

Abstract
NYX‐2925, a new chemical entity, acts as a co‐agonist to glutamate at the N‐methyl‐D‐aspartate receptor (NMDAR). At low concentrations of endogenous agonists (glycine/D‐serine), NYX‐2925 partially activates NMDARs, modulating neural pathways relevant for chronic pain. NYX‐2925 is being developed for the treatment of chronic pain conditions, including painful diabetic peripheral neuropathy and fibromyalgia. In this first‐in‐human, phase I, single‐ascending dose (50–1,200 mg) and multiple‐ascending dose (150–900 mg) study, the safety, tolerability, and pharmacokinetics (PKs) of NYX‐2925 were evaluated in 84 healthy adult volunteers. No safety concerns emerged, including no dissociative side effects. NYX‐2925 exhibited dose‐proportional PKs and minimal accumulation following once‐daily dosing for 7 days. Cerebrospinal fluid (CSF) measurements confirmed that NYX‐2925 crosses the blood brain barrier, with maximum CSF concentrations approximating 6–9% of maximum plasma concentrations at the same dose level. NYX‐2925 was safe and well‐tolerated in healthy volunteers, and the study results support the continued clinical development for chronic pain conditions.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
NMDARs have been studied as therapeutic targets for chronic pain conditions. NYX‐2925, a novel agent designed using rapastinel as a template, is a co‐agonist to glutamate at the NMDAR, and acts at a site distinct from known NMDAR agonists/antagonists.
what question did this study address?
This first‐in‐human study assessed the safety, tolerability, and PKs of ascending doses of NYX‐2925 in healthy volunteers.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
NYX‐2925 is safe and well‐tolerated, exhibits dose‐proportional PKs in single (≤ 1200 mg) and multiple once‐daily doses (≤ 900 mg × 7 days), and adequately penetrates the blood brain barrier (concentrations are 6–9% of maximum plasma concentrations). The studied doses approximated and far exceeded anticipated therapeutic doses. The NMDAR can be targeted without apparent central nervous system or other toxicity.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
Current chronic pain therapies remain suboptimal, often with intolerable adverse reactions. NYX‐2925 is a non‐opioid, selective, and central‐acting NMDAR modulator with a demonstrated safety profile. These characteristics support clinical investigation of NYX‐2925 for chronic pain treatment.
NYX‐2925 ((2S, 3R)‐3‐hydroxy‐2‐((R)‐5‐isobutyryl‐1‐oxo‐2,5‐diazaspiro(3.4)octan‐2‐yl)butanamide; Figure 1) is a new chemical entity, and the first in a series of novel, oral, highly soluble, and bioavailable (class 1 Biopharmaceutics Classification System compound)1 N‐methyl‐D‐aspartate receptor (NMDAR) modulators to be investigated in humans. These small bicyclic molecules modulate NMDAR activity in a manner similar to the tetrapeptide rapastinel, and distinct from known NMDAR agonists, including D‐cycloserine, or antagonists, such as ketamine, MK‐801 (dizocilpine), kynurenic acid, or ifenprodil.2 NYX‐2925 is being developed for the treatment of chronic pain conditions, such as neuropathic pain and fibromyalgia. It acts as a co‐agonist to glutamate at the NMDAR, and at low concentrations of endogenous agonist (glycine or D‐serine), NYX‐2925 partially activates NMDARs.
Figure 1.
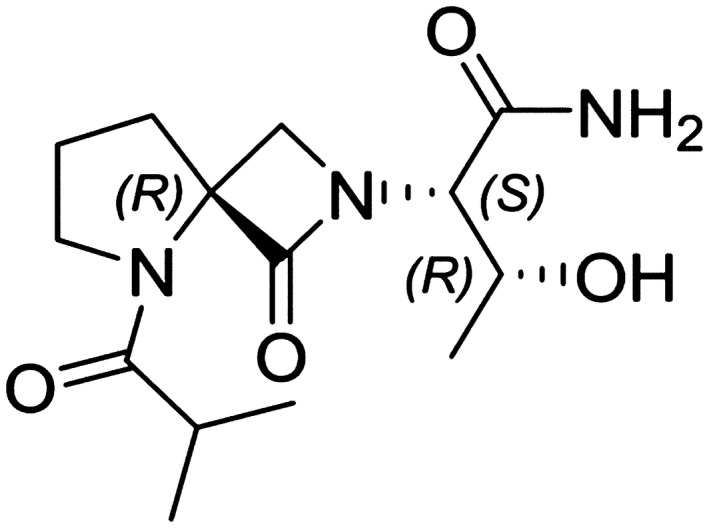
Chemical structure of NYX‐2925.
