

An intermolecular dialkylation of alkenes via a radical process with two distinct C(sp3)─H bonds is described.
Abstract
The functionalization of unactivated C(sp3)─H bonds represents one of the most powerful and most atom-economical tools for the formation of new carbon-based chemical bonds in synthesis. Although cross-dehydrogenative coupling reactions of two distinct C─H bonds for the formation of carbon-carbon bonds have been well investigated, controlled functionalizations of two or more different C(sp3)─H bonds across a functional group or a molecule (e.g., an alkene or alkyne) in a single reaction remain challenging. Here, we present a three-component dialkylation of alkenes with common alkanes and 1,3-dicarbonyl compounds via synergistic photoredox catalysis and iron catalysis for the synthesis of two functionalized 1,3-dicarbonyl compounds. Mechanistic studies suggest that the photoredox catalysis serves as a promotion system to allow the dialkylation to proceed under mild conditions by reducing the oxidation and reduction potentials of the iron intermediates and the reaction partners.
INTRODUCTION
The development of a mild, economical, and practical catalytic process that can selectively and rapidly increase molecular complexity starting from readily available materials, especially petroleum-based feedstocks (e.g., alkenes, alkanes, and arenes), is a critical objective in both the academic and industrial communities. In this field, the functionalization of a molecule through transformations of an alkene and/or C─H bonds is a particularly fascinating but challenging goal, which has therefore attracted substantial attention from chemical researchers. Typical technologies include alkene dicarbofunctionalizations that enable the concomitant incorporation of two vicinal carbon-based functional groups to lengthen carbon chains and increase structural complexity (1–4). However, the vast majority of alkene dicarbofunctionalization approaches focus on the merger of the classical cross-couplings (1–18), which suffer from requiring expensive nucleophilic and electrophilic functional reagents, such as organometallic species and organohalides, for inserting the C═C bonds in the presence of noble transition metals and/or ligands. Moreover, examples that allow the addition of alkyl groups across alkenes to achieve dicarbofunctionalization are rare; this is partly because competitive side reactions, such as the Heck-type β-hydride elimination, homocoupling, isomerization, and/or protodemetalation, are common in these types of reactions (5–18).
Alternatively, alkene dicarbofunctionalizations via the oxidative radical functionalization of one or two C─H bonds has attracted increasing interest in the past few years because it features excellent selectivity, high atom economy, and cost efficiency by avoiding the use of expensive functional reagents and noble metals (19–21). However, the number of available methods is much lower, and methods are limited to two modes, alkylarylation (22–26) and acylarylation (27, 28), which are accomplished via an intramolecular inherent aryl C(sp2)─H cyclization process to prepare cyclic compounds at high reaction temperatures. Furthermore, to the best of our knowledge, examples of using two distinct C(sp3)─H bonds in the intermolecular dialkylation of alkenes has not been reported, as controlled functionalizations of two or more distinct C(sp3)─H bonds across a functional group or a molecule (e.g., an alkene or alkyne) in a single reaction is challenging.
Merging visible light photoredox catalysis with transition metal catalysis has become a conceptually powerful strategy for the formation of chemical bonds owing to its extraordinary catalytic activity and mild conditions (29–32), and this method has also been applied to the functionalization of C(sp3)─H bonds (29–37). In these radical processes, selectivity and reactivity could be manipulated mainly through tuning the oxidation and reduction potentials of the reaction partners and catalysts (29–32). We envisioned that using photoredox and transition metal catalysts cooperatively might allow the controlled and simultaneous transformation of an alkene with two or more C(sp3)─H bonds under mild conditions by regulating the oxidation and reduction potentials. Here, we report a new radical-mediated intermolecular 1,2-dialkylation of terminal alkenes with two distinct C(sp3)─H bonds enabled by synergistic photoredox catalysis and iron catalysis (Fig. 1). The activation of two distinct C(sp3)─H bonds for insertion across C═C bonds is disclosed for the first time, and in this reaction, a C(sp3)─H bond of a cycloalkane, linear alkane, or ether serves as an sp3-hybridized carbon-centered radical precursor to initiate this reaction, and the other C(sp3)─H bond, α to a 1,3-dicarbonyl fragment, terminates this reaction. Notably, such multifunctional 1,3-dicarbonyl compounds are among the most common and versatile building blocks in synthesis, and they are privileged structural units in natural products, bioactive molecules, and materials.
Fig. 1. 1,2-Dialkylation of alkenes with two distinct C(sp3)─H bonds.
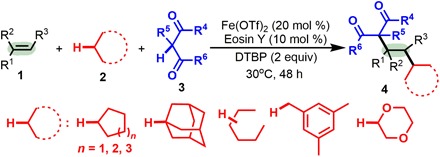
Synergistic photoredox catalysis and iron catalysis for the intermolecular dialkylation of alkenes with alkanes and 1,3-dicarbonyl compounds to synthesize two functionalized 1,3-dicarbonyl compounds.
