

Abstract
Dorsal root ganglia, dorsal roots (DR), and dorsal root entry zones (DREZ) are vulnerable to frataxin deficiency in Friedreich ataxia (FA). A previously unrecognized abnormality is the intrusion of astroglial tissue into DR. Segments of formalin-fixed upper lumbar spinal cord of 13 homozygous and 2 compound heterozygous FA patients were sectioned longitudinally to represent DREZ and stained for glial fibrillary acidic protein (GFAP), S100, vimentin, the central nervous system (CNS)-specific myelin protein proteolipid protein, the peripheral nervous system (PNS) myelin proteins PMP-22 and P0, and the Schwann cell proteins laminin, alpha-dystroglycan, and periaxin. Normal DREZ showed short, sharply demarcated, dome-like extensions of CNS tissue into DR. The Schwann cell-related proteins formed tight caps around these domes. In FA, GFAP-, S100-, and vimentin-reactive CNS tissue extended across DREZ and into DR over much longer distances by breaching the CNS-PNS barrier. The transition between PNS and CNS myelin proteins was disorganized. During development, neural-crest derived boundary cap cells provide guidance to dorsal root ganglia axons growing into the dorsal spinal cord and at the same time block the inappropriate intrusion of CNS glia into DR. It is likely that frataxin is required during a critical period of permissive (axons) and nonpermissive (astroglia) border-control.
Keywords: Boundary cap, Dorsal root entry zone, Dorsal root ganglion, Friedreich ataxia, Glia, Schwann cell
INTRODUCTION
Friedreich ataxia (FA), the most common autosomal recessive ataxia, affects central and peripheral nervous systems (CNS, PNS), heart, and the insulin-producing β-cells of the pancreas. The underlying mutation in most cases is a pathogenic homozygous guanine-adenine-adenine (GAA) trinucleotide repeat expansion in intron 1 of the frataxin gene (FXN) (chromosome 9q21.11) that causes frataxin deficiency. Dorsal root ganglia (DRG), dorsal roots (DR), DR entry zones (DREZ), and sensory nerves are highly vulnerable to FA. At the level of the spinal cord, “classic” lesions are subtotal depletion of myelinated fibers in the dorsal columns, more moderate loss of axons in the lateral corticospinal and dorsal spinocerebellar tracts, and the absence of neurons in the dorsal nuclei. These abnormalities have been interpreted as “atrophy” though recent systematic measurements of neuronal sizes in DRG and cross-sectional areas of the thoracic spinal cord were more consistent with hypoplasia than with atrophy (1). Under this hypothesis, frataxin deficiency affects the normal growth of the spinal cord and neural crest-derived DRG neurons. Sural nerves of patients with FA show loss of larger axons but no numerical reduction in total fiber counts per unit cross-sectional area (2). While myelinated fibers in sural nerves of FA patients are sparse (2), the degree of myelination in DR is actually higher than normal (3). The differential involvement of sensory nerves and DR is unexplained, but an obvious anatomical difference is the length of their axons within the PNS. An additional difference is that during development centrally projecting DRG axons must clear the boundary cap (BC). BC cells (BCC) derive from the neural crest and have a 2-fold role: (i) they constitute a second wave of developing DRG neurons and Schwann cell precursors (4); and (ii) they serve as a barrier that permits DRG-derived axons to grow into the developing spinal cord for a limited period of time during prenatal and postnatal development (5–7) while blocking the invasion of the DR by CNS glial cells. Immunohistochemical stains of the DREZ for a PNS-specific myelin protein, PMP-22, and a CNS myelin protein, proteolipid protein (PLP), showed highly irregular transitions in FA (1). In this report, we present evidence that frataxin deficiency causes incomplete demarcation of the DREZ between CNS and PNS. These observations lend further support to the conclusion that the lesions of DRG, spinal cord, and DR in FA are developmental.
MATERIALS AND METHODS
Spinal Cord Specimens
The research described herein was approved by the Institutional Review Board of the Veterans Affairs Medical Center in Albany, New York. Formalin-fixed spinal cord samples came from among 50 archival cases of FA. The Table shows basic genetic and clinical data of 15 patients with FA, including 2 compound heterozygotes, from whom 2-cm-long segments of lumbar spinal cord were available for longitudinal sectioning to represent DREZ. DREZ were defined as showing nerve fiber continuity between DR and spinal cord. Sectioning of the fixed specimens differed from the method described by Obersteiner and Redlich (8) in that the section plane was parallel to the anteroposterior axis of the spinal cord. In addition to longitudinal sections of the DREZ, transverse sections were made of the adjacent spinal cord.
Controls consisted of upper lumbar spinal cord segments of 11 persons (6 females, 5 males) (mean age in years ± standard deviation: 46.7 ± 18.6; range 9–68 years) without nervous system illness.