The NMDARs are a large family of ionotropic glutamate receptors found predominantly in the central nervous system (CNS).3, 4, 5 These receptors consist of multiple subunits and, alone or together, have been explored previously as a drug development target due to their key function as a “salience detector” involved in critical roles in normal neuronal function, including activity‐dependent synaptic plasticity associated with learning and memory.4, 5, 6, 7, 8, 9 Glutamate is the major excitatory neurotransmitter within the CNS3, 5 and exerts it signaling function through activation of glutamate receptors.3, 10 Unlike other ionotropic glutamate receptors found in the brain, such as α‐amino‐3‐hydroxy‐5‐methyl‐4‐isoxazolepropionic acid or kainic acid receptors, the NMDARs are unique in that they have distinct binding sites for both glutamate and glycine, and binding of both ligands is required for receptor activation.6 In addition to their pivotal role in physiological CNS‐related processes, NMDARs seem to be crucially involved in numerous CNS disorders, including anxiety,11, 12 schizophrenia,6, 13, 14, 15 ischemic stroke,6, 13, 16 Parkinson disease,6, 12 and chronic pain conditions, such as neuropathic pain4, 10, 16 and fibromyalgia.17, 18
The potency (half‐maximal effective concentration) of NYX‐2925 was 0.028–55 pM when measured against NMDAR subtypes, NR2A‐D, in human embryonic kidney cells expressing human recombinant NMDA receptors.2 In both hippocampal CA1 pyramidal neurons and medial prefrontal cortex, NYX‐2925 facilitated NMDA current and NMDA receptor‐dependent long‐term potentiation.2 Moreover, a single dose of NYX‐2925 (1 mg/kg) was shown to persistently enhance long‐term potentiation at 24 hours and 1 week postdose, demonstrating induction of long‐term metaplasticity.2 NYX‐2925 did not significantly inhibit or stimulate any of 81 neuroactive receptors tested in a panel of binding assays, suggesting low potential for off‐target activity.2 It also did not produce ketamine‐associated lever responding at dose of 1–100 mg/kg in a drug‐discrimination model used to predict the abuse potential of psychoactive drugs in humans.2
NYX‐2925 has been studied in several animal models of neuropathic pain, including neuropathic pain induced by sciatic nerve ligation19 and painful diabetic neuropathy‐induced pain with streptozotocin. In the Bennett chronic constriction injury model of neuropathic pain, rats given a single dose of NYX‐2925 at 1–2 weeks post‐sciatic nerve ligation demonstrated analgesia at 1 hour, 24 hours, and 1 week postdrug administration.20 Similar rapid (1 hour) and long‐lasting (1 week) analgesia was observed in the rat model of streptozotocin‐induced painful diabetic neuropathy.20 NYX‐2925 modulates NMDARs in a highly specific and selective manner to enhance synaptic plasticity, which is a probable mechanism for the long‐duration effects seen in these pain models. Rather than fully turning the receptor “on” (agonist) or “off” (antagonist), this compound likely normalizes NMDAR function, enhancing communication between neural cells and avoiding the issues associated with excessive unidirectional activation or inhibition that have plagued NMDAR‐targeted drug development historically.
This first‐in‐human study was designed to characterize the safety, tolerability, and pharmacokinetics (PKs) of NYX‐2925 over a range of single and multiple doses in healthy volunteers. The doses selected for this study were based on active doses in translational‐pain models and toxicology studies on animals; the aim of this study was to explore doses near and well above the anticipated therapeutic doses.
METHODS
Study design
This was a single‐center, phase I, randomized, double‐blind, sponsor‐open, placebo‐controlled study conducted in two phases, a single ascending dose (SAD) phase and a multiple ascending dose (MAD) phase. Each dose group consisted of eight subjects who were randomly assigned (3:1) to receive NYX‐2925 (n = 6) or placebo (n = 2), which is common for phase I studies of this type. Additionally, each of the dose phases had a cohort from whom cerebrospinal fluid (CSF) samples were collected, and these cohorts consisted of six subjects, all of whom received open‐label NYX‐2925. As this was the first‐in‐human trial, a Data Review Committee (DRC) met periodically to review safety and preliminary PK data relevant to proceeding to the next dose group, cohort, and/or phase of the study. The randomization schedule was produced by the investigative site and provided to the sponsor for DRC meetings.
In the SAD phase, subjects received a single 50‐mg, 150‐mg, 300‐mg, 400‐mg, 800‐mg, or 1200‐mg dose of NYX‐2925 or placebo. The CSF cohort received a single 50‐mg dose of NYX‐2925. Doses were administered in the morning following an overnight fast. Subjects in the 300‐mg group were identified as the elderly cohort, and these subjects received study drug or placebo after all SAD and MAD groups had completed the study and relevant data were reviewed by the DRC. Subjects in the 400‐mg group (the food‐effect group) first received NYX‐2925 under fasting conditions and then, after data review by the DRC, received a second single 400‐mg dose 7 days later following a standard high‐calorie, high‐fat meal. Subjects enrolled in the SAD phase were confined to the study center the night before dosing and remained confined until completion of the 24‐hour postdose assessments.
In the MAD phase, subjects received 7 once‐daily doses of 150‐mg, 600‐mg, or 900‐mg of NYX‐2925 or placebo. Subjects in the CSF cohort received NYX‐2925 300 mg once daily for 7 days. Doses were administered in the morning following an overnight fast. Dosing was initiated in the MAD phase following completion of the second SAD dose group (150 mg) and after the DRC reviewed the data from the first two SAD dose groups. For the MAD phase, subjects reported to the clinic the night before dosing and remained confined for the entire 7 days of dosing and through completion of the 48‐hour post‐dose assessments on day 9.
The protocol and amendments were approved by an independent institutional review board, and all subjects provided written informed consent prior to the performance of any study‐related procedures. The study was conducted in accordance with Good Clinical Practice, including International Council for Harmonisation Guidelines, and according to the principles of the Declaration of Helsinki. The study is registered on clinicaltrials.gov (identifier: NCT02834741).
Study participants
Eligible subjects were healthy men or nonpregnant women aged 18–55 years (or 55–80 years for the elderly cohort) with a body mass index between 18 and 30 kg/m2. Subjects had to have normal hepatic and renal functions, with calculated creatinine clearance ≥ 90 mL/minute (or ≥ 60 mL/minute for the elderly cohort), using Cockcroft‐Gault Formula.
Subjects with a history of allergy, sensitivity, or intolerance to NMDAR ligands, including ketamine, dextromethorphan, memantine, methadone, dextropropoxyphene, or ketobemidone, were not eligible for study enrollment, and current use was prohibited. Other key exclusion criteria included prolonged corrected QT Fridericia's formula (QTcF) interval at screening (> 450 msec (men) or ≥ 470 msec (women)), heart rate ≤45 bpm, history of excessive bleeding, use of blood thinners within 6 months before dosing, or antibiotics or infection within 3 months before dosing (CSF cohort only). Subjects also could not have had a lumbar spine abnormality, history of elevated intracranial pressure, normal pressure hydrocephalus, or other neurological conditions considered clinically significant by the investigator, or history of seizures or any type of psychosis (including but not limited to bipolar disease or schizophrenia).