RESULTS
Optimization of the reaction conditions
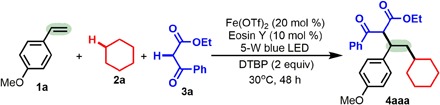
Initial experiments were performed using three reaction partners, namely, 1-methoxy-4-vinylbenzene (1a), cyclohexane (2a), and ethyl 3-oxo-3-phenylpropanoate (3a), by merging photoredox catalysis and iron catalysis (Table 1). The use of Fe(OTf)2 as the metal catalyst and Eosin Y as the visible light catalyst in the presence of 2 equiv of di-tert-butyl peroxide (DTBP) and a 5-W blue light-emitting diode (LED) light at 30°C successfully allowed the 1,2-dialkylation of alkene 1a with 2a and 3a, giving the desired product 4aaa in 82% yield (entry 1). However, in the absence of Fe(OTf)2, only trace gas chromatography (GC) yield of 4aaa was observed at either 30° or 120°C (entry 2). We found that a lower [10 mole percent (mol %)] or a higher (30 mol %) loading of Fe(OTf)2 had a negative effect (entries 3 and 4). Other Fe catalysts, including FeCl2, Fe(acac)2, Fe(OTf)3, and FeCl3, were examined (entries 1 and 5 to 7), and each of them was less efficient than Fe(OTf)2, and the Fe(II) catalysts were more active than the Fe(III) catalysts. Both Eosin Y (entry 8) and DTBP (entry 9) are necessary for the dialkylation to proceed below 100°C (entries 2 to 7), as in the absence of either, the reaction did not proceed. It is noteworthy that without Eosin Y, the reaction can proceed when the reaction temperature is increased to 110°C (entry 9; 36% yield at 110°C and 80% yield at 120°C), but without DTBP, the reaction does not occur even at 120°C (entry 12). Furthermore, the reaction did not proceed in the dark at 30°C but did generate 4aaa in 78% yield (entry 15). These results suggest that the photoredox catalyst promotes the reaction, probably by reducing the oxidation and reduction potentials of the iron intermediates. However, other visible light metal catalysts [e.g., Ru(bpy)3Cl2 or Ir(ppy)3] and peroxides [e.g., tert-Butyl hydroperoxide (TBHP) or tert-Butyl peroxybenzoate (TBPB)] all proved to be less reactive (entries 10 and 11 and 13 and 14). The reaction proceeded smoothly on scales up to 1 mmol of 1a (80% yield; entry 16).
Table 1. Optimization of reaction conditions.
Experiments were performed with 1a (0.2 mmol), 2a (2 ml), 3a (2 equiv), Fe(OTf)2 (20 mol %), Eosin Y (10 mol %), DTBP (2 equiv), 5-W blue LED, argon, 30y°C, and 48 hours. The dr value is 1.1:1, as determined by 1H nuclear magnetic resonance (NMR) analysis of the crude product.
| Entry | Variation from the optimal conditions | Isolated yield (%) |
| 1 | None | 82 |
| 2 | Without Fe(OTf)2 | Trace* |
| 3 | Fe(OTf)2 (10 mol %) at 60°C | 40 |
| 4 | Fe(OTf)2 (30 mol %) | 53 |
| 5 | FeCl2 instead of Fe(OTf)2 | 71 |
| 6 | Fe(acac)2 instead of Fe(OTf)2 | 62 |
| 7 | Fe(OTf)3 instead of Fe(OTf)2 | 8 |
| 8 | FeCl3 instead of Fe(OTf)2 | 21 |
| 9 | Without Eosin Y | Trace†‡/36%§/80%║ |
| 10 | Ru(bpy)3Cl2 instead of Eosin Y | <5 |
| 11 | Ir(ppy)3 instead of Eosin Y | <5/51%¶ |
| 12 | Without DTBP | Trace* |
| 13 | TBHP instead of DTBP | 22 |
| 14 | TBPB instead of DTBP | 57 |
| 15 | Without visible light (in the dark) | Trace/31%‡/78%║ |
| 16# | None | 80 |
*At 30° or 120°C.
†At 30° or 100°C.
‡At 60°C.
§At 110°C.
║At 120°C.
¶At 80°C.
#1a (1 mmol).
Substrate scope with respect to alkenes
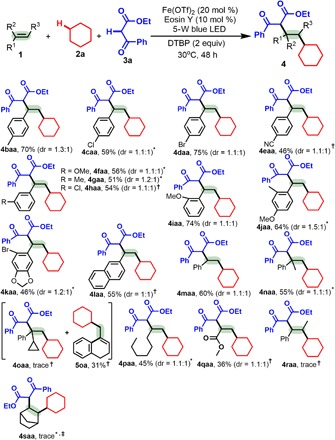
With the optimal reaction conditions in hand, we then investigated the generality of this three-component dialkylation protocol with respect to alkenes (Table 2), alkanes, and 1,3-dicarbonyl compounds (Table 3). Under the optimal conditions, this protocol proved amenable to a wide range of terminal alkenes, including arylalkenes 1b to 1l, 1,1-disubstituted alkene 1m, alkyalkene 1n, and methyl acrylate 1o but unsuitable for internal alkene 1r (Table 2). For arylalkenes 1b to 1l, several substituents, including Me, Cl, Br, CN, and MeO, on the aryl ring were well tolerated (4baa to 4jaa), and their electronic and steric properties influenced the yields. While alkene 1b with an electron-donating 4-Me group gave 70% yield of 4baa, alkene 1e with an electron-withdrawing 4-CN group afforded 46% yield of 4eaa. Unexpectedly, o-MeO–substituted arylalkene 1i is more reactive (4iaa) than m-MeO–substituted alkene 1f (4faa), probably due to the electronic effect more so than the steric effects. Notably, halogen groups, including Cl and Br, are compatible with the optimal conditions and thus provide easy handles for further synthetic elaboration of the halogenated positions (4caa to 4daa and 4kaa). Using polysubstituted arylalkenes 1j and 1k provided 4jaa to 4kaa. Both 2-vinylnaphthalene 1l and styrene 1m are suitable substrates for constructing 4laa to 4maa. Prop-1-en-2-ylbenzene 1n, a 1,1-disubstituted alkene, was smoothly transformed into 4naa. However, (1-cyclopropylvinyl)benzene 1o did not afford 4oaa and instead generated 4-(cyclohexylmethyl)-1,2-dihydronaphthalene (5oa) via ring opening, which supports a radical process (24). Notably, alkylalkene 1p and electron-poor methyl acrylate 1q successfully delivered 4paa to 4qaa, albeit in reduced yields. Unfortunately, attempts at the 1,2-dialkylation of internal alkene 1r failed to provide 4raa.
