

Abstract
Objective
The ubiquitin-proteasome system pathway has been recognised as a crucial cellular mechanism for the proper function of photoreceptor cells. In particular, ubiquitin ligases (E3s) recognise and ubiquitinate specific proteins for degradation. The KLHL7 protein (a BTB-Kelch protein) has been found to play an important role in this process. There have been several reports that heterozygous mutations in the KLHL7 gene in adults are responsible for a rare cause of late-onset autosomal dominant retinitis pigmentosa with preservation of central vision and homozygous mutations in two young children, with Crisponi syndrome (CS)/cold-induced sweating syndrome type 1, result in a recessive form of early-onset peripheral retinal dystrophy type changes. The majority of children do not survive through to adulthood. The objective of this study is to report the visual symptoms and signs of two young adults clinically diagnosed with overlapping BOS/Cisproni syndrome, expanding the phenotypic presentation of KLHL7 gene mutations.
Methods and analysis
This is a case report of the ophthalmic findings of two siblings with biallelic KLHL7 gene mutations. Siblings born to a non-consanguineous family and diagnosed with the overlapping clinical phenotype of Bohring-Opitz and and confirmed biallelic KLHL 7 gene mutation by whole exome sequencing were identified. Ophthlamic history and fundal examination was performed and analysed.
Results
Both patients had similar retinal findings. The fundus shows confluent hypopigmented/pale yellow lesions in the mid-periphery. The optic disc appears to be pale with a ring of atrophy and vessels appear attenuated. The macular of the younger patient shows a depigmented area around the fovea giving a bull’s-eye appearance while the older sibling shows a fibrotic ring around the fovea suggesting a more advanced pathology.
Conclusion
This paper expands the retinal phenotype to include a distinctive maculopathy in a recently described homozygous mutation in the KLHL7 gene in two young adults presenting with features that overlap the Bohring-Opitz syndrome and CS.
Keywords: retina, genetics
Key messages.
What is already known about this subject?
Ubiquitin-proteasome system (UPS) pathway is recognised to be implicated in maintaining proper function of photoreceptor cells and the KLHL7 protein (a BTB-Kelch protein) has been found to play an important role in this process. However, there had been a paucity of literature with a handful of case reports suggesting associations between the KLHL7 gene and autosomal dominant retinitis pigmentosa. In homozygous KLHL7 mutation, mid-periphery retinal changes were previously described.
What are the new findings?
In this study, we report novel macular findings observed in two young adults with homozygous KLHL7 gene mutation. The macular of the younger patient shows a depigmented area around the fovea giving a bull’s-eye appearance while the older sibling shows a fibrotic ring around the fovea suggesting a more advanced pathology.
How might these results change the focus of research or clinical practice?
This further strengthens the evidence of the implication and role of UPS in photoreceptor health. The newly described findings will add to the list of features for KLHL7 gene mutation, which will suggest the need for ophthalmic referrals as part of the clinical management of cases.
Introduction
Heterozygous variants in the KLHL7 gene have been causally associated with autosomal dominant retinitis pigmentosa (adRP) after linkage analysis in several families.1–3 KLHL7 encodes a 586 amino acid BTB-Kelch protein, which contains a BTB, BACK and a Kelch domain (with six Kelch motifs). It has earlier been identified to be a substrate recognition subunit of a CUL3-based ubiquitin ligase complex, through formation of a dimer via its BTB and BACK domains. The resultant E3 (cullin ubiquitin ligating enzyme) activity then polyubiquitinates target proteins for proteasome-mediated degradation. Although the exact biological function of KLHL7 is currently unknown, it is widely expressed in rod photoreceptor cells.4 It is proposed that failure to form the E3 ligase complexes could result in accumulation of substrate proteins in photoreceptor cells, with their eventual demise and consequential retinal degeneration.4
The adRP caused by heterozygous KLHL7 variants is late onset (>50 years of age), with better preservation of vision and rod function (demonstrated by the lower annual rate of decline in 31 Hz Flicker) than that of a typical patient with RP (3% vs 10%).3
Biallelic KLHL7 gene variants have been reported to cause a severe multisystem disorder which overlaps Bohring-Opitz syndrome (BOS) and Crisponi syndrome. To date, 11 patients have been reported in two papers. Briefly, reported systemic features include severe to profound learning difficulties, microcephaly, feeding and swallowing difficulties, hypotonia and joint contractures including camptodactyly. The facial gestalt is somewhat variable but often includes exophthalmos, nevus flammeus, micro/retrognathia, anteverted nares, hypertelorism and a high-arched narrow palate.5 6 In 2016, Angius et al described the effect of biallelic KLHL7 mutations on the retina in two young children: aged 4 (c.1258C>T/p.Arg420Cys) and 6 (c.1022delT/p.Leu341Trpfs*9) years, both showed signs of early-onset RP.7 In the same paper, it was noted that other causes for a Crisponi syndrome phenotype such as variants in the CRLF1 or L1FR genes were not found to cause retinal changes.
