

Abstract
Growing evidence supports involvement of low-affinity/high-capacity organic cation transporters (OCTs) and plasma membrane monoamine transporter (PMAT) in regulating clearance of monoamines. Currently decynium-22 (D22) is the best pharmacological tool to study these transporters, however it does not readily discriminate among them, underscoring a need to develop compounds with greater selectivity for each of these transporters. We developed seven D22 analogs, and previously reported that some have lower affinity for α1-adrenoceptors than D22 and showed antidepressant-like activity in mice. Here, we extend these findings to determine the affinity of these analogs for OCT2, OCT3 and PMAT, as well as serotonin, norepinephrine and dopamine transporters (SERT, NET and DAT) using a combination of uptake competition with [3H]methyl-4-phenylpyridinium acetate in overexpressed HEK cells and [3H]citalopram, [3H]nisoxetine and [3H]WIN 35428 displacement binding in mouse hippocampal and striatal preparations. Like D22, all analogs showed greater binding affinities for OCT3 than OCT2 and PMAT. However, unlike D22, some analogs also showed modest affinity for SERT and DAT. Dual OCT3/SERT and/or OCT3/DAT actions of certain analogs may help explain their ability to produce antidepressant-like effects in mice and help account for our previous findings that D22 lacks antidepressant-like effects unless SERT function is either genetically or pharmacologically compromised. Though these analogs are not superior than D22 in discriminating among OCTs/PMAT, our findings point to development of compounds with combined ability to inhibit both low-affinity/high-capacity transporters, such as OCT3, and high-affinity/low-capacity transporters, such as SERT, as therapeutics with potentially improved efficacy for treatment of psychiatric disorders.
Keywords: organic cation transporters, plasma membrane monoamine transporter, monoamine uptake, antidepressants, transporter inhibition, decynium-22
1. Introduction
Low-affinity/high-capacity monoamine transporters, including plasma membrane monoamine transporter (PMAT) and three isoforms of organic cation transporters (OCT1–3), contribute to monoamine clearance. Monoamines, like dopamine, serotonin, and norepinephrine, are among the many substrates for this transporter family, widely expressed in brain and periphery (Koepsell, 2013). However, few selective ligands exist to map the spatial distribution and functional capacity of low-affinity/high-capacity transporters in regulation of monoamine clearance.
The quinolone analog decynium-22 (D22) non-selectively inhibits OCTs and PMAT and is currently the best available pharmacological tool to probe their contribution to monoamine uptake. For example, D22 inhibits substrate uptake in rat CNS neurons (Hill, Makky, Shrestha, Hillard, & Gasser, 2011); blocks serotonin uptake in SERT knockout mouse brain synaptosomes (Hagan, Schenk, & Neumaier, 2011); increases hypothalamic extracellular serotonin (5-HT) in rats (Feng et al., 2005; Feng et al., 2010); potentiates the antidepressant-like effect of a selective serotonin reuptake inhibitor (SSRI) in wildtype mice (Horton et al., 2013); and produces antidepressant-like effects in a rat model of depression (Marcinkiewcz & Devine, 2015). Additionally, D22 produces antidepressant-like effects in heterozygous and homozygous SERT knockout mice where OCT3 expression is increased, an effect that is absent in wildtype counterparts (Baganz et al., 2008); and the OCT3 inhibitor corticosterone, potentiates fenfluramine-induced increases in hypothalamic extracellular serotonin levels in rats (Feng et al., 2009). Thus, while the latter point to OCT3 as perhaps the primary low-affinity/high-capacity transporter mediating these effects of D22, selective pharmacological tools are sorely needed to untangle specific contributions of OCTs and PMAT.
To this end, we evaluated novel D22 analogs for selectivity to inhibit substrate transport in OCT2, OCT3, and PMAT heterologous cell expression systems, and in mouse hippocampal and striatal preparations. Chosen analogs were based upon availability of essential chemical precursors (e.g. 6-substituted-2-methyl quinolines). Five analogs contained varying isocyanine ring substituents (halogen and methoxy groups), two were based upon the structurally related cyanine heterocyclic ring system (Table 1). Initial characterization showed that the analogs were significantly less potent inhibitors at alpha-1 adrenergic receptors (compounds 2, 5, 6 & 7), giving these analogs potential for lesser in vivo off-target effects than D22 (Krause-Heuer, A., Fraser-Spears, R. et al., 2017). Moreover, some analogs were less potent inhibitors of spontaneous locomotor activity (compounds 6 & 7), and independently, showed antidepressant-like activity (i.e. reduced time spent immobile) in the mouse tail suspension test (compounds 5 & 6) (Krause-Heuer, A., Fraser-Spears, R. et al., 2017). This independent antidepressant-like effect is unique given our previous studies showed D22 to produce antidepressant-like effects in wildtype mice only when given with a sub-effective dose of a selective serotonin reuptake inhibitor (SSRI) (Horton et al., 2013), or when given to mice with genetically reduced SERT expression (Baganz et al., 2008). However, it remains unclear if D22, or its analogs, have any activity at the high-affinity/low-capacity transporters DAT, NET, and SERT. Likewise, which of the low-affinity/high-capacity transporter(s) mediate the antidepressant-like effects of D22, and those certain analogs, remains unclear.
Table 1.
Name and structures for reference compounds and decynium 22 analogs.
| Compound Name | Chemical Structure |
|---|---|
| Corticosterone | 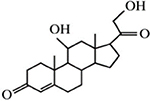 |
| Decynium 22 | 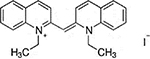 |
| MPP+ | 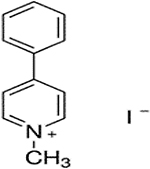 |
| Compound 1 | 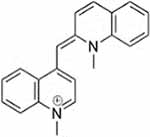 |
| Compound 2 | 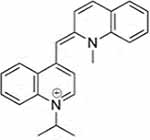 |
| Compound 3 | 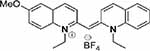 |
| Compound 4 | 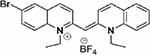 |
| Compound 5 | 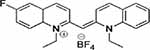 |
| Compound 6 | 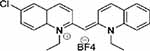 |
| Compound 7 | 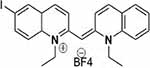 |
Corticosterone, MPP+, and decynium 22 were purchased commercially; D22 analogs (compounds 1–7) were custom synthesized as previously published (Krause-Heuer, A., Fraser-Spears, R. et al., 2017).
We found that these analogs are not superior to D22 in discriminating among OCTs/PMAT. However, importantly, we found that analogs with stand-alone antidepressant-like activity (previously reported (Krause-Heuer, A., Fraser-Spears, R. et al., 2017)) support the development of compounds with combined ability to inhibit both low-affinity/high-capacity transporters, such as OCT3, and high-affinity/low-capacity transporters, such as SERT, as therapeutics with potentially improved efficacy for treatment of psychiatric disorders.
2. Materials and Methods
2.1. Chemicals and supplies
All chemicals were purchased from Sigma Aldrich (St. Louis, MO), unless otherwise noted. Tritium conjugated radioligands: [3H]5-HT, [3H]citalopram, [3H]MPP+, [3H]nisoxetine hydrochloride, and [3H]WIN 35428 were purchased from Perkin Elmer (Waltham, MA) or American Radiolabeled Chemical (St. Louis, MO). Decynium-22 analogs were synthesized as previously published (Krause-Heuer, A., Fraser-Spears, R. et al., 2017) and provided by the Australian Nuclear Science and Technology Organization (ANSTO). Glass microfiber filter mats used in radioligand experiments were supplied by Brandel Inc. (Gaithersburg, MD).
2.2. Animal use and brain tissue preparation
Male, wild type C57BL/6 mice were cared for and used through IACUC approved university protocols in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. Mice of ages 4 – 12 months were rapidly killed via cervical dislocation and decapitation. Whole hippocampi or striata were dissected, on ice, from the left and right hemispheres, and quickly frozen in a 1.5 ml microfuge tube on dry ice. Brain tissue from two mice per experiment were combined and stored at −80°C until further use. To prepare tissue homogenate, dissected brain regions were thawed then homogenized twice for 10 s then kept on ice, using a Polytron homogenizer instrument (PT 3100 Model with generator PT-DA 3005/2EC), at 25,000 rpm (shear rate 4333 s−1). In between homogenizations, the tissue suspension was centrifuged at 25,000 × g for 10 min at 4°C in a Beckman Coulter floor centrifuge (Avanti J-E series, rotor JA 25.5). The final tissue pellet was suspended in ~5 ml of appropriate uptake buffer containing protease inhibitor (Roche Diagnostics complete cocktail tablets, Indianapolis, IN) and homogenized with the Polytron instrument. Tissue homogenate solution was kept on ice until used in radioligand assays. A 50 μl aliquot of tissue homogenate was reserved to determine protein concentration using the BCA protein assay kit (Pierce Thermo Scientific). The average protein concentration of tissue homogenate used in binding assays ranged from 0.6 – 0.8 mg protein/100 μl.
