

Abstract
Background:
Stress is associated with an increased prevalence of anxiety and depression. Repeated social defeat (RSD) stress in mice increases the release of monocytes from the bone marrow that are recruited to the brain by microglia. These monocytes enhance inflammatory signaling and augment anxiety. Moreover, RSD promotes “stress-sensitization”, in which exposure to acute stress 24 days after cessation of RSD causes anxiety recurrence. The purpose of this study was to determine if microglia were critical to stress-sensitization and exhibited increased reactivity to subsequent acute stress or immune challenge.
Methods:
Mice were exposed to RSD, microglia were eliminated by CSF1R-antagonism (PLX5622), allowed to repopulate, and responses to acute stress or immune challenge (lipopolysaccharide) were determined 24 days after RSD-sensitization.
Results:
Microglia maintained a unique mRNA signature 24 d after RSD. Moreover, elimination of RSD-sensitized microglia prevented monocyte accumulation in the brain and blocked anxiety recurrence following acute stress (24 d). When microglia were eliminated prior to RSD, repopulated and mice were subjected to acute stress, there was monocyte accumulation in the brain and anxiety in RSD-sensitized mice. These responses were unaffected by microglial elimination/repopulation. This may be related to neuronal sensitization that persisted 24 d after RSD. Following immune challenge, there was robust microglial reactivity in RSD-sensitized mice associated with prolonged sickness behavior. Here, microglial elimination/repopulation prevented the amplified immune reactivity ex vivo and in vivo in RSD-sensitized mice.
Conclusions:
Microglia and neurons remain “sensitized” weeks after RSD and only the immune reactivity component of RSD-sensitized microglia was prevented by elimination/repopulation.
Keywords: Repeated Social Defeat, Stress, Anxiety, Microglia, Monocytes, CSF1R antagonist
Introduction
Psychosocial stressors are associated with an increased prevalence of anxiety and depression (1, 2). Moreover, individuals exposed to chronic stressors are vulnerable to subsequent adversity, known as ‘stress-sensitization’ (3, 4). The immune system contributes to chronic stress responses and is implicated in poor mental health outcomes (5). Mounting clinical evidence indicates that chronic stress increases circulating “inflammatory” (CD14 +/CD16−) monocytes in humans (6–10). CD14+/CD16− monocytes have a higher inflammatory capacity and display increased ability to traffic into tissue (9, 11–13). These immune alterations may be maladaptive, increase inflammation, and contribute to psychiatric complications associated with stress (14, 15).
Repeated social defeat (RSD) is a preclinical model of stress that drives the sympathetic-mediated production and release of ‘inflammatory’ Ly6C hi monocytes into circulation (6, 16,17). Notably, rodent Ly6Chi monocytes are the functional counterpart to human CD14+/CD16−monocytes (6, 18). These monocytes have a “primed” profile characterized by glucocorticoid-insensitivity, elevated expression of receptors for PAMPS and higher expression of pro-inflammatory cytokines (IL-1β) (6, 19–23). RSD causes prolonged anxiety-like behavior that persists for 8 days and is dependent on recruitment of inflammatory monocytes to brain regions associated with fear circuitry (24, 25). Notably, monocyte recruitment is microglial and chemokine dependent. Furthermore, recruited monocytes produce IL-1β, which is required for induction of anxiety-like behavior (26). Overall, inflammatory monocytes augment IL-1β signaling to endothelial cells, thereby causing anxiety following RSD (26).
RSD also promotes “stress-sensitization”, in which subsequent exposure to acute (sub-threshold) stress caused anxiety recurrence (25, 27). This acute stress is one cycle of RSD and is defined as a “subthreshold stress” because it does not cause monocyte trafficking or anxiety-like behavior in naïve mice (25, 27). In RSD-sensitization, we reported that the spleen served as a unique reservoir of inflammatory monocytes that were released and trafficked to the brain following acute stress (25). There were also longer-lasting changes in the CNS 24 days after RSD. For example, isolated microglia from RSD-sensitized mice had elevated CD14 mRNA and were more reactive to ex vivo LPS stimulation 24 days after RSD (17, 28). A persistent alteration in resident microglia after RSD is relevant because microglia are involved with the recruitment of monocytes to the brain (26). In addition, primed or sensitized profiles of microglia with stress, injury or age conferred hyper-reactivity to peripheral immune challenges (29–33). Therefore, the purpose of this study was to determine if microglia were a critical component of stress-sensitization and increased reactivity to acute stress or innate immune challenge.
Methods
Mice:
Male C57BL/6 (6–8 weeks old) and CD-1 (12 months) mice were used (Charles River Laboratory). CX3CR1CreER/ROSA26-STOPflfl-tdTomato were generated by crossing CX3CR1CreER and Ai9-ROSA26-STOPflfl-tdTomato mice (Jackson Laboratories). All procedures were performed in accordance with the NIH and OSU Guidelines.
Repeated Social Defeat (RSD) and Stress-Sensitization:
Mice were subjected to RSD as described (16) and outlined in the supplemental methods. An aggressive CD-1 mouse was introduced to an established cohort of three resident mice 2 hours daily for six days. Naïve controls were left undisturbed in their home cages. For stress sensitization (SS), mice were exposed to control (naïve) or RSD and exposed to acute social defeat 24d later (25, 27).
RNA-sequencing of FAC-sorted Microglia:
Microglia were enriched using Percoll separation, labeled with anti-CD11b and CD45 antibodies, and FAC-sorted. RNA was extracted, synthesized to cDNA, and 20 million 75bp reads were sequenced on an Illumina NextSeq 500. Sequences were aligned to the mm10 mouse reference genome using STAR Aligner (34). Factors of unwanted variance were controlled with RUVseq (35) and normalization and differential expression was determined using DESeq2 in R (36). Genes with p<0.05 and fold changes over 1.5 (log2FC>0.585) were considered differentially expressed.
Plexxikon Administration:
PLX5622 was provided by Plexxikon Inc., and formulated in standard AIN-76A rodent chow at a concentration of 1200 mg/kg and provided ad libitum.
Tamoxifen Injections:
Postnatal day 21 mice received 20 mg/kg tamoxifen per day for 4 consecutive days by intraperitoneal injection (i.p.) as described (37).
Flow Cytometry:
CD11b+ cells were isolated from brain homogenates as reported (16) and described in Supplementary Methods. In brief, brains were mechanically dissociated and CD11b+ cells were enriched by Percoll separation. Cells from the blood, spleen, and brain labeled with appropriate antibodies. Cell surface antigen expression was determined using a DxP9 cytometer (Cytek). Data were analyzed using FlowJo software and positive labeling for each antibody was determined based on isotype-labeled controls.
RNA Isolation and RT-PCR:
RNA was isolated from Percoll-enriched CD11b+ cells and coronal brain sections. Real-time qPCR was performed using the Applied Biosystems Assay-on-Demand Gene Expression protocol. mRNA expression was determined on an ABI PRISM 7300-sequence detection system, converted to CT, and results are expressed as fold change.
Ex vivo stimulation of Enriched CD11b+ cells from Brain:
As reported (28, 38), CD11b cells were isolated by Percoll separation, counted, plated, and stimulated with vehicle or LPS (100 ng/ml).
Immunohistochemistry:
Mice were transcardially perfuse-fixed with paraformaldehyde. Brain samples were post-fixed and cryosectioned. Labeling for Iba1, c-Fos, or Phospho-CREB was performed as described in the supplemental methods.
Behavioral Analyses:
Anxiety-like behavior in the open field was determined as described (16, 25). Social exploratory behavior of a juvenile conspecific mouse was determined at baseline and again 4, 8 and 24 h after LPS injection as described (25, 26, 33). Both behaviors are detailed in the supplemental methods.
Statistical Analysis:
To determine significant main effects and interactions between main factors, data were analyzed using one-, two-, or three-way ANOVA using the General Linear Model procedures of SPSS statistical software (IBM). In the event of a main effect of treatment, differences between group-means were evaluated by Post hoc analyses (Fisher’s LSD) and are graphically presented in figures (p<0.05)
Results
Evidence for Primed Microglial Profile 24 days after RSD.