Immunohistochemistry and Immunofluorescence
The fixed specimens were embedded in paraffin and sectioned serially at 6-μm thickness. After preliminary identification of DREZ by staining with hematoxylin and eosin, adjacent sections were processed for immunohistochemistry and double-label laser scanning confocal microscopy to visualize 2 PNS myelin proteins, PMP-22 and P0; the CNS myelin protein PLP; the Schwann cell-related proteins laminin, α-dystroglycan, and periaxin (l-isoform); the astrocytic proteins glial fibrillary acidic protein (GFAP), vimentin, and S100; and the neurofilament proteins class-III-β-tubulin and phosphorylated neurofilament protein. Sources of commercial antibodies; catalog numbers; host species and clonality; final protein concentrations or, in case of ascites fluid, dilution; and antigen retrieval methods were as follows: anti-PMP-22 (Abcam, Cambridge, MA, ab-31851, rabbit polyclonal, 2 μg protein/mL, incubation in 80% ethanol at 4 °C overnight); anti-P0 (Abcam, ab15508, rabbit polyclonal, 1 μg protein/mL, incubation in 80% ethanol at 4 °C overnight, followed by 0.01 M citric acid-sodium citrate buffer, pH 6, for 20 minutes at 95 °C); anti-PLP (Abcam, ab-105784, rabbit polyclonal, 2 μg protein/mL, incubation in 80% ethanol at 4 °C overnight, followed by 0.01 M citric acid-sodium citrate buffer, pH 6, for 20 minutes at 95 °C); anti-α-dystroglycan (Novus Biologicals, Littleton, CO, NBP1-49634, mouse monoclonal, 0.4 μg protein/mL, 0.01 M citric acid-sodium citrate buffer, pH 6, for 20 minutes at 95 °C); anti-periaxin (Novus, NBP1-89598, rabbit polyclonal, 2 μg protein/mL, 0.01 M citric acid-sodium citrate buffer, pH 6, 20 minutes at 95 °C); anti-laminin (Sigma-Aldrich, St. Louis, MO, L8271, mouse monoclonal, ascites fluid, diluted 1:1000, proteinase K [Sigma, P-6556], 0.1 μg/mL in tris buffer, pH 8, containing 0.01 M NaCl for 30 minutes at 37 °C); anti-GFAP (BioLegend, San Diego, CA, 837201, mouse monoclonal, ascites fluid, diluted 1:500, DIVA [a proprietary decloaking solution sold by Biocare Medical, Pacheco, CA] 1× for 30 minutes at 95 °C); anti-vimentin (Santa Cruz Biotechnology, Santa Cruz, CA, sc-66002, mouse monoclonal, 2 μg protein/mL, 0.01 M citric acid-sodium citrate buffer, pH 6, 20 minutes at 95 °C); anti-nestin (Santa Cruz, sc-23927, mouse monoclonal, 2 μg protein/mL, 0.01 M citric acid-sodium citrate buffer, pH 6, for 20 minutes at 95 °C, or DIVA for 30 minutes at 95 °C, or trypsin [DIFCO Laboratories, Detroit, MI, 0152-13] 1 mg/mL in 0.07 M CaCl2 at pH 7.8 for 30 minutes at 37 °C); anti-S100 (Santa Cruz, sc-53438, mouse monoclonal, 0.8 μg protein/mL, 0.01 M citric acid-sodium citrate buffer, pH 6, 20 minutes at 95 °C); anti-class-III-β-tubulin (R&D Systems, Minneapolis, MN, MAB1195, mouse monoclonal, 2 μg protein/mL, 0.01 M citric acid-sodium citrate buffer, pH 6, 20 minutes at 95 °C); anti-phosphorylated neurofilament protein (Covance [now BioLegend] SMI-31 R, mouse monoclonal, ascites fluid, diluted 1:500, 0.01 M citric acid-sodium citrate buffer, pH 6, 20 minutes at 95 °C).
The pairs for double-label immunofluorescence were PLP/GFAP and periaxin/GFAP. Technical details of immunohistochemistry and immunofluorescence are available from a prior publication from this laboratory (3). The fluorophores attached to secondary anti-rabbit IgG and anti-mouse IgG were Cy3 (red) and Alexa Fluor 488 (green), respectively (Jackson Immunoresearch, West Grove, PA, Cy3-conjugated donkey anti-rabbit IgG, 711-165-152, 0.375 μg protein/mL; Alexa Fluor 488-conjugated donkey anti-mouse IgG, 715-545-150, 0.375 μg protein/mL).
The antigen-antibody reaction of the myelin proteins (PMP-22, P0, PLP), GFAP, and vimentin was validated by sodium dodecylsulfate polyacrylamide gel electrophoresis (SDS-PAGE) and Western blotting. Samples of normal spinal cord and spinal nerve were extracted as described by Condò et al (9) and delipidated (10) prior to routine SDS-PAGE (5%–10% acrylamide) and Western blotting. The apparent molecular weights of the main reactive bands were compared with protein mass information in UniProt (11). On Western blots, α-dystroglycan and periaxin reaction products appeared in bands below the expected location of these proteins (97 kDa for α-dystroglycan and 150 kDa for l-periaxin). We interpreted the atypical migration as evidence of postmortem proteolysis. Periaxin immunoreactivity was greatly reduced or eliminated by preincubation of the antibody solution in an 80-fold excess of periaxin recombinant protein (by weight of protein) (Novus, NBP1-89598PEP). The epitope recognized by monoclonal anti-laminin has not been determined, and Western blots of this protein did not reveal bands in the expected high-molecular weight range (195–393 kDa). The specificity of S100 reaction product in tissue sections was supported by absorption of the antibody with recombinant S100α and full-length S100β-peptide as described previously (12).