2.3. Cell culture
Human embryonic kidney (HEK) cells that stably express human organic cation transporter 2 or 3 (hOCT2-HEK and hOCT3-HEK) were kindly provided by Dr. Dirk Grundemann (University of Cologne, Germany). The HEK cell line expressing human plasma membrane monoamine transporter (hPMAT-HEK) was generously provided by Dr. Joanne Wang (University of Washington, USA). Cells were maintained at 37°C/5% CO2 in DMEM high glucose media (Gibco™) supplemented with 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin. Selection of expression vectors were maintained by the addition of 150 μg/ml of G418 (hOCT2-& hOCT3-HEK cells) or hygromycin B (hPMAT-cells) to the growth media. Cells were passaged up to 20 times or less and used for experimentation at approximately 80–90 % growth confluency.
2.4. Radioligand assays
2.4.1. [3H]Radioligand binding displacement in mouse hippocampal or striatal preparations
Mouse brain tissue homogenate preparations were used to determine binding of the D22 analogs to DAT with [3H]WIN 35428 displacement in striatum, to NET with [3H]nisoxetine displacement, and SERT with [3H]citalopram binding displacement in hippocampus. Radiolabeled and unlabeled ligands were prepared at 10X concentrations in a total reaction volume of 250 μl. The concentration of radioligand remained constant while increasing the concentration of competing ligands that included parent D22, the D22 analogs, and appropriate high-affinity reference ligands for DAT, NET, or SERT. Both nisoxetine and citalopram binding assays were performed in Tris-HCl buffer with the following composition [final mM], pH 7.4: 50 Tris-HCl; 120 NaCl; 5 KCl; and 10 Tris-base. On the other hand, WIN 35428 binding assays were conducted in a sodium-phosphate buffer composed of [final mM], pH 7.4: 39.86 mM NaH2PO4·H2O (~40 mM) and 24.31 Na2HPO4. Displacement of radioligand was used at halfmaximal concentration or near their reported Kd values and saturated time course. A final 4 nM [3H]WIN 35428 in striatum was measured for 60 min at room temperature (Reith & Selmeci, 1992; Scheffel, Pogun, Stathis, Boja, & Kujar, 1991; Tejani-Butt, 1992). Binding displacement of a final 5 nM [3H]nisoxetine (Tejani-Butt, 1992) or 0.5 nM [3H]citalopram (D’Amato, Largent, Snowman, & Snyder, 1987; Mitchell, Gould, Koek, & Daws, 2016) in hippocampus was, respectively, measured for 120 min on ice or 60 min at room temperature. The 8 final concentrations of unlabeled, competing ligands ranged from 0 – 10 mM. Non-specific binding was determined in the presence of 10 μM GBR12935 (DAT, striatum); 10 μM desipramine (NET, hippocampus); or 10 μM citalopram (SERT, hippocampus) as the final experiment concentrations.
At the end of the incubation period, binding was terminated with four rapid washes with ice-cold binding buffer via vacuum filtration onto a glass fiber, 48-well mat to trap transporter bound radioligand. Each sample filter was transferred to individual scintillation vials with 4 ml of scintillation fluid. Radioactive counts were measured as described for uptake assays.
2.4.2. [3H]MPP+ uptake competition in transient expressing, murine DAT-, NET-, and SERT-HEK cells
Uptake inhibition assays in human embryonic kidney (HEK293) cells expressing murine (m) DAT, mNET or mSERT were performed as previously described (Mayer et al., 2017) with minor modifications. Briefly, HEK293 cells were cultured in a humidified atmosphere (5 % CO2, 37°C) in Dulbecco’s modified Eagle’s medium (DMEM, containing high glucose 4,500 mg per L, sodium bicarbonate and L-Glutamine) supplemented with 10 % FBS and maintained at a sub-confluent state. HEK293 cells were transiently transfected with the desired transporter with Lipofectamine® LTX reagent with PLUS™ reagent (Invitrogen™), strictly following the supplier’s protocol 48h prior to the experiment. On the day preceding the uptake inhibition assay, cells were seeded onto PDL-coated 96-well plates at a density of 40,000 cells per well in a 200 μl volume. On the next day, DMEM was aspirated and replaced with 200 μl Krebs-HEPES buffer (KHB) per well: 120 mM NaCl, 5 mM KCl, 1.2 mM CaCl2, 1.2 mM MgSO4, 25 mM HEPES, 5 mM D-glucose, pH 7.3. At the beginning of the experiment, the cells were incubated with increasing concentrations of the drug of interest (in KHB, 50 μl per well) for 5 min to achieve equilibrium. Subsequently, tritiated substrate was added ([3H]5-HT (28.3 μCi/mmol, Perkin Elmer) for SERT, final concentration 100 nM; [3H]MPP+ (80–85 μCi/mmol, American Radiolabelled Chemicals) for DAT and NET, final concentration 20 nM). Uptake was terminated after 1 min (SERT) or 3 mins (DAT and NET) by aspirating the tritiated substrate and washing each well of cells with 200 μl ice-cold KHB. Finally, the cells were lysed in 100 μl of 1% SDS. The amount of tritium within the cells was determined by use of a beta scintillation counter. Nonspecific uptake was determined in the presence of mazindol, desipramine or paroxetine (10 μM each) for DAT, NET or SERT, respectively, and was subtracted from total uptake data. Uptake in the absence of inhibitors or test drugs was defined as 100% control uptake and all other data points were expressed as percentage thereof. Data were subjected to non-linear regression fits (Y = Bottom + (Top - Bottom) / (1 + 10^((LogIC50 - X)*Hill Slope) to determine the half-maximal inhibitory concentration (IC50).
2.4.3. [3H]MPP+ uptake competition in stable, human OCT2-, OCT3-, and PMAT-HEK cells
Human OCT2, OCT3 and PMAT were used for these studies to provide data that is clinically relevant. [3H]MPP+ uptake assays were performed in attached, whole hOCT2-, hOCT3-, or hPMAT-HEK cells on poly-d-lysine (PDL) coated 24-well plates. Cells were plated at a density of 150,000 cells per well 48-hours prior to the experiment to achieve 80–90% growth confluency. Each well was washed once with Krebs Ringer HEPES (KRH) uptake buffer, then kept in 250 μl of KRH buffer until the assay began. KRH buffer composition (mM), pH 7.4: 125 NaCl; 4.8 KCl; 1.2 K2HPO4; 1.3 CaCl2·2H2O; 1.2 MgSO4·7H2O; 5.6 glucose; 25 HEPES.
To initiate the uptake assay, KRH wash buffer was gently aspirated and replaced with 250 μl of KRH containing the appropriate of 6 competing ligand concentrations ranging from 0 – 10 mM, and 10 nM [3H]MPP+ [specific activity 80–82 Ci/mmol]. Due to low water solubility of D22 and its analogs, stock solutions of all drugs were prepared in 100% DMSO, with final reaction solutions containing 1% of DMSO. The uptake reaction was measured for 10 min at room temperature. Total uptake was measured in triplicate; one representative well for each concentration of competing ligand contained a high concentration (10 or 100 μM) of an inhibitor to determine non-specific uptake. Specific uptake was taken as the subtraction of non-specific from total uptake measurements. To terminate the uptake assay, the radioactive solution was gently aspirated. The cells were quickly washed, three times, with ice-cold phosphate buffered saline, then lysed in 250 μl 1% sodium dodecyl sulfate (SDS) to release transported [3H]MPP+ from inside the cells. Samples were transferred to individual scintillation vials with 4 ml of EcoLume scintillation fluid, capped and vortexed to mix well. The amount of radioactivity in disintegrations per min within each sample was measured for 5-min readings by a TriCarb 2910 counter (Perkin Elmer).
2.5. Statistical analysis
Rests from radioligand assays were analyzed using GraphPad Prism 7 software. Specific uptake or binding was taken as the averaged non-specific measurements subtracted from the total measurements at each concentration. The dependent variable, the specific uptake or binding measured, is reported as a percent of the control (zero concentration, in the absence of competing ligand) as a function of competing ligand concentrations plotted on a log-scale. Percent of control values are plotted as the mean of at least three independent experiments performed in duplicate or triplicate ± S.E.M. Human transporter uptake inhibition and mouse binding displacement data were fitted by non-linear regression analysis to determine the drug concentration to inhibit half the uptake/binding response (IC50). A least-squares fitting method was applied for a one-site fit, logIC50 model using the equation: Y = Bottom + (Top-Bottom)/(1+10^(X-LogIC50)). The best-fit IC50 values were used to compare drug affinities among transporter subtypes. An extra sum-of-squares F test was applied to compare the fit of nested models, the null-hypothesis model of a common IC50 between two compounds/transporters or the alternative hypothesis model of two different IC50 values between comparisons. A p-value ≤ 0.05 of determined whether to reject the null hypothesis that IC50 values between data sets were the same and conclude the alternative hypothesis model as a better fit (i.e. the IC50 values significantly differed). IC50 values are reported with their 95% confidence interval to provide information about the precision of each estimation.
