increased suppressive function.
The long-lasting therapeutic benefit of alemtuzumab may involve a shift to an anti-inflammatory cytokine balance.19,20,23
The rate and patterns of lymphocyte reconstitution are currently not thought to correlate with subsequent re-emergence of disease activity.24,25
Efficacy
One full course of alemtuzumab consists of 12 mg of alemtuzumab administered intravenously daily for five consecutive days and again one year later for three consecutive days.
Alemtuzumab is approved in the EU for treatment of active MS, while in the USA the FDA has advised: ‘Because of its safety profile, the use of Lemtrada should generally be reserved for patients who have had an inadequate response to two or more drugs indicated for the treatment of MS.’
Alemtuzumab has never been tested in large placebo-controlled trials, but has been compared with subcutaneous interferon-beta (INFβ)-1a and was found to be superior to INFβ-1a 44 μg three times weekly in one phase II study and two phase III studies, one with de novo treated patients and one in patients with breakthrough disease on a first-line drug. In these studies, assessment of the treatment effects was performed by a blinded neurologist, while the treating neurologist and the patient were not blinded.
In the phase II trial, alemtuzumab reduced 3-month confirmed worsening on the Expanded Disability Status Scale (EDSS) by 71% and the annualized relapse rate (ARR) by 74% compared with INFβ (both p < 0.001).26 All comparisons between drugs regarding effects on relapses and disease worsening are shown as relative differences. The proportion of relapse-free patients treated with alemtuzumab was 80% and the proportion free of worsening on the EDSS was 91%. The mean EDSS score decreased by 0.39. The study was extended up to 60 months, at which time 87% were still free of worsening on the EDSS, the mean EDSS had decreased by 0.30 and the proportion of relapse-free patients was 72%.27
The CARE-MS I study included treatment-naïve RRMS patients, who were randomized to receive either alemtuzumab (N = 376) or subcutaneous INFβ-1a 44 μg three times weekly (N = 187).28 Alemtuzumab reduced the ARR by 55% (alemtuzumab 0.18 versus IFNβ-1a 0.39, p < 0.0001). Six-month confirmed disability worsening on the EDSS was 8% in the alemtuzumab group and 11% in the IFNβ1a group (p = 0.22). The median change in the Multiple Sclerosis Functional Composite (MSFC) score was significantly decreased by alemtuzumab (p = 0.01). Compared with IFNβ-1a, alemtuzumab significantly reduced the proportion of patients with gadolinium-enhancing lesions and new or enlarging T2-hyperintense lesions, and the median parenchymal brain volume loss. Disease-free status [no evidence of disease activity (NEDA-3): no relapse, no 3-month sustained change in EDSS score, and no new MRI lesions] after 2 years was achieved in 39% of the patients.28
The CARE-MS II study was performed in patients, who previously had been treated with a disease-modifying drug, with similar study design, but slightly different inclusion criteria as those of CARE-MS I. A total of 426 patients were randomized to receive alemtuzumab and 202 to receive subcutaneous INFβ-1a 44 μg three times weekly.29 An additional third arm in CARE-MS II using infusion of 24 mg alemtuzumab was terminated early. Alemtuzumab significantly reduced the ARR by 49% (alemtuzumab 0.26 versus IFNβ-1a 0.52, p < 0.0001) and 6-month confirmed disability worsening on the EDSS by 42% (alemtuzumab 13% versus IFNβ-1a 21%; p = 0.0084) (Table 1). Also gadolinium-enhancing lesions, new or enlarging T2-hyperintense lesions and parenchymal brain volume loss were significantly reduced by alemtuzumab compared with IFNβ-1a. NEDA-3 after 2 years was achieved in 39% of the patients.29
Table 1.
Efficacy of alemtuzumab and cladribine.
| ARR (relative reduction of relapses) | Relative reduction of 6-month EDSS worsening | 2-year NEDA-3 | ARR in years 3–4 | Long-term EDSS worsening | |
|---|---|---|---|---|---|
| Alemtuzumab* | 0.18; (55%)†§§ | 30%† (NS) | 30% | 0.17 | 18%a |
| Alemtuzumab** | 0.26; (49%)†§§ | 42%†§ | 32% | 0.23 | 23%a |
| Cladribine# | 0.14; (58%)§§ | 47%§ | 47% | 0.15 | 13%b |
ARR: annualized relapse rate; NEDA: No evidence of disease activity; EDSS: Expanded Disability Status Scale; *CARE-MS I study; **CARE-MS II study; #CLARITY study (placebo-controlled); † Compared with IFN-β1a; aafter 5 years; bafter 4 years; §p < 0.001; §§p < 0.01; NS: not significant.
Additional analyses of the data from the CARE-MS II study showed that more alemtuzumab-treated patients achieved 6-month confirmed disability improvement on the EDSS compared to IFNβ-1a.30
Quality-of-life (QoL) using the Functional Assessment of Multiple Sclerosis (FAMS), European Quality of Life-5 Dimensions (EQ-5D) and its visual analogue scale (EQ-VAS), and the 36-Item Short-Form Survey (SF-36), was improved in patients treated with alemtuzumab compared with IFNβ-1a.31
In the 5-year extension of the CARE-MS I study, the majority of patients (68.5%) received no additional treatment after their initial two courses of alemtuzumab. However, 31.5% of the patients received additional alemtuzumab cycles: 22.1% received one additional cycle, 8.0% two additional cycles and 1.4% received three additional cycles.
ARR remained low (0.14–0.19). After 5 years, 80% of patients were free from 6-month confirmed disability worsening and 33% achieved 6-month confirmed disability improvement. The majority of patients attained NEDA-3 annually in the extension (62% in year 3, 60% in year 4 and 62% in year 5).32
In the 5-year extension of CARE-MS II, 58% had only received the initial two courses of alemtuzumab. However, 42.0% of the patients received additional alemtuzumab cycles: 30.1% received one additional cycle, 10.4% two additional cycles and 1.6% received three additional cycles.
ARR remained low (0.18–0.23). After 5 years, 77% were stable and 26% achieved 6-month confirmed disability improvement. A large proportion of patients attained NEDA-3 annually in the extension (53% in year 3, 54% in year 4, and 58% in year 5), and 27% of the patients had NEDA-3 after 5 years.33
In an observational study of 87 patients treated with alemtuzumab with a median follow-up of 7 years, most patients only required two cycles of alemtuzumab. After 87 months 68% of the patients had either improved or stable EDSS scores, while 32% had worsened one point in EDSS score sustained over 6 months.34
Safety
Infusion reactions occurred in the majority of patients, of whom 3% experienced serious reactions,28,29 and in the first clinical trials patients experienced transient increases in MS symptoms associated with cytokine release,35 which can be avoided by pretreatment with corticosteroids and antihistamines.