Here, we report novel visual symptoms and signs in two young adults clinically diagnosed with the overlapping BOS/Crisponi syndrome expanding the phenotypic presentation of homozygous KLHL7 gene variants to include a distinctive maculopathy.
Case presentation
Research procedures were performed in accord with institutional guidelines and the Declaration of Helsinki and informed consent was obtained from the patient’s parents.
Patients 1 and 2 are siblings born to a non-consanguineous family with no significant family history including that of RP. They were diagnosed with the overlapping clinical phenotype of Bohring-Opitz and Crisponi syndromes with features reported by Bruel et al.5 Genetic testing identified homozygous truncating variants NM_001031710.2:c.1051C>T (p.Arg351*) in the KLHL7 gene by whole exome sequencing in both siblings while each parent was found to be heterozygous for the truncating variant.
Clinical presentation
At the time of presentation to the ophthalmology department, patient 1 was a 21-year-old man and patient 2 was a 19-year-old woman. Both siblings developed respiratory distress requiring intubation and surfactant at birth, and had significant feeding difficulties, with reflux, requiring fundoplication and gastrotomy. They have similar facial features, including a glabellar nevus flammeus, high-arched pallet with broad alveolar ridges, hypertelorism, thin upper lip, low-set dysplastic ears and microcephaly. They also have a BOS posture with fixed contractions at the elbows and fingers and poor vision.
Ophthalmic symptoms and signs
The parents reported that their 21-year-old son had difficulties with depth perception, in particular, reaching out for objects at a closer distance than they were actually placed. He could fix and follow an object but not read any of the Cardiff Cards. He appeared to see better if he turned his head to view an object held directly in front of him. His younger sister could read the Cardiff Card D held at 50 cm but appeared to struggle more with her peripheral field on a functional level. Both patients had severe mental disabilities prohibiting any detailed testing of visual function. Both patients had normal cornea, lens and anterior chamber examination. Peripheral retinal signs were remarkably similar and symmetrical in both patients, with bilateral confluent hypopigmented/pale yellow lesions (reminiscent of, but larger than, deposits seen in Stargardt disease) in the mid-periphery. The far periphery was unaffected in both siblings (figure 1). In the macular of the younger sibling there was a depigmented area around the fovea giving a bull’s-eye appearance. In the older male there was a fibrotic ring around the fovea suggesting a more advanced pathology (figure 2). The optic discs were again a paler colour in the older sibling with a ring of peripapillary atrophy. Retinal arterioles were generally attenuated in both siblings. No pigmented bone spicules were noted and there was no evidence of inflammation or vitritis.
Figure 1.
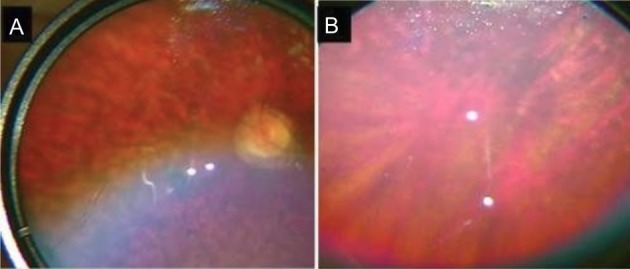
Fundal images of patient 2. (A) The fundal image of patient 2, which shows confluent hypopigmented/pale yellow lesions in the mid-periphery. The optic disc appears to be pale with a ring of atrophy and vessels appear attenuated. (B) The far periphery of retina of the same eye of the patient, note that the hypopigmented lesions do not extend to the far periphery.
Figure 2.
