

Abstract
Atrial fibrillation (AF) is the most common cardiac arrhythmia. Here, we show the identification and functional characterization of one AF-associated mutation p.Arg399Cys in lamin A/C. Co-immunoprecipitation and GST pull-down assays demonstrate that lamin A/C interacts with NUP155, which is a nucleoporin and causes AF when mutated. Lamin A/C mutation p.Arg399Cys impairs the interaction between lamin A/C and NUP155, and increases extractability of NUP155 from the nuclear envelope (NE). Mutation p.Arg399Cys leads to aggregation of lamin A/C in the nucleus, although it does not impair the integrity of NE upon cellular stress. Mutation p.Arg399Cys inhibits the export of HSP70 mRNA and the nuclear import of HSP70 protein. Electrophysiological studies show that mutation p.Arg399Cys decreases the peak cardiac sodium current by decreasing the cell surface expression level of cardiac sodium channel Nav1.5, but does not affect IKr potassium current. In conclusion, our results indicate that lamin A/C mutation p.Arg399Cys weakens the interaction between nuclear lamina (lamin A/C) and the nuclear pore complex (NUP155), leading to the development of AF. The findings provide a novel molecular mechanism for the pathogenesis of AF.
Keywords: atrial fibrillation (AF), LMNA, mutation, NUP155, SCN5A/Nav1.5
1 |. INTRODUCTION
Atrial fibrillation (AF) is the most common sustained cardiac arrhythmia at the clinical setting, and associated with significant mobility and mortality (Fuster et al., 2001). An estimated 2.2 million people in the United States have AF and 160,000 new cases are diagnosed each year (Kannel, Wolf, Benjamin, & Levy, 1998; P. A. Wolf et al., 1996). Common AF is a complex disease, but AF can also occur as isolated AF when it develops in the absence of known risk factors such as structural heart disease and congestive heart failure (Benjamin et al., 1994). Beyond the traditionally recognized sporadic, non-genetic disorder, a series of studies have shown the contribution of genetic factors to the familiar and sporadic forms of AF. To date, about 30 AF-associated genes have been identified (Tucker & Ellinor, 2014). Mutations associated with AF were initially identified and functionally validated in genes encoding cardiac potassium or sodium ion channels, including KCNQ1, KCNJ2, KCNA5, KCNH2, SCN5A, and SCN3B. (Remme, 2013; Tsai, Lai, Hwang, Lin, & Chiang, 2008). However, we and others found that mutations in non-ion-channel genes such as NUP155 and NPPA also cause AF (Hodgson-Zingman et al., 2008; Ren et al., 2010; Zhang et al., 2008), although the non-ion-channel genes and the related molecular mechanisms involved in AF remain to be further explored.
The nuclear envelope (NE) is a bilayer membrane structure enclosing the nucleus. The NE is composed of inner and outer nuclear membranes, the nuclear lamina, and the nuclear pore complexes (NPCs). Nuclear lamins are the major components of the nuclear lamina, and shared a common primary sequence consisting of a central α-helical rod domain flanked by globular N- and C-terminal domain (Gruenbaum, Margalit, Goldman, Shumaker, & Wilson, 2005; Hutchison, 2002). The basic lamin unit is a two-stranded parallel coiled-coil dimer, and the head to tail association of these dimer units ultimately yields a higher-order filamentous meshwork and plays a fundamental role in the nuclear physical architecture, chromatin organization, and gene expression (Gruenbaum et al., 2005; Hutchison, 2002).
In mammals, lamins are classified into A- and B-types. The A-type lamins are encoded by the LMNA gene. Lamins A and C are the major alternative spliced products in most differentiated cells. The B-type lamins are encoded by two separate genes LMNB1 and LMNB2 (Shumaker, 2003). Since the first disease-causing LMNA mutation was identified in 1999, more than 400 LMNA mutations have been identified to date, and most of these LMNA mutations are scattered throughout the entire coding region of the LMNA gene. LMNA-related human hereditary diseases encompass a wide range of phenotypes, including muscular dystrophies, cardiomyopathies, peripheral neuropathies, lipodystrophies, and premature aging syndromes. This heterogeneous group of inherited disorders is now collectively known as “laminopathies” (Capell & Collins, 2006; Schreiber & Kennedy, 2013). It remains unclear how different mutations in the LMNA gene cause different types of laminopathies. To date, two main hypotheses including the “structure” model hypothesis and the “gene expression” model hypothesis have been proposed to explain the etiology of the diseases, but the detailed pathogenic mechanisms of laminopathies are still poorly understood (Capell & Collins, 2006).
Human genetic studies have revealed that patients carrying LMNA mutations face a significant burden of atrial arrhythmias, especially in patients with muscular dystrophy (MD) or dilated cardiomyopathy (DCM) (Fatkin et al., 1999; Sanna, 2003; van Tintelen et al., 2007). In one meta-analysis of 299 LMNA mutation positive patients suffering from isolated DCM or MD-associated DCM, the incidence of AF accounted for 16% of total patients (van Berlo et al., 2005). In a cohort of 269 patients with LMNA mutations, van Rijsingen et al. found that the frequency of atrial tachyarrhythmias (AT) was 43% and 45% in men and women, respectively (van Rijsingen et al., 2013). Kumar et al. investigated the long-term dysrhythmia outcome of a cohort of 122 LMNA mutation carriers, and found that the prevalence of atrial arrhythmias (AA) increased with the increasing age, and the incidence of AA increased from 39% (presentation) to 63% (7-years follow-up; Kumar et al., 2016). Interestingly, in some cases, mutations in LMNA manifested as familiar or sporadic forms of AF (Beckmann et al., 2010; Madej-Pilarczyk et al., 2008; Marsman et al., 2011; Pan et al., 2009; Saj et al., 2012; Sebillon et al., 2003; Zhao et al., 2016). In addition, heterozygous LMNA-knockout (LMNA+/−) mice developed AF after rapid atrial pacing (C. M. Wolf et al., 2008). But to date, the molecular mechanism of LMNA mutations linked to AF remains unknown.
Considering the fact that LMNA mutation carriers have a high risk of AF, here, we examined the possibility that LMNA mutations are associated with AF. By sequencing the LMNA gene in 610 patients with lone AF, we identified one point mutation p.Arg399Cys in one sporadic case. We also analyzed the functional consequences of this identified mutation. Our results identified multiple functional effects of mutation p.Arg399Cys, which may underlie the pathogenesis of AF. Most importantly, during the process in identifying the molecular mechanism by which LMNA mutations cause AF, we found that lamin A/C interacted with NUP155, and the weakening of the interaction by mutation p.Arg399Cys was an important cause for the pathogenesis of AF.
2 |. MATERIALS AND METHODS
2.1 |. Study subjects and mutational screening of the LMNA gene
We studied >600 unrelated patients with lone AF by direct DNA sequence analysis as described previously (S. Chen et al., 2015; Huang et al., 2015; Ren et al., 2010; Shi et al., 2009; Wang et al., 2016; Zhang et al., 2008). Lone AF was defined as AF occurring in individuals <65 years of age without significant structural heart disease, no significant valve disease, and no known significant CAD. The diagnosis was established on the basis of clinical history, physical examinations, ECG, and echocardiographic data. The study was approved by the Ethics Committee of Huazhong University of Science and Technology and the Cleveland Clinic Institutional Review Board on Human Subject Research. The study adhered to the guidelines of the Declaration of Helsinki. Informed written consent was obtained from each participant.
Human genomic DNA was extracted from peripheral blood samples by the DNA Isolation Kit (Roche Diagnostic Co.). The 12 coding exons of LMNA were amplified by polymerase chain reactions (PCR) from patient DNA samples. Mutation screening was carried out using direct DNA sequence analysis performed using the BigDye Terminator Cycle Sequencing v1.1 kit and on an ABI PRISM 3100 Genetic Analyzer (Applied Biosystems Inc.).
2.2 |. Whole exome sequencing analysis
We performed whole-exome sequencing (WES) for the patient with lamin A/C mutation p.Arg399Cys as described previously (Wang et al., 2016). In brief, genomic DNA was captured using the SOLiD Fragment Library Core Kit. After the exome capture, massively parallel sequencing was carried out using the SOLiD 5500 Genetic Analysis System. Variant detection, filtering, and annotation were described in detail previously (Wang et al., 2016).
2.3 |. Construction of plasmids and antibodies
The wild-type LMNA cDNA (NM_170707.3, NP_733821.1) was cloned into the mammalian expression vector pEGFP-C1 (EGFP-LMNA-WT for abbreviation), and generously provided by Dr. Juliet A. Ellis. For immuno-fluorescence analysis, the wild-type LMNA cDNA was amplified by PCR using plasmid EGFP-LMNA-WT as the template and subcloned into the pcDNA3.1-RFP mammalian expression vector between the Nhe 1 and Xho 1 site, resulting in expression plasmid RFP-LMNA-WT.
For co-immunoprecipitation (Co-IP) analysis, LMNA cDNA was amplified and subcloned into the p3×FLAG-CMV mammalian expression vector between the EcoR 1 and Sal 1 site, resulting in expression plasmid FLAG-LMNA-WT. For GST pull-down analysis, LMNA cDNA was amplified and subcloned into the pGEX4T-1 prokaryotic expression vector between the BamH 1 and Sal 1 site, resulting in expression plasmid pGEX-LMNA-WT. The cDNA sequences encoding the N-terminal (encoding residues 1–380) and C-terminal (encoding residues 381–646) lamin A fragments were amplified, respectively, and subcloned into the pGEX4T-1 expression vector between the BamH 1 and Sal 1 site, resulting in two truncated plasmids GST-LMNA-N and GST-LMNA-C.
The p.Arg399Cys mutation in LMNA (NM_170707.3, NP_733821.1) was created into EGFP-LMNA-WT, RFP-LMNA-WT, FLAG-LMNA-WT, and pGEX-LMNA-WT plasmids using a PCR-based mutagenesis method as described previously (Du et al., 2005).
For nucleocytoplasmic trafficking assays, the pEGFP-NLS (containing the insert SV40 nuclear localization signal (NLS)) plasmid was generously provided by Dr. Qian Qian Liu. The NES (nuclear export signal) sequence residues 367–427 of human STAT1 was amplified and subcloned into the pEGFP-N1 expression vector between the Xho 1 and Sal 1 site, resulting in the pEGFP-NES plasmid.
Antibodies for lamin A/C (Proteintech, 10298-1-AP), NUP155 (Abcam, ab199528), NUP205 (Proteintech, 24439-1-AP), lamin B1 (Proteintech, 12987-1-AP), emerin (Abcam, ab156871), and Na+–K+ ATPase (Cell signaling technology, 3010) were all rabbit polyclonal antibodies and used by a dilution factor of 1:1000. The NUP153 antibody (Abcam, ab24700) was a mouse monoclonal antibody and diluted by 1:1000. A rabbit Nav1.5 antibody (Alomone, ASC-005) and an HERG antibody (Alomone, APC-016) were diluted by 1:600. The anti-HSP70 antibody (Assay Designs, no. SPA-810) was a mouse antibody and diluted by 1:100 in immunofluorescence assays. The FLAG antibody (MBL, M185–3L) was a mouse monoclonal antibody and diluted by 1:5000 in Western blotting and 2.5 μg in co-immunoprecipitation (Co-IP) assay. The dilution factors for other antibodies were 1:5000 for an anti-α-tubulin (Proteintech, 11224-1-AP), and 1:200 for rabbit and mouse IgG from Proteintech, fluorescein (FITC)-conjugated goat anti-mouse/rabbit IgG (H+L) (Proteintech, SA00003–1/2), and Texas Red-conjugated Affinipure goat anti-rabbit IgG (H+L) (Proteintech, SA00005–2).
