

Abstract
Since the identification of the elusive endothelium-derived relaxing factor as nitric oxide (NO), much attention has been devoted to understanding its physiological effects. NO is a free radical with many roles, and owing to its neutral charge and high diffusion capacity, it appears NO is involved in every mammalian biological system. Most attention has been focused on the NO generating pathways within the endothelium; however, the recent discovery of a NO synthase (NOS)-like enzyme residing in red blood cells (RBC) has increased our understanding of the blood flow and oxygen delivery modulation by RBC. In the present review, pathways of NO generation are summarized, with attention to those residing within RBC. While the bioactivity of RBC-derived NO is still debated due to its generation within proximity of NO scavengers, current theories for NO export from RBC are explored, which are supported by recent findings demonstrating an extracellular response to RBC-derived NO. The importance of NO in the active regulation of RBC deformability is discussed in the context of the subsequent effects on blood fluidity, and the complex interplay between blood rheology and NO are summarized. This review provides a summary of recent advances in understanding the role played by RBC in NO equilibrium and vascular regulation.
Keywords: Erythrocyte, nitric oxide synthase, nitrite, vascular function
1. Nitric oxide
1.1. Introduction
The field of nitric oxide (NO) biology has expanded considerably over the past decades. Many works demonstrated the key role of NO in the cardiovascular system, and the importance of this discovery led to the 1998 Nobel Prize Award in Physiology/Medicine to Robert Furchgott, Louis Ignarro and Fred Murad. NO is a free radical, which can diffuse at a rate of approximately 50 μm/s in aqueous solution [76]. In vivo, the direct action of NO is regionally limited due to its half-life of ~10 s [60], although several longer-lived NO-related species exist that increase the temporal and spatial bioactivity of NO.
1.2. NO and vasodilation
Although the functions of NO are numerous, including beta-adrenergic dependent modulation of myocardial contractility [1], inhibition of platelet activation and adhesion, action as a neurotransmitter and involvement in host defense reactions, it is best known as a powerful modulator of vasomotor tone and blood flow resistance [44,59]. Generation of NO occurs endogenously by a family of structurally similar enzymes (nitric oxide synthase; NOS), in addition to NOS-independent reactions that involve NO-containing compounds and/or the inorganic anions nitrate and nitrite. The NOS isoforms are classified by the tissue within which they were first identified, and not surprisingly also indicates their principle functions: endothelial NOS (eNOS), for example, is critically involved in maintaining vasomotor tone, and is sensitive to free calcium. Consequently, upon increased intracellular calcium concentration and/or an association with the calcium/calmodulin complex, eNOS is phosphorylated and generates NO from L-arginine. The diffusion of NO into adjacent smooth muscle cells results in its binding of the ferrous (Fe2+) heme group of soluble Guanylyl Cyclase (sGC). This activated form of sGC enzymatically converts guanosine triphosphate (GTP) into the second messenger cyclic guanosine monophosphate (cGMP), which interacts with and activates cGMP-dependent protein kinases (PKGs). Activated PKGs induce smooth muscle relaxation by multiple mechanisms, including the modulation of myosin light chain kinase, decreased intracellular free calcium, as well as hyperpolarization of the cell membrane by regulating the activity of potassium channels or Na+/K+ ATPase [15,46,70]. Increased cGMP may also act to regulate intracellular calcium concentration and dephosphorylate myosin light chain, and thus in doing so, reduce its calcium sensitivity [44,89].
1.3. Shear stress modulation of endothelial NO production
Endogenous production of NO within endothelial cells may occur both with, and without, the involvement of calcium, and is mainly regulated by shear stress (the product of shear rate times blood viscosity). G protein–phospholipase interactions that involve tyrosine-kinase (TK)-dependent phosphorylation result in transient inositol tri-phosphate (IP3) evoked calcium release from internal stores. The subsequent depletion of these stores promotes influx of calcium via store-operated channels (SOC) that appear to be activated by mechanisms involving TK-dependent phosphorylation and epoxyeicosatrienoic acids derived from arachidonic acid via cytochrome P-450 monooxygenase (CYP450). Shear stress and elevations in internal calcium availability also stimulate K+ channel activity, and the associated more negative membrane potential promotes calcium entry by increasing the electrochemical gradient for influx. Elevated intracellular calcium concentration is of significance, given eNOS has a calmodulin binding site on its reductase domain; the binding of the calcium/calmodulin complex to eNOS is believed to initiate electron exchange between the domains of the eNOS dimer, resulting in NO synthesis [28].
