

Abstract
Purpose
Children and young adults with hypodiploid B-lymphoblastic leukemia (B-ALL) fare poorly and hematopoietic stem-cell transplantation (HSCT) is often pursued in first complete remission (CR1). We retrospectively reviewed the outcomes of children and young adults with hypodiploid B-ALL who were enrolled in recent Children’s Oncology Group (COG) trials to evaluate the impact of HSCT on outcome.
Patients and Methods
Cytogenetic analyses and DNA index were performed at COG-approved laboratories, and hypodiploidy was defined as modal chromosome number less than 44 and/or DNA index less than 0.81. Minimal residual disease (MRD) was determined centrally using flow cytometry at two reference laboratories. Patients with hypodiploid ALL came off protocol therapy postinduction and we retrospectively collected details on their subsequent therapy and outcomes. Event-free survival (EFS) and overall survival (OS) were estimated for the cohort.
Results
Between 2003 and 2011, 8,522 patients with National Cancer Institute standard-risk and high-risk B-ALL were enrolled in COG AALL03B1 (ClinicalTrials.gov identifier: NCT00482352). Hypodiploidy occurred in 1.5% of patients (n = 131), 98.3% of whom achieved CR after induction therapy. Five-year EFS and OS were 52.2% ± 4.9% and 58.9% ± 4.8%, respectively. Outcomes for patients undergoing CR1 HSCT were not significantly improved: 5-year EFS and OS were 57.4% ± 7.0% and 66.2% ± 6.6% compared with 47.8% ± 7.5% and 53.8% ± 7.6%, respectively (P = .49 and .34, respectively) for those who did not undergo transplantation. Patients with MRD of 0.01% or greater at the end of induction had 5-year EFS and OS of 26.7% ± 9.3% and 29.3% ± 10.1%, respectively, and HSCT had no significant impact on outcomes.
Conclusion
Children and young adults with hypodiploid B-ALL continue to fare poorly and do not seem to benefit from CR1 HSCT. This is especially true for patients with MRD of 0.01% or greater at the end of induction. New treatment strategies are urgently needed for these patients.
INTRODUCTION
Using risk-adapted therapy, 5-year overall survival (OS) rates for pediatric B-lymphoblastic leukemia (B-ALL) exceed 90%.1,2 Clinical factors, lymphoblast genetic features, and early response to therapy determine postinduction therapy intensity. Favorable cytogenetic abnormalities include hyperdiploidy and the ETV6-RUNX1 fusion.3,4 Unfavorable abnormalities include the Philadelphia chromosome [t(9;22)], which results in BCR-ABL1 fusion,5,6 KMT2A (formerly MLL) rearrangements,7,8 intrachromosomal amplification of chromosome 21,9-11 genetic alterations that lead to Philadelphia chromosome-like acute lymphoblastic leukemia (ALL),12,13 and hypodiploidy.14-17
Hypodiploid ALL affects less than 5% of pediatric patients.14,17,18 Early studies defined hypodiploid leukemic clones as having fewer than 46 chromosomes, with 45 being the most common modal chromosome number.14,19 Modal numbers as low as 24 have been described,14,17,19 as well as hypodiploid clones that double, which results in a hyperdiploid or triploid complement of chromosomes (masked hypodiploidy).20 Outcomes of patients whose blasts harbor 44 or 45 chromosomes are significantly better than those with lower modal numbers, and outcomes worsen as modal number decreases.15-17 A study from St. Jude Children’s Research Hospital reported 5-year event-free survival (EFS) rates of 78% ± 5.9% with a modal number of 45 versus less than 30% with fewer than 45 chromosomes (P < .001).15 In a series of Medical Research Council protocols, 3-year EFS with 42 to 45 chromosomes was 66% compared with 29% with 25 to 39 chromosomes (P = .0002).16 The Ponte di Legno group reported an 8-year EFS of 52.2% ± 9.3% with 44 chromosomes compared with 30.1% ± 7% with fewer than 44 chromosomes (P = .01).17
Recurrent genetic alterations have been identified in hypodiploid ALL.2,21,22 In near-haploid ALL, alterations in receptor tyrosine kinase and Ras signaling, deletions of IKZF3, and focal deletions of a histone gene cluster at 6p22 are recurrent. Mutations in NF1, NRAS, KRAS, MAPK1, FLT3, and PTPN11 account for more than two thirds of receptor tyrosine kinase or Ras pathway aberrations.21 In low-hypodiploid ALL, the frequency of TP53 alterations is remarkable. Mutations are present in the leukemic blasts of more than 90% of patients21,22 and nearly one half are germline, which renders hypodiploid ALL a manifestation of Li-Fraumeni syndrome.2,21 Another IKAROS family gene, IKZF2, is deleted in approximately one half of low-hypodiploid ALL, and alterations of RB1 are also common.21 Preclinical studies suggest that targeting Ras and related signaling pathways may yield therapeutic options for this difficult-to-treat population.21
In addition to such features as National Cancer Institute (NCI) risk group and lymphoblast genetics, disease response is essential in B-ALL risk stratification. Bone marrow (BM) minimal residual disease (MRD) at the end of induction (EOI) is the most significant prognosticator.23-28 Standard chemotherapy regimens for B-ALL are well established, but there is no unified treatment approach for hypodiploid ALL. As a result of poor outcomes, treatment is generally intensified and frequently includes hematopoietic stem-cell transplantation (HSCT) in first complete remission (CR1); however, the benefit of CR1 HSCT has yet to be elucidated. We report the outcomes of 131 children, adolescents, and young adults with hypodiploid B-ALL who were enrolled in Children’s Oncology Group (COG) classification study, AALL03B1, 61 of whom underwent CR1 HSCT. We correlated outcomes with NCI risk group and MRD and examined the impact of CR1 HSCT on EFS and OS.
PATIENTS AND METHODS
Patient Characteristics and Risk Stratification
From December 2003 to September 2011, 8,522 patients with B-ALL were enrolled in AALL03B1, a companion biology study for therapeutic studies AALL0331 (ClinicalTrials.gov identifier: NCT00103285; N = 5,434) and AALL0232 (ClinicalTrials.gov identifier: NCT00075725; N = 3,088)29 for children, adolescents, and young adults with NCI standard-risk (SR) B-ALL (age > 1 year and < 10 years, with initial WBC < 50,000/μL) or NCI high-risk (HR) B-ALL (age 10 to 30 years or initial WBC ≥ 50,000/μL and any age), respectively. Institutional review board approval and informed consent according to the Declaration of Helsinki were obtained. AALL0331 and AALL0232 treatment regimens have been previously described.29,30 Routine evaluations on AALL03B1 included blast karyotype, fluorescence in situ hybridization, and DNA index (DI). Hypodiploidy was defined as blasts containing fewer than 44 chromosomes or DI less than 0.81.
