

Abstract
The spread of antimicrobial resistance is a major threat to human health and patients requiring prolonged antibiotic exposure are in desperate need of new therapeutic strategies. It has been hypothesized that tailoring our antibiotics to inhibit molecular targets specific to pathogens may stem the spread of resistance. A prime candidate for such a strategy is Pseudomonas aeruginosa, which can be found in the lungs of nearly all adult cystic fibrosis patients and, due to chronic exposure to antibiotics, has a high rate of multi-drug resistant strains. While much research has been done on P. aeruginosa virulence factors as narrow-spectrum targets, less attention has been paid to primary carbon metabolism being leveraged for pathogen specific mechanisms. However, early studies show that primary metabolic pathways, while shared amongst all organisms, contain intricacies specific to Pseudomonas species that have potential for antibiotic exploitation. Here we lay out some of this work in the hopes that it inspires researchers to continue developing a knowledge base that future antibiotic discovery can be built upon and include a case study of a Pseudomonas primary metabolic pathway that has been targeted by small molecules in a species-specific manner.
Keywords: antibiotics, narrow-spectrum, primary metabolism, Pseudomonas sp., tricarboxylic acid cycle
Graphical Abstract
Introduction
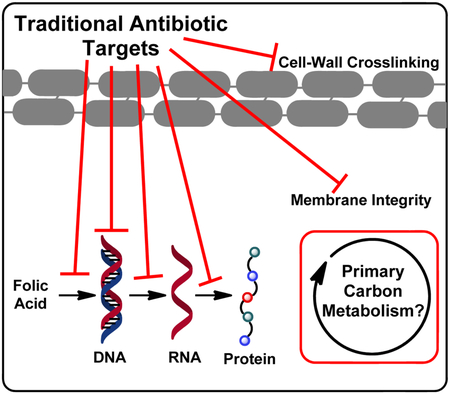
The development of antibiotics and the “Golden Age of antibiotic discovery” has been heralded as one of the great scientific achievements of the modern era; penicillin alone has saved millions of lives.[1] However, increased exposure to broad-spectrum drugs creates an evolutionary pressure for resistance development, thereby exacerbating the spread of multi-drug resistant pathogens.[1] Almost all antibiotics in the clinic target the same handful of universal microbial functions[2] (Figure 1, left) and while a “cure-all” may be useful under many circumstances, this wipes out all susceptible bacteria whether harmful or not.[3] The consequences of this are (1.) The entire resource pool is open for resistant microbes to consume, proliferate uninhibited, and spread resistance genes[4] (2.) Collateral damage to the host microbiome, the proper function of which is implicated in every realm of human physiology including immunity, metabolism, cancer, and cognition.[5] This presents a challenge; creating narrow-spectrum antibiotics to specifically target the lethal bacteria while sparing non-pathogenic host commensals, thereby decreasing the likelihood of resistance development and reducing physical burden on the patient.
Figure 1:

(Left) Targets of current clinical antibiotics: Folic acid biosynthesis inhibitors, DNA gyrase inhibitors, RNA polymerase inhibitors, ribosomal protein synthesis inhibitors, membrane disruptors, and inhibitors of bacterial cell wall biosynthesis. (Middle) New targets being explored for their potential to diversify our antibiotic arsenal and help stem the spread of resistance: Efflux pumps, excreted toxins, quorum signaling, micronutrient acquisition, surface adhesion, virulence-associated proteases (e.g. ClpA), adjuvation to restore potency to current drugs. (Right) Generalized schematic of primary metabolic pathways. While common to all organisms, early studies suggest that subtle differences in the utilization of these pathways between organisms may have potential for exploitation as narrow spectrum activity.
