

Abstract
The drug‐induced liver injury (DILI)‐sim Initiative is a public‐private partnership involving scientists from industry, academia, and the US Food and Drug Administration (FDA). The Initiative uses quantitative systems toxicology (QST) to build and refine a model (DILIsym) capable of understanding and predicting liver safety liabilities in new drug candidates and to optimize interpretation of liver safety biomarkers used in clinical studies. Insights gained to date include the observation that most dose‐dependent hepatotoxicity can be accounted for by combinations of just three mechanisms (oxidative stress, interference with mitochondrial respiration, and alterations in bile acid homeostasis) and the importance of noncompetitive inhibition of bile acid transporters. The effort has also provided novel insight into species and interpatient differences in susceptibility, structure‐activity relationships, and the role of nonimmune mechanisms in delayed idiosyncratic hepatotoxicity. The model is increasingly used to evaluate new drug candidates and several clinical trials are underway that will test the model's ability to prospectively predict liver safety. With more refinement, in the future, it may be possible to use the DILIsym predictions to justify reduction in the size of some clinical trials. The mature model could also potentially assist physicians in managing the liver safety of their patients as well as aid in the diagnosis of DILI.
The drug‐induced liver injury (DILI)‐sim Initiative is a public‐private partnership applying quantitative systems toxicology (QST) methods to understand and predict liver safety liability in new drug candidates. The effort has provided new insights into mechanisms underlying hepatotoxicity, which have implications for species differences and patient risk factors. The modeling has also helped optimize interpretation of serum liver chemistries routinely used to assess liver safety.
The DILI‐sim Initiative
The DILI‐sim Initiative officially began in 2011 as a public‐private partnership that has involved scientists from academia, 19 major pharmaceutical companies, and the US Food and Drug Administration (FDA).1 The Initiative addresses the concern that new drug candidates continue to be abandoned late in clinical development due to liver safety concerns that were not suspected in preclinical models. The Initiative partners feel that a consortium is an appropriate way to pool resources, unpublished data, and expertise to solve problems with liver safety. The priorities and direction of the Initiative are determined by partner vote assuring that the effort addresses the most relevant needs in drug development. Current industry commitments to the Initiative will ensure its existence until at least 2021. The Initiative applies QST to understand and predict DILI liability in new drug candidates. When the DILI‐sim Initiative was started, QST was a relatively new field but applications have greatly expanded since then.2
QST uses differential equations to recapitulate relevant pathways whereby drugs or other chemicals can cause stress and death to cells, tissues, and organs. In the QST model developed by DILI‐sim Initiative, these pathways are built as submodels, and the submodels are connected with the outcome of hepatocyte death and release of biomarkers into serum. Figure 1 gives the overview of the submodels. Hepatocyte regeneration in response to injury is also built into the model, which has been named DILIsym (DILIsym is a trademark registered to DILIsym Services Inc.). The modeling approach used to develop DILIsym has been termed “middle out” because the modeling initially focuses on the organ toxicity rather than subcellular events that might lead to toxicity (i.e., “bottom up” modeling). Only cellular mechanisms necessary to account for the observed liver toxicity are incorporated into the model. Model parameters are varied to create simulated patient populations. There are mouse, rat, and dog, as well as human versions of the model.3, 4 The first drug modeled was acetaminophen, in which glutathione depletion and oxidative stress could account for toxicity observed with overdose in rodents and humans. The modeling was used to propose the optimal protocol for treatment of acetaminophen overdoses with N‐acetyl cysteine.5 The modeling was also used to evaluate several hypotheses for why an isomer of acetaminophen, which also generates reactive metabolites, is much less toxic than acetaminophen in mice.6
Figure 1.
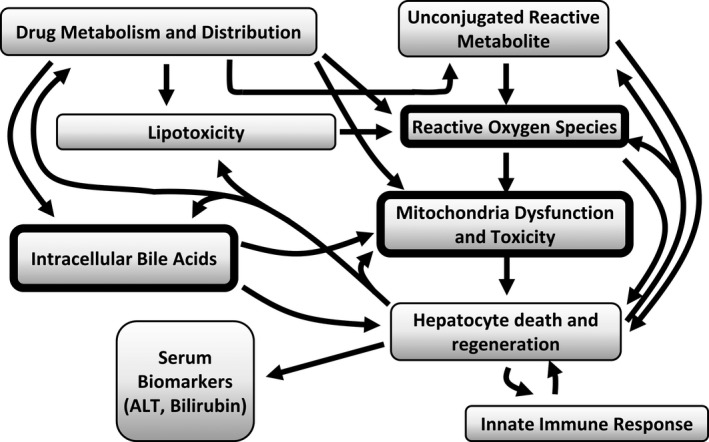
The major submodels that currently comprise the DILIsym software. The model includes production of reactive metabolites, generation of reactive oxygen species (ROS; oxidative stress), mitochondrial dysfunction, accumulation of toxic bile acids within the hepatocytes, lipotoxicity, and activation of an innate immune response. These processes are integrated with the potential outcome of hepatocyte death by either apoptosis or necrosis. This results in release into circulation of traditional biomarkers, including alanine aminotransferase (ALT), as well as nontraditional biomarkers, such as glutamate dehydrogenase and microRNA 122. Hepatocyte regeneration in response to hepatocyte loss is incorporated in the model and the functioning hepatocyte mass determines global liver function at any point in time. When loss of hepatocyte mass reaches 30%, serum bilirubin rises due to loss of global liver function. One of the surprising observations that has evolved from application of the software is that just three mechanisms, which can be commercially assessed (those within the thick‐lined boxes), can account for hepatotoxicity in rats and humans for > 80% of the drugs in the validation cohort tested to date. The model also includes some adaptation mechanisms that reduce injury, including farnesoid X receptor activation by bile acids, mitochondrial biogenesis initiated by adenosine tri‐phosphate (ATP) reduction, nuclear factor erythroid 2–related factor 2 (NRF‐2) response leading to faster glutathione synthesis, faster ROS clearance, and also faster liver regeneration with increased injury. Mitochondrial biogenesis and ROS clearance upregulation by NRF‐2 are still being optimized in the model.