3. Results
3.1. Evaluations of D22 analogs at high-affinity/low-capacity transporters in mouse brain tissue
While literature reports have shown D22 to be a potent inhibitor of the low-affinity/high-capacity transporter family, its action at the high-affinity monoamine transporters, DAT, NET and SERT, remains questionable. It is also unknown if the novel analogs have activity at these transporters. It was therefore crucial to determine if behavioral effects of D22 and its analogs in mice could be attributed to their actions at these high-affinity monoamine transporters. Chemical structures for all compounds tested are provided in Table 1. The D22 analogs include two isocyanine analogs, compounds 1 and 2 (Buettner, 1967), and five pseudoisocyanines, compounds 3–7 (Table 1). While 3 and 7 have been previously made (Russ, H., Engel, & Schömig, 1993), compounds 4, 5, and 6 are completely novel analogs; the full chemical characterization of all seven analogs has been recently published (Krause-Heuer, A., Fraser-Spears, R. et al., 2017). To align with our use of mice in our published in vivo study (Krause-Heuer, A., Fraser-Spears, R. et al., 2017), we assessed the selectivity of D22 and novel analogs to inhibit high-affinity/low-capacity transporters by measuring displacement of DAT, NET, and SERT specific, high-affinity radioligands. An appropriate transporter substrate and inhibitor was also included in the binding assessment. For ease of comparison, the concentration-response curve for a reference substrate and inhibitor, along with the parent D22, are repeated in the subfigure for each D22 analog in Figs. 1–3. A complete summary of IC50 values and statistical comparisons for all compounds tested in DAT, NET and SERT binding is provided in Table 2. Displacement of the highly selective DAT radioligand, [3H]WIN 35428 was assessed in mouse striatum homogenate (Fig. 1). The unlabeled (cold) substrate, dopamine, and the unlabeled cocaine analog, WIN 35428, fully displaced [3H]WIN 35428 binding in striatum. Cold WIN 35428 had the highest binding affinity for DAT with a 3-log, leftward shift in the IC50 value in comparison to cold dopamine and D22 analogs. The parent D22 compound was capable of displacing [3H]WIN 35428 binding in a significantly more potent manner than the dopamine substrate (Fig. 1, Table 2). Nearly all D22 analogs shared similar binding potencies, and their IC50 values were not statistically different from the parent D22 compound. The only pseudoisocyanine to show highaffinity binding displacement at DAT and significantly more so than D22 was compound 4, with an IC50 value trending towards that of cold WIN 35428 (Fig. 1D, Table 2) (P =0.06). The rank order for tested compounds to displace binding at mDAT is as follows, from highest affinity (lowest IC50 values) to lowest affinity (highest IC50 value): WIN 35428 >>> analog 4 >> D22 = analog 1, 2, 3, 6, 7 ≥ dopamine > analog 5.
Fig. 1A-G. DAT specificity of D22 analogs by displacement of [3H]WIN 35428 binding in mouse striatum.

Compounds 1–7 (A-G, black, open circles) were tested for their ability to displace the high-affinity DAT blocker [3H]WIN 35428 in 60 min binding assays performed on ice. For comparison purposes, concentration-response curves obtained for unlabeled reference compounds are repeated in gray for each subfigure and include the DAT substrate dopamine (gray, closed square), inhibitor WIN 35428 (gray, closed circle), and decynium-22 (D22, gray, open square). Non-linear regression analyses allowed for the determination of IC50 values provided in Table 2.
Fig. 3A-G. SERT specificity of D22 analogs by displacement of [3H]citalopram binding in mouse hippocampus.

Compounds 1–7 (A-G, black, open circles) were tested for their ability to displace the high-affinity SERT blocker [3H]citalopram in 60 min binding assays performed at room temperature. Concentration-response curves obtained for unlabeled reference compounds are shown in gray. SERT substrate serotonin (5-HT, gray, closed squares), inhibitors citalopram (gray, closed circle), and decynium-22 (D22, gray, open square). The IC50 determinations for each compound obtained from non-linear regression analyses can be found in Table 2.
Table 2.
Radioligand displacement in tissue homogenate from mouse brain striatum ([3H]WIN 35428 at DAT) or hippocampus ([3H]Nisoxetine at NET & [3H]Citalopram at SERT).
| Compound Name | DAT [3H]WIN 35428 IC50 in μM [95% CI] (n) |
NET [3H]Nisoxetine IC50 in μM [95% CI] (n) |
SERT [3H]Citalopram IC50 in μM [95% CI] (n) |
|---|---|---|---|
|
Substrate: Dopamine (DAT) Dopamine (NET) Serotonin (SERT) |
99.8
aa [28.6 – 347.9] (5) |
12.7
aaa [8.2 – 20.2] (4) |
14.2 [4.1 – 49.2] (4) |
|
Inhibitor: WIN 35428 (DAT) Desipramine (NET) Citalopram (SERT) |
0.06
aaa, bbb [0.0.25 – 0.14] (3) |
0.01
aaa, bbb [0.005 – 0.002] (3) |
0.01
aaa, bbb [0.008 – 0.015] (4) |
| Decynium 22 |
10.8
bb [5.4 – 21.4] (4) |
100.7
bbb [34 – 245] (5) |
26.2 [16 – 42.9] (4) |
| Compound 1 |
20.9
b [11.8 – 36.8] (3) |
61.3
bb [24.1 – 134.1] (4) |
15.6 [7.3 – 33.2] (4) |
| Compound 2 |
15.0
b [6.8 – 33.2] (3) |
23.1 [8.1 – 74.3] (4) |
18.4 [14.6 – 23.3] (4) |
| Compound 3 |
30.9 [5.9 – 162.9] (2) |
18.0 [2.67 – 99.4] (4) |
10.3
aa [7.7 – 13.8] (4) |
| Compound 4 |
0.32
aaa, bbb [0.048 – 2.2] (3) |
11.9
aa [5.52 – 25.6] (4) |
1.04
aaa, bbb [0.75 – 1.46] (4) |
| Compound 5 |
223.8
a [0.21 – 0.24M] (3) |
31.9 [8.05 – 126.6] (5) |
13.9 [8.04 – 23.9] (4) |
| Compound 6 |
10.74
b [6.05 – 19.1] (3) |
55.5
b [10.9 – 282.5] (3) |
0.75
aaa, bbb [0.52 – 1.1] (3) |
| Compound 7 |
12.9
b [4.98 – 33.2] (3) |
222.9
bb [0.43– 0.11M] (3) |
1.14
aaa, bbb [0.84 – 1.5] (3) |
IC50 estimations determined from concentration response curves (Figs. 1–3) fit to a nonlinear regression.
P < 0.05
P < 0.01
P < 0.001 indicates significance of difference for the compound versus decynium 22 (D22) within each radioligand.
P < 0.05
P < 0.01
P < 0.001 denotes significant difference between the compound and reference transporter substrate, either dopamine (DAT and NET) or serotonin (SERT).
Displacement of [3H]nisoxetine binding to NET by D22 analogs was measured in mouse hippocampal homogenate. The selective, high-affinity NET inhibitor desipramine displaced radioligand binding with the greatest affinity as seen by its estimated IC50 value in the low nanomolar range (Fig. 2, Table 2). We found the known NET substrate, dopamine, to have an IC50 value near 13 μM (Table 2). This is consistent with the long-standing establishment of dopamine as a favored substrate for NET when compared to DAT (Giros, Jaber, Jones, Wightman, & Caron, 1996; Gu, Wall, & Rudnick, 1994; Moron, Brockington, Wise, Rocha, & Hope, 2002). The parent D22 compound bound weakly to NET and was significantly less potent than dopamine (P = 0.0006). Some of the analogs, specifically compounds 2–5 were sub-potent inhibitors at NET, as demonstrated by similar binding profiles to dopamine’s at displacing [3H]nisoxetine (Fig. 2B-E, Table 2). On the other hand, compounds 1, 6, and 7 (Figs. 2A, 4F, and 4G) displaced [3H]nisoxetine at NET with a lower affinity that of dopamine. With the exception of compound 4 (P =0.0027), the six other analogs were not statistically different from D22 in their ability to displace [3H]nisoxetine binding to NET (Table 2). In summary, the apparent rank order for displacement binding at the mNET is as follows: desipramine >>> dopamine = analogs 2, 3, 4, 5 > analog 1, 6, 7 = D22.
Fig. 2A-G. NET specificity of D22 analogs by displacement of [3H]Nisoxetine binding in mouse hippocampus.