Patients who were treated on AALL0331 and AALL0232 had BM morphology at day 8 and/or 15 and at EOI (day 29), as well as EOI BM MRD using flow cytometry in one of two COG reference laboratories.23,31,32 Rapid early responders achieved an M1 marrow (< 5% blasts by morphology) by day 15 and had EOI BM MRD less than 0.1%. Patients were otherwise identified as slow early responders. Patients with an EOI M2 marrow (5% to 25% blasts) or EOI MRD level of 1% or greater received 2 weeks of extended induction therapy. These patients were deemed slow early responders if they had an M1 marrow with MRD less than 1% after extended induction.
Patients who were classified as very high risk (VHR), including those with hypodiploidy, among others, were removed from AALL0331 and AALL0232 at EOI. VHR ALL patients were eligible to enroll in a separate COG study, AALL0031 (ClinicalTrials.gov identifier: NCT00022737).33,34 AALL0031 included intensive consolidation blocks followed by allogeneic HSCT for patients with an identified donor. The conditioning regimen included total body irradiation, cyclophosphamide, and etoposide. Those patients without a donor or who did not meet HSCT criteria received reinduction and intensification blocks followed by prolonged maintenance therapy.33 Sixteen patients from AALL03B1 enrolled in AALL0031, but after study accrual goals were met in 2006, there was no open postinduction COG clinical trial for hypodiploid ALL and therapy was delivered at the discretion of treating physicians. A case report form was created post hoc to collect data on CR1 HSCT.
Molecular, Cytogenetics, and Statistical Methods
Routine karyotyping and fluorescence in situ hybridization to detect trisomies of chromosomes 4, 10, and 17, ETV6-RUNX1, KMT2A-R, and BCR-ABL1 were performed either at COG central or COG-approved local cytogenetic laboratories, with central review of all results (A.J.C. or N.A.H.). DI was determined in the COG Molecular Reference Laboratory. Details of techniques have been described previously.9,23,35
EFS was defined as the time from study enrollment to first event (induction failure, relapse, second malignant neoplasm [SMN], or death) or date of last contact. OS was defined as the time from study enrollment to death or date of last of contact. For all comparisons of outcomes for HSCT versus no HSCT, EFS and OS time started at transplantation for the HSCT cohort and at the median time to transplantation (137 days) for the non-HSCT cohort.
Data current as of June 30, 2017, are included in this work. We estimated survival rates using the Kaplan-Meier method36 with SEs of Peto et al.37 Survival rates are presented as estimate ± SE. We used log-rank tests to compare survival curves, and proportions were compared between groups using χ2 or Fisher’s exact test. Cumulative incidence rates were computed using the cumulative incidence function for competing risks, and comparisons were made using the K-sample test.38 P values < .05 were considered significant. All analyses were performed using SAS (SAS/STAT User’s Guide, Version 9.4; SAS Institute, Cary, NC). Graphics were generated with R (version 3.0.1; http://www.R-project.org).
RESULTS
Patient Characteristics
Of the 8,522 patients with NCI SR and HR B-ALL who were enrolled in AALL03B1, 131 (1.5%) were identified as having hypodiploid ALL (Fig 1), with a median age of 10 years. Of these patients, 83 (63.4%) were males and 48 (36.6%) were NCI SR (Table 1). The higher percentage of NCI HR patients is reflected in the older median age of hypodiploid patients. Extramedullary disease was uncommon. There were 109 patients (83.2%) without evidence of CNS disease (CNS1)30 and only two patients (1.5%) were CNS3. One patient had no CNS status reported. No males had testicular leukemia. Median number of chromosomes was 28 (range, 25 to 43), and median DI was 0.8 (range, 0.5 to 1.5). Of the patients with modal chromosome numbers reported, 55 had 25 to 29 chromosomes, 47 had 30 to 39 chromosomes, and three had 40 to 43 chromosomes. Twenty-six patients had masked hypodiploidy (Appendix Table A1, online only).
FIG 1.

CONSORT diagram for patients on AALL03B1 with hypodiploidy. B-ALL, hypodiploid B-lymphoblastic leukemia; CR1, first complete remission; HSCT, hematopoietic stem-cell transplantation.
TABLE 1.
Characteristics of Patients Enrolled in AALL03B1 With Hypodiploid B-Lymphoblastic Leukemia
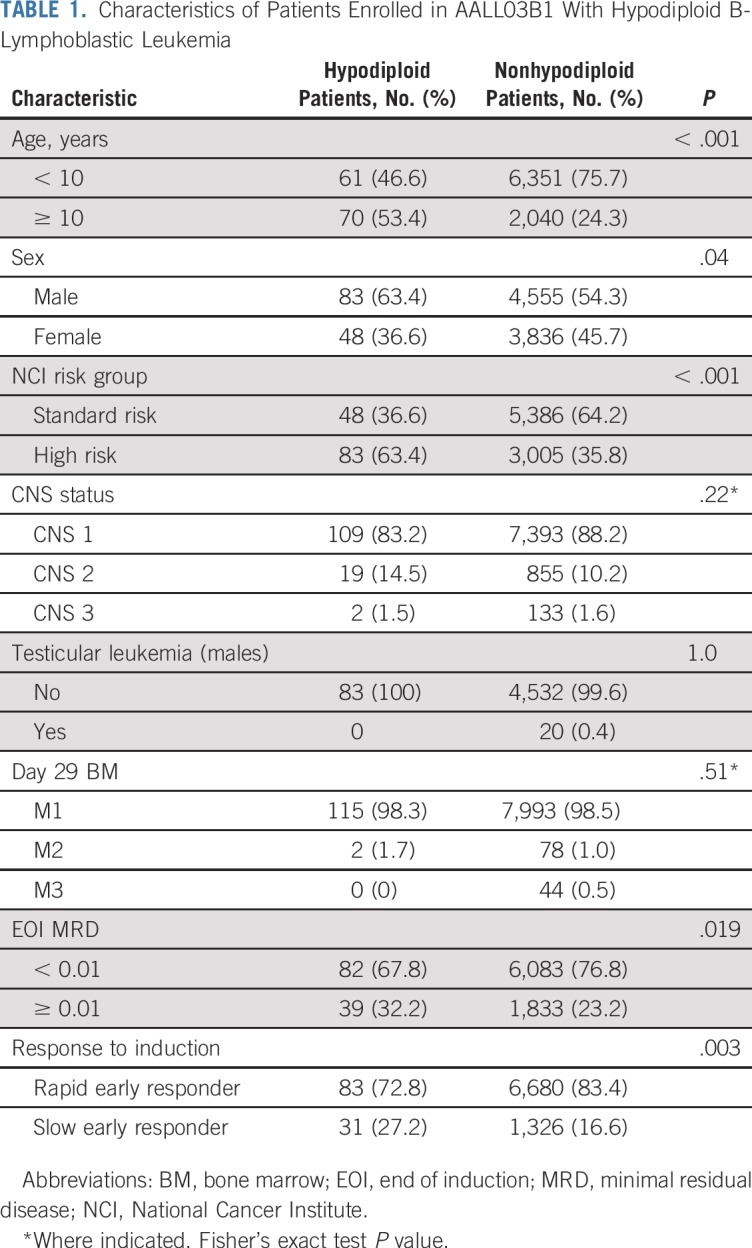
Response to Induction Therapy
After induction therapy, 115 patients (98.3%) had an M1 BM and two patients (1.7%) had an M2 BM (Table 1). There were no reported induction failures and 14 patients had no day 29 BM morphologic response reported. Hypodiploid patients experienced an inferior response to induction therapy with a lower rate of rapid early responders (72.8% v 83.4%; P = .003) and a higher rate of EOI MRD of 0.01% or greater (32.2% v 23.3%; P = .019). Of the 48 NCI SR patients, 38 had day-29 BM morphology reported, 36 (94.7%) of whom had an M1 marrow. Of the 41 NCI SR patients with EOI MRD reported, 27 (65.9%) had MRD of less than 0.01%.23 EOI BM morphology data were available in 79 of the 83 NCI HR patients, all of whom were M1. MRD was available in 81 of the NCI HR patients, 55 (67.9%) of whom had MRD of less than 0.01% (Appendix Fig A1, online only). When response was stratified by modal group, 47 patients (95.8%) out of 49 with 25 to 29 chromosomes and available data had an M1 BM at EOI. Fifty-two had EOI MRD reported, with 34 patients (65.4%) having MRD of less than 0.01%. All patients with 30 to 39 chromosomes and available morphology data (n = 41) had an M1 BM. In this group, 42 patients had MRD data and 29 patients (69%) had MRD of less than 0.01%.