2.4 |. Cell culture
The HL-1 adult mouse cardiac muscle cell line was kindly provided by Dr. William C. Claycomb, and the cells were maintained in Claycomb medium (JRH Biosciences) supplemented with 10% fetal bovine serum (FBS; Life Technologies). Human SCN5A cDNA was cloned into vector pcDNA3 as described previously and used for establishing a stable HEK293 cell line with constant expression of Nav1.5 (referred to as HEK293/Nav1.5; Wu et al., 2008). HEK293/Nav1.5 and HeLa cells were cultured in DMEM (GIBICO) with 10% FBS (GIBICO) at 37°C with 5% CO2. Cells were transfected with plasmid DNA using Lipofectamine 2000 (Invitrogen) according to the manufacturer’s instructions as previously described (Wu et al., 2008).
2.5 |. Immunostaining assays
Cells were transfected for 48 hr, washed with PBS buffer, fixed with 4% PFA for 20 min at room temperature (RT), perforated with 0.5% TritonX-100 for 15 min at room temperature, and immunostained as described previously (Wu et al., 2008). The cells were then stained for the nucleus with DAPI for 15 min at RT. All microscopic images were obtained using an Olympus confocal microscope (Olympus). Images were captured and fluorescence intensity was quantified using FV10-ASW 3.1 Viewer software and Image-Pro Plus.
2.6 |. Stress experiments
HL-1 cells were co-transfected with the LMNA expression vector (RFP-LMNA-WT or RFP-LMNA-R399C) and a plasmid expressing EGFP-NLS. After 48 hr, cells were treated with 300 mM mannitol (Sigma) for 2 hr or 300 μM H2O2 (Sigma) for 4 hr as described previously (Forleo et al., 2015). The structural integrity of the NE and its resistance to stress were examined under a confocal microscope.
2.7 |. Nucleocytoplasmic trafficking assays for the NPC at a steady state
In order to investigate the effects of the p.Arg399Cys mutation on nuclear import or export, HL-1 cells were co-transfected with the LMNA expression vector (RFP-LMNA-WT or RFP-LMNA-R399C) and the plasmid expressing EGFP-NLS or EGFP-NES. After 48 hr, subcellular distribution of EGFP fusion proteins and the ratio of nuclear over cytosolic fluorescence was analyzed under a confocal microscope as previously described (Busch, Kiel, Heupel, Wehnert, & Hubner, 2009; Stochaj, Rassadi, & Chiu, 2000).
2.8 |. Extraction of nuclear envelope proteins
Extraction of NE proteins was carried out as described previously by Hawryluk et al. (Hawryluk-Gara, Shibuya, & Wozniak, 2005). Briefly, NE proteins were extracted with 1% Triton X-100 in buffer containing 20 mM TEA (pH 7.5), 0.1 mM MgCl2, 1 mM DTT, and protease inhibitors to solubilize the nuclear membrane. The suspension was centrifuged at 4000 × g for 10 min at 4°C, and the pellet containing the extracted NE fraction was collected. The NE pellet was re-extracted with the same buffer containing 0.1, 0.25, or 0.5 M NaCl, but lacking Triton X-100, and proteins were analyzed by SDS-PAGE and Western blotting analysis.
2.9 |. Cell membrane fraction separation
The transfected HEK293/Nav1.5 cells were cultured for 48 hr. Cells were surface-biotinylated using EZLink Sulfo-NHS-SS-Biotin (Pierce), and then lysed in lysis buffer as described by us previously (Huang et al., 2016). Lysates were centrifuged and the supernatants were incubated with NeutrAvidin-agarose resins (Pierce) at 4°C. The agarose beads were washed, and bound proteins were resuspended and analyzed by SDS-PAGE and Western blotting analysis.
2.10 |. Assays for export of HSP70 mRNA and nuclear import for HSP70 protein
Heat-shock experiments were conducted 48 hr after transfection as described by us previously (Zhang et al., 2008). HL-1 cells in six-well plate were rinsed with PBS, and then cultured in the media containing 10% FBS and pre-warmed to 42°C. The cells were incubated at 42°C and under 5% CO2 for 3 hr, washed with PBS, and lysed with 0.1% NP40/PBS buffer. After centrifugation at 5000 × g for 30 s, cytoplasmic and nuclear RNA samples were isolated using the Cytoplasmic & Nuclear RNA Purification Kit (Norgen) following the manufacturer’s protocols. Quantitative real time PCR was performed with SYBR Green (Roche) using an ABI QuantStudio™12K Flex real time PCR System. The specific primers used for mouse HSP70 were 5′-TGGTGCTGACGAAGATGAAG-3′ (F) and 5′-AGGTCGAAGATGAGCACGTT-3′ (R). All the samples were analyzed in triplicate and subsequently quantified by normalization with mouse GAPDH (primer sequence: 5′-AGGTCGGTGTGAACGGATTTG-3′ (F), 5′-TGTAGACCATGTAGTTGAGGT CA-3′ (R)) in three independent experiments. The ratios of cytoplasmic/nuclear expression levels of HSP70 were calculated as we described previously (Zhang et al., 2008). Nuclear import of HSP70 protein was measured using immunostaining analyses as previously described (Zhang et al., 2008).
2.11 |. Electrophysiological recordings of ionic currents
For patch-clamping experiments, HEK/Nav1.5 cells were transfected with 0.8 μg of LMNA expression plasmid EGFP-LMNA-WT or EGFP-LMNA-R399C for 48 hr. The cells expressing an approximately equal amount of EGFP were selected for electrophysiological recordings of sodium currents (INa) as described previously by us (Huang et al., 2016). The HERG K+ channel current in HEK293 cells transfected with the KCNH2 expression plasmid was recorded as described (Han, Jing, Yang, Zhang, & Zhang, 2016).
2.12 |. Statistical Analysis
Data are represented as mean ± SEM. Statistical analysis was carried out using two-tailed Student’s t test. A P-value of ≤0.05 was considered to be statistically significant.
3. |. RESULTS
3.1 |. Genetic study identified LMNA mutation p.Arg399Cys in a patient with lone AF
More than 600 sporadic lone AF patients were screened for mutations in the LMNA gene. The entire coding region of the LMNA gene spans approximately 24 kb and contains 12 exons. All 12 coding exons and exon-intron boundaries were PCR amplified and directly sequenced. A heterozygous missense mutation p.Arg399Cys was identified in one patient (Figure 1a). The patient with p.Arg399Cys mutation was a male patient affected with paroxysmal AF and lone AF without any other cardiac or systemic abnormalities. Echocardiography revealed normal structure and left ejection fraction. The p.Arg399Cys mutation occurs in exon 7 at an evolutionally conserved arginine residue (Figure 1b). The mutated residue is located between the middle helical rod domain and the C-terminal globular domain (Figure 1c). The allele frequency of p.Arg399Cys was 0 in the 1000 Genomes (http://www.internationalgenome.org/1000-genomes-browsers) and 0.000008342 in the Exome Aggregation Consortium (ExAC, http://exac.broadinstitute.org).
FIGURE 1.
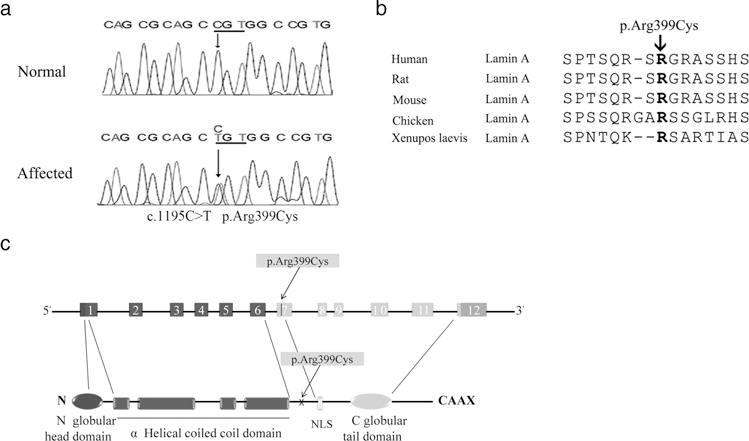
Identification of the p.Arg399Cys mutation in LMNA associated with AF. (a) DNA sequences of LMNA from an affected patient and a normal individual. A heterozygous missense mutation (c.1195C>T, p.Arg399Cys) was identified in the affected patient. (b) Multiple amino acid sequence alignment of lamin A proteins. The p.Arg399Cys mutation occurs at an arginine (R) residue that is evolutionarily conserved. (c) Schematic structure of the LMNA gene and lamin A protein. The p.Arg399Cys mutation was found in exon 7. NLS, nuclear localization signal
To make sure that the AF phenotype identified in this patient was not caused by another pathogenic mutation in cardiomyopathy-or arrhythmia-associated genes, we performed whole-exome sequencing (WES) for this patient, but we failed to find any other mutations within well-known cardiovascular disease-causing genes (data not shown). These data suggest that the phenotype in this AF patient is unlikely to be caused by a mutation in another known cardiovascular disease gene. Together, these results suggest that mutation p.Arg399Cys is the likely genetic cause of AF.
3.2 |. LMNA mutation p.Arg399Cys causes abnormal nuclear localization of the lamin A protein in HL-1 cells
Lamin A/C is critical to nuclear assembly, and LMNA mutations may cause NE (nuclear envelope)-associated proteins to be mis-localized in the cell, and disrupt nuclear assembly. In order to determine the pathogenic consequence of LMNA mutation p.Arg399Cys, we constructed mammalian expression constructs for wild-type (WT) and mutant EGFP-tagged LMNA with p.Arg399Cys, and each construct was transiently transfected into atrial cardiac HL-1 cells. We counted 300 transfected cells expressing WT or mutant lamin A with green EGFP signals. Normal ovoid nuclear morphology was observed when cells were transfected with WT or mutant LMNA constructs. In transfected cells, the WT lamin A EGFP fusion protein was normally localized at the whole nuclear periphery in the vast majority of the cells, and only 3.9% cells exhibited a “foci” phenotype, which would be indicative of the overexpressed, misfolded lamin A protein aggregates. Compared to the WT, mutant lamin A with p.Arg399Cys showed a slightly but significantly higher percentage of aberrant protein aggregates in 7.6% cells, accumulating in intranuclear or NE-associated “foci” (P < 0.05; Figure 2a). These results suggest that LMNA mutation p.Arg399Cys results in a mild increase of “foci” in the nucleus, suggesting a moderate defect in lamin A assembly and function.
FIGURE 2.
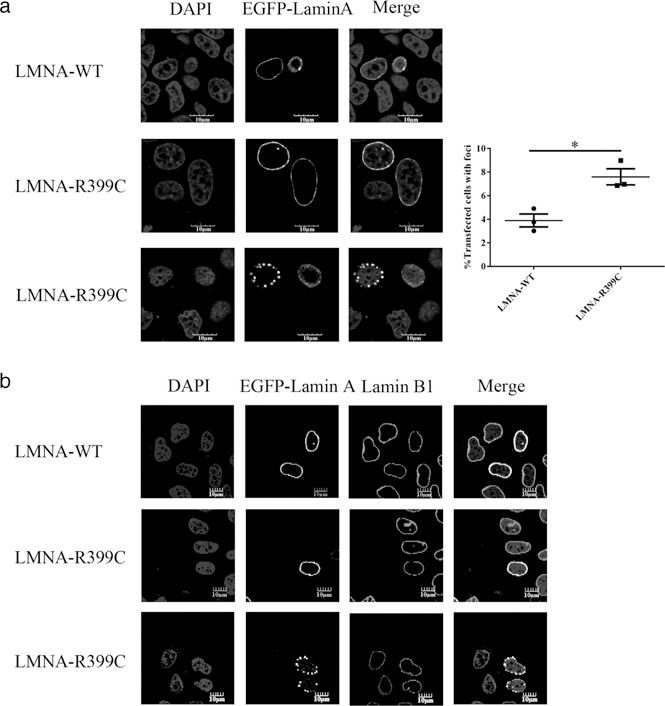
Mutation p.Arg399Cys affects the localization of lamin A. (a) Immunofluorescence analysis of EGFP-tagged WT or p.Arg399Cys lamin A expressed in HL-1 cells. WT lamin A was correctly localized to the nuclear periphery in the majority of the cells, while p.Arg399Cys showed a higher frequency of a “foci” phenotype. Data are presented as mean ± SEM, *P < 0.05. (b) Analysis of lamin B1 localization in cells expressing WT or mutant lamin A with p.Arg399Cys. EGFP-tagged WT or p.Arg399Cys mutant LMNA plasmid was transfected into HL-1 cells. The p.Arg399Cys mutation did not disturb the distribution of endogenous lamin B1 on the nuclear rim. Scale bar: 10 μm
The localization of the endogenous B-type lamin (lamin B1), a marker for the nuclear lamina and NE, appears to be normal in the presence of either WT or mutant lamin A (Figure 2b). The lamin A binds to emerin and ensure its nuclear localization, and mutations in emerin were associated with AF in X-linked EDMD or AF in X-linked sinus node dysfunction (Karst, Herron, & Olson, 2008; Sakata et al., 2005). LMNA mutation p.Arg399Cys is also located in the emerin-binding domain (residues 384–566 amino acids) of lamin A (Stierle et al., 2003), however, the nuclear localization and immunostaining intensity of endogenous emerin were not affected by the LMNA mutation (Supporting Information Figure S1a). Western blot analysis showed that the mutation p.Arg399Cys did not affect the expression levels of total emerin, cytoplasmic emerin, or emerin on NE. (Supporting Information Figure S1b).