Study assessments
Pharmacokinetics
Blood samples for PK analysis were collected on day 1 in the SAD phase and on days 1 and 7 in the MAD phase. Samples were collected predose and then at 0.5, 1, 1.5, 2, 3, 4, 6, 8, 12, and 24 hours postdose (day 1) in the SAD phase and at these same times on days 1 and 7 in the MAD phase with 1 additional collection at 48 hours postdose of day 7 in the MAD phase. Twenty‐four hour PK sampling for subjects in the food‐effect group was on day 1 (fasting) and day 8 (fed).
Urine samples for measurement of urinary excretion of NYX‐2925 were collected predose and at intervals of at 0–4, 4–8, 8–12, 12–16, and 16–24 hours postdose on day 1 in the SAD phase and on days 1 and 7 in the MAD phase.
Safety
Safety assessments included a review of adverse events, clinical laboratory evaluations, vital signs, physical examinations, 12‐lead safety electrocardiograms (ECGs), and continuous 12‐lead triplicate ECGs for thorough ECG analysis. Triplicate ECGs were recorded at the same times as the PK sampling (i.e., 24‐hour recordings on day 1 of the SAD and MAD phases and 48‐hour recordings on day 7 of the MAD phase). The study was designed to meet US Food and Drug Administration requirements for assessment of electrocardiographic repolarization effects using concentration‐effect modeling in lieu of a standard E‐14 thorough QT study. Other safety assessments included dissociative rating scale questionnaires (Brief Psychiatric Rating Scale – positive symptoms) the Clinician Administered Dissociative States Scale, and the Columbia‐Suicide Severity Rating Scale (C‐SSRS). In the SAD phase, the dissociative scale questionnaires were completed on day 1 at 2, 3, and 24 hours postdose and the C‐SSRS at 2 and 24 hours postdose. In the MAD phase, the dissociative scale questionnaires were completed at 2 and 3 hours postdose on days 1 and 7, and the C‐SSRS at 2 hours postdose on days 1 and 7.
Cerebrospinal fluid
The concentration of NYX‐2925 in CSF was measured only in the CSF cohorts, which did not include placebo subjects. In the 50‐mg single‐dose CSF cohort, CSF samples were collected on day 1 at 1 and 4 hours postdose in three subjects and at 2 and 8 hours postdose in three subjects. For subjects in the 300‐mg multiple‐dose CSF cohort, CSF sample collection occurred on day 6 at 1.5 and 3 hours postdose.
Measurement of NYX‐2925 concentrations in plasma and urine
Plasma and urine NYX‐2925 concentrations were measured using validated high‐performance liquid chromatography/tandem mass spectroscopy methods. The lower limit of quantitation in plasma was 1.00 ng/mL and the lower limit of quantitation for urine was 50.0 ng/mL.
Statistical methods
Descriptive statistics were used for baseline and demographic characteristics, safety data, PK parameter estimates, and CSF concentrations. Plasma and urine PK parameters were calculated using noncompartmental methods with Phoenix WinNonlin version 6.3 (Pharsight, St. Louis, MO). Statistical analyses were performed with SAS version 9.4 using procedures appropriate for the particular analysis. All statistical tests were two‐sided, with α = 0.05, unless otherwise stated. Using the same methods of analysis, a post hoc analysis of maximum plasma concentration (Cmax) and area under the plasma concentration‐time curve (AUC) by gender was performed on the PK population.
Dose proportionality was evaluated visually with plots for dose‐normalized Cmax, dose‐normalized AUC from 0 to last collection time after drug administration (AUC0–last), dose‐normalized AUC from 0‐24 hours (AUC0‐24; MAD groups only), AUC from zero to infinity (AUC0–∞; SAD groups only), and oral clearance (CL/F).
The effect of food was evaluated with a mixed‐effects model using natural log (ln)‐transformed values for Cmax, AUC0–last, and AUC0–∞ for subjects who completed both periods (fasted and fed) in the SAD phase food‐effect group. The model included treatment as a fixed effect and subject as a random effect. Treatment least squares mean differences and 90% confidence intervals for the difference (fed minus fasted) were constructed for the ln‐scale values of each parameter and back transformed and expressed as percent geometric mean ratio.
The potential effect of age was explored by visually comparing the single‐dose PK results from the healthy elderly cohort (300 mg) with those from the nonelderly cohort (remaining SAD groups) across dose groups.
RESULTS
Study population
Overall, in the combined SAD and MAD groups, 84 subjects met inclusion/exclusion criteria and were randomized, 66 to NYX‐2925 and 18 to placebo. Of these, 80 subjects (95.2%) completed the study and 4 withdrew early (2 NYX‐2925 subjects and 2 placebo subjects). The reasons for withdrawal were noncompliance (n = 1) and protocol deviation (n = 1) in NYX‐2925 subjects and lost to follow‐up (n = 1) and physician decision (n = 1) in the placebo subjects. The study was initiated on June 29, 2016, and completed on March 27, 2017. All 84 subjects were included in the safety population, and all 66 subjects who received NYX‐2925 were included in the PK population.
Demographic data are summarized in Table 1. For the overall study population (N = 84), mean age was 36.9 years and slightly more men (52.4%) than women (47.6%) were enrolled. Subjects in the elderly cohort were between 59 and 64 years of age (mean 61.3 years).
Table 1.