We reported that microglia were required for RSD-induced recruitment of monocytes to the brain and prolonged anxiety-like behavior (16, 24, 26). Moreover, anxiety reoccurred in RSD-sensitized mice following acute stress 24 days later and was dependent on the release and recruitment of splenic-derived monocytes to the brain (25, 27). Thus, we hypothesize that microglia are a critical cellular component of stress-sensitization and are hyper-reactive to subsequent stressors and immune challenges.
First, we determined the microglial mRNA profile 24 days after RSD-sensitization. Microglia were FAC-sorted and RNA was sequenced (Fig. 1A). Volcano plots show genes in microglia that were increased or decreased by 1.5 fold (p<0.05) 24 d after RSD compared to controls (Fig. 1B). Of the 137 genes that were differentially expressed, 87 genes were increased and 50 were decreased. For instance, several immune-related genes including Mmp9, Cd200, Cxcl10, Cxcr4, and Ccl24 were increased over 1.5 fold (p<0.05) in microglia. Several other genes including Tgfbr3, Il1r2, Aqp1, and Cadm2, were decreased in microglia (p<0.05). Next, significantly altered upstream regulators were determined by Ingenuity Pathway Analysis (39). The six pathways with the highest activation z-scores between microglia from RSD mice versus controls (p<0.05) were IL-1β, Receptor tyrosine for glial derived neurotrophic factor ligands (Ret), Interferon−γ, MyD88, IL-4, and TLR4 (Fig. 1C). These findings confirm that significantly regulated genes are in pathways associated with microglial immune priming. Collectively, these data provide evidence that microglia develop a distinct immune profile that persists 24 days after RSD.
Figure 1: Evidence for Primed Microglial profile 24 days after RSD.
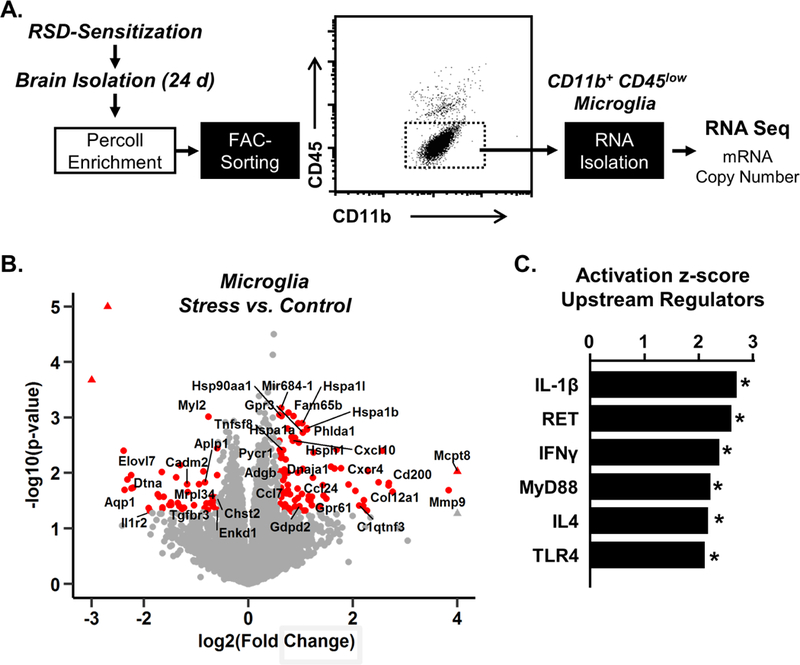
A) Male C57BL/6 mice were stress-sensitized (SS) by RSD or left undisturbed as controls (Naϊve). At 24 d after stress, microglia were Percoll-enriched, FAC-sorted, and RNA was collected for RNASeq (n=6). B) Volcano plots of RSD vs control of differentially expressed genes. Red points indicate differentially expressed genes (p<0.05, absolute fold change>1.5). C) Ingenuity Pathway Analysis of significantly altered upstream regulators in microglia. Graphs with (*) are significantly different from controls.
Monocyte Recruitment to the Brain and Recurrence of Anxiety in Stress-Sensitized Mice (24 d later) was Abrogated by the Elimination of Microglia.
The microglial mRNA signature remained altered 24 days after the cessation of RSD. Thus, we next eliminated microglia 10 days after RSD-sensitization and maintained their absence throughout the exposure to acute stress 24 days later (26). Mice were stress-sensitized by RSD (SS) or undisturbed (Naïve) and microglia were depleted using a CSF1R antagonist (PLX5622). All mice were exposed to acute defeat 24 d after RSD (Fig. 2A) and several immune and behavioral parameters were determined. Acute defeat does not cause monocyte trafficking or anxiety-like behavior in naïve mice, therefore this group served as the control for these experiments (25, 27). Consistent with a subthreshold stressor, acute defeat increased Ly6Chi monocytes in circulation and in the spleen of stress-sensitized (SS) mice compared to naïve mice (Fig.2B&C, p<0.002 for each). Monocyte release and accumulation in the spleen following acute defeat was dependent on stress-sensitization (Fig.2B&C). PLX5622 neither affected the number of circulating monocytes nor monocyte accumulation in the spleen (Fig.2B&C). Acute defeat also increased IL-1β mRNA levels in the brain of stress-sensitized mice compared to controls (p<0.001) and this effect was abrogated by microglial elimination (Fig. 2D, intervention x SS, p<0.04). Acute defeat increased monocyte accumulation in the brain of stress-sensitized (SS) mice (Fig.2E&F, p<0.05), and this was prevented by microglial elimination (Fig.2E&F, intervention x SS, p<0.004). Thus, microglia were essential for monocyte accumulation in the brain of RSD-sensitized mice exposed to acute stress.
Figure 2: Monocyte Recruitment to the Brain and Recurrence of Anxiety in Stress-Sensitized Mice was Abrogated by the Elimination of Microglia.

A) Male C57BL/6 mice were stress-sensitized (SS) by RSD or left undisturbed as controls (Naϊve). Ten days later, mice were provided diets formulated with vehicle (Veh) or a CSF1R antagonist (PLX5622). 24 days after stress-sensitization (SS), all mice were subjected to one cycle of social defeat (Acute Defeat) and blood, spleen, and brain samples were collected 14 h later (n=9–10). The percentage of monocytes (CD11b+/Ly6Chi) in the B) blood (main effect of SS, F1,39=21.45, p<0.0001) and spleen (main effect of SS, F1,39=28.63, p<0.002) were determined 14 h after acute defeat. D) IL-1β mRNA expression in a coronal brain section was determined after acute defeat (SS x intervention, F1,38=4.35, p<0.04). E) Representative bivariate dot plots of CD11b and CD45 labeling of Percoll-enriched microglia (CD11b+/CD45low) and macrophages (CD11b+/CD45high) in the brain after acute defeat. F) Number of CD45+ macrophages in the brain (SS x intervention, F1,36=10.68, p<0.003). G) Representative heat maps of activity during open-field testing. H) Time to enter center of the open field (SS x intervention, F1,40=5.20, p<0.02). I) Time spent in center in the open field after acute defeat (SS x intervention, F1,40=7.03, p<0.01). J) Total distance traveled in the open field (not significant). Bars represent the mean ± SEM. Means with (*) are significantly different from Naïve-Vehicle controls.
Acute defeat caused anxiety-like behavior in stress-sensitized (SS) mice compared to controls (Fig. 2G). This was associated with increased time to enter the center of the open field (Fig. 2H, p<0.03) and decreased time spent in the center for stress-sensitized mice (Fig. 2I, p<0.05) compared to controls. Moreover, acute defeat-associated anxiety in stress-sensitized (SS) mice was prevented by microglial elimination (Fig. 2H, SS x intervention, p<0.02 and Fig. 2I, SS x intervention, p<0.01). Notably, there were no differences in total distance traveled between groups (Fig. 2J). Taken together, the recurrence of anxiety in RSD-sensitized mice with acute stress was blocked by microglial elimination.