Table 2. Variation of the alkene (1).
Experiments were performed with 1 (0.2 mmol), 2a (2 ml), 3a (2 equiv), Fe(OTf)2 (20 mol %), Eosin Y (10 mol %), DTBP (2 equiv), 5-W blue LED, argon, 30°C, and 48 hours. The dr value is given in the Supplementary Materials and was determined by 1H NMR or GC–mass spectrometry (MS) analysis of the crude product.
*At 80°C.
†At 100°C.
‡At 130°C without the photocatalyst.
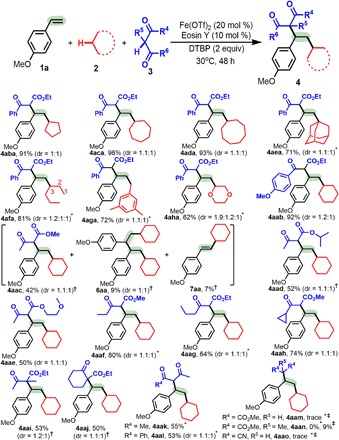
Table 3. Variation of the alkane (2) and 1,3-dicarbonyl compounds (3).
Experiments were performed with 1a (0.2 mmol), 2 (2 ml), 3 (2 equiv), Fe(OTf)2 (20 mol %), Eosin Y (10 mol %), DTBP (2 equiv), 5-W blue LED, argon, 30°C, and 48 hours. The dr value is given in the Supplementary Materials and was determined by 1H NMR or GC-MS analysis of the crude product.
*At 80°C.
†At 60°C
‡At 130°C without the photocatalyst.
Substrate scope with respect to alkanes and 1,3-dicarbonyl compounds
As shown in Table 3, a broad scope was also found for the alkane 2 and 1,3-dicarbonyl 3 reaction partners. A number of cycloalkanes, including cyclopentane 2b, cycloheptane 2c, cyclooctane 2d, and bulky adamantane 2e, underwent 1,2-dialkylation with alkene 1a and 1,3-dicarbonyl 3a in good to excellent yields (4aba to 4aea). Using n-hexane 2f, a linear alkane, delivered 4afa in 81% yield, albeit as a mixture of regioisomers. Notably, the benzyl C(sp3)─H bond of 2g and the C(sp3)─H bond adjacent to an oxygen atom in 2h could react to afford corresponding products 4aga and 4aha.
The optimized conditions also proved to be tolerant of a wide array of 1,3-keto esters 3b to 3j and 1,3-diketones 3k to 3l (4aab to 4aal). Ethyl 3-(4-methoxyphenyl)-3-oxopropanoate (3b) was highly reactive and afforded 92% yield of 4aab. However, the reactivity of 3-oxobutanoates 3c to 3e was lower, and the products were generated in moderate yields (4aac to 4aae). Notably, two by-products, 6aa and 7aa, were observed in the reaction of 3c. Using 3-oxopentanoate 1f, 3-oxohexanoate 1g, or 3-cyclopropyl-3-oxopropanoate 1h successfully delivered 4aaf to 4aah in 64 to 80% yields. Both 2-methyl-3-oxobutanoate 3i and 2-oxocyclohexane-1-carboxylate 3j provided challenging quaternary carbon centers, making this an attractive method for synthesis (4aai to 4aaj). 1,3-Diketones 3k and 3l were also suitable substrates and provided 4aak to 4aal. Unfortunately, diethyl malonate 3m had no reactivity for accessing 4aam.
DISCUSSION
Control experiments and mechanistic studies
Control experiments (Table 1 and fig. S2) and the reaction progress with the light on or off over time (fig. S3) show that this alkene 1,2-dialkylation protocol is achieved through cooperative photoredox and iron catalysis (38). Consequently, the mechanisms of this alkene dialkylation reaction were proposed on the basis of the current results and the reported processes (Fig. 3). To further verify it, the loadings of Eosin Y were examined (Fig. 2A). The results demonstrated that the yield of 4aaa decreased with reducing the Eosin Y loadings. While in the absence of Eosin Y the dialkylation protocol cannot occur at 30°, 60°, or 100°C under blue light (entry 9; Table 1), using 10 mol % Eosin Y at 60°C led to 31% yield of 4aaa in the dark (entry 15; Table 1). According to these results and the half-lift period data of DTBP (39, 40), the Eosin Y and iron cooperating catalysis might activate DTBP to make DTBP decomposition at lower temperature. Moreover, the cyclic voltammogram experiments indicated that the photocatalyst could modulate the redox potential of the Fe(OTf)2 catalyst (figs. S4 to S6). It was noted that in the presence of TEMPO, a radical inhibitor, the reaction of alkene 1a with cyclohexane 2a and dicarbonyl compound 3a was completely inhibited, and another product (10) from the reaction of cyclohexane 2a with TEMPO was observed (Fig. 2A). The results suggest that the radical process first generates cyclohexyl sp3-hybridized carbon-centered radical A (Fig. 3), which was also supported by the formation of 5oa (Table 2). In the absence of dicarbonyl compounds 3, both the Heck-type β-H elimination product 6aa and the two radicals homocoupling product 7aa were obtained (Fig. 2C): With enhancing temperatures, yield of 6aa increased slowly, but yield of 7aa increased sharply. However, the reaction could not take place without Eosin Y at 30° or 100°C (entry 9 in Table 1 and Fig. 2C). The results support the formation of the 2-cyclohexyl-1-(4-methoxyphenyl)ethyl radical B and carbocation C. Moreover, the higher temperatures are conducive to the radical B forming, and the conversion of the radical B into the carbocation C is far slower than the radicals homocoupling. In contrast to the results in Table 1, addition of dicarbonyl nucleophiles 3 obviously promotes the generation of the carbocation C.