Baseline demographics (safety population)
| Parameter | All SAD NYX‐2925 (N = 42) | All MAD NYX‐2925 (N = 24) | All NYX‐2925 (N = 66) | All placebo (N = 18) |
|---|---|---|---|---|
| Age, mean (SD), year | 39.0 (13.8) | 32.7 (10.0) | 36.7 (12.9) | 37.6 (13.8) |
| Sex, n (%) | ||||
| Male | 23 (54.8) | 12 (50.0) | 35 (53.0) | 9 (50.0) |
| Female | 19 (45.2) | 12 (50.0) | 31 (47.0) | 9 (50.0) |
| Race, n (%) | ||||
| White | 31 (73.8) | 12 (50.0) | 43 (65.2) | 13 (72.2) |
| Black | 10 (23.8) | 12 (50.0) | 22 (33.3) | 4 (22.2) |
| Asian | 1 (2.4) | 0 | 1 (1.5) | 1 (5.6) |
| Not Hispanic or Latino, n (%) | 39 (92.9) | 22 (91.7) | 61 (92.4) | 17 (94.4) |
| BMI, mean (SD), kg/m2 | 25.9 (2.8) | 25.9 (2.7) | 25.9 (2.7) | 25.9 (3.3) |
BMI, body mass index; MAD, multiple ascending dose; SAD, single ascending dose.
Single‐dose pharmacokinetics
Mean plasma‐concentration time profiles for NYX‐2925 after single ascending doses are shown in Figure 2 a. Under fasted conditions, the increases in Cmax and AUC were dose‐proportional following single doses of NYX‐2925 from 50–1200 mg (Table 2). NYX‐2925 was rapidly absorbed (median time to maximum plasma concentration (Tmax) ranged from 1.0–1.5 hours across dose levels) and declined in a roughly monophasic manner over 24 hours. The geometric percent coefficient of variation (CV%) for Cmax was relatively low and ranged from 24.3–31.5% across the cohorts. The geometric CV% for AUC0–last and AUC0–∞ ranged from 7.7–23.4%. Mean oral clearance (CL/F) values were consistent across the single‐dose levels. Mean renal clearance ranged from 6.0–8.1 L/hours following a single dose, and the amount of NYX‐2925 excreted over 24 hours (Ae0–24) was 56–69% of the dose.
Figure 2.
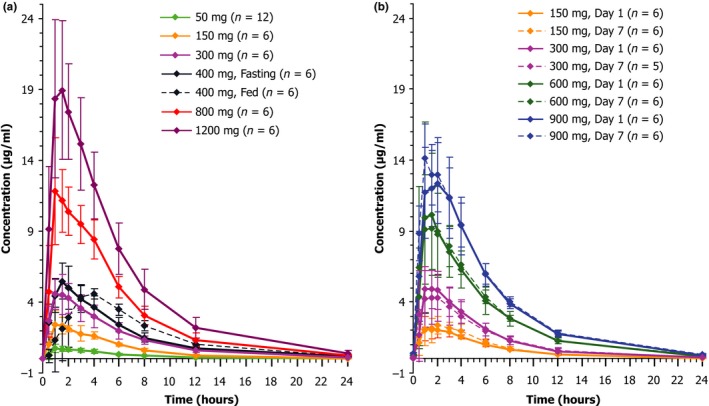
NYX‐2925 exposures over time. Mean plasma concentration profile of NYX‐2925 (linear scale) — (a) single ascending dose groups; (b) multiple ascending dose groups, day 1 and day 7. The error bars represent SD.
Table 2.
Plasma pharmacokinetics of NYX‐2925: single ascending dose groups
| Parameter (units) | Group 1 & CSF cohort 50 mg (n = 12) | Group 2 150 mg (n = 6) | Elderly cohort 300 mg (n = 6) | Group 3 400 mg fasting (n = 6) | Group 3 400 mg fed (n = 6) | Group 4 800 mg (n = 6) | Group 5 1,200 mg (n = 6) |
|---|---|---|---|---|---|---|---|
| Cmax (μg/mL) | 0.8 (31.5) | 2.7 (24.3) | 5.0 (26.3) | 5.5 (25.1) | 4.9 (14.0) | 12.4 (26.3) | 20.1 (24.8) |
| Tmax (h) | 1.0 (0.5–2.8) | 1.0 (1.0–2.0) | 1.25 (1.0–2.0) | 1.5 (1.0–2.0) | 3.0 (1.5–4.0) | 1.25 (1.0–4.0) | 1.5 (1.0–3.0) |
| AUC0–last (h*μg/mL) | 4.4 (17.8) | 14.1 (11.1) | 28.5 (23.4) | 34.2 (13.7) | 35.6 (15.5) | 71.5 (7.7) | 113.7 (17.6) |
| AUC0–24 (h*μg/mL) | 4.4 (17.8) | 14.1 (11.1) | 28.5 (23.4) | 34.2 (13.7) | 35.6 (15.5) | 71.5 (7.7) | 113.7 (17.6) |
| AUC0–∞ (h*μg/mL) | 4.5 (16.7) | 14.3 (11.2) | 29.0 (23.1) | 35.1 (13.2) | 36.4 (15.6) | 72.7 (7.7) | 116.2 (17.6) |
| t1/2 (hour) | 3.9 (20.5) | 3.6 (7.8) | 4.0 (11.2) | 4.4 (23.0) | 3.9 (10.5) | 3.9 (19.7) | 4.3 (12.7) |
| CL/F (L/h) | 11.0 (16.7) | 10.5 (11.3) | 10.3 (23.1) | 11.4 (13.4) | 11.0 (15.5) | 11.0 (7.6) | 10.3 (17.8) |
| Vd/F (L) | 62.1 (31.0) | 54.1 (10.1) | 60.0 (29.1) | 71.6 (28.2) | 61.2 (15.2) | 61.9 (21.1) | 64.7 (20.1) |
AUC0–24, area under the plasma concentration‐time curve from 0–24 hours; AUC0‐∞, area under the plasma concentration‐time curve from 0 to infinity; AUC0–last, area under the plasma concentration‐time curve from 0 to last collection time after drug administration; CL/F, apparent oral clearance; Cmax, maximum plasma concentration; CSF, cerebrospinal fluid; t1/2, apparent terminal elimination half‐life; Tmax, time to maximum plasma concentration; Vd/F, apparent volume of distribution.