Microglia Repopulated from Non-Progenitor CX3CR1+ cells after CSF1R antagonist-Mediated Elimination.
The time course of repopulation and the origin of microglia after CSF1R antagonist-mediated elimination was assessed (Fig. 3A). As expected, Iba-1+ microglia repopulated after elimination in a time-dependent manner (Fig. 3B, p<0.0001). CD11b+/CD45lo microglia were reduced below baseline 0 d, 7 d, and 14 d after removal of the CSF1R antagonist (Fig.3B&C, p<0.05, for each). Microglia returned to baseline within 21 d, which was in line with previous studies (26, 40, 41). Next, CX3CR1CreER/+/R26tdTOM mice (42) were used to assess the origin of repopulated microglia. In these mice, CX3CR1+ microglia express yellow fluorescent protein (YFP) at baseline. Following tamoxifen injection, Cre-recombination in CX3CR1+ cells induces tdTomato expression (Fig. 3E). In this experiment, CX3CR1CreER-YFP/R26tdTOM mice (3-week) were injected with tamoxifen, such that all CX3CR1+ cells became YFP+/tdTom+. After 28 d, mice were subjected to microglial elimination and subsequent repopulation. Microglia that Repopulated from a CX3CR1neg progenitor cell would be YFP+/tdTomneg while microglia repopulating from CX3CR1+ microglia that remained following elimination would be YFP+/tdTom+. Indeed, the majority of microglia (99% of CD11b+/CD45lo) were YFP+/tdTom+ after tamoxifen induction, elimination, and subsequent repopulation (Fig.3F&G, p<0.0001).While CNS macrophages (CD11b+/CD45hi) were approximately 50% YFP+, none were YFP+/tdTom+ weeks after tamoxifen injection. Notably, 50% of circulating monocytes were YFP+/tdTom+ 7 d after tamoxifen injection (data not shown). This loss of tdTom with time in BM-derived monocyte/macrophages is consistent with turnover from the bone marrow (43–45).Also, 60% of the microglia from control and repopulated mice (Tamneg) were also YFP+/tdTom+ in the absence of tamoxifen (Fig. 3G). This may reflect a “leaky” Cre-recombinase with spontaneous tdTom induction over time. Nonetheless, all of the microglia were YFP+/tdTom+ after tamoxifen induction, elimination, and subsequent repopulation (Fig. 3H). These data indicate self-renew of microglia following elimination. The YFP+/tdTom+ microglia after elimination/repopulation indicates that these originate from the 3–5% of microglia that remained after CSF1R antagonism.
Figure 3: Microglia Repopulated from Non-Progenitor CX3CR1+ cells after CSF1R Antagonist-Mediated Elimination.

A) Male C57BL/6 mice were provided diets formulated with vehicle or CSF1R antagonist (PLX5622) for 14 days. Next, the CSF1R antagonist diet was removed and all mice were provided vehicle diets for 1, 7, 14 or 21 days to allow for repopulation of microglia. B) Representative images of Iba-1 labeling in the cortex 1, 7, 14 or 21 days after the cessation of the CSF1R antagonist. C) Representative bivariate dot plots of CD11b/CD45 labeling of Percoll-enriched cells at each time point. D) Number of microglia (CD11b+/CD45low) in the after brain 1, 7, 14, or 21 days of repopulation (main effect of time, F4,16= 8.57, p<0.001). E) Schematic representation of the experimental design using CX3CR1CreER/+/R26tdTOM/+ mice, which were administered 4 daily injections of control or tamoxifen (20 mg/kg, i.p.) at 3 weeks of age. Mice were left undisturbed for 28 d, provided diets formulated with a CSF1R antagonist (PLX5622) for 14 d and then provided vehicle diets for an additional 21 days to allow for repopulation of microglia. F) Representative bivariate dot plots of YFP and tdTom expression in microglia (CD11b+/CD45low) and macrophages (CD11b+/CD45high) isolated from +/− tamoxifen-injected CX3CR1CreER/+/R26tdTOM/+ mice Subjected to microglial elimination/repopulation. G) Percentage of tdTom+ microglia (CD11b+/CD45low) and macrophages (CD11b+/CD45high) in the brain isolated from +/− tamoxifen-injected CX3CR1CreER/+/R26tdTOM/+ mice subjected to microglial elimination/repopulation. H) Representative images of YFP and tdTom expression in tamoxifen-injected CX3CR1CreER/+/R26tdTOM/+ mice subjected to microglial elimination/repopulation. Inset shows YFP+/tdTom+ microglia identified by white arrows. Bars represent the mean ± SEM. Means with (*) are significantly different from control (p<0.05).
Stress-Sensitization to Acute Stress was Maintained after Elimination and Repopulation of Microglia.
The next objective was to determine if microglia elimination/repopulation prevented the recurrence of anxiety after acute defeat in RSD-sensitized mice. Microglia were eliminated using a CSF1R antagonist prior to RSD (Fig. 4A). Microglial elimination prevented anxiety-like behavior 14 h after RSD (Fig.4B&C) in the time to enter the center of the open field (Fig. 4 B, intervention x SS, p=0.06) and time spent in the center (Fig. 4C, intervention x SS, p<0.003). Next PLX5622 diet was removed to allow for microglia repopulation. After 24 d repopulation, naive and stress-sensitized (SS) mice were subjected to acute defeat (Fig. 4A). Acute defeat increased monocytes in the blood of SS mice, but not in naïve mice (Fig. 4D, p<0.0002). This monocyte induction in SS mice with acute defeat, however, was independent of microglial elimination/repopulation (Fig. 4D). In addition, acute defeat increased monocyte accumulation in the brain of stress-sensitized mice (Fig.4E&F, p<0.0002) and was associated with increased IL-1β mRNA levels (Fig. 4G, p<0.001). Again these events induced by acute stress in SS mice were independent of microglial elimination/repopulation. Similar effects were evident in anxiety-like behavior (Fig.4H&I). While the time to enter the center of the open field was unaffected by acute stress (Fig. 4H), total time spent in the center was decreased by acute defeat (Fig. 4G, p<0.001). This behavior was independent of microglia elimination/repopulation. Thus, stress-induced monocyte trafficking to the brain, IL-1β induction, and anxiety were dependent on the presence of microglia but was not prevented by microglial repopulation.
Figure 4: Microglia Repopulation in Stress-Sensitized Mice Re-established Monocyte Trafficking and Anxiety-Like Behavior Induced by Acute Defeat.

A) Male C57BL/6 mice were provided diets formulated with vehicle or CSF1R antagonist (PLX5622) for 14 days. Next, mice were stress-sensitized (SS) by RSD or left undisturbed as controls (Naϊve). Anxiety-like behavior was determined 14 h after the last cycle of RSD in the open field (n=9–10) by B) time to enter the center (Intervention x SS interaction; F1,38=10.36, p<0.003) and C) time spent in the center (Intervention x SS interaction; F1,38=3.675, p=0.06). After RSD, all mice were provided vehicle diets for an additional 24 d to allow for repopulation of microglia. After 24 days of repopulation, all mice were exposed to one cycle of social defeat (acute defeat). D) Percentage of monocytes (CD11b+/CD45high) in circulation 14 h after acute defeat (main effect of SS, F1,37=16.46, p<0.0002). E) Representative bivariate dot plots of CD11b and CD45 labeling on enriched microglia and macrophages. F) Number of brain macrophages (CD11b+/CD45high) 14 h after acute defeat (main effect of SS, F1,37=16.46, p<0.0002). G) mRNA levels of IL-1β were determined in a coronal brain section (n=4) collected 14 h after acute defeat (main effect of SS; F1,14=20.1, p<0.001). Anxiety-like behavior (n=10) was determined by H) Time to enter (not significant) and I) time spent in the center of the open field 0.5 d after acute defeat (main effect of SS; F1,39=14.85, p<0.001). Bars represent the mean ± SEM. Means with (*) are significantly different from Control-Naïve (p<0.05).
Evidence for Neuronal Sensitization with RSD.