Fig. 3. Possible mechanism.
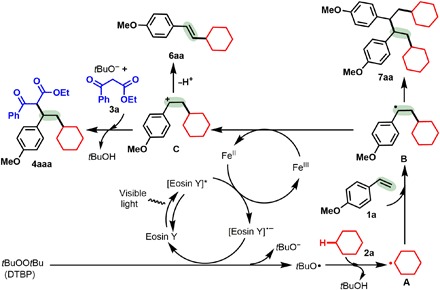
The generation of cyclohexyl sp3-hybridized carbon-centered radical A and new alkyl radical intermediate B is supported by experimental evidence, and subsequent single-electron oxidation and nucleophilic reaction with 1,3-keto ester 3a afford 4aaa.
Fig. 2. Control experiments.
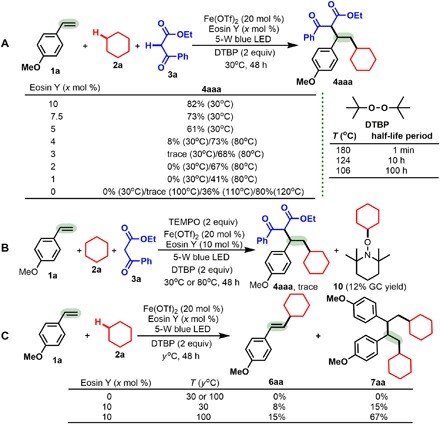
(A) Loadings of Eosin Y on the reaction. (B) Trapping experiment with a stoichiometric amount of radical inhibitor. (C) Reaction between alkene 1a and cyclohexane 2a in the absence of dicarbonyl compounds.
To further understand the mechanism, the cyclic voltammetry experiments of the catalytic system and substrates were conducted to study the redox potential (figs. S4 to S6). Two oxidation peaks of Fe(OTf)2 were observed at −1.560 VSCE and −0.790 VSCE in CH3CN, while two reduction peaks appeared on the reverse scan at −0.203 VSCE and −1.000 VSCE (fig. S4). The first oxidation peak of Fe(OTf)2 was quasi-reversible, and only one electron was transferred in the oxidation process. Ethyl 3-oxo-3-phenylpropanoate (3a) (three oxidation peaks) exhibited a higher oxidation potential than cyclohexane (2a), implying that 2a was likely to first undergo oxidative C─H cleavage. As shown in fig. S5, the cyclic voltammogram curve from a mixture of Fe(OTf)2 and Eosin Y featured an onset oxidation potential of −0.255 VSCE and an oxidation peak at 0.871 VSCE for the oxidation of Fe(II) to Fe(III) with the aid of Eosin Y*, while it featured a reduction peak at −0.416 VSCE and a notable reduction peak at −1.725 VSCE, which makes the Fe(III) reduction very easy. A mixture of multiple component experiments outlined in fig. S6 indicated that both the mixed 1a/2a/3a/Eosin Y and 1a/2a/3a/Eosin Y/Fe(OTf)2/DTBP have one oxidation peak at about 0.763 VSCE, whereas the mixed 1a/2a/3a/Eosin Y/DTBP has two higher oxidation peaks (0.436 VSCE and 0.742 VSCE). However, the mixed 1a/2a/3a/Fe(OTf)2/Eosin Y shows a rather low oxidation peak at 0.845 VSCE, presumably signifying a one-electron reduction event of Fe(OTf)2 to form the Eosin Y* radical anion. Notably, the Stern-Volmer fluorescence quench experiments were investigated (fig. S7). The emission intensity of Eosin Y is obviously diminished, which is affected by the concentration of Fe(OTf)2, and is slightly influenced when using DTBP or cyclohexane (2a), suggesting that Fe(OTf)2 provides the electron for the reductive quenching of the Eosin Y* excited state.
Consequently, DTBP may be readily split into the tert-butoxyl radical (38) with the aid of photoredox (29–37) and iron (19–28) synergistic catalysis [the Fenton chemistry (38)] (39, 40), and the radical then abstracts a hydrogen from cyclohexane (2a) to afford sp3 carbon-centered radical intermediate A (Fig. 3). Subsequently, addition of intermediate A across the C═C bond of alkene 1a gives new alkyl radical intermediate B, which is supported by these results, including the formation of product 5oa from the reaction of alkene 1o with cyclohexane 2a (Table 2) (24) and two other by-products 6aa and 7aa from the reaction of alkene 1a with cyclohexane 2a at 60°C in the presence of methyl 3-oxobutanoate (Table 3 and Fig. 2C). Last, intermediate B undergoes single-electron oxidation via the cooperative effects of the active Fe(III) species (19–28), which is generated from the oxidation of the Fe(II) species by the [Eosin Y]* intermediate (29–37) to deliver carbocation intermediate C followed by nucleophilic reaction with 1,3-keto ester 3a to access 4aaa. Among these processes, the photoredox catalysis might assist in the single-electron oxidation step by regulating the oxidation and reduction potentials of the iron intermediates and the reaction partners (29–40).