For electron microscopy, DR tissue samples were fixed in a mixture of 2% paraformaldehyde and 2% glutaraldehyde in 0.1 M sodium cacodylate buffer (pH 7.2) and, after a period of 6 hours at room temperature, postfixed in 1% osmium tetroxide in the same buffer. Tissue samples were stained en bloc with 2% aqueous uranyl acetate, dehydrated in ethanol and embedded in Poly-Bed 812 resin (Polysciences, Warrington, PA). One μm-thick sections were stained with Toluidine Blue. After suitable trimming, 85-nm-thick sections were viewed in a CM100 Philips electron microscope. Images were collected with a Gatan digital camera (University of Texas Medical Branch).
RESULTS
Oblong Expansions in the DR of FA
Figure 1A shows a transverse section of the DR of an FA patient with early onset and death (FA4, Table). The hematoxylin and eosin stain reveals eosinophilic spherical inclusions with diameters ranging from 20 to 50 μm. A longitudinal DR section from the same case shows that the expansions are oblong and contain multiple nuclei of which some are characteristic of Schwann cell nuclei (Fig. 1B). One nucleus, however, displays a different morphology with a more elliptical shape and less compact karyoplasm (Fig. 1B, arrow). The expansions are strongly reactive with antibodies to GFAP (Fig. 1C), S100 (Fig. 1D) and vimentin (Fig. 1E). Anti-nestin yielded no reaction product despite the application of 3 separate antigen retrieval methods. The expansions are negative for class-III-β-tubulin and phosphorylated neurofilament protein. At the ultrastructural level, the expansions consist of heterogeneous bundles of filaments measuring 12.5–25 nm in thickness (Fig. 1F).
FIGURE 1.

Transverse and longitudinal sections of DR in FA. (A–D) Compound heterozygous FA (FA4, Table, GAA 896/c.11_12TCdel); (E, F) Homozygous FA (FA1, Table, GAA 1016/1016). (A) The transverse section shows multiple spherical eosinophilic expansions within the DR, measuring 20–50 µm in diameter. Some of the expansions contain nuclei. (B) The longitudinal section of this case shows a number of nuclei that are embedded in a fibrillary matrix. Most are consistent with Schwann cell nuclei, but one (arrow) lacks crenation and displays a clearer nucleoplasm, suggesting a non-Schwannian origin. (C–E) The expansions are strongly reactive with anti-GFAP (C), anti-S100 (D), and anti-vimentin (E), respectively. (F) An electron micrograph of an expansion reveals a heterogeneous collection of fibrillary bundles with variable main directions. The transverse diameter of the constituent filaments is 12.5–25 nm. Hematoxylin and eosin (A, B); GFAP (C); S100 (D); vimentin (E); electron micrograph (F). Scale bars: (A–E), 100 µm; (F), 200 nm.
TABLE.
Basic Clinical Data of 15 Patients With Friedreich Ataxia
| Identifier | Sex | Age of Onset | Age of Death | Disease Duration | GAA1 | GAA2 or Other Mutation |
|---|---|---|---|---|---|---|
| FA1 | M | 2 | 10 | 8 | 1016 | 1016 |
| FA2 | M | 3 | 23 | 20 | 1200 | 1200 |
| FA3 | F | 5 | 25 | 20 | 800 | 1100 |
| FA4 (compound heterozygous) | M | 5 | 11 | 6 | 896 | c.11_12TC del |
| FA5 (compound heterozygous) | M | 6 | 28 | 22 | 744 | exon 5 del |
| FA6 | M | 7 | 35 | 28 | 750 | 1000 |
| FA7 | F | 7 | 28 | 21 | 681 | 837 |
| FA8 | M | 8 | 27 | 19 | 700 | 1070 |
| FA9 | M | 9 | 33 | 24 | 925 | 925 |
| FA10 | M | 10 | 39 | 29 | 557 | 662 |
| FA11 | F | 10 | 47 | 37 | 613 | 613 |
| FA12 | F | 15 | 69 | 54 | 568 | 568 |
| FA13 | M | 16 | 45 | 29 | 599 | 599 |
| FA14 | F | 17 | 50 | 33 | 512 | 1122 |
| FA15 | F | 18 | 67 | 49 | 621 | 766 |
| Mean±SD | 9.2±5.1 | 35.8±17.5 | 26.6±13.1 | 745±193 |
GAA, guanine-adenine-adenine trinucleotide repeats; SD, standard deviation.
