

Abstract
Impaired mitochondrial function has been associated with the etiopathogenesis of Parkinson's disease (PD). Sustained inhibition of complex I produces mitochondrial dysfunction, which is related to oxidative injury and nigrostriatal dopamine (DA) neurodegeneration. This study aimed to identify disease-modifying treatments for PD. Unsubstituted phenothiazine (PTZ) is a small and uncharged aromatic imine that readily crosses the blood-brain barrier. PTZ lacks significant DA receptor-binding activity and, in the nanomolar range, exhibits protective effects via its potent free radical scavenging and anti-inflammatory activities. Given that DAergic neurons are highly vulnerable to oxidative damage and inflammation, we hypothesized that administration of PTZ might confer neuroprotection in different experimental models of PD. Our findings showed that PTZ rescues rotenone (ROT) toxicity in primary ventral midbrain neuronal cultures by preserving neuronal integrity and reducing protein thiol oxidation. Long-term treatment with PTZ improved animal weight, survival rate, and behavioral deficits in ROT-lesioned rats. PTZ protected DA content and fiber density in the striatum and DA neurons in the SN against the deleterious effects of ROT. Mitochondrial dysfunction, axonal impairment, oxidative insult, and inflammatory response were attenuated with PTZ therapy. Furthermore, we have provided a new insight into the molecular mechanism underlying the neuroprotective effects of PTZ.
Keywords: Phenothiazine, Parkinson's disease, Rotenone, Mitochondria, Oxidative stress, Inflammation
Graphical abstract
Highlights
-
•
Rotenone recapitulates many of the behavioral, neurochemical, and neuropathological features of PD in rats.
-
•
Phenothiazine preserves neuronal integrity in primary ventral midbrain neuronal cultures incubated with ROT.
-
•
PTZ prevents PD-like features in ROT-injected rats, including nigrostriatal DA degeneration and mitochondrial dysfunction.
-
•
We have provided new insights into the molecular mechanism underlying the neuroprotective effects of PTZ.
1. Introduction
Although functional efficiency of mitochondria declines with age, experimental outcomes support the notion of mitochondrial dysfunction as a key mechanism in the pathogenesis of Parkinson's disease (PD), characterized, in part, by a depletion of dopamine (DA) content in the caudate-putamen and progressive and selective degeneration of DA-containing neurons in the substantia nigra (SN) [1,2].
Fusion and fission events are responsible for regulating the dynamic features of mitochondria and serve as important quality control mechanism for preserving a healthy mitochondrial network. The mitochondrial fusion machinery is regulated by large GTPase dynamin-related proteins, such as the inner membrane optic atrophy protein 1 (Opa1) and the outer mitochondrial membrane mitofusins 1 and 2 (Mfn1 and Mfn2). The fission mechanism is governed by four integral membrane proteins of the outer mitochondrial membrane: fission protein 1 (Fis1), fission factor, and 49 and 51 kDa mitochondrial dynamic proteins (MiD49 and MiD51) that recruit dynamin-related protein 1 (Drp1) from the cytosol to outer mitochondrial membrane [3,4]. Fis1 only translocates to the outer mitochondrial membrane when fission is activated.
Axonal transport is a physiological process critical to neuronal viability and function, regulated by the kinesin and dynein motor proteins. Kinesins are heterotetrametric protein complexes composed of two heavy chain (KHC) and two light chain (KLC) subunits that move cargoes unidirectionally in the anterograde direction; contrarily, dynein (DYNLT3) is the major motor protein driving retrograde transport. Mitochondria undergo bidirectional transport along both microtubule and actin filaments [5]. Subtle defects in mitochondrial dynamics and/or trafficking along axons may play a prominent role in the pathophysiology and progression of PD [[6], [7], [8]].
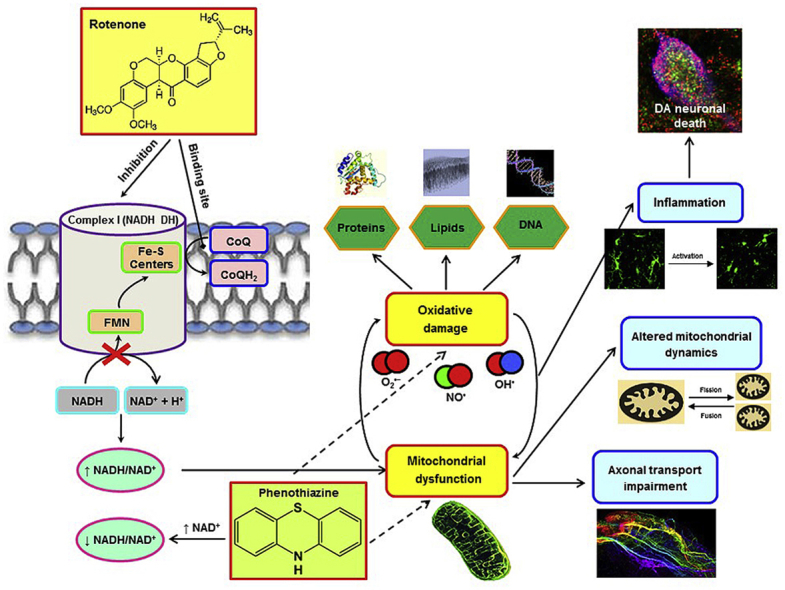
Persistent oxidative stress leads to impaired mitochondrial function by disrupting the electron transport chain and subsequent electron leakage from donor redox centers to molecular oxygen, constituting a major source of reactive oxygen species (ROS) [9]. Cellular membrane lipids, especially polyunsaturated fatty acids, are prone to ROS attack which culminates in the generation of lipid peroxides. Elevated immunoreactivity of 4-hydroxy-trans-2-nonenal (4-HNE), a lipid peroxidation-derived aldehyde, has been reported in SN DA neurons of neurotoxin-lesioned animal models of PD and patients with PD [10,11]. Levels of nitric oxide (NO•) are substantially increased in response to inducible NO• synthase (iNOS) induction, leading to overproduction of reactive nitrogen species (RNS). NO•-derived RNS react with tyrosine residues in proteins to form nitrotyrosine (3-NT), which is significantly increased in SN post-mortem parkinsonian specimens and experimental animal models of PD [8,12,13]. Dysfunctional mitochondria can contribute to free radical formation initiating a vicious cycle that amplifies the burden of oxidative stress.
Mutations in certain PD-linked genes, such as alpha-synuclein (α-syn), leucine-rich repeat kinase 2 (LRRK2), PTEN-induced putative kinase1 (PINK1), Parkin, and DJ-1 have detrimental effects on oxidative stress and mitochondrial function, dynamics, and movement [7,14]. Systemic administration of rotenone (ROT) to rats causes specific inhibition of mitochondrial complex I and concomitant oxidative stress, and recapitulates many of the behavioral, neurochemical, and neuropathological features of PD [11,[15], [16], [17], [18], [19]]. In addition, ROT treatment results in synaptic pathology, altered expression of axonal transport- and mitochondrial dynamic-related proteins, DNA damage, an inflammatory response, and selective DA cell death in primary ventral midbrain neuronal cultures [8,11,19].
Therapeutic approaches targeting mitochondrial dysfunction and associated oxidative injury are an attractive and promising direction of ongoing neuroprotective research. Unsubstituted phenothiazine (PTZ) is the parent molecule of a myriad drugs, including methylene blue, and undergoes metabolic transformation by oxidation and conjugation, producing leucophenothiazone sulphate as its major metabolite [20]. PTZ has low toxicity in mammals and does not evoke CNS depression, carcinogenicity or genotoxicity [21,22]. PTZs can cross the blood-brain barrier because they are uncharged, lipophilic, and small molecules and exert a protective effect against cell death at low concentrations in vitro and in transgenic C. elegans [23,24]. Even though PTZs are used as antipsychotic agents with strong binding affinity for DA receptors, unsubstituted PTZ does not show any significant binding activity for D1 (Ki: 15.6 μM) and D2 (Ki > 20 μM) receptors [25]. PTZ and its derivatives are more powerful antioxidants than phenols. We therefore anticipate that chronic treatment with PTZ would have a beneficial effect against the neurotoxic effects of ROT. Our results demonstrate that PTZ confers protection in vitro and prevents the development of PD-like behavioral deficits and preserves the nigrostriatal DA system against ROT intoxication in rats. Our findings also provide new insights into the molecular mechanisms underlying the neuroprotective actions of PTZ.
2. Materials and methods
2.1. Animals
All the experiments were carried out in seven to eight-month-old male Lewis rats purchased from Hilltop (Scottdale, PA, USA) weighing ∼425–475 g upon arrival. Animals were maintained under standard conditions of 12 h light/dark cycle, 22 ± 1 °C temperature-controlled room, and 50–70% humidity. Subjects were given ad libitum access to food and water and were allowed to acclimate to the vivarium conditions for 2 weeks prior experimentation. All procedures were performed with the approval of the University of Pittsburgh Animal Care and Use Committee.
2.2. Rat ventral midbrain neuronal culture
Cell cultures were obtained from Sprague-Dawley rat embryos on gestational day 17 and were prepared as previously described [8,18,19]. Briefly, the ventral midbrain region (nuclei A8, A9, and A10) was dissected following removal of meninges and trypsin enzymatic digestion. Cells were seeded on a 24-well plate and incubated at 37 °C in a tri-gas incubator containing 5% CO2, 5% O2, and 90% N2 in 0.5 mL/well of MEM with 2% FBS, 2% HS, 1 g/L glucose, 2 mM GlutaMax, 1 mM sodium pyruvate, 100 μM non-essential amino acids, 50 U/mL penicillin, and 50 μg/mL streptomycin. To improve survival, 50 ng/mL of glial cell line-derived neurotrophic factor (GDNF) per well was added to the cultures. About 10–11% of cells were TH-immunoreactive.
2.3. Experimental design and treatment protocol
A series of dose-response assays were carried out to determine the optimal concentration of PTZ (≥98%, Sigma-Aldrich). Cells were seeded at a density of 5 × 105 cells/well (Fig. S1 A). On the second day in vitro (2 DIV), MEM was replaced to serum-free Neurobasal medium containing 2% B27 supplement, 2 mM GlutaMax, 0.5 mg/mL albumax I, 50 U/mL penicillin, and 50 μg/mL streptomycin and supplemented with GDNF. At 5 DIV, cells were incubated with 50 nM ROT whereas PTZ (10, 20 or 50 nM) was added 1 h later for a period of 5 days. Drugs were freshly prepared in DMSO and diluted with cell culture medium to the desired final concentration. Seven days after initial seeding, half of the medium was removed and replenished with fresh serum-free Neurobasal medium. Ten-day-old cultures were fixed and processed for cell counting, 3D neurite reconstruction, and thiol staining analyses.