In summary, we have established the first intermolecular unsymmetrical 1,2-dialkylation of alkenes with two distinct C(sp3)─H bonds through cooperative photoredox and iron catalysis. The reaction allows concomitant incorporation of two vicinal alkyl groups across the C═C bond via dual C(sp3)─H functionalization under mild conditions, and the method features high atom economy, excellent functional group tolerance, and broad substrate scopes with respect to both the sp3 carbon-centered radical precursors (such as cycloalkanes, linear alkanes, and 1,4-dioxane) and the 1,3-dicarbonyl nucleophiles. This alkene dicarbofunctionalization reaction represents a novel and powerful strategy for the controlled functionalization of two or more different C(sp3)─H bonds across an alkene or alkyne in a single reaction to access complex functionalized molecules through regulating the oxidation and reduction potentials of the iron intermediates and the reaction partners. Further studies on the applications of this new strategy for transforming petroleum-based feedstocks are currently underway.
MATERIALS AND METHODS
General procedure for intermolecular dialkylation of alkenes with two distinct C(sp3)─H bonds enabled by synergistic photoredox catalysis and iron catalysis
Alkene 1 (0.2 mmol), alkane 2 (2 ml), 1,3-dicarbonyl 3 (0.3 mmol), Fe(OTf)2 (20 mol %; 0.02 mmol), Eosin Y (10 mol %; 0.02 mmol), and DTBP (2 equiv; 0.4 mmol) were added to a Schlenk tube. Then the tube was charged with argon, and the mixture was stirred at the indicated reaction temperature (30° to 130°C) for 48 hours until complete consumption of starting material, as monitored by thin-layer chromatography and/or GC–mass spectrometry (MS) analysis. After cooling to room temperature, the mixture was filtered through a small plug of silica gel to remove the precipitate, and the plug was washed with EtOAc (3 × 10 ml). The solvent was then removed in vacuo, and the residue was further purified by silica gel flash column chromatography (10 to 40% ethyl acetate in hexane) to afford the desired product 4.
Supplementary Material
Acknowledgments
Funding: We thank the National Natural Science Foundation of China (nos. 21625203 and 21472039), the Jiangxi Province Science and Technology Project (nos. 20171ACB20015 and 20165BCB18007), and the Opening Fund of Key Laboratory of Chemical Biology and Traditional Chinese Medicine Research (Hunan Normal University), Ministry of Education (no. KLCBTCMR18-02) for financial support. Author contributions: X.-H.O., Y.L., R.-J.S., M.H., S.L., and J.-H.L. conceived the project, wrote the manuscript, and analyzed the data. X.-H.O., Y.L., R.-J.S., and M.H. performed the experiments. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. Additional data related to this paper may be requested from the authors.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/3/eaav9839/DC1
Section S1. General information and procedures
Section S2. Preliminary synthetic utilization
Section S3. Control experiments and mechanistic studies
Section S4. Analytical data for 4aaa to 4naa, 4paa to 4qaa, 4aba to 4aha, 4aab to 4aan, 5oa, 6aa, 7aa, 8aaa, and 9aaa
Fig. S1. Preliminary synthetic utilization.
Fig. S2. Control experiments.
Fig. S3. Profile of 4aaa with the light on or off over time.
Fig. S4. Possible mechanisms.
Fig. S5. Cyclic voltammogram curves.
Fig. S6. Cyclic voltammogram curves.
Fig. S7. Cyclic voltammogram curves.
Fig. S8. Stern-Volmer fluorescence quenching experiments.
Fig. S9. 1H, 13C-NMR spectra of product 4aaa.
Fig. S10. 1H, 13C-NMR spectra of product 4baa.
Fig. S11. 1H, 13C-NMR spectra of product 4caa.
Fig. S12. 1H, 13C-NMR spectra of product 4daa.
Fig. S13. 1H, 13C-NMR spectra of product 4eaa.
Fig. S14. 1H, 13C-NMR spectra of product 4faa.
Fig. S15. 1H, 13C-NMR spectra of product 4gaa.
Fig. S16. 1H, 13C-NMR spectra of product 4haa.
Fig. S17. 1H, 13C-NMR spectra of product 4iaa.
Fig. S18. 1H, 13C-NMR spectra of product 4jaa.
Fig. S19. 1H, 13C-NMR spectra of product 3kaa.
Fig. S20. 1H, 13C-NMR spectra of product 4laa.
Fig. S21. 1H, 13C-NMR spectra of product 4maa.
Fig. S22. 1H, 13C-NMR spectra of product 4naa.
Fig. S23. 1H, 13C-NMR spectra of product 4oaa.
Fig. S24. 1H, 13C-NMR spectra of product 3paa.
Fig. S25. 1H, 13C-NMR spectra of product 4aba.
Fig. S26. 1H, 13C-NMR spectra of product 4aca.
Fig. S27. 1H, 13C-NMR spectra of product 4ada.
Fig. S28. 1H, 13C-NMR spectra of product 4aea.
Fig. S29. 1H, 13C-NMR spectra of product 4afa.
Fig. S30. 1H, 13C-NMR spectra of product 4aga.