Data are geometric mean (% coefficient of variation), except Tmax is median (minimum‐maximum).
When a single 400‐mg dose of NYX‐2925 was administered after a high‐calorie, high‐fat meal, Tmax was delayed by 1.5 hours (3.0 vs. 1.5 hours, fed vs. fasted) and Cmax was lowered by ~10% (4.9 vs. 5.5 μg/mL, fed vs. fasted); however, AUC values were comparable (AUC0–∞: 36.4 vs. 35.1 h*μg/mL) in the fed vs. fasted states. Terminal elimination half‐life (t½) in the fed state was marginally shorter (3.9 vs. 4.4 hours).
The PK of a 300 mg dose of NYX‐2925 in the elderly cohort was in proportion to dose, such that the observed values for Cmax and AUC were approximately two times those of the 150 mg dose, and Tmax and t½ were similar to the values observed across all dose groups (Table 2). Although the estimated glomerular filtration rate was ~15% lower in the elderly cohort (mean = 99.0 mL/minutes) compared with the remaining SAD dose groups (mean = 115.6 mL/minutes), there were no clinically meaningful PK differences.
A post hoc analysis demonstrated dose proportionality for Cmax (Figure 3 a) and AUC (Figure 3 b) in men and women in the SAD groups as well as in the MAD groups (data not shown).
Figure 3.
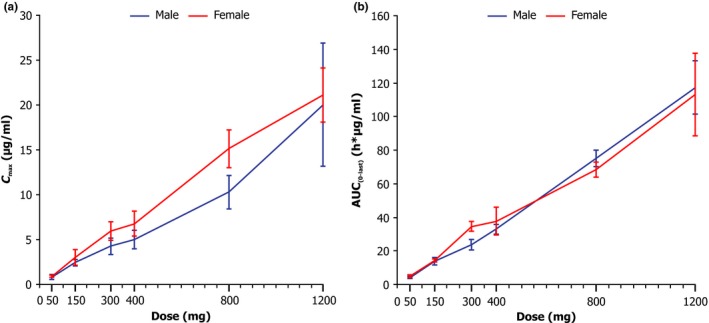
Dose proportionality plot by gender and dose (single ascending dose groups) of (a) maximum plasma concentration (Cmax) and (b) area under the plasma concentration‐time curve from 0 to last collection time after drug administration (AUC 0‐last). The error bars represent SD.
Multiple‐dose pharmacokinetics
The PK parameters of NYX‐2925 following once‐daily dosing with 150–900 mg/day for 7 days are provided in Table 3, and the day 1 and day 7 plasma‐concentration time profiles for NYX‐2925 are shown in Figure 2 b. Mean Cmax and overall plasma exposures increased in proportion to dose, with minimal accumulation after multiple doses across the four dosing groups (accumulation ratio (rAUC0–24) ranged from 0.97–1.13). Median Tmax (1.00–1.75 hours) and mean CL/F (9.3–10.8 L/hours) were similar among all doses and days. Values for t½ trended higher on day 7 (4.8–7.3 hours) compared with day 1 (3.9–4.1 hours). The geometric CV% for Cmax on day 1 ranged from 25.1–45.6% and on day 7 from 14.1–60.4% across cohorts. The geometric CV% for AUC0–last was 11.8–34.1% (day 1) and 11.6–29.6% (day 7). After multiple daily doses, mean renal clearance ranged from 6.5–7.6 L/hours, with no association of dose, and Ae0–24 was 66–73% of the administered dose.
Table 3.
Plasma pharmacokinetics of NYX‐2925: multiple ascending dose groups
| Parameter (units) | Group 1 150 mg day 1 (n = 6) | Group 1 150 mg day 7 (n = 6) | CSF cohort 300 mg day 1 (n = 6) | CSF cohort 300 mg day 7 (n = 5)a | Group 2 600 mg day 1 (n = 6) | Group 2 600 mg day 7 (n = 6) | Group 3 900 mg day 1 (n = 6) | Group 3 900 mg day 7 (n = 6) |
|---|---|---|---|---|---|---|---|---|
| Cmax (μg/mL) | 2.3 (27.3) | 2.6 (25.3) | 5.4 (26.4) | 4.5 (60.4) | 10.7 (45.6) | 10.0 (33.1) | 13.0 (25.1) | 14.3 (14.1) |
| Tmax (hour) | 1.25 (1.0–2.0) | 1.5 (1.0–3.0) | 1.25 (1.0–2.0) | 1.5 (0.5–2.0) | 1.25 (1.0–2.0) | 1.5 (0.5–3.0) | 1.75 (0.5–3.0) | 1.0 (1.0–2.0) |
| AUC0–last (h*μg/mL) | 14.3 (12.0) | 16.8 (20.3) | 26.2 (34.1) | 29.2 (29.6) | 59.8 (22.6) | 64.8 (18.7) | 83.9 (11.8) | 93.7 (11.6) |
| AUC0–24 (h*μg/mL) | 14.3 (12.0) | 16.2 (18.6) | 28.8 (27.3) | 27.9 (32.4) | 59.9 (22.6) | 62.3 (20.1) | 83.9 (11.8) | 89.7 (10.3) |
| AUC0–∞ (h*μg/mL) | 14.5 (11.8) | NA | 29.2 (27.1) | NA | 61.2 (21.6) | NA | 85.4 (11.3) | NA |
| t1/2 (hour) | 4.0 (13.4) | 4.8 (14.3) | 3.9 (14.9) | 6.1 (48.7) | 4.1 (18.3) | 5.8 (13.0) | 4.0 (13.8) | 7.3 (31.8) |
| CL/F (L/hour) | 10.3 (11.7) | 9.3 (18.5) | 10.3 (27.1) | 10.8 (32.4) | 9.8 (21.6) | 9.6 (20.2) | 10.5 (11.2) | 10.0 (10.4) |
| Vd/F (L) | 59.7 (18.5) | 68.4 (14.8) | 57.7 (32.4) | 99.4 (104.3) | 57.8 (36.9) | 83.7 (12.1) | 60.8 (21.0) | 109.0 (26.0) |
| rAUC0–24 (ratio)b | NA | 1.1 (12.3) | NA | 0.97 (9.6) | NA | 1.0 (7.1) | NA | 1.1 (5.8) |
AUC0–24, area under the plasma concentration‐time curve from 0–24 hours; AUC0‐∞, area under the plasma concentration‐time curve from 0 to infinity; AUC0–last, area under the plasma concentration‐time curve from 0 to last collection time after drug administration; CL/F, apparent oral clearance; Cmax, maximum plasma concentration; CSF, cerebrospinal fluid; NA, not applicable; rAUC, accumulation ratio area under the plasma concentration‐time curve; t1/2, apparent terminal elimination half‐life; Tmax, time to maximum plasma concentration; Vd/F, apparent volume of distribution.