Patient Outcomes
Five-year EFS and OS for all 131 hypodiploid patients were 52.2% ± 4.9% and 58.9% ± 4.8%, respectively (Fig 2A), which was inferior to the 85.2% ± 0.4% and 91.8% ± 0.3%, respectively, observed for nonhypodiploid patients (P < .001 for both EFS and OS). There were no significant differences in EFS and OS according to NCI risk group (P = .26 and .32, respectively) for hypodiploid patients, with 5-year EFS and OS of 60.1% ± 7.9% and 67.3% ± 7.6%, respectively, for NCI SR, and 47.4% ± 6.1% and 54.0% ± 6.1%, respectively, for NCI HR patients (Figs 2B and 2C). Outcomes for nonhypodiploid patients were significantly different between NCI risk groups. For nonhypodiploid patients, 5-year EFS of NCI SR and NCI HR were 90.1% ± 0.4% and 76.3% ± 0.8%, respectively (P < .001), and 5-year OS was 95.8% ± 0.3% and 84.5% ± 0.7%, respectively (P < .001).
FIG 2.

Outcomes for all patients with hypodiploid B-lymphoblastic leukemia (B-ALL). (A) All patients with hypodiploid B-ALL. (B) National Cancer Institute (NCI) standard-risk patients with hypodiploid B-ALL. (C) NCI high-risk patients with hypodiploid B-ALL. EFS, event-free survival; OS, overall survival.
CR1 HSCT data were available for 113 (86.3%) of 131 hypodiploid patients. Sixty-one underwent CR1 HSCT, and 5-year EFS and OS for these 113 patients were 53.0% ± 5.1% and 60.5% ± 5.0%, respectively, which is similar to the entire cohort of 131 hypodiploid patients. Presenting features and disease responses were no different between those who underwent HSCT and those who did not (Table 2). There was a nonsignificant trend for the use of HSCT among those < 10 years (63.5% v 45.9%; P = .06). The 5-year EFS with and without CR1 HSCT were 56.4% ± 7.3% and 48.8% ± 7.8%, respectively (P = .62). The 5-year OS with and without HSCT was 65.6% ± 6.9% and 53.8% ± 7.8%, respectively (P = .32; Fig 3). Most of the 61 CR1 HSCT patients (n = 56; 91.8%) were conditioned with a total body irradiation–based regimen. Most donors were matched-related (n = 31; 50.8%) or matched-unrelated (n = 25; 41.0%), and BM was the stem-cell source for 34 patients (55.7%; Appendix Table A2, online only).
TABLE 2.
Comparison of Clinical Characteristics and Disease Response in Patients Who Underwent HSCT Compared With Those Treated With Chemotherapy Alone
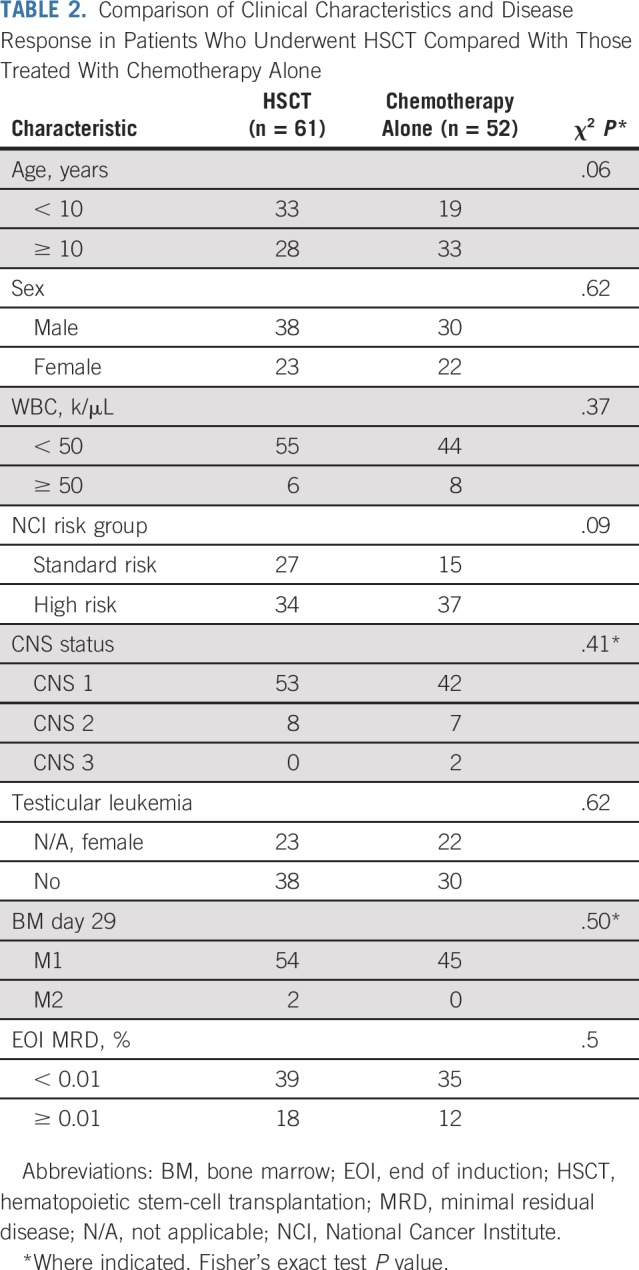
FIG 3.