Compounds 1–7 (A-G, black, open circles) were tested for their ability to displace the high-affinity NET blocker [3H]nisoxetine in 120 min binding assays performed on ice. Concentration-response curves obtained for unlabeled reference compounds are repeated in gray for each subfigure and include NET substrate dopamine (gray, closed squares), inhibitor desipramine (gray, closed circle), and decynium-22 (D22, gray, open square). The IC50 determinations obtained from non-linear regression curve fits can be found in Table 2.
Fig. 4A-C. High-affinity/low-capacity transporter competitive uptake inhibition by cocaine, corticosterone, and decynium-22.

Inhibition of [3H]MPP+ uptake was measured in HEK cells transiently transfected with mouse DAT (A), NET (B) for 3 min, or inhibition of [3H]5-HT uptake measured in SERT-HEK cells (C) for 1 min. Concentrations of unlabeled cocaine (Coc, ●), corticosterone (CORT, ■), and decynium-22 (D22, □) ranged from 0 nM to 1 mM. Concentration-response curves were fit to a non-linear regression analysis and IC50 estimations determined as follows in μM [95% CI]: DAT (A) Coc = 0.18 [0.09–0.32], CORT = 364.6 [154.7–859.4], D22 = 12.9 [9.8–16.9]; NET (B) Coc = 1.2 [0.8–1.8], CORT = 840 [334–2113], D22 = 35.0 [25.0–49.2]; and SERT (C) Coc = 0.51 [0.22–1.2], CORT = 444.7 [128.6–153.8], D22 = 7.9 [6.4–9.7]. Data represents an n of 3 independent experiments performed in duplicate fit to a non-linear regression analysis to determine estimated IC50 values (refer to Table 3).
In a similar fashion, binding of D22 analogs to SERT was evaluated through displacement of the high-affinity ligand [3H]citalopram in mouse hippocampal homogenate preparations. Unlabeled citalopram, used as a reference inhibitor, displaced [3H]citalopram at SERT with the greatest affinity as indicated by its IC50 value of about 10 nM. The native substrate to SERT, serotonin, expectedly displaced [3H] citalopram binding to the transporter with a greater IC50 value of 14 μM (Fig. 3, Table 2). D22 analogs showed variable affinities for SERT being either equipotent or more potent than serotonin or parent D22 to displace binding of [3H]citalopram. D22’s IC50 of 26 μM was similar (P = 0.27) to that of serotonin’s binding displacement at SERT (Fig. 3, Table 2). Several analogs including: compounds 1 (Fig. 3a), 2 (Fig. 3b), 3 (Fig. 3c), and 5 (Fig. 3e) shared a similar affinity for SERT as serotonin, with compounds 1, 2, and 5 also not significantly different from D22. On the other hand, compounds 4 (Fig. 3d), 6 (Fig. 3f), and 7 (Fig. 3g), showed much greater affinity to displace binding at SERT in comparison to serotonin or D22 as demonstrated by their lower IC50 values near 1 μM (Table 2). The following rank order can best summarize the affinities for tested compounds to displace binding at SERT: citalopram >>> analog 4,6, 7 > 5-HT = D22, analog 1, 2, 3, 5.
3.2. Comparison of cocaine, corticosterone, and D22 at murine high-affinity/low-capacity transporters in transient expressing cell lines
Extending our characterization of D22, we turned to HEK cells transiently expressing mouse (m)DAT, mNET and mSERT to evaluate inhibition of [3H]MPP+ uptake by D22 in comparison to cocaine (known DAT, NET and SERT blocker) or corticosterone (OCT3 blocker) (Fig. 4). Cocaine was, expectedly, the most potent of the inhibitors to block [3H]MPP+ uptake in all three high-affinity transporter cell lines. The IC50 for cocaine to inhibit uptake at mDAT, mNET, and mSERT was determined to be 0.18, 1.2, and 0.5 μM, respectively (Figs. 4A-C). On the other hand, the known OCT3 inhibitor, corticosterone, was not a potent blocker of uptake via mDAT, mNET or mSERT, as seen by the rightward shifts in concentration-response analyses (Figs. 4A-C). Corticosterone’s IC50 to inhibit [3H]MPP+ uptake was 364 μM for mDAT, 840 μM for mNET, and 445 μM for mSERT. In the case of all three high-affinity transporters, the uptake inhibition profile with D22 was in between that of cocaine and corticosterone. At mDAT, D22 could inhibit [3H]MPP+ uptake with an IC50 value of 13 μM, 35 μM at mNET, and 8 μM at mSERT (Table 3). These data show modest activity of D22 at high-affinity/low-capacity transporters in the rank order of SERT > DAT > NET.
Table 3.
Radioligand uptake inhibition by cocaine, corticosterone, and decynium-22 at mouse DAT, NET, and SERT transiently expressed in HEK cells.
| Drug Name | mDAT-HEK IC50 in μM [95% CI] (n) |
mNET-HEK IC50 in μM [95% CI] (n) |
mSERT-HEK IC50 in μM [95% CI](n) |
|---|---|---|---|
| Cocaine |
0.18
aaa [0.095–0.32] (6) |
1.18
aaa, bbb [0.8–1.8] (6) |
0.51
aaa [0.22–1.16] (6) |
| Corticosterone |
364.6
aaa [154.7–859.4] (6) |
839.8
aaa [334–2113] (6) |
444.7
aaa [128.6–153.8] (6) |
| Decynium 22ccc |
12.9 [9.8–16.9] (6) |
35.0bbb [25–49.2] (6) |
7.9
bb [6.4–9.7] (6) |
IC50 estimations determined from concentration response curves (Fig. 4) fit to a nonlinear regression.
P < 0.05
P < 0.01
P < 0.001 indicates significance of difference for the drug versus decynium 22 within a cell line.
P < 0.05
P < 0.01
P < 0.001 indicates significance of difference for the compound compared to mDAT
P < 0.001 indicates significant difference between mNET and mSERT.
3.3. Evaluations of D22 analogs at human low-affinity/high-capacity transporter stable expressing cell lines
Finally, to increase translational relevance of our findings in mice to humans, we evaluated the ability of D22 and its analogues to inhibit [3H]MPP+ uptake in HEK cell lines expressing human (h)OCT2, hOCT3 and hPMAT. Concentration-response curves for reference compounds, MPP+, parent D22, and the OCT3 blocker, corticosterone, were tested in each individual transporter expressing HEK cell line (Fig. 5). The IC50 value for MPP+ to inhibit [3H]MPP+ uptake was similar for hOCT2 and hOCT3 (P = 0.77) whereas the potency of MPP+ blockade was significantly reduced and rightward-shifted in hPMAT cells (P = 0.004 vs. OCT2 and P = 0.002 vs. OCT3) (Fig. 5A, Table 4). The potency of D22 inhibition at hPMAT was significantly greater than hOCT2 (P < 0.0001). However, D22 most potently inhibited uptake of [3H]MPP+ at hOCT3 as indicated by the leftward shift in the concentration-response (Fig. 5B) when compared to hOCT2 or hPMAT (P < 0.0001). The final reference compound tested was the known OCT3 inhibitor, corticosterone. As expected, corticosterone potently inhibited [3H]MPP+ uptake in hOCT3-HEK cells with an IC50 of 0.6 μM. However, corticosterone was a poor inhibitor of uptake at both hOCT2 and hPMAT (Fig. 5C). D22 was a more potent blocker of [3H]MPP+ uptake than corticosterone in hOCT3 cells (Table 4, P = 0.02).
Fig. 5A-C. Low-affinity/high-capacity transporter cell line comparison of [3H]MPP+ uptake inhibition with reference compounds. A. MPP+, B. Decynium (D22), and C. Corticosterone (CORT).

Competition of 10 nM [3H]MPP+ uptake inhibition in human PMAT- (⭕, solid line), OCT2- (■, solid line), and OCT3-HEK (Ɥ, dotted line) expressing cell lines. Concentrations of unlabeled competing ligands ranged from 0 nM (control) to 1 or 10 mM for MPP+, or 100 μM for decynium-22, and 1 mM for corticosterone and are plotted on a log-scale. Uptake inhibition assays were performed at room temperature for 10 min. Data are fit to a non-linear regression analysis to determine IC50 estimations (refer to Table 4).
Table 4.