Using DILIsym
Data put into the model
The way DILIsym is typically used to assess the liver safety liability of a drug is illustrated in Figure 2. The exposure of the drug inside the hepatocyte during dosing is obviously a key variable, and this is estimated by physiologically based pharmacokinetic (PBPK) modeling based on available data. If blood level monitoring data are available, they can be used to validate and/or further refine the constructed PBPK model. To predict hepatic concentrations of drugs, the partitioning to the liver can be estimated using physiochemical properties of the drug or preclinical tissue distribution data. If hepatic uptake of drugs by transporters is known, this is also taken into consideration.
Figure 2.
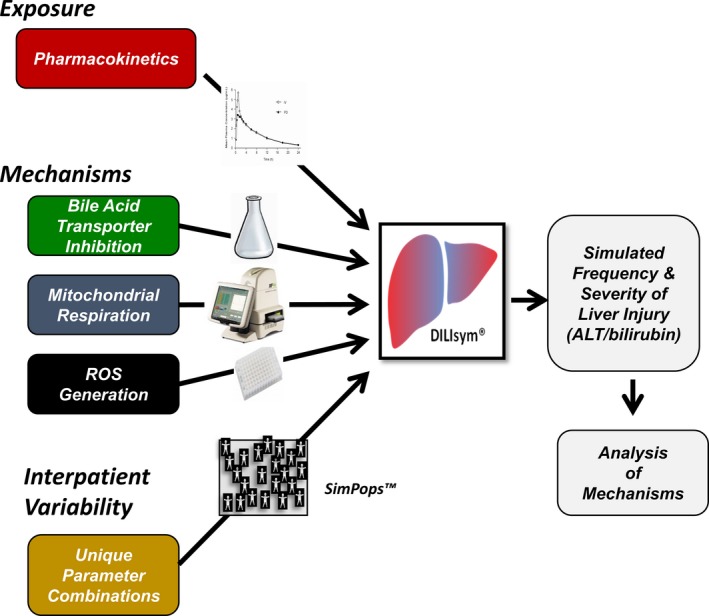
Application of the DILIsym model to predict hepatotoxicity. Extrahepatocyte and intrahepatocyte exposure to study drug is predicted by physiologically based pharmacokinetic modeling (see text). The three mechanisms listed are typically assessed from the dose‐dependent effects of drug and major metabolites on: (i) bile acid transporters using membrane vesicles, cell lines overexpressing transporters, or hepatocytes; (ii) mitochondrial respiration using the Seahorse instrument; and (iii) reactive oxygen species (ROS) generation measured with high content imaging. The collected exposure estimates and mechanistic data are put into the model, which will then predict the time‐dependent death of hepatocytes and, hence, the time‐dependent release of biomarkers into serum. Simulated patient populations have been created by changing parameters in the model to capture interpatient variation due to genetic or nongenetic factors. This permits estimates of the frequency as well as the extent of liver injury that can be anticipated in a real patient population receiving the drug. It is often possible to vary dosing and liver chemistry monitoring parameters to define protocols predicted to prevent serious DILI. ALT, alanine aminotransferase.
The drug is then tested for its ability to interfere with the key processes in the submodels, specifically the concentration‐dependent ability to: (i) inhibit bile acid transporters and thereby raise hepatocyte bile acid concentration, (ii) inhibit mitochondrial respiration, and (iii) cause oxidative stress. There are multiple hepatocyte transporters that can influence the intrahepatocyte concentration of bile acids,7 and the ability of a drug to inhibit each of these transporters (generally expressed in vesicles) is assayed. The ability to inhibit mitochondrial respiration and to generate oxidative stress has been typically measured in HepG2 cells using the Seahorse instrument and high content imaging, respectively. The predictions of the current model are dependent on these methods, which have been chosen by the DILI‐sim Initiative partners because they are commercially available if not already up and running in their organizations. In addition to assessing the effect of the drug as a function of media concentration, the intracellular drug concentration is also assessed using mass spectroscopy. If major metabolites are available, these typically also undergo these assays.
Data output from the model
The collected data together with estimates of hepatocyte concentration of the drug and major metabolites are input, and the model will then predict the time‐dependent death of hepatocytes, and, hence, the time‐dependent release of certain biomarkers into serum. The biomarker of most interest is generally serum alanine aminotransferase (ALT) because this is the most sensitive and specific traditional biomarker for hepatocyte death. Serum total bilirubin is also assessed, as this is an important parameter of global liver function. Nontraditional biomarkers,8 such as glutamate dehydrogenase, microRNA 122, full length and the caspase cleaved fragment of cytokeratin 18, are also incorporated in the model. Simulated patient populations have been created by changing parameters related to each mechanism to capture interpatient variation due to genetic or nongenetic factors. Simulated populations (SimPops (trademark registered to DILIsym Services Inc.)) currently include healthy volunteers, patients with nonalcoholic steatohepatitis (NASH), and patients with diabetes. Where possible, the extent of variation in each parameter is based on literature data, such as mitochondrial enzyme activities measured in liver biopsies from patients with NASH.9 In addition, some of the parameters in simulated patient populations have been modified over time to fit the data obtained in actual clinical trials. Pharmacokinetic variability is not built into the SimPops because this will be largely drug‐specific. However, pharmacokinetic variability can be added to the PBPK model inputs. A typical simulated population contains roughly 300 individuals, but the variation in susceptibility in the simulated population is designed to exceed what would be encountered in a far larger population of patients.