Outcomes for patients with hypodiploid B-lymphoblastic leukemia according to transplantation status, corrected for median time to hematopoietic stem-cell transplantation (HSCT). (A) Event-free survival [EFS] (B) overall survival (OS).
Stratifying by NCI risk group, CR1 HSCT did not significantly affect outcomes for hypodiploid SR patients (n = 27); 5-year EFS was 68.8% ± 10.3% and OS was 77.3% ± 9.2% compared with 57.1% ± 13.2% and 64.3% ± 12.8% in those who did not undergo transplantation (n = 15; Figs 4A and 4B; P = .64 for EFS and P = .62 for OS). The 5-year EFS of HR patients who underwent HSCT (n = 34) was 48.3% ± 9.0% versus 44.4% ± 9.2% in those who did not (n = 37; P = .75; Fig 4C), and there was also no difference in OS in HR patients by CR1 HSCT (57.6% ± 8.8% v 49.9% ± 9.4%; P = .56; Fig 4D).
FIG 4.

Outcomes for patients with hypodiploid B-lymphoblastic leukemia according to National Cancer Institute (NCI) risk group and transplantation status. (A) Event-free survival (EFS) and (B) overall survival (OS) in NCI standard-risk patients. (C) EFS and (D) OS in NCI high-risk patients. HSCT, hematopoietic stem-cell transplantation.
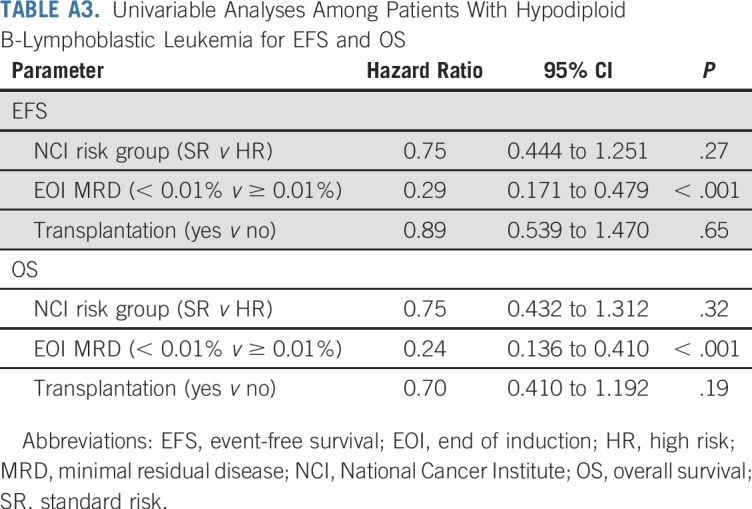
Given the strength of MRD as a prognostic factor, we evaluated outcomes by EOI MRD and allocation to HSCT. Hypodiploid patients with EOI MRD of less than 0.01% had 5-year EFS and OS of 63.7% ± 5.8% and 73.2% ± 5.3%, respectively. Those with EOI MRD of 0.01% or greater had inferior 5-year EFS and OS (26.2% ± 8.5% and 27.7% ± 8.9%, respectively). HSCT had no significant effect on outcomes regardless of EOI MRD. Patients with MRD of less than 0.01% at EOI had 5-year EFS of 66.3% ± 7.9% with HSCT (n = 39) and 60.3 ± 9.2% without (n = 35; P = .77). Five-year OS was 79.5% ± 6.7% with HSCT and 66.7% ± 8.8% without (P = .39; Figs 5A and 5B). Furthermore, cumulative incidence of relapse did not differ significantly between chemotherapy and HSCT groups (Appendix Fig A2, online only). Patients with MRD of 0.01% or greater at EOI had dismal outcomes regardless of CR1 HSCT. EFS and OS at 5 years were 29.4% ± 14.3% and 29.4% ± 14.3%, respectively, with HSCT (n = 18), whereas EFS and OS were 16.7% ± 10.8% and 22.2% ± 13.9%, respectively, without (n = 12; Figs 5C and 5D; P = .67 for EFS and P = .86 for OS). Univariable analyses, including NCI risk group, EOI MRD, and HSCT, revealed EOI MRD of less than 0.01% as the only significant prognostic factor (hazard ratio, 0.29 [95% CI, 0.171 to 0.479; P < .001] for EFS and 0.24 [95% CI, 0.136 to 0.410; P < .001] for OS [Appendix Table A3, online only]).
FIG 5.

Outcomes for patients with hypodiploid B-lymphoblastic leukemia according to end-of-induction minimal residual disease (MRD) and transplantation status. (A ) Event-free survival (EFS) and (B) overall survival in patients deemed MRD-negative (MRD < 0.01%). (C) EFS and (D) OS in patients deemed MRD-positive (MRD ≥ 0.01%). HSCT, hematopoietic stem-cell transplantation.
While not specifically investigated in these patients, we presume that many low-hypodiploid patients harbored germline TP53 mutations. Twelve patients developed SMNs, seven after HSCT and three after chemotherapy (remainder unknown; Appendix Fig A3, online only). All HSCT patients developed solid tumors, whereas two of the three non-HSCT patients developed myelodysplastic syndromes. The 5-year cumulative incidences of SMN for chemotherapy versus HSCT were 4.0% ± 2.8% and 7.3% ± 3.6%, respectively (P = .13).
DISCUSSION
Hypodiploidy is a rare, established adverse prognostic factor for children and young adults with B-ALL.15,17 This study of 131 patients with B-ALL and fewer than 44 chromosomes, to our knowledge, constitutes the largest series to date of hypodiploid B-ALL treated by a single cooperative group. Patients underwent a three- or four-drug induction on the basis of initial NCI risk group.30 After induction, they were ineligible to continue on upfront clinical trials for B-ALL but had the option of enrolling in the companion VHR trial AALL0031 until study closure.33,34 Consistent with previous reports, we confirm poor outcomes for patients with hypodiploidy. The 5-year EFS was 52.1% and 5-year OS was 58.9%, despite 98.3% of patients achieving CR after induction. Whereas NCI risk group did not significantly predict outcome, NCI SR patients fared better than did NCI HR patients. The only significant prognostic factor was EOI MRD, which underscores the importance of disease response in predicting outcomes. Hypodiploid patients with EOI MRD less than 0.01% had better outcomes, though still inferior compared with nonhypodiploid patients. For those patients with MRD of 0.01% or greater, EFS and OS were dismal, with only one quarter surviving long term. Mullighan et al39 similarly reported on adverse outcomes for hypodiploid patients with positive EOI MRD. For 20 patients with hypodiploid ALL who were treated at St. Jude Children’s Research Hospital, 5-year EFS was 85.1% with EOI MRD less than 0.01% (n = 14) compared with 44.4% with detectable MRD (P = .03).39 Only two patients underwent CR1 HSCT, one of whom had no detectable EOI MRD.39 We recognize that the EFS for EOI MRD-negative patients treated at St. Jude Children’s Research Hospital was higher than our cohort, likely due to the fact that the COG cohort is substantially larger, representing a multi-institution consortium.