3.3 |. LMNA mutation p.Arg399Cys does not impair NE integrity upon cell stress
As lamin A/C plays a fundamental role in the nuclear physical architecture, LMNA knockout or LMNA mutations have been reported to be associated with reduced NE integrity under mechanical strain or various cellular stresses (Forleo et al., 2015; Lammerding et al., 2004). Because the p.Arg399Cys mutation induced a mild defect in lamin A assembly, we determined whether the p.Arg399Cys mutation affects the integrity of the NE, and increases NE fragility under cellular stress. We constructed mammalian expression constructs for WT and p.Arg399Cys mutant RFP-tagged LMNA, and a construct for an EGFP-tagged nuclear transport reporter EGFP-NLS (nuclear localization signal). HL-1 atrial cells were transiently co-transfected with the RFP-tagged LMNA (WT or mutant p.Arg399Cys) construct and the EGFP-NLS construct, and the nuclear morphology and the subcellular distribution of EGFP-NLS were analyzed under a confocal immunofluorescence microscope. Cells with overexpression of the WT or mutant p.Arg399Cys lamin A showed slightly compromised rounded morphology of nuclei after either hyperosmosis stress (300 mM d-mannose) or oxidative stress (300 μM H2O2). The nucleus-targeted EGFP-NLS fusion protein was almost exclusively localized and confined in the nucleus in cells transfected with the WT or mutant p.Arg399Cys construct either under control or challenged conditions (Supporting Information Figure S2a–d). These data suggest that the p.Arg399Cys mutation does not make the nucleus more susceptible to leakage under stress. The TUNEL assay was also performed to examine apoptosis in response to cellular stress. The number of TUNEL positive cells was not significantly changed by mutation p.Arg399Cys (data not shown). Taken together, these results suggest that the LMNA p.Arg399Cys mutation does not significantly weaken the nucleus or render it more fragile under cellular stress.
3.4 |. LMNA mutation p.Arg399Cys does not affect importin-dependent nuclear import and export, localization of NUP153, or the nucleocytoplasmic RAN gradient under a steady state condition
The nuclear lamina provides a scaffold for spatial organization and function of NPCs. It is now recognized that a deficiency of lamin A assembly may cause not only the impairment of the nuclear architecture, but also mis-compartmentalization of nuclear pore proteins such as NUP153 (Busch et al., 2009; Kelley et al., 2011). Therefore, we investigated the functional consequences of the LMNA p.Arg399Cys mutation on localization of NUP153. HL-1 cells were transiently cotransfected with an RFP-tagged LMNA (WT or mutant p.Arg399Cys) mammalian expression construct and the nuclear transport reporter EGFP-NLS construct, and the subcellular distribution of EGFP-NLS was analyzed by confocal immunofluorescence microscopy to investigate the importin-dependent nuclear import. The EGFP-NLS signal was almost exclusively localized to the nucleus in cells transfected with either WT or mutant p.Arg399Cys construct (Supporting Information Figure S3a). We also used the EGFP-NES construct to analyze the CRM-1 mediated nuclear export, but the relative levels of cytoplasmic EGFP-NES did not change in cells co-transfected with either WT or mutant p.Arg399Cys construct (Supporting Information Figure S3b). Furthermore, endogenous NUP153 and RAN were analyzed by indirect immunofluorescence. LMNA mutants induced mislocalization of NUP153, and dysfunctional NUP153 was associated with deregulated nucleocytoplasmic RAN gradient and reduction of nuclear import (Busch et al., 2009; Kelley et al., 2011). However, in our study, NUP153 assembled normally into NE in cells transfected with either WT or mutant p.Arg399Cys construct (Supporting Information Figure S3c). The cellular distribution of RAN was preserved as well (Supporting Information Figure S3d). Western blot analysis showed that the p.Arg399Cys mutation did not affect the expression level of NUP153 (Supporting Information Figure S3e). These results suggest that the LMNA p.Arg399Cys mutation does not disrupt localization of NUP153 or the nucleocytoplasmic RAN gradient.
3.5 |. Lamin A interacts with NUP155, a key component of the NPC
Nucleoporins are the critical molecular components for the structure, assembly, and function of the NPC (Raices & D’Angelo, 2012). Among about 30 different nucleoporins that have been identified, our previous study found that a genetic mutation in NUP155 was associated with familial AF and sudden death (Zhang et al., 2008). Recently, Roux et al. developed the BioID (proximity-dependent biotin identification) method to identify lamin A-interacting or vicinal endogenous proteins. Among the unique 122 proteins identified in the report, NUP155 was found to be one potential lamin A-interacting or neighboring protein (Roux, Kim, Raida, & Burke, 2012). However, the interaction between lamin A/C and NUP155 was not detected and validated by the standard co-immunoprecipitation (Co-IP) and GST pull-down assays.
The physical interaction between lamin A and NUP155 was characterized. First, we carried out a Co-IP assay. We constructed a FLAG-tagged WT LMNA mammalian expression construct, and transiently transfected it into HeLa cells. Whole-cell extracts were immunoprecipitated using an anti-FLAG antibody, and proteins that bound FLAG-lamin A were detected by Western blot analysis with an anti-NUP155 antibody. The anti-FLAG antibody, but not the control IgG, precipitated the endogenous NUP155 protein (Figure 3a). Emerin was used as a positive control because it interacts with lamin A (Stierle et al., 2003). Consistently, reciprocal Co-IP showed that an anti-NUP155 antibody, but not the control IgG, precipitated the FLAG-tagged lamin A (Figure 3b). NUP205 was used as a positive control because it interacts with NUP155 (Hawryluk-Gara et al., 2005). The Co-IP demonstrated the interaction between lamin A and NUP155 (Figure 3a,b).
FIGURE 3.
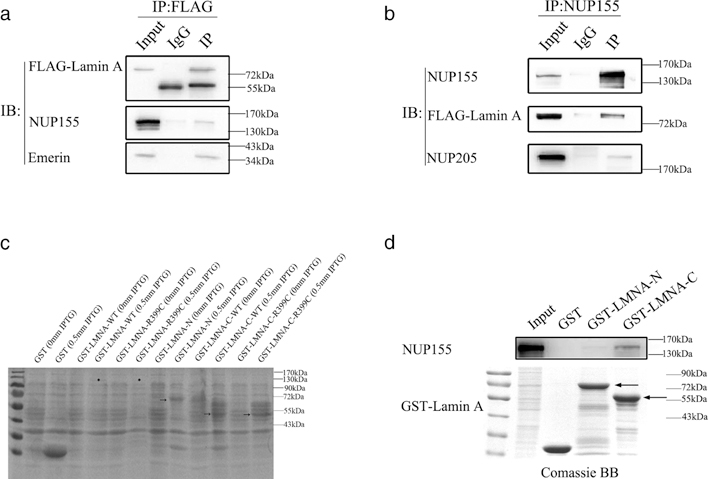
Lamin A interacts with NUP155. (a) Co-immunoprecipitation (Co-IP) assays. HeLa cells were transiently transfected with a FLAG-LMNA-WT plasmid, and cell lysates were immunoprecipitated with anti-FLAG antibody, followed by immunoblotting analysis (IB) with an anti-NUP155 antibody. Emerin was used as a positive control, and normal IgG were used as negative control. (b) Reciprocal co-immunoprecipitation assays. HeLa cells were transiently transfected with a FLAG-LMNA-WT plasmid. Cell lysates were immunoprecipitated with anti-NUP155 antibody and analyzed by immunoblotting analysis (IB) with an anti-FLAG antibody. NUP205 was used as a positive control, and normal IgG were used as negative control. (c) Bacterial expression of recombinant GST-lamin A. Protein expression was induced by 0 or 0.5 mM IPTG, the recombinant protein was analyzed by SDS-PAGE and Coomassie brilliant blue staining. The result indicated that the N-terminal or the C-terminal truncated lamin A protein was expressed. The arrow indicated the estimated size of the truncated fusion proteins. Asterisk indicated the location of the full length fusion proteins. (d) GST pull-down assays. The cell lysates extracted from HeLa cells were incubated with a purified GST fusion N-terminal or C-terminal truncated lamin A protein or negative control GST. GST-LMNA-C-WT, but not GST-LMNA-N-WT or control GST, pulled down the endogenous NUP155. Coomassie brilliant blue staining showed the amount of protein. The arrow indicated the estimated size of fusion proteins
We also performed a glutathione S-transferase (GST) pull-down assay to further demonstrate the interaction between lamin A and NUP155. We constructed a prokaryotic expression plasmid that expresses the GST-fused lamin A protein. Under several different conditions, the protein of interest was not detected through Coomassie blue staining (Figure 3c). Therefore, two constructs for truncated GST-lamin A were generated, including a plasmid referred to as GST-LMNA-N (encodes residues 1–380, contains N-terminal head and rod domain of lamin A), and another plasmid GST-LMNA-C (encodes residues 381–646, contains C-terminal tail domain of lamin A). Both truncated forms of GST-lamin A were successfully expressed in Escherichia coli (Figure 3c). We then carried out GST pull-down assays using purified, truncated GST-lamin A after incubation with the whole HeLa cells extracts. The proteins that were bound to GST-lamin A were detected by Western blot analysis with an anti-NUP155 antibody. As shown in Figure 3d, the N-terminal domain of lamin A possessed weak endogenous NUP155-interacting capability, while the C-terminal domain of lamin A is the critical region for the interaction with NUP155.
To analyze the co-localization of lamin A and NUP155, HeLa cells were transiently transfected with an RFP-tagged WT LMNA construct, and immunofluorescent staining of HeLa cells showed that endogenous NUP155 was enriched at nuclear NE, and co-localized with lamin A (Supporting Information Figure S4a). Collectively, these data unequivocally demonstrate the interaction between lamin A and NUP155.
3.6 |. LMNA mutation p.Arg399Cys weakens the binding of lamin A to NUP155
The p.Arg399Cys mutation in lamin A is located at the C-terminal region of lamin A, which was shown to interact with NUP155 above. Thus, we hypothesized that the p.Arg399Cys mutation might affect the interaction between lamin A and NUP155. To test the hypothesis, we preformed GST pull-down assays with GST-LMNA-C with the C-terminal lamin A residues 381–646 and HeLa cell extracts. The interaction between NUP155 and lamin A was significantly reduced by the LMNA p.Arg399Cys mutation (58% of the WT) compared with the WT allele (P < 0.05; Figure 4a). Our results suggest that the p.Arg399Cys mutation weakens the interaction between lamin A and NUP155.
FIGURE 4.