One useful output of the model is an eDISH plot.10 The eDISH stands for evaluation of drug‐induced serious hepatotoxicity and is a way the FDA evaluates liver safety of new drug candidates in clinical trials. The eDISH graphs the peak serum ALT value and peak serum bilirubin value observed in each patient in a clinical trial (along the x and y‐axis, respectively). DILIsym creates this same graph for the peak serum ALT and bilirubin values predicted for each simulated subject in the SimPops.
DILIsym and decision making in drug development
The DILIsym model is increasingly used to help decision making within pharmaceutical companies and, to date, DILIsym modeling has been presented in 20 different communications with the regulatory agencies (Brett Howell, personal communication). The FDA has also recently licensed the software (https://www.businesswire.com/news/home/20180508005079/en/FDA-Procures-DILIsym-Software-License-Package). When the model predicts serum ALT elevations, it is possible to determine a dosing protocol that would not cause or minimize ALT elevations. If dosing cannot be reduced to eliminate ALT elevations, the frequency of liver chemistry monitoring and stopping criteria based on ALT value can also be varied in the model to determine the optimal protocol to avoid serious liver injury. The use of DILIsym in this way has been applied to several new drugs, including an antibiotic.11 Another application of DILIsym has been to assess the safety of next‐in‐class drugs when first‐in‐class drugs had liver safety liability (e.g., tolcapone and entacapone,12 troglitazone and pioglitazone,13 and tolvaptan and lixivaptan14). There are prospective clinical trials now well underway of novel, next‐in‐class drugs with dosing and/or monitoring protocols predicted by DILIsym to be safe. The outcome of these trials will be an important test for the model.15
Insights into Mechanisms Causing DILI
Three mechanisms account for most dose‐dependent DILI
The DILIsym model was built by using data available in the literature, unpublished data provided by partners in the Initiative, and from new studies sponsored by the Initiative. These data were from drugs that had preclinical liver safety signals as well as those that did not and from drugs that had liver safety signals in the clinical trials and those that did not. To recapitulate the known safety profile of each “exemplar” drug, the model parameters were optimized. Once the model was optimized in this way, the Initiative began testing a new “validation” set of drugs where the preclinical and clinical safety profiles were known. As of December 2018, 59 molecules have been prospectively tested with 80% success in identifying the presence or absence of a liver safety liability at the administered dosing (Brett Howell, personal communication). Among the 20% failure rate, only one molecule was predicted to cause liver toxicity when none was observed in clinical trials (i.e., a false‐positive); all other failures were predictions of safety when toxicity was observed (i.e., false‐negatives).
It was unexpected that just three mechanisms account for prediction success: bile acid accumulation, interference with mitochondrial respiration, and oxidative stress (Figure 1 ). Once liver safety liability is identified, it is possible to use the model to identify which of the three mechanisms is most contributing to the predicted toxicity. This is done by simply turning off in the model each of the three mechanisms, one at a time, and observing what this does to the predicted frequency of serum ALT elevations in the simulated population. Typically, no one mechanism accounts for the observed toxicity, and there are instances where at least two mechanisms must be operative to produce any toxicity.16 There are as yet unpublished examples of where knowing the major mechanism underlying the toxicity of a drug has explained drug–drug interactions causing increased frequency of elevations in serum ALT in clinical trials (Brett Howell, personal communication).
The prominence of the three mechanisms in accounting for toxicity is remarkable because none directly take into account some DILI mechanisms that are generally recognized to be important, such as reactive metabolite production17 or endoplasmic reticulum stress.18 Such mechanisms may account for the roughly 20% failure rate of the current model predictions and addition of new mechanisms to DILIsym is likely in the future. It is also possible that there exist correlations with the three mechanisms in the model, such that those “left out” are indirectly taken into account. For example, a reactive metabolite may produce oxidative stress and oxidative stress can result in endoplasmic reticulum stress. It should also be noted that parent and major metabolites have been routinely tested in HepG2 cells, which lack most of the drug metabolism capability of hepatocytes. The role of unrecognized metabolites may, therefore, account in part for the 20% prediction failure rate. The Initiative has begun to collect mitochondrial inhibition and oxidative stress data in culture systems containing spheroids of HepaRG cells and primary human hepatocytes with and without nonparenchymal cells. In one case involving a molecule that was a false‐negative with traditional DILIsym inputs (Figure 2), time‐dependent appearance of oxidative stress was noted in these spheroids, likely reflecting a role for unrecognized metabolites (Merrie Mosedale, personal communication). Modifications of DILIsym to incorporate data from these culture systems are being explored.