For the in vivo study, rats received one intraperitoneal (i.p.) injection of ROT daily given as a 1 mL/kg solution (Fig. S1 B). ROT was prepared as a 50× stock dissolved in DMSO and diluted with Miglyol 812 N and 2% DMSO [11,[16], [17], [18], [19]]. PTZ was also dissolved in Miglyol and DMSO and administered 25 h following the first ROT injection (1 h after the second dose of ROT). Animals were i.p. injected with PTZ bis in die (BID) at 10-h intervals within 24 h. A volume of 1 mL/kg body weight was necessary to dissolve 1, 5, and 10 mg/kg PTZ while 2 mL/kg body weight were required to dissolve 20 and 50 mg/kg PTZ. Rats were divided into 3 different cohorts, which were randomly assigned to one of four groups: (i) VEH + VEH; (ii) PTZ + VEH; (iii) VEH + ROT; and (iv) PTZ + ROT. For dose-response experiments (cohort #1), 21 rats were received 3.0 mg/kg of ROT and different doses of PTZ (1, 5, 10, 20 or 50 mg/kg/day) for 16 days, when the last ROT-intoxicated rat died. Subjects were monitored daily for weight loss and motor signs. One rat in the 5 mg/kg PTZ + ROT group died at 11 days during the study.
For the time-window studies (cohorts #2 and #3), 58 rats were daily exposed to 3.0 mg/kg ROT – defined by bradykinesia, rigidity, and other parkinsonian symptoms – followed by two doses of 20 mg/kg PTZ every day at 10-h intervals until the rats reached the endpoint. Importantly, a few PTZ-treated animals were euthanized before the endpoint due to development of skin laceration around the injection area over the time.
2.4. Immunocytochemistry
Neuron cultures were fixed in 4% paraformaldehyde (PFA), 0.02% Triton X-100, and 1 mM magnesium chloride in 1× PBS for 30 min. Cells were washed trice in PBS for 10 min each and blocked with a solution consisting in 10% normal donkey serum (NDS) in PBS for 1 h. Unless otherwise specified, all incubations were carried out at RT. Following overnight incubation with primary antibodies (Table S1 for detailed information) in PBS with 1% NDS at 4 °C, cultures were rinsed 3 times in PBS for 10 min intervals and sequentially incubated with conjugated secondary antibodies (as listed in Table S1) in PBS with 1% NDS for 2 h. After washing once in PBS, cells were counterstained with Hoechst (H) 33,342 for 5 min to reveal nuclei. Three additional rinses in PBS were carried out before the coverslips were mounted directly onto plus-coated slides using gelvatol mounting media.
2.5. Thiol redox labeling
Oxidative stress was determined by thiol staining as previously described [26]. To initially label reduced thiol groups (SH), neuron cultures were fixed for 30 min in 4% PFA solution containing 0.02% Triton X-100, 10 μM NEM, and 1 μM Alexa-Fluor 680C2-maleimide, prepared in 1× PBS pH 7.0. To diminish disulfide bonds, cells were rinsed 3 times in PBS followed by incubation with 5 mM tris(2-carboxyethyl)phosphine hydrochloride solution. Cells were quickly washed in PBS (to prevent air-induced reoxidation of hitherto reduced thiol groups) and then incubated in a solution prepared in PBS with 10 μM NEM and 1 μM Alexa-Fluor 546C5-maleimide for 30 min to label the oxidized thiols (SS). After 3 additional washing steps in PBS, cells were ready for further immunocytochemical staining. The overall SS/SH ratio is the result of the combined action of ROS and the subsequent antioxidants defenses.
2.6. Assessment of functional outcome
To evaluate the deterioration of the nigrostriatal DA system, instinctive exploratory behavior was assessed as described elsewhere [11,17]. Rats were placed individually in a Plexiglas cylinder (30 × 20 cm) and examined under red-light (10 lx), to encourage movement. Video recording of rearing behavior analysis within 5 min was performed. A rear was scored when the animal raised the forelimb(s) above shoulder level and contact the enclosing walls of the cylinder and the bottom surface. The postural instability test was used to assess forelimb motor function [11,17]. Subjects were held almost vertically facing downward and one forelimb was gently restrained against the torso of the rat, allowing the animal to stand on its unrestrained forelimb. The rat was then moved forward until it made a “catch-up” step to regain the center of gravity. Tests were carried out at baseline and after 5, 8, 10, and 12 days of ROT injection.
2.7. Tissue harvesting and processing for histology
Rats were sacrificed by CO2 asphyxiation and brains were immediately removed. The left hemibrain was immersion-fixed in 4% PFA in pH 7.4 PBS for 1 week and then cryoprotected in 30% sucrose in PBS for a minimum of 3 days until infiltration was complete. Brains were then sectioned in the coronal plane at 35 μm. Free-floating sections were collected in cryoprotectant and maintained at −20 °C until used. Several brain areas, including striatum and SN, were immediately dissected from the right hemibrain, snap frozen in liquid nitrogen, and kept at −80 °C until experiment.
2.8. Immunohistochemistry
For chromogenic detection, striatum and SN sections were washed 6 times in 1× PBS for 10 min each and incubated overnight at 4 °C in a 1% Triton X-100 solution, to increase permeabilization. Next, sections were quenched with 3% H2O2 for 10 min, rinsed in 3 changes of PBS, and incubated in blocking solution consisting of 10% NDS in 0.3% Triton X-100/PBS for 1 h. To label DA neurons, tissue sections were stained with a primary mouse anti-tyrosine hydroxylase (TH+) antibody for 72 h at 4 °C plus 1 h at RT to obtain optimal antibody penetration. Following 3 additional washing steps, brain sections were placed in biotinylated secondary antibody prepared in PBS containing 0.3% Triton X-100 and 1% blocking serum for 1 h. Brain sections were then rinsed, incubated with Avidin-Biotin Complex reagent for 1 h, and washed 3 times again. The reaction was developed in 3,3′-diaminobenzidine substrate for 3–5 min. Sections were allowed to dry overnight, dehydrated in an alcohol gradient for 3 min each and Histoclear for 5 min, and coverslipped using Histomount mounting solution.
Brain sections used for immunofluorescence labeling were rinsed in PBS 6 times for 10 min each and incubated in 10% NDS blocking solution for 1 h. Sections were incubated in primary antibodies directed against the protein of interest in the presence of 0.3% Triton X-100 to facilitate antibody access to the epitope for 72 h at 4 °C. Tissue sections were washed 3 times and the staining was revealed with appropriate secondary antibodies for 2 h. Next, tissue sections were rinsed twice in PBS and nuclear counterstain was carried out using the H 33342 reagent (1:3000) for 5 min. After 3 rinses in PBS, sections were mounted onto plus-coated slides, and coverslipped using gelvatol mounting media. Primary and secondary antibodies used and working dilutions are detailed in Table S1.
For near-infrared fluorescence detection, tissue sections were washed 6 times in PBS for 10 min each and incubated in 10% NDS blocking solution for 1 h. Next, sections were stained with primary antibodies for 48 h at 4 °C. After 3 additional washing steps, tissue sections were covered with IRDye secondary antibodies for 2 h. Brain sections were rinsed in PBS and mounted using gelvatol.
2.9. Neurochemical analysis
Striatal levels of DA hydrochloride, 3,4-dihydroxyphenylacetate (DOPAC), and homovanillic acid (HVA) were measured by high-performance liquid chromatography (HPLC) with electrochemical detection [11,17]. Tissue homogenates were prepared by microtip sonication on ice in 0.1 N perchloric acid and centrifuged at 16,100 g for 30 min at 4 °C. The supernatant was filtered through 0.22-μM nylon membrane tubes and centrifuged at 1000 g for 5 min. The supernatant was injected into a Waters 2695 HPLC (Milford, MA, USA) separation module. The mobile phase consisted of 0.06 M sodium phosphate monobasic, 1.125 mM citric acid 1-octanesulfonic acid, 0.03 M citric acid, 0.5 mM EDTA, 2 mM NaCl, and 8% methanol. All components were adjusted to a pH 3.5 with phosphoric acid, pumped at a flow rate of 0.8 mL/min. A reversed-phase C18 column (150 × 4.6 mm) with a 3.5 μm particle size was used to separate the different analytes, which were detected on an electrochemical detector (Waters 2465) with a glassy carbon electrode set at 750 mV referenced to an ISAAC electrode at 28 °C. Values obtained were normalized to total homogenate protein (ng/mg) and quantified by comparison with high purity standards.
2.10. Quantitative near-infrared fluorescence detection
An IR-tagged secondary antibody combined with image capture on a LI-COR Odyssey scanning system (Lincoln, NE, USA) was used to quantify DA nerve terminal density in the striatum as previously described [11,17]. Serial TH immunolabeled sections were scanned at a wavelength of 680 and 800 nm at highest resolution (v. 3.0). The dorsolateral region of the striatum of 5 sections per animal was outlined and the average pixel intensity for each section was generated.
2.11. Unbiased neuronal counts in vitro and in vivo
The motorized stage approach yields identical results to classic stereology and has a lower coefficient of error (<0.05 for each animal) owing to the much greater number of neurons sampled [18,19]. Assessment of TH+ neuron survival in primary ventral midbrain neuronal cultures was performed in a large area of the coverslip, excluding the edges to avoid saturation issues and debris. Series of every sixth section of whole rat SN (from bregma −4.36 mm to −6.72 mm) were processed for immunofluorescence staining. High-resolution images were acquired using an Eclipse Ni-E wide-field microscope equipped with a linear-encoded motorized stage and a QImaging Retiga CCD camera. Quantitative analysis was performed by determining the colocalization between the MAP2, TH+, and H 33342 channels using the NIS-Elements software (v. 4.30). Image capture settings and quantification criteria were maintained throughout the entire experiment.
2.12. High-resolution confocal laser scanning microscopy
Cell cultures were analyzed using confocal (Olympus BX61) ratiometric measurements of reduced and oxidized thiol groups in both DA and non-DA neurons (MAP2) with an UPlanSApo 40× (N.A. 0.90) objective lens. Confocal laser microscopy was also used to examine alterations in axonal and mitochondrial dynamic proteins as well as variations in tyrosine nitration, lipid peroxidation, and iNOS levels. Photomicrographs of stained SN sections were acquired under constant power and pinhole aperture with a 40× lens. Quantification was carried out using the Fluoview Viewer Olympus software (v. 4.2c). Activated microglia images were taken at 20× (N.A. 0.75). Microglial activation was assessed using the NIH ImageJ software (v. 1.48).
2.13. Assessment of neuritic architecture in primary culture of DA neurons and rat SN sections
An efficient strategy for accurate neuron reconstruction must operate in 3D. Neurite arborization was determined using the automated FilamentTracer module of the Imaris software (Bitplane, v. 7.1.1.). For in vitro analysis, an area of 10 mm2 was consistently selected in the center of the image to remain unbiased. In rats, the SN pars compacta was outlined and subjected to analysis. Total neurite length and both number of segments and branches were standardized to the number of TH+ neurons.