Fig. S31. 1H, 13C-NMR spectra of product 4aha.
Fig. S32. 1H, 13C-NMR spectra of product 4aab.
Fig. S33. 1H, 13C-NMR spectra of product 4aac.
Fig. S34. 1H, 13C-NMR spectra of product 4aad.
Fig. S35. 1H, 13C-NMR spectra of product 4aae.
Fig. S36. 1H, 13C-NMR spectra of product 4aaf.
Fig. S37. 1H, 13C-NMR spectra of product 4aag.
Fig. S38. 1H, 13C-NMR spectra of product 4aah.
Fig. S39. 1H, 13C-NMR spectra of product 4aai.
Fig. S40. 1H, 13C-NMR spectra of product 4aaj.
Fig. S41. 1H, 13C-NMR spectra of product 4aak.
Fig. S42. 1H, 13C-NMR spectra of product 4aal.
Fig. S43. 1H, 13C-NMR spectra of product 4aan.
Fig. S44. 1H, 13C-NMR spectra of product 5oa.
Fig. S45. (E)-1-(2-cyclohexylvinyl)-4-methoxybenzene (6aa).
Fig. S46. 4,4′-(1,4-Dicyclohexylbutane-2,3-diyl)bis(methoxybenzene) (7aa).
Fig. S47. 1H, 13C-NMR spectra of product 8aaa.
Fig. S48. 1H, 13C-NMR spectra of product 8aaa.
REFERENCES AND NOTES
- 1.McDonald R. I., Liu G., Stahl S. S., Palladium(II)-catalyzed alkene functionalization via nucleopalladation: Stereochemical pathways and enantioselective catalytic applications. Chem. Rev. 111, 2981–3019 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Merino E., Nevado C., Addition of CF3 across unsaturated moieties: A powerful functionalization tool. Chem. Soc. Rev. 43, 6598–6608 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tang S., Liu K., Liu C., Lei A., Olefinic C–H functionalization through radical alkenylation. Chem. Soc. Rev. 44, 1070–1082 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Dhungana R. K., KC S., Basnet P., Giri R., Transition metal-catalyzed dicarbofunctionalization of unactivated olefins. Chem. Rec. 18, 1314–1340 (2018). [DOI] [PubMed] [Google Scholar]
- 5.Basnet P., KC S., Dhungana R. K., Shrestha B., Boyle T. J., Giri R., Synergistic bimetallic Ni/Ag and Ni/Cu catalysis for regioselective γ,δ-diarylation of alkenyl ketimines: Addressing β-H elimination by in situ generation of cationic Ni(II) Catalysts. J. Am. Chem. Soc. 140, 15586–15590 (2018). [DOI] [PubMed] [Google Scholar]
- 6.Shrestha B., Basnet P., Dhungana R. K., KC S., Thapa S., Sears J. M., Giri R., Ni-catalyzed regioselective 1,2-dicarbofunctionalization of olefins by intercepting heck intermediates as imine-stabilized transient metallacycles. J. Am. Chem. Soc. 139, 10653–10656 (2017). [DOI] [PubMed] [Google Scholar]
- 7.Basnet P., Dhungana R. K., Thapa S., Shrestha B., KC S., Sears J. M., Giri R., Ni-catalyzed regioselective β,δ-diarylation of unactivated olefins in ketimines via ligand-enabled contraction of transient nickellacycles: Rapid access to remotely diarylated ketones. J. Am. Chem. Soc. 140, 7782–7786 (2018). [DOI] [PubMed] [Google Scholar]
- 8.McCammant M. S., Liao L., Sigman M. S., Palladium-catalyzed 1,4-difunctionalization of butadiene to form skipped polyenes. J. Am. Chem. Soc. 135, 4167–4170 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Cong H., Fu G. C., Catalytic enantioselective cyclization/cross-coupling with alkyl electrophiles. J. Am. Chem. Soc. 136, 3788–3791 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.You W., Brown M. K., Diarylation of alkenes by a Cu-catalyzed migratory insertion/cross-coupling cascade. J. Am. Chem. Soc. 136, 14730–14733 (2014). [DOI] [PubMed] [Google Scholar]
- 11.Thapa S., Basnet P., Giri R., Copper-catalyzed dicarbofunctionalization of unactivated olefins by tandem cyclization/cross-coupling. J. Am. Chem. Soc. 139, 5700–5703 (2017). [DOI] [PubMed] [Google Scholar]
- 12.García-Domínguez A., Li Z., Nevado C., Nickel-catalyzed reductive dicarbofunctionalization of alkenes. J. Am. Chem. Soc. 139, 6835–6838 (2017). [DOI] [PubMed] [Google Scholar]
- 13.Derosa J., Tran V. T., Boulous M. N., Chen J. S., Engle K. M., Nickel-catalyzed β,γ-Dicarbofunctionalization of alkenyl carbonyl compounds via conjunctive cross-coupling. J. Am. Chem. Soc. 139, 10657–10660 (2017). [DOI] [PubMed] [Google Scholar]
- 14.KC S., Dhungana R. K., Shrestha B., Thapa S., Khanal N., Basnet P., Lebrun R. W., Giri R., Ni-catalyzed regioselective alkylarylation of vinylarenes via C(sp3)−C(sp3)/C(sp3)−C(sp2) bond formation and mechanistic studies. J. Am. Chem. Soc. 140, 9801–9805 (2018). [DOI] [PubMed] [Google Scholar]