Particularly vulnerable to antibiotic resistant bacteria are those with compromised immune systems due to their inability to ward off infection without heavy antibiotic use. In the lungs of cystic fibrosis (CF) patients a mutation of the CFTR membrane protein causes thick layers of mucus buildup which provide a perfect environment for colonizing pathogens, particularly the Gram-negative Pseudomonas aeruginosa.[6] Once colonized, CF patients will likely take high doses of antibiotics for their entire lives to keep bacteria at bay, but almost all patients will eventually die from chronic pulmonary P. aeruginosa infection rather than from the disease itself.[6] Pseudomonas is also highly relevant in wound infections, where early intervention and prevention of reinfection can make the difference between full recovery and amputation/sepsis.[7] The study of narrow-spectrum, species specific targets in Pseudomonas is of high clinical importance and while in its infancy, exploratory work has revealed great opportunity.
The study of narrow-spectrum antibiotics has largely focused on secondary metabolites and virulence factors[8] (Figure 1, middle), the notion being that targeting these types of molecules biases a drug towards bacteria that are actively trying to establish infection. Conversely, broad spectrum antibiotics generally target essential cellular processes, often enzymatic pathways that are common to all microbes. However, early studies suggest potential for primary carbon metabolism as a target for narrow-spectrum antibiotic activity. While pathways shared amongst all organisms do not sound narrow-spectrum at first blush, this seeming contradiction is resolved by understanding that there are subtle differences in how individual organisms are able to utilize their primary metabolic pathways to respond to environmental stress. The focus of this article is to detail, from a molecular perspective, efforts to probe these subtleties in Pseudomonas using biochemical tools. We will conclude by showcasing small molecules that leverage these differences in Pseudomonas primary metabolism to exhibit species-specific antibiotic activity.
Due to wide availability of genomic data, research on Pseudomonas largely focuses on four key organisms: the previously mentioned P. aeruginosa, the causative agent of tobacco wildfire disease Pseudomonas syringae, the soil bacteria Pseudomonas putida, and the plant-symbiotic Pseudomonas fluorescens. As with many organisms, the carbon metabolism of Pseudomonas can be broadly categorized into a handful of intersecting major pathways which are detailed further later in the article (Figure 1, right, Figure 2).
Figure 2:

Schematic of the combined EDEMP pathway in Pseudomonas putida. Enzymes from the pentose phosphate pathway (Zwf, Pgi), Entner-Doudoroff pathway (Edd, Eda), and gluconeogenic Embden-Meyerhof-Parnas pathway (Fba, Tpi, Fbp, and Pgi) act in concert to create a loop that recycles triose phosphates and generates an excess of NADPH reducing equivalents. TCA = Tricarboxylic Acid Cycle
Metabolic Intermediates as Probes into Antibiotic Adjuvation in Pseudomonas
CF patients under prolonged exposure to antibiotics are at increased risk for Pseudomonas infections that, due to resistance, cannot be treated with clinically approved drugs.[6] In addition to the need for new molecular targets, there is great interest in breathing life back into old antibiotics with molecules that themselves have no killing effect but restore the efficacy of outdated drugs. This strategy of antibiotic adjuvation can in fact be found in the clinically approved β-lactam/β-lactamase inhibitor combination therapy Augmentin,[9] the success of which has inspired researchers of Pseudomonas (non-Augmentin susceptible) to search for other such synergistic combinations.
Recently, Collins and coworkers were able to show that by forcing tobramycin-tolerant P. aeruginosa to subsist on the primary metabolite fumarate, they were able to restore tobramycin activity by kick-starting primary metabolic pathways.[10] Tobramycin is an aminoglycoside antibiotic that is primarily administered via inhalation for the treatment of CF but has seen diminished efficacy. A major mechanism is exclusion; Pseudomonas goes into a stasis mode wherein membrane permeability is lowered. As a result, Pseudomonas must decrease flux through primary metabolism due to a lack of energy sources able to enter the cell. By creating an artificial buildup of fumarate, the authors forced the resistant microbes to turn these pathways back on, and as a result trick the cells into expressing upstream import pathways. With these transporters back in place tobramycin is able to penetrate the membrane and the cells are once again susceptible. Since both tobramycin and fumarate (Formoterol, a supplementary therapy for asthma) are already clinically approved as inhaled medicines, the regulatory hurdles to a combination therapy may be decreased. This molecular-level understanding of Pseudomonas metabolism lays the foundation for novel antibacterial strategies.