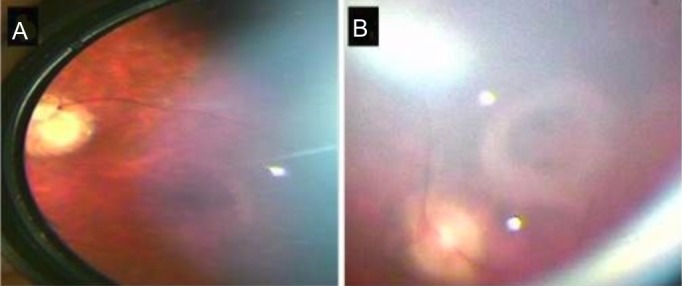
Fundal images demonstrating macular features in biallelic KLHL7 mutation. (A) The fundal image of patient 2 (younger sibling) which shows a depigmented area around the fovea giving a bull’s-eye appearance. (B) (patient 1) A fibrotic ring around the fovea which may suggest a more advanced pathology.
Ophthalmic examination of both parents, aged 60 years, demonstrated good visual acuities. The mother had diabetes and changes in association with diabetic retinopathy, while the father had a completely normal eye examination. There were no signs of RP in either parent.
Discussion
Here, we described the retinal findings in a sibling pair of young adults with homozygous truncating mutations NM_001031710.2:c.1051C>T (p.Arg351*) in the KLHL7 gene and a systemic phenotype matching the BOS-Crisponi syndrome. There has only been one other report of early-onset RP in two children aged 4 and 6 years with biallelic mutations in the KLHL7 gene.7
There appears to be a phenotype-genotype correlation in the KLHL7 gene.4 The three substitutions resulting in late-onset adRP were found in the BACK domain, which is required for CUL3 binding with BTB domain. In these cases, the central macula area was well preserved. The biallelic mutations causing the more severe early-onset form of RP in both the previously described cases and our two older siblings were in the Kelch domain (hypothesised to form salt bridges with nearby proteins), resulting in either a premature termination codon producing an incomplete Kelch domain from the KLHL7 protein, or missense mutations resulting in loss of stabilisation, with diminished substrate binding. There are no published studies which demonstrate the functional effect of the current mutation (pArg351), but we hypothesise similar loss of function of KLHL7 protein as a possible mechanism due to its position in the Kelch domain and the premature termination of translation (figure 3).
Figure 3.
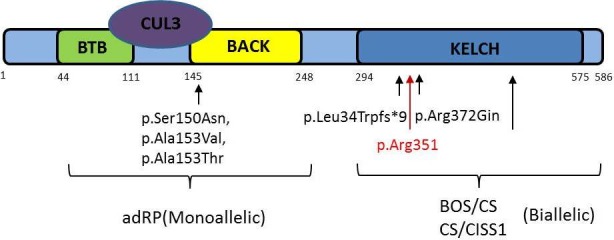
Figure demonstrating KLHL7 protein monoallelic gene mutations are located at the BACK domain, which with BTB domain will interfere with CUL3 binding, which usually polyubiquitinates target proteins for proteasome-mediated degradation. This has been found to be associated with autosomal dominant retinitis pigmentosa (RP). Biallelic gene mutations (gene mutation in current paper highlighted in red, p.Arg351) are found in the Kelch domain with clinical phenotypes of BOS/CS and CS/CSS1 syndrome. adRP, autosomal dominant retinitis pigmentosa; BOS, Bohring-Opitz syndrome; CISS1, old-induced sweating syndrome type 1; CS, Crisponi syndrome.
The observed retinal changes in our two cases had some overlap with those described in the earlier reports with well-circumscribed hypopigmented lesions that appeared clinically reminiscent of lipofuscin deposits, mainly in the mid-periphery area (table 1). However, we also observed a distinctive maculopathy with a bull’s-eye appearance progressing to a fibrotic/atrophic ring in the older sibling (figure 2). Macular features were present in both siblings bilaterally. This is the first report of the ocular phenotype of adults with recessive KLHL7 variants and it is possible that the older age may have contributed to these differences and represent a more advanced phenotype. Such changes may be hypothesised to be due to the accumulation of toxic substrates due to the lack of E3 activity which polyubiquitinates target proteins for proteasome-mediated degradation. Interestingly, the heterozygous or monoallelic mutation of the same gene did not show any evidence of retinal dysfunction in either of the parents in this study or in previous studies.1–3 7 It appears therefore that haploinsufficiency is not sufficient alone to disrupt the ubiquitin-proteasome pathway and produce a clinical phenotype but complete lack of functional protein is necessary.
Table 1.