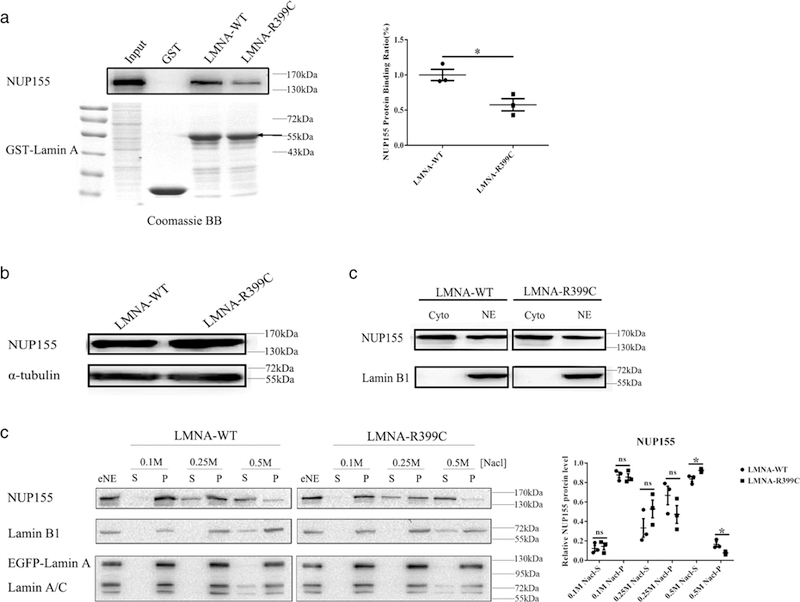
LMNA mutation p.Arg399Cys weakens binding of lamin A to NUP155, and increased extractability of NUP155 from NE. (a) GST pull-down assays. Nuclear extracts from HeLa cells were incubated with GST-LMNA-C-WT or GST-LMNA-C- p.Arg399Cys. Coomassie brilliant blue staining showed the approximately equal amount of WT and mutant fusion proteins. The interaction between endogenous NUP155 and mutant lamin A with p.Arg399Cys was significantly reduced by 42% compared with WT lamin A. The arrow indicated the estimated size of fusion proteins. Data are presented as mean ± SEM, *P < 0.05. (b) Mutant lamin A with p.Arg399Cys did not affect the expression level of endogenous NUP155 protein. EGFP-LMNA-WT or EGFP-LMNA-R399C was transferred into HeLa cells, and cell lysates were analyzed by Western blotting with an anti-NUP155 antibody or a control anti-α-tubulin antibody. (c) Mutant lamin A with p.Arg399Cys did not affect the NUP155 localization at NE (nuclear envelope). EGFP-LMNA-WT or EGFP-LMNA-R399C was transferred into HeLa cells, and the cytoplasmic and NE extracts were analyzed by Western blotting with antibodies for NUP155 and lamin B1. Cyto: cytoplasm; NE: nuclear envelope. (d) Mutant lamin A with p.Arg399Cys increased the extractability of NUP155 from NE. EGFP-LMNA-WT or EGFP-LMNA-R399C was transferred to HeLa cells, and the insoluble NE fraction was prepared by treatment with TrionX-100. The extracted NE (eNE) was further re-extracted with increasing concentrations of NaCl, and analyzed by Western blotting with an anti- NUP155 antibody or an anti-lamin B1 as a positive control. S, supernatant; P, precipitate. The relative NUP155 protein level at the Y-axis was expressed as either S/S+P or P/S+P. Data are presented as mean ± SEM, *P < 0.05
3.7 |. LMNA mutation p.Arg399Cys increases extractability of NUP155 from NE
We used immunostaining analysis to investigate the effect of mutation p.Arg399Cys on nuclear localization of NUP155. The majority of endogenous NUP155 was specifically located at the NPC, and no significant difference was observed between WT and mutation p.Arg399Cys (Supporting Information Figure S5a). Western-blot analysis showed that p.Arg399Cys mutation did not affect the expression level of endogenous NUP155 in cells (Figure 4b). At the same time, the p.Arg399Cys mutation did not affect the localization of NUP155 at NE (Figure 4c). Interestingly, when we analyzed the extractability or release of NUP155 from NE membranes using differential extraction procedures, we found a significant difference between WT and p.Arg399Cys mutant lamin A. After transient transfection of WT or mutant p.Arg399Cys LMNA into HeLa cells, we isolated NE and analyzed the extraction of NUP155 from NE with increasing concentrations of NaCl. As shown in Figure 4d, although 82% of NUP155 was efficiently extracted from NE by 0.5 M NaCl in cells with WT lamin A, a small fraction of NUP155 remained to be associated with NE. Compared to cells with WT lamin A, significantly more NUP155 (93%) was released into the soluble fraction form NE at the similar concentration of NaCl in cells with mutant lamin A with p.Arg399Cys. (P < 0.05) These data suggest that mutation p.Arg399Cys in lamin A increases the extractability or release of NUP155 from NE, although it does not affect the protein level and NPC localization of NUP155. In contrast, the LMNA mutation did not affect the extractability of lamin B1, endogenous lamin A or the EGFP tagged lamin A from NE (Supporting Information Figure S6a–c).
3.8 |. LMNA mutation p.Arg399Cys inhibits the nuclear export of the HSP70 mRNA and the nuclear import of the HSP70 proteins
We previously reported that NUP155 regulated nuclear-cytoplasmic trafficking of HSP70, and both NUP155 haploinsufficiency (NUP155+/−) and AF-causing NUP155 mutation (p.Arg391His) affected HSP70 mRNA export and HSP70 protein import (Zhang et al., 2008). Because LMNA p.Arg399Cys mutation weakens the interaction of lamin A with NUP155 and increases the release NUP155 from NE, we investigated whether LMNA mutation p.Arg399Cys affected HSP70 mRNA export and HSP70 protein import. The WT or mutant LMNA expression construct was transfected into HL-1 cells, and the expression level and subcellular localization of the HSP70 protein were analyzed after exposure of cells to heat shock. Immunostaining with an anti-HSP70 antibody showed that not only the total expression level of the HSP70 protein was reduced, but also the nuclear import of the HSP70 protein was inhibited in cells expressing mutant p.Arg399Cys lamin A (P < 0.05) (Figure 5a; Supporting Information Figure S7a). We also analyzed the subcellular distribution of the HSP70 mRNA after heat shock. RT-PCR analysis showed that HL-1 cells with the mutant LMNA decreased the export of the HSP70 mRNA (P < 0.05; Figure 5b). Our results indicate that the LMNA p.Arg399Cys mutation inhibits both the export of the HSP70 mRNA and the nuclear import of the HSP70 protein. These data are similar to the AF-causing mutation in NUP155 (Zhang et al., 2008).
FIGURE 5.
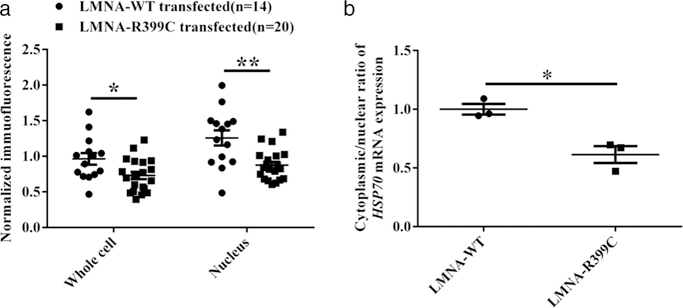
LMNA mutation p.Arg399Cys reduces the export of HSP70 mRNA, and nuclear import of HSP70 protein. (a) Analysis of nuclear import of HSP70 protein in HL-1 cells transiently transfected using a EGFP-LMNA-WT or EGFP-LMNA-R399C LMNA expression plasmid for 48 hr. Data are presented as mean ± SEM, *P < 0.05, **P < 0.01; n, number of images analyzed as indicated. (b) Analysis of the nuclear export of HSP70 mRNA in HL-1 cells. After heat shock, total RNA from nuclear or cytoplasmic fractions was isolated and reverse-transcribed with random primers. Quantitative real time RT-PCR analysis was used to estimate the levels of HSP70 mRNA in the cytoplasm and nucleus. LMNA mutation p.Arg399Cys inhibits HSP70 mRNA export into the cytoplasm. Data are presented as mean ± SEM, *P < 0.05
3.9 |. LMNA mutation p.Arg399Cys decreases the density of cardiac sodium current in HEK293/Nav1.5 cells
Genetic studies have identified cardiac ion channel mutations associated with AF (Andalib, Brugada, & Nattel, 2008). The cardiac sodium channel Nav1.5 encoded by the SCN5A gene is responsible for depolarization and conduction of the action potential in the heart. Patients with mutations or variants in SCN5A presented with a high incidence of isolated AF or common forms of AF (L. Y. Chen, Ballew, Herron, Rode-heffer, & Olson, 2007; Darbar et al., 2008; Rivera-Torres et al., 2016). Interestingly, sodium channel dysfunction was linked to DCM-related LMNA mutation p.Asn195Lys (Markandeya et al., 2016). Thus, we performed electrophysiological studies to assess the effect of the LMNA p.Arg399Cys mutation on the cardiac sodium current generated from SCN5A (Nav1.5). EGFP-tagged WT or mutant LMNA with p.Arg399Cys were overexpressed in HEK293/Nav1.5 stable cells by transient transfection and sodium currents (INa) were recorded by patch-clamping. Cells with overexpression of the mutant LMNA showed a significant decrease (37%) of INa density compared to cells with WT LMNA (P < 0.05; Figure 6a–c). The effects of the LMNA mutation on steadystate activation and inactivation of sodium currents were also studied, however, the LMNA mutation did not cause significant changes in the activation or inactivation kinetics of INa (Figure 6d,e). In addition, the LMNA mutation did not affect the late sodium current (data not shown).
FIGURE 6.
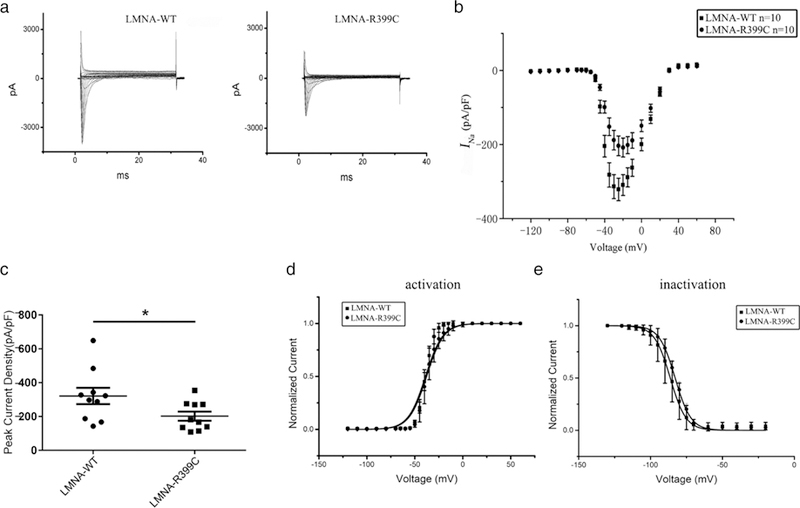
LMNA mutation p.Arg399Cys decreases cardiac sodium current density in HEK293/Nav1.5 cells. (a) Representative whole-cell sodium currents recorded from HEK/Nav1.5 cells transfected with a EGFP-LMNA-WT or p.Arg399Cys mutant expression plasmid. (b) The relationship of average current densities (current normalized to cell capacitance) and voltages. (c) Peak sodium current densities at –25 mV. Data are presented as mean ± SEM, *P < 0.05. (d) Steady-state activation curve. (e) Steady-state inactivation curve
Recently, it has been reported that a high prevalence of variants in potassium channel encoding genes such as KCNH2 (HERG) may also modulate atrial repolarization and predispose individuals to AF (Hayashi et al., 2015; Sinner et al., 2008). We transiently cotransfected HEK293 cells with a WT or mutant FLAG-tagged LMNA mammalian expression construct and an EGFP-tagged HERG construct, and recorded the IKr potassium current by patch-clamping. Mutation p.Arg399Cys did not have significant effects on the IKr potassium current and activation or inactivation kinetics of IKr (Supporting Information Figure S8a–d) or the expression level of the HERG protein (Supporting Information Figure S8e).