Clinicians frequently recommend CR1 HSCT when a poor outcome is anticipated with chemotherapy, and the advantages of this approach for some have been confirmed.40,41 Outcomes have improved with advances in donor selection, supportive care, and with improved management of post-HSCT complications.42,43 In this study, more than one half of patients with hypodiploid ALL underwent CR1 HSCT, providing an opportunity to examine the impact of HSCT on outcome. No subgroup significantly benefitted from HSCT, although trends toward improved outcomes were noted. Likewise, patients with hypodiploid ALL who were treated with intensive chemotherapy (n = 28) were compared with those who underwent HSCT (n = 12) in AALL0031 for VHR ALL.34 There was no significant improvement in disease-free survival, but numbers were small. Finally, the Center for International Blood and Marrow Transplant Research reported leukemia-free survival of 51% and OS of 56% in 78 patients who underwent transplantation for hypodiploid ALL from 1990 to 2010, with a 5-year OS of 38% (95% CI, 24% to 52%) in those harboring 43 or fewer chromosomes.44 Knowing the predictive power of MRD, these poor outcomes could be a result of residual disease that was present, but not detectable, at the time of HSCT. TP53 alterations could be another contributing factor, given the high rate of treatment failure in patients with acute myeloid leukemia and abnormalities of chromosome 17p undergoing HSCT.21,22,45-47 SMNs are also of concern in hypodiploid patients because of the correlation with germline TP53 alterations and low hypodiploidy. Although the difference was not statistically significant, there was a worrisome incidence of SMNs in HSCT patients. DNA damage from conditioning regimens potentially compounds the already increased risk in patients with Li-Fraumeni syndrome, but, of interest, SMNs were not limited to low-hypodiploid patients.
A limitation of our analysis is that the final decision for CR1 HSCT is subjective, made in conjunction with the patient (when appropriate) and family. The role of MRD in guiding treatment decisions has become standard, but when these patients were treated MRD was not routinely assessed at any point after EOI. The technical quality of later MRD assessments is unknown, as is the precise pre-HSCT treatment of these patients. Finally, a higher likelihood of HSCT in younger patients may have favorably influenced outcomes, another potential bias. It is possible that with more patients, subgroups that benefit from HSCT might be identified; however, taken together, our data strongly support alternative approaches for these patients. Chemotherapy for VHR ALL cannot be further intensified; therefore, novel approaches must be considered.48,49 For example, the previously dismal outcomes for patients with Philadelphia chromosome-positive ALL have substantially improved with the incorporation of targeted therapies.33,50-52
As noted above, genomic profiling has revealed biologic drivers of hypodiploid B-ALL. Phosphatidylinositol 3-kinase pathway and Bcl-2 inhibitors reduce the proliferation of hypodiploid cell lines and xenograft cells in vitro, with Bcl-2 inhibition also leading to apoptosis.21,53 However, despite Ras pathway alterations in near-haploid ALL, data are lacking for specific therapeutic benefit from tyrosine kinase inhibitors.21,53 In addition to small-molecule inhibitors, there are exciting immunotherapeutic options for ALL, such as monoclonal antibodies (blinatumomab and inotuzumab), as well as chimeric antigen receptor T-cell therapy. These provide impressive response rates, including cures that were impossible not long ago.54-56 Small-molecule and immunotherapies could potentially be a bridge to HSCT to deepen remissions and improve post-HSCT cure rates. Chimeric antigen receptor T-cell therapy has potential as a definitive therapy. With our ability to identify patients early on who are at high risk of relapse, we must enhance therapy without incurring unnecessary risks from nonbeneficial treatments.
Hypodiploid B-ALL remains an adverse subtype of childhood acute leukemia, but it is not clear that CR1 HSCT is of benefit. Among hypodiploid patients in aggregate, those with residual disease at EOI fare especially poorly. The advent of genomic discovery has brought an understanding of the genetic drivers of hypodiploid ALL, and additional work may reveal targetable signaling pathways. The role of immunotherapy in the treatment of ALL is expanding rapidly and may prove to be beneficial as well. Regardless, this is a group of patients in urgent need of new treatment strategies.
ACKNOWLEDGMENT
The authors thank the Henry Schueler 41 & 9 Foundation for inspiring research on hypodiploid ALL. The authors also acknowledge the late James Nachman, MD (1948-2011), who had a particular interest in hypodiploid ALL; he was a mentor, colleague, and friend, and his dedication to the care of children with leukemia continues to inspire us all.
Appendix
FIG A1.

Disease response of patients with hypodiploid B-lymphoblastic leukemia stratified by (A and B) National Cancer Institute risk group and (C and D) modal chromosome number. BM, bone marrow; EOI, end of induction; HR, high risk; MRD, minimal residual disease; NCI, National Cancer Institute; SR, standard risk.
FIG A2.

Cumulative incidence of relapse for chemotherapy versus transplantation in patients with end-of-induction minimal residual disease < 0.01%. HSCT, hematopoietic stem-cell transplantation.
FIG A3.

Cumulative incidence of second malignant neoplasms (SMNs) for chemotherapy versus hematopoietic stem-cell transplantation (HSCT).
TABLE A1.
NCI Risk Group, Cytogenetics, and DNA Index of Hypodiploid Patients Enrolled in AALL03B1

TABLE A2.
Summary of Conditioning Regimens, Transplantation Types, and Cell Sources for Patients Undergoing First Complete Remission Hematopoietic Stem-Cell Transplantation
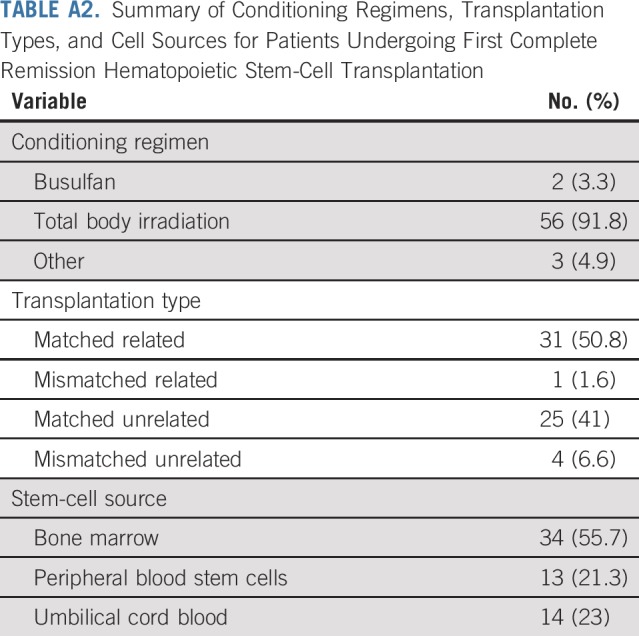
TABLE A3.