Data are geometric mean (percent coefficient of variation), except Tmax is median (minimum‐maximum).
aOne subject discontinued early and, therefore, did not have day 7 pharmacokinetic blood draws; brAUC0–24 is the accumulation ratio, calculated as day 7 AUC0–24 divided by day 1 AUC0–24.
Cerebrospinal fluid concentrations
The geometric mean CSF concentrations following a single 50 mg dose of NYX‐2925 ranged from 3 ng/mL at 1 hour postdose to 71 ng/mL at 8 hours postdose. After 6 days of once‐daily dosing with 300 mg of NYX‐2925, the geometric mean CSF concentrations were 131 ng/mL at 1.5 hours postdose and 292 ng/mL at 3 hours postdose. Time to maximum CSF concentration was not evaluated.
Safety
NYX‐2925 was well‐tolerated with no reported serious or severe adverse events, and no study discontinuations due to adverse events. There were no clinically meaningful changes in any clinical laboratory value over time, and no laboratory result was reported as a treatment‐emergent adverse event (TEAE). The maximum tolerated dose was not reached in this study. Overall, at least one TEAE was reported by 24.2% (16/66) of subjects administered NYX‐2925 and by 16.7% (3/18) of subjects administered placebo. The most common TEAEs were procedural headache (n = 8), procedural pain (n = 8), and vomiting (n = 3); these events occurred only in the CSF cohorts, all of whom received NYX‐2925. A total of six events occurring in four subjects (n = 3 NYX‐2925; n = 1 placebo) were considered by the investigator to be treatment‐related (NYX‐2925: abdominal distension and flatulence (n = 1), headache (n = 1), procedural headache (n = 1); placebo: fatigue and lethargy (n = 1)). Additionally, urinary retention was reported for one subject in the NYX‐2925 treatment group; this event was considered unlikely related to treatment by the investigator, but possibly treatment‐related by the study sponsor. One TEAE (nasopharyngitis) was reported in the elderly cohort, and this was not considered related to the study drug.
There were no changes from baseline in the dissociative rating scale questionnaires (Brief Psychiatric Rating Scale – positive symptoms and Clinician Administered Dissociative States Scale), and no subject receiving NYX‐2925 or placebo reported any suicidal tendencies. No individual ECG finding was reported as a TEAE, and there were no clinically significant changes in HR, or PR and QRS intervals, or in ECG diagnostic statements. No subject had a QTcF interval > 480 msec at any post‐baseline visit, and there were no instances of a change from baseline > 60 msec. The PK/triplicate ECG analyses confirmed that NYX‐2925 does not result in QT prolongation. Further, there were no dose‐response relationships with HR, or PR and QRS, or QTcF.
DISCUSSION
This article reports on the first‐in‐human administration of NYX‐2925, an NMDAR modulator that is in development for the treatment of chronic pain conditions, including painful diabetic peripheral neuropathy (DPN) and fibromyalgia. The study evaluated single and multiple ascending doses of NYX‐2925 for safety, tolerability, and PK in healthy male and female volunteers. At single doses up to and including 1,200 mg and after multiple dosing up to and including 900 mg/day for 7 days, NYX‐2925 was well‐tolerated and no safety concerns emerged, including no increase in QTc interval. NYX‐2925 exhibited dose‐proportional PK and minimal accumulation following once‐daily dosing for 7 days. Renal clearance was found to be the predominant elimination route for NYX‐2925, with ~60–70% of the dose excreted unchanged in the urine.
Data from the single ascending dose phase of the study showed approximate dose proportional increases in NYX‐2925 exposure (Cmax and AUC) across a 24‐fold range of orally administered doses of NYX‐2925. Absorption was relatively rapid and consistent across the doses studied. The geometric mean estimates for clearance were consistent across doses. Likewise, volume of distribution was comparable across cohorts.
The PK profile of NYX‐2925 following multiple ascending doses was similar to that of single ascending doses, with Cmax and AUC demonstrating dose‐proportionality. Repeat dose administration over 7 days resulted in minimal accumulation across cohorts, as shown by rAUC0–24 comparing day 7 with day 1. Renal clearance over 7 days showed no dose relationship, and the percentage of NYX‐2925 recovered in the urine was comparable across doses.
Dose proportionality of Cmax and AUC by gender was demonstrated. Although overall exposures may have been somewhat greater in women than men at any given dose, the significance of this finding is unknown considering the limited data set.
Half‐life was ~4 hours after single‐dose administration and trended higher on day 7 vs. day 1 in the MAD phase. However, although single doses in preclinical studies have shown efficacy in models of chronic pain, once‐daily doses were also able to convey and maintain analgesia, with long‐lasting (1 week) effects, far exceeding that of NYX‐2925 half‐life.2, 20 Therefore, a once‐daily dosing regimen is being evaluated in the phase II studies.