Retinal findings of families with KLHL7 mutation
| Literature | Systemic associations | Patient cohort | Retinal findings | Genetic mutation |
| Friedman et al 1 | Not reported | 24 patients from a six-generation Scandinavian family | Changes of retinitis pigmentosa: retinal degeneration and bone spicules | Heterogeneous c.449G/A (p.S150N), in exon 6 of the KLHL7 gene |
| Hugosson et al 2 | Not reported | 11 patients from a single family from the Swedish retinitis pigmentosa register |
|
Heterozygous exon 6 change (c.458C>T) |
| Wen et al 3 | Nil | 5 unrelated families referred to the Retina Foundation of the Southwest |
|
Heterozygous c.458C>T (p.Ala153Val), c.449G>A (p.Ser150Asn) and c.457G>A (p.Ala153Thr) |
| Angius et al 7 | Crisponi syndrome (CS)/cold-induced sweating syndrome type 1 (CISS1) | 2 children who has been diagnosed with CS/CISS1 syndrome, 4 and 6 years of age | Fundi: abnormal appearance of the macula, attenuated arteriolar vessels, retinal pigmentary changes | Homozygous c.1258C>T/p.Arg420Cys and c.1022delT/p.Leu341 |
| Heng et al (current paper) | BOS-Crisponi syndrome | 2 siblings from non-consanguineous parents from the UK | Fundi: soft confluent pale yellow lesions in mid-periphery, attenuated vessels, waxy optic disc, bull’s-eye maculopathy | Homozygous c.1051C>T (p.Arg351*) |
BOS, Bohring-Opitz syndrome; ERG, electroretinography; RP, retinitis pigmentosa.
In conclusion, this paper is the first to describe the ocular phenotype in adults with a biallelic KLHL7 stop mutation (Arg 351) and expands the clinical findings to include a distinctive maculopathy with associated loss of visual function.
Footnotes
Contributors: LZH and AC had substantial contributions to the conception or design of the work, or the acquisition, analysis or interpretation of data; drafting the work or revising it critically for important intellectual content; gave final approval of the version published. JK, SS and RNE had substantial contributions to the acquisition and interpretation of genetic data; drafting and revising of the paper; gave final approval of the version submitted.
Funding: The authors have not declared a specific grant for this research from any funding agency in the public, commercial or not-for-profit sectors.
Competing interests: None declared.
Patient consent for publication: Obtained.
Provenance and peer review: Not commissioned; externally peer reviewed.
References
- 1. Friedman JS, Ray JW, Waseem N, et al. Mutations in a BTB-Kelch protein, KLHL7, cause autosomal-dominant retinitis pigmentosa. Am J Hum Genet 2009;84:792–800. 10.1016/j.ajhg.2009.05.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Hugosson T, Friedman JS, Ponjavic V, et al. Phenotype associated with mutation in the recently identified autosomal dominant retinitis pigmentosa KLHL7 gene. Arch Ophthalmol 2010;128:772–8. 10.1001/archophthalmol.2010.98 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Wen Y, Locke KG, Klein M, et al. Phenotypic characterization of 3 families with autosomal dominant retinitis pigmentosa due to mutations in KLHL7. Arch Ophthalmol 2011;129:1475–82. 10.1001/archophthalmol.2011.307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Kigoshi Y, Tsuruta F, Chiba T. Ubiquitin ligase activity of Cul3-KLHL7 protein is attenuated by autosomal dominant retinitis pigmentosa causative mutation. J Biol Chem 2011;286:33613–21. 10.1074/jbc.M111.245126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Bruel AL, Bigoni S, Kennedy J, et al. Expanding the clinical spectrum of recessive truncating mutations of KLHL7 to a Bohring-Opitz-like phenotype. J Med Genet 2017;54:830–5. 10.1136/jmedgenet-2017-104748 [DOI] [PubMed] [Google Scholar]
- 6. Crisponi L, Crisponi G. Meloni aet al. Crisponi syndrome is caused by mutations in the CRLF1 gene and is allelic to cold-induced sweating syndrome type 1. Am. J. Hum. Genet 2016;99:236–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Angius A, Uva P, Buers I, et al. Bi-allelic mutations in KLHL7 cause a Crisponi/CISS1-like phenotype associated with early-onset retinitis pigmentosa. Am J Hum Genet 2016;99:236–45. 10.1016/j.ajhg.2016.05.026 [DOI] [PMC free article] [PubMed] [Google Scholar]