- 15.Wu Z., Wang D., Liu Y., Huan L., Zhu C., Chemo- and regioselective distal heteroaryl ipso-migration: A general protocol for heteroarylation of unactivated alkenes. J. Am. Chem. Soc. 139, 1388–1391 (2017). [DOI] [PubMed] [Google Scholar]
- 16.Thapa S., Dhungana R. K., Magar R. T., Shrestha B., KC S., Giri R., Ni-catalysed regioselective 1,2-diarylation of unactivated olefins by stabilizing Heck intermediates as pyridylsilyl-coordinated transient metallacycles. Chem. Sci. 9, 904–909 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Giri R., KC S., Strategies toward dicarbofunctionalization of unactivated olefins by combined heck carbometalation and cross-coupling. J. Org. Chem. 83, 3013–3022 (2018). [DOI] [PubMed] [Google Scholar]
- 18.Liu Z., Zeng T., Yang K. S., Engle K. M., β,γ-Vicinal dicarbofunctionalization of alkenyl carbonyl compounds via directed nucleopalladation. J. Am. Chem. Soc. 138, 15122–15125 (2016). [DOI] [PubMed] [Google Scholar]
- 19.McCarroll A. J., Walton J. C., Programming organic molecules: Design and management of organic syntheses through free-radical cascade processes. Angew. Chem. Int. Ed. 40, 2224–2248 (2001). [DOI] [PubMed] [Google Scholar]
- 20.Chen Z.-M., Zhang X.-M., Tu Y.-Q., Radical aryl migration reactions and synthetic applications. Chem. Soc. Rev. 44, 5220–5245 (2015). [DOI] [PubMed] [Google Scholar]
- 21.Yi H., Zhang G., Wang H., Huang Z., Wang J., Singh A. K., Lei A., Recent advances in radical C−H activation/radical cross-coupling. Chem. Rev. 117, 9016–9085 (2017). [DOI] [PubMed] [Google Scholar]
- 22.Wei W.-T., Zhou M.-B., Fan J.-H., Liu W., Song R.-J., Liu Y., Hu M., Xie P., Li J.-H., Synthesis of oxindoles by iron-catalyzed oxidative 1,2-alkylarylation of activated alkenes with an aryl C(sp2)−H bond and a C(sp3)−H bond adjacent to a heteroatom. Angew. Chem. Int. Ed. 52, 3638–3641 (2013). [DOI] [PubMed] [Google Scholar]
- 23.Fan J.-H., Wei W.-T., Zhou M.-B., Song R.-J., Li J.-H., Palladium-catalyzed oxidative difunctionalization of alkenes with α-carbonyl alkyl bromides initiated through a heck-type insertion: A route to indolin-2-ones. Angew. Chem. Int. Ed. 53, 6650–6654 (2014). [DOI] [PubMed] [Google Scholar]
- 24.Bunescu A., Wang Q., Zhu J., Copper-catalyzed cyanomethylation of allylic alcohols with concomitant 1,2-aryl migration: Efficient synthesis of functionalized ketones containing an α-quaternary center. Angew. Chem. Int. Ed. 54, 3132–3135 (2015). [DOI] [PubMed] [Google Scholar]
- 25.Ouyang X.-H., Song R.-J., Hu M., Yang Y., Li J.-H., Silver-mediated intermolecular 1,2-alkylarylation of styrenes with α-carbonyl Alkyl bromides and indoles. Angew. Chem. Int. Ed. 55, 3187–3191 (2016). [DOI] [PubMed] [Google Scholar]
- 26.Li Z., Zhang Y., Zhang L., Liu Z.-Q., Free-radical cascade alkylarylation of alkenes with simple alkanes: Highly efficient access to oxindoles via selective (sp3)C-H and (sp2)C-H bond functionalization. Org. Lett. 16, 382–385 (2014). [DOI] [PubMed] [Google Scholar]
- 27.Wang H., Guo L.-N., Duan X.-H., Silver-catalyzed decarboxylative acylarylation of acrylamides with α-oxocarboxylic acids in aqueous media. Adv. Synth. Catal. 355, 2222–2226 (2013). [Google Scholar]
- 28.Zhou M.-B., Song R.-J., Ouyang X.-H., Liu Y., Wei W.-T., Deng G.-B., Li J.-H., Metal-free oxidative tandem coupling of activated alkenes with carbonyl C(sp2)-H bonds and aryl C(sp2)-H bonds using TBHP. Chem. Sci. 4, 2690–2694 (2013). [Google Scholar]
- 29.Prier C. K., Rankic D. A., MacMillan D. W. C., Visible light photoredox catalysis with transition metal complexes: Applications in organic synthesis. Chem. Rev. 113, 5322–5363 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Hopkinson M. N., Sahoo B., Li J.-L., Glorius F., Dual catalysis sees the light: Combining photoredox with organo-, acid, and transition-metal catalysis. Chem. Eur. J. 20, 3874–3886 (2014). [DOI] [PubMed] [Google Scholar]
- 31.Chen J.-R., Hu X.-Q., Lu L.-Q., Xiao W.-J., Visible light photoredox-controlled reactions of N-radicals and radical ions. Chem. Soc. Rev. 45, 2044–2056 (2016). [DOI] [PubMed] [Google Scholar]