Myelin Proteins in the DREZ of FA
Stains for the peripheral myelin proteins PMP-22 and P0 show sharp transitions to CNS-derived PLP-ensheathed axons in the normal DREZ (Fig. 2A–C). PMP-22 (Fig. 2A) and P0 (Fig. 2B) form a tight cap of peripheral myelin about a short segment of CNS-derived PLP (Fig. 2C). In FA, the immunohistochemical stains reveal a paucity of PNS myelin and an irregular, poorly defined, transition to CNS myelin (Fig. 2D–F). The ventral root (VR) entry zones in FA (Fig. 2D–F, insets) do not differ from normal (Fig. 2A–C, insets).
FIGURE 2.

DR and DREZ in a normal control and a patient with FA shown by immunohistochemistry of 2 PNS myelin proteins and one CNS myelin protein. (A–C) Normal control; (D–F) Homozygous FA (FA15, Table, GAA 621/766). The insets in the upper left corner of each composite represent VR on the same section. (A, B) In the normal control, the DREZ shows a sharp transition of PMP-22- and P0-reactive PNS tissue to PMP-22- and P0-negative CNS tissue. The dome-like extension of CNS tissue into the DR measures approximately 500 µm, and the PNS extends in a cap-like manner over the dome of CNS tissue ([A], PMP-22; [B], P0). (C) The matching composite of the DREZ stained for PLP reveals a similar sharp transition between the tissue types. (D–F) DR in FA show severe deficiency of PMP-22 and P0, and the transition between PNS and CNS is not sharply demarcated. The junction of PNS and CNS myelin in VR ([D–F], insets) does not differ from normal ([A–C]). PMP-22 (A, D); P0 (B, E); PLP (C, F). Scale bars: 200 µm.
GFAP in DREZ of FA
Figure 3 displays composites of DREZ stained for GFAP in a normal control and 2 patients with FA. In the normal control (Fig. 3A), reaction product extends into the DR over a distance of approximately 400 μm. A thin GFAP-reactive extension reaches as far as 0.8 mm from the spinal cord. In FA, GFAP-reactive oblong expansions extend over much larger distances into the DR: Figure 3B (FA3, Table), 2.8 mm; Figure 3C (FA13, Table), 2.4 mm. Near the DREZ, several expansions show continuity with the CNS portion of the DR (Fig. 3B, arrow).
FIGURE 3.
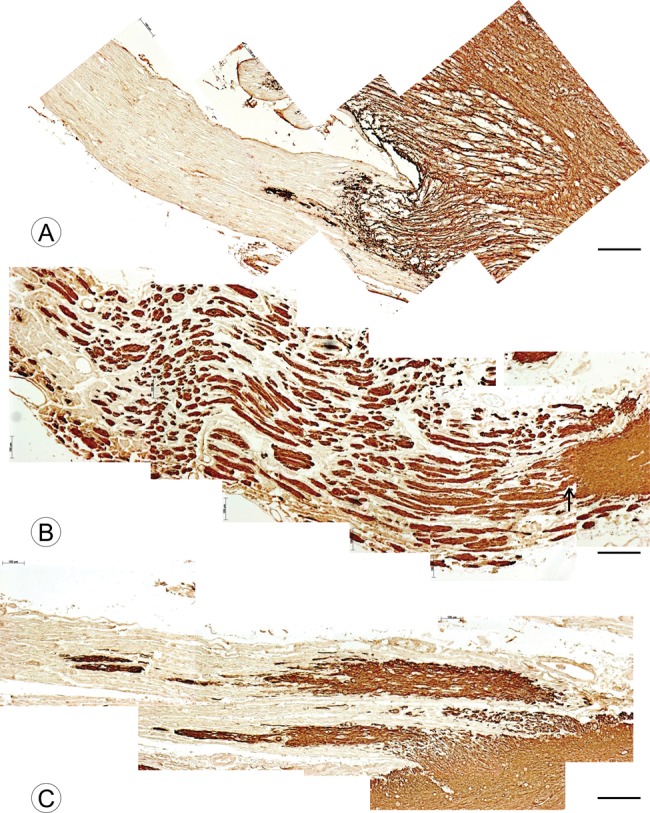
DR and DREZ in a normal control and 2 patients with FA shown by GFAP immunohistochemistry. (A) Normal control; (B) FA (FA2, Table, GAA 1200/1200); (C) FA (FA14, Table, GAA 512/1122). (A) In the normal DREZ, GFAP-reactive CNS tissue enters the DR over an approximate distance of 500 µm. A narrow extension of GFAP reaction product reaches further into the root for another 400 µm. (B) In FA2, numerous segments of GFAP-reactive tissue are present in the DR over a distance of 3 mm. The connection to the CNS portion of the DREZ is apparent for some (arrow). (C) In FA11, broader “fingers” of GFAP-reactive tissues enter the DR and taper to thinner segments at an approximate distance of 2 mm. GFAP (A–C). Scale bars: 200 µm.