Univariable Analyses Among Patients With Hypodiploid B-Lymphoblastic Leukemia for EFS and OS
Footnotes
Presented at the 2018 Annual Meeting of the American Society of Pediatric Hematology and Oncology and at the 2018 Annual Meeting of the International Society of Pediatric Oncology.
Supported by National Institutes of Health Grants No. U10-CA98543, U10-CA98413, U10-CA180886, 1U24-CA196173, and U10-CA180899, and by St. Baldrick’s Foundation. Becton Dickinson Biosciences provided in-kind support. M.L.L. is the University of California, San Francisco Benioff Chair of Children’s Health and Deborah and Arthur Ablin Endowed Chair in Pediatric Molecular Oncology. E.A.R. is a KiDS of New York University (NYU) Foundation Professor at NYU Langone Health. S.P.H. is the Jeffrey E. Perelman Distinguished Chair in the Department of Pediatrics at the Children’s Hospital of Philadelphia.
Clinical trial information: NCT00482352.
See accompanying Editorial on page 763
AUTHOR CONTRIBUTIONS
Conception and design: Jennifer L. McNeer, Meenakshi Devidas, Michael J. Borowitz, Eric Larsen, Kelly W. Maloney, Naomi J. Winick, Kirk R. Schultz, Stephen P. Hunger, William L. Carroll, Mignon L. Loh, Elizabeth A. Raetz
Administrative support: Stephen P. Hunger
Provision of study materials or patients: Michael J. Borowitz, Brent L. Wood
Collection and assembly of data: Meenakshi Devidas, Andrew J. Carroll, Nyla A. Heerema, Julie M. Gastier-Foster, Samir B. Kahwash, Michael J. Borowitz, Brent L. Wood, Leonard Mattano, Stephen P. Hunger, Mignon L. Loh, Elizabeth A. Raetz
Data analysis and interpretation: Jennifer L. McNeer, Meenakshi Devidas, Yunfeng Dai, Julie M. Gastier-Foster, Michael J. Borowitz, Eric Larsen, Kelly W. Maloney, Leonard Mattano, Naomi J. Winick, Kirk R. Schultz, Stephen P. Hunger, William L. Carroll, Mignon L. Loh, Elizabeth A. Raetz
Manuscript writing: All authors
Final approval of manuscript: All authors
Accountable for all aspects of the work: All authors
AUTHORS' DISCLOSURES OF POTENTIAL CONFLICTS OF INTEREST
Hematopoietic Stem-Cell Transplantation Does Not Improve the Poor Outcome of Children With Hypodiploid Acute Lymphoblastic Leukemia: A Report From Children’s Oncology Group
The following represents disclosure information provided by authors of this manuscript. All relationships are considered compensated. Relationships are self-held unless noted. I = Immediate Family Member, Inst = My Institution. Relationships may not relate to the subject matter of this manuscript. For more information about ASCO's conflict of interest policy, please refer to www.asco.org/rwc or ascopubs.org/jco/site/ifc.
Meenakshi Devidas
Honoraria: Pfizer, Novartis, PSI
Travel, Accommodations, Expenses: PSI
Julie M. Gastier-Foster
Research Funding: Bristol-Myers Squibb (Inst), Incyte (Inst)
Michael J. Borowitz
Honoraria: Beckman Coulter
Research Funding: Becton Dickinson, Amgen
Brent L. Wood
Honoraria: Amgen, Seattle Genetics, AbbVie, Janssen Pharmaceuticals
Research Funding: Amgen (Inst), Seattle Genetics (Inst), Pfizer (Inst), Juno Therapeutics (Inst), BiolineRx (Inst), Biosight (Inst), Stemline Therapeutics (Inst)
Travel, Accommodations, Expenses: Amgen
Leonard Mattano
Stock and Other Ownership Interests: Pfizer, Pfizer (I), Amgen, Amgen (I), Monsanto, Monsanto (I)
Consulting or Advisory Role: Pfizer, Mylan, Novartis, Melinta Therapeutics (I), Melinta Therapeutics
Kirk R. Schultz
Honoraria: Shire
Consulting or Advisory Role: MesoScale Discovery, Juno Therapeutics
Travel, Accommodations, Expenses: Jazz Pharmaceuticals
Stephen P. Hunger
Stock and Other Ownership Interests: Amgen, Merck (I), Amgen (I), Pfizer (I)
Honoraria: Amgen
Consulting or Advisory Role: Novartis
William L. Carroll
Other Relationship: Amgen
Elizabeth A. Raetz
Research Funding: Pfizer (Inst)
No other potential conflicts of interest were reported.
REFERENCES
- 1.Hunger SP, Lu X, Devidas M, et al. Improved survival for children and adolescents with acute lymphoblastic leukemia between 1990 and 2005: A report from the Children’s Oncology Group. J Clin Oncol. 2012;30:1663–1669. doi: 10.1200/JCO.2011.37.8018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Pui CH, Yang JJ, Hunger SP, et al. Childhood acute lymphoblastic leukemia: Progress through collaboration. J Clin Oncol. 2015;33:2938–2948. doi: 10.1200/JCO.2014.59.1636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Schultz KR, Pullen DJ, Sather HN, et al. Risk- and response-based classification of childhood B-precursor acute lymphoblastic leukemia: A combined analysis of prognostic markers from the Pediatric Oncology Group (POG) and Children’s Cancer Group (CCG) Blood. 2007;109:926–935. doi: 10.1182/blood-2006-01-024729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Moorman AV, Enshaei A, Schwab C, et al. A novel integrated cytogenetic and genomic classification refines risk stratification in pediatric acute lymphoblastic leukemia. Blood. 2014;124:1434–1444. doi: 10.1182/blood-2014-03-562918. [DOI] [PubMed] [Google Scholar]
- 5.Aricò M, Valsecchi MG, Camitta B, et al. Outcome of treatment in children with Philadelphia chromosome-positive acute lymphoblastic leukemia. N Engl J Med. 2000;342:998–1006. doi: 10.1056/NEJM200004063421402. [DOI] [PubMed] [Google Scholar]
- 6.Aricò M, Schrappe M, Hunger SP, et al. Clinical outcome of children with newly diagnosed Philadelphia chromosome-positive acute lymphoblastic leukemia treated between 1995 and 2005. J Clin Oncol. 2010;28:4755–4761. doi: 10.1200/JCO.2010.30.1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Pui CH, Gaynon PS, Boyett JM, et al. Outcome of treatment in childhood acute lymphoblastic leukaemia with rearrangements of the 11q23 chromosomal region. Lancet. 2002;359:1909–1915. doi: 10.1016/S0140-6736(02)08782-2. [DOI] [PubMed] [Google Scholar]
- 8.Pui CH, Chessells JM, Camitta B, et al. Clinical heterogeneity in childhood acute lymphoblastic leukemia with 11q23 rearrangements. Leukemia. 2003;17:700–706. doi: 10.1038/sj.leu.2402883. [DOI] [PubMed] [Google Scholar]