Competition of [3H]MPP+ uptake inhibition in hOCT2-, hOCT3-, and hPMAT-HEK cells.
| Compound Name | hOCT2-HEK IC50 in μM [95% CI] (n) |
hOCT3-HEK IC50 in μM [95% CI] (n) |
hPMAT-HEK IC50 in μM [95% CI] (n) |
|---|---|---|---|
| Corticosterone |
79.6 [0.39 – 0.016M] (3) |
0.62
a [0.28 – 1.37] (4) |
1059
aaa, bbb [0.57 – 1.99M] (4) |
| Decynium 22 ddd |
10.0
bbb [4.1 – 25.1] (4) |
0.2 [0.11 – 0.33] (8) |
1.1
bbb 0.57 – 2.1] (5) |
| MPP+ddd |
110
aa [37.8 – 320.7] (3) |
91
aaa [32.3 – 256.2] (6) |
1035
aaa, bbb [580 – 3144] (5) |
| Compound 1 |
10.02
bb [3.75 – 26.7] (4) |
2.55
aaa, cc [1.63 – 3.99] (4) |
14.78
aaa, bb [4.38 – 49.9] (3) |
| Compound 2 |
6.16 [2.99 – 12.67] (3) |
2.64
aaa, cc [1.14 – 6.11] (3) |
4.70 [0.91 – 24.2] (3) |
| Compound 3 ddd |
11.7
bbb [5.78 – 23.7] (3) |
0.72
aaa [0.45 – 1.15] (3) |
1.85
bbb [1.04 – 3.32] (4) |
| Compound 4 |
7.76
bbb [2.5 – 24.1] (3) |
0.73
aa [0.48 – 1.11] (3) |
4.23
b [0.88 – 20.4] (3) |
| Compound 5 |
1.71
aaa, bb [0.67 – 4.35] (3) |
0.45
aa [0.28 – 0.72] (7) |
2.40
bb [0.64 – 8.97] (3) |
| Compound 6 |
2.53
a, bbb [1.14 – 5.73] (4) |
0.77
aaa [0.57 – 1.03] (6) |
2.11
bbb [1.38 – 3.22] (3) |
| Compound 7 |
11.4
bbb [4.05 – 32.3] (5) |
1.3
aaa [0.85 – 1.98] (6) |
5.97
b [2.07 – 17.2] (4) |
IC50 estimations determined from concentration response curves (Figs. 5 and 6) fit to a nonlinear regression. All values are shown as micromolar unless otherwise noted.
P < 0.05
P < 0.01
P < 0.001 indicates significance of difference, within each cell line, for the compound versus decynium 22.
P < 0.05
P < 0.01
P < 0.001 indicates significant differences for the compound versus hOCT3.
P < 0.01 denotes significance compared to corticosterone
P < 0.001 denotes significance of comparison between hOCT2 and hPMAT.
After establishing inhibition potencies for the three reference compounds, we evaluated the ability for each of the seven D22 analogs to block [3H]MPP+ uptake (Fig. 6). Compound 1, the methyl isocyanine, had a significantly lower IC50 to inhibit [3H]MPP+ uptake at hOCT3 in comparison to both hOCT2 (P = 0.009) and hPMAT (P = 0.002) (Fig. 6A). Inhibition potency of compound 1 did not differ between hOCT2 and hPMAT (P = 0.61). In contrast, the IC50 for the isopropyl isocyanine, compound 2, was the same for all three transporters (Fig. 6B).
Fig. 6A-G. Low-affinity/high-capacity transporter cell line comparison of [3H]MPP+ uptake inhibition by compounds 1–7 (A-G.).

Competition of 10 nM [3H]MPP+ uptake inhibition in human PMAT- (⭕, solid line), OCT2- (■, solid line), and OCT3-HEK (□, dotted line) expressing cell lines. Concentrations of unlabeled competing D22 analogs ranged from 0 nM (control) up to 100 μM and are plotted on a log-scale. Uptake inhibition assays were performed at room temperature for 10 min. Data are fit to a non-linear regression analysis to determine IC50 estimations (see Table 4).
All five pseudoisocyanines were potent uptake inhibitors at hOCT3 compared to either hOCT2 or hPMAT. The IC50 values for [3H]MPP+ uptake inhibition by compounds 4, 5, 6, and 7 were the same between hOCT2 and hPMAT (Fig. 6D-G). In contrast, compound 3, was a more potent uptake inhibitor at hPMAT in comparison to hOCT2, but still significantly less effective than blockade at hOCT3 (Fig. 6C). Apart from compound 2, all other D22 analogs were more potent inhibitors of uptake via hOCT3 versus either hOCT2 or hPMAT, as indicated by their significantly reduced IC50 values (Table 4). Within cell line comparisons between D22 and the analogs revealed significantly reduced potencies with compound 5 (P = 0.045 vs. D22) and compound 6 (P = 0.02 vs. D22), but similar potencies with the remaining compounds, 1–5, to inhibit uptake at hOCT2. Most analogs shared the same potency as D22 to inhibit uptake via hPMAT, except compounds 1 and 7 that were significantly less potent blockers (P = 0.002 and 0.02, respectively). The IC50 values for all seven analogs were significantly greater than that of parent D22. Therefore, none of the compounds were more potent than the parent D22 to inhibit hOCT3-mediated uptake. Despite the analogs being less potent than the parent D22, their binding preference for hOCT3 is demonstrated in their compared potencies to the known OCT3 blocker, corticosterone. Here, we found both isocyanine compounds 1 and 2 were statistically less potent inhibitors of [3H]MPP+ uptake at hOCT3 compared to corticosterone. However, all the pseudoisocyanines, compounds 3 through 7, shared similar potencies as corticosterone, with the lowest IC50 value being compound 5 at 0.4 μM (Table 4).
3.4. Transporter binding preference for parent and D22 analogs
The observed transporter binding preference for parent and D22 analogs is summarized in Fig. 7. Using the IC50 values for each compound to inhibit [3H]MPP+ uptake, we generated a visual representation of the relative potency of corticosterone, parent D22, and the analogs at hOCT3 compared to hOCT2 or hPMAT (Fig. 7A). Similarly, the IC50 values for D22 or each analog to displace binding of radiolabeled inhibitors at mSERT was compared to mDAT or mNET (Fig. 7B). All compounds shared a preference to inhibit hOCT3 mediated [3H]MPP+ uptake versus hOCT2 or hPMAT as indicated by their relative potency ratios falling below 1. In contrast, there was variation in the relative preference to bind mSERT. All the compounds favored binding to mSERT over binding to mNET (ratios less than 1). However, in comparison to mDAT, D22, compound 2, and compound 4 had ratio values above 1, suggesting a preference for mSERT versus mDAT.
Fig. 7A-C.

Relative potency of corticosterone (Cort), parent D22, and D22 analogs (C1-C7). A. shows the IC50 ratio for the comparison of hOCT3 to hOCT2 (black, closed circles) and hPMAT (open circles); and B. shows the IC50 potency ratio for the comparison of mSERT to mDAT (open squares) and mNET (black, closed squares). A. The IC50 values, in μM (Table 4), for the competitive inhibition of [3H]MPP+ uptake by each compound at hOCT3 were divided by that of hOCT2 (●) or hPMAT (◯) to give a unitless ratio. Ratio values < 1 represent greater uptake inhibition at hOCT3, and values > 1 indicate greater potency at hPMAT or hOCT2. A lower range of the y-axis in depicted since all compounds had a ratio value that did not exceed 1. B. The IC50 values, in μM (Table 2), for each compound to displace [3H]citalopram at SERT were divided by the [3H]WIN 35428 displacement at DAT (⬜) and [3H]nisoxetine binding at NET (■) from mouse brain tissue preparations (as described earlier). For the unitless ratio obtained, values < 1 were more potent at mSERT and >1 more potent at mDAT or mNET. An IC50 ratio value of 1 indicates equipotency of the compound for both transporters of the comparison. A wider range of the y-axis in depicted to reflect the compounds with ratio values greater than 1.
4. Discussion
We previously characterized antidepressant-like effects of seven D22 analogs (Krause-Heuer, A., Fraser-Spears, R. et al., 2017). Here, we tested their binding specificity at high-affinity/lowcapacity (SERT, DAT, NET) and low-affinity/high-capacity (OCT2, OCT3, PMAT) transporters to understand the relationship between their affinity and ability to produce antidepressant-like behavioral effects in mice. All analogs bound with low affinity to mSERT, mDAT and mNET, determined by [3H]citalopram, [3H]WIN 35428, or [3H]nisoxetine binding displacement, respectively (Figs. 1–3). D22 and a subset of the analogs (1, 2, 4, 6, 7) showed higher affinity for mDAT than the endogenous neurotransmitter dopamine (Table 2, DAT column). Likewise, a smaller subset of the analogs (4, 6, 7) showed higher affinity for mSERT than the endogenous neurotransmitter 5-HT (Table 2-SERT column). Consistent with D22, all analogs were more potent inhibitors of [3H]MPP+ uptake via hOCT3 than hOCT2 or hPMAT (Fig. 6, Table 4). Together with our previous behavioral characterization of these D22 analogs (Krause-Heuer, A., Fraser-Spears, R. et al., 2017), and published findings (Horton et al., 2013), our results suggest that multimodal inhibition at both low- and high-affinity transporter families may be a strategy for compound development with improved therapeutic efficacy for disorders like depression. Several investigations have elegantly demonstrated the contribution of corticosterone- or D22-sensitive transporters to monoamine clearance (Daws, 2009; Feng et al., 2009; Gasser, Orchinik, Raju, & Lowry, 2009; Hill et al., 2011; Shaskan & Snyder, 1970; Takeda, Inazu, & Matsumiya, 2002; Yoshikawa et al., 2013; Zwart, Verhaagh, Buitelaar, Popp-Snijders, & Barlow, 2001). Mouse models are an essential preclinical resource to study the action of low-affinity transporters. Functional similarities have been observed between the uptake kinetics of substrates like dopamine, MPP+, and serotonin by human and mouse PMAT (Miura et al., 2017; Shirasaka et al., 2016). On the other hand, hOCT3 and mOCT3 were found to share similar affinities for MPP+, but not serotonin (Massmann et al., 2014). Others have found corticosterone to either have enhanced potency for hOCT3 (Gründemann et al., 2002) or equipotent blockade at hOCT3 and mOCT3 (Massmann et al., 2014). Of the few studies that have evaluated species-specific transporter functions, observed differences can depend on the competing ligand, as well as experimental conditions (e.g. temperature, type of radioligand). Our experiments in mouse tissue and high-affinity transporter expressing cell lines help expand functional information of these transporters both in a commonly used model of preclinical relevance, and commonly used cell line.