Based on this model we speculate that there are other ways to modulate behavior in Pseudomonas with primary metabolic intermediates to achieve antibiotic effects. In 2017, Malone and coworkers discovered a new transcriptional regulator in P. fluorescens which they dubbed RccR.[11] While this enzyme bares structural similarity to the known Entner-Duodoroff pathway (discussed further in the next section) regulator HexR, and both are themselves controlled by concentration of the Entner-Duodoroff intermediate 2-keto-3-deoxy-phosphogluconate (KDPG, Figure 2), they are responsible for regulating entirely different aspects of Pseudomonas primary metabolism. In the presence of high KDPG concentrations, RccR has high affinity for glyoxylate shunt and gluconeogenesis genes, thereby downregulating transcription of these pathways, and low affinity for pyruvate metabolism genes, thereby upregulating them. This dual mode of action is enabled by two distinct “pseudo-palindromic” binding sites, which are differentially active depending on KDPG binding. This allows P. fluorescens to respond rapidly to changes in carbon source availability in response to concentration of a single metabolite. Future exploratory studies should search for RccR in multi-drug resistant pathogenic Pseudomonas, determine the effect of KDPG on fitness, screen antibiotic-KDPG combinations for adjuvation effects, and search for RccR agonists/antagonists.
The work by Collins emphasized the importance of nutrient transporter expression in Pseudomonas antibiotic susceptibility. In 2013, Valantini and Lapouge illuminated a mechanism by which exogenous succinate controls two enzyme cascades in Pseudomonas aeruginosa, the Crc (Carbon-Catabolite Repressor) and the Cbr two-component system, that regulate the expression of two different succinate transporters.[12] Under millimolar succinate concentrations, DctA is expressed as the preferred succinate transporter, whereas under micromolar succinate concentrations DctPQM fills this role. This finding is highly relevant because DctA, a sodium:dicarbozylate symporter, and DctPQM, a tripartite ATP-independent periplasmic (TRAP) transporter, belong to entirely distinct enzyme families and mechanistically behave in completely different ways. To expend so much biosynthetic energy on this interwoven series of regulatory systems suggests that Pseudomonas gains a specific competitive advantage by importing succinate by two different enzymes depending on the concentration and invites further study as a target for behavioral modulation and potentially Trojan-horse drug import (a la the Collins model). The Crc-cascade has also been shown to regulate production of the secondary metabolite pyocyanin.[13] Pyocyanin is an important virulence factor in P. aeruginosa, playing roles in host toxicity, nutrient acquisition, and redox chemistry.[13] Because the Crc-cascade is regulated by succinate concentration, there is opportunity to explore the effects of nutrient availability and metabolic regulation on downstream virulence effects.
Ultimately, knowledge gained from studies of primary metabolic intermediates, their modulation of Pseudomonas nutrient flux, and resultant effects on antibiotic resistance and virulence will be important in exploiting these pathways as Pseudomonas specific targets. Although efforts in this field are still preliminary, the following sections detail a template for how such exploratory research can lead to new potential targets.