2.14. NAD+/NADH redox index
The content of NAD+ and NADH was determined using the Promega NAD/NADH-Glo Assay kit according to manufacturer instructions (Madison, WI, USA). Cells were plated at density of 80,000 cells/well in 96-well plates and maintained at 37 °C in 0.2 mL/well MEM. After 48 h, the culture medium was replaced with serum-free Neurobasal medium. On the 7 DIV, half of the medium was removed and replenished with fresh Neurobasal medium (0.1 mL/well). Ten-day-old cultures were incubated with 50 nM PTZ 1 h before administration of 100 nM ROT. Culture media was replaced with PBS and both the base solution (0.2 N NaOH) and 1% DTAB were added to lyse cells and preserve the stability of the dinucleotides. NAD+ and NADH bioluminescence was obtained individually following acid or base treatment and recorded using a spectrofluorometer.
2.15. Statistics
Statistical analyses were generated using GraphPad Prism software v. 7.0. (La Jolla, CA, USA). The Mann-Whitney U test (Wilcoxon rank sum test) was utilized to assess for significant differences in animal weight. Survival curves were compared with the Log-rank (Mantel-Cox) test. Two-way ANOVA with Tukey's post-hoc test was undertaken to compare motor function of diverse subjects from different treatment groups. Thiol redox assessment, neuronal cell counting, neurite morphometric evaluation, neurochemistry, near-infrared detection, and NAD+/NADH measurements were analyzed using ANOVA with Newman-Keuls post-hoc correction to determine pairwise comparisons amongst multiple data sets. Data were expressed as mean values ± SEM. For all tests, p < 0.05 was deemed significant.
3. Results
3.1. Neuroprotective effects of PTZ on primary DA neuron cultures
Administration of 50 nM ROT to 5-day-old primary rat DA cell cultures over a period of 5 days leads to neuron degeneration [8,11,17]. We therefore sought to investigate the potential neuroprotective properties of PTZ (Fig. 1). We first examined whether unsubstituted PTZ improves cell survival (Fig. 1 A). PTZ alone did not show either neurotoxic or neurotrophic effects while ROT treatment resulted in a dramatic decrease in the number of TH+ neurons (∼71%, Fig. 1 A11), MAP2 neurons (∼48%, Fig. 1 A12), and the percentage of DA-producing neurons (∼45%, Fig. 1 A13) as compared to the control group (VEH). Administration of PTZ (10, 20 or 50 nM) 1 h after ROT incubation significantly protected TH+ and MAP2 neurons from ROT toxicity (p < 0.0001 for all doses of PTZ tested). We further evaluated the efficacy of PTZ in preserving neurite morphometry (Fig. 1 B). Total neurite length (∼32%, Fig. 1 B6) and both number of segments (∼59%, Fig. 1 B7) and branches (∼57%, Fig. 1 B8) were significantly reduced in response to ROT. However, PTZ was able to abrogate ROT-induced neurite shrinkage and fragmentation.
Fig. 1.

Protective effects of PTZ against ROT-induced toxicity on primary ventral midbrain cultures. (A) Representative scans from immunocytochemical preparations acquired with a 20× objective lens. PTZ protects TH+ (A11) and MAP2 neurons (A12) against the deleterious effects of ROT with ensuing increase in the percentage of DA-containing neurons (A13). Insets correspond to high magnification images. Scale bar: 200 μm. Data are representative of 20–40 neurons per group obtained from seven independent cultures. (B) Morphometric neurite reconstruction in 3D. PTZ treatment preserved neurite length (B6), number of segments (B7), and branch points (B8) in ROT-exposed cells. Insets represent a 4× zoomed image. Scale bar: 200 μm. Two images for each condition and experiment were averaged. Data from four independent cultures were combined to determine means ± SEM (C) Ratiometric measurements of reduced and oxidized thiol groups. High resolution confocal images obtained with a 40× objective and image analysis demonstrated that administration of PTZ restores the SS/SH ratio altered in ROT-treated cells. Scale bar: 20 μm. On an average, 25–30 neurons per condition were tested from three independent cultures. ****p < 0.0001, ***p < 0.001, **p < 0.01 and * p < 0.05 compared to VEH + VEH. ####p < 0.0001 and ###p < 0.001 relative to PTZ + VEH (one-way ANOVA followed by Newman-Keuls multiple comparisons test).
To investigate the putative effect of PTZ on oxidative damage, a redox immunohistochemistry approach was used for the sensitive quantification of intracellular redox state in situ in both DA and non-DA neurons (Fig. 1 C). The thiol (SH)/disulfide (SS) redox balance system shifted to toward disulfide bound generation in ROT-stimulated cells relative to VEH group (Fig. 1 C5 vs C10; 0.58 ± 0.01 vs 1.01 ± 0.03 TH+ neurons and 0.59 ± 0.02 vs 1.10 ± 0.05 MAP2 neurons, p < 0.0001). In contrast, the SS/SH ratio was reduced near to basal levels in the PTZ + ROT groups in comparison to cells incubated with ROT alone (Fig. 1 C10 vs C15, C20 and C25; p < 0.0001). Ratiometric analysis of disulfide and thiol species in DA (C26) and non-DA (C27) neurons. Taken together, these findings provide strong support for the concept that PTZ has a potent protective effect against ROT-mediated neuron degeneration and oxidative impairment in vitro.
3.2. PTZ attenuates ROT-induced PD-like pathology in a dose-dependent manner
We conducted a dose-response study (cohort #1) in which several doses of PTZ (1, 5, 10, 20 or 50 mg/kg) were given to rats. A decrease in body weight (Fig. 2 A) and survival rate (Fig. 2 B) was found in rats that received ROT. PTZ alone (50 mg/kg, PTZ + VEH) did not affect animal weight or lifespan. Likewise, PTZ did not reverse the loss of weight elicited by ROT. Since the remaining alive PTZ + ROT rats were sacrificed when the last ROT-injected rat died, changes in overall survival could not be determined.
Fig. 2.

PTZ preserves striatal DA projections and SN DA neurons in a dose-response manner. ROT intoxicated rats (cohort #1) received i.p. injections of PTZ (1, 5, 10, 20 or 50 mg/kg) BID at 10-h intervals within 24 h for sixteen days. Animal body weight (A) and survival (B) were reduced in ROT-lesioned subjects and PTZ did not show any significant effect during this experimental period. Coronal sections at the level of the striatum were immunostained for TH using DAB as the chromogen (C1–C4). Near-infrared images depicting TH+ immunoreactive signal (C5–C8). Quantitative analysis of TH+ fiber density (C9). Data from 4–6 sections per animal were pooled and presented as the average mean optical density ±SEM. Experimental groups comprised 3 rats. Scale bars: 500 μm for DAB staining and 1 mm for immunofluorescence labeling. **p < 0.01 and * p < 0.05 compared to VEH + VEH. ####p < 0.0001, ###p < 0.001 and ##p < 0.01 relative to VEH + ROT (one-way ANOVA followed by Newman-Keuls post-hoc test). Midbrain sections were stained with an antibody against TH+. Representative micrographs of DAB-based immunohistochemical staining captured at low (2× objective, D1-D4) and high magnification (10× objective, D5-D8). Fluorescent images acquired using a 20× objective (D9-D12). Insets correspond to high magnification images of the dorsolateral region (D13-D16). Quantitative assessment of the number of TH+-immunoreactive neurons (D17). DAB scale bar: 500 μm for low magnification and 50 μm for high magnification. Fluorescence scale bar: low magnification, 500 μm; high magnification, 40 μm. Ten to thirteen SN sections per animal were analyzed. Values expressed are mean ± SEM of three different rats per group and analyzed using one-way ANOVA followed by Newman-Keuls multiple comparisons test. ***p < 0.001 and **p < 0.01 vs VEH + VEH. ##p < 0.01 compared to VEH + ROT.
As an objective measurement of the integrity of the nigrostriatal DA system, coronal sections of both striatum and SN were immunostained for TH antibody and visualized using either DAB chromogenic or fluorescent detection. Control groups receiving vehicle or PTZ (50 mg/kg) were found to be statistically equivalent to each other. ROT led to significant reduction in striatal TH fiber density relative to control rats (Fig. 2 C; 966 ± 23 vs 623 ± 39 O.D, p < 0.01). Diverse PTZ dose-response profiles were examined. Low doses of PTZ (1 and 5 mg/kg) did not show neuroprotection upon ROT administration. However, 10 mg/kg PTZ displayed a notable degree of protection in ROT-treated animals (623 ± 39 vs 982 ± 88 O.D., p < 0.01). Importantly, 20 mg/kg PTZ was the most effective dosage in preserving DA nerve terminals (623 ± 39 vs 1311 ± 94 O.D, p < 0.0001) whereas the highest dose of PTZ tested (50 mg/kg) was less effective than 20 mg/kg PTZ but still within the therapeutic range (623 ± 39 vs 1054 ± 89 O.D., p < 0.001).
Unbiased stereological cell counting demonstrated that ROT per se provokes a marked loss of nigral DA neurons (Fig. 2 D; 19,649 ± 615 vs 10,278 ± 1395, p < 0.001), representing ∼48% decrease. In response to PTZ, the number of DA neurons in the SN varied in a dose-dependent fashion. Low doses of PTZ (1 or 5 mg/kg) failed to attenuate ROT-induced DA neuron demise. A trend toward neuroprotection was seen following 10 mg/kg PTZ administration (10,278 ± 1395 vs 14,167 ± 304), which reflects ∼27% increase. Consistent with the effects observed on striatal TH fiber density, 20 mg/kg PTZ provided the greatest protective action against DA cell death (10,278 ± 1395 vs 17,099 ± 1068, p < 0.01), equivalent to an ∼40% increase. 50 mg/kg PTZ also protected DA neurons against the deleterious effects of ROT (10,278 ± 1395 vs 13,671 ± 680, p < 0.01) but was less efficient in comparison to 20 mg/kg PTZ.
Based on these observations, PTZ exhibits a typical bell-shaped dose-response curve with a peak effect at 20 mg/kg dose (BID), resulting in a robust preservation of both striatal DA fiber density and number of SN DA neurons from ROT-induced toxicity.
3.3. PTZ prevents progressive motor deficits and phenotypic abnormalities in ROT-lesioned rats
For the time-window study, the optimal dose of PTZ (20 mg/kg) was delivered twice daily at 10-h interval until animals reached the endpoint or were sacrificed. After ROT injection, rats showed a significant decrease in weight mass and survival rate. Animals on the PTZ + ROT treatment exhibited greater body weight between days 17 and 24 (Fig. 3 A; p < 0.001) and higher survival rate (Fig. 3 B, hazard ratios of 0.35 [95% CI: 0.17 to 0.72] vs 2.86 [95% CI: 1.38 to 5.91]) in comparison to VEH + ROT group.