The major concern with the use of alemtuzumab is the high risk of developing secondary autoimmunity (Table 2), which occurred in 48% over a median 7-year follow-up (range 33–144 months).34 More than 40% of patients developed thyroid disorders and the rate of immune thrombocytopenia was 3.4%.34 A few patients have encountered immune-mediated nephropathies, including cases of Goodpasture syndrome (anti-glomerular basement membrane) and membranous nephropathy, two of whom needed a renal transplant.27–29 Autoimmune disorders peak 2–3 years after the first dose of alemtuzumab,28,29 but may occur more than 5 years after the last dose.34
Table 2.
Tolerability of alemtuzumab and cladribine.
| Administration-related adverse effects | Secondary autoimmune disorders | Herpes zoster infections | PML | Secondary cancer development | Long-term monitoring of adverse effects | |
|---|---|---|---|---|---|---|
| Alemtuzumab | >90%* | Thyroid disease >40% ITP: 1–3% Nephropathy <1% |
6%§ | PML not reported in MS | Not increased | 4 years after last administration |
| Cladribine | 0 | 0 | 1.9% | PML not reported in MS | Not increased | No |
Manageable with appropriate premedication and modification of infusion; § a proportion of patients (~50%) received aciclovir prophylaxis.
ITP: autoimmune thrombocytopenia; MS, multiple sclerosis; PML, progressive multifocal leukoencephalopathy.
Profound disease activity has been reported in a few patients 3–6 months after administration of alemtuzumab and has been ascribed to early B-cell repopulation and peripheral expansion following alemtuzumab treatment.36
Because of the drug safety profile, its prescription is restricted by the Risk Evaluation and Mitigation Strategy (REMS) Program, which includes not only patients, but also physicians. Moreover, as rare fatal cases have occurred, a continuous evaluation of possible signs and symptoms of thrombocytopenia is mandatory. As part of the programme, a monthly laboratory assessment of complete blood counts, testing of serum creatinine and urine analysis is carried out, and determination of thyroid functionality is recommended every 3 months until 48 months after the last dose.
Biomarkers that would allow the identification of patients at risk for the development of secondary autoimmune diseases are not yet available. Initial claims that secondary autoimmunity was associated with increased pretreatment blood levels of interleukin-21 were not confirmed in subsequent studies using different immunoassays.37,38
There is also an increased risk of infection, especially for herpes zoster, and antiviral prophylaxis with acyclovir is recommended at least one month after alemtuzumab therapy. Rare serious adverse event reported postmarketing include patients with Listeria meningitis, one fatal, occurring a few days after the first infusion cycle of alemtuzumab,39,40 and one lethal case of disseminated necrotizing leukoencephalopathy occurring 8 months after alemtuzumab treatment.41 Haemophagocytic lymphohistiocytosis was reported in two patients with MS treated with alemtuzumab.42 Other rare adverse effects are haemolytic anaemia43 and alopecia.44
Progressive multifocal leukoencephalopathy (PML) was not reported in any MS patient treated with alemtuzumab but has occurred in patients treated with alemtuzumab for malignant diseases.45,46 Another adverse event after alemtuzumab administration is acute acalculous cholecystitis, but whether this has an infectious pathogenesis is still uncertain.47
Female patients need to practice effective contraception and avoid pregnancy until 4 months after treatment with alemtuzumab.
After alemtuzumab treatment, serum antibodies against common viruses remained detectable, and vaccine responses were normal to both novel and recall antigens within 6 months of alemtuzumab administration.48
Anti-alemtuzumab antibodies developed in 86% and 81% of patients after the second treatment in CARE-MS I and CARE-MS II, respectively, and the prevalence and mean peak anti-alemtuzumab antibody titres were higher after the second than after the first treatment course.28,29 Anti-alemtuzumab antibodies did not appear to influence efficacy, safety or lymphocyte depletion and repopulation.49 However, anti-alemtuzumab antibodies may still have an effect in some patients, in whom lymphocyte depletion after alemtuzumab therapy may be less complete even after the first alemtuzumab cycle.50 This is most likely explained by individual differences in the efficiency of effector mechanisms or trogocytosis, which is a process in which antibodies are stripped from the cell surface by other cell types.51
Cladribine
Mechanism of action
Cladribine (2-chloro-2′deoxy-β-D-adenosine) is a synthetic deoxyadenosine analogue with substitution of a hydrogen atom with chlorine at the 2-position of the purine ring. This substitution makes the nucleoside analogue resistant to degradation by adenosine deaminase, an enzyme that metabolizes and clears the naturally occurring deoxynucleosides.
The dosage of cladribine tablets is 3.5 mg/kg over 2 years, with 1.75 mg/kg being administered each year. The yearly treatment includes two treatment periods, one at the start of the first month and the other starting in the second month, each comprising 4 or 5 days in which a single 10 or 20 mg dose of cladribine tablets is taken, depending on bodyweight.
Oral bioavailability of cladribine varies between 37% and 55%.52 Cladribine has biphasic half-life elimination. The mean terminal half-life with normal renal function is 5.6–7.6 h.53
Cladribine enters the cell via nucleoside transporter proteins. Inside the cell, cladribine is activated through phosphorylation by the enzyme deoxycytidine kinase (DCK) to 2-chloro-2′deoxy-β-D-adenosine monophosphate and can be inactivated through de-phosphorylation by the enzyme 5′-nucleotidase (5′-NTase).53,54 The selective effect on lymphocytes is explained by a high concentration of DCK and a low concentration of 5′-NTase compared with other cells, resulting in intracellular accumulation of activated cladribine.53
The exact mechanism of action of cladribine in dividing and non-dividing cells is still unknown, but it has immunosuppressive effects and has been approved for the treatment of hairy cell leukaemia since 1980.55 Furthermore, 2-chloro-2′deoxy-β-D-adenosine monophosphate is converted to active triphosphate deoxynucleotides (CdATP) by DCK and this accumulation interferes with DNA repair of single-stranded breaks, rendering cell death.56 In dividing cells, CdATP can also be incorporated into the DNA, impairing transcription. Cladribine causes apoptosis through the caspase system, in which the cytochrome C and apoptotic protease-activating factor activate caspase-3 and damage DNA.57 These cytotoxic mechanisms interfere with the synthesis and repair of DNA and, therefore, target both resting and dividing lymphocytes.
Work on a human leukaemia cell line showed that cladribine inhibits global DNA methylation.58 There is growing evidence that cladribine may have epigenetic properties by turning off oncogenic signalling.59 Cladribine may also influence the cytokine milieu independently of the effect on lymphocytes, inhibiting pro-inflammatory cells and cytokines.53
Treatment with cladribine leads to a preferential and sustained reduction in lymphocytes, resulting in long-lasting partial depletion of circulating CD4+ T-cells.53,54 Cladribine has also induced a rapid depletion of CD56+ NK cells that, however, rapidly recovered and were even slightly above baseline at 6 and 12 months.60
Within the B-cell population the magnitude and kinetics of depletion vary substantially. The ratio of DCK to 5′-NTase expression was particularly high in mature, memory and notably germinal centre B-cells, but not in plasma cells. Thus, cladribine depletes class-switched and unswitched memory B-cells to levels comparable with alemtuzumab.15
Median CD4+ T-cell counts in patients treated with a cumulative dose of cladribine of 3.5 mg/kg reached a nadir at 4 months and then gradually increased. After treatment in year 2, a nadir was reached at 60 weeks and then gradually recovered, exceeding the threshold of 0.350 × 109 cells/L by 120 weeks. At the end of year 2 some patients had still not reached the lower limit, but reconstitution continued and at 4 years almost all patients had reached the lower limit of normal. The decrease in CD8+ T-cells is less pronounced compared with the decrease in CD4+ T-cells, and overall did not drop below the lower limit of normal. Furthermore, the recovery is faster, meaning that the CD4+/CD8+ ratio is temporarily decreased.