In experiments using D22 to displace binding of specific high-affinity radioligands for DAT, NET, and SERT in mouse brain tissue, we observed a ranking with D22 displacement efficiency in the order of DAT > SERT > NET (IC50 values being ~ 11 (DAT), 26 (SERT), and 101 (NET) μM; Figs. 1–3, Table 2). Although to a significantly lower extent than the high-affinity blocker cocaine, our data in heterologous, transiently expressed cells lines, show that D22, in contrast to corticosterone, can inhibit [3H]MPP+ uptake in mDAT and mNET, and [3H]5-HT uptake through mSERT, with a similar binding potency rank order of SERT > DAT > NET (IC50 values being 8 (SERT), 13 (DAT), and 35 (NET) μM; Fig. 4, Table 3). However, given that 1) the affinity of psychotherapeutic drugs used clinically to target SERT, NET, and DAT (e.g. SSRIs, selective NET inhibitors) is in the nanomolar range (Frazer, 2001), 2) plasma levels of these drugs in patients are typically in the ng/ml range (Boulton et al., 2010; Kaye et al., 1989), and 3) doses of D22 required to produce behavioral effects in rodents are in the sub-milligram/kg range (Baganz et al., 2008; Horton et al., 2013), it is highly unlikely that behaviorally relevant concentrations of D22 in extracellular fluid would reach the micromolar range required to block SERT, NET or DAT.
In general, most of the D22 analogs were more potent inhibitors of SERT and NET than D22. As depicted by dividing the IC50 values for [3H]citalopram by the values for [3H]nisoxetine binding (Fig. 7), all 7 analogs had a potency ratio below 1 and thus favor binding to mSERT over mNET. Compounds 3 (P = 0.0019), 4, 6, and 7 (P < 0.0001) each have a markedly lower IC50 value to inhibit SERT (mean IC50 range 0.75 – 10 μM) than the parent D22, with the latter three compounds (4, 6, and 7, P < 0.0001) also being statistically more potent at SERT binding than the native substrate, serotonin (Table 2). Compounds 4 (P < 0.001), 6 (P = 0.01), and 7 (P < 0.02) showed a significantly greater affinity to displace binding at DAT when compared to dopamine, as well. Based on having potency ratio values above 1 (Fig. 7), parent D22, along with compounds 2 and 4 prefer binding at mDAT over binding to mSERT. All analogs either shared a similar or had a higher IC50 value (thus, were less potent) than D22 to displace nisoxetine binding at NET.
Earlier work established D22 as a potent inhibitor of low-affinity/high-capacity transporters, including OCT2, OCT3, and PMAT (Gasser et al., 2009; Hayer-Zillgen, Brüss, & Bönisch, 2002; Russ, H., Gliese, Sonna, & Schömig, 1992; Russ, H. et al., 1993; Russ, H., Sonna, Keppler, Baunach, & Schömig, 1993; Russ, Hermann, Staudt, Martel, Gliese, & Schomig, 1996; Schomig, Babin-Ebell, & Russ, 1993; Shirasaka et al., 2016; Sun et al., 2014; Wang, Sun, Li, Tu, & Jiang, 2014; Zhang, Schaner, & Giacomini, 1998). In line with prior D22 action at hOCT3 (Hayer-Zillgen et al., 2002), we found that D22 was a more potent inhibitor of [3H]MPP+ uptake at hOCT3 (IC50 ≈ 0.2 μM) than hOCT2 (IC50 ≈ 10 μM) and extended this finding to hPMAT (IC50 ≈ 1 μM). The glucocorticoid corticosterone also inhibits OCT3-mediated transport and is extremely useful to probe OCT3 function. Our corticosterone competition data (Fig. 5C) are similar to prior studies (Hayer-Zillgen et al., 2002), where corticosterone preferentially bound to hOCT3 (IC50 = 0.6 μM) relative to hOCT2 (IC50 ≈ 80 μM). Corticosterone is also a poor blocker of hPMAT-driven transport (Engel, Zhou, & Wang, 2004; Russ, H. et al., 1993), as supported by its observed IC50 ≈ 1 mM in our experiments, and in the literature (Engel et al., 2004; Miura et al., 2017). The consistency of our results with the literature establishes an important platform on which to perform comparative analyses of D22 analogs in human transporter expressing cell lines.
As seen in concentration-response analyses (Fig. 6) and summarized IC50 data (Table 4), all D22 analogs favored hOCT3 binding in comparison to hPMAT or hOCT2, apart from compound 2 where there was not a significant difference between the IC50 values for hOCT2, hOCT3, or hPMAT. The inhibition potency at hOCT2 and hPMAT was equal for most analogs, except compound 3, which had a significantly higher IC50 value, thus lower potency, to inhibit [3H]MPP+ uptake at hOCT2 (P = 0.0001 vs. hPMAT). Therefore, structural changes among many of the analogs do not appear to impart differences in binding affinities for hOCT2 or hPMAT. Taken together with data for D22 (IC50 = 0.2 μM) and corticosterone (IC50 = 0.6 μM), D22 analogs are potent hOCT3-prefering inhibitors with IC50 values ranging from ~0.4 – 2.6 μM (Table 4). Analogs with IC50 values under or near 1 μM (compounds 3–7) were not statistically different from corticosterone; nor did the lowest of those, being compound 5, have a lower IC50 value than the parent D22 compound. Therefore, even the most potent of D22 analogs was not as potent as the parent compound in blocking hOCT3-mediated [3H]MPP+ uptake.
Our overall goal is to develop selective compounds for low-affinity/high-capacity transporters inhibitors (i.e. OCT1–3 and PMAT), and provide the field with much needed, specific pharmacological tools to study these transporters. In our initial efforts, we synthesized halogen substituted analogs and cyanine analogs of D22 yielding compounds that had greater affinity at hOCT3 rather than hOCT2 and hPMAT, however, none with improved selectivity for OCT3 over D22. Several compounds also had modest affinity for high-affinity/low-capacity monoamine transporters, SERT in particular, which may contribute to their “stand-alone” antidepressant-like effects we previously reported (Krause-Heuer, A., Fraser-Spears, R. et al., 2017). For example, compound 6, which produced the most robust antidepressant-like effect (Krause-Heuer, A., Fraser-Spears, R. et al., 2017), had the greatest affinity for SERT (0.75 μM), combined with affinity for OCT3 equivalent to corticosterone (0.77 μM). This contrasts to D22 with affinity values of 26.2 μM and 0.2 μM at SERT and OCT3, respectively. Thus, a possible reason for a lack of antidepressant-like effect of D22 on its own, in the absence of an SSRI (Horton et al., 2013), is its relatively low affinity for SERT, thereby supporting our contention that combined SERT/OCT3 pharmacotherapeutics may have improved efficacy for treating depression, and related serotonin-linked pathologies.