Fig. 3.

PTZ reverses PD-like phenotype and alleviates progressive locomotor deficits. (A) Animal weight was monitored daily. (B) Survival curve. PTZ significantly improved phenotypic outcome in ROT-injected rats. The experimental groups were comprised of 8 rats for VEH and 21 rats for ROT. ***p < 0.001 vs VEH + ROT (Mann-Whitney U and Log-rank (Mantel-Cox) tests, respectively). (C) Spontaneous exploratory behavior. Higher number of independent wall placements were detected for both forelimbs following PTZ treatment in ROT-injected animals. Data were represented as the mean ± SEM number of rearing movements per 5 min. (D) The postural instability test. Administration of PTZ reduced the distance to trigger a compensatory forelimb movement in response to ROT. The measurements were performed in triplicate and values were expressed as mean ± SEM with both sides averaged together. VEH groups, n = 7; ROT groups, n = 14. ****p < 0.0001, **p < 0.01 and * p < 0.05 vs VEH + ROT (Two-way ANOVA with Tukey's post-hoc multiple comparisons test).
Administration of ROT results in severe bradykinesia, rigidity, and postural instability [11,16,17]. In our study, long-lasting exposure to ROT reduced spontaneous rearing behavior (Fig. 3C). Cylinder exploration was significantly increased when animals were treated with PTZ and ROT together relative to ROT + VEH rats, especially at days 5 and 8. The displacement of the rat necessary to provoke a compensatory forelimb movement was symmetrically and progressively augmented after ROT exposure (Fig. 3 D). In contrast, ROT-injured rats treated with PTZ performed much better on the postural instability test at any of the time points evaluated. Taken together, our data demonstrate that PTZ can prevent parkinsonian features and restore progressive loss of functional outcome produced by ROT (Movies S1–3)
Supplementary video related to this article can be found at https://doi.org/10.1016/j.redox.2019.101164.
The following are the supplementary data related to this article:
Movie S1PTZ ameliorates motor activity and exploratory behavior after 15 days of ROT administration. Video corresponds to one representative animal from each experimental group.
Movie S2PTZ improves motor activity and exploratory behavior in rats treated with ROT for 20 days. Video of three ROT-lesioned rats injected either with vehicle or PTZ.
Movie S3Rats injected with PTZ and ROT after 40 days of treatment. Video monitoring the motor function of four PTZ + ROT-treated subjects. .
3.4. PTZ renders neuroprotection against ROT-mediated nigrostriatal DA degeneration
It has been reported that ROT causes alterations in DA distribution, vesicular storage, and metabolism and reduces the expression of VMAT2 and DAT [27,28]. TH catalyzes the conversion of tyrosine into L-3,4-dihydroxyphenylalanine (l-DOPA), the precursor of the neurotransmitter DA, which can be metabolized into DOPAC and HVA. We first investigated whether PTZ can modulate the amount of striatal catecholamines (Fig. 4). HPLC analysis indicated that ROT intoxication leads to DA depletion (Fig. 4 A; 90 ± 9 vs 57 ± 6 ng/mg, p < 0.01) and increased DOPAC/DA (used as an index to mainly describe presynaptic processes), HVA/DA (postsynaptic processes), and DOPAC + HVA/DA turnover ratios (Fig. 4 D-F). Importantly, PTZ treatment abolishes ROT-induced adverse effects. No differences were detected in the levels of serotonin (data not shown). We also examined the putative effects of PTZ on striatal DA nerve fiber density (Fig. 4 G-H). Using near-infrared detection we found that ROT causes a robust reduction in the amount of TH processes in the dorsolateral region of the striatum, which were significantly preserved in rats injected with PTZ (963 ± 70 vs 1515 ± 77 O.D., p < 0.001).
Fig. 4.

Striatal catecholamine levels and DA terminals. Levels of (A) DA, (B) DOPAC, (C) HVA and (D–F) the ratio of DA metabolites to DA as an index of DA turnover. Treatment with PTZ abrogated ROT-induced alterations in the concentrations of DA and turnover ratios. The experimental groups were comprised of 7 rats for VEH and 10 rats for ROT. (G–H) Striatal DA fiber density. Near-infrared immunofluorescence scan denoted a loss of striatal TH nerve terminal network in ROT-injected rats which was significantly protected upon PTZ exposure. Scale bar: 1 mm. Results are expressed as mean ± SEM VEH groups, n = 8; ROT groups, n = 18. ****p < 0.0001 and * p < 0.05 relative to VEH + VEH. ####p < 0.0001, ##p < 0.01 and #p < 0.05 vs PTZ + VEH. ***p < 0.001 and **p < 0.01 compared to VEH + ROT (one-way ANOVA followed by Newman-Keuls multiple comparisons test).
The morphological and morphometric characteristics of axons and neurites are a crucial feature of neuronal phenotype and function. Axon pathology and DA neuron death are prominent features of PD [8,18,19,29]. In this study we used an algorithm that generates automated 3D evaluation of neurite architecture (Fig. 5 A). A severe reduction in total neurite length (304 ± 16 vs 167 ± 10 μm, p < 0.0001), number of intermediate segments (9 ± 0.6 vs 5 ± 0.4, p < 0.0001), and branch points (4 ± 0.3 vs 2 ± 0.2, p < 0.0001) was detected in response to ROT. DA processes were significantly protected in ROT-injured rats treated with PTZ relative to ROT rats that did not receive PTZ.
Fig. 5.

PTZ preserves SN DA neurons and their processes from ROT-mediated toxicity. (A) Representative 3D neuritic arborization in the rat SN. Severe loss of neuronal processes was clearly evident in ROT-intoxicated animals whereas subjects that received both PTZ and ROT treatments showed a notable preservation of neurite morphometric features, with an increase in overall length (A5), number of intermediate segments (A6) and branches (A7). Insets correspond to 4× zoomed images. Scale bar: 200 μm. Bar graphs depict mean values ± SEM of four sections per animal (n = 4 rats). (B) Unbiased stereology. Micrographs were acquired at 20× and processed for colocalization analysis between TH+ (green), MAP2 (red), and nuclei (blue). PTZ treatment resulted in a robust protection against SN DA neuron death in ROT-treated rats (B9). Scale bar: 250 μm. Each value represents the mean ± SEM of 9–12 sections per animal; n = 4 for vehicle groups and 7–10 for ROT-lesioned groups. ****p < 0.0001, ***p < 0.001 and * p < 0.05 compared to VEH + VEH. ####p < 0.0001, ###p < 0.001 and #p < 0.05 relative to PTZ + VEH. ***p < 0.001 and **p < 0.01 vs VEH + ROT. One-way ANOVA followed by post-hoc Newman-Keuls was used to compare mean differences between groups. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
To further examine the effects that PTZ can elicit on the integrity of the nigrostriatal DA system, unbiased stereological estimation of the number of DA-containing neurons was carried out in the rat SN using the motorized stage technique (Fig. 5 B). Prolonged treatment with ROT resulted in a significant loss of TH+ neurons in comparison with the control group (19,649 ± 744 vs 12,883 ± 722, p < 0.05). However, the loss of nigral DA neurons was significantly attenuated in the PTZ + ROT group (12,883 ± 722 vs 17,839 ± 1885, p < 0.01). The immunoreactivity of TH remained unchanged between groups, suggestive that ROT does not induce complete TH downregulation (Fig. S2). Together, these findings show that PTZ treatment rescues striatal DA content and nerve terminal networks as well as SN DA neurons and axonal arborizations from ROT-induced degeneration.
3.5. PTZ modulates ROT-associated alterations in mitochondrial dynamic protein expression profiles
Mitochondrial content, localization, and function are dependent on the dynamic balance between fusion and fission. Fusion results in the formation of elongated and interconnected mitochondria and diminishes the load of defective mtDNA gene products or accumulation of misfolded proteins while fission triggers mitochondrial fragmentation and excessive mitophagy, regulates the release of pro-apoptotic proteins, and increases ROS production [3,4]. To assess quantitative changes in mitochondrial dynamic-related protein concentrations, midbrain sections were immunostained using antibodies against Opa1, Mfn2, and Drp1 epitopes (Fig. 6). Chronic exposure to ROT reduced the immunoreactive signal of Opa1 (∼48%, p < 0.0001) and Mfn2 (∼36%, p < 0.01) fusion proteins whereas it caused a significant increase in the expression of the fission protein Drp1 (∼48%, p < 0.01) in comparison to vehicle-treated rats. When PTZ was combined with ROT, the values of Opa1 (Fig. 6 U; 1160 ± 21 vs 2079 ± 33 F.U., p < 0.0001) and Mfn2 (Fig. 6 V; 1267 ± 34 vs 1908 ± 112 F.U., p < 0.01) returned to baseline levels. Although not statistically significant, there was a trend toward lower Drp1 immunoreactivity in the PTZ + ROT group relative to VEH + ROT-treated animals (Fig. 6 W). Our results imply that PTZ administration counteracts variations in the levels of mitochondrial fusion and fission proteins in nigral DA neurons of ROT-lesioned rats.
Fig. 6.

PTZ administration restores mitochondrial dynamic protein levels. (A–T) Series of high-resolution confocal microscopy images of SN sections stained for TH+ (gray), Opa1 (green), Mfn2 (red), and Drp1 (blue). ROT reduced Opa1 (U) and Mfn2 (V) immunoreactivity and upregulated Drp1 protein levels (W). PTZ was able to readjust the derangements of the mitochondrial dynamic machinery provoked by ROT. Scale bar: 30 μm. Values are mean ± SEM representative of the average of ∼200–300 TH+ neurons comprised in 3–4 different SN sections per animal; n = 4 per group. ****p < 0.0001, **p < 0.01 and * p < 0.05 relative to VEH + VEH. ####p < 0.0001, ##p < 0.01 and #p < 0.05 compared to PTZ + VEH. ****p < 0.0001 and **p < 0.01 vs VEH + ROT (one-way ANOVA followed by Newman-Keuls multiple comparisons test). (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
3.6. PTZ blocks the axonal transport-associated protein deficits provoked by ROT
Active transport between the cell body and synaptic terminals is critical for neuronal function. A variety of observations support the hypothesis that altered axonal transport is involved in the pathogenesis of neurodegenerative diseases, including PD [8,29,30]. We therefore assessed the effects of PTZ on the levels of axonal transport proteins after ROT intoxication. Midbrain sections were subjected to immunostaining with antibodies against KLC1, KHC and DYNLT3 (Fig. 7). Representative confocal micrographs and quantitative analysis indicated that ROT significantly diminishes KLC1 (Fig. 7 L and U; ∼40%, p < 0.001), KHC (Fig. 7 M and V; ∼49%, p < 0.01) and DYNLT3 (Fig. 7 N and W; ∼51%, p < 0.001) fluorescence intensity. PTZ per se did not modify the immunoreactivity of any axonal transport-related protein but when combined with ROT, there was an increase in the levels of KLC1 (1020 ± 61 vs 1319 ± 79 F.U., p < 0.05), KHC (401 ± 39 vs 788 ± 41 F.U., p < 0.01), and DYNLT3 (584 ± 39 vs 894 ± 48 F.U.; p < 0.01) as compared to ROT-injected rats. These data suggest that PTZ lessens ROT-induced perturbations on trafficking along axons in the SN.