In addition, cladribine treatment led to changes in the absolute cell count and relative distribution of many lymphocyte subsets, but the relationship to clinical effects is not known.
Cladribine also depleted various innate immune cells, including NK cells and monocytes, although to a lesser extent than lymphocytes. Reductions in neutrophils, platelets and erythrocytes were modest, and mean levels of these cells remained within the normal range throughout the treatment period.60
Cladribine can cross the blood–brain barrier (BBB) and the concentration in the cerebrospinal fluid is 25% of that in plasma.53 It is plausible that cladribine reduces disease activity in MS, at least in part, by depleting circulating memory B-cells and thus reducing the influx of these cells into the central nervous system (CNS),60 and that cladribine may deplete CNS-resident immune cells in vivo; however, the effects of cladribine in the CNS are virtually unknown.
Efficacy
In the CLARITY trial, treatment with cladribine tablets significantly reduced the ARR (0.14 for the group receiving 3.5 mg/kg and 0.15 for the 5.25 mg/kg versus 0.33 for the placebo group; relative reductions were 58% and 55%, respectively; p < 0.001).61 Treated patients had a higher relapse-free rate (80% and 79%, respectively) versus placebo-treated patients (60.9%) (p < 0.001) and had a significantly lower risk of 3-month and 6-month sustained disability worsening.62 NEDA-3 after 2 years was achieved in 178 (44%) of 402 patients in the cladribine 3.5 mg/kg group, 189 (46%) of 411 patients in the cladribine 5.25 mg/kg group and 60 (16%) of 379 patients in the placebo group (OR 4.28, 3.05–6.02 for the 3.5 mg/kg group; 4.62, 3.29–6.48 for the 5.25 mg/kg group; both p < 0.0001).62 Subgroup analysis of patients with high disease activity (⩾2 relapses in de novo treated patients or ⩾1 relapse plus MRI changes in patients on a DMT) indicated greater responsiveness to cladribine 3.5 mg/kg in this patient subgroup.63 The clinical findings were underscored by the results of MRI studies.64 Patients in the 3.5 mg/kg or 5.25 mg/kg cladribine groups had fewer lesions per patient per scan for: T1 gadolinium-enhancing lesions (mean 0.12 and 0.11 versus 0.91), active T2 lesions (mean 0.38 and 0.33 versus 1.43) and combined unique lesions (mean 0.43 and 0.38 versus 1.72); all p < 0.001 versus placebo.65
Of the 1184 patients that completed the CLARITY trial, 867 (73%) were enrolled in the 2-year CLARITY EXTENSION study. AAR remained low in patients who in the CLARITY trial were treated with cladribine 3.5 mg/kg (ARR 0.14; 95% CI: 0.12–0.17), independently of whether patients were randomized to placebo (N = 98) (ARR 0.15; 95% CI: 0.09–0.21) or cladribine 3.5 mg/kg (N = 186) (ARR 0.10; 95% CI: 0.06–0.13) in the CLARITY EXTENSION (NS). The proportions of relapse-free patients were 76% in the placebo group and 81% in the cladribine 3.5 mg/kg group (NS), and the proportions of patients who remained free of confirmed 3-month EDSS worsening were 72% in the placebo group and 77% in the cladribine 3.5 mg/kg group (NS). Of importance, the efficacy was maintained even after the lymphocyte count had reached the lower limit of normal.66
The mean number of T1 gadolinium-enhancing lesions was significantly higher and the proportion of patients with no active T2 lesions was significantly lower in the placebo group compared with the cladribine 3.5 mg/kg group.67
In another placebo-controlled trial, cladribine was studied in 903 patients with the first demyelinating episode, of whom 37% fulfilled the McDonald 2010 criteria for MS, while 63% were clinically isolated syndrome (CIS) patients. The primary endpoint was conversion to clinically definite MS. The risk reduction for time to conversion to clinically definite MS was 67% for 3.5 mg/kg and 62% for 5.25 mg/kg [hazard ratio (HR) 0.33, 0.21–0.51 and 0.38, 95% CI: 0.25–0.58, respectively, both p < 0.0001], which in fact is the highest risk reduction reported in any placebo-controlled trial in patients with a first demyelinating episode.68
Cladribine 3.5 mg/kg was also compared with placebo as add-on therapy in relapsing patients with clinical activity despite IFN-β treatment in a 96-week phase II study. The study was, however, discontinued because of frequent (64%) grade 3/4 lymphopenia (lymphocyte count < 500 cells/mm3) in patients treated with the combination of cladribine and IFNβ. Add-on of cladribine reduced the risk of relapse by 63% compared to placebo.69
Safety and tolerability
Cladribine tablets were well tolerated with no symptoms in relation to drug administration. Across all studies cladribine showed a favourable safety profile. As a reflection of the mechanism of action of cladribine,53 lymphopenia was more frequent in the cladribine groups (combined cladribine group 27% versus 1.8% in placebo) in the CLARITY trial.61
Following treatment with cladribine 3.5 mg/kg, 86–89% of patients recovered to grade 0 or 1 lymphopenia by week 48 in each treatment year. At the end of year 2, severe lymphopenia (grade 3 or 4) was only seen in 2.3%.70 In patients with a normal baseline absolute lymphocyte count (ALC), only 0.5% had lymphopenia grade 3 at the end of the year; in patients who at the start of year 2 had lymphopenia grade 0 or 1, only 0.8% had grade 3 lymphopenia at the end of year 2.