eNOS is expressed in several intracellular compartments, and also at the plasma membrane where it co-localizes and is negatively regulated by the structural coat protein caveolin. Moreover, TK-dependent phosphorylation and G proteins may stimulate phosphatidylinositol 3-kinase (PI3K), which is an upstream activator of protein kinase B (Akt/PKB) that in turn phosphorylates eNOS to increase its activity in a calcium-independent manner in response to shear stress. Increased shear stress favors the activation of eNOS via phosphorylation at various sites [23,30,31] although phosphorylation of a serine residue at 1177 (Ser1177) has been accepted as the activated form of the enzyme under various conditions [23,32]. Once activated via this shear stress induced pathway, eNOS consumes L-arginine to produce NO. It may also be noted that shear stress may increase NO production by releasing endothelium-dependent agonists such as ATP, bradykinin or substance P [57]. Moreover, the level of shear stress has been shown to regulate eNOS expression and therefore is critically involved in the production of NO by endothelial cells [65]. The enhancement of eNOS mRNA expression in bovine aortic endothelial cells by increasing shear stress is not inhibited by dexamethasone, inhibitors of tyrosine kinases (TK), or inhibition of G-protein signaling. In contrast, chelation of intracellular calcium in endothelial cells reduced shear stress induction of eNOS mRNA by 70% [100].
Increased shear rate associated with change in local blood flow, is known to increase the production of NO through shear stress-dependent mechanisms in different vascular beds: conduit arteries [75], resistance arteries [39] and arterioles [50]. However, few studies addressed the effects of viscosity (another component of shear stress). Several authors reported that high-molecular-weight dextran solutions increased NO release from isolated vessels, although the mechanism appeared to be independent of flow rate [43]. The group of Intaglietta [88] also found that increased plasma viscosity in anemic animals stimulated the expression of eNOS, increased NO production and resulted in vasodilation. Blood viscosity-related increases in NO production are likely to contribute to the increased organ blood flow found in high viscosity states, such as in polycythemia [98], although it is clear that other disorders associated with hyperviscosity (e.g., hypertension, hypercholesterolemia) result in reduced eNOS activity and decreased NO bioavailability [68]. Finally, the increase in blood flow and blood viscosity during acute exercise are presently thought to result in increased NO production through shear stress-dependent mechanisms [19].
1.4. RBC as a source of NO
The interactions between RBC- and endothelial-derived NO, and the subsequent physiological responses are provided in Fig. 1. Plasma free-hemoglobin scavenges NO by reducing NO to nitrate in the bloodstream and by depositing itself at the basement membrane of endothelial cells [33]. Furthermore, NO entering into RBC is rapidly inactivated by oxyhemoglobin (HbO2) via conversion to methemoglobin (MetHb) and nitrate, which cannot be converted back to nitrite or other NO equivalents except in oral/GI bacteria; hence, this outcome is a virtual “dead end” of nitric oxide activity [97]. The production of NO within such close proximity to the scavenging effects of free-hemoglobin at the endothelium level, in the presence of free heme or within RBC which contain a high concentration of hemoglobin, raises the question regarding the associated bioactivity given the inherent challenge for NO escaping this environment [33]. However, the unstirred diffusional barrier that exists around RBC limits the rate of NO–hemoglobin reactions by approximately 600-fold [52]. Moreover, and perhaps of more biological significance, S-nitrosylated hemoglobin or nitrite-treated deoxyhemoglobin may behave as NO donorsinstead of NO scavengers [34]. Consequently, there is increased interest regarding the role that RBC could play in the regulation of NO bioavailability, including:
Fig. 1.

Nitric oxide (NO) generation pathways in endothelial cells and red blood cells (RBC). Similarities exist between activation of NO synthase (NOS) in endothelial cells and RBC, including sensitivity to shear stress and the involvement of Ca2+ (see in-text for more detail). With respect to blood flow modulation, NO-bioactivity includes decreasing aggregation of platelets, inhibiting cell adhesion, increasing RBC deformability, and vascular smooth muscle relaxation. Whereas endothelium-generated NO can diffuse into adjacent smooth muscle cells and induce vasodilation, for RBC-derived NO to induce vasodilation several NO “sinks” (e.g., hemoglobin in RBC and plasma) must be avoided due to its short half-life and reactivity. While some RBC-NOS generated NO appears to directly modulate blood flow (e.g., increased RBC deformability; vasodilation), it appears that this NO may also substantially contribute to RBC-pools of S-nitrosylated proteins and nitrite, and therefore may provide an increased spatial and temporal effect beyond the RBC membrane. (Colors are visible in the online version of the article; http://dx.doi.org/10.3233/BIR-140653.)