The concept of targeting multiple transporters to achieve greater concentrations of extracellular monoamines is not new. However, tactics have largely focused on combined blockade of DAT, NET, SERT, monoamine receptors, and/or metabolism inhibitors (Moret, 2005; Sambunaris, Hesselink, Pinder, Panagides, & Stahl, 1997). Current selective reuptake inhibitors for the high-affinity/low-capacity transporters (SERT, NET, DAT) have no appreciable activity at OCT2, OCT3, and PMAT (Emberger et al., 2011; Haenisch & Bönisch, 2010; Matthaeus, Schloss, & Lau, 2015; Wang et al., 2014; Zhou, Engel, & Wang, 2007). Work by us and others support the idea for greater therapeutic potential by blockade of the low-affinity/high-capacity transporter system, in tandem with blockers of high-affinity/low capacity transporters, especially in instances of resistance to high-affinity monoamine transporter therapies (Daws, Koek, & Mitchell, 2013; Hagan et al., 2011; Horton et al., 2013). Our comparative analysis of this small library of D22 analogs identified select compounds that, unlike parent D22, lack activity at alpha-1 adrenoceptors, have independent antidepressant-like effects, and reduced potency to suppress spontaneous locomotion (Krause-Heuer, A., Fraser-Spears, R. et al., 2017). The present study extends these findings to show that most analogs have significant binding preference for hOCT3 versus hOCT2 or hPMAT. Moreover, compounds 4, 6 and 7 show potential as drugs with selective action at the low-affinity transport system (OCT3), but also SERT and/or DAT binding ability.
Unlike D22, select D22 analogs were bimodal inhibitors of high-affinity transporters determined by their binding affinity at SERT and DAT. Since combination inhibitors can often be more efficacious than monotherapies (Moret, 2005), this study supports the notion that the pharmacological method of dual blockade of both the low-affinity/high-capacity and highaffinity/low-capacity transporter families can be of future value. Development of drugs with low- and high-affinity transporter blockade activity in a single compound warrants further investigation towards discovering improved treatments for disorders sub-optimally managed by currently available medications.
Acknowledgements
We gratefully acknowledge the Australian Institute of Nuclear Science and Engineering for the provision of a 2017 postgraduate research award to Jeremy Dobrowolski.
Funding Sources
This research was supported by National Institutes of Health grants awards: R01 MH093320 (LCD and WK); T32 DA031115 and K12 GM111726 (RFS). All authors declare no conflict of interest.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Baganz NL, Horton RE, Calderon AS, Owens WA, Munn JL, Watts LT, Daws LC (2008). Organic cation transporter 3: Keeping the brake on extracellular serotonin in serotonin-transporter-deficient mice. Proceedings of the National Academy of Sciences of the United States of America, 105(48), 18976–18981. doi: 10.1073/pnas.0800466105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulton D, Balch A, Royzman K, Patel C, Berman R, Mallikaarjun S, & Reeves R (2010). The pharmacokinetics of standard antidepressants with aripiprazole as adjunctive therapy: Studies in healthy subjects and in patients with major depressive disorder. Journal of Psychopharmacology, 24(4), 537–546. doi: 10.1177/0269881108096522 [DOI] [PubMed] [Google Scholar]
- Buettner AV (1967). Radiationless transitions in cyanine dyes. The Journal of Chemical Physics, 46(4), 1398–1401. doi: 10.1063/1.1840863 [DOI] [Google Scholar]
- D’Amato RJ, Largent BL, Snowman AM, & Snyder SH (1987). Selective labeling of serotonin uptake sites in rat brain by [3H]citalopram contrasted to labeling of multiple sites by [3H]imipramine. Journal of Pharmacology and Experimental Therapeutics, 242(1), 364 Retrieved from http://jpet.aspetjournals.org/content/242/1/364.abstract [PubMed] [Google Scholar]
- Daws LC (2009). Unfaithful neurotransmitter transporters: Focus on serotonin uptake and implications for antidepressant efficacy. Pharmacology and Therapeutics, 121(1), 89–99. doi: 10.1016/j.pharmthera.2008.10.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daws LC, Koek W, & Mitchell NC (2013). Revisiting serotonin reuptake inhibitors and the therapeutic potential of “uptake-2” in psychiatric disorders. ACS Chemical Neuroscience, 4(1), 16 Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/23336039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emberger M, Koller J, Laimer M, Hell M, Oender K, Trost A, Lechner AM (2011). Interaction of antidepressant and antipsychotic drugswith the human organic cation transporters hOCT1, hOCT2and hOCT3. Journal of the European Academy of Dermatology and Venereology, 25(2), 227–231. doi: 10.1111/j.1468-3083.2010.03766.x [DOI] [PubMed] [Google Scholar]
- Engel K, Zhou M, & Wang J (2004). Identification and characterization of a novel monoamine transporter in the human brain. Journal of Biological Chemistry, 279(48), 50042–50049. doi: 10.1074/jbc.M407913200 [DOI] [PubMed] [Google Scholar]
- Feng N, Lowry CA, Lukkes JL, Orchinik M, Forster GL, & Renner KJ (2010). Organic cation transporter inhibition increases medial hypothalamic serotonin under basal conditions and during mild restraint. Brain Research, 1326, 105–113. doi: 10.1016/j.brainres.2010.02.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng N, Mo B, Johnson PL, Orchinik M, Lowry CA, & Renner KJ (2005). Local inhibition of organic cation transporters increases extracellular serotonin in the medial hypothalamus. Brain Research, 1063(1), 69–76. doi: 10.1016/j.brainres.2005.09.016 [DOI] [PubMed] [Google Scholar]
- Feng N, Telefont M, Kelly KJ, Orchinik M, Forster GL, Renner KJ, & Lowry CA (2009). Local perfusion of corticosterone in the rat medial hypothalamus potentiates d-fenfluramine-induced elevations of extracellular 5-HT concentrations. Hormones and Behavior, 56(1), 149–157. doi: 10.1016/j.yhbeh.2009.03.023 [DOI] [PubMed] [Google Scholar]
- Frazer A (2001). Serotonergic and noradrenergic reuptake inhibitors: Prediction of clinical effects from in vitro potencies. The Journal of Clinical Psychiatry, 62 Suppl 12, 16 Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/11430614 [PubMed] [Google Scholar]
- Gasser PJ, Orchinik M, Raju I, & Lowry CA (2009). Distribution of organic cation transporter 3, a corticosterone-sensitive monoamine transporter, in the rat brain. The Journal of Comparative Neurology, 512(4), 529–555. doi: 10.1002/cne.21921 [DOI] [PubMed] [Google Scholar]
- Giros B, Jaber M, Jones SR, Wightman RM, & Caron MG (1996). Hyperlocomotion and indifference to cocaine and amphetamine in mice lacking the dopamine transporter. Nature, 379(6566), 606–612. doi: 10.1038/379606a0 [doi] [DOI] [PubMed] [Google Scholar]
- Gründemann D, Koschker A, Haag C, Honold C, Zimmermann T, & Schömig E (2002). Activation of the extraneuronal monoamine transporter (EMT) from rat expressed in 293 cells. British Journal of Pharmacology, 137(6), 910–918. doi: 10.1038/sj.bjp.0704926 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu H, Wall SC, & Rudnick G (1994). Stable expression of biogenic amine transporters reveals differences in inhibitor sensitivity, kinetics, and ion dependence. The Journal of Biological Chemistry, 269(10), 7124–7130. [PubMed] [Google Scholar]
- Haenisch B, & Bönisch H (2010). Interaction of the human plasma membrane monoamine transporter (hPMAT) with antidepressants and antipsychotics. Naunyn-Schmiedeberg’s Archives of Pharmacology, 381(1), 33–39. doi: 10.1007/s00210-009-0479-8 [DOI] [PubMed] [Google Scholar]
- Hagan CE, Schenk JO, & Neumaier JF (2011). The contribution of low-affinity transport mechanisms to serotonin clearance in synaptosomes. Synapse, 65(10), 1015–1023. doi: 10.1002/syn.20929 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayer-Zillgen M, Brüss M, & Bönisch H. (2002). Expression and pharmacological profile of the human organic cation transporters hOCT1, hOCT2 and hOCT3. British Journal of Pharmacology, 136(6), 829–836. doi: 10.1038/sj.bjp.0704785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill JE, Makky K, Shrestha L, Hillard CJ, & Gasser PJ (2011). Natural and synthetic corticosteroids inhibit uptake 2-mediated transport in CNS neurons. Physiology & Behavior, 104(2), 306–311. doi: 10.1016/j.physbeh.2010.11.012 [DOI] [PubMed] [Google Scholar]