Microglial elimination prior to RSD and then subsequent repopulation did not prevent the exaggerated immune and behavioral responsiveness to acute defeat in the stress-sensitized mice. Thus, there are likely other CNS components involved in stress-sensitization besides microglia. To address this, neuronal activation (c-Fos) and reactivity (phospho-CREB) to acute defeat were determined in two relevant fear and threat appraisal areas (prelimbic cortex and hippocampus) of naïve and stress-sensitized (SS) mice immediately following acute defeat (46). As expected, there was no c-Fos activation 24 d after RSD (Control-SS group). After acute stress, there was a robust increase in c-Fos+ neurons of prelimbic cortex (Fig. 5A&B, p<0.001) and dentate gyrus (Fig. 5A&C, p<0.001) in both naïve and stress-sensitized mice. There was no difference after acute defeat in the number of c-Fos+ cells between naïve and SS groups (Fig.5B&C).
Figure 5: Evidence of Neuronal Sensitization with RSD.
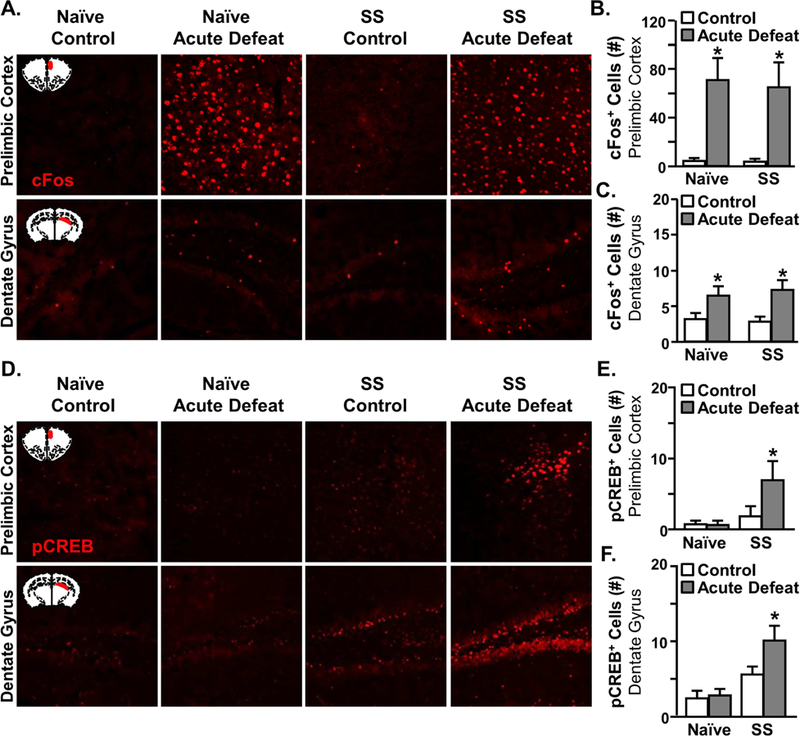
Male C57BL/6 mice were stress-sensitized (SS) by RSD or left undisturbed as controls (Naϊve). At 24 d after stress, all mice were exposed to one cycle of social defeat (acute defeat). Immediately after acute defeat, brains were perfused, fixed, sectioned, and labeled for c-Fos or pCREB (n=6). A) Representative images of c-Fos expression in the prelimbic cortex (top panel) and dentate gyrus (bottom panel). The number of c-Fos+ cells in the B) prelimbic cortex (F1,23=27.6,p<0.001) and C) dentate gyrus of the hippocampus in control and SS mice 14 h after acute defeat (F1,23=75.8,p<0.001). D) Representative images of pCREB expression in the prelimbic cortex (top panel) and dentate gyrus (bottom panel). Number of pCREB+ cells in the E) prelimbic cortex (SS, F1,24=4.7 p<0.04), SS x acute stress interaction (F1,24=2.9, p=0.1) and F) dentate gyrus of control and SS mice 0.5 d after acute defeat (SS, F1,24=17.7, p<0.001). Bars represent the mean ± SEM. Means with (*) are significantly different from Control-Naïve (p<0.05).
Increased expression of phospho-CREB is implicated in learning-induced synaptic plasticity and therefore may indicate altered neuronal reactivity to threatening stimuli following stress-sensitization (47–49). Similar to the c-Fos induction, there were few pCREB+ cells present 24 d after RSD (Fig. 5D). There tended to be an interaction between stress-sensitization and acute defeat for pCREB in the prelimbic cortex (interaction, p=0.1). Only the SS mice exposed to acute defeat had increased pCREB activity in the prelimbic cortex (p<0.04, Fig.5D&E). Similar interactions were detected in the dentate gyrus (Fig. 5D&F, p<0.001). Again, the SS mice exposed to acute defeat had the most pCREB+ neurons compared to all other groups (p<0.03). These data provide evidence of neuronal sensitization after RSD within fear and threat appraisal regions.
Microglial Hyperactivity to Innate Immune Challenge in Stress-Sensitized Mice was Attenuated by Microglia Elimination and Repopulation.
To further delineate the role of microglial sensitization after RSD, the response to an immune challenge with lipopolysaccharide (LPS) was determined. LPS challenge activates microglia by a different pathway than the one elicited by acute stress. Acutely following RSD, microglia respond to LPS challenge with an exaggerated immune and neuroinflammatory response (33).
Here, microglia were eliminated prior to RSD-sensitization (SS) and then allowed to repopulate for 24 d before assessing sensitization to LPS challenge. Microglia were collected,cultured ex vivo with saline or LPS and mRNA levels of TLR-4, CD14, IL-6, and IL-1β were determined (Fig. 6A). There was no effect of SS or LPS on TLR-4 expression ex vivo (Fig. 6B). CD14 mRNA was higher at baseline in SS-Repop mice compared to controls (Fig. 6C, p<0.05). LPS increased CD14 mRNA in microglia (p<0.05). The SS-Control-LPS group had the highest levels of CD14 mRNA (p<0.05) and these levels were attenuated by repopulation (Fig. 6C, p<0.03). LPS also increased IL-6 mRNA in microglia (p<0.06) and the SS-Control-LPS group had the highest levels of IL-6 mRNA compared to all other groups (Fig. 6D, p<0.03). LPS increased IL-1β mRNA in microglia. Again, the SS-Control-LPS group had the highest IL-1β mRNA expression compared to controls (p<0.05) and these levels were attenuated by elimination/repopulation (Fig. 6E, p<0.05). Taken together the higher microglial reactivity to ex vivo LPS challenge in stress-sensitized mice was attenuated by microglial elimination/repopulation.
Figure 6: Microglial Hyperactivity to ex vivo LPS Stimulation in Stress-Sensitized Mice was Attenuated by Microglial Elimination and Repopulation.
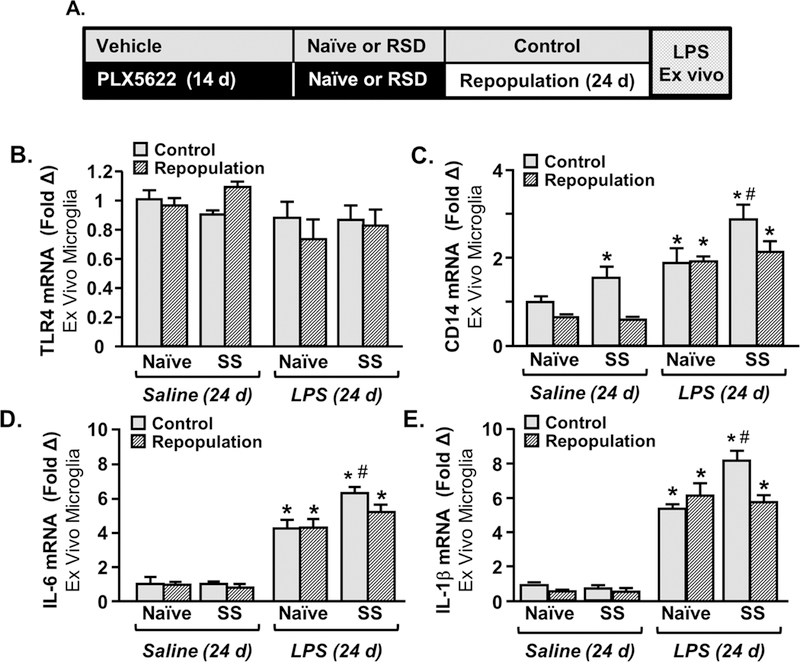
A) Male C57BL/6 mice were provided diets formulated with vehicle or CSF1R antagonist (PLX5622) for 14 days. Next, mice were stress-sensitized (SS) by RSD or left undisturbed as controls (Naϊve). After RSD-sensitization, all mice were provided vehicle diets for an additional 24 days to allow for repopulation of microglia. After 24 days of repopulation, microglia were collected by Percoll-enrichment and were cultured ex vivo with saline or LPS (100 ng/ml). mRNA levels of B) TLR4, CD14, D) IL-6, and E) IL-1β were determined in ex vivo microglia 4 h after LPS stimulation. Means with (*) are significantly different from Control-Naïve (p<0.05) and means with (#) are significantly different from SS-Naïve (p<0.05).