Together, these data indicate that mutation p.Arg399Cys in lamin A had a major effect on reduction of cardiac INa density, but not the IKr potassium current.
3.10 |. LMNA mutation p.Arg399Cys decreases the amount of Nav1.5 on the surface of HEK293/Nav1.5 cells
To determine whether the p.Arg399Cys mutation reduces INa density by decreasing the Nav1.5 protein level, we carried out Western blot analysis of total protein extracts from HEK293/Nav1.5 cells. Compared with WT LMNA, overexpression of mutant LMNA did not significantly affect the total protein expression level of Nav1.5 (Figure 7a). Given the important role of the level of cell surface expression of Nav1.5 in INa density, cell surface biotinylation assays were further performed. LMNA mutation p.Arg399Cys significantly decreased the cell surface expression level of Nav1.5 by 38% (P < 0.05; Figure 7b). These results suggest that the p.Arg399Cys mutation may inhibit sodium channel trafficking to reduce the INa density.
FIGURE 7.
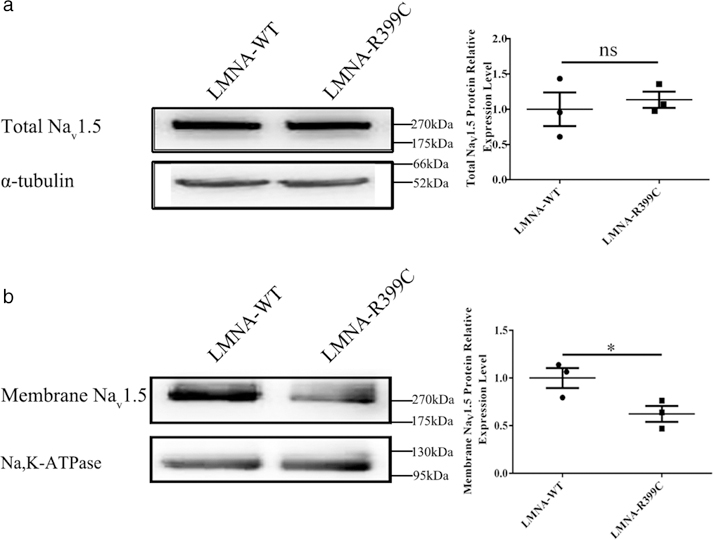
LMNA mutation p.Arg399Cys decreases the cell surface expression level of Nav1.5 without affecting the expression of total Nav1.5 in HEK293/Nav1.5 cells. (a) Western blotting analysis of total Nav1.5 protein in HEK293/Nav1.5 cells transiently transfected with a EGFP-LMNA-WT or EGFP-LMNA-R399C LMNA expression plasmid for 48 hr. Data are presented as mean ± SEM. ns, not significant. (b) Western blotting analysis of cell surface expression levels of Nav1.5 in HEK293/Nav1.5 cells. Data are presented as mean ± SEM, *P < 0.05
4 |. DISCUSSION
Roux et al. (2012) previously identified NUP155 as one of the 122 candidate proteins that may interact with lamin A using BioID. In this study, we unequivocally demonstrated the interaction between lamin A/C and nucleoporin NUP155. Both Co-IP and GST pull-down assays showed that lamin A/C interacted with NUP155 (Figure 3a–d). Immunostaining of HeLa cells showed that endogenous NUP155 colocalized with lamin A/C at the NE (Supporting Information Figure S4a). Most interestingly, lamin A/C mutation p.Arg399Cys, which causes AF, impaired the interaction between lamin A/C and NUP155 (Figure 4a), and increased the release of NUP155 from the NE (Figure 4d). Lamin A/C mutation p.Arg399Cys also impaired the core function of the NPC, the nucleocytoplasmic transport of macromolecules. In a model system, lamin A/C mutation p.Arg399Cys inhibited the nuclear export of the HSP70 mRNA and the nuclear import of the HSP70 proteins (Figure 5a,b). These studies with AF-causing mutation p.Arg399Cys in lamin A/C, therefore, identified a novel molecular mechanism involving the interaction between the nuclear lamina and nuclear pore complexes for the pathogenesis of AF.
To date, our understanding of the genetic basis of AF is mostly limited to genes encoding ion channels as a series of previous studies investigated the prevalence and functional properties of ion channel mutations (Giudicessi & Ackerman, 2012; Remme, 2013). Recently, unexpected findings were made on genetic alterations of non-ion-channel genes linked to familial or sporadic cases of AF, which provided a novel non-ion-channel mechanism underlying the pathogenesis of AF (Hodgson-Zingman et al., 2008; Zhang et al., 2008). Mutations in the LMNA gene showed variable phenotypic expression, and atrial arrhythmia was found in approximate 10–39% of LMNA mutation carriers suffering from cardiomyopathies or cardiomyopathies associated with muscular dystrophy (Kumar et al., 2016; van Berlo et al., 2005; van Rijsingen et al., 2013). It is of note that AF may precede the onset of structural heart disease in laminopathy patients, however, in some cases, isolated AF was reported. To date, eight AF-associated LMNA mutations have been identified (Beckmann et al., 2010; Madej-Pilarczyk et al., 2008; Marsman et al., 2011; Pan et al., 2009; Saj et al., 2012; Sebillon et al., 2003; Zhao et al., 2016; Supporting Information Table S2). Despite a few publications implying LMNA mutations in AF, the exact prevalence of LMNA mutations in AF remains unknown.
LMNA mutation p.Arg399Cys was previously reported to be pathological (http://www.dmd.nl/nmdb2/variants.php?select_db=LMNA&action=search_all&search_Variant%2FDNA=c.1195C>T). Lanktree et al. described one patient with partial lipodystrophy (FPLD2) and the p.Arg399Cys mutation, but no evidence of AF or other cardiac disease were found in this patient (Lanktree, Cao, Rabkin, Hanna, & Hegele, 2007). Cowan et al. identified the same mutation in a patient with early onset DCM (Cowan, Li, Gonzalez-Quintana, Morales, & Hershberger, 2010). Our study indicated that the patient carrying this previously reported FPLD2- or DCM-associated mutation presented with lone AF, but no other cardiac or systemic abnormalities. Such phenotypic heterogeneity for a single mutation has been described in LMNA-linked laminopathies already (Supporting Information Table S1). We summarized the information of the reported eight LMNA-mutations associated with AF in Supporting Information Table S2. It is of note that three AF-associated mutations, including p.Ser143Phe, p.Glu161Lys, and p.Thr528Met, were also associated with other different types of laminopathies, suggesting that the environmental factors or disease modifier genes may contribute to variable clinical phenotype. It is also possible that our AF patient with the p.Arg399Cys mutation did not present with FPLD2 or DCM at the time of enrollment (52 years of age) in our study due to the late onset of these clinical phenotypes.
The functional and molecular consequences of LMNA mutation p.Arg399Cys were characterized in great details in this study. The mutation affects the Arg399 residue, which is located between the middle helical rod domain and the C-terminal globular domain. It is noteworthy that the positively charged arginine cluster (R397SRGR401), in which the p.Arg399Cys mutation is located, was proposed to mediate the longitudinal association of lamin dimers (Strelkov, Schumacher, Burkhard, Aebi, & Herrmann, 2004). The p.Arg399Cys mutation leads to the replacement of charged residue arginine 399 by a cysteine residue, which is uncharged at physiological pH. The mutation is likely to weaken the electrostatic interactions between the dimers. In fact, in our study, we found that the p.Arg399Cys mutation was assembly incompetent and prone to formation of lamin A aggregates in the nucleus of cardiac HL-1 cells (Figure 2a). Cowan et al. identified the same p.Arg399Cys mutation in a patient with DCM, and showed that it exhibited no obvious evidence of aberrant localization in COS-7 cells (Cowan et al., 2010). In addition, Cowan et al. observed that the retraction of DNA from a clearly lamin A-demarcated nuclear lamina, but the detachment of DNA from NE was not found in our study. This conflict may be a reflection on the different cell types used in these experiments. It is of note that the proportion of lamin A aggregates may depend on the different cell culture conditions (Holt, Nguyen, Wehnert, & Morris, 2006). Our results are consistent with previous reports that compared with C-terminal mutations, mutations in the N-terminal rod domain may cause more dramatic intranuclear aggregation and defective lamin assembly (Raharjo, Enarson, Sullivan, Stewart, & Burke, 2001).
The molecular mechanism underlying LMNA-related AF remains unclear to date. Two pathogenicity models of laminopathies, including a “structural” model and a “gene-expression” model, were proposed before. The “structural” model proposed that the disruption of lamina integrity caused by LMNA mutation could impair nuclear mechanics, decrease nuclear stiffness, and result in cell fragility and cell death in mechanically stressed cardiac or muscle tissues (Hutchison, 2002). In our study, unlike cardiomyopathy-related or progeria-related LMNA mutations, LMNA mutation p.Arg399Cys caused a moderate defect in the lamin A assembly, whereas most cells transfected with mutant lamin A with p.Arg399Cys showed normally shaped nuclei, and maintained normal nuclear stability even under various cellular stress conditions (Forleo et al., 2015; Gupta et al., 2010; McClintock, Gordon, & Djabali, 2006). Thus, the “structural” model is not likely to be the main mechanism underlying the pathogenesis of the AF-related LMNA p.Arg399Cys mutation.
In 2008, our group reported that a genetic mutation in NUP155 caused familial human AF associated with sudden death, and moreover, NUP155 haploinsufficiency caused AF in mice (Zhang et al., 2008). Interestingly, in this study, both Co-IP and GST pull-down studies demonstrated the interaction between lamin A and NUP155, and we also found that LMNA mutation p.Arg399Cys impaired the lamin A/NUP155 interaction. These data suggest that LMNA mutation p.Arg399Cys causes AF by weakening the interaction of lamin A with NUP155, another well-established protein that causes AF when mutated. NUP155 has an essential function in the formation of the NE (Franz et al., 2005) and in the regulation of the expression of downstream genes by controlling mRNA or protein nucleocytoplasmic transport (Zhang et al., 2008), although the specific target genes of NUP155 remain to be identified. Here, we showed that the LMNA p.Arg399Cys mutation was associated with the reduced export of HSP70 mRNA and the decreased nuclear import of the HSP70 protein, suggesting that AF caused by the LMNA p.Arg399Cys mutation may be associated with impaired nucleocytoplasmic transport across NPC as by NUP155 mutations. Multiple reports linked HSP70 to AF. Under conditions of various stress, the induced HSP70 expression likely represents the essential early survival response of cardiomyocytes, and provides cardiac protection against the AF (Mace et al., 2009). HSP70 upregulation was associated with a low risk of postoperative AF (Kampinga, Henning, van Gelder, & Brundel, 2007; St Rammos et al., 2002). HSP70 may also play an antithrombotic role in AF patients (Allende et al., 2016). Interestingly, a p.Met439Thr substitution in HSP70 was associated with an increased risk of postoperative AF (Afzal et al., 2008). A series of cellular and animal experimental models revealed that heat shock proteins (HSPs) not only prevent atrial remodeling and attenuate the promotion of AF, but also play a protective role in the development of AF (Chang et al., 2013; Hoogstra-Berends et al., 2012). The lamin A/C protein can also interact directly with HSPs, which may be the part of intranuclear lamin A/C network or factors stabilizing this specific network (Adhikari, Sridhar Rao, Rangaraj, Parnaik, & Mohan Rao, 2004; Willsie & Clegg, 2002). Considering the cardioprotective and anti-arrhythmia role of HSPs, we propose that the LMNA p.Arg399Cys mutation impairs the nuclear-cytoplasmic trafficking of HSP70 or other unidentified critical atrial genes by disrupting the interaction with NUP155, which results in the pathogenesis of AF.