Structurally similar drugs can have markedly different mechanisms underlying DILI
It seems likely that sometime in the future it will be possible to examine the three‐dimensional structure of a drug and accurately predict its effects on the three major pathways in DILIsym. However, when drugs with nearly identical structures have been modeled, the major mechanisms accounting for liver safety signals have often not been the same. This has been best shown with the macrolide antibiotics clarithromycin and erythromycin, which have only very small differences in structure. However, the major mechanism contributing to clarithromycin toxicity in the model was inhibition of mitochondrial respiration, whereas the major mechanism contributing to erythromycin toxicity was bile acid accumulation.19 Likewise, solithromycin and telithromycin are ketolide antibiotics with nearly identical structure, but the dominant mechanisms of toxicity in the model for solithromycin was inhibition of mitochondrial respiration whereas the model failed to predict the liver safety liability known for telithromycin19 (i.e., telithromycin is among the ~20% of model prediction failures in the validation cohort).
The importance of bile salt export pump inhibition as a DILI mechanism
Although bile salt export pump (BSEP) is the major transporter of bile acids into bile, there has been some recent controversy regarding the role of BSEP inhibition as a cause for liver toxicity.20 One argument against a role for BSEP inhibition is that the half‐maximal inhibitory concentration (IC50; i.e., the concentration of drug that results in a 50% reduction in bile acid transport in BSEP expressed in vesicles) is a poor predictor of liver safety liability when considered in isolation. DILIsym modeling has provided new insights into the multiple factors in addition to the BSEP IC50 that determine intrahepatocyte concentration of bile acids. For example, drugs that inhibit BSEP often also inhibit Na+‐taurocholate cotransporting polypeptide, the major uptake pump for bile acids, resulting in reduced bile acid uptake, which may offset the effects of BSEP inhibition. Alternatively, inhibition of the basolateral efflux pumps, Multidrug resistance‐associated protein (MRP) 3 and MRP4, can significantly contribute to bile acid accumulation when inhibition of BSEP is modest. The success of DILIsym in predicting hepatotoxicity by integrating drug effects on multiple bile acid transporters is the strongest support for the role of bile acid accumulation as a cause of human hepatotoxicity.13, 16, 21, 22 Importantly, the modeling has identified key data gaps to help prioritize research in the field.23
Another issue brought to light by the modeling was the importance of the mechanism whereby a drug inhibits BSEP. If the mechanism is simple competitive inhibition, the accumulation of bile acids within the hepatocyte may not reach toxic levels. This is because as the concentration of bile acids rises within the hepatocyte, the bile acids may “out compete” (displace) the inhibitor, maintaining bile acid transport into bile. In this case, the toxic threshold concentration in the hepatocyte may not be reached. On the other hand, if the mechanism of inhibition is noncompetitive, the rising intrahepatocyte concentration of bile acids will not displace the inhibitor, making achievement of toxic concentrations of bile acids more likely. The importance of determining the mechanism of BSEP inhibition has been illustrated by several drugs modeled in DILIsym, including TAK‐875.16
Finally, it has recently been proposed that if the IC50 for inhibition of the BSEP is > 25 μM, bile acid accumulation can be generally excluded as a relevant mechanism.24 This is probably the case with most drugs and where oxidative stress and mitochondrial impairment are not involved. However, DILIsym modeling has shown with certain drugs and metabolites with BSEP IC50 values > 25 μM, bile acid accumulation can still contribute significantly to toxicity if one or both of the other mechanisms are operative. This was the case with modeling tolvaptan hepatotoxicity.22
Identification of mechanisms underlying species differences in susceptibility to DILI
It has long been appreciated that species differences in metabolism may underlie differences in susceptibility to hepatotoxicity. DILIsym modeling has also provided additional explanations for species differences in susceptibility on the basis of the three mechanisms. It has been proposed that rats have low susceptibility to bile acid–mediated hepatotoxicity because their profile of bile acids is inherently less toxic than is the case in humans. DILIsym modeling has provided further support for this idea.13 In addition, a recent study indicated that rats were more sensitive to inhibition of mitochondrial respiration from a chemokine receptor antagonist and that this contributed to the drug causing liver toxicity in rats but not in humans.25
Mechanisms underlying idiosyncratic DILI
It is increasingly appreciated that delayed, idiosyncratic hepatotoxicity is frequently the result of an adaptive immune attack on the liver.26 However, it is important to note that DILIsym successfully predicted the liver safety liability of three drugs that cause delayed idiosyncratic DILI, troglitazone,13 tolvaptan,22 and TAK‐875.16 This supports the generally accepted concept that drug‐induced hepatocyte stress is necessary to stimulate an adaptive immune attack on the liver26 and that this stress may be generally caused by the three mechanisms assayed in DILIsym.27 It is also interesting that the DILIsym predicted the 3–4‐month delay in achieving peak serum ALT values during troglitazone treatment, mirroring the results in the clinical trials.13 This may suggest that delayed presentation of hepatotoxicity can occur without involving an adaptive immune response. Nonetheless, until adaptive immune mechanisms are incorporated into DILIsym, the model cannot be considered reliable to assess idiosyncratic DILI liability.
Optimization of DILI Biomarker Interpretation
Assessing hepatocyte loss during DILI
Current FDA guidelines and treatment modification guidelines in clinical protocols are based on the peak serum ALT and bilirubin values observed. In some instances, use of DILIsym has improved interpretation of elevations in serum ALT and bilirubin.28 DILIsym predicts the time‐dependent death of hepatocytes and from that can predict the time‐dependent concentration of serum biomarkers, typically ALT (Figure 1). The model can be used in reverse. That is, when elevations in serum ALT are observed in the clinic (or in animals), the model can be optimized to recapitulate the observed ALT vs. time curve, and the percent of hepatocytes that died (and released ALT) can therefore be estimated. Regeneration of hepatocytes in response to hepatocyte death is built into the model such that the mass of viable and functioning hepatocytes is predicted at any point in time. The regeneration rate does not influence the ALT kinetics but can be very important in determining liver function (and potentially fatal outcome), especially for prolonged liver injuries.