Using a similar design, we next investigated if elimination/repopulation of microglia reduced the reactivity of stress-sensitized mice to an in vivo challenge with LPS. Social exploratory behavior of a juvenile conspecific is a measure of sickness behavior following peripheral LPS challenge (29, 33). For example, peripheral LPS injection elicits a transient sickness behavior response in control mice associated with reduced social exploratory behavior that returns to baseline within 24 h (29, 33). Following microglial elimination/repopulation, mice were injected with LPS and social exploratory behavior was determined at baseline and 4, 8, and 24 h after LPS (Fig. 7A). LPS caused a significant reduction in social exploratory behavior (p<0.03) that tended to be dependent on time (Fig. 7B, p=0.07). LPS-injected naïve-control mice returned to baseline social interaction by 24 h after LPS, but the Con-SS mice had reduced social behavior 24 h after LPS (Fig.7B&C, p<0.05). This protracted sickness behavior after LPS in stress-sensitized mice was prevented by microglia elimination/repopulation. SS-Repop-LPS returned to baseline behavior by 24 h and were not different from control groups (Fig. 7B).
Figure 7: Microglial Hyperactivity to in vivo LPS Challenge in Stress-Sensitized Mice was Attenuated by Microglial Elimination and Repopulation.
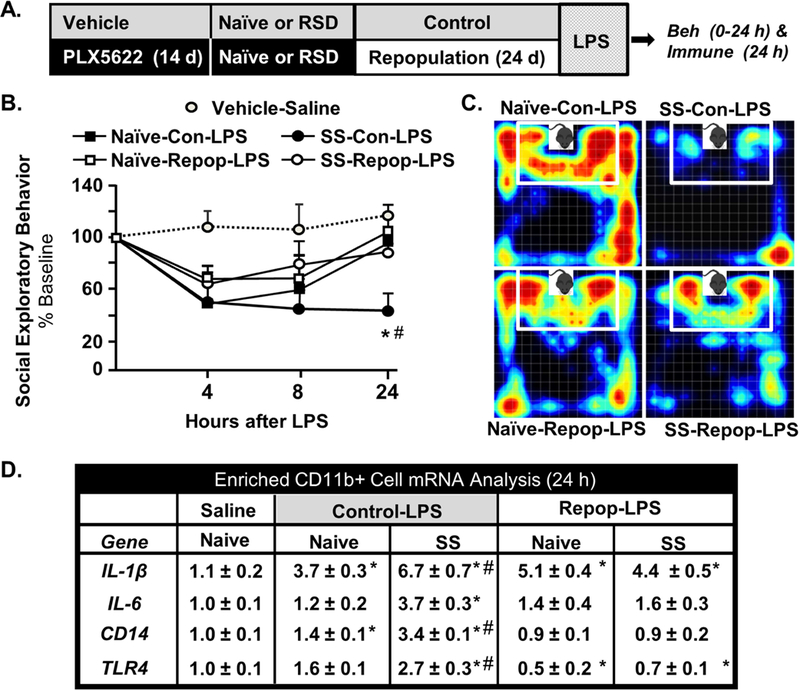
A) Male C57BL/6 mice were provided diets formulated with vehicle or CSF1R antagonist (PLX5622) for 14 days. Next, mice were stress-sensitized (SS) by RSD or left undisturbed as controls (Naϊve). After RSD-sensitization, all mice were provided vehicle diets for an additional 24 days to allow for repopulation of microglia. After 24 days of repopulation, mice were injected with LPS (0.5 mg/kg; i.p.) and B) social exploratory behavior (percent of baseline) was determined at baseline and 4, 8, and 24 h after LPS challenge (main effect of SS: F1, 83=18.59, p<0.03, SS x Time F3,83=3.42, p=.066). C) Representative heat maps of social exploratory behavior 24 h after LPS.D) mRNA levels of IL-1β (SS x Repop interaction; F1,18=14.6, p<0.001), IL-6 (SS x Repop interaction; F1,18=14.2, p<0.001, CD14 (SS x Repop interaction; F1,18=10.1, p<0.005), and TLR4 SS x Repop interaction; F1,18=5.715, p<0.03) were determined in Percoll-enriched microglia collected 24 h after LPS challenge. Bars represent the mean ± SEM. Means with (*) are significantly different from Control-Naive (p<0.05) and means with (#) are significantly different from saline control (p<0.05).
Next, the mRNA expression of several inflammatory genes (IL-1β, IL-6, CD14, and TLR4) was determined in enriched microglia/macrophages (Fig. 7D). LPS increased IL-1β mRNA levels in microglia/macrophages (p<0.05, for each) with the highest expression in the microglia of stress-sensitized (SS) mice injected with LPS (SS-Con-LPS) (p<0.05). Moreover, this exaggerated IL-1β mRNA response to LPS in SS mice was attenuated by elimination/repopulation (SS x Repop, p<0.001). This exaggerated response to LPS in microglia of stress-sensitized (SS-Con-LPS) was also evident in IL-6, CD14, and TLR4. Each mRNA level was highest in the Control-SS-LPS group (p<0.05) and was attenuated by elimination/repopulation (SS x Repop, p<0.03, for each). Collectively, removal of microglia prior to stress-sensitization and subsequent repopulation ablated the amplified immune reactivity to peripheral LPS challenge at 24 d.
Discussion
Chronic stress may elicit “stress-sensitization” in which individuals become more vulnerable to subsequent stressful stimuli (3, 4). We provide mRNA and functional evidence that microglia remain “primed” or “sensitized” weeks after RSD. For instance, acute defeat in RSD-sensitized mice caused significant monocyte accumulation in the brain (microglia-dependent) and promoted the recurrence of anxiety. Immune challenge (LPS) also elicited microglial reactivity in RSD-sensitized mice that corresponded with prolonged sickness behavior. Stress reactivity to acute defeat, however, remained when microglia were eliminated and repopulated after RSD. Immune reactivity to LPS (ex vivo and in vivo) was prevented when microglia were eliminated and repopulated after RSD. Collectively, RSD-sensitization is a complex process in which microglia play a role in the recurrence of anxiety with acute defeat and are essential for the increased reactivity to immune challenge.
Two important aspects of this study were that microglia from RSD-sensitized mice were primed and were critical for recruitment of monocytes to the brain after acute stress 24 d later. The RNA signature of microglia at 24 d after RSD had 137 differentially expressed genes which were associated with pathways consistent with inflammatory and microglial priming. Also, our previous studies demonstrated that the recurrence of anxiety-like behavior in stress-sensitized mice was dependent on recruitment of splenic monocytes to the brain (25, 27). Notably, anxiety-like behavior following RSD is well-validated and is evident in exploratory based measures (e.g., open field, light/dark preference, and elevated plus maze (16, 24, 50). Moreover, anxiety after RSD is also evident in other tests including the Morris water maze (51, 52) and fear conditioning paradigms (53). A potential limitation of these studies is that anxiety like behavior was only measured using the open field test. Anxiety-like behavior in the open field (fully automated analyses) is the most consistent and reproducible of the tests we have used. Furthermore, exploratory based tests can confound one another when used in the same mice. Thus, we selectively used the open field test for these experiments. Here, we confirmed our previous work that monocyte recruitment to the brain was dependent on presence of microglia. Moreover, the increased accumulation of inflammatory monocytes within the brain vasculature of the stress-sensitized mice was associated with the recurrence of anxiety. It is important to note that CSF1R-antagonism does not influence circulating monocyte numbers (26). Collectively, we interpreted these data to indicate that monocytes were recruited to the brain by microglia, and these monocytes augmented neuroinflammatory signaling that reinforced the recurrence of anxiety in RSD-sensitized mice.