- Horton RE, Apple DM, Owens WA, Baganz NL, Cano S, Mitchell NC, Daws LC (2013). Decynium-22 enhances SSRI-induced antidepressant-like effects in mice: Uncovering novel targets to treat depression. The Journal of Neuroscience : The Official Journal of the Society for Neuroscience, 33(25), 10534 Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/23785165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaye CM, Haddock RE, Langley PF, Mellows G, Tasker TCG, Zussman BD, & Greb WH (1989). A review of the metabolism and pharmacokinetics of paroxetine in man. Acta Psychiatrica Scandinavica, 80(S350), 60–75. doi: 10.1111/j.16000447.1989.tb07176.x [DOI] [PubMed] [Google Scholar]
- Koepsell H (2013). The SLC22 family with transporters of organic cations, anions and zwitterions. England: Elsevier B.V. doi: 10.1016/j.mam.2012.10.010 [DOI] [PubMed] [Google Scholar]
- Krause-Heuer A, Fraser-Spears R, Dobrowolski J, Ashford ME, Wyatt NA, Roberts MP, Gould GG,.Fraser BH. (2017). Evaluation of the antidepressant therapeutic potential of isocyanine and pseudoisocyanine analogues of the organic cation decynium-22. European Journal of Medicinal Chemistry, 137, 476–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcinkiewcz CA, & Devine DP (2015). Modulation of OCT3 expression by stress, and antidepressant-like activity of decynium-22 in an animal model of depression. Pharmacology, Biochemistry, and Behavior, 131, 33–41. doi: 10.1016/j.pbb.2015.01.004 [DOI] [PubMed] [Google Scholar]
- Massmann V, Edemir B, Schlatter E, Al-Monajjed R, Harrach S, Klassen P, Ciarimboli G (2014). The organic cation transporter 3 (OCT3) as molecular target of psychotropic drugs: Transport characteristics and acute regulation of cloned murine OCT3. Pflügers Archiv - European Journal of Physiology, 466(3), 517–527. doi: 10.1007/s00424-013-1335-8 [DOI] [PubMed] [Google Scholar]
- Matthaeus F, Schloss P, & Lau T (2015). Differential uptake mechanisms of fluorescent substrates into stem-cell-derived serotonergic neurons. ACS Chemical Neuroscience, 6(12), 1906 Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/26503837 [DOI] [PubMed] [Google Scholar]
- Mayer FP, Luf A, Nagy C, Holy M, Schmid R, Freissmuth M, & Sitte HH (2017). Application of a combined approach to identify new psychoactive street drugs and decipher their mechanisms at monoamine transporters. Current Topics in Behavioral Neurosciences, 32, 333–350. doi: 10.1007/7854_2016_63 [doi] [DOI] [PubMed] [Google Scholar]
- Mitchell NC, Gould GG, Koek W, & Daws LC (2016). Ontogeny of SERT expression and antidepressant-like response to escitalopram in wild-type and SERT mutant mice. The Journal of Pharmacology and Experimental Therapeutics, 358(2), 271–281. doi: 10.1124/jpet.116.233338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miura Y, Yoshikawa T, Naganuma F, Nakamura T, Iida T, Kárpáti A, Yanai K (2017). Characterization of murine polyspecific monoamine transporters. FEBS Open Bio, 7(2), 237–248. doi: 10.1002/2211-5463.12183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moret C (2005). Combination/augmentation strategies for improving the treatment of depression. Neuropsychiatric Disease and Treatment, 1(4), 301 Retrieved from http://www.ncbi.nlm.nih.gov/pubmed/18568111 [PMC free article] [PubMed] [Google Scholar]
- Moron JA, Brockington A, Wise RA, Rocha BA, & Hope BT (2002). Dopamine uptake through the norepinephrine transporter in brain regions with low levels of the dopamine transporter: Evidence from knock-out mouse lines. The Journal of Neuroscience: The Official Journal of the Society for Neuroscience, 22(2), 389–395. doi:22/2/389 [pii] [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reith ME, & Selmeci G (1992). Radiolabeling of dopamine uptake sites in mouse striatum: Comparison of binding sites for cocaine, mazindol, and GBR 12935. Naunyn-Schmiedeberg’s Archives of Pharmacology, 345(3), 309–318. doi: 10.1007/BF00168692 [DOI] [PubMed] [Google Scholar]
- Russ H, Engel W, & Schömig E (1993). Isocyanines and pseudoisocyanines as a novel class of potent noradrenaline transport inhibitors: Synthesis, detection, and biological activity. Journal of Medicinal Chemistry, 36(26), 4208–4213. doi: 10.1021/jm00078a010 [DOI] [PubMed] [Google Scholar]
- Russ H, Gliese M, Sonna J, & Schömig E (1992). The extraneuronal transport mechanism for noradrenaline (uptake2) avidly transports 1-methyl-4-phenylpyridinium (MPP+). Naunyn-Schmiedeberg’s Archives of Pharmacology, 346(2), 158–165. doi: 10.1007/BF00165297 [DOI] [PubMed] [Google Scholar]
- Russ H, Sonna J, Keppler K, Baunach S, & Schömig E (1993). Cyanine-related compounds: A novel class of potent inhibitors of extraneuronal noradrenaline transport. Naunyn-Schmiedeberg’s Archives of Pharmacology, 348(5), 458. doi: 10.1007/BF00173203 [DOI] [PubMed] [Google Scholar]
- Russ H, Staudt K, Martel F, Gliese M, & Schomig E (1996). The extraneuronal transporter for monoamine transmitters exists in cells derived from human central nervous system glia [DOI] [PubMed] [Google Scholar]
- Sambunaris A, Hesselink JK, Pinder R, Panagides J, & Stahl SM (1997). Development of new antidepressants. Journal of Clinical Psychiatry, 58, 40–53. [PubMed] [Google Scholar]
- Scheffel U, Pogun S, Stathis M, Boja JW, & Kujar MJ (1991). In vivo labeling of cocaine binding sites on dopamine transporters with [3H]WIN 35,428. Journal of Pharmacology and Experimental Therapeutics, 257(3), 954 Retrieved from http://jpet.aspetjournals.org/content/257/3/954.abstract [PubMed] [Google Scholar]
- Schomig E, Babin-Ebell J, & Russ H (1993). 1,1’-diethyl-2,2’-cyanine (decynium22) potently inhibits the renal transport of organic cations. Naunyn-Schmiedeberg’s Archives of Pharmacology, 347, 379–383. [DOI] [PubMed] [Google Scholar]
- Shaskan EG, & Snyder SH (1970). Kinetics of serotonin accumulation into slices from rat brain: Relationship to catecholamine uptake [PubMed] [Google Scholar]
- Shirasaka Y, Lee N, Duan H, Ho H, Pak J, & Wang J (2016). Interspecies comparison of the functional characteristics of plasma membrane monoamine transporter (PMAT) between human, rat and mouse. Journal of Chemical Neuroanatomy, doi: 10.1016/j.jchemneu.2016.09.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun S, Wang K, Lei H, Li L, Tu M, Zeng S, Jiang H (2014). Inhibition of organic cation transporter 2 and 3 may be involved in the mechanism of the antidepressant-like action of berberine. Progress in Neuro-Psychopharmacology & Biological Psychiatry, 49, 1–6. doi: 10.1016/j.pnpbp.2013.11.005 [DOI] [PubMed] [Google Scholar]
- Takeda H, Inazu M, & Matsumiya T (2002). Astroglial dopamine transport is mediated by norepinephrine transporter. Naunyn-Schmiedeberg’s Archives of Pharmacology, 366(6), 620–623. doi: 10.1007/s00210-002-0640-0 [DOI] [PubMed] [Google Scholar]
- Tejani-Butt SM (1992). [3H]nisoxetine: A radioligand for quantitation of norepinephrine uptake sites by autoradiography or by homogenate binding. Journal of Pharmacology and Experimental Therapeutics, 260(1), 427 Retrieved from http://jpet.aspetjournals.org/content/260/1/427.abstract [PubMed] [Google Scholar]
- Wang K, Sun S, Li L, Tu M, & Jiang H (2014). Involvement of organic cation transporter 2 inhibition in potential mechanisms of antidepressant action. Progress in Neuro-Psychopharmacology & Biological Psychiatry, 53, 90–98. doi: 10.1016/j.pnpbp.2014.03.005 [DOI] [PubMed] [Google Scholar]
- Yoshikawa T, Naganuma F, Iida T, Nakamura T, Harada R, Mohsen AS,.Yanai K (2013). Molecular mechanism of histamine clearance by primary human astrocytes. Glia, 61(6), 905–916. doi: 10.1002/glia.22484 [DOI] [PubMed] [Google Scholar]
- Zhang L, Schaner ME, & Giacomini KM (1998). Functional characterization of an organic cation transporter (hOCT1) in a transiently transfected human cell line (HeLa). Journal of Pharmacology and Experimental Therapeutics, 286(1), 354 Retrieved from http://jpet.aspetjournals.org/content/286/1/354.abstract [PubMed] [Google Scholar]
- Zhou M, Engel K, & Wang J (2007). Evidence for significant contribution of a newly identified monoamine transporter (PMAT) to serotonin uptake in the human brain. Biochemical Pharmacology, 73(1), 147–154. doi: 10.1016/j.bcp.2006.09.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zwart R, Verhaagh S, Buitelaar M, Popp-Snijders C, & Barlow DP (2001). Impaired activity of the extraneuronal monoamine transporter system known as uptake-2 in Orct3/Slc22a3-deficient mice. Molecular and Cellular Biology, 21(13), 4188–4196. doi: 10.1128/MCB.21.13.4188-4196.2001 [DOI] [PMC free article] [PubMed] [Google Scholar]