One example of the use of DILIsym in the interpretation of serum ALT elevations involved entolimod, a Toll‐like receptor 5 agonist shown to reduce mortality from radiation in monkeys.29 This observation in monkeys satisfied the “animal rule” for FDA approval because a clinical trial in people exposed to lethal radiation is not an option. However, the safety of the drug still had to be established in healthy volunteers. When this trial was undertaken, some subjects experienced alarmingly high elevations in serum ALT (one subject's serum ALT exceeded 1,000 U/L); these elevations were interpreted as severe liver injury, and the trial was halted. However, in each subject, the serum ALT rose almost immediately to the peak value and then fell at approximately the published half‐life of ALT in serum, suggesting a very short duration of hepatocyte death. Using DILIsym,30 it was estimated that in the most affected subject, only 2.6–4.9% of the total liver hepatocytes had been lost (the range reflects variation in published values for both the half‐life of serum ALT and estimates of hepatocyte content of ALT built into the model). The modeling with DILIsym, therefore, indicated that the elevations in serum ALT produced by entolimod, although far above typical criteria for treatment discontinuation, were in fact likely to reflect relatively minor liver injury.
Another example where DILIsym modeling of hepatocyte loss reduced liver safety concern involved cimaglermin alfa,31 a proposed biological treatment for patients with heart failure. An early clinical trial was put on hold because two patients experienced concomitant elevations in both serum ALT and serum bilirubin to levels satisfying FDA criteria for liver injury sufficient to cause global liver dysfunction (serum values for ALT and bilirubin exceeding 3 and 2 times the upper limits of normal (ULN), respectively). Clinical trial subjects who experienced concomitant elevations of serum ALT and bilirubin to these levels due to study drug are termed “Hy's Law Cases,” which are considered the most reliable indicators that the drug can cause acute liver failure. In this case, the serial serum samples from these patients were archived and assayed for cytokeratin 18 and caspase‐cleaved K18. The results suggested that the predominant mode of hepatocyte death was apoptosis rather than necrosis.31 Because ALT is partially digested during apoptosis, the predominance of apoptosis is taken into account in DILIsym, and the hepatocyte loss in these two patients was estimated to be 6.6–12.4%. Based on a liver biopsy study performed in patients with severe liver injuries due to acetaminophen overdose,32 at least 30% of hepatocytes must be lost in a DILI event before the serum bilirubin will rise to greater than 2X ULN. The conclusion from DILIsym modeling was that the greatest liver injury produced by cimaglermin alfa in the clinical trial was insufficient to cause the degree of global liver dysfunction required to raise serum bilirubin to 2X ULN (Figure 3). Subsequent toxicogenomic studies performed in cultured human hepatocytes suggested that effects of the drug on gene expression of bilirubin transporters may account for the rise in serum bilirubin observed.33 It should also be noted that factors influencing bilirubin homeostasis have been incorporated into DILIsym.34 This has allowed prediction of serum bilirubin elevations due to inhibition of bilirubin transporters or inhibition of uridine 5′‐diphosphate glucuronosyltransferase 1A1 in the absence of overt liver injury. In summary, DILIsym modeling combined with the toxicogenomic studies supported the conclusion that the rise in serum bilirubin observed in the two patients was not due to global liver dysfunction and that these two subjects should not be considered Hy's Law Cases.
Figure 3.
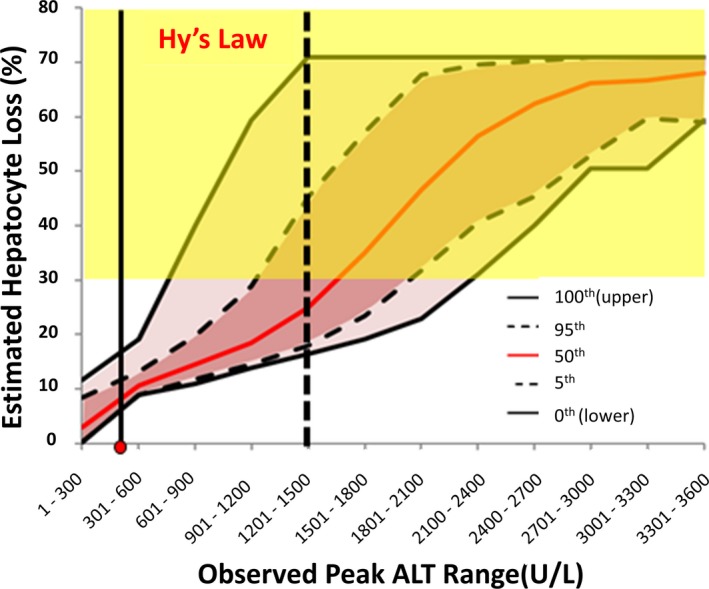
Modeled percent hepatocyte loss vs. peak serum alanine aminotransferase (ALT) caused by cimaglermin alfa. The relationship between peak serum ALT and estimated range of hepatocyte loss was determined using DILIsym in a simulated population receiving the drug. Because the pattern of rise and fall in serum ALT was characteristic among all subjects experiencing ALT rises, peak ALT elevations much higher than actually observed in the clinical trial were modeled. The modeling incorporates variability in factors relevant to ALT dynamics and hepatocyte regeneration creating the confidence intervals for percent hepatocyte loss observed in the simulated subjects as a function of peak serum ALT value observed. A rise in serum total bilirubin (TBIL) > 2X upper limits of normal (ULN; a criteria for a “Hy's Law Case”) requires at least a 30% reduction in hepatocytes (range shown in yellow). It can be seen that the maximum peak serum ALT values observed in the clinical trial (shown as the red dot on the X‐axis) cannot alone account for the rise in serum TBIL > 2X ULN that was observed in this subject and supports that this subject should not be considered as Hy's Law Case. The figure also indicates that the ability of a drug to cause a Hy's Law Case can be estimated from the probability that a given peak serum ALT would reflect a 30% reduction in functioning hepatocytes. For example, a subject with a peak serum ALT value of between 1,201 and 1,500 IU/L (vertical dotted black line) is predicted by DILIsym to have a < 50% chance (as indicated by 50th percentile red line) of exhibiting a rise in serum TBIL > 2X ULN. However, the 95% confidence interval clearly includes sufficient hepatocyte loss to result in a rise in serum TBIL > 2X ULN. It may, therefore, be appropriate for this subject to be considered a Hy's Law Case even if the currently accepted biochemical criteria were not achieved. It should be noted that the ALT elevations caused by many drugs have similar characteristics across the affected population, so this approach should be applicable to other drugs causing serum ALT elevations in clinical trials. Reproduced with permission from ref. 28. Copyright © 2017 SAGE Publications. https://doi.org/10.1177/1535370217740853.