Besides cardiac structural remodeling, a few studies revealed that cardiac ion channel remodeling or electrical remolding is also involved in laminopathies. Rivera-Torres et al. generated a mouse model of progeria syndrome by creating a knockout line of mice deficient in the ZMPSTE24 gene encoding ZMPSTE24, the specific protease that is required to remove the C-terminus of prelamin A. ZMPSTE24−/− hearts displayed after depolarizations. They assessed the expression levels of ion channel genes between ZMPSTE24−/− and control mice. Among the six genes studied, including KCNA5, KCND3, KCNH2, KCNJ2, KCNQ1, and SCN5A, the KCNH2 mRNA was upregulated in ZMPSTE24−/− mice compared with control mice (Rivera-Torres et al., 2016). In two different studies with the mouse model for DCM with the LMNA p.Asn195Lys mutation, LMNA p.Asn195Lys enhanced both peak and late INa current in addition to decreased expression and mis-localization of gap junction proteins connexin 40 and connexin 43. Augmentation of late INa prolongs action potential duration in ventricular myocytes, was suggested to be considered as a potential antiarrhythmic target in patients carrying the LMNA p.Asn195Lys mutation (Markandeya et al., 2016; Mounkes, Kozlov, Rottman, & Stewart, 2005). Several studies have reported altered mRNA and protein expression levels of KCNH2 and SCN5A in AF (Brundel et al., 2001; Yue, Melnyk, Gaspo, Wang, & Nattel, 1999). Mutations or common variants in KCNH2 or SCN5A were associated with inherited susceptibility to AF (L. Y. Chen et al., 2007; Darbar et al., 2008; Hayashi et al., 2015; Sinner et al., 2008). In this study, electrophysiological studies showed that the peak INa current density for the AF-associated LMNA p.Arg399Cys mutation was significantly smaller than that for WT LMNA (Figure 6a–c), although no abnormalities were found for late INa. Moreover, the LMNA p.Arg399Cys mutation did not have significant effect on the IKr potassium current. As both decreased and increased peak INa was suggested as a possible mechanism for AF by slowing local conduction and promoting reentry (Makiyama et al., 2008; Nattel, Maguy, Le Bouter, & Yeh, 2007; Olesen et al., 2012), the decreased INa density is a potential molecular mechanism by which the LMNA p.Arg399Cys mutation promotes AF.
Several reports revealed that the dysfunction of nucleoporins is a pathological feature of laminopathies. Dialynas et al. reported that muscular dystrophy-related LMNA mutations may be associated with the small percentage (0.5%) rupture of the general nuclear envelope and mis-compartmentalization of nuclear pore proteins in cytoplasm (Dialynas et al., 2012). Busch et al. presented some evidence of potential link between LMNA mutations (causing prenatal skin disease restrictive dermopathy RD and the Progeria Syndrome) and impairment of NUP153-related nuclear protein import. They found that mutant lamin A induced redistribution of NUP153 into nuclear aggregates and impaired lamina dynamics, which may possibly account for the compromised nuclear import efficiency (Busch et al., 2009). However, we did not find any evidence that the LMNA mutation p.Arg399Cys associated with AF affected the function of NUP153 (Supporting Information Figure S3c,e). Kelley et al. further showed that oxidative stress (ROS) caused by progerin (the mutant form of lamin A associated with progeria) reduced the function of the RAN GTPase, and further inhibited nuclear import of TPR and UBC9, which may contribute to disease-associated phenotypes in progeria (Datta, Snow, & Paschal, 2014; Kelley et al., 2011; Snow, Dar, Dutta, Kehlenbach, & Paschal, 2013). Observations in several mouse models of LMNA-linked DCM also suggested that the critical NPC-related mRNA/protein nuclear transport dysfunction contributed to the pathophysiology of DCM. LMNA knockout mice developed DCM, and Nikolova et al. observed the selectively decreased SREBP1 nuclear import in left ventricular myocytes in LMNA–/– mice, which may exacerbate cardiac contractile dysfunction (Nikolova et al., 2004). Mice carrying the p.His222Pro LMNA mutation developed muscular dystrophy (MD) and DCM. Arimura et al. found an increased nuclear accumulation of phosphorylated SMAD2/3 in cardiac and skeletal muscles, and the activation of SMAD proteins may explain the marked increase of fibrosis in the heart (Arimura et al., 2005). Arimura et al. also investigated the gender-specific differences in the development of DCM, and found that the nuclear accumulation of androgen receptor (AR) complex in LMNA p.His222Pro mice was associated with gender differences and disease progression (Arimura et al., 2013). In both LMNA KO mouse model and LMNA p.Asn195Lys mice model (also developed DCM with conduction system disease), Ho et al. revealed impaired nuclear translocation and downstream signaling of the mechanosensitive transcription factor megakaryoblastic leukemia 1 (MKL1), which is the critical co-activator regulating cardiovascular development and adaptation (Ho, Jaalouk, Vartiainen, & Lammerding, 2013). It should be interesting to determine whether all these effects identified for LMNA mutations on TPR, UBC9, SREBP1, SMAD2/3, AR, and MKL1 are affected by the AF-associated mutation p.Arg399Cys in the future.
In summary, it is well known that the majority of arrhythmias including AF can be accounted for by mutations in genes encoding ion channels (Andalib et al., 2008), however, in some patients AF was caused by mutations in non-ion channels including NUP155, NPPA, and LMNA. To date, the functional consequences and the underlying mechanism of AF-causing LMNA mutations are unclear. In our study, we showed that LMNA p.Arg399Cys mutation weakened the interaction between lamin A and NUP155, and caused abnormalities in the nuclear export of HSP70 mRNA and the nuclear import of HSP70 protein. It is likely that in addition to HSP70 mRNA and HSP70 protein, LMNA p.Arg399Cys mutation may affect the nucleocytoplasmic transport of other mRNAs and proteins, some of which may also be causes for AF. Meanwhile, we showed that LMNA mutation p.Arg399Cys significantly reduced cardiac INa density, which may contribute to electrical remodeling and cause AF. Future studies can focus on studies on the molecular mechanism on linking the nucleocytoplasmic transport mechanism and electrical remodeling mechanism together by LMNA mutation p.Arg399Cys.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank Dr. Michael Sanguinetti for his help with analysis of our data on HERG and IKr, and other members of the Center for Human Genome Research for help and assistance. This work was supported by grants from the National Natural Science Foundation of China (no. 81070080, 81370206, 81270162, 81630002, 91439129, and 31430047), 2016 Top-Notch Innovative Talent Development Project from the Bureau of Human Resources and Social Security of Wuhan City, NIH/NHLBI grants R01 HL094498, R01 HL121358, and R01 HL126729, Hubei Province Natural Science Key Program (2014CFA074), the Chinese National Basic Research Programs (973 Programs 2013CB531101 and 2012CB517801), Hubei Province’s Outstanding Medical Academic Leader Program, Specialized Research Fund for the Doctoral Program of Higher Education from the Ministry of Education, and the “Innovative Development of New Drugs” Key Scientific Project (2011ZX09307-001-09).
Funding information National Natural Science Foundation of China (no. 81070080, 81370206, 81270162, 81630002, 91439129, and 31430047), 2016 Top-Notch Innovative Talent Development Project from the Bureau of Human Resources and Social Security of Wuhan City, NIH/NHLBI grants R01 HL094498, R01 HL121358 and R01 HL126729, Hubei Province Natural Science Key Program (2014CFA074), the Chinese National Basic Research Programs (973 Programs 2013CB531101 and 2012CB517801), Hubei Province’s Outstanding Medical Academic Leader Program Specialized Research Fund for the Doctoral Program of Higher Education from the Ministry of Education, and the “Innovative Development of New Drugs” Key Scientific Project (2011ZX09307-001-09).
Footnotes
CONFLICTS OF INTEREST
The authors declare that they have no conflict of interest.
SUPPORTING INFORMATION
Additional supporting information may be found online in the Supporting Information section at the end of the article.
REFERENCES
- Adhikari AS, Sridhar Rao K, Rangaraj N, Parnaik VK, & Mohan Rao C (2004). Heat stress-induced localization of small heat shock proteins in mouse myoblasts: Intranuclear lamin A/C speckles as target for alphaB-crystallin and Hsp25. Experimental Cell Research, 299(2), 393–403. 10.1016/j.yexcr.2004.05.032 [DOI] [PubMed] [Google Scholar]
- Afzal AR, Mandal K, Nyamweya S, Foteinos G, Poloniecki J, Camm AJ, … Xu Q (2008). Association of Met439Thr substitution in heat shock protein 70 gene with postoperative atrial fibrillation and serum HSP70 protein levels. Cardiology, 110(1), 45–52. 10.1159/000109406 [DOI] [PubMed] [Google Scholar]
- Allende M, Molina E, Guruceaga E, Tamayo I, Gonzalez-Porras JR, Gonzalez-Lopez TJ, … Hermida J (2016). Hsp70 protects from stroke in atrial fibrillation patients by preventing thrombosis without increased bleeding risk. Cardiovascular Research, 110(3), 309–318. 10.1093/cvr/cvw049 [DOI] [PubMed] [Google Scholar]
- Andalib A, Brugada R, & Nattel S (2008).Atrial fibrillation: Evidence for genetically determined disease. Current Opinion in Cardiology, 23(3), 176–183. 10.1097/HCO.0b013e3282fa7142 [DOI] [PubMed] [Google Scholar]
- Arimura T, Helbling-Leclerc A, Massart C, Varnous S, Niel F, Lacene E, … Bonne G (2005). Mouse model carrying H222P-Lmna mutation develops muscular dystrophy and dilated cardiomyopathy similar to human striated muscle laminopathies. Human Molecular Genetics, 14(1), 155–169. 10.1093/hmg/ddi017 [DOI] [PubMed] [Google Scholar]
- Arimura T, Onoue K, Takahashi-Tanaka Y, Ishikawa T, Kuwahara M, Setou M, … Kimura A (2013). Nuclear accumulation of androgen receptor in gender difference of dilated cardiomyopathy due to lamin A/C mutations. Cardiovascular Research, 99(3), 382–394. 10.1093/cvr/cvt106 [DOI] [PubMed] [Google Scholar]
- Beckmann BM, Holinski-Feder E, Walter MC, Haseruck N, Reithmann C, Hinterseer M, … Kaab S (2010). Laminopathy presenting as familial atrial fibrillation. International Journal of Cardiology, 145(2), 394–396. 10.1016/j.ijcard.2010.04.024 [DOI] [PubMed] [Google Scholar]
- Benjamin EJ, Levy D, Vaziri SM, D’Agostino RB, Belanger AJ, & Wolf PA (1994). Independent risk factors for atrial fibrillation in a population-based cohort. The framingham heart study. JAMA, 271(11), 840–844. [PubMed] [Google Scholar]
- Brundel BJ, Van Gelder IC, Henning RH, Tieleman RG, Tuinenburg AE, Wietses M, … Crijns HJ (2001). Ion channel remodeling is related to intraoperative atrial effective refractory periods in patients with paroxysmal and persistent atrial fibrillation. Circulation, 103(5), 684–690. [DOI] [PubMed] [Google Scholar]
- Busch A, Kiel T, Heupel WM, Wehnert M, & Hubner S (2009). Nuclear protein import is reduced in cells expressing nuclear envelopathy- causing lamin A mutants. Experimental Cell Research, 315(14), 2373–2385. 10.1016/j.yexcr.2009.05.003 [DOI] [PubMed] [Google Scholar]