Another relevant finding was that microglia repopulated after removal of PLX5622, returning to baseline by 21 d. Several other studies show similar kinetics of microglial repopulation (40, 41, 54, 55). Here, elimination/repopulation resulted in 99% of repopulated microglia that were double-positive for YFP and tdTom. Notably, if microglia from CX3CR1CreER/+/R26tdTOM/+ mice repopulated from CNS myeloid progenitor cells, then these cells would be YFP+ and tdTomneg. This finding is consistent with several recent studies into the origin of the repopulated microglia (42, 56). Thus, it is likely that repopulation of microglia after PLX6522 removal originated from the 3–4% of the microglia that remained throughout PLX5622 administration.
A critical question was to determine the extent to which microglia underlie maintenance of stress-sensitization and the recurrence of anxiety with acute stress. We hypothesized that preventing the microglial sensitization by eliminating prior to RSD and allowing for repopulation would prevent recurrence of anxiety following subsequent acute stress. Nonetheless, microglia elimination/repopulation did not affect hypersensitivity of RSD-sensitized mice to acute stress.We show that the presence of microglia at the time of acute stress was essential for the expression of stress sensitization, but depletion and repopulation did not prevent the sensitized stress response. Thus, these data indicate that microglia are not essential for the sensitization per se, but are essential for the expression of that sensitization.
One explanation for the above finding is that the “priming” of microglia represents only one component of RSD-sensitization. Our previous and current data implicate RSD-sensitization of neurons and myeloid cells in the spleen. For example, we reported a splenic population of myeloid cells that persisted in the spleen 24 days after RSD (27). Indeed, acute stress increased trafficking of Ly6Chi monocytes from the spleen to the brain in SS mice (25, 27). Furthermore, removal of the spleen blocked the acute stress-induced recurrence of anxiety (26, 57). Thus, the spleen acts as a unique reservoir for maintaining inflammatory monocytes that are readily releasable into circulation after acute stress in SS mice (27). Here, microglial elimination/repopulation did not diminish the number of Ly6Chi monocytes in circulation in SS mice. Notably, PLX5622 does not affect circulating monocyte survival (26). Thus, the splenic release of monocytes in stress-sensitized mice was independent of microglial priming and corresponded with increased monocyte accumulation in the brain. In addition, acute defeat increased pCREB expression in neurons within two key threat appraisal areas, the prelimbic cortex and dentate gyrus, only in the stress-sensitized mice. pCREB activity in the hippocampus has been implicated in associative learning (58), suggesting a mechanism for altered neuronal reactivity following stress-sensitization. We interpret these findings to indicate that stress-sensitized mice have a sensitized neuronal interpretation of acute stress compared to otherwise naïve mice. These sensitized neurons may in-turn cause microglial activation (24, 26). Moreover, other murine stressors elicited microglial activation that was dependent on neuronal activity (59–62). Thus, heightened fear and threat appraisal in stress-sensitized mice may explain the higher reactivity to a subsequent acute stressor. In addition, monocyte release is dependent on the sympathetic nervous system (6, 21, 24, 27), so neuronal sensitization may also persist weeks after RSD. These data indicate there are multiple CNS cell types that contribute to RSD-sensitization.
A critical finding was that the elimination/repopulation of microglia after RSD attenuated the amplified response to LPS. For example, microglia from stress-sensitized mice had increased inflammatory cytokine expression following direct ex vivo stimulation with LPS. This response was associated with a higher level of CD14 mRNA, a co-receptor for LPS (33), which persisted in microglia 24 days after RSD. This direct responsiveness of microglia to LPS and the higher CD14 mRNA from SS mice was consistent with our previous reports (24, 33). The results from the RNASeq and ex vivo experiments support the conclusion of “microglial priming” 24 d after RSD (25). Here we extend our previous work and show that this reactivity to LPS ex vivo was prevented when microglia were absent during RSD and then repopulated. Parallel with the ex vivo data, LPS injection was associated with prolonged sickness behavior and exaggerated pro-inflammatory cytokine mRNA expression in RSD-sensitized mice compared to controls. Importantly, the elimination/repopulation of microglia in RSD-sensitized mice prevented the prolonged sickness behavior induced by LPS and attenuated the cytokine responses in enriched microglia/macrophages. Despite renewing from microglia present at the time of stress, the repopulated microglia were no longer primed to peripheral immune challenge. These findings indicate that microglial elimination/repopulation was a successful strategy to prevent microglial priming to immune challenge. Therefore, microglial priming in response to RSD can be prevented by elimination/repopulation with PLX5622.
Stress-sensitization differentially affected microglia-mediated immune response to acute stress and LPS challenge because of intrinsic differences in the responses to these stimuli. For instance, LPS challenge activates the peripheral innate immune response first prior to microglial activation (63). In this context, peripheral cytokine production initiates a transient sickness response, characterized by lethargy, anorexia, and reduced social interaction. Notably, mice with evidence of primed microglia (e.g., aged, stressed, or injured) have prolonged sickness behavior following LPS compared to controls (29, 31–33, 64). Thus, microglial priming has a role in exacerbating social withdrawal following peripheral LPS challenge. The mechanism of behavioral perturbation differs following acute stress. In acute stress, there is rapid increase in neuronal activation in brain regions associated with fear and threat appraisal, followed by subsequent activation of microglia (26, 62, 65, 66). Thus, stress causes brain-to-immune communication. Microglial repopulation did not affect neuronal sensitization to acute stress, evidenced by persistent increases in pCREB 24 d following RSD. Neuronal activation in regions associated with fear and threat appraisal is likely not a critical modulator of the inflammatory response to LPS (31, 63, 67). Therefore, microglia play a more central role in initiating the behavioral and immune responses to LPS challenge compared to acute defeat.
In summary, we highlight the complex immune and CNS cellular interactions that occur during stress-sensitization. Our findings reinforce the idea of long lasting priming of microglia after exposure to RSD (68). Moreover our findings continue to show a critical role of inflammatory monocytes, which are actively recruited to brain by microglia, in the augmentation and recurrence of anxiety. While microglial elimination/repopulation in RSD-sensitized mice did not affect hyperactivity to acute stress, it was effective in reversing microglial reactivity to LPS challenge (ex vivo and in vivo). Taken together, microglia and neurons remain “sensitized” weeks after RSD and only the immune reactivity component of primed microglia was prevented by elimination/repopulation.
Supplementary Material
Acknowledgements:
This research was supported by NIH grants R01-MH-093473 and R01-MH093472 to J.F.S. and R01-AG051902 to J.P.G. MDW, CMS, KGW were supported by NIDCR Training Grant T32-DE014320. DBM was supported by F31-MH109234 and CMS was supported by F30-DE026075. WY and KGW are supported by University Fellowships (OSU). The authors thank Plexxikon Inc. for the use of PLX5622. The authors also thank The Ohio State University Comprehensive Cancer Center’s (OSUCCC) Analytical Cytometry and Nucleic Acid Shared Resources. We also thank the Campus Microscopy and Imaging Facility, supported in part by National Cancer Institute (NCI) grant P30-CA016058, for the instruments and services to generate confocal images presented in this report. The authors would like to acknowledge the Genomic Shared Resources and the Center for Genome Technology at the University of Miami for their help with the RNAseq. Last, this work was supported, in part, by an allocation of computing time from the Ohio Supercomputing Center.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
The authors report no biomedical financial interests or potential conflicts of interest.