The EU Summary of Product Characteristics (SMPC) for Cladribine Tablets states that the treatment should only be initiated in patients who have a normal ALC, and the treatment in year 2 should only be administered to patients with grade 0 or 1 lymphopenia (ALC ⩾ 0.8 × 109 cells/L). The course in year 2 can be delayed for up to 6 months to allow for recovery of lymphocytes, but if recovery takes more than 6 months, the patient should not receive further treatment.71
In the cladribine group, severe neutropenia was reported in three patients, of whom one also had severe thrombocytopenia and pancytopenia; the patient turned out to have a reactivation of latent tuberculosis and subsequently died.70 As it was conceivable that cladribine had contributed to this reactivation, screening measures for tuberculosis have been implemented before treatment and retreatment. The maximum decrease in ALC of 45–64% compared to baseline, and the median reduction in ALC from baseline to the end of the second treatment period was 43–48%.61 The total incidence of serious adverse events was 8.4% in the cladribine 3.5 mg/kg group, 9.0% in the 5.25 mg/kg group and 6.4% in the placebo group.61 Infections or infestations were reported as serious adverse events in 2.3% (cladribine 3.25 mg/kg), 2.9% (cladribine 5.25 mg/kg) and 1.6% (placebo), and the incidence of infections in the cladribine groups showed an inverse correlation with the lowest lymphocyte count. Herpes zoster infections, all dermatomal restricted, developed in 20 patients who received cladribine, compared to none in the placebo group.61 Herpes zoster infections were most frequently seen in patients suffering lymphopenia grade 3 or 4,61 although this was not evident when serial lymphocyte counts were applied.70 Nevertheless, prophylactic treatment for herpes is recommended in patients with lymphopenia grade 3 or 4.
PML has not been reported in MS patients treated with cladribine, but has been observed in patients treated with cladribine for malignant diseases.72
In the CLARITY study, neoplasms were found only in the cladribine group (four cancers and five benign uterine leiomyomas) versus none in the placebo group.61 However, the observed number of cancers during the study did not differ from the expected number obtained from a reference population standardized for country, gender and age [standard incidence rate (SIR)],70 and comparison with other DMTs shows that the number of malignancies in CLARITY was comparable.73 Accordingly, observations from all clinical trials and observational studies of cladribine in MS do not support any suspicion of an increase in rate of cancers.
There were four deaths during the CLARITY study and two after discontinuation, equally distributed across the three study groups.61
Data on human teratogenicity are sparse, but cladribine inhibits DNA synthesis and should therefore not be administered during pregnancy.53 Pregnancies have occurred during the clinical studies of cladribine in MS with various outcomes, most frequently induced abortion, but also delivery of healthy babies.70 Female patients need to practice effective contraception and avoid pregnancy until 6 months after treatment with cladribine. Male patients must take precautions to prevent pregnancy of their female partner during cladribine treatment and for at least 6 months after the last dose.
There is only limited experience with use of other DMTs after treatment with cladribine, and knowledge of long-term safety in MS patients treated with cladribine is sparse.
After treatment with a pulsed immune reconstitution therapy it is advisable to avoid cell-depleting treatment until the lymphocyte count has reached lower limits of normal and use, for example, the injectable platform therapies. With a normal cell count and no signs of immune deficiency, treatment with other disease-modifying drugs such as dimethyl fumarate, natalizumab or ocrelizumab would appear to be well tolerated.
Haematopoietic stem cell transplantation
Intense immunosuppression followed by autologous HSCT has been used to treat patients with MS during the last 2–3 decades.74 The most commonly used immunosuppressive, conditioning regimen is the so-called BEAM, including treatment with carmustine, etoposide, cytarabine and melphalan. A less intensive conditioning regimen has consisted of cyclophosphamide, sometimes combined with fludarabine. Surviving cells are further depleted by infusion of polyclonal anti-thymocyte globulin (ATG).74 After HSCT there is extensive regeneration of the circulating T-cell pool with emergence of a novel repertoire of CD4+ T-cells, whereas CD8+ predominantly emerge from expansion from the preexisting T-cell repertoire.75 As a result, there are increased numbers of regulatory T-cells, diminished pro-inflammatory T-helper type 17 responses and loss of the subset of mucosal-associated invariant T-cells after HSCT.76,77 Interestingly, disease activity after HSCT has been associated with emergence of a more clonally restricted CD4+ T-cell repertoire after HSCT.75
Whereas HSCT was initially used primarily in patients with progressed disease, the trend during the last 10–15 years has been to use HSCT in patients with less-progressed disease, primarily patients with RRMS, and to use less intense conditioning regimens. Because of this, the mortality has decreased from 5–7% to 1.3%,74 and in the most recent years the reported mortality after HSCT has been below 1%.
A study of 19 patients, four of whom had RRMS with an average EDSS of 6.4, followed for a mean observation time of 102 months after HSCT, found that 95% of the patients were free of disease progression in EDSS and 64% experienced neither relapses nor worsening on the EDSS.78
In a study in 21 patients with RRMS using non-myeloablative conditioning with cyclophosphamide, with a median observation time of 37 months, none of the patients progressed one point on the EDSS, 76% were free of relapses and 62 had NEDA-3.79
An Italian multi-centre collaboration using BEAM/ATG included 74 patients between 1996 and 2008, of whom 33 had RRMS with a mean EDSS of 6.3. After a mean observation time of 48 months, 74% of the patients remained free of worsening in EDSS, and of 18 patients followed for more than 7 years 44% remained stable or had an improvement in EDSS. The mortality was 2.7%.80
In a Swedish study of 48 patients, efficacy assessment was performed in 41 patients, of whom 34 patients (83%) had RRMS, with a mean follow-up time of 47 months, 4 patients (10%) experienced relapses after HSCT and the ARR was 0.03. Eight patients progressed in EDSS score, while one patient improved. At 5 years NEDA-3 was 68%.81 No mortality was recorded. Acute toxicity during hospitalization included the well-known and expected side-effects of alopecia, anaemia, thrombocytopenia and leukopenia, and were seen in almost all patients. A little less than half of the patients experienced fever with bacteraemia, neutropenic fever was seen in about one-third and one patient was diagnosed with invasive fungal infection. Eight patients (17%) experienced herpes zoster reactivation, four patients (8.3%) developed thyroid disease, one patient developed Crohn’s disease and one patient developed alopecia areata. No patient developed malignancy during the follow-up period.81
Recently, a study of 24 patients with RRMS with EDSS 3.0–5.5 were treated with HSCT using BEAM. Median follow-up was 62 months (range 12–72). Progression-free survival was 91% (90% CI: 75–97%), relapse-free survival was 87% (90% CI: 70–95%), and no evidence of disease activity (NEDA-3) was reported in 69% (90% CI: 50–82%) (Figure 2). No HSCT-related deaths occurred.82
Figure 2.

Proportion of patients with disease-free status (NEDA-3) in patients treated with cladribine,58 alemtuzumab,32 and three recent trials of HSCT (HALT-MS77; Swedish trial76; and Burt74).
A meta-analysis of 15 studies comprising 764 patients undergoing HSCT for MS integrated results of both efficacy and safety. They found a pooled disability progression rate of 23% and a pooled proportion of patients maintaining NEDA-3 of 68% at 5 years after the procedure. The treatment-related mortality was 2.1%, with a decrease in most recent years.83
Conclusion
The advantages and potential disadvantages of continuous immunosuppression and pulsed immune reconstitution therapy are shown in Table 3. Advantages include infrequent administration and efficacy beyond the period of active treatment. Long-term remission is seen in a considerable proportion of patients after one full course of alemtuzumab, and in fact 58.0–68.5% did not need additional cycles of alemtuzumab for 5 years.32,33 After cladribine 3.5 mg/kg in the 2-year randomized placebo-controlled study, 75.1% of patients remained relapse-free in years 3 and 4 without further treatment, but the effect after year 4 is not known.84
Table 3.