- Capell BC, & Collins FS (2006). Human laminopathies: Nuclei gone genetically awry. Nature Reviews Genetics, 7(12), 940–952. 10.1038/nrg1906 [DOI] [PubMed] [Google Scholar]
- Chang SL, Chen YC, Hsu CP, Kao YH, Lin YK, Lai YJ, … Chen YJ (2013). Heat shock protein inducer modifies arrhythmogenic substrate and inhibits atrial fibrillation in the failing heart. International Journal of Cardiology, 168(4), 4019–4026. 10.1016/j.ijcard.2013.06.072 [DOI] [PubMed] [Google Scholar]
- Chen LY, Ballew JD, Herron KJ, Rodeheffer RJ, & Olson TM (2007). A common polymorphism in SCN5A is associated with lone atrial fibrillation. Clinical Pharmacology and Therapeutics, 81(1), 35–41. 10.1038/sj.clpt.6100016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Wang C, Wang X, Xu C, Wu M, Wang P, … Wang QK (2015). Significant association between CAV1 variant rs3807989 on 7p31 and atrial fibrillation in a Chinese han population. Journal of the American Heart Association, 4(5), e001980 10.1161/JAHA.115.001980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cowan J, Li D, Gonzalez-Quintana J, Morales A, & Hershberger RE (2010). Morphological analysis of 13 LMNA variants identified in a cohort of 324 unrelated patients with idiopathic or familial dilated cardiomyopathy. Circulation Cardiovascular Genetics, 3(1), 6–14. 10.1161/circgenetics.109.905422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darbar D, Kannankeril PJ, Donahue BS, Kucera G, Stubblefield T, Haines JL, … Roden DM(2008). Cardiac sodium channel (SCN5A) variants associated with atrial fibrillation. Circulation, 117(15), 1927–1935. 10.1161/circulationaha.107.757955 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Datta S, Snow CJ, & Paschal BM (2014). A pathway linking oxidative stress and the Ran GTPase system in progeria. Molecular Biology of the Cell, 25(8), 1202–1215. 10.1091/mbc.E13-07-0430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dialynas G, Flannery KM, Zirbel LN, Nagy PL, Mathews KD, Moore SA, & Wallrath LL (2012). LMNA variants cause cytoplasmic distribution of nuclear pore proteins in Drosophila and human muscle. Human Molecular Genetics, 21(7), 1544–1556. 10.1093/hmg/ddr592 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du W, Bautista JF, Yang H, Diez-Sampedro A, You SA, Wang L, … Wang QK(2005). Calcium-sensitive potassium channelopathy in human epilepsy and paroxysmal movement disorder. Nature Genetics, 37(7), 733–738. 10.1038/ng1585 [DOI] [PubMed] [Google Scholar]
- Fatkin D, MacRae C, Sasaki T, Wolff MR, Porcu M, Frenneaux M, … McDonough B (1999). Missense mutations in the rod domain of the lamin A/C gene as causes of dilated cardiomyopathy and conductionsystem disease. New England Journal of Medicine, 341(23), 1715–1724. 10.1056/nejm199912023412302 [DOI] [PubMed] [Google Scholar]
- Forleo C, Carmosino M, Resta N, Rampazzo A, Valecce R, Sorrentino S, … Favale S(2015). Clinical and functional characterization of a novel mutation in lamin a/c gene in a multigenerational family with arrhythmogenic cardiac laminopathy. PLoS One, 10(4), e0121723 10.1371/journal.pone.0121723 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franz C, Askjaer P, Antonin W, Iglesias CL, Haselmann U, Schelder M, … Mattaj IW (2005). Nup155 regulates nuclear envelope and nuclear pore complex formation in nematodes and vertebrates. EMBO Journal, 24(20), 3519–3531. 10.1038/sj.emboj.7600825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fuster V, Ryden LE, Asinger RW, Cannom DS, Crijns HJ, Frye RL, … Torbicki A (2001). ACC/AHA/ESC guidelines for the management of patients with atrial fibrillation: Executive summary. A report of the American college of cardiology/American heart association task force on practice guidelines and the European society of cardiology committee for practice guidelines and policy conferences (committee to develop guidelines for the management of patients with atrial fibrillation): Developed in collaboration with the North American society of pacing and electrophysiology. Journal of the American College of Cardiology, 38(4), 1231–1266. [DOI] [PubMed] [Google Scholar]
- Giudicessi JR, & Ackerman MJ (2012). Potassium-channel mutations and cardiac arrhythmias–diagnosis and therapy. Nature Reviews Cardiology, 9(6), 319–332. 10.1038/nrcardio.2012.3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruenbaum Y, Margalit A, Goldman RD, Shumaker DK, & Wilson KL (2005). The nuclear lamina comes of age. Nature Reviews Molecular Cell Biology, 6(1), 21–31. 10.1038/nrm1550 [DOI] [PubMed] [Google Scholar]
- Gupta P, Bilinska ZT, Sylvius N, Boudreau E, Veinot JP, Labib S, … Tesson F (2010). Genetic and ultrastructural studies in dilated cardiomyopathy patients: A large deletion in the lamin A/C gene is associated with cardiomyocyte nuclear envelope disruption. Basic Research in Cardiology, 105(3), 365–377. 10.1007/s00395-010-0085-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han SN, Jing Y, Yang LL, Zhang Z, & Zhang LR (2016). Propofol inhibits hERG K(+) channels and enhances the inhibition effects on its mutations in HEK293 cells. European Journal of Pharmacology, 791, 168–178. 10.1016/j.ejphar.2016.08.028 [DOI] [PubMed] [Google Scholar]
- Hawryluk-Gara LA, Shibuya EK, & Wozniak RW (2005). Vertebrate Nup53 interacts with the nuclear lamina and is required for the assembly of a Nup93-containing complex. Molecular Biology of the Cell, 16(5), 2382–2394. 10.1091/mbc.E04-10-0857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi K, Konno T, Tada H, Tani S, Liu L, Fujino N, … Yamagishi M (2015). Functional characterization of rare variants implicated in susceptibility to lone atrial fibrillation. Circulation: Arrhythmia and Electrophysiology, 8(5), 1095–1104. 10.1161/circep.114.002519 [DOI] [PubMed] [Google Scholar]
- Ho CY, Jaalouk DE, Vartiainen MK, & Lammerding J (2013). Lamin A/C and emerin regulate MKL1-SRF activity by modulating actin dynamics. Nature, 497(7450), 507–511. 10.1038/nature12105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodgson-Zingman DM, Karst ML, Zingman LV, Heublein DM, Darbar D, Herron KJ, … Olson TM (2008). Atrial natriuretic peptide frameshift mutation in familial atrial fibrillation. New England Journal of Medicine, 359(2), 158–165. 10.1056/NEJMoa0706300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holt I, Nguyen TM, Wehnert M, & Morris GE (2006). Lamin A/C assembly defects in Emery–Dreifuss muscular dystrophy can be regulated by culture medium composition. Neuromuscular Disorders, 16(6), 368–373. 10.1016/j.nmd.2006.03.014 [DOI] [PubMed] [Google Scholar]
- Hoogstra-Berends F, Meijering RA, Zhang D, Heeres A, Loen L, Seerden JP, … Brundel BJ (2012). Heat shock protein-inducing compounds as therapeutics to restore proteostasis in atrial fibrillation. Trends in Cardiovascular Medicine, 22(3), 62–68. 10.1016/j.tcm.2012.06.013 [DOI] [PubMed] [Google Scholar]
- Huang Y, Wang C, Yao Y, Zuo X, Chen S, Xu C, … Wang QK (2015). Molecular basis of gene-gene interaction: Cyclic crossregulation of gene expression and post-GWAS gene-gene interaction involved in atrial fibrillation. PLoS Genetics, 11(8), e1005393 10.1371/journal.pgen.1005393 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y, Wang Z, Liu Y, Xiong H, Zhao Y, Wu L, … Wang QK (2016). alphaB-crystallin interacts with Nav1.5 and regulates ubiquitination and internalization of cell surface Nav1.5. Journal of Biological Chemistry, 291(21), 11030–11041. 10.1074/jbc.M115.695080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hutchison CJ (2002). Lamins: Building blocks or regulators of gene expression? Nature Reviews Molecular Cell Biology, 3(11), 848–858. 10.1038/nrm950 [DOI] [PubMed] [Google Scholar]
- Kampinga HH, Henning RH, van Gelder IC, & Brundel BJ (2007). Beat shock proteins and atrial fibrillation. Cell Stress & Chaperones, 12(2), 97–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannel WB, Wolf PA, Benjamin EJ, & Levy D (1998). Prevalence, incidence, prognosis, and predisposing conditions for atrial fibrillation: Population-based estimates. American Journal of Cardiology, 82(8a), 2n–9n. [DOI] [PubMed] [Google Scholar]
- Karst ML, Herron KJ, & Olson TM (2008). X-linked nonsyndromic sinus node dysfunction and atrial fibrillation caused by emerin mutation. Journal of Cardiovascular Electrophysiology, 19(5), 510–515. 10.1111/j.1540-8167.2007.01081.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelley JB, Datta S, Snow CJ, Chatterjee M, Ni L, Spencer A, … Paschal BM (2011). The defective nuclear lamina in Hutchinson-gilford progeria syndrome disrupts the nucleocytoplasmic Ran gradient and inhibits nuclear localization of Ubc9. Molecular and Cellular Biology, 31(16), 3378–3395. 10.1128/mcb.05087-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar S, Baldinger SH, Gandjbakhch E, Maury P, Sellal JM, Androulakis AF, … Lakdawala NK (2016). Long-term Arrhythmic and nonarrhythmic outcomes of lamin A/C mutation carriers. Journal of the American College of Cardiology, 68(21), 2299–2307. 10.1016/j.jacc.2016.08.058 [DOI] [PubMed] [Google Scholar]
- Lammerding J, Schulze PC, Takahashi T, Kozlov S, Sullivan T, Kamm RD, … Lee RT (2004). Lamin A/C deficiency causes defective nuclear mechanics and mechanotransduction. Journal of Clinical Investigation, 113(3), 370–378. 10.1172/jci19670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanktree M, Cao H, Rabkin SW, Hanna A, & Hegele RA (2007). Novel LMNA mutations seen in patients with familial partial lipodystrophy subtype 2 (FPLD2; MIM 151660). Clinical Genetics, 71(2), 183–186. 10.1111/j.1399-0004.2007.00740.x [DOI] [PubMed] [Google Scholar]
- Mace LC, Yermalitskaya LV, Yi Y, Yang Z, Morgan AM, & Murray KT (2009). Transcriptional remodeling of rapidly stimulated HL-1 atrial myocytes exhibits concordance with human atrial fibrillation. Journal of Molecular and Cellular Cardiology, 47(4), 485–492. 10.1016/j.yjmcc.2009.07.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madej-Pilarczyk A, Kmiec T, Fidzianska A, Rekawek J, Niebroj-Dobosz I, Turska-Kmiec A, … Hausmanowa-Petrusewicz I (2008). Progeria caused by a rare LMNA mutation p.S143F associated with mild myopathy and atrial fibrillation. European Journal of Paediatric Neurology, 12(5), 427–430. 10.1016/j.ejpn.2007.11.011 [DOI] [PubMed] [Google Scholar]
- Makiyama T, Akao M, Shizuta S, Doi T, Nishiyama K, Oka Y, … Horie M (2008). A novel SCN5A gain-of-function mutation M1875T associated with familial atrial fibrillation. Journal of the American College of Cardiology, 52(16), 1326–1334. 10.1016/j.jacc.2008.07.013 [DOI] [PubMed] [Google Scholar]
- Markandeya YS, Tsubouchi T, Hacker TA, Wolff MR, Belardinelli L, & Balijepalli RC (2016). Inhibition of late sodium current attenuates ionic arrhythmia mechanism in ventricular myocytes expressing LaminA-N195K mutation. Heart Rhythm, 13(11), 2228–2236. 10.1016/j.hrthm.2016.08.007 [DOI] [PubMed] [Google Scholar]