- 9.Heerema NA, Carroll AJ, Devidas M, et al. Intrachromosomal amplification of chromosome 21 is associated with inferior outcomes in children with acute lymphoblastic leukemia treated in contemporary standard-risk children’s oncology group studies: A report from the Children’s Oncology Group. J Clin Oncol. 2013;31:3397–3402. doi: 10.1200/JCO.2013.49.1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Harrison CJ, Moorman AV, Schwab C, et al. An international study of intrachromosomal amplification of chromosome 21 (iAMP21): Cytogenetic characterization and outcome. Leukemia. 2014;28:1015–1021. doi: 10.1038/leu.2013.317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Moorman AV, Richards SM, Robinson HM, et al. Prognosis of children with acute lymphoblastic leukemia (ALL) and intrachromosomal amplification of chromosome 21 (iAMP21) Blood. 2007;109:2327–2330. doi: 10.1182/blood-2006-08-040436. [DOI] [PubMed] [Google Scholar]
- 12.Den Boer ML, van Slegtenhorst M, De Menezes RX, et al. A subtype of childhood acute lymphoblastic leukaemia with poor treatment outcome: A genome-wide classification study. Lancet Oncol. 2009;10:125–134. doi: 10.1016/S1470-2045(08)70339-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mullighan CG, Su X, Zhang J, et al. Deletion of IKZF1 and prognosis in acute lymphoblastic leukemia. N Engl J Med. 2009;360:470–480. doi: 10.1056/NEJMoa0808253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Heerema NA, Nachman JB, Sather HN, et al. Hypodiploidy with less than 45 chromosomes confers adverse risk in childhood acute lymphoblastic leukemia: A report from the Children’s Cancer Group. Blood. 1999;94:4036–4045. [PubMed] [Google Scholar]
- 15.Raimondi SC, Zhou Y, Mathew S, et al. Reassessment of the prognostic significance of hypodiploidy in pediatric patients with acute lymphoblastic leukemia. Cancer. 2003;98:2715–2722. doi: 10.1002/cncr.11841. [DOI] [PubMed] [Google Scholar]
- 16.Harrison CJ, Moorman AV, Broadfield ZJ, et al. Three distinct subgroups of hypodiploidy in acute lymphoblastic leukaemia. Br J Haematol. 2004;125:552–559. doi: 10.1111/j.1365-2141.2004.04948.x. [DOI] [PubMed] [Google Scholar]
- 17.Nachman JB, Heerema NA, Sather H, et al. Outcome of treatment in children with hypodiploid acute lymphoblastic leukemia. Blood. 2007;110:1112–1115. doi: 10.1182/blood-2006-07-038299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pui CH, Crist WM, Look AT. Biology and clinical significance of cytogenetic abnormalities in childhood acute lymphoblastic leukemia. Blood. 1990;76:1449–1463. [PubMed] [Google Scholar]
- 19.Pui CH, Williams DL, Raimondi SC, et al. Hypodiploidy is associated with a poor prognosis in childhood acute lymphoblastic leukemia. Blood. 1987;70:247–253. [PubMed] [Google Scholar]
- 20.Charrin C, Thomas X, Ffrench M, et al. A report from the LALA-94 and LALA-SA groups on hypodiploidy with 30 to 39 chromosomes and near-triploidy: 2 possible expressions of a sole entity conferring poor prognosis in adult acute lymphoblastic leukemia (ALL) Blood. 2004;104:2444–2451. doi: 10.1182/blood-2003-04-1299. [DOI] [PubMed] [Google Scholar]
- 21.Holmfeldt L, Wei L, Diaz-Flores E, et al. The genomic landscape of hypodiploid acute lymphoblastic leukemia. Nat Genet. 2013;45:242–252. doi: 10.1038/ng.2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mühlbacher V, Zenger M, Schnittger S, et al. Acute lymphoblastic leukemia with low hypodiploid/near triploid karyotype is a specific clinical entity and exhibits a very high TP53 mutation frequency of 93% Genes Chromosomes Cancer. 2014;53:524–536. doi: 10.1002/gcc.22163. [DOI] [PubMed] [Google Scholar]
- 23.Borowitz MJ, Devidas M, Hunger SP, et al. Clinical significance of minimal residual disease in childhood acute lymphoblastic leukemia and its relationship to other prognostic factors: A Children’s Oncology Group study. Blood. 2008;111:5477–5485. doi: 10.1182/blood-2008-01-132837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Borowitz MJ, Wood BL, Devidas M, et al. Prognostic significance of minimal residual disease in high risk B-ALL: A report from Children’s Oncology Group study AALL0232. Blood. 2015;126:964–971. doi: 10.1182/blood-2015-03-633685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Coustan-Smith E, Sancho J, Hancock ML, et al. Clinical importance of minimal residual disease in childhood acute lymphoblastic leukemia. Blood. 2000;96:2691–2696. [PubMed] [Google Scholar]
- 26.Conter V, Bartram CR, Valsecchi MG, et al. Molecular response to treatment redefines all prognostic factors in children and adolescents with B-cell precursor acute lymphoblastic leukemia: Results in 3184 patients of the AIEOP-BFM ALL 2000 study. Blood. 2010;115:3206–3214. doi: 10.1182/blood-2009-10-248146. [DOI] [PubMed] [Google Scholar]
- 27.Campana D, Pui CH. Minimal residual disease-guided therapy in childhood acute lymphoblastic leukemia. Blood. 2017;129:1913–1918. doi: 10.1182/blood-2016-12-725804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Berry DA, Zhou S, Higley H, et al. Association of minimal residual disease with clinical outcome in pediatric and adult acute lymphoblastic leukemia: A meta-analysis. JAMA Oncol. 2017;3:e170580. doi: 10.1001/jamaoncol.2017.0580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Larsen EC, Devidas M, Chen S, et al. Dexamethasone and high-dose methotrexate improve outcome for children and young adults with high-risk B-acute lymphoblastic leukemia: A report from Children’s Oncology Group study AALL0232. J Clin Oncol. 2016;34:2380–2388. doi: 10.1200/JCO.2015.62.4544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Winick N, Devidas M, Chen S, et al. Impact of initial CSF findings on outcome among patients with National Cancer Institute standard- and high-risk B-cell acute lymphoblastic leukemia: A report from the Children’s Oncology Group. J Clin Oncol. 2017;35:2527–2534. doi: 10.1200/JCO.2016.71.4774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Borowitz MJ, Pullen DJ, Shuster JJ, et al. Minimal residual disease detection in childhood precursor-B-cell acute lymphoblastic leukemia: Relation to other risk factors. A Children’s Oncology Group study. Leukemia. 2003;17:1566–1572. doi: 10.1038/sj.leu.2403001. [DOI] [PubMed] [Google Scholar]
- 32.Weir EG, Cowan K, LeBeau P, et al. A limited antibody panel can distinguish B-precursor acute lymphoblastic leukemia from normal B precursors with four color flow cytometry: Implications for residual disease detection. Leukemia. 1999;13:558–567. doi: 10.1038/sj.leu.2401364. [DOI] [PubMed] [Google Scholar]