Correlative Localization of PLP and GFAP in the DREZ of FA
Figure 4 shows double-label immunofluorescence of PLP and GFAP in a control DREZ (Fig. 4A) and the DREZ of an FA patient (Fig. 4B; FA2, Table). In the normal specimen (Fig. 4A), red PLP immunofluorescence extends into the DREZ over an approximate distance of 330 μm. The accompanying CNS glial tissue (green) ends a few micrometers beyond PLP fluorescence. In FA, the DREZ shows multiple parallel expansions of GFAP-containing tissue, but PLP-reactive myelin sheaths are absent (Fig. 4B).
FIGURE 4.

DREZ in a normal control and a patient with FA shown by double-label immunofluorescence of PLP and GFAP. (A) Normal control; (B) FA (FA2, Table, GAA 1200/1200). (A) In the control case, the DREZ contains myelinated CNS axons (PLP, red) and astrocytes (GFAP, green). Myelinated fibers end in close proximity to the GFAP-reactive tissue. (B) In FA, the DREZ contains oblong segments of GFAP-reactive tissue, but PLP-positive fibers are absent. (A, B) PLP (red) and GFAP (green). Scale bars: 100 µm.
Schwann Cell Proteins in the DREZ of FA
Figure 5A shows the sharp demarcation of Schwann cell-related laminin from the CNS portion of the DREZ. Laminin reaction product in the normal DR is very compact. In FA, laminin surrounds oblong expansions in the DR (Fig. 5B, arrows; FA2, Table), giving the DR a much looser texture. Breach points at the junction between CNS and PNS are shown by asterisks (Fig. 5B).
FIGURE 5.
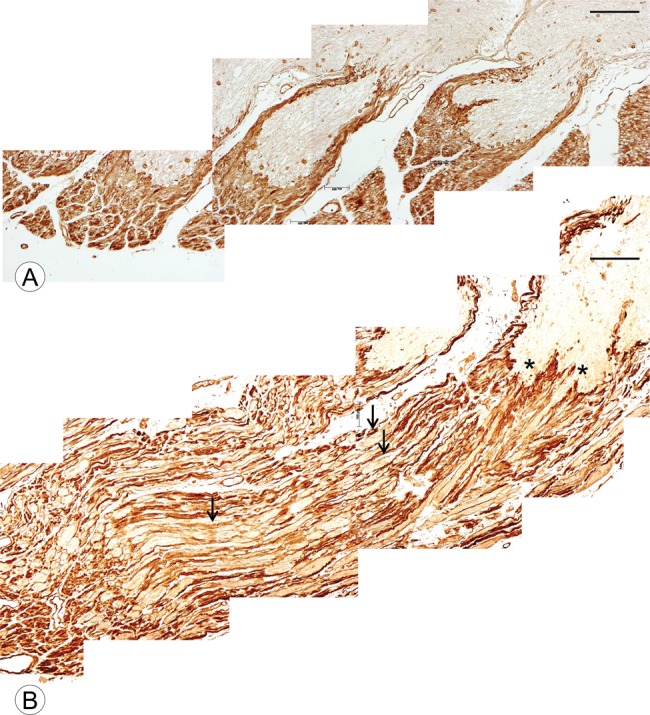
DR and DREZ in a normal control and a patient with FA shown by laminin immunohistochemistry. (A) Normal control; (B) FA (FA2, Table, GAA 1200/1200). (A) In the normal control, compact laminin-positive tissue of the PNS surrounds the DREZ and is sharply demarcated from CNS tissue. (B) In FA2, the laminin-negative CNS nerve tissue protrudes into the DR, making laminin reaction product less compact than normal (arrows). The asterisks show the locations where CNS tissue “breaches” the boundary between CNS and PNS. Laminin (A, B). Scale bars: 200 µm.
Positive-contrast immunohistochemistry of α-dystroglycan in a normal control (Fig. 6A) and an FA case (Fig. 6C; FA2, Table) resembles the reaction product of laminin. Figure 6C shows 2 breach points at the junction of CNS and PNS tissue of the DREZ in FA (arrows). Double-label immunofluorescence images of periaxin (red) and GFAP (green) confirm the relatively sharp transition between myelinating Schwann cells and GFAP in the normal DREZ (Fig. 6B). A few PNS axons penetrate into the GFAP-reactive cone of CNS tissue (Fig. 6B). In contrast, comparable immunofluorescence in FA shows a haphazard admixture of periaxin and GFAP (Fig. 6D).
FIGURE 6.

DREZ in a normal control and a patient with FA shown by α-dystroglycan immunohistochemistry and double-label immunofluorescence of periaxin and GFAP. (A, B) Normal control; FA (C, D) (FA2, Table, GAA 1200/1200). (A) In the normal control, compact α-dystroglycan reaction product surrounds the dome of the CNS portion of the DREZ. (B) Double-label immunofluorescence of periaxin and GFAP shows a relatively sharp transition of myelinating Schwann cells (red) to GFAP-reactive CNS tissue (green) in the DREZ. (C) In FA, the demarcation between α-dystroglycan-positive and negative regions of the DREZ is interrupted by short breaks where CNS tissue penetrates into the DR (arrows). (D) In FA, the normal demarcation between Schwann cells (periaxin) and CNS (GFAP) is indistinct. (A, C) α-dystroglycan; (B, D) double-label immunofluorescence of periaxin (red) and GFAP (green). Scale bars: (A, C) 200 µm; (B, D) 100 µm.