Predicting severe liver toxicity before it occurs
Because the rates of rise and fall in serum ALT observed in the cimaglermin alfa clinical trial were similar in all patients experiencing ALT elevations, it is reasonable to assume that if patients experienced more serious liver injuries due to this drug, the rise and fall of serum ALT would be similar. It was therefore possible to use DILIsym to estimate the peak serum ALT value that would correspond to sufficient hepatocyte loss to result in an elevation in serum bilirubin > 2X ULN (Figure 3). Furthermore, because of variation of the relevant parameters in the simulated populations (e.g., range of published serum half‐life of ALT), it is possible to estimate for any peak serum ALT value the probability that a given patient would (on the basis of global liver dysfunction) experience a rise in serum bilirubin > 2X ULN. It is often the case that, like cimaglermin alfa, a given drug will cause a characteristic pattern of ALT elevations in terms of the rates of rise and fall such that the approach shown in Figure 3 could be used to assess liver safety. It may, therefore, not be necessary to actually observe a Hy's Law Case in a clinical trial to indicate a new drug's potential to produce liver failure. For example, if modeling serial serum ALT values observed in a real patient indicate that 25% of simulated patients with those values would experience a concomitant rise in serum total bilirubin > 2X ULN, it may not be necessary to actually observe a Hy's Law Case in the clinical trial to assume that a drug could cause liver failure. This modeling approach could be important because true Hy's Law Cases have serious liver injury that despite discontinuing treatment with the study drug may rarely progress to liver failure, and this has happened in clinical trials.35 Recognizing potential for causing liver failure early in the course of the injury would be an important advance.
Future Directions
To date, applications of DILIsym have been primarily directed at dose‐dependent toxicity to hepatocytes. The just‐released DILIsym version 8A incorporates some aspects of dose‐dependent toxicity to cholangiocytes, which results in release of alkaline phosphatase into serum. This type of injury is increasingly recognized as a cause of prolonged illness and, rarely, liver failure.36 Drug‐induced cholangiocyte toxicity can result from direct toxicity to these cells by the drug or its metabolites secreted into bile.37 It is also believed to occur indirectly as a result of drugs interfering with biliary transport of micelle components that encapsulate bile acids in bile. One mechanism of particular interest is drug inhibition of the canalicular transporter MDR3 resulting in reduced phospholipid secretion into bile.38 Poor micellation of bile acids should lead to the presence of “naked” bile acids in bile concentrations that would be toxic to cholangiocytes. Onging DILIsym modeling should provide fresh insight into these mechanisms.
Kupffer cell and recruited macrophage activation (the innate immune response in Figure 1) are built into the DILIsym.39 This activation can promote injury and affect regeneration rates. Modeling of adaptive immune responses to the liver has begun and is an area of emphasis going forward. This modeling should provide novel insights into mechanisms underlying idiosyncrasy as well as hepatotoxicity observed with immune modulators, such as the check‐point inhibitors.40
Another area of focus will be creation of additional simulated patient populations in DILIsym, including pediatric and elderly patients and patients with liver or kidney dysfunction. At some point in the future, liver safety of a new drug candidate may not have to be tested in these populations if the modeling result is accepted by regulators.
Finally, DILIsym modeling may ultimately be useful to clinicians beyond translating serum ALT levels into percent hepatocyte loss and risk of global liver dysfunction. For example, if DILIsym modeling has already been performed for a specific drug, including identification of the operative mechanism(s) causing DILI, this information could qualitatively indicate patient risk factors for DILI due to that drug, such as underlying diseases (e.g., NASH) or concomitant medications with the same mechanisms for DILI. Availability of the DILIsym model for that drug, perhaps through a web‐based application, could potentially allow a quantitative prediction of DILI risk for that patient based on the patient‐specific information and may also indicate a safe dosing regimen for that patient despite the presence of risk factors. Furthermore, when assessing a patient with suspected DILI receiving multiple drugs that have been modeled in DILIsym, such a web‐based application could potentially assess the likelihood that the patient had DILI as well as which drug was the most likely culprit.