Fig. 7.

Unsubstituted PTZ increases the concentration of axonal transport-related proteins. Representative confocal laser scanning micrographs acquired at 60× illustrating the immunoreactivity of TH+ (gray), KLC1 (green), KHC (red), and DYNLT3 (blue) (A–T). Quantitative analysis showed that PTZ restores the content of axonal transport-linked proteins in the SN of rats injected with ROT. Bar graphs summarizing the fluorescence intensity of KLC1 (U), KHC (V), and DYNLT3 (W). Scale bar: 30 μm. An average of 3–4 SN sections per animal were analyzed and data from each tissue section were combined to determine means. Results are expressed as mean ± SEM of four different animals per group and analyzed using one-way ANOVA followed by Newman-Keuls multiple comparisons test. ***p < 0.001, **p < 0.01 and * p < 0.05 vs VEH + VEH. ###p < 0.001 and ##p < 0.01 compared to PTZ + VEH. **p < 0.01 and * p < 0.05 relative to VEH + ROT. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
3.7. PTZ mitigates protein tyrosine nitration and lipid peroxidation following mitochondrial complex I impairment
Neurons are particularly sensitive to mitochondrial dysfunction due to their high-energy demand that causes depletion of the ATP pool and generation of ROS/RNS. There is solid evidence showing an anomalous increase of oxidative damage in SN DA neurons in PD [8,11,31]. This fact prompted us to examine whether PTZ would be protective against ROT-induced oxidative injury. To assess the extent of NO•-related oxidative damage and lipid modification, we quantified the immunoreactivity of 3-NT and 4-HNE, respectively (Fig. 8). Repeated application of ROT increased the fluorescence intensity of both 3-NT and 4-HNE in SN DA neurons whereas their levels were significantly diminished after administration of PTZ (Fig. 8 E, 3-NT; 1299 ± 90 vs 1033 ± 66 F.U., p < 0.05 and Fig. 8 F, 4-HNE; 1076 ± 126 vs 491 ± 82 F.U., p < 0.01). Thus, PTZ can act as potent antioxidant agent abrogating the oxidative stress burden in SN DA neurons of ROT-injected rats.
Fig. 8.

PTZ rescues SN DA neurons from oxidative stress damage. Midbrain sections were immunostained using antibodies against TH+ (gray), 3-NT (red), 4-HNE (green), and nuclei (blue) and processed for confocal microscopy analysis (A–D). The immunoreactive signal of 3-NT and 4-HNE was visibly reduced in PTZ + ROT subjects compared to ROT alone treated rats, similar to control levels. Graphical representation of 3-NT levels (E) and 4-HNE (F) within SN DA neurons. Insets represent a 4× zoomed images. Scale bar: 40 and 10 μm for low and high magnification, respectively. An average of 300–400 SN DA-containing neurons was examined and data from each nigral section were combined to calculate means. Four-five SN sections per animal were used. Each treatment group was comprised of 4–5 rats and analyzed using one-way ANOVA followed by Newman-Keuls post-hoc test. *p < 0.05 relative to VEH + VEH. ##p < 0.01 vs PTZ + VEH. **p < 0.01 and * p < 0.05 compared to VEH + ROT. (For interpretation of the references to colour in this figure legend, the reader is referred to the Web version of this article.)
3.8. PTZ moderates the neuroinflammatory response induced by ROT
A sustained production of large amounts of NO• has been reported in PD, which is synthesized from l-arginine in a reaction catalyzed by the NO• synthase. iNOS gene expression and enzyme activity become readily upregulated under pathological conditions and play an important role in orchestrating inflammation in PD [8,13,32]. We therefore investigated whether iNOS levels were altered in the rat SN upon PTZ treatment (Fig. 9 A). In control sections, there is a lack of iNOS staining but ROT exposure resulted in a ∼4-fold increase in iNOS immunoreactive signal (Fig. 9 A1-2 and A5-6; 245 ± 25 vs 923 ± 43 F.U., p < 0.0001). Similar to controls, rats treated with PTZ + ROT exhibit sparse iNOS immunoreactivity as compared to ROT alone group (Fig. 9 A5-6 and A7-8; 923 ± 43 vs 317 ± 31 F.U., p < 0.0001).
Fig. 9.

PTZ dampens ROT-induced inflammatory response. (A) Representative micrographs acquired at 60× depicting the immunoreactivity of iNOS over the neuropil and perikarya of DA neurons in midbrain sections. PTZ administration abrogated ROT-mediated neuroinflammation (A5-A6 vs A7-A8). Scale bar: 40 μm. An average of 4–5 SN sections per animal was examined. Data are shown as mean values for groups of 4–5 animals and error bars represent SEM. (B) Microglial stimulation was determined in the SN of rat brain. Confocal image acquisition was performed at 20×. The number of Iba1-positive cells (B9) and the total area occupied by Iba1 (B10) were significantly reduced upon PTZ treatment in ROT-injured rats. Insets represent a 4× zoomed images. Scale bar: 100 for low magnification and 50 μm for high magnification. An average of 4–5 sections per animal were assessed. All values were the means ± SEM obtained from 4 to 5 animals per group. ****p < 0.0001 and **p < 0.01 vs VEH + VEH. ####p < 0.0001, ###p < 0.001 and ##p < 0.01 relative to PTZ + VEH. ****p < 0.0001 and **p < 0.01 compared to VEH + ROT.
To further evaluate the anti-inflammatory activity of PTZ, microglial activation was assessed in our experimental paradigm (Fig. 9 B). Midbrain sections were immunolabeled using an antibody against the ionized calcium binding adaptor molecule 1 (Iba1) epitope. A significant increase in the number of microglia-positive cells (Fig. 9 B9; ∼45%) and the total area occupied by microglial particles (Fig. 9 B10; ∼95%) was detected upon ROT exposure, which was considerably reduced in PTZ + ROT rats (8358 ± 922 vs 5300 ± 513 Iba+ cells, p < 0.01 and 534,028 ± 51,844 vs 275,285 ± 193,014 overall area covered, p < 0.0001). These experiments indicate that PTZ can modulate ROT-induced inflammatory molecule expression profiles in the rat SN.
3.9. PTZ exhibits neuroprotective properties by preserving NAD+ levels
To elucidate a potential molecular mechanism underpinning the neuroprotective effects of PTZ against ROT-induced nigrostriatal DA degeneration, we measured the intracellular levels of NAD+ and NADH (Fig. 10). ROT binds to the ubiquinone site and inhibits NADH-ubiquinone oxidoreductase activity [33]. Spectrofluorimetric analysis showed that the increment of NAD+ fluorescence was 1.02 ± 0.01 for vehicle-treated cells (VEH + VEH) whereas the value was 0.77 ± 0.01 following ROT injection (Fig. 10 A, p < 0.0001). Conversely, NADH fluorescence value was 0.99 ± 0.01 for vehicle group and 1.1 ± 0.01 for ROT exposed cells (Fig. 10 B, p < 0.0001). The NADH/NAD+ redox index value was significantly elevated in ROT-treated neurons in comparison to the vehicle group (Fig. 10C; 0.98 ± 0.01 vs 1.44 ± 0.03, p < 0.0001), representing ∼47% increase. PTZ per se (50 nM) did not generate any type of effect on the levels of the coenzymes. However, PTZ + ROT co-treatment restored the concentration of NAD+ (p < 0.0001) but found no difference in the levels of the reduced form compared to ROT-stimulated cells. As a result, PTZ + ROT cells displayed a diminished NADH/NAD+ ratio than cells incubated with ROT alone.
Fig. 10.

PTZ restores the NAD+/NADH ratio. Bioluminescent analysis was performed to monitor the levels of NAD+ (A), NADH (B), and the NADH/NAD+ ratio (C). ROT perturbs the redox balance between NAD+ and NADH. Administration of PTZ did not modulate NADH levels in ROT-treated cells but preserved the content of NAD+, leading to normalization of the NADH/NAD+ redox index. Data are presented as the fold change in the mean intensity of NAD+ or NADH fluorescence. Results are presented as mean ± SEM from 4 separate experiments performed in sextuplicate. ****p < 0.0001 relative to VEH + VEH. ####p < 0.0001 compared to PTZ + VEH. ****p < 0.0001, ***p < 0.001 and **p < 0.01 vs VEH + ROT (one-way ANOVA followed by Newman-Keuls multiple comparisons test).
3.10. Discussion
Despite a considerable effort has been devoted towards the synthesis of numerous PTZ derivatives, very little research has been conducted in studying the efficacy of unsubstituted PTZ as a neuroprotective compound. Interesting data from a same group show that several imines, including PTZ, are beneficial in SH-SY5Y human neuroblastoma cells and transgenic C. elegans treated with MPP+ and ROT through its potent antioxidant activity [23,24]. Because DA neurons are substantially susceptible to oxidative damage and mitochondrial impairment, we surmised that administration of PTZ might confer protection against the pathological and phenotypic features of PD in ROT-treated rats. Our findings provide proof-of-concept evidence that PTZ displays a classical bell-shaped dose-response profile with a partial protective effect against the deleterious effects of ROT at a dose of 10 mg/kg, optimal at 20 mg/kg, and then diminishes at higher doses (50 mg/kg). Doses of PTZ above 50 mg/kg can be out of the therapeutic range and might cause overt toxicity.
Treatment with methylene blue, a thiazine dye that transfers electrons from NADH to cytochrome c, improves cognitive performance in subjects with mild/moderate AD and attenuates nigrostriatal DA degeneration in ROT-injected rats [34,35]. However, methylene blue can cause important adverse effects on the CNS, which are associated with serotonin noxiousness [36,37]. Unsubstituted PTZ has low toxicity in mammals; overdose and accidental exposure to PTZ can cause hepatitis, hemolytic anemia, pruritus, and skin photosensitivity though there is no evidence for CNS depression, genotoxicity or carcinogenicity [21,22]. Pharmacological studies showed that radiolabeled 35S-PTZ is selectively taken up by diverse brain regions in cats, monkeys, and humans and is predominantly concentrated in the brain stem and to a lesser extent in cerebral cortex > thalamus/hypothalamus > cerebellum [38]. Upon oral administration, the plasma half-life of PTZ may range between 14 and 78 h [39]. Like most xenobiotics, urine and feces are the major elimination routes of PTZ. The highest amount of drug is excreted over a 24-h interval at considerably low rates due to its elevated protein binding and low aqueous solubility [20].