Potential advantages and disadvantages comparing continuous immunosuppression and pulsed immunosuppression.
| Continuous immunosuppression (e.g. dimethyl fumarate, teriflunomide, fingolimod, natalizumab) | Pulsed immunosuppression (e.g. alemtuzumab, cladribine) | |
|---|---|---|
| Clinical efficacy duration | Only during active treatment | Extends beyond period of active treatment |
| Infections: • PML • Reactivation of latent infections, e.g. tuberculosis • Herpes zoster |
Risk of PML with some treatments Low risk of reactivation Low to moderate risk |
Low risk of PML Higher risk of reactivation Higher risk during lymphopenia |
| Live vaccines | Contraindicated | May not be contraindicated after immune reconstitution |
| Risk of cancer | Long-term immunosuppression may increase risk of cancer | No indication of increased risk of cancer |
| Pregnancy | Not recommended* | Pregnancy safe after drug elimination (4 months after alemtuzumab and 6 months after cladribine administration) |
| Lactation | Not recommended | Lactation safe after drug elimination (see above) |
| Treatment escalation in case of suboptimal treatment response | Well-documented and generally well tolerated using risk-mitigation strategies | Only limited experience |
| Risk of adverse effects after discontinuation of treatment | Short-term risk only | Long-term risk even after immune reconstitution |
| Long-term tolerability | Well-known and generally good. Serious infections occur, and PML has been reported for natalizumab and in rare cases for fingolimod and dimethyl fumarate | Uncertain |
The general rule is that pregnancy is not recommended. However, for some drugs, such as natalizumab, use during pregnancy should be based on a benefit–risk evaluation taking into account the patient’s clinical condition and the possible return of disease activity after stopping the medicinal product.
PML: progressive multifocal leukoencephalopathy.
Another advantage is the possibility to become pregnant and lactate 4 months (alemtuzumab) or 6 months (cladribine) after the last administration of the drug.
Potential disadvantages of pulsed immune reconstitution therapy include reactivation of latent infections such as tuberculosis, and risk of herpes zoster which is associated with severe lymphopenia. Also, several rare infections such as Listeria meningitis have been reported. The main disadvantage in alemtuzumab-treated patients is the risk of secondary immune-mediated disorders, which, however, has not been reported with cladribine. Also, the long-term monitoring required after the last administration of alemtuzumab is a drawback.
The development of secondary cancers, which initially was suspected for cladribine, does not seem to be related to pulsed immune reconstitution therapy.
Pulsed immune reconstitution therapy is an option as initial therapy in RRMS patients with high disease activity, which in untreated patients may be defined as two or more relapses within the last year, and in patients on treatment with another DMT as two or more relapses or one relapse and significant MRI activity. However, pulsed immune reconstitution therapy might also be considered as the initial therapy in early RRMS with negative prognostic factors and active disease from onset (not only patients with two relapses in the previous year, but also patients with a relapse with incomplete recovery associated with new or enhancing lesions in MRI). If pulsed immune reconstitution therapy has been decided, the choice between alemtuzumab and cladribine should be based on efficacy balanced against the risks. Figure 3 depicts the authors’ opinion of the efficacy and tolerability of cladribine, alemtuzumab and HSCT. The potentially higher efficacy of alemtuzumab compared to cladribine, although still only hypothetical as no head-to-head studies have been performed, comes at the costs of more frequent and severe adverse events. In our opinion, based on the published data, the evidence of long-term efficacy is increasing from cladribine to alemtuzumab and is best for HSCT.
Figure 3.
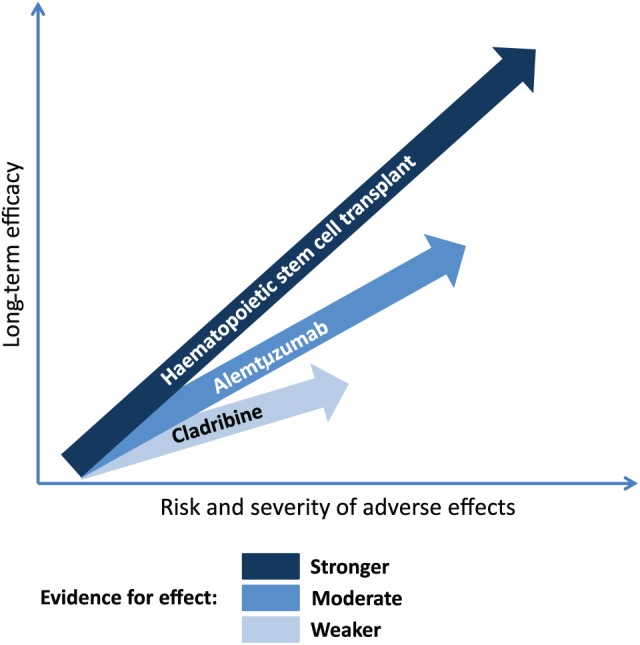
Long-term efficacy and safety of pulsed immune reconstitution therapies and haematopoietic stem cell transplantation (HSCT). The figure depicts the authors’ opinion of the efficacy and tolerability of cladribine, alemtuzumab and HSCT, but no head-to-head studies have been performed.
Whereas treatment with intense immunosuppression supported with autologous HSCT can establish long-term disease-free status and, in some patients, possibly even permanent remission, it still needs to be shown that pulsed immune reconstitution therapy with alemtuzumab or cladribine can also induce long-term or even permanent drug-free remission.85 However, as the most encouraging results with HSCT have been reported in very early RRMS, induction of long-term NEDA-3 with pulsed immune reconstitution therapy could at least be an exciting possibility in some patients, if therapy is given early in the disease course.
Presently, the experience with long-term outcome for these therapies is missing and must be addressed in long-term follow-up studies.
Footnotes
Funding: This research received no specific grant from any funding agency in the public, commercial or not-for-profit sectors.
Conflict of interest statement: PSS has received personal compensation for serving on scientific advisory boards, steering committees or independent data monitoring boards for Biogen, Merck, Novartis, TEVA, GlaxoSmithKline, MedDay Pharmaceuticals, Genzyme, Celgene and Forward Pharma, and has received speaker honoraria from Biogen, Merck, Teva, Genzyme and Novartis. FS has served on scientific advisory boards, been on the steering committees of clinical trials, served as a consultant, received support for congress participation, received speaker honoraria or received research support for his laboratory from Biogen, EMD Serono, Merck, Novartis, Roche and Sanofi Genzyme.
Contributor Information
Per Soelberg Sorensen, Department of Neurology 2082, Danish Multiple Sclerosis Center, University of Copenhagen, Rigshospitalet, 9, Blegdamsvej, DK-2100 Copenhagen, Denmark.
Finn Sellebjerg, Department of Neurology, Danish Multiple Sclerosis Center, University of Copenhagen, Rigshospitalet, Copenhagen, Denmark.