DISCUSSION
Previous Descriptions of DR in FA
Systematic studies of DR in FA are few ([3], 19 cases; [13], 1 case; [14], 1 case; [15], 18 cases). All authors agreed that DR show loss of large myelinated fibers, but it is peculiar that they did not detect the intraradicular expansions reported here. The oblong voids in a longitudinal section of a fifth cervical DR illustrated by Hughes et al ([15], Fig. 3) are similar to the expansions in the DR in our cases of FA. While the balloon- or sausage-shaped structures are readily overlooked on routine stains, an additional explanation is the anatomy of DREZ and DR (Fig. 5A): DREZ in the normal lumbar spinal cord have an approximate rostrocaudal diameter of 250 μm and are separated by 600 μm. Taking into consideration these measurements and a section thickness of 6 μm, the probability of successful representation of DREZ and proximal DR in random transverse sections of the spinal cord is 0.3082 or ∼31% ([250 + 6 + 6]/[600 + 250] = 0.3082).
Nature and Origin of the DR Expansions in FA
GFAP, S100, and vimentin (Fig. 1C–E) immunoreactivity of the expansions and their continuity with the short segment of glial tissue in the proximal DR (Fig. 3B, C) strongly support an origin from the CNS. S100 immunoreactivity in the expansions (Fig. 1D) derives from the CNS though this protein is also a marker of Schwann cells. In DR of FA, S100 expression in Schwann cell cytoplasm is deficient (3), and Figure 1D confirms this original observation. The fibrillary nature of the expansions and the nuclear morphology shown at the light level (Fig. 1B) also favor an astrocytic origin. The electron micrograph in Figure 1F is more difficult to interpret because the constituent filaments are rather thick at 12.5–25 nm. Astroglial filaments are normally formed in partnership of two or more proteins, and the compactness of the bundles in Figure 1F may be the result of predominantly GFAP-containing filaments (16). The ultrastructure resembles the junction of CNS and PNS of a human DREZ illustrated by Schlaepfer et al (17). The negative neurofilament stains exclude an axonal origin. None of the 11 controls revealed similar expansions in their DR.
Disruption of DREZ and the Effect on DR
Stains for laminin (Fig. 5B) and α-dystroglycan (Fig. 6C) directly illustrate the breach of the boundary between CNS and PNS of the DREZ in FA. While some of the expansions are connected to the CNS portion of the DREZ, most of them lie seemingly free in the substance of the DR. They may have found their way into this location by “CNS seeding” during development, having lost their original connection to the spinal cord. Periaxin is an excellent marker of myelinating Schwann cells (18), and double-label immunofluorescence shows interspersion of GFAP-positive expansions and myelinated axons in DREZ of FA (Fig. 6D). It is likely that the admixture of CNS-derived glia and PNS-derived Schwann cells (“peripheral glia”) constitutes an unfavorable environment for the myelination of DR axons. Immunohistochemistry for the PNS-specific myelin proteins PMP-22 (Fig. 2D) and P0 (Fig. 2E) confirms impoverished myelination, and the paucity of PLP reaction product implies impairment of axonal signals also in the CNS portion of the DR (Fig. 2F). The deficit in myelination of DR and DREZ must be reconciled with the normal appearance of VR in FA (Fig. 2D–F). The difference points to an abnormality of axonal signals arising from DRG, adding to the putative untoward effects of CNS seeding in DREZ.
In normal proximal DREZ, GFAP, and PLP are expressed in a comparable territory, which is not the case in the DREZ of FA. We conclude that the GFAP-reactive expansions in DR do not represent mature CNS tissue because they lack PLP (Fig. 4B), and an attempt to visualize Olig2 in DREZ was unsuccessful.
It was of interest to compare DREZ with the root entry zone of the trigeminal nerve in FA. Suitable specimens were available from 6 patients. GFAP staining confirmed the known highly variable extent of CNS tissue in normal sensory trigeminal roots, ranging from 0.1 to 2.5 mm in the medial portion and from 0.17 to 6.75 mm in the lateral portion (19). In only one FA case did the most distal extent of the entry zone of the trigeminal nerve show a continuation of GFAP immunoreactivity in sausage-like expansions. There is a remarkable dearth of information on the Gasserian ganglion in FA. Spiller reported proliferation of “cells of the capsules”, presumably satellite cells, and an “increase of round cells”, presumably inflammatory cells, between neurons of the ganglion (20). He also commented on the degeneration of the sensory trigeminal root.