- 33.Schultz KR, Bowman WP, Aledo A, et al. Improved early event-free survival with imatinib in Philadelphia chromosome-positive acute lymphoblastic leukemia: A Children’s Oncology Group study. J Clin Oncol. 2009;27:5175–5181. doi: 10.1200/JCO.2008.21.2514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Schultz KR, Devidas M, Bowman WP, et al. Philadelphia chromosome-negative very high-risk acute lymphoblastic leukemia in children and adolescents: Results from Children’s Oncology Group study AALL0031. Leukemia. 2014;28:964–967. doi: 10.1038/leu.2014.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Look AT, Roberson PK, Williams DL, et al. Prognostic importance of blast cell DNA content in childhood acute lymphoblastic leukemia. Blood. 1985;65:1079–1086. [PubMed] [Google Scholar]
- 36.Kaplan E, Meier P. Nonparametric estimation from incomplete observations. J Am Stat Assoc. 1958;53:457–481. [Google Scholar]
- 37.Peto R, Pike MC, Armitage P, et al. Design and analysis of randomized clinical trials requiring prolonged observation of each patient. II. Analysis and examples. Br J Cancer. 1977;35:1–39. doi: 10.1038/bjc.1977.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Gray RJ. A class of K-sample tests for comparing the cumulative incidence of a competing risk. Ann Stat. 1988;16:1141–1154. [Google Scholar]
- 39.Mullighan CG, Jeha S, Pei D, et al. Outcome of children with hypodiploid ALL treated with risk-directed therapy based on MRD levels. Blood. 2015;126:2896–2899. doi: 10.1182/blood-2015-09-671131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Balduzzi A, Valsecchi MG, Uderzo C, et al. Chemotherapy versus allogeneic transplantation for very-high-risk childhood acute lymphoblastic leukaemia in first complete remission: Comparison by genetic randomisation in an international prospective study. Lancet. 2005;366:635–642. doi: 10.1016/S0140-6736(05)66998-X. [DOI] [PubMed] [Google Scholar]
- 41.Satwani P, Sather H, Ozkaynak F, et al. Allogeneic bone marrow transplantation in first remission for children with ultra-high-risk features of acute lymphoblastic leukemia: A Children’s Oncology Group study report. Biol Blood Marrow Transplant. 2007;13:218–227. doi: 10.1016/j.bbmt.2006.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mateos MK, O’Brien TA, Oswald C, et al. Transplant-related mortality following allogeneic hematopoeitic stem cell transplantation for pediatric acute lymphoblastic leukemia: 25-Year retrospective review. Pediatr Blood Cancer. 2013;60:1520–1527. doi: 10.1002/pbc.24559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Harvey J, Green A, Cornish J, et al. Improved survival in matched unrelated donor transplant for childhood ALL since the introduction of high-resolution matching at HLA class I and II. Bone Marrow Transplant. 2012;47:1294–1300. doi: 10.1038/bmt.2012.8. [DOI] [PubMed] [Google Scholar]
- 44.Mehta PA, Zhang MJ, Eapen M, et al. Transplantation outcomes for children with hypodiploid acute lymphoblastic leukemia. Biol Blood Marrow Transplant. 2015;21:1273–1277. doi: 10.1016/j.bbmt.2015.04.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Middeke JM, Fang M, Cornelissen JJ, et al. Outcome of patients with abnl(17p) acute myeloid leukemia after allogeneic hematopoietic stem cell transplantation. Blood. 2014;123:2960–2967. doi: 10.1182/blood-2013-12-544957. [DOI] [PubMed] [Google Scholar]
- 46.Mori J, Yanada M, Uchida N, et al. Outcomes of allogeneic hematopoietic cell transplantation in acute myeloid leukemia patients with abnormalities of the short arm of chromosome 17. Biol Blood Marrow Transplant. 2017;23:1398–1404. doi: 10.1016/j.bbmt.2017.04.020. [DOI] [PubMed] [Google Scholar]
- 47.Poiré X, Labopin M, Maertens J, et al. Allogeneic stem cell transplantation in adult patients with acute myeloid leukaemia and 17p abnormalities in first complete remission: A study from the Acute Leukemia Working Party (ALWP) of the European Society for Blood and Marrow Transplantation (EBMT) J Hematol Oncol. 2017;10:20. doi: 10.1186/s13045-017-0393-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Salzer WL, Burke MJ, Devidas M, et al. Toxicity associated with intensive postinduction therapy incorporating clofarabine in the very high-risk stratum of patients with newly diagnosed high-risk B-lymphoblastic leukemia: A report from the Children’s Oncology Group study AALL1131. Cancer. 2018;124:1150–1159. doi: 10.1002/cncr.31099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Burke M, Salzer W, Chen S, et al. Substitution with cyclophosphamide and etoposide does not improve outcome for children and young adults with very high risk B-lymphoblastic leukemia: Children’s Oncology Group study AALL1131. Pediatr Blood Cancer. 2017;64:19. [Google Scholar]
- 50.Schultz KR, Carroll A, Heerema NA, et al. Long-term follow-up of imatinib in pediatric Philadelphia chromosome-positive acute lymphoblastic leukemia: Children’s Oncology Group study AALL0031. Leukemia. 2014;28:1467–1471. doi: 10.1038/leu.2014.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Biondi A, Schrappe M, De Lorenzo P, et al. Imatinib after induction for treatment of children and adolescents with Philadelphia-chromosome-positive acute lymphoblastic leukaemia (EsPhALL): A randomised, open-label, intergroup study. Lancet Oncol. 2012;13:936–945. doi: 10.1016/S1470-2045(12)70377-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Slayton WB, Schultz KR, Jones T, et al. Continuous dose dasatinib is safe and feasible in combination with intensive chemotherapy in pediatric Philadelphia chromosome positive acute lymphoblastic leukemia (Ph+ ALL): Children’s Oncology Group (COG) trial AALL0622. Blood. 2012;120:137. [Google Scholar]
- 53.Diaz-Flores E, Comeaux EQ, Kim K, et al. BCL-2, a therapeutic target for high risk hypodiploid B-cell acute lymphoblastic leukemia. Blood. 2016;128(abstr):280. [Google Scholar]
- 54.Davila ML, Riviere I, Wang X, et al. Efficacy and toxicity management of 19-28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med. 2014;6:224ra25. doi: 10.1126/scitranslmed.3008226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Maude SL, Frey N, Shaw PA, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N Engl J Med. 2014;371:1507–1517. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee DW, Kochenderfer JN, Stetler-Stevenson M, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: A phase 1 dose-escalation trial. Lancet. 2015;385:517–528. doi: 10.1016/S0140-6736(14)61403-3. [DOI] [PMC free article] [PubMed] [Google Scholar]