Metabolic Flexibility in Pseudomonas
The studies previously mentioned show that we can probe and exploit the distinct carbon source and concentration-based regulation of metabolic pathways in Pseudomonas species in order to tease out potential antibiotic targets. Similarly, it could be possible to deduce a target based on the subtle differences in the flux of these metabolic pathways between Pseudomonas and other bacterial species, as well as between individual Pseudomonas species. Primary metabolic pathways intersect to redirect carbon-flux depending on a variety of factors including nutrient availability, environmental stress, and energy requirements. Briefly, the Embden-Meyerhof-Parnas (EMP) pathway can run in a glycolytic (break down glucose) or gluconeogenic (regenerate glucose) direction.[14] At the end of glycolysis, metabolites can be utilized in fermentation or in the tricarboxylic acid cycle (which intersects with respiration).[15] Certain stages of glycolysis can be bypassed in the Entner-Doudoroff (ED) or Pentose-Phosphate (PP) pathways.[14]
It is generally known that Pseudomonas does not use the EMP pathway in a glycolytic fashion. Recently, Nikel, Lorenzo and coworkers were able to show that P. putida utilizes ED pathway enzymes to catabolize glucose, and couples this with the gluconeogenic action of the upper EMP and PP pathways to recycle triose phosphates (i.e. glyceraldehyde-3-phosphate) in a combined pathway dubbed the EDEMP cycle[16] (Figure 2). It was originally postulated that this somewhat convoluted pathway emerged due to the lack of a functional PFK,[17] an enzyme responsible for irreversibly converting fructose-6-phosphate (F6P) into fructose-1,6-biphosphate (FBP) in the glycolytic direction,[18] in P putida. However, despite the availability of enzymes to direct fructose into the EMP pathway, P. putida still preferentially catabolizes it through the ED pathway (52%) over the EMP (34%) and PP (14%) pathways.[19] Additionally, the full EMP pathway generates more ATP per molecule of glucose than does the ED pathway.[20] Therefore, the ED pathway must be essential for something other than glucose catabolism in P. putida. To elucidate what benefit this combined pathway could provide, a P. putida eda mutant strain (lacking an enzyme critical to the ED pathway) was transformed with pfkA, theoretically restoring the mutant’s glycolytic competence. Somewhat surprisingly, the growth was still impaired and was accompanied by a drastic drop in the production of NADPH, a critical reducing agent for counteracting oxidative stress in P. putida (and other redox-sensitive species).[21]
The evolution of this distinctive pathway could presumably be linked to the conditions of the rhizosphere, home to a diverse array of bacteria, including relatively benign species (P. putida) and pathogenic species (P. aeruginosa).[22, 23] Both species utilize the ED pathway for carbon catabolism, but P. aeruginosa lacks the gluconeogenic activity observed in the EDEMP cycle and utilizes the ED pathway exclusively for glycolysis and the PP pathway exclusively for anabolic purposes, as demonstrated by 13C-metabolic flux analysis.[24] Hence, both species metabolize glucose through pathways that yield optimal amounts of NADPH to combat environmental oxidative stress, doing so in subtly different ways. A more comprehensive understanding of these differences could serve as inspiration for new species-specific targets.
Also important in the discovery of antibiotic targets is an understanding of how carbon metabolism can differ within a single species. Fluxomic studies by Berger and coworkers, which quantify differences in metabolic flux by tracking 13C in vivo, have demonstrated that carbon flux through the TCA cycle and the glyoxylate shunt differ greatly between P. aeruginosa clinical isolates from the urinary tract and the surface of catheter infections.[24] Despite having highly conserved upstream metabolic pathways, these strains displayed highly varied flux-partitioning ratios at isocitrate, which can either continue through the TCA cycle, or be directed toward the glyoxylate shunt.[24] In fact, flux through the glyoxylate shunt was as high as 44% in certain strains, and undetectable in others.[24] These results combined with rigorous statistical analysis demonstrated that although highly variable, flux through the glyoxylate shunt in P. aeruginosa is infection-site-specific.
Infection site could also be linked to pathogenesis of P. aeruginosa, as higher flux through the shunt has been correlated with higher levels of virulence. In light of Malone’s work (detailed in the previous section), high KDPG concentrations could conversely be linked to reduced levels of virulence in Pseudomonas, as this enables RccR to bind glyoxylate shunt genes more strongly, thereby reducing their transcription and reducing flux through the shunt.[11] The glyxoxylate shunt also plays an important role in the ability of P. putida to resist its own endogenously produced primary metabolism targeting antibiotics (detailed in next section). These assertions along with Lorenzo’s demonstration of the effect that the ED pathway has on P. putida’s ability to combat oxidative stress illustrates the important role KDPG may play, not only as a metabolic regulator, but also in the survival of Pseudomonas.