Citations
- 1.Kessler RC (1997): The effects of stressful life events on depression. Annual review of psychology 48:191–214. [DOI] [PubMed] [Google Scholar]
- 2.Gilman SE, Trinh NH, Smoller JW, Fava M, Murphy JM, Breslau J (2013): Psychosocial stressors and the prognosis of major depression: a test of Axis IV. Psychol Med 43:303–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Post RM (1992): Transduction of Psychosocial Stress into the Neurobiology of Recurrent Affective-Disorder. Am J Psychiat 149:999–1010. [DOI] [PubMed] [Google Scholar]
- 4.McLaughlin KA, Conron KJ, Koenen KC, Gilman SE (2010): Childhood adversity, adult stressful life events, and risk of past-year psychiatric disorder: a test of the stress sensitization hypothesis in a population-based sample of adults. Psychol Med 40:1647–1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Setiawan E, Wilson AA, Mizrahi R, Rusjan PM, Miler L, Rajkowska G, et al. (2015): Role of Translocator Protein Density, a Marker of Neuroinflammation, in the Brain During Major Depressive Episodes. Jama Psychiat 72:268–275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Powell ND, Sloan EK, Bailey MT, Arevalo JM, Miller GE, Chen E, et al. (2013): Social stress up-regulates inflammatory gene expression in the leukocyte transcriptome via beta-adrenergic induction of myelopoiesis. Proc Natl Acad Sci U S A 110:16574–16579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Cole SW, Hawkley LC, Arevalo JM, Cacioppo JT (2011): Transcript origin analysis identifies antigen-presenting cells as primary targets of socially regulated gene expression in leukocytes. Proc Natl Acad Sci U S A 108:3080–3085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Miller GE, Chen E, Sze J, Marin T, Arevalo JM, Doll R, et al. (2008): A functional genomic fingerprint of chronic stress in humans: blunted glucocorticoid and increased NF-kappaB signaling. Biol Psychiatry 64:266–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miller GE, Murphy ML, Cashman R, Ma R, Ma J, Arevalo JM, et al. (2014): Greater inflammatory activity and blunted glucocorticoid signaling in monocytes of chronically stressed caregivers. Brain Behav Immun 41:191–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bierhaus A, Wolf J, Andrassy M, Rohleder N, Humpert PM, Petrov D, et al. (2003): A mechanism converting psychosocial stress into mononuclear cell activation. Proceedings of the National Academy of Sciences of the United States of America 100:1920–1925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Miller AH, Raison CL (2015): The role of inflammation in depression: from evolutionary imperative to modern treatment target. Nat Rev Immunol 16:22–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Beumer W, Gibney SM, Drexhage RC, Pont-Lezica L, Doorduin J, Klein HC, et al. (2012): The immune theory of psychiatric diseases: a key role for activated microglia and circulating monocytes. J Leukoc Biol [DOI] [PubMed]
- 13.Starikova EA, Lebedeva AM, Freidlin IS (2010): [CD14++CD16- and CD14+CD16+ human monocytes adhesion to endothelial cells]. Tsitologiia 52:380–383. [PubMed] [Google Scholar]
- 14.McEwen BS (1998): Stress, adaptation, and disease. Allostasis and allostatic load. Annals of the New York Academy of Sciences 840:33–44. [DOI] [PubMed] [Google Scholar]
- 15.Maier SF, Watkins LR (1998): Cytokines for psychologists: implications of bidirectional immune-to-brain communication for understanding behavior, mood, and cognition. Psychological review 105:83–107. [DOI] [PubMed] [Google Scholar]
- 16.Wohleb ES, Powell ND, Godbout JP, Sheridan JF (2013): Stress-induced recruitment of bone marrow-derived monocytes to the brain promotes anxiety-like behavior. J Neurosci 33:13820–13833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ramirez K, Shea DT, McKim DB, Reader BF, Sheridan JF (2015): Imipramine attenuates neuroinflammatory signaling and reverses stress-induced social avoidance. Brain Behav Immune 46:212–220 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Yang J, Zhang L, Yu C, Yang XF, Wang H (2014): Monocyte and macrophage differentiation: circulation inflammatory monocyte as biomarker for inflammatory diseases. Biomarker research 2:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Quan N, Avitsur R, Stark JL, He LL, Lai WM, Dhabhar F, et al. (2003): Molecular mechanisms of glucocorticoid resistance in splenocytes of socially stressed male mice. J Neuroimmunol 137:51–58. [DOI] [PubMed] [Google Scholar]
- 20.Avitsur R, Stark JL, Dhabhar FS, Padgett DA, Sheridan JF (2002): Social disruption-induced glucocorticoid resistance: kinetics and site specificity. J Neuroimmunol 124:54–61. [DOI] [PubMed] [Google Scholar]
- 21.Hanke ML, Powell ND, Stiner LM, Bailey MT, Sheridan JF (2012): Beta adrenergic blockade decreases the immunomodulatory effects of social disruption stress. Brain Behav Immun 26:1150–1159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Powell ND, Bailey MT, Mays JW, Stiner-Jones LM, Hanke ML, Padgett DA, et al. (2009): Repeated social defeat activates dendritic cells and enhances Toll-like receptor dependent cytokine secretion. Brain Behav Immun 23:225–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weber MD, Godbout JP, Sheridan JF (2017): Repeated Social Defeat, Neuroinflammation, and Behavior: Monocytes Carry the Signal. Neuropsychopharmacology 42:46–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Wohleb ES, Hanke ML, Corona AW, Powell ND, Stiner LM, Bailey MT, et al. (2011): beta-Adrenergic receptor antagonism prevents anxiety-like behavior and microglial reactivity induced by repeated social defeat. The Journal of neuroscience : the official journal of the Society for Neuroscience 31:6277–6288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Wohleb ES, McKim DB, Shea DT, Powell ND, Tarr AJ, Sheridan JF, et al. (2014): Re-establishment of anxiety in stress-sensitized mice is caused by monocyte trafficking from the spleen to the brain. Biol Psychiatry 75:970–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McKim DB, Weber MD, Niraula A, Sawicki CM, Liu X, Jarrett BL, et al. (2017): Microglial recruitment of IL-1beta-producing monocytes to brain endothelium causes stress-induced anxiety. Mol Psychiatry [DOI] [PMC free article] [PubMed]
- 27.McKim DB, Patterson JM, Wohleb ES, Jarrett BL, Reader BF, Godbout JP, et al. (2016): Sympathetic Release of Splenic Monocytes Promotes Recurring Anxiety Following Repeated Social Defeat. Biol Psychiatry 79:803–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ramirez K, Niraula A, Sheridan JF (2016): GABAergic modulation with classical benzodiazepines prevent stress-induced neuro-immune dysregulation and behavioral alterations. Brain Behav Immun 51:154–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Godbout JP, Chen J, Abraham J, Richwine AF, Berg BM, Kelley KW, et al. (2005): Exaggerated neuroinflammation and sickness behavior in aged mice following activation of the peripheral innate immune system. FASEB journal : official publication of the Federation of American Societies for Experimental Biology 19:1329–1331. [DOI] [PubMed] [Google Scholar]
- 30.Godbout JP, Moreau M, Lestage J, Chen J, Sparkman NL, O’Connor J, et al. (2008): Aging exacerbates depressive-like behavior in mice in response to activation of the peripheral innate immune system. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology 33:2341–2351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Norden DM, Trojanowski PJ, Villanueva E, Navarro E, Godbout JP (2016): Sequential activation of microglia and astrocyte cytokine expression precedes increased Iba-1 or GFAP immunoreactivity following systemic immune challenge. Glia 64:300–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Fenn AM, Gensel JC, Huang Y, Popovich PG, Lifshitz J, Godbout JP (2014): Immune activation promotes depression 1 month after diffuse brain injury: a role for primed microglia. Biological psychiatry 76:575–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wohleb ES, Fenn AM, Pacenta AM, Powell ND, Sheridan JF, Godbout JP (2012): Peripheral innate immune challenge exaggerated microglia activation, increased the number of inflammatory CNS macrophages, and prolonged social withdrawal in socially defeated mice. Psychoneuroendocrinology 37:1491–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. (2013): STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Risso D, Ngai J, Speed TP, Dudoit S (2014): Normalization of RNA-seq data using factor analysis of control genes or samples. Nature biotechnology 32:896–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Love MI, Huber W, Anders S (2014): Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome biology 15:550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sohal DS, Nghiem M, Crackower MA, Witt SA, Kimball TR, Tymitz KM, et al. (2001): Temporally regulated and tissue-specific gene manipulations in the adult and embryonic heart using a tamoxifen-inducible Cre protein. Circulation research 89:20–25. [DOI] [PubMed] [Google Scholar]