The effect of food and age were explored in two separate dose groups in the SAD phase. Compared with the administration of a single 400‐mg dose of NYX‐2925 in a fasted state, a high‐fat, high‐calorie meal minimally slowed absorption (Tmax) and reduced Cmax, but did not affect overall extent of exposure (AUC), suggesting that NYX‐2925 can be administered with or without food. A single 300‐mg dose of NYX‐2925 in the elderly cohort did not result in meaningful PK differences compared with the remaining SAD dose groups, despite an ~15% lower estimated glomerular filtration rate in the elderly, suggesting that the absorption, exposure, and elimination of NYX‐2925 may not be impacted by age. These findings are limited by the small sample sizes of the populations and will need to be confirmed.
The concentration of NYX‐2925 in CSF was investigated in two dose cohorts (50‐mg single dose and 150‐mg multiple dose). NYX‐2925 was found to cross the blood brain barrier in a dose‐proportional fashion, with maximum CSF concentrations 6–9% of plasma Cmax, indicating adequate penetration into the target effect compartment. These data are in accord with preclinical findings in a rat study in which CSF concentrations were 7–8% of plasma Cmax (unpublished data).
The maximum doses studied in the SAD and MAD phases were determined to be at least 5 to 10‐times the anticipated therapeutic doses, and were within the safety margin of exposure established by preclinical studies. No serious or severe adverse events were reported and no laboratory value changes were considered clinically meaningful or reported as TEAEs. Most TEAEs occurred in the CSF cohorts and were common sequelae of lumbar punctures. In this study, safety and tolerability were demonstrated at doses of up to and including 1,200 mg, and a maximum tolerated dose was not reached. Importantly, there were no dissociative side effects or ECG findings, including QTc, at any dose. These data confirm the safety findings observed in preclinical studies, including no observed toxicity in rats administered up to 1,000 mg/kg/day for 14 days.2
Neuropathic pain (“pain caused by a lesion or disease of the somatosensory nervous system”)21 has an estimated prevalence of around 10%22, 23, 24 and is distinct from nociceptive pain (“pain that arises from actual or threatened damage to non‐neural tissue and is due to activation of nociceptors” and occurs with a normally functioning somatosensory nervous system).21 Somatosensory abnormalities causing neuropathic pain can occur in both the peripheral and central nervous systems.25 One common neuropathic pain condition is painful DPN, which is estimated to occur in 11–24% of individuals who develop DPN.26
Fibromyalgia is a syndrome of chronic widespread musculoskeletal pain with no identifiable cause, and is estimated to affect around 4 million adults in the United States.27 Although fibromyalgia is not considered a neuropathic condition, there seems to be a CNS component to the pain and symptoms experienced by most individuals with fibromyalgia.17 Elevated glutamate levels have been observed in both the pain‐related regions of the brain and in the CSF of patients with fibromyalgia.18 The CSF of patients with fibromyalgia has also been shown to have elevated glycine levels.18 In a recent systematic literature review, a significant correlation was found between elevated glutamate levels and risk for fibromyalgia symptoms.28
Treatment for DPN and fibromyalgia has relied upon agents originally developed to treat conditions, including seizures, depression, and generalized pain. Current therapies include pregabalin, duloxetine, and opioids. Unfortunately, these treatments are only modestly effective and often associated with intolerable side effects.29, 30, 31, 32 Thus, there is a substantial need to develop novel agents that are well tolerated and provide effective pain relief for these conditions.
NYX‐2925 is being developed for the treatment of chronic pain conditions, including painful DPN and fibromyalgia. In this first‐in‐human study in healthy volunteers, in which the range of doses approximated and far exceeded the expected therapeutic doses, there were no safety or tolerability issues. NYX‐2925 demonstrated dose‐proportional PK and adequately crossed the blood brain barrier. These data, along with the current understanding of the pivotal role that the CNS plays in neuropathic pain, support the continued evaluation of NYX‐2925 in chronic pain conditions.
Funding
This study was funded by Aptinyx Inc.
Conflict of Interest
D. Houck, L. Sindelar, M. Krueger, M. Suh, and T. Madsen are employees and stock and option holders of Aptinyx Inc. C. Sanabria and S. Stanworth are employees of Spaulding Clinical Research, LLC.
Author Contributions
T.M., D.H., and L.S. wrote the manuscript. T.M., D.H., and L.S. designed the research. C.R.S. performed the research. T.M., D.H., L.S., S.H.S., M.K., and M.S. analyzed the data.
Acknowledgments
This study was sponsored by Aptinyx Inc. Medical writing support was provided by Lorraine R. Baer, PharmD (Baer PharMed Consulting, Ltd.), on behalf of and funded by Aptinyx Inc.