- 32.Chen J.-R., Hu X.-Q., Lu L.-Q., Xiao W.-J., Exploration of visible-light photocatalysis in heterocycle synthesis and functionalization: Reaction design and beyond. Acc. Chem. Res. 49, 1911–1923 (2016). [DOI] [PubMed] [Google Scholar]
- 33.Shi L., Xia W., Photoredox functionalization of C–H bonds adjacent to a nitrogen atom. Chem. Soc. Rev. 41, 7687–7697 (2012). [DOI] [PubMed] [Google Scholar]
- 34.Hu X.-Q., Chen J.-R., Xiao W.-J., Controllable remote C−H bond functionalization by visible-light photocatalysis. Angew. Chem. Int. Ed. 56, 1960–1962 (2017). [DOI] [PubMed] [Google Scholar]
- 35.Zhang J., Li Y., Zhang F., Hu C., Chen Y., Generation of alkoxyl radicals by photoredox catalysis enables selective C(sp3)−H functionalization under mild reaction conditions. Angew. Chem. Int. Ed. 55, 1872–1875 (2016). [DOI] [PubMed] [Google Scholar]
- 36.Choi G. J., Zhu Q., Miller D. C., Gu C. J., Knowles R. R., Catalytic alkylation of remote C–H bonds enabled by proton-coupled electron transfer. Nature 539, 268–271 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chu J. C. K., Rovis T., Amide-directed photoredox-catalysed C–C bond formation at unactivated sp3 C–H bonds. Nature 539, 272–275 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Walling C., Fenton’s reagent revisited. Acc. Chem. Res. 8, 125–131 (2002). [Google Scholar]
- 39.Zou S., Shi Y., Organic peroxide TBPV & DTBP and their applications. Petrochem. Ind. Trends 5, 39 (1997). [Google Scholar]
- 40.E. T. Denisov, T. G. Denisova, T. S. Pokidova, Handbook of Free Radical Initiators (John Wiley & Sons Inc., 2003), chap. 4, pp. 61–97. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/5/3/eaav9839/DC1
Section S1. General information and procedures
Section S2. Preliminary synthetic utilization
Section S3. Control experiments and mechanistic studies
Section S4. Analytical data for 4aaa to 4naa, 4paa to 4qaa, 4aba to 4aha, 4aab to 4aan, 5oa, 6aa, 7aa, 8aaa, and 9aaa
Fig. S1. Preliminary synthetic utilization.
Fig. S2. Control experiments.
Fig. S3. Profile of 4aaa with the light on or off over time.
Fig. S4. Possible mechanisms.
Fig. S5. Cyclic voltammogram curves.
Fig. S6. Cyclic voltammogram curves.
Fig. S7. Cyclic voltammogram curves.
Fig. S8. Stern-Volmer fluorescence quenching experiments.
Fig. S9. 1H, 13C-NMR spectra of product 4aaa.
Fig. S10. 1H, 13C-NMR spectra of product 4baa.
Fig. S11. 1H, 13C-NMR spectra of product 4caa.
Fig. S12. 1H, 13C-NMR spectra of product 4daa.
Fig. S13. 1H, 13C-NMR spectra of product 4eaa.
Fig. S14. 1H, 13C-NMR spectra of product 4faa.
Fig. S15. 1H, 13C-NMR spectra of product 4gaa.
Fig. S16. 1H, 13C-NMR spectra of product 4haa.
Fig. S17. 1H, 13C-NMR spectra of product 4iaa.
Fig. S18. 1H, 13C-NMR spectra of product 4jaa.
Fig. S19. 1H, 13C-NMR spectra of product 3kaa.
Fig. S20. 1H, 13C-NMR spectra of product 4laa.
Fig. S21. 1H, 13C-NMR spectra of product 4maa.
Fig. S22. 1H, 13C-NMR spectra of product 4naa.
Fig. S23. 1H, 13C-NMR spectra of product 4oaa.
Fig. S24. 1H, 13C-NMR spectra of product 3paa.
Fig. S25. 1H, 13C-NMR spectra of product 4aba.
Fig. S26. 1H, 13C-NMR spectra of product 4aca.
Fig. S27. 1H, 13C-NMR spectra of product 4ada.
Fig. S28. 1H, 13C-NMR spectra of product 4aea.
Fig. S29. 1H, 13C-NMR spectra of product 4afa.
Fig. S30. 1H, 13C-NMR spectra of product 4aga.
Fig. S31. 1H, 13C-NMR spectra of product 4aha.
Fig. S32. 1H, 13C-NMR spectra of product 4aab.
Fig. S33. 1H, 13C-NMR spectra of product 4aac.
Fig. S34. 1H, 13C-NMR spectra of product 4aad.
Fig. S35. 1H, 13C-NMR spectra of product 4aae.
Fig. S36. 1H, 13C-NMR spectra of product 4aaf.
Fig. S37. 1H, 13C-NMR spectra of product 4aag.
Fig. S38. 1H, 13C-NMR spectra of product 4aah.
Fig. S39. 1H, 13C-NMR spectra of product 4aai.
Fig. S40. 1H, 13C-NMR spectra of product 4aaj.
Fig. S41. 1H, 13C-NMR spectra of product 4aak.
Fig. S42. 1H, 13C-NMR spectra of product 4aal.
Fig. S43. 1H, 13C-NMR spectra of product 4aan.
Fig. S44. 1H, 13C-NMR spectra of product 5oa.
Fig. S45. (E)-1-(2-cyclohexylvinyl)-4-methoxybenzene (6aa).
Fig. S46. 4,4′-(1,4-Dicyclohexylbutane-2,3-diyl)bis(methoxybenzene) (7aa).
Fig. S47. 1H, 13C-NMR spectra of product 8aaa.
Fig. S48. 1H, 13C-NMR spectra of product 8aaa.