Case Study: Succinate Dehydrogenase and Potential for Narrow-Spectrum Inhibition
An intriguing case study of the how the subtle differences in primary metabolism lend to intragenus specificity is that of promysalin, an amphipathic, salicylic acid-containing small molecule isolated in 2011 from P. putida RW10S1 by De Mot and coworkers.[25] Controlled by the Gac regulatory system, P. putida produces this secondary metabolite, which promotes its own swarming and biofilm formation and exhibits species-specific growth inhibitory activity against other Pseudomonads,[25] including human-pathogenic P. aeruginosa (IC50 = 67 nM against PA14; IC50 = 4.1 μM against PA01).[26] The initial hypothesis that the mechanism of action may be through chelation of iron, preventing P. aeruginosa from sequestering sufficient iron for growth,[27] was refuted when promysalin’s potency was seemingly unaffected by varying iron concentration.[28] Ultimately, the succinate dehydrogenase C-subunit (SdhC) was identified as the biological target through an affinity-based protein profiling approach, and further validated through computational molecular docking as well as genome sequencing of a promysalin-resistant PA14 mutant (Figure 3).[28]
Figure 3:

Schematic of the tricarboxylic acid cycle and the glyoxylate shunt. Succinate dehydrogenase (Sdh) is a shared enzyme of the TCA cycle and the electron transport chain and thus is important in respiration. On the left are shown known inhibitors of the ubiquinone binding pocket of SdhC.
Succinate dehydrogenase serves a critical role in both the electron transport chain and the TCA cycle by coupling the reduction of ubiquinone to ubiquinol with the oxidation of succinate to fumarate.[29] By binding to SdhC, the ubiquinone binding site, promysalin prevents these coupled reactions from occurring. However, growth inhibition by promysalin was only observed in P. aeruginosa and not P. putida, which prompted conduction of selected feeding studies in order to rationalize the species-specific growth inhibition. While promysalin inhibited the growth of P. aeruginosa strains grown on minimal media supplemented with glucose, but not P. putida strains, it inhibited the growth of both organisms grown on minimal media supplemented with succinate.[28] This indicates that when Sdh is rendered inactive, P. putida simply redirects carbon metabolism, presumably through the glyoxylate shunt, and its growth is only inhibited when it is forced to utilize Sdh such as when succinate is its only carbon source. This presumption is supported by RNA-sequencing data, which showed that expression of several ED pathway genes are down-regulated in the presence of promysalin.[28] If RccR is present in P. putida as in P. fluorescens, a down-regulated ED pathway should yield lower levels of KDPG, decreasing the binding of RccR to glyoxylate shunt genes, increasing flux through the shunt. The downstream effects of down-regulated expression of ED pathway genes could explain why promysalin inhibits the growth of P. aeruginosa and not P. putida. Hence, promysalin exhibits narrow-spectrum effects despite its seemingly ubiquitous target.
Another small molecule antibiotic that targets the ubiquinone binding site of Sdh is siccanin, isolated from the fungus Helminthosposium siccans in 1962 and highly similar in structure to ubiquinone.[30, 31, 32, 33] Exhibiting potent antifungal activity against pathogenic fungi such as Trichophyton mentagrophytes (IC50 = 87 nM), siccanin was originally used as a fungicidal topical agent in the treatment of skin infections like eczema.[31, 32] It was not until recently that siccanin was rediscovered as an inhibitor of Sdh in Gram-negative bacteria such as P. aeruginosa (IC50 = 150 μM) as part of a screen of natural antibiotics in the Kitasato Institute for Life Sciences Chemical Library.[33] Other recent studies have shown that siccanin inhibits Sdh in other bacteria like T. cruzi (IC50 = 1.48 μM), T. brucei (IC50 = 0.368 μM), and L. donovani (IC50 = 1.17 μM), associated with Chagas disease, African sleeping sickness, and black fever, respectively.[31, 32] Although it is not proven to be species-specific like promysalin, siccanin does have markedly higher selectivity for Sdh in the previously mentioned pathogens than for porcine Sdh.[31, 32] Therefore, it could potentially serve as a probe molecule to study Sdh in various pathogens, as well as a structural template in the design of species-specific Sdh inhibitors.