Neurons degenerate through a protracted dying-back phenotype, in which the earliest pathological feature is the loss of distal processes. The triad of cardinal motor symptoms appears when approximately 80% of putaminal DA levels and 70% of nerve terminals are depleted, which correlates with a 50% loss of SN DA neurons [1]. Our lab has established that long-term i.p. administration of ROT causes about 40–45% DA depletion in the striatum, ∼35–45% loss of striatal DA fiber density, ∼40–45% neurite shrinkage, and ∼40–45% SN DA neuron death in rats [11,[16], [17], [18], [19]]. Our findings demonstrate that chronic administration of PTZ prevents ROT-induced progressive PD-like motor abnormalities, reduces DA turnovers, preserves the intrinsic morphometric features of DA processes, and mitigates DA neuron demise.
Evidence for a causative role of mitochondrial dysfunction in the pathogenesis of PD is compelling. Mitochondria undergo constant cycles of fusion and fission but disrupted balance in this machinery promotes neurodegenerative diseases, such as PD. Individuals with mutations in Opa1 provide further support for the notion that defective mitochondrial dynamics contribute to PD pathology [40,41]. Axonal transport is controlled by the motor proteins kinesin – that powers the movement of cargoes towards the plus end of the microtubule – and dynein – that is responsible for the transport toward the minus end. Bidirectional trafficking of mitochondria in neurons is mediated through the actions of several motor-adaptor proteins or mitochondrial receptors [5]. The role of deficient axonal transport in PD has become increasingly evident. Variations in the concentration of different populations of motor proteins were found in DA neurons of parkinsonian patients and cellular and animal models of PD [8,29,30].
A growing number of observations suggest important functional connections between mitochondrial dynamics and axonal transport integrity. Gene mutations or knockdown of Opa1 produced dramatic changes in the mass, morphology, and distribution of the mitochondrial network in distal axons and synaptic terminals of motor neurons in rat cortical neuron cultures and fibroblasts and skeletal muscle biopsies from subjects with autosomal dominant optic atrophy [42,43]. Axonal degeneration and aberrant movement of mitochondria were reported in human induced pluripotent stem cell-derived cortical neurons and DA neurons of mice lacking the Mfn2 gene [44,45]. Genetic ablation of Drp1 in motor neurons perturbed mitochondrial trafficking toward presynaptic nerve terminals [46]. Decreased mitochondrial motility and number was reported in neuronal processes of Drp1 KO mice [47]. ROT treatment decreased the immunoreactivity of Opa1 and Drp1 in primary ventral midbrain neuronal cultures [8]. The combination of ROT and α-syn fibrils (PFFs) resulted in an additive depletion of Opa1 and Drp1 levels. In this work, we have demonstrated for the first time that the levels of mitochondrial dynamic and axonal transport proteins were compromised upon ROT injection in vivo. Besides the restoration of the derangements among mitochondrial fusion and fission, post-treatment with PTZ also re-established axonal transport balance.
The first evidence for mitochondrial dysfunction in PD came from the discovery of a complex I deficiency in post-mortem analysis of SN tissue [48,49]. Persistent inhibition of complex I activity leads to inefficient scavenging or excessive formation of ROS and results in oxidative damage to biomolecules, which is considered a major culprit in the degenerative process of DA neurons. Nitration of tyrosine residues is a covalent RNS-mediated post-translational modification responsible for causing protein structural and functional alterations. The immunoreactivity of 3-NT was significantly increased in midbrain DA neurons of BAC transgenic rats expressing the human E46K α-syn mutation, which exhibited heightened sensitivity to ROT [50]. Elevated 3-NT content was detected in the rat SN following systemic administration of ROT [11]. DA cell population become selectively immunoreactive for 3-NT upon incubation with ROT and/or PFFs [8]. 4-HNE, a product derived from oxidized ω-6 polyunsaturated fatty acids, can react with several targets – mainly cysteine residues of thiol-containing proteins – to form Michael adducts. Mechanistically, 4-HNE can reduce mitochondrial respiration and ETC complex activities, deplete the glutathione pool, and lead to caspase activation, PARP cleavage, and DNA fragmentation [51,52]. Marked increase in the immunoreactive signal of 4-HNE was found in DA neurons of the rat SN and SH-SY5Y cells in response to ROT [11,53]. In this work, we found increased protein nitration and lipid peroxidation in SN DA neurons of ROT-lesioned rats but administration of PTZ reduced the oxidative/nitrosative stress burden.
Sustained activation of microglial cells is an important contributor to the pathogenic processes in PD. Data from post-mortem studies showed the existence of reactive Iba1 microglia in the SN of individuals with PD [54,55]. A combination between genomic and proteomic assays suggested that aggregation of nitrated α-syn promotes microglial stimulation and oxidative stress, which can hasten DA neurodegeneration [56]. Increased number of either activated microglial cells or mRNA Iba1 gene expression levels was detected in the SN of transgenic mice [57]. Iba1 activated microglia underwent proliferation in the SN of ROT-treated rats [11,58].
Evidence indicates that upon stimulation of microglia there is a NO• overproduction via iNOS induction, which is directly implicated in midbrain DA neuron degeneration [8,13]. Stereotaxic delivery of rAAV6 expressing α-syn into the SN of Nrf2-null mice led to microglial activation and upregulated iNOS mRNA expression [59]. Exogenous addition of PFFs significantly increased iNOS concentration in DA cells, which was further augmented following repeated application of ROT; interestingly, downregulation of iNOS dampened the concentration of 3-NT and protected DA neurons from ROT and PFFs-induced toxicity [8]. Increased iNOS immunoreactive signal and gene expression levels were observed in the striatum and SN of ROT-intoxicated rats [11,60]. Taken together, our results indicate that ROT elicits a neuroinflammatory and microglial response, which is mitigated by PTZ treatment.
We attempted to unravel the molecular mechanisms underlying the neuroprotective effect of PTZ against ROT-induced progressive neurodegeneration. Complex I oxidizes NADH to NAD+ and transfers two electrons to ubiquinone, thus creating an electrochemical proton gradient from the mitochondrial matrix to the intermembrane space used to drive ATP synthesis. ROT binds to the ubiquinone-specific site of complex I and blocks the electron flow from NADH to ubiquinone, whereupon there is an increase in the NADH/NAD+ redox index and concomitant ROS generation [33]. The NADH/NAD+ ratio plays a central role in regulating the intracellular redox state and is tightly linked to mitochondrial disorders and neurodegeneration [61,62]. High levels of mitochondrial NADH leads to a complete flavin mononucleotide reduction, which stimulates superoxide formation [63,64]. Interventions aimed to boost intracellular NAD+ availability are protective against neurodegenerative diseases [65,66]. Increased availability of NAD+ following dietary nicotinamide supplementation or by mutations in the PARP gene rescued evidence for dysfunctional mitochondria and prevented DAergic neurodegeneration in a Drosophila model of PD [67]. Treatment with the NAD+ precursor nicotinamide riboside partially preserved mitochondrial function and protected against DA cell death in β-glucocerebrosidase mutant flies and iPSC-derived neurons from individuals with glucocerebroside mutations [61]. Accumulating evidence supports the beneficial effect of sirtuins, a NAD+-dependent protein deacetylases, in different experimental models of PD [68,69]. Our findings established that ROT is associated with decreased intracellular NAD+ levels and increased NADH content, leading to a higher NADH/NAD+ ratio. PTZ treatment did not change the pool of NADH but maintains the rate of NAD+ production and normalized the NADH/NAD+ balance. This effect is directly related to an alleged mitochondrial complex I activity improvement.
4. Conclusions
Long-term administration of PTZ prevents PD-like features in ROT-treated rats, including progressive loss of functional outcome, nigrostriatal DA degeneration, mitochondrial dysfunction, axonopathy, oxidative damage, and neuroinflammation. The ability of PTZ to reduce the oxidative stress burden, preserve NAD+ concentration, and maintain mitochondrial integrity provides a plausible mechanism that might account for its neuroprotective actions. PTZ could become a promising neuroprotective agent and a safety candidate for future clinical studies in PD.
Author contributions
VT participated in the design and conceptualization of the study, performed the experiments, analyzed the data, prepared the figures and wrote the manuscript. JLM performed the histopathologic studies. JTG participated in the data interpretation and revision of the manuscript.
Conflicts of interest
The authors declare no competing interests.
Summary
PTZ prevents PD-like features in rats. The ability of PTZ to normalize the NAD+/NADH ratio and maintain mitochondrial integrity provides a plausible mechanism that might account for its neuroprotective actions. PTZ could become a promising neuroprotective agent for future clinical studies in PD.
Acknowledgements
This work was supported by research grants from the NIH (R21ES027470, NS095387) and the American Parkinson Disease Association.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2019.101164.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
Primary and secondary antibodies. Antibodies used for immunohistochemical staining and Western blotting.
Fig. S1.

Schematic representation of the experimental design and pharmacological treatment. (A) Schematic diagram depicts the experimental design followed for the in vitro study. Ventral midbrain cultures were plated at a density of 5 × 105 cells/well and maintained in a tri-gas incubator. Drug treatment was added to five-day-old cultures. On the 10 DIV, cells were harvested and processed for cell counting, 3D neurite morphometric analysis, and thiol staining. The number of biological replicates is also itemized. (B) General experimental plan for the in vivo study. Rats were divided into three cohorts and four different groups. In the dose-response experiments (cohort #1) 21 animals received i.p. injections of ROT and different concentrations of PTZ for 16 days, when the last ROT injured rat died. Animals were monitored for weight loss, motor signs, and nigrostriatal DA system impairment. For the time-window experiments (cohorts #2 and #3) 58 subjects were treated with ROT and 20 mg/kg/day of PTZ until endpoint (44 days) when brains were collected and subjected to subsequent processing
Fig. S2.

PTZ does not significantly alter TH expression in SN DA neurons. ROT did not induce overall TH downregulation in nigral DAergic cells compared to the vehicle group. Likewise, PTZ did not alter TH immunoreactivity. Four-five SN sections per animal were used and data from each nigral section were combined to calculate means. Each treatment group was comprised of 12–13 rats and analyzed using one-way ANOVA followed by Newman-Keuls multiple comparisons test.