Mechanism of DREZ Disorganization
The hypothesis is that the extension of CNS tissue beyond the regular DREZ into DR occurs during embryonal and perinatal development. Observations on human autopsy tissues offer limited insight into the possible time frame of the process. Animal experiments, however, provide an analog to the development of human neural tube, neural crest, BCC, DRG, DR, and DREZ (7). The evidence favors that BCC constitute a bidirectional barrier in the DREZ (6, 21). Some of the experimental designs are very elegant and reveal that the open period for axonal growth across the BCC barrier is short (6). Several proteins involved in the barrier have been identified (semaphorin 6 A [21]; Krox20/Egr2 [22]; Sox10 [23]). In a 7-month-old Krox20-deficient patient, Coulpier et al observed passage of CNS tissue across the CNS-PNS boundary at the level of the spinal cord, as shown by GFAP, Olig2, and PLP immunohistochemistry (22). The illustration of this phenomenon, however, pertains to VR and is not directly comparable to the DREZ in FA. In a Krox20-deficient mouse model, however, oligodendroglia and astrocytes grew into DR and generated normal CNS myelin (22). This last observation is at variance with the DR in FA because in this disease, the GFAP-positive expansions occur without CNS myelin. In mouse models of FA, a small percentage of DRG neurons contains vacuoles (24, 25), but the authors did not report comparable expansions in DR.
It is unknown how frataxin deficiency in FA leads to seeding of CNS tissues into DR. This small mitochondrial protein may have a role in growth and integrity of BC, and adequate levels of frataxin may be needed for border control during the critical phase of axonal growth into the dorsal columns and horns of the spinal cord. Jiralerspong et al detected strong frataxin expression in thoracic spinal cord and DRG in mouse embryos at the gestational age of 16.5 days (26). It is relevant that deletion of frataxin is fatal to the murine embryo (27). Incompetence of the BC and related seeding of CNS tissue into DR across DREZ do not fully explain hypoplasia of DRG neurons (1) and progression of sensory neuropathy in FA (2). Additional pathological mechanisms are likely. Lack of neuregulin 1 type III is a potential downstream effect of frataxin deficiency. Neuregulin 1 type III is required for Schwann cell differentiation and PNS myelination (28) though this protein may not be essential for maintenance of myelin in the adult PNS (29).
Clinical Correlation and Possible Impact on Therapy
Regression analysis of age of onset and the shorter GAA trinucleotide repeat expansion in the 13 homozygous FA patients (FA1-FA3 and FA6-FA15, Table) confirms the expected significant negative correlation (using log of age; R2 = 0.71, p < 0.001). Cardiac and nervous system pathology, and frataxin levels, in the 2 compound heterozygous FA patients (FA4 and FA5, Table) were reported previously (30). While the extent of the DREZ breach was not quantifiable on immunohistochemical images, the lesion was most readily detected in patients with very young age of onset (FA1–FA5, Table). The observations do not provide sufficient evidence that DREZ disruption occurs in all FA patients, irrespective of their GAA trinucleotide repeat expansion, or at all levels of the spinal cord, Nevertheless, our findings support the conclusion that modification of DREZ and DR occurs during embryonic and perinatal life. It is unlikely that frataxin replacement after clinical onset of FA will reverse the intrusion of CNS tissues into DR or repair hypoplastic neural tissues (1). Inflammation of DRG in adults with FA (12), however, may be a signal that tissue damage continues, perhaps correlating with the progression of sensory neuropathy (2).
ACKNOWLEDGMENTS
The authors are grateful to the families who allowed autopsies to advance research in Friedreich ataxia. Paul Feustel, PhD, Department of Neuroscience and Experimental Therapeutics, Albany Medical College, Albany, NY, USA, provided the probability calculations.
REFERENCES
- 1. Koeppen AH, Becker AB, Qian J, et al. Friedreich ataxia: hypoplasia of spinal cord and dorsal root ganglia. J Neuropathol Exp Neurol 2017; 76:101–8 [DOI] [PubMed] [Google Scholar]
- 2. Morral JA, Davis AN, Qian J, et al. Pathology and pathogenesis of sensory neuropathy in Friedreich's ataxia. Acta Neuropathol 2010; 120:97–108 [DOI] [PubMed] [Google Scholar]
- 3. Koeppen AH, Morral JA, Davis AN, et al. The dorsal root ganglion in Friedreich's ataxia. Acta Neuropathol 2009; 118:763–76 [DOI] [PubMed] [Google Scholar]
- 4. Maro GS, Vermeren M, Voiculescu O, et al. Neural crest boundary cap cells constitute a source of neuronal and glial cells of the PNS. Nat Neurosci 2004; 7:930–8 [DOI] [PubMed] [Google Scholar]
- 5. Golding JP, Cohen J.. Border controls at the mammalian spinal cord: late-surviving neural crest boundary cap cells at dorsal root entry sites may regulate sensory afferent ingrowth and entry zone morphogenesis. Mol Cell Neurosci 1997; 9:381–96 [DOI] [PubMed] [Google Scholar]