Both of these molecules highlight the potential to exploit differences in primary metabolism in Pseudomonas in an effort to design molecules with species-specific antibiotic activity. While promysalin shows promise in its species-specific inhibition of P. aeruginosa, siccanin represents an opportunity to further study small-molecule interactions with the ubiquinone binding domain of Sdh in various species. Ultimately, these molecules could open the door to development of highly potent and species-specific Sdh inhibitors.
Concluding Remarks
Novel solutions to the problem of bacterial pathogenesis increasingly require molecular level understanding of complex biological systems. While much of the work highlighted herein is preliminary, we hope that it sheds new light on the nuances of Pseudomonas primary metabolism and inspires research into its potential as a target of next generation antibiotics. The development of new molecules that help combat the spread of antibiotic resistance and protect the host against depletion of microbiota is a complex endeavor that requires a multi-pronged approach to explore a variety of potential new targets. With further study, the primary metabolism could be another tool in this arsenal. Of course the ultimate obstacle such potential therapeutic targets face, as with many innovative antibiotic strategies being explored today, is the ability to analyze the role of pathways and efficacy/scope of potential inhibitors under laboratory conditions that mimic infection. We anticipate that advances in whole genome sequencing, metabolimics, and proteomics will shed light on this burgeoning research area and enable the evaluation of potentially pathogen-specific therapeutics, as well as the use of variable metabolic activity for biomarker diagnostic purposes in human infection.
Acknowledgements
The authors thank the National Institutes of Health for NIGMS grant no. GM119426, and the National Science Foundation for grant no. CHE1755698.
References
- [1].Pilla G, Lobanovska M, Yale J Biol. Med, 2017, 90, 135–145. [PMC free article] [PubMed] [Google Scholar]
- [2].Lewis K, Nature, 2013, 12, 371–387. [DOI] [PubMed] [Google Scholar]
- [3].Garland M, Loscher S, Bogyo M, Chem. Rev, 2017, 117, 4422–4461. [DOI] [PubMed] [Google Scholar]
- [4].Cantón R, Morosini M, FEMS Microbiol. Rev, 2011, 35, 977–991. [DOI] [PubMed] [Google Scholar]
- [5].Langdon A, Crook N, Dantes G, Genome Med, 2016, 8, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Bhagirath AY, Li Y, Somayajula D, Dadashi M, Badr S, Duan K, BMC Pulm. Med, 2016, 16, 174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Serra R, Grande R, Butrico L, Rossi A, Settimio UF, Caroleo B, Amato B, Galleilli L, de Franciscis S, Expert. Rev. Anti. Infect. Ther, 2015, 13, 605–613. [DOI] [PubMed] [Google Scholar]
- [8].Johnson BK, Abramovitch RB, Trends Pharmacol. Sci, 2017, 38, 339–362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Deforges L, le Van Thoi J, Soussy CJ, Duval J, Pathol. Biol. (Paris), 1985, 33, 301–308. [PubMed] [Google Scholar]