References
- 1.Cheng H.C., Ulane C.M., Burke R.E. Clinical progression in Parkinson disease and the neurobiology of axons. Ann. Neurol. 2010;67(6):715–725. doi: 10.1002/ana.21995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Langston J.W., Forno L.S., Tetrud J., Reeves A.G., Kaplan J.A., Karluk D. Evidence of active nerve cell degeneration in the substantia nigra of humans years after 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine exposure. Ann. Neurol. 1999;46(4):598–605. doi: 10.1002/1531-8249(199910)46:4<598::aid-ana7>3.0.co;2-f. [DOI] [PubMed] [Google Scholar]
- 3.Mishra P., Chan D.C. Mitochondrial dynamics and inheritance during cell division, development and disease. Nat. Rev. Mol. Cell Biol. 2014;15(10):634–646. doi: 10.1038/nrm3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Youle R.J., van der Bliek A.M. Mitochondrial fission, fusion, and stress. Science. 2012;337(6098):1062–1065. doi: 10.1126/science.1219855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sheng Z.H., Cai Q. Mitochondrial transport in neurons: impact on synaptic homeostasis and neurodegeneration. Nat. Rev. Neurosci. 2012;13(2):77–93. doi: 10.1038/nrn3156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Koch J.C., Bitow F., Haack J., d'Hedouville Z., Zhang J.N., Tonges L. Alpha-Synuclein affects neurite morphology, autophagy, vesicle transport and axonal degeneration in CNS neurons. Cell Death Dis. 2015;6:e1811. doi: 10.1038/cddis.2015.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pozo Devoto V.M., Dimopoulos N., Alloatti M., Pardi M.B., Saez T.M., Otero M.G. alphaSynuclein control of mitochondrial homeostasis in human-derived neurons is disrupted by mutations associated with Parkinson's disease. Sci. Rep. 2017;7(1):5042. doi: 10.1038/s41598-017-05334-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tapias V., Hu X., Luk K.C., Sanders L.H., Lee V.M., Greenamyre J.T. Synthetic alpha-synuclein fibrils cause mitochondrial impairment and selective dopamine neurodegeneration in part via iNOS-mediated nitric oxide production. Cell. Mol. Life Sci. : CMLS. 2017;74(15):2851–2874. doi: 10.1007/s00018-017-2541-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lin M.T., Beal M.F. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature. 2006;443(7113):787–795. doi: 10.1038/nature05292. [DOI] [PubMed] [Google Scholar]
- 10.Castellani R.J., Perry G., Siedlak S.L., Nunomura A., Shimohama S., Zhang J. Hydroxynonenal adducts indicate a role for lipid peroxidation in neocortical and brainstem Lewy bodies in humans. Neurosci. Lett. 2002;319(1):25–28. doi: 10.1016/s0304-3940(01)02514-9. [DOI] [PubMed] [Google Scholar]
- 11.Tapias V., Cannon J.R., Greenamyre J.T. Pomegranate juice exacerbates oxidative stress and nigrostriatal degeneration in Parkinson's disease. Neurobiol. Aging. 2014;35(5):1162–1176. doi: 10.1016/j.neurobiolaging.2013.10.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Giasson B.I., Duda J.E., Murray I.V., Chen Q., Souza J.M., Hurtig H.I. Oxidative damage linked to neurodegeneration by selective alpha-synuclein nitration in synucleinopathy lesions. Science. 2000;290(5493):985–989. doi: 10.1126/science.290.5493.985. [DOI] [PubMed] [Google Scholar]
- 13.Liberatore G.T., Jackson-Lewis V., Vukosavic S., Mandir A.S., Vila M., McAuliffe W.G. Inducible nitric oxide synthase stimulates dopaminergic neurodegeneration in the MPTP model of Parkinson disease. Nat. Med. 1999;5(12):1403–1409. doi: 10.1038/70978. [DOI] [PubMed] [Google Scholar]
- 14.McCoy M.K., Cookson M.R. Mitochondrial quality control and dynamics in Parkinson's disease. Antioxid. Redox Signal. 2012;16(9):869–882. doi: 10.1089/ars.2011.4019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Betarbet R., Sherer T.B., MacKenzie G., Garcia-Osuna M., Panov A.V., Greenamyre J.T. Chronic systemic pesticide exposure reproduces features of Parkinson's disease. Nat. Neurosci. 2000;3(12):1301–1306. doi: 10.1038/81834. [DOI] [PubMed] [Google Scholar]
- 16.Cannon J.R., Tapias V., Na H.M., Honick A.S., Drolet R.E., Greenamyre J.T. A highly reproducible rotenone model of Parkinson's disease. Neurobiol. Dis. 2009;34(2):279–290. doi: 10.1016/j.nbd.2009.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tapias V., Cannon J.R., Greenamyre J.T. Melatonin treatment potentiates neurodegeneration in a rat rotenone Parkinson's disease model. J. Neurosci. Res. 2010;88(2):420–427. doi: 10.1002/jnr.22201. [DOI] [PubMed] [Google Scholar]
- 18.Tapias V., Greenamyre J.T. A rapid and sensitive automated image-based approach for in vitro and in vivo characterization of cell morphology and quantification of cell number and neurite architecture. Paul Robinson J., editor. Curr. Protoc. Cytom./Ed. Board. 2014;68 doi: 10.1002/0471142956.cy1233s68. 12 33 1-12 33 22. [DOI] [PubMed] [Google Scholar]
- 19.Tapias V., Greenamyre J.T., Watkins S.C. Automated imaging system for fast quantitation of neurons, cell morphology and neurite morphometry in vivo and in vitro. Neurobiol. Dis. 2013;54:158–168. doi: 10.1016/j.nbd.2012.11.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mitchell S.C. Mammalian metabolism of orally administered phenothiazine. Drug Metab. Rev. 1982;13(2):319–343. doi: 10.3109/03602538209030001. [DOI] [PubMed] [Google Scholar]
- 21.Mitchell S.C. Phenothiazine: the parent molecule. Curr. Drug Targets. 2006;7(9):1181–1189. doi: 10.2174/138945006778226552. [DOI] [PubMed] [Google Scholar]
- 22.Yamamoto R.S., Weisburger J.H., Weisburger E.K. Controlling factors in urethan carcinogenesis in mice: effect of enzyme inducers and metabolic inhibitors. Cancer Res. 1971;31(5):483–486. [PubMed] [Google Scholar]
- 23.Hajieva P., Mocko J.B., Moosmann B., Behl C. Novel imine antioxidants at low nanomolar concentrations protect dopaminergic cells from oxidative neurotoxicity. J. Neurochem. 2009;110(1):118–132. doi: 10.1111/j.1471-4159.2009.06114.x. [DOI] [PubMed] [Google Scholar]
- 24.Mocko J.B., Kern A., Moosmann B., Behl C., Hajieva P. Phenothiazines interfere with dopaminergic neurodegeneration in Caenorhabditis elegans models of Parkinson's disease. Neurobiol. Dis. 2010;40(1):120–129. doi: 10.1016/j.nbd.2010.03.019. [DOI] [PubMed] [Google Scholar]
- 25.Andersen P.H., Gronvald F.C., Jansen J.A. A comparison between dopamine-stimulated adenylate cyclase and 3H-SCH 23390 binding in rat striatum. Life Sci. 1985;37(21):1971–1983. doi: 10.1016/0024-3205(85)90028-1. [DOI] [PubMed] [Google Scholar]
- 26.Horowitz M.P., Milanese C., Di Maio R., Hu X., Montero L.M., Sanders L.H. Single-cell redox imaging demonstrates a distinctive response of dopaminergic neurons to oxidative insults. Antioxid. Redox Signal. 2011;15(4):855–871. doi: 10.1089/ars.2010.3629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lawal H.O., Chang H.Y., Terrell A.N., Brooks E.S., Pulido D., Simon A.F. The Drosophila vesicular monoamine transporter reduces pesticide-induced loss of dopaminergic neurons. Neurobiol. Dis. 2010;40(1):102–112. doi: 10.1016/j.nbd.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Watabe M., Nakaki T. Mitochondrial complex I inhibitor rotenone-elicited dopamine redistribution from vesicles to cytosol in human dopaminergic SH-SY5Y cells. J. Pharmacol. Exp. Ther. 2007;323(2):499–507. doi: 10.1124/jpet.107.127597. [DOI] [PubMed] [Google Scholar]
- 29.Chu Y., Morfini G.A., Langhamer L.B., He Y., Brady S.T., Kordower J.H. Alterations in axonal transport motor proteins in sporadic and experimental Parkinson's disease. Brain. 2012;135(Pt 7):2058–2073. doi: 10.1093/brain/aws133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chung C.Y., Koprich J.B., Siddiqi H., Isacson O. Dynamic changes in presynaptic and axonal transport proteins combined with striatal neuroinflammation precede dopaminergic neuronal loss in a rat model of AAV alpha-synucleinopathy. J. Neurosci. 2009;29(11):3365–3373. doi: 10.1523/JNEUROSCI.5427-08.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lotharius J., Brundin P. Pathogenesis of Parkinson's disease: dopamine, vesicles and alpha-synuclein. Nat. Rev. Neurosci. 2002;3(12):932–942. doi: 10.1038/nrn983. [DOI] [PubMed] [Google Scholar]
- 32.Tapias V., Escames G., Lopez L.C., Lopez A., Camacho E., Carrion M.D. Melatonin and its brain metabolite N(1)-acetyl-5-methoxykynuramine prevent mitochondrial nitric oxide synthase induction in parkinsonian mice. J. Neurosci. Res. 2009;87(13):3002–3010. doi: 10.1002/jnr.22123. [DOI] [PubMed] [Google Scholar]
- 33.Kushnareva Y., Murphy A.N., Andreyev A. Complex I-mediated reactive oxygen species generation: modulation by cytochrome c and NAD(P)+ oxidation-reduction state. Biochem. J. 2002;368(Pt 2):545–553. doi: 10.1042/BJ20021121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Wen Y., Li W., Poteet E.C., Xie L., Tan C., Yan L.J. Alternative mitochondrial electron transfer as a novel strategy for neuroprotection. J. Biol. Chem. 2011;286(18):16504–16515. doi: 10.1074/jbc.M110.208447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Wischik C.M., Staff R.T., Wischik D.J., Bentham P., Murray A.D., Storey J.M. Tau aggregation inhibitor therapy: an exploratory phase 2 study in mild or moderate Alzheimer's disease. J. Alzheimer's Dis. 2015;44(2):705–720. doi: 10.3233/JAD-142874. [DOI] [PubMed] [Google Scholar]
- 36.Gillman P.K. Methylene blue implicated in potentially fatal serotonin toxicity. Anaesthesia. 2006;61(10):1013–1014. doi: 10.1111/j.1365-2044.2006.04808.x. [DOI] [PubMed] [Google Scholar]