- 38.Lisboa SF, Niraula A, Resstel LB, Guimaraes FS, Godbout JP, Sheridan JF (2018): Repeated social defeat-induced neuroinflammation, anxiety-like behavior and resistance to fear extinction were attenuated by the cannabinoid receptor agonist WIN55,212–2. Neuropsychopharmacology : official publication of the American College of Neuropsychopharmacology [DOI] [PMC free article] [PubMed]
- 39.Kramer A, Green J, Pollard J Jr., Tugendreich S (2014): Causal analysis approaches in Ingenuity Pathway Analysis. Bioinformatics 30:523–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Elmore MR, Najafi AR, Koike MA, Dagher NN, Spangenberg EE, Rice RA, et al. (2014): Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron 82:380–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Elmore MR, Lee RJ, West BL, Green KN (2015): Characterizing newly repopulated microglia in the adult mouse: impacts on animal behavior, cell morphology, and neuroinflammation. PloS one 10:e0122912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Parkhurst CN, Yang G, Ninan I, Savas JN, Yates JR 3rd, Lafaille JJ, et al. (2013): Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell 155:1596–1609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ajami B, Bennett JL, Krieger C, Tetzlaff W, Rossi FM (2007): Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat Neurosci 10:1538–1543. [DOI] [PubMed] [Google Scholar]
- 44.Yona S, Kim KW, Wolf Y, Mildner A, Varol D, Breker M, et al. (2013): Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity 38:79–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lawson LJ, Perry VH, Gordon S (1992): Turnover of resident microglia in the normal adult mouse brain. Neuroscience 48:405–415. [DOI] [PubMed] [Google Scholar]
- 46.Nie X, Kitaoka S, Tanaka K, Segi-Nishida E, Imoto Y, Ogawa A, et al. (2018): The Innate Immune Receptors TLR2/4 Mediate Repeated Social Defeat Stress-Induced Social Avoidance through Prefrontal Microglial Activation. Neuron 99:464–479 e467. [DOI] [PubMed] [Google Scholar]
- 47.Bilodeau J, Schwendt M (2016): Post-cocaine changes in regulator of G-protein signaling (RGS) proteins in the dorsal striatum: Relevance for cocaine-seeking and protein kinase C-mediated phosphorylation. Synapse 70:432–440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kivinummi T, Kaste K, Rantamaki T, Castren E, Ahtee L (2011): Alterations in BDNF and phospho-CREB levels following chronic oral nicotine treatment and its withdrawal in dopaminergic brain areas of mice. Neuroscience letters 491:108–112. [DOI] [PubMed] [Google Scholar]
- 49.Tropea TF, Kosofsky BE, Rajadhyaksha AM (2008): Enhanced CREB and DARPP-32 phosphorylation in the nucleus accumbens and CREB, ERK, and GluR1 phosphorylation in the dorsal hippocampus is associated with cocaine-conditioned place preference behavior. Journal of neurochemistry 106:1780–1790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kinsey SG, Bailey MT, Sheridan JF, Padgett DA, Avitsur R (2007): Repeated social defeat causes increased anxiety-like behavior and alters splenocyte function in C57BL/6 and CD-1 mice. Brain Behav Immun 21:458–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jianhua F, Wei W, Xiaomei L, Shao-Hui W (2017): Chronic social defeat stress leads to changes of behaviour and memory-associated proteins of young mice. Behavioural brain research 316:136–144. [DOI] [PubMed] [Google Scholar]
- 52.McKim DB, Niraula A, Tarr AJ, Wohleb ES, Sheridan JF, Godbout JP (2016): Neuroinflammatory Dynamics Underlie Memory Impairments after Repeated Social Defeat. J Neurosci 36:2590–2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Yu T, Guo M, Garza J, Rendon S, Sun XL, Zhang W, et al. (2011): Cognitive and neural correlates of depression-like behaviour in socially defeated mice: an animal model of depression with cognitive dysfunction. The international journal of neuropsychopharmacology 14:303–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rice RA, Pham J, Lee RJ, Najafi AR, West BL, Green KN (2017): Microglial repopulation resolves inflammation and promotes brain recovery after injury. Glia 65:931–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Szalay G, Martinecz B, Lenart N, Kornyei Z, Orsolits B, Judak L, et al. (2016): Microglia protect against brain injury and their selective elimination dysregulates neuronal network activity after stroke. Nat Commun 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Bruttger J, Karram K, Wortge S, Regen T, Marini F, Hoppmann N, et al. (2015): Genetic Cell Ablation Reveals Clusters of Local Self-Renewing Microglia in the Mammalian Central Nervous System. Immunity 43:92–106. [DOI] [PubMed] [Google Scholar]
- 57.Feng X, Valdearcos M, Uchida Y, Lutrin D, Maze M, Koliwad SK (2017): Microglia mediate postoperative hippocampal inflammation and cognitive decline in mice. JCI insight 2:e91229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brightwell JJ, Smith CA, Neve RL, Colombo PJ (2007): Long-term memory for place learning is facilitated by expression of cAMP response element-binding protein in the dorsal hippocampus. Learning & memory 14:195–199. [DOI] [PubMed] [Google Scholar]
- 59.Johnson JD, Campisi J, Sharkey CM, Kennedy SL, Nickerson M, Greenwood BN, et al. (2005): Catecholamines mediate stress-induced increases in peripheral and central inflammatory cytokines. Neuroscience 135:1295–1307. [DOI] [PubMed] [Google Scholar]
- 60.Nair A, Bonneau RH (2006): Stress-induced elevation of glucocorticoids increases microglia proliferation through NMDA receptor activation. J Neuroimmunol 171:72–85. [DOI] [PubMed] [Google Scholar]
- 61.Sugama S, Takenouchi T, Fujita M, Conti B, Hashimoto M (2009): Differential microglial activation between acute stress and lipopolysaccharide treatment. J Neuroimmunol 207:24–31. [DOI] [PubMed] [Google Scholar]
- 62.Wohleb ES, Terwilliger R, Duman CH, Duman RS (2018): Stress-Induced Neuronal Colony Stimulating Factor 1 Provokes Microglia-Mediated Neuronal Remodeling and Depressive-like Behavior. Biological psychiatry 83:38–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dantzer R, O’Connor JC, Freund GG, Johnson RW, Kelley KW (2008): From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci 9:46–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Cunningham C, Campion S, Lunnon K, Murray CL, Woods JF, Deacon RM, et al. (2009): Systemic inflammation induces acute behavioral and cognitive changes and accelerates neurodegenerative disease. Biological psychiatry 65:304–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shin LM, Liberzon I (2010): The neurocircuitry of fear, stress, and anxiety disorders. Neuropsychopharmacology 35:169–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wohleb ES, Franklin T, Iwata M, Duman RS (2016): Integrating neuroimmune systems in the neurobiology of depression. Nature reviews Neuroscience 17:497–511. [DOI] [PubMed] [Google Scholar]
- 67.Lacroix S, Feinstein D, Rivest S (1998): The bacterial endotoxin lipopolysaccharide has the ability to target the brain in upregulating its membrane CD14 receptor within specific cellular populations. Brain Pathol 8:625–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Niraula A, Sheridan JF, Godbout JP (2017): Microglia Priming with Aging and Stress. Neuropsychopharmacology 42:318–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.