References
- 1. Sorensen PS. New management algorithms in multiple sclerosis. Curr Opin Neurol 2014; 27: 246–259. [DOI] [PubMed] [Google Scholar]
- 2. Comi G, Radaelli M, Soelberg Sorensen P. Evolving concepts in the treatment of relapsing multiple sclerosis. Lancet 2017; 389: 1347–1356. [DOI] [PubMed] [Google Scholar]
- 3. Sorensen PS, Koch-Henriksen N, Petersen T, et al. Recurrence or rebound of clinical relapses after discontinuation of natalizumab therapy in highly active MS patients. J Neurol 2014; 261: 1170–1177. [DOI] [PubMed] [Google Scholar]
- 4. Wiendl H. Cladribine: an old newcomer for pulsed immune reconstitution in MS. Nat Rev Neurol 2017; 13: 573–574. [DOI] [PubMed] [Google Scholar]
- 5. Muraro PA, Pasquini M, Atkins HL, et al. Long-term outcomes after autologous hematopoietic stem cell transplantation for multiple sclerosis. JAMA Neurol 2017; 74: 459–469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hauser SL, Bar-Or A, Comi G, et al. Ocrelizumab versus interferon beta-1a in relapsing multiple sclerosis. N Engl J Med 2017; 376: 221–234. [DOI] [PubMed] [Google Scholar]
- 7. Le PE, Deburghgraeve V, Lester MA, et al. Alemtuzumab as rescue therapy in a cohort of 16 aggressive multiple sclerosis patients previously treated by mitoxantrone: an observational study. J Neurol 2015; 262: 1024–1034. [DOI] [PubMed] [Google Scholar]
- 8. Edan G, Comi G, Le PE, et al. Mitoxantrone prior to interferon beta-1b in aggressive relapsing multiple sclerosis: a 3-year randomised trial. J Neurol Neurosurg Psychiatry 2011; 82: 1344–1350. [DOI] [PubMed] [Google Scholar]
- 9. Le PE, Leray E, Taurin G, et al. Mitoxantrone as induction treatment in aggressive relapsing remitting multiple sclerosis: treatment response factors in a 5 year follow-up observational study of 100 consecutive patients. J Neurol Neurosurg Psychiatry 2008; 79: 52–56. [DOI] [PubMed] [Google Scholar]
- 10. Martinelli V, Cocco E, Capra R, et al. Acute myeloid leukemia in Italian patients with multiple sclerosis treated with mitoxantrone. Neurology 2011; 77: 1887–1895. [DOI] [PubMed] [Google Scholar]
- 11. Rao SP, Sancho J, Campos-Rivera J, et al. Human peripheral blood mononuclear cells exhibit heterogeneous CD52 expression levels and show differential sensitivity to alemtuzumab mediated cytolysis. PLoS One 2012; 7: e39416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hale G. The CD52 antigen and development of the CAMPATH antibodies. Cytotherapy 2001; 3: 137–143. [DOI] [PubMed] [Google Scholar]
- 13. Ruck T, Bittner S, Wiendl H, et al. Alemtuzumab in multiple sclerosis: mechanism of action and beyond. Int J Mol Sci 2015; 16: 16414–16439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Baker D, Giovannoni G, Schmierer K. Marked neutropenia: significant but rare in people with multiple sclerosis after alemtuzumab treatment. Mult Scler Relat Disorder 2017; 18: 181–183. [DOI] [PubMed] [Google Scholar]
- 15. Ceronie B, Jacobs BM, Baker D, et al. Cladribine treatment of multiple sclerosis is associated with depletion of memory B cells. J Neurol 2018; 265: 1199–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Freedman MS, Kaplan JM, Markovic-Plese S. Insights into the mechanisms of the therapeutic efficacy of alemtuzumab in multiple sclerosis. J Clin Cell Immunol 2013; 4: pii: 1000152. [PMC free article] [PubMed] [Google Scholar]
- 17. Hartung HP, Aktas O, Boyko AN. Alemtuzumab: a new therapy for active relapsing-remitting multiple sclerosis. Mult Scler 2015; 21: 22–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. von Essen MR, Ammitzboll C, Hansen RH, et al. Proinflammatory CD20+ T cells in the pathogenesis of multiple sclerosis. Brain 2019;142: 120–132. [DOI] [PubMed] [Google Scholar]
- 19. Thompson SA, Jones JL, Cox AL, et al. B-cell reconstitution and BAFF after alemtuzumab (Campath-1H) treatment of multiple sclerosis. J Clin Immunol 2010; 30: 99–105. [DOI] [PubMed] [Google Scholar]
- 20. Robertson NP, Scolding NJ. Immune reconstitution and treatment response in multiple sclerosis following alemtuzumab. Neurology 2014; 82: 2150–2151. [DOI] [PubMed] [Google Scholar]
- 21. Menge T, Stuve O, Kieseier BC, et al. Alemtuzumab: the advantages and challenges of a novel therapy in MS. Neurology 2014; 83: 87–97. [DOI] [PubMed] [Google Scholar]
- 22. Jones JL, Thompson SA, Loh P, et al. Human autoimmunity after lymphocyte depletion is caused by homeostatic T-cell proliferation. Proc Natl Acad Sci USA 2013; 110: 20200–20205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. De Mercanti S, Rolla S, Cucci A, et al. Alemtuzumab long-term immunologic effect: Treg suppressor function increases up to 24 months. Neurol Neuroimmunol Neuroinflamm 2016; 3: e194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Cossburn MD, Harding K, Ingram G, et al. Clinical relevance of differential lymphocyte recovery after alemtuzumab therapy for multiple sclerosis. Neurology 2013; 80: 55–61. [DOI] [PubMed] [Google Scholar]
- 25. Kousin-Ezewu O, Azzopardi L, Parker RA, et al. Accelerated lymphocyte recovery after alemtuzumab does not predict multiple sclerosis activity. Neurology 2014; 82: 2158–2164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Coles AJ, Compston DA, Selmaj KW, et al. Alemtuzumab vs. interferon beta-1a in early multiple sclerosis. N Engl J Med 2008; 359: 1786–1801. [DOI] [PubMed] [Google Scholar]
- 27. Coles AJ, Fox E, Vladic A, et al. Alemtuzumab more effective than interferon beta-1a at 5-year follow-up of CAMMS223 clinical trial. Neurology 2012; 78: 1069–1078. [DOI] [PubMed] [Google Scholar]
- 28. Cohen JA, Coles AJ, Arnold DL, et al. Alemtuzumab versus interferon beta 1a as first-line treatment for patients with relapsing-remitting multiple sclerosis: a randomised controlled phase 3 trial. Lancet 2012; 380: 1819–1828. [DOI] [PubMed] [Google Scholar]
- 29. Coles AJ, Twyman CL, Arnold DL, et al. Alemtuzumab for patients with relapsing multiple sclerosis after disease-modifying therapy: a randomised controlled phase 3 trial. Lancet 2012; 380: 1829–1839. [DOI] [PubMed] [Google Scholar]