- 37.Ramsay R.R., Dunford C., Gillman P.K. Methylene blue and serotonin toxicity: inhibition of monoamine oxidase A (MAO A) confirms a theoretical prediction. Br. J. Pharmacol. 2007;152(6):946–951. doi: 10.1038/sj.bjp.0707430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tadros F.W., Wahab F.A. The site of action of phenothiazines 35 in the central nervous system. J. Egypt. Med. Assoc. 1962;45:164–171. [PubMed] [Google Scholar]
- 39.Sweetman S.C. 34th Ed. Pharmaceutical Press; London: 2005. The Complete Drug Reference. p. 675, 99, 703, 17. [Google Scholar]
- 40.Carelli V., Musumeci O., Caporali L., Zanna C., La Morgia C., Del Dotto V. Syndromic parkinsonism and dementia associated with OPA1 missense mutations. Ann. Neurol. 2015;78(1):21–38. doi: 10.1002/ana.24410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Lynch D.S., Loh S.H.Y., Harley J., Noyce A.J., Martins L.M., Wood N.W. Nonsyndromic Parkinson disease in a family with autosomal dominant optic atrophy due to OPA1 mutations. Neurol. Genet. 2017;3(5):e188. doi: 10.1212/NXG.0000000000000188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bertholet A.M., Millet A.M., Guillermin O., Daloyau M., Davezac N., Miquel M.C. OPA1 loss of function affects in vitro neuronal maturation. Brain. 2013;136(Pt 5):1518–1533. doi: 10.1093/brain/awt060. [DOI] [PubMed] [Google Scholar]
- 43.Spinazzi M., Cazzola S., Bortolozzi M., Baracca A., Loro E., Casarin A. A novel deletion in the GTPase domain of OPA1 causes defects in mitochondrial morphology and distribution, but not in function. Hum. Mol. Genet. 2008;17(21):3291–3302. doi: 10.1093/hmg/ddn225. [DOI] [PubMed] [Google Scholar]
- 44.Fang D., Yan S., Yu Q., Chen D., Yan S.S. Mfn2 is required for mitochondrial development and synapse formation in human induced pluripotent stem cells/hiPSC derived cortical neurons. Sci. Rep. 2016;6:31462. doi: 10.1038/srep31462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lee S., Sterky F.H., Mourier A., Terzioglu M., Cullheim S., Olson L. Mitofusin 2 is necessary for striatal axonal projections of midbrain dopamine neurons. Hum. Mol. Genet. 2012;21(22):4827–4835. doi: 10.1093/hmg/dds352. [DOI] [PubMed] [Google Scholar]
- 46.Verstreken P., Ly C.V., Venken K.J., Koh T.W., Zhou Y., Bellen H.J. Synaptic mitochondria are critical for mobilization of reserve pool vesicles at Drosophila neuromuscular junctions. Neuron. 2005;47(3):365–378. doi: 10.1016/j.neuron.2005.06.018. [DOI] [PubMed] [Google Scholar]
- 47.Berthet A., Margolis E.B., Zhang J., Hsieh I., Zhang J., Hnasko T.S. Loss of mitochondrial fission depletes axonal mitochondria in midbrain dopamine neurons. J. Neurosci. 2014;34(43):14304–14317. doi: 10.1523/JNEUROSCI.0930-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Mann V.M., Cooper J.M., Daniel S.E., Srai K., Jenner P., Marsden C.D. Complex I, iron, and ferritin in Parkinson's disease substantia nigra. Ann. Neurol. 1994;36(6):876–881. doi: 10.1002/ana.410360612. [DOI] [PubMed] [Google Scholar]
- 49.Schapira A.H., Cooper J.M., Dexter D., Clark J.B., Jenner P., Marsden C.D. Mitochondrial complex I deficiency in Parkinson's disease. J. Neurochem. 1990;54(3):823–827. doi: 10.1111/j.1471-4159.1990.tb02325.x. [DOI] [PubMed] [Google Scholar]
- 50.Cannon J.R., Geghman K.D., Tapias V., Sew T., Dail M.K., Li C. Expression of human E46K-mutated alpha-synuclein in BAC-transgenic rats replicates early-stage Parkinson's disease features and enhances vulnerability to mitochondrial impairment. Exp. Neurol. 2013;240:44–56. doi: 10.1016/j.expneurol.2012.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Liu W., Kato M., Akhand A.A., Hayakawa A., Suzuki H., Miyata T. 4-hydroxynonenal induces a cellular redox status-related activation of the caspase cascade for apoptotic cell death. J. Cell Sci. 2000;113(Pt 4):635–641. doi: 10.1242/jcs.113.4.635. [DOI] [PubMed] [Google Scholar]
- 52.Picklo M.J., Amarnath V., McIntyre J.O., Graham D.G., Montine T.J. 4-Hydroxy-2(E)-nonenal inhibits CNS mitochondrial respiration at multiple sites. J. Neurochem. 1999;72(4):1617–1624. doi: 10.1046/j.1471-4159.1999.721617.x. [DOI] [PubMed] [Google Scholar]
- 53.Chiu C.C., Yeh T.H., Lai S.C., Wu-Chou Y.H., Chen C.H., Mochly-Rosen D. Neuroprotective effects of aldehyde dehydrogenase 2 activation in rotenone-induced cellular and animal models of parkinsonism. Exp. Neurol. 2015;263:244–253. doi: 10.1016/j.expneurol.2014.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Doorn K.J., Moors T., Drukarch B., van de Berg W., Lucassen P.J., van Dam A.M. Microglial phenotypes and toll-like receptor 2 in the substantia nigra and hippocampus of incidental Lewy body disease cases and Parkinson's disease patients. Acta Neuropathol. Commun. 2014;2:90. doi: 10.1186/s40478-014-0090-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shin W.H., Jeon M.T., Leem E., Won S.Y., Jeong K.H., Park S.J. Induction of microglial toll-like receptor 4 by prothrombin kringle-2: a potential pathogenic mechanism in Parkinson's disease. Sci. Rep. 2015;5:14764. doi: 10.1038/srep14764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Reynolds A.D., Glanzer J.G., Kadiu I., Ricardo-Dukelow M., Chaudhuri A., Ciborowski P. Nitrated alpha-synuclein-activated microglial profiling for Parkinson's disease. J. Neurochem. 2008;104(6):1504–1525. doi: 10.1111/j.1471-4159.2007.05087.x. [DOI] [PubMed] [Google Scholar]
- 57.Su X., Maguire-Zeiss K.A., Giuliano R., Prifti L., Venkatesh K., Federoff H.J. Synuclein activates microglia in a model of Parkinson's disease. Neurobiol. Aging. 2008;29(11):1690–1701. doi: 10.1016/j.neurobiolaging.2007.04.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Huang C., Zhu L., Li H., Shi F.G., Wang G.Q., Wei Y.Z. Adulthood exposure to Lipopolysaccharide exacerbates the neurotoxic and inflammatory effects of rotenone in the substantia nigra. Front. Mol. Neurosci. 2017;10:131. doi: 10.3389/fnmol.2017.00131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Lastres-Becker I., Ulusoy A., Innamorato N.G., Sahin G., Rabano A., Kirik D. alpha-Synuclein expression and Nrf2 deficiency cooperate to aggravate protein aggregation, neuronal death and inflammation in early-stage Parkinson's disease. Hum. Mol. Genet. 2012;21(14):3173–3192. doi: 10.1093/hmg/dds143. [DOI] [PubMed] [Google Scholar]
- 60.Michel H.E., Tadros M.G., Esmat A., Khalifa A.E., Abdel-Tawab A.M. Tetramethylpyrazine ameliorates rotenone-induced Parkinson's disease in rats: involvement of its anti-inflammatory and anti-apoptotic actions. Mol. Neurobiol. 2017;54(7):4866–4878. doi: 10.1007/s12035-016-0028-7. [DOI] [PubMed] [Google Scholar]
- 61.Schondorf D.C., Ivanyuk D., Baden P., Sanchez-Martinez A., De Cicco S., Yu C. The NAD+ precursor nicotinamide riboside rescues mitochondrial defects and neuronal loss in iPSC and fly models of Parkinson's disease. Cell Rep. 2018;23(10):2976–2988. doi: 10.1016/j.celrep.2018.05.009. [DOI] [PubMed] [Google Scholar]
- 62.Verdin E. NAD(+) in aging, metabolism, and neurodegeneration. Science. 2015;350(6265):1208–1213. doi: 10.1126/science.aac4854. [DOI] [PubMed] [Google Scholar]
- 63.Keeney P.M., Xie J., Capaldi R.A., Bennett J.P., Jr. Parkinson's disease brain mitochondrial complex I has oxidatively damaged subunits and is functionally impaired and misassembled. J. Neurosci. 2006;26(19):5256–5264. doi: 10.1523/JNEUROSCI.0984-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Kussmaul L., Hirst J. The mechanism of superoxide production by NADH:ubiquinone oxidoreductase (complex I) from bovine heart mitochondria. Proc. Natl. Acad. Sci. U. S. A. 2006;103(20):7607–7612. doi: 10.1073/pnas.0510977103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Katsyuba E., Auwerx J. Modulating NAD(+) metabolism, from bench to bedside. EMBO J. 2017;36(18):2670–2683. doi: 10.15252/embj.201797135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhou M., Ottenberg G., Sferrazza G.F., Hubbs C., Fallahi M., Rumbaugh G. Neuronal death induced by misfolded prion protein is due to NAD+ depletion and can be relieved in vitro and in vivo by NAD+ replenishment. Brain. 2015;138(Pt 4):992–1008. doi: 10.1093/brain/awv002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lehmann S., Costa A.C., Celardo I., Loh S.H., Martins L.M. Parp mutations protect against mitochondrial dysfunction and neurodegeneration in a PARKIN model of Parkinson's disease. Cell Death Dis. 2016;7:e2166. doi: 10.1038/cddis.2016.72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gleave J.A., Arathoon L.R., Trinh D., Lizal K.E., Giguere N., Barber J.H.M. Sirtuin 3 rescues neurons through the stabilisation of mitochondrial biogenetics in the virally-expressing mutant alpha-synuclein rat model of parkinsonism. Neurobiol. Dis. 2017;106:133–146. doi: 10.1016/j.nbd.2017.06.009. [DOI] [PubMed] [Google Scholar]
- 69.Outeiro T.F., Kontopoulos E., Altmann S.M., Kufareva I., Strathearn K.E., Amore A.M. Sirtuin 2 inhibitors rescue alpha-synuclein-mediated toxicity in models of Parkinson's disease. Science. 2007;317(5837):516–519. doi: 10.1126/science.1143780. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Movie S1PTZ ameliorates motor activity and exploratory behavior after 15 days of ROT administration. Video corresponds to one representative animal from each experimental group.
Movie S2PTZ improves motor activity and exploratory behavior in rats treated with ROT for 20 days. Video of three ROT-lesioned rats injected either with vehicle or PTZ.
Movie S3Rats injected with PTZ and ROT after 40 days of treatment. Video monitoring the motor function of four PTZ + ROT-treated subjects. .
Primary and secondary antibodies. Antibodies used for immunohistochemical staining and Western blotting.