Conclusion
The DILI‐sim Initiative has advanced our understanding of mechanisms underlying DILI and this has provided insight into species differences in susceptibility to hepatotoxicity as well as potential risk factors in patient populations. The model that is continuing to evolve from the Initiative, DILIsym, is becoming a useful tool in decision making within the pharmaceutical industry both in terms of assessing the liver safety liabilities of new drug candidates and in the optimal interpretation of serum biomarkers. In the future, the model may reduce the need for some clinical trials and assist physicians in managing the liver safety of their patients. The DILI‐sim Initiative provides an example of the potential for precompetitive collaborations to develop useful QST models.
Funding
No funding was received for this work.
Conflict of Interest
Dr Watkins chairs the Scientific Advisory Committee for the DILI‐sim Initiative and receives compensation for this role. He also has a financial interest in the spinoff company DILIsym Services, Inc., which is a subsidiary of Simulations Plus.
References
- 1. Howell, B.A. et al Development of quantitative systems pharmacology and toxicology models within consortia: experiences and lessons learned through DILIsym development. Drug Discov. Today 22 (suppl. C), 5–13 (2016).27634342 [Google Scholar]
- 2. Bloomingdale, P. et al Quantitative systems toxicology. Curr. Opin. Toxicol. 4, 79–87 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Howell, B.A. et al In vitro to in vivo extrapolation and species response comparisons for drug‐induced liver injury (DILI) using DILIsym™: a mechanistic, mathematical model of DILI. J. Pharmacokinet. Pharmacodyn. 39, 527–541 (2012). [DOI] [PubMed] [Google Scholar]
- 4. Shoda, L.K.M. , Woodhead, J.L. , Siler, S.Q. , Watkins, P.B. & Howell, B.A. Linking physiology to toxicity using DILIsym®, a mechanistic mathematical model of drug‐induced liver injury. Biopharm. Drug Dispos. 35, 33–49 (2014). [DOI] [PubMed] [Google Scholar]
- 5. Woodhead, J.L. et al An analysis of N‐acetylcysteine treatment for acetaminophen overdose using a systems model of drug‐induced liver injury. J. Pharmacol. Exp. Ther. 342, 529–540 (2012). [DOI] [PubMed] [Google Scholar]
- 6. Howell, B.A. , Siler, S.Q. & Watkins, P.B. Use of a systems model of drug‐induced liver injury (DILIsym®) to elucidate the mechanistic differences between acetaminophen and its less‐toxic isomer, AMAP, in mice. Toxicol. Lett. 226, 163–172 (2014). [DOI] [PubMed] [Google Scholar]
- 7. Guo, C. , Yang, K. , Brouwer, K.R. , St. Claire, R.L. 3rd & Brouwer, K.L. Prediction of altered bile acid disposition due to inhibition of multiple transporters: an integrated approach using sandwich‐cultured hepatocytes, mechanistic modeling, and simulation. J. Pharmacol. Exp. Ther. 358, 324–333 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Church, R.J. & Watkins, P.B. The transformation in biomarker detection and management of drug‐induced liver injury. Liver Int. 37, 1582–1590 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Perez‐Carreras, M. et al Defective hepatic mitochondrial respiratory chain in patients with nonalcoholic steatohepatitis. Hepatology (Baltimore, MD) 38, 999–1007 (2003). [DOI] [PubMed] [Google Scholar]
- 10. Watkins, P.B. et al Evaluation of drug‐induced serious hepatotoxicity (eDISH): application of this data organization approach to phase III clinical trials of rivaroxaban after total hip or knee replacement surgery. Drug Saf. 34, 243–252 (2011). [DOI] [PubMed] [Google Scholar]
- 11. Woodhead, J.L. et al Prediction of safety margin and optimization of dosing protocol for a novel antibiotic using quantitative systems pharmacology modeling. Clin. Transl. Sci. 11, 498–505 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Longo, D.M. , Yang, Y. , Watkins, P.B. , Howell, B.A. & Siler, S.Q. Elucidating differences in the hepatotoxic potential of tolcapone and entacapone with DILIsym®, a mechanistic model of drug‐induced liver injury. CPT Pharmacometrics Syst. Pharmacol. 5, 31–39 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yang, K. , Woodhead, J.L. , Watkins, P.B. , Howell, B.A. & Brouwer, K.L. Systems pharmacology modeling predicts delayed presentation and species differences in bile acid‐mediated troglitazone hepatotoxicity. Clin. Pharmacol. Ther. 96, 589–598 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Howell, B.A. , Woodhead, J.L. , Pellegrini, L. , Siler, S.Q. & Shoda, L.K.M. Liver Safety Comparison of Two Treatments for Autosomal‐Dominant Polycystic Kidney Disease (ADPKD) Using Quantitative Systems Toxicology Software (DILIsym). (American Association of Pharmceutical Scientists, Baltimore, MD, 2018). [Google Scholar]
- 15. Shon, J. & Abernethy, D.R. Application of systems pharmacology to explore mechanisms of hepatotoxicity. Clin. Pharmacol. Ther. 96, 536–537 (2014). [DOI] [PubMed] [Google Scholar]
- 16. Longo, D.M. et al Quantitative systems toxicology analysis of in vitro mechanistic assays reveals importance of bile acid accumulation and mitochondrial dysfunction in TAK‐875‐induced liver injury. Toxicol. Sci. 67, 458–467 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Park, B.K. , Kitteringham, N.R. , Maggs, J.L. , Pirmohamed, M. & Williams, D.P. The role of metabolic activation in drug‐induced hepatotoxicity. Annu. Rev. Pharmacol. Toxicol. 45, 177–202 (2005). [DOI] [PubMed] [Google Scholar]