When NO enters into RBC, it can bind to the highly conserved β-globin chain cysteine 93 residue to form bioactive S-nitrosohemoglobin (SNO–Hb) [71]. SNO–Hb associates with the membrane primarily through the interaction with Band 3 (i.e. the transmembrane anion-exchanger protein 1; AE1) via its N-terminal cytoplasmic domain (CDAE1). Upon deoxygenation, transfer of the NO group from β-Cys-93 of SNO–Hb to a cysteine thiol within CDAE1 favors generation and release of significant portion of RBC vasodilator activity; i.e., release of S-nitrosothiols (SNO) [71,72]. The role of circulating SNO–Hb on vasodilation has been challenged with results suggesting a minor role of SNO–Hb and SNO in the regulation of basal vascular tone [35]. Indeed, a knock-in mouse model that contained human hemoglobin modified to express an alanine in place of cysteine 93 residue on the β-chain resulted in no change to the level of RBC-induced hypoxic vasodilation [45]. These results supported a limited bioactive role for SNO–Hb, although this mouse model still expressed typical SNO abundance. Pawloski et al. [72] investigated the possibility of sickle RBC to mediate hypoxic vasodilation through the involvement of SNO-Hemoglobin S (SNO–HbS). The authors demonstrated that both SNO–HbS formation, and transfer of NO groups from SNO–HbS to a cysteine thiol within the CDAE1, are deficient in sickle RBC. The authors concluded that RBC-induced vasodilation is impaired in sickle cell disease.
NO may be formed from nitrite entering RBC, mediated by the reductive potential of deoxyhemoglobin [34]. Nitrite reduction by hemoglobin results in a large fraction of the NO generated being retained in the intermediate state where NO is bound to MetHb and is in equilibrium with the nitrosonium bound to hemoglobin [67]. This pool of NO, unlike heme-nitrosylated hemoglobin, is weakly bound and can be released from the heme, particularly during hypoxic conditions to provide compensatory vasodilation (i.e., hypoxic vasodilation) to increase local blood flow and reestablish oxygen supply to metabolically active tissues [21,42,58,62]. Huang et al. [42] demonstrated that the nitrite reduction rate is maximal when hemoglobin is 40–60% saturated with oxygen. Supporting this hypothesis was the in vivo observation that there is a significant and positive arteriovenous nitrite gradient, consistent with the oxygen uptake (and RBC deoxygenation) across active tissue [22].
A third method of NO production by RBC has been recently described in humans by Kleinbongard et al. [47]. They demonstrated that RBC express an active and functional eNOS-like enzyme (RBC NOS), which is localized in the RBC membrane and cytoplasm. RBC NOS has similar properties than eNOS in terms of phosphorylation sites controlling enzymatic activity and the dependence of its activity on intracellular calcium and L-arginine concentrations [47]. Extracorporeal circulation has been reported to increase RBC NOS activity [29]. More recently, Ulker et al. demonstrated that RBC exposed to shear stress resulted in RBC NOS activation (increased immunostaining for Ser1177 phosphorylated NOS), and subsequent production and exportation of NO (increased NO levels in RBC suspensions as measured by electrochemical NO probes) [91–93]. Similar to the vasculature, shear stress appears to stimulate mechanosensitive channels in the RBC plasma membrane that facilitate calcium flux into the cell. Ulker et al. [93] demonstrated that extracellular calcium availability (1 mM extracellular [Ca2+]) and importantly, shear stress-mediated influx of calcium, was requisite for RBC NOS activation. Chelation of extracellular calcium inhibited intra-cellular NO formation, and thus emphasized the involvement of calcium in shear stress activation of RBC NOS. Basal free intracellular [Ca2+] in the RBC is between 30–60 nM and total intracellular [Ca2+] (bound +free) may be as high as 5.7 μM, whereas extracellular [Ca2+] may approach 1.8 mM, thus creating a large gradient by which RBC influx via Ca2+ channels may increase intracellular [Ca2+] > 10-fold [89]. It must be acknowledged that while it seems shear stress is the main stimulus for RBC NOS activation, the amount of NO exported from RBC is higher in hypoxic than in normoxic conditions [92,93], supporting the complementary roles that SNO–Hb and nitrite reduction may contribute to RBC-derived NO generation.