- 6. Golding J, Shewan D, Cohen J.. Maturation of the mammalian dorsal root entry zone-from entry to no entry. Trends Neurosci 1997; 20:303–8 [DOI] [PubMed] [Google Scholar]
- 7. Altman J, Bayer SA, Development of the Human Spinal Cord. An Interpretation Based on Experimental Studies in Animals. Oxford: Oxford University Press; 2001 [Google Scholar]
- 8. Obersteiner H, Redlich E.. Ueber Wesen und Pathogenese der tabischen Hinterstrangsdegeneration. Arbeit Neurol Inst Wien Univ 1894; 1–3:158–72 [Google Scholar]
- 9. Condò I, Ventura N, Malisan F, et al. A pool of extramitochondrial frataxin that promotes cell survival. J Biol Chem 2006; 281:16750–6 [DOI] [PubMed] [Google Scholar]
- 10. Wessel D, Flügge UIA.. method for the quantitative recovery of protein in dilute solution in the presence of detergents and lipids. Anal Biochem 1984; 138:141–3 [DOI] [PubMed] [Google Scholar]
- 11. UniProt: the universal protein knowledgebase. Nucl Acids Res 2017; 45:D158–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Koeppen AH, Ramirez RL, Becker AB, et ak. Dorsal root ganglia in Friedreich ataxia: satellite cell proliferation and inflammation. Acta Neuropathol Commun 2016; 4:46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Mott FW. Case of Friedreich's disease, with autopsy and systematic microscopical examination of the nervous system. Arch Neurol Psychiat 1907; 3:180–200 [Google Scholar]
- 14. Lambrior AA. Un cas de maladie der Friedreich avec autopsie. Rev Neurol 1911; 21:525–40 [Google Scholar]
- 15. Hughes JT, Brownell B, Hewer RL.. The peripheral sensory pathway in Friedreich's ataxia. Brain 1968; 91:803–18 [DOI] [PubMed] [Google Scholar]
- 16. Eliasson C, Sahlgren C, Berthold C-H, et al. Intermediate filament protein partnership in astrocytes. J Biol Chem 1999; 274:23996–4006 [DOI] [PubMed] [Google Scholar]
- 17. Schlaepfer WW, Freeman LA, Eng LF.. Studies of human and bovine spinal nerve roots and the outgrowth of CNS tissues into the nerve root entry zone. Brain Res 1979; 177:219–29 [DOI] [PubMed] [Google Scholar]
- 18. Scherer SS, Xu Y-t, Bannerman PGC, et al. Periaxin expression in myelinating Schwann cells: modulation by axon–glial interactions and polarized localization during development. Development 1995; 121:4265–73 [DOI] [PubMed] [Google Scholar]
- 19. Peker S, Kurtkaya Ö, Üzün I, Pamir MN.. Microanatomy of the central myelin-peripheral myelin transition zone of the trigeminal nerve. Neurosurgery 2006; 59:354–9 [DOI] [PubMed] [Google Scholar]
- 20. Spiller WG. Friedreich's ataxia. J Nerv Ment Dis 1910; 37:411–35 [Google Scholar]
- 21. Mauti O, Domanitskaya E, Andermatt I, et al. Semaphorin6A acts as a gate keeper between the central and the peripheral nervous system. Neural Dev 2007; 2:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Coulpier F, Decker L, Funalot B, et al. CNS/PNS boundary transgression by central glia in the absence of Schwann cells or Krox20/Egr2 function. J Neurosci 2010; 30:5958–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Fröb F, Bremer M, Finzsch M, et al. Establishment of myelinating Schwann cells and barrier integrity between central and peripheral nervous systems depend on Sox10. Glia 2012;60:806–19. [DOI] [PubMed] [Google Scholar]
- 24. Simon D, Seznec H, Gansmuller A, et al. Friedreich ataxia mouse models with progressive cerebellar and sensory ataxia reveal autophagic neurodegeneration in dorsal root ganglia. J Neurosci 2004; 24:1987–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Al-Mahdawi S, Pinto RM, Varshney D, et al. GAA repeat expansion mutation mouse models of Friedreich ataxia exhibit oxidative stress leading to progressive neuronal and cardiac pathology. Genomics 2006; 88:580–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Jiralerspong S, Liu Y, Montermini L, et al. Frataxin shows developmentally regulated tissue-specific expression in the mouse embryo. Neurobiol Dis 1997; 4:103–13 [DOI] [PubMed] [Google Scholar]
- 27. Cossée M, Puccio H, Gansmuller A, et al. Inactivation of the Friedreich ataxia mouse gene leads to early embryonic lethality without iron accumulation. Hum Mol Genet 2000; 9:1219–26 [DOI] [PubMed] [Google Scholar]
- 28. Michailov BG, Sereda MW, Brinkmann BG, et al. Axonal neuregulin-1 regulates myelin-sheath thickness. Science 2004; 304:700–3 [DOI] [PubMed] [Google Scholar]
- 29. Fricker FR, Zhu N, Tsantoulas C, et al. Sensory axon-derived neuregulin-1 is required for axoglial signaling and normal sensory function but not for long-term axon maintenance. J Neurosci 2009; 29:7667–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Becker AB, Qian J, Gelman BB, et al. Heart and nervous system pathology in compound heterozygous Friedreich ataxia. J Neuropathol Exp Neurol 2017; 76:665–75 [DOI] [PMC free article] [PubMed] [Google Scholar]