- 30. Giovannoni G, Cohen JA, Coles AJ, et al. Alemtuzumab improves preexisting disability in active relapsing-remitting MS patients. Neurology 2016; 87: 1985–1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Arroyo Gonzalez R, Kita M, Crayton H, et al. Alemtuzumab improves quality-of-life outcomes compared with subcutaneous interferon beta-1a in patients with active relapsing-remitting multiple sclerosis. Mult Scler 2017; 23: 1367–1376. [DOI] [PubMed] [Google Scholar]
- 32. Havrdova E, Arnold DL, Cohen JA, et al. Alemtuzumab CARE-MS I 5-year follow-up: durable efficacy in the absence of continuous MS therapy. Neurology 2017; 89: 1107–1116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Coles AJ, Cohen JA, Fox EJ, et al. Alemtuzumab CARE-MS II 5-year follow-up: efficacy and safety findings. Neurology 2017; 89: 1117–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tuohy O, Costelloe L, Hill-Cawthorne G, et al. Alemtuzumab treatment of multiple sclerosis: long-term safety and efficacy. J Neurol Neurosurg Psychiatry 2015; 86: 208–215. [DOI] [PubMed] [Google Scholar]
- 35. Moreau T, Coles A, Wing M, et al. Transient increase in symptoms associated with cytokine release in patients with multiple sclerosis. Brain 1996; 119: 225–237. [DOI] [PubMed] [Google Scholar]
- 36. Haghikia A, Dendrou CA, Schneider R, et al. Severe B-cell-mediated CNS disease secondary to alemtuzumab therapy. Lancet Neurol 2017; 16: 104–106. [DOI] [PubMed] [Google Scholar]
- 37. Azzopardi L, Thompson SA, Harding KE, et al. Predicting autoimmunity after alemtuzumab treatment of multiple sclerosis. J Neurol Neurosurg Psychiatry 2014; 85: 795–798. [DOI] [PubMed] [Google Scholar]
- 38. Jones JL, Phuah CL, Cox AL, et al. IL-21 drives secondary autoimmunity in patients with multiple sclerosis, following therapeutic lymphocyte depletion with alemtuzumab (Campath-1H). J Clin Invest 2009; 119: 2052–2061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rau D, Lang M, Harth A, et al. Listeria meningitis complicating alemtuzumab treatment in multiple sclerosis: report of two cases. Int J Mol Sci 2015; 16: 14669–14676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Canham LJW, Manara A, Fawcett J, et al. Mortality from Listeria monocytogenes meningoencephalitis following escalation to alemtuzumab therapy for relapsing-remitting multiple sclerosis. Mult Scler Relat Disord 2018; 24: 38–41. [DOI] [PubMed] [Google Scholar]
- 41. Metz I, Rieckmann P, Kallmann BA, et al. Disseminated necrotizing leukoencephalopathy eight months after alemtuzumab treatment for multiple sclerosis. Acta Neuropathol Commun 2016; 4: 81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Saarela M, Senthil K, Jones J, et al. Hemophagocytic lymphohistiocytosis in 2 patients with multiple sclerosis treated with alemtuzumab. Neurology 2018; 90: 849–851. [DOI] [PubMed] [Google Scholar]
- 43. Meunier B, Rico A, Seguier J, et al. Life-threatening autoimmune warm hemolytic anemia following treatment for multiple sclerosis with alemtuzumab. Mult Scler 2018; 24: 811–813. [DOI] [PubMed] [Google Scholar]
- 44. Zimmermann J, Buhl T, Muller M. Alopecia universalis following alemtuzumab treatment in multiple sclerosis: a barely recognized manifestation of secondary autoimmunity – report of a case and review of the literature. Front Neurol 2017; 8: 569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Isidoro L, Pires P, Rito L, et al. Progressive multifocal leukoencephalopathy in a patient with chronic lymphocytic leukaemia treated with alemtuzumab. BMJ Case Rep 2014. Epub ahead of print 8 January 2014. DOI: 10.1136/bcr-2013-201781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Martin SI, Marty FM, Fiumara K, et al. Infectious complications associated with alemtuzumab use for lymphoproliferative disorders. Clin Infect Dis 2006; 43: 16–24. [DOI] [PubMed] [Google Scholar]
- 47. Croteau D, Flowers C, Kulick CG, et al. Acute acalculous cholecystitis: a new safety risk for patients with MS treated with alemtuzumab. Neurology 2018; 90: e1548–e1552. [DOI] [PubMed] [Google Scholar]
- 48. McCarthy CL, Tuohy O, Compston DA, et al. Immune competence after alemtuzumab treatment of multiple sclerosis. Neurology 2013; 81: 872–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Sorensen PS, Arnold D, Cohen J, et al. Immunogenicity of alemtuzumab treatment in relapsing-remitting multiple sclerosis (RRMS) patients in the CARE-MS II study. Neurology 2013; 80(suppl. 7): PO7.101. [Google Scholar]
- 50. Dubuisson N, Baker D, Kang AS, et al. Alemtuzumab depletion failure can occur in multiple sclerosis. Immunology 2018; 154: 253–260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Taylor RP, Lindorfer MA. Fcgamma-receptor-mediated trogocytosis impacts mAb-based therapies: historical precedence and recent developments. Blood 2015; 125: 762–766. [DOI] [PubMed] [Google Scholar]
- 52. Liliemark J. The clinical pharmacokinetics of cladribine. Clin Pharmacokinet 1997; 32: 120–131. [DOI] [PubMed] [Google Scholar]
- 53. Leist TP, Weissert R. Cladribine: mode of action and implications for treatment of multiple sclerosis. Clin Neuropharmacol 2011; 34: 28–35. [DOI] [PubMed] [Google Scholar]
- 54. Beutler E. Cladribine (2-chlorodeoxyadenosine). Lancet 1992; 340: 952–956. [DOI] [PubMed] [Google Scholar]
- 55. Cheson BD. The purine analogs: a therapeutic beauty contest. J Clin Oncol 1992; 10: 868–871. [DOI] [PubMed] [Google Scholar]
- 56. Brousil JA, Roberts RJ, Schlein AL. Cladribine: an investigational immunomodulatory agent for multiple sclerosis. Ann Pharmacother 2006; 40: 1814–1821. [DOI] [PubMed] [Google Scholar]
- 57. Genini D, Budihardjo I, Plunkett W, et al. Nucleotide requirements for the in vitro activation of the apoptosis protein-activating factor-1-mediated caspase pathway. J Biol Chem 2000; 275: 29–34. [DOI] [PubMed] [Google Scholar]
- 58. Wyczechowska D, Fabianowska-Majewska K. The effects of cladribine and fludarabine on DNA methylation in K562 cells. Biochem Pharmacol 2003; 65: 219–225. [DOI] [PubMed] [Google Scholar]