- Marsman RF, Bardai A, Postma AV, Res JC, Koopmann TT, Beekman L, … Bezzina CR (2011). A complex double deletion in LMNA underlies progressive cardiac conduction disease, atrial arrhythmias, and sudden death. Circulation Cardiovascular Genetics, 4(3), 280–287. 10.1161/CIRCGENETICS.110.959221 [DOI] [PubMed] [Google Scholar]
- McClintock D, Gordon LB, & Djabali K (2006). Hutchinson-Gilford progeria mutant lamin A primarily targets human vascular cells as detected by an anti-Lamin A G608G antibody. Proceedings of the National Academy of Sciences of the United States of America, 103(7), 2154–2159. 10.1073/pnas.0511133103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mounkes LC, Kozlov SV, Rottman JN, & Stewart CL (2005). Expression of an LMNA-N195K variant of A-type lamins results in cardiac conduction defects and death in mice. Human Molecular Genetics, 14(15), 2167–2180. 10.1093/hmg/ddi221 [DOI] [PubMed] [Google Scholar]
- Nattel S, Maguy A, Le Bouter S, & Yeh YH (2007). Arrhythmogenic ion-channel remodeling in the heart: Heart failure, myocardial infarction, and atrial fibrillation. Physiological Reviews, 87(2), 425–456. 10.1152/physrev.00014.2006 [DOI] [PubMed] [Google Scholar]
- Nikolova V, Leimena C, McMahon AC, Tan JC, Chandar S, Jogia D, … Fatkin D (2004). Defects in nuclear structure and function promote dilated cardiomyopathy in lamin A/C-deficient mice. Journal of Clinical Investigation, 113(3), 357–369. 10.1172/jci19448 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olesen MS, Yuan L, Liang B, Holst AG, Nielsen N, Nielsen JB, … Svendsen JH (2012). High prevalence of long QT syndrome-associated SCN5A variants in patients with early-onset lone atrial fibrillation. Circulation Cardiovascular Genetics, 5(4), 450–459. 10.1161/circgenetics.111.962597 [DOI] [PubMed] [Google Scholar]
- Pan H, Richards AA, Zhu X, Joglar JA, Yin HL, & Garg V (2009). A novel mutation in LAMIN A/C is associated with isolated early-onset atrial fibrillation and progressive atrioventricular block followed by cardiomyopathy and sudden cardiac death. Heart Rhythm, 6(5), 707–710. 10.1016/j.hrthm.2009.01.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raharjo WH, Enarson P, Sullivan T, Stewart CL, & Burke B (2001). Nuclear envelope defects associated with LMNA mutations cause dilated cardiomyopathy and Emery-Dreifuss muscular dystrophy. Journal of Cell Science, 114(Pt 24), 4447–4457. [DOI] [PubMed] [Google Scholar]
- Raices M, & D’Angelo MA (2012). Nuclear pore complex composition: A new regulator of tissue-specific and developmental functions. Nature Reviews Molecular Cell Biology, 13(11), 687–699. 10.1038/nrm3461 [DOI] [PubMed] [Google Scholar]
- Remme CA (2013). Cardiac sodium channelopathy associated with SCN5A mutations: Electrophysiological, molecular and genetic aspects. Journal of Physiology, 591(17), 4099–4116. 10.1113/jphysiol.2013.256461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren X, Xu C, Zhan C, Yang Y, Shi L, Wang F, … Wang QK (2010). Identification of NPPA variants associated with atrial fibrillation in a Chinese GeneID population. Clinica Chimica Acta, 411(7–8), 481–485. 10.1016/j.cca.2009.12.019 [DOI] [PubMed] [Google Scholar]
- Rivera-Torres J, Calvo CJ, Llach A, Guzman-Martinez G, Caballero R, Gonzalez-Gomez C, … Jalife J (2016). Cardiac electrical defects in progeroid mice and Hutchinson-Gilford progeria syndrome patients with nuclear lamina alterations. Proceedings of the National Academy of Sciences of the United States of America, 113(46), E7250–E7259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux KJ, Kim DI, Raida M, & Burke B (2012). A promiscuous biotin ligase fusion protein identifies proximal and interacting proteins in mammalian cells. Journal of Cell Biology, 196(6), 801–810. 10.1083/jcb.201112098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saj M, Dabrowski R, Labib S, Jankowska A, Szperl M, Broda G, … Ploski R (2012). Variants of the lamin A/C (LMNA) gene in non- valvular atrial fibrillation patients: A possible pathogenic role of the Thr528Met mutation. Molecular Diagnosis & Therapy, 16(2), 99–107. 10.2165/11597540-000000000-00000 [DOI] [PubMed] [Google Scholar]
- Sakata K, Shimizu M, Ino H, Yamaguchi M, Terai H, Fujino N, … Mabuchi H (2005). High incidence of sudden cardiac death with conduction disturbances and atrial cardiomyopathy caused by a nonsense mutation in the STA gene. Circulation, 111(25), 3352–3358. 10.1161/CIRCULATIONAHA.104.527184 [DOI] [PubMed] [Google Scholar]
- Sanna T (2003). Cardiac features of Emery–Dreifuss muscular dystrophy caused by lamin A/C gene mutations. European Heart Journal, 24(24), 2227–2236. 10.1016/j.ehj.2003.09.020 [DOI] [PubMed] [Google Scholar]
- Schreiber KH, & Kennedy BK (2013). When lamins go bad: Nuclear structure and disease. Cell, 152(6), 1365–1375. 10.1016/j.cell.2013.02.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sebillon P, Bouchier C, Bidot LD, Bonne G, Ahamed K, Charron P, … Komajda M (2003). Expanding the phenotype of LMNA mutations in dilated cardiomyopathy and functional consequences of these mutations. Journal of Medical Genetics, 40(8), 560–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi L, Li C, Wang C, Xia Y, Wu G, Wang F, … Wang QK(2009). Assessment of association of rs2200733 on chromosome 4q25 with atrial fibrillation and ischemic stroke in a Chinese Han population. Human Genetics, 126(6), 843–849. 10.1007/s00439-009-0737-3 [DOI] [PubMed] [Google Scholar]
- Shumaker D (2003). The nucleoskeleton: Lamins and actin are major players in essential nuclear functions. Current Opinion in Cell Biology, 15(3), 358–366. 10.1016/s0955-0674(03)00050-4 [DOI] [PubMed] [Google Scholar]
- Sinner MF, Pfeufer A, Akyol M, Beckmann BM, Hinterseer M, Wacker A, … Kaab S (2008). The non-synonymous coding IKr-channel variant KCNH2-K897T is associated with atrial fibrillation: Results from a systematic candidate gene-based analysis of KCNH2 (HERG). European Heart Journal, 29(7), 907–914. 10.1093/eurheartj/ehm619 [DOI] [PubMed] [Google Scholar]
- Snow CJ, Dar A, Dutta A, Kehlenbach RH, & Paschal BM (2013). Defective nuclear import of Tpr in Progeria reflects the Ran sensitivity of large cargo transport. Journal of Cell Biology, 201(4), 541–557. 10.1083/jcb.201212117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rammos K, Koullias GJ, Hassan MO, Argyrakis NP, Voucharas CG, Scarupa SJ, & Cowte TG (2002). Low preoperative HSP70 atrial myocardial levels correlate significantly with high incidence of postoperative atrial fibrillation after cardiac surgery. Cardiovascular Surgery, 10(3), 228–232. [DOI] [PubMed] [Google Scholar]
- Stierle V, Couprie J, Ostlund C, Krimm I, Zinn-Justin S, Hossenlopp P, … Duband-Goulet I(2003). The carboxyl-terminal region common to lamins A and C contains a DNA binding domain. Biochemistry, 42(17), 4819–4828. 10.1021/bi020704g [DOI] [PubMed] [Google Scholar]
- Stochaj U, Rassadi R, & Chiu J (2000). Stress-mediated inhibition of the classical nuclear protein import pathway and nuclear accumulation of the small GTPase Gsp1p. Faseb Journal, 14(14), 2130–2132. 10.1096/fj.99-0751fje [DOI] [PubMed] [Google Scholar]
- Strelkov SV, Schumacher J, Burkhard P, Aebi U, & Herrmann H (2004). Crystal structure of the human lamin A coil 2B dimer: Implications for the head-to-tail association of nuclear lamins. Journal of Molecular Biology, 343(4), 1067–1080. 10.1016/j.jmb.2004.08.093 [DOI] [PubMed] [Google Scholar]
- Tsai CT, Lai LP, Hwang JJ, Lin JL, & Chiang FT (2008). Molecular genetics of atrial fibrillation. Journal of the American College of Cardiology, 52(4), 241–250. 10.1016/j.jacc.2008.02.072 [DOI] [PubMed] [Google Scholar]
- Tucker NR, & Ellinor PT (2014). Emerging directions in the genetics of atrial fibrillation. Circulation Research, 114(9), 1469–1482. 10.1161/CIRCRESAHA.114.302225 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Berlo JH, de Voogt WG, van der Kooi AJ, van Tintelen JP, Bonne G, Yaou RB, … Pinto YM (2005). Meta-analysis of clinical characteristics of 299 carriers of LMNA gene mutations: Do lamin A/C mutations portend a high risk of sudden death? Journal of Molecular Medicine (Berlin, Germany), 83(1), 79–83. 10.1007/s00109-004-0589-1 [DOI] [PubMed] [Google Scholar]
- van Rijsingen IA, Nannenberg EA, Arbustini E, Elliott PM, Mogensen J, Hermans-van Ast JF, … Pinto YM (2013). Gender-specific differences in major cardiac events and mortality in lamin A/C mutation carriers. European Journal of Heart Failure, 15(4), 376–384. 10.1093/eurjhf/hfs191 [DOI] [PubMed] [Google Scholar]
- van Tintelen JP, Hofstra RM, Katerberg H, Rossenbacker T, Wiesfeld AC, du Marchie Sarvaas GJ, … Working Group on Inherited Cardiac Disorders, line 27/50, Interuniversity Cardiology Institute of The Netherlands (2007) High yield of LMNA mutations in patients with dilated cardiomyopathy and/or conduction disease referred to cardiogenetics outpatient clinics. American Heart Journal, 154(6), 1130–1139. 10.1016/j.ahj.2007.07.038 [DOI] [PubMed] [Google Scholar]
- Wang C, Wu M, Qian J, Li B, Tu X, Xu C, … Wang QK (2016). Identification of rare variants in TNNI3 with atrial fibrillation in a Chinese GeneID population. Molecular Genetics and Genomics, 291(1), 79–92. 10.1007/s00438-015-1090-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willsie JK, & Clegg JS (2002). Small heat shock protein p26 associates with nuclear lamins and HSP70 in nuclei and nuclear matrix fractions from stressed cells. Journal of Cellular Biochemistry, 84(3), 601–614. [PubMed] [Google Scholar]
- Wolf CM, Wang L, Alcalai R, Pizard A, Burgon PG, Ahmad F, … Seidman JG (2008). Lamin A/C haploinsufficiency causes dilated cardiomyopathy and apoptosis-triggered cardiac conduction system disease. Journal of Molecular and Cellular Cardiology, 44(2), 293–303. 10.1016/j.yjmcc.2007.11.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf PA, Benjamin EJ, Belanger AJ, Kannel WB, Levy D, & D’Agostino RB (1996). Secular trends in the prevalence of atrial fibrillation: The framingham study. American Heart Journal, 131(4), 790–795. [DOI] [PubMed] [Google Scholar]
- Wu L, Yong SL, Fan C, Ni Y, Yoo S, Zhang T, … Wang QK (2008). Identification of a new co-factor, MOG1, required for the full function of cardiac sodium channel Nav 1.5. Journal of Biological Chemistry, 283(11), 6968–6978. 10.1074/jbc.M709721200 [DOI] [PubMed] [Google Scholar]
- Yue L, Melnyk P, Gaspo R, Wang Z, & Nattel S (1999). Molecular mechanisms underlying ionic remodeling in a dog model of atrial fibrillation. Circulation Research, 84(7), 776–784. [DOI] [PubMed] [Google Scholar]
- Zhang X, Chen S, Yoo S, Chakrabarti S, Zhang T, Ke T, … Wang QK (2008). Mutation in nuclear pore component NUP155 leads to atrial fibrillation and early sudden cardiac death. Cell, 135(6), 1017–1027. 10.1016/j.cell.2008.10.022 [DOI] [PubMed] [Google Scholar]
- Zhao J, Yao H, Li Z, Wang L, Liu G, Wang DW, … Liang Z (2016). A novel nonsense mutation in LMNA gene identified by Exome Sequencing in an atrial fibrillation family. The European Journal of Medical Genetics, 59(8), 396–400. 10.1016/j.ejmg.2016.06.006 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.