- 18. Iorga, A. , Dara, L. & Kaplowitz, N. Drug‐induced liver injury: cascade of events leading to cell death, apoptosis or necrosis. Int. J. Mol. Sci. 18, pii: E1018 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Woodhead, J.L. et al Analyzing the mechanisms behind macrolide antibiotic‐induced liver injury using quantitative systems toxicology modeling. Pharm. Res. 36, 48 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Chan, R. & Benet, L.Z. Measures of BSEP inhibition in vitro are not useful predictors of DILI. Toxicol. Sci. 162, 499–508 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Woodhead, J.L. et al Exploring BSEP inhibition‐mediated toxicity with a mechanistic model of drug‐induced liver injury. Front. Pharmacol. 5, 240 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Woodhead, J.L. et al Application of a mechanistic model to evaluate putative mechanisms of Tolvaptan drug‐induced liver injury and identify patient susceptibility factors. Toxicol. Sci. 155, 61–74 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Woodhead, J.L. et al Mechanistic modeling reveals the critical knowledge gaps in bile acid‐mediated DILI. CPT Pharmacometrics Syst. Pharmacol. 3, e123 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kenna, J.G. et al Can bile salt export pump inhibition testing in drug discovery and development reduce liver injury risk? An International Transporter Consortium Perspective. Clin. Pharmacol. Ther. 104, 916–932 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Battista, C. et al Using quantitative systems toxicology to investigate observed species differences in CKA‐mediated hepatotoxicity. Toxicol. Sci. 166, 123–130 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Mosedale, M. & Watkins, P.B. Drug‐induced liver injury: advances in mechanistic understanding that will inform risk management. Invited “state‐of‐the‐art” review. Clin. Pharmacol. Ther. 101, 469–480 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Woodhead, J.L. , Watkins, P.B. , Howell, B.A. , Siler, S.Q. & Shoda, L.K.M. The role of quantitative systems pharmacology modeling in the prediction and explanation of idiosyncratic drug‐induced liver injury. Drug Metab. Pharmacokinet. 32, 40–45 (2017). [DOI] [PubMed] [Google Scholar]
- 28. Church, R.J. & Watkins, P.B. In silico modeling to optimize interpretation of liver safety biomarkers in clinical trials. Exp. Biol. Med. (Maywood, NJ) 243, 300–307 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Liu, Z. , Lei, X. , Li, X. , Cai, J.M. , Gao, F. & Yang, Y.Y. Toll‐like receptors and radiation protection. Eur. Rev. Med. Pharmacol. Sci. 22, 31–39 (2018). [DOI] [PubMed] [Google Scholar]
- 30. Howell, B.A. , Siler, S.Q. , Shoda, L.K. , Yang, Y. , Woodhead, J.L. & Watkins, P.B. A mechanistic model of drug‐induced liver injury AIDS the interpretation of elevated liver transaminase levels in a phase I clinical trial. CPT Pharmacometrics Syst. Pharmacol. 3, e98 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Longo, D.M. et al Refining liver safety risk assessment: application of mechanistic modeling and serum biomarkers to cimaglermin alfa (GGF2) clinical trials. Clin. Pharmacol. Ther. 102, 961–969 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Portmann, B. , Talbot, I.C. , Day, D.W. , Davidson, A.R. , Murray‐Lyon, I.M. & Williams, R. Histopathological changes in the liver following a paracetamol overdose: correlation with clinical and biochemical parameters. J. Pathol. 117, 169–181 (1975). [DOI] [PubMed] [Google Scholar]
- 33. Mosedale, M. et al Transient changes in hepatic physiology that alter bilirubin and bile acid transport may explain elevations in liver chemistries observed in clinical trials of GGF2 (cimaglermin alfa). Toxicol. Sci. 161, 401–411 (2018). [DOI] [PubMed] [Google Scholar]
- 34. Yang, K. et al Systems pharmacology modeling of drug‐induced hyperbilirubinemia: differentiating hepatotoxicity and inhibition of enzymes/transporters. Clin. Pharmacol. Ther. 101, 501–509 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Watkins, P.B. Drug safety sciences and the bottleneck in drug development. Clin. Pharmacol. Ther. 89, 788–790 (2011). [DOI] [PubMed] [Google Scholar]
- 36. Hayashi, P.H. et al Death and liver transplantation within 2 years of onset of drug‐induced liver injury. Hepatology (Baltimore, MD) 66, 1275–1285 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Roth, R.A. & Dahm, L.J. Neutrophil‐ and glutathione‐mediated hepatotoxicity of alpha‐naphthylisothiocyanate. Drug Metab. Rev. 29, 153–165 (1997). [DOI] [PubMed] [Google Scholar]
- 38. He, K. , Cai, L. , Shi, Q. , Liu, H. & Woolf, T.F. Inhibition of MDR3 activity in human hepatocytes by drugs associated with liver injury. Chem. Res. Toxicol. 28, 1987–1990 (2015). [DOI] [PubMed] [Google Scholar]
- 39. Shoda, L.K. , Battista, C. , Siler, S.Q. , Pisetsky, D.S. , Watkins, P.B. & Howell, B.A. Mechanistic modelling of drug‐induced liver injury: investigating the role of innate immune responses. Gene Regulat. Syst. Bio. 11, 1177625017696074 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Suzman, D.L. , Pelosof, L. , Rosenberg, A. & Avigan, M.I. Hepatotoxicity of immune checkpoint inhibitors: an evolving picture of risk associated with a vital class of immunotherapy agents. Liver Int. 38, 976–987 (2018). [DOI] [PubMed] [Google Scholar]