- [10].Meylan S, Porter CBM, Yang JH, Belenky P, Guiterrez A, Lobritz MA, Park J, Kim SH, Moskowitz SM, Collins JJ, Cell Chem. Biol, 2017, 24, 195–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Campilongo R, Fung RKY, Little RH, Grenga L, Trampari E, Pepe S, Chandra G, Stevenson CEM, Roncarati D, Malone JG, PLOS Genetics, 2017, 13, e1006839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Valentini M, Lapouge K, Environ. Microbiol, 2013, 15, 1707–1716. [DOI] [PubMed] [Google Scholar]
- [13].Price-Whelan A, Dietrich LEP, Newman DK, J. Bacteriol, 2007, 189, 6372–6381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Conway T, Microbiol. Revl, 1992, 9, 1–27. [DOI] [PubMed] [Google Scholar]
- [15].Thauer RK, Eur. J. Biochem, 1988, 176, 497–508. [DOI] [PubMed] [Google Scholar]
- [16].Nikel P, Chavarría M, Fuhrer T, Sauer U, Lorenzo V. de., J. Biol. Chem 2015, 290, 25920–25932 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Velázquez F, Bartolo I. di., Lorenzo V. de., J. Bacteriol 2004, 186, 8267–8275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Ronimus RS, Morgan HW, Extremophiles. 2001, 5, 357–373 [DOI] [PubMed] [Google Scholar]
- [19].Chavarría M, Kleijn RJ. Sauer U, Plüger-Grau K, Lorenzo V. de., mBio. 2012, 3, e00028–e00012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Bar-Even A, Flamholz A, Noor E, Milo R, Nat. Chem. Biol 2012, 8, 509–517 [DOI] [PubMed] [Google Scholar]
- [21].Chavarría M, Nikel P, Pérez-Pantoja D, Lorenzo V. de., Environ. Microbiol 2012, 15, 1772–1785 [DOI] [PubMed] [Google Scholar]
- [22].Lugtenberg BJJ, Dekkers L, Environ. Microbiol 1999, 1, 9–13 [DOI] [PubMed] [Google Scholar]
- [23].Keohane CE, Steele AD, Wuest WM, Synlett. 2015, 26, 2739–2744 [Google Scholar]
- [24].Berger A, Dohnt K, Tielen P, Jahn D, Becker J, Wittmann C, PLoS. ONE. 2014, 9, e88368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Li W, Santos PE, Matthijs S, Xie G, Busson R, Cornelis P, Rozenski J, De Mot R, Chem. Biol 2011, 18, 1320–1330 [DOI] [PubMed] [Google Scholar]
- [26].Steele AD, Knouse KW, Keohane CE, Wuest WM, J. Am. Chem. Soc 2015, 137, 7314–7317 [DOI] [PubMed] [Google Scholar]
- [27].Steele AD, Keohane CE, Knouse KW, Rossiter SE, Williams SJ, Wuest WM, J. Am. Chem. Soc. 2016, 138, 5833–5836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Keohane CE, Steele AD, Fetzer C, Khowsathit J, Tyne DV, Moynie L, Gilmore MS, Karanicolas J, Sieber SA, Wuest WM, J. Am. Chem. Soc 2018, 140, 1774–1782 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Hederstedt L, Ruthberg L, Microbiol. Rev 1981, 45, 542–555 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Nose K, Endo A, J. Bacteriol. 1970, 105, 176–184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kiyoshi K, Inaoka KD, Yamamoto M, Antiparasitic Agent, 2016. [Google Scholar]
- [32].Nihashi N, Inaoka DK, Tsuge C, Balogun EO, Osada Y, Goto Y, Matsumoto Y, Nara T, Mogi T, Harada S, Kiyoshi K, 2016, Siccanin Is a Novel Selective Inhibitor of Trypanosomatid Complex II (Succinate-Ubiquinone Reductase) and a Potent Broad-Spectrum Anti-trypanosomatid Drug Candidate In Kala Azar in South Africa (pp. 101–122). Springer, Cham: Doi: 10.1007/978-3-319-47101-3_9. [DOI] [Google Scholar]
- [33].Mogi T, Kawakami T, Arai H, Igarashi Y, Matsushita K, Mori M, Shiomi K, Omura S, Harada S, Kita K, J. Biochem 2009, 146, 383–387 [DOI] [PubMed] [Google Scholar]