References
- 1. Amidon, G.L. , Lennernäs, H. , Shah, V.P. & Crison, J.R. A theoretical basis for a biopharmaceutic drug classification: the correlation of in vitro drug product dissolution and in vivo bioavailability. Pharm. Res. 12, 413–420 (1995). [DOI] [PubMed] [Google Scholar]
- 2. Khan, M.A. et al NYX‐2925 is a novel NMDA receptor‐specific spirocyclic‐β‐lactam that modulates synaptic plasticity processes associated with learning and memory. Int. J. Neuropsychopharmacol. 21, 242–254 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Hollmann, M. & Heinemann, S. Cloned glutamate receptors. Annu. Rev. Neurosci. 17, 31–108 (1994). [DOI] [PubMed] [Google Scholar]
- 4. Zhou, H.Y. , Chen, S.R. & Pan, H.L. Targeting N‐methyl‐D‐aspartate receptors for treatment of neuropathic pain. Expert Rev. Clin. Pharmacol. 4, 379–388 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Paoletti, P. , Bellone, C. & Zhou, Q. NMDA receptor subunit diversity: Impact on receptor properties, synaptic plasticity and disease. Nat. Rev. Neurosci. 14, 383–400 (2013). [DOI] [PubMed] [Google Scholar]
- 6. Hansen, K.B. , Yi, F. , Perszyk, R.E. , Menniti, F.S. & Traynelis, S.F. NMDA receptors in the central nervous system. Methods Mol. Biol. 1677, 1–80 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Bliss, T.V. & Collingridge, G.L. A synaptic model of memory: long‐term potentiation in the hippocampus. Nature 361, 31–39 (1993). [DOI] [PubMed] [Google Scholar]
- 8. Yashiro, K. & Philpot, B.D. Regulation of NMDA receptor subunit expression and its implications for LTD, LTP, and metaplasticity. Neuropharmacology 55, 1081–1094 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Morris, R.G. NMDA receptors and memory encoding. Neuropharmacology 74, 32–40 (2013). [DOI] [PubMed] [Google Scholar]
- 10. Traynelis, S.F. et al Glutamate receptor ion channels: structure, regulation, and function. Pharmacol. Rev. 62, 405–496 (2010). Erratum in: Pharmacol Rev 66, 1141 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Zafra, F. , Ibáñez, I. , Bartolomé‐Martín, D. , Piniella, D. , Arribas‐Blázquez, M. & Giménez, C. Glycine transporters and its coupling with NMDA receptors. Adv. Neurobiol. 16, 55–83 (2017). [DOI] [PubMed] [Google Scholar]
- 12. Gonzalez, J. , Jurado‐Coronel, J.C. , Ávila, M.F. , Sabogal, A. , Capani, F. & Barreto, G.E. NMDARs in neurological diseases: a potential therapeutic target. Int. J. Neurosci. 125, 315–327 (2015). [DOI] [PubMed] [Google Scholar]
- 13. Dore, K. , Stein, I.S. , Brock, J.A. , Castillo, P.E. , Zito, K. & Sjöström, P.J. Unconventional NMDA receptor signaling. J. Neurosci. 37, 10800–10807 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Balu, D.T. et al Multiple risk pathways for schizophrenia converge in serine racemase knockout mice, a mouse model of NMDA receptor hypofunction. Proc. Natl. Acad. Sci. 110, E2400–E2409 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Coyle, J.T. NMDA receptor and schizophrenia: A brief history. Schizophr. Bull. 38, 920–926 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kalia, L.V. , Kalia, S.K. & Salter, M.W. NMDA receptors in clinical neurology: excitatory times ahead. Lancet Neurol. 7, 742–755 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Sluka, K.A. & Clauw, D.J. Neurobiology of fibromyalgia and chronic widespread pain. Neuroscience 338, 114–129 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Littlejohn, G. & Guymer, E. Modulation of NMDA receptor activity in fibromyalgia. Biomedicines 5, piiE15 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Bennett, G.J. & Xie, Y.K. A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain 33, 87–107 (1988). [DOI] [PubMed] [Google Scholar]
- 20. Ghoreishi‐Haack, N. et al NYX‐2925 is a novel NMDA receptor modulator that induces rapid and long‐lasting analgesia in rat models of neuropathic pain. J. Pharmacol. Exp. Ther. 366, 485–497 (2018). [DOI] [PubMed] [Google Scholar]
- 21. International Association for the Study of Pain. IASP Terminology, Pain Terms <https://www.iasp-pain.org/terminology?navItemNumber=576> (1994). Accessed May 22, 2018.
- 22. DiBonaventura, M.D. et al The prevalence of probable neuropathic pain in the US: results from a multimodal general‐population health survey. J. Pain Res. 10, 2525–2538 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. van Hecke, O. , Austin, S.K. , Khan, R.A. , Smith, B.H. & Torrance, N. Neuropathic pain in the general population: a systematic review of epidemiological studies. Pain 155, 654–662 (2014). Erratum in: Pain 155, 1907 (2014). [DOI] [PubMed] [Google Scholar]
- 24. Yawn, B.P. , Wollan, P.C. , Weingarten, T.N. , Watson, J.C. , Hooten, W.M. & Melton, L.J. 3rd The prevalence of neuropathic pain: clinical evaluation compared with screening tools in a community population. Pain Med. 10, 586–593 (2009). Erratum in: Pain Med 12, 1294 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Jones, R.C. 3rd , Lawson, E. & Backonja, M. Managing neuropathic pain. Med. Clin. North Am. 100, 151–167 (2016). [DOI] [PubMed] [Google Scholar]
- 26. Pruitt, J. 3rd , Moracho‐Vilrriales, C. , Threatt, T. , Wagner, S. , Wu, J. & Romero‐Sandoval, E.A. Identification, prevalence, and treatment of painful diabetic neuropathy in patients from a rural area in South Carolina. J. Pain Res. 10, 833–843 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Centers for Disease Control and Prevention. Fibromyalgia. <https://www.cdc.gov/arthritis/basics/fibromyalgia.htm> (2018). Accessed April 17, 2018.
- 28. Pyke, T.L. , Osmotherly, P.G. & Baines, S. Measuring glutamate levels in the brains of fibromyalgia patients and a potential role for glutamate in the pathophysiology of fibromyalgia symptoms: a systematic review. Clin. J. Pain 33, 944–954 (2017). [DOI] [PubMed] [Google Scholar]
- 29. Macfarlane, G.J. et al EULAR revised recommendations for the management of fibromyalgia. Ann. Rheum. Dis. 76, 318–328 (2017). [DOI] [PubMed] [Google Scholar]
- 30. Pop‐Busui, R. et al Diabetic neuropathy: a position statement by the American Diabetes Association. Diabetes Care 40, 136–154 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bril, V. et al Evidence‐based guideline: treatment of painful diabetic neuropathy: report of the American Academy of Neurology, the American Association of Neuromuscular and Electrodiagnostic Medicine, and the American Academy of Physical Medicine and Rehabilitation. Neurology 76, 1758–1765 (2011). Erratum in: Neurology 77, 603 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Finnerup, N.B. et al Pharmacotherapy for neuropathic pain in adults: a systematic review and meta‐analysis. Lancet Neurol. 14, 162–173 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]