- 59. Tan J, Yang X, Zhuang L, et al. Pharmacologic disruption of Polycomb-repressive complex 2-mediated gene repression selectively induces apoptosis in cancer cells. Genes Dev 2007; 21: 1050–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Baker D, Herrod SS, Alvarez-Gonzalez C, et al. Both cladribine and alemtuzumab may effect MS via B-cell depletion. Neurol Neuroimmunol Neuroinflamm 2017; 4: e360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Giovannoni G, Comi G, Cook S, et al. A placebo-controlled trial of oral cladribine for relapsing multiple sclerosis. N Engl J Med 2010; 362: 416–426. [DOI] [PubMed] [Google Scholar]
- 62. Giovannoni G, Cook S, Rammohan K, et al. Sustained disease-activity-free status in patients with relapsing-remitting multiple sclerosis treated with cladribine tablets in the CLARITY study: a post-hoc and subgroup analysis. Lancet Neurol 2011; 10: 329–337. [DOI] [PubMed] [Google Scholar]
- 63. Giovannoni G, Soelberg Sorensen P, Cook S, et al. Efficacy of cladribine tablets in high disease activity subgroups of patients with relapsing multiple sclerosis: a post hoc analysis of the CLARITY study. Mult Scler 2018. Epub ahead of print 1 April 2018. DOI: 10.1177/1352458518771875. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. De Stefano N, Giorgio A, Battaglini M, et al. Reduced brain atrophy rates are associated with lower risk of disability progression in patients with relapsing multiple sclerosis treated with cladribine tablets. Mult Scler 2018; 24: 222–226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Comi G, Cook SD, Giovannoni G, et al. MRI outcomes with cladribine tablets for multiple sclerosis in the CLARITY study. J Neurol 2012; 260: 1136–1146. [DOI] [PubMed] [Google Scholar]
- 66. Giovannoni G, Soelberg Sorensen P, Cook S, et al. Safety and efficacy of cladribine tablets in patients with relapsing-remitting multiple sclerosis: results from the randomized extension trial of the CLARITY study. Mult Scler 2017: 1352458517727603. [DOI] [PubMed] [Google Scholar]
- 67. Comi G, Cook S, Rammohan K, et al. Long-term effects of cladribine tablets on MRI activity outcomes in patients with relapsing-remitting multiple sclerosis: the CLARITY extension study. Ther Adv Neurol Disord 2018; 24: 1594–1604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Leist TP, Comi G, Cree BA, et al. Effect of oral cladribine on time to conversion to clinically definite multiple sclerosis in patients with a first demyelinating event (ORACLE MS): a phase 3 randomised trial. Lancet Neurol 2014; 13: 257–267. [DOI] [PubMed] [Google Scholar]
- 69. Montalban X, Cohen BA, Jeffery DR, et al. Oral cladribine added to interferon beta-1a for active multiple sclerosis: a 96-week, double-blind, placebo-controlled phase IIb study. Mult Scler 2007; 13: S245–S245. [Google Scholar]
- 70. Cook S, Vermersch P, Comi G, et al. Safety and tolerability of cladribine tablets in multiple sclerosis: the CLARITY (CLAdRIbine Tablets treating multiple sclerosis orallY) study. Mult Scler 2011; 17: 578–593. [DOI] [PubMed] [Google Scholar]
- 71. European Medicines Agency. Annex I: summary of product characteristics, www.ema.europa.eu/en/documents/product-information/mavenclad-epar-product-information_en.pdf.
- 72. Alstadhaug KB, Fykse Halstensen R, Odeh F. Progressive multifocal leukoencephalopathy in a patient with systemic mastocytosis treated with cladribine. J Clin Virol 2017; 88: 17–20. [DOI] [PubMed] [Google Scholar]
- 73. Pakpoor J, Disanto G, Altmann DR, et al. No evidence for higher risk of cancer in patients with multiple sclerosis taking cladribine. Neurol Neuroimmunol Neuroinflamm 2015; 2: e158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Mancardi G, Saccardi R. Autologous haematopoietic stem-cell transplantation in multiple sclerosis. Lancet Neurol 2008; 7: 626–636. [DOI] [PubMed] [Google Scholar]
- 75. Muraro PA, Robins H, Malhotra S, et al. T cell repertoire following autologous stem cell transplantation for multiple sclerosis. J Clin Invest 2014; 124: 1168–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Darlington PJ, Touil T, Doucet JS, et al. Diminished Th17 (not Th1) responses underlie multiple sclerosis disease abrogation after hematopoietic stem cell transplantation. Ann Neurol 2013; 73: 341–354. [DOI] [PubMed] [Google Scholar]
- 77. Darlington PJ, Stopnicki B, Touil T, et al. Natural killer cells regulate Th17 cells after autologous hematopoietic stem cell transplantation for relapsing remitting multiple sclerosis. Front Immunol 2018; 9: 834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Saccardi R, Mancardi GL, Solari A, et al. Autologous HSCT for severe progressive multiple sclerosis in a multicenter trial: impact on disease activity and quality of life. Blood 2005; 105: 2601–2607. [DOI] [PubMed] [Google Scholar]
- 79. Burt RK, Loh Y, Cohen B, et al. Autologous non-myeloablative haemopoietic stem cell transplantation in relapsing-remitting multiple sclerosis: a phase I/II study. Lancet Neurol 2009; 8: 244–253. [DOI] [PubMed] [Google Scholar]
- 80. Mancardi GL, Sormani MP, Di GM, et al. Autologous haematopoietic stem cell transplantation with an intermediate intensity conditioning regimen in multiple sclerosis: the Italian multi-centre experience. Mult Scler 2012; 18: 835–842. [DOI] [PubMed] [Google Scholar]
- 81. Burman J, Iacobaeus E, Svenningsson A, et al. Autologous haematopoietic stem cell transplantation for aggressive multiple sclerosis: the Swedish experience. J Neurol Neurosurg Psychiatry 2014; 85: 1116–1121. [DOI] [PubMed] [Google Scholar]
- 82. Nash RA, Hutton GJ, Racke MK, et al. High-dose immunosuppressive therapy and autologous HCT for relapsing-remitting MS. Neurology 2017; 88: 842–852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Sormani MP, Muraro PA, Schiavetti I, et al. Autologous hematopoietic stem cell transplantation in multiple sclerosis: a meta-analysis. Neurology 2017; 88: 2115–2122. [DOI] [PubMed] [Google Scholar]
- 84. Giovannoni G, Soelberg Sorensen P, Cook S, et al. Safety and efficacy of cladribine tablets in patients with relapsing-remitting multiple sclerosis: results from the randomized extension trial of the CLARITY study. Mult Scler 2018; 24: 1594–1604. [DOI] [PubMed] [Google Scholar]
- 85. Wiendl H, Bourdette D, Ciccarelli O. Can immune reprogramming with alemtuzumab induce permanent remission in multiple sclerosis? Neurology 2017; 89: 1098–1100. [DOI] [PubMed] [Google Scholar]