The manner of NO export from RBC is controversial and depends on which species is being discussed – SNO–Hb, nitrite or NO – and each pathway must overcome barriers to successfully contribute to bioactivity. Transfer of NO from SNO–Hb to membrane-bound AE1 in order to transfer NO out of the RBC is dependent on SNO–Hb state (T or R) and SNO–Hb concentration; therefore, SNO–Hb ability to transfer NO to AE1 or other proteins (e.g., glutathione) appear to be crucial limiting factors. The kinetics and allosteric regulation of hemoglobin nitrosylation by oxygen and pH are consistent with physiologic mechanisms to modulate tissue blood flow whereby acidosis, hypoxemia and tissue hypoxia lead to NO generation by the RBC via SNO–protein transfer of NO activity [24,83]. A barrier to NO export specific to the reduction of nitrite is the potential for irreversible oxidization of nitrite to nitrate [33]. Moreover, when nitrite is converted to NO by deoxyhemoglobin, diffusion of NO out of RBC without being reduced by the now relatively abundant unoccupied Fe2+ within heme, or glutathione, poses a significant barrier to bioactivity. RBC NOS production of NO would also be prone to the same diffusion barriers as NO produced from nitrite reduction. While membranes and hydrophobic structures do not present diffusion barriers per se due to NO’s high solubility [52], it is suggested that NO auto-oxidation may be accelerated up to 300-times in the presence of a hydrophobic phase [53] – consequently, the lipid bilayer membrane of RBC may also represent a significant barrier to bioactivity. Despite these findings, however, recent observations support that RBC suspensions show a rise in extracellular NO concentration following exposure to shear stress and deoxygenation [91].
The efficiency of NO produced by RBC NOS to promote vasodilation was unknown and debated until the recent work of Ulker et al. [90]. The authors observed that perfusion of vessel segments with pre-sheared RBC suspensions caused a significant dilation response under hypoxic conditions, but not at high oxygen levels (a small but not-significant vasodilation was observed under this condition). Incubation of RBC suspension with L-NAME prior to shear stress application abolished vasodilation under both conditions. These findings indicate that shear stress activation of RBC NOS, leading to NO release, results in vasodilation of vessel segments under hypoxic conditions, and supports that RBC-derived NO has a functional role in local blood flow regulation [90]. When these findings are viewed alongside other studies which demonstrated that shear stress was able to induce ATP release from hypoxic RBC (a potent vasodilator also capable of stimulating vascular eNOS), it is well-founded that RBC are now considered O2 sensors [25]. This hypothesis is built upon the pivotal role that RBC play in vasoregulation when O2 tension is low, as is the case within metabolically active tissues.
Polymorphisms and allele number variation of the eNOS gene have been implicated in abnormal responses to exercise, impaired RBC rheology, and elevated disease risk. Kojda [48] demonstrated that mice heterozygous for eNOS (eNOS+/eNOS−) have normal endothelial dependent vasodilation; however, in response to exercise, the increase in eNOS expression and activation was absent [48]. Fatini demonstrated that T-786C and G894T polymorphisms in the eNOS gene independently affect RBC deformability [26] and increase the risk for acute coronary syndromes [27], while others reported that G894T is associated with idiopathic sudden sensorineural hearing loss after adjusting for other vascular risk factors [54]. T-786C polymorphism is associated with RBC membrane fluidic changes [63], coronary artery disease in some Caucasians groups [79] but not those in Australia [37], vasoocclusive events and acute chest syndrome in sickle cell disease [13,80]. Therefore, the mechanism underlying decreased RBC-deformability and vascular disease in healthy as well as chronically ill patients may depend on eNOS polymorphisms, which affect both endothelial cells and RBC.
2. NO and blood rheology
2.1. Short introduction on RBC rheology
RBC deformability and aggregation are known to affect blood viscosity in a shear-dependent fashion with increased RBC aggregation increasing blood viscosity at low shear rate, and elongation and alignment of RBC decreasing blood viscosity at high shear rate [3]. In addition, these two RBC rheological properties may affect blood flow independent of their effects on blood viscosity. Decreased RBC deformability may cause mechanical obstruction at the entry of capillaries and decrease tissue perfusion. Interventions to decrease RBC deformability have demonstrated significant decrements in blood flow for a given perfusion pressure indicating increased vascular resistance [69]. Moreover, increased RBC rigidity has been demonstrated to cause increased mechanical stress applied on endothelial cells promoting dysfunction and over-expression of vascular cell adhesion molecules, even in the absence of ischemia and oxidative stress [55]. Increased RBC aggregation may increase vascular resistance, particularly at the microcirculation level where RBC aggregates need to be dispersed to facilitate blood flow through narrow apertures [101,103]. Several studies have investigated the RBC rheological responses to metabolic factors such as lactate [17,18,81], oxidative stress [4,82], hydrogen ions [51], etc.; however, evidence is also accumulating that NO is an important modulator of RBC rheology.
2.2. NO and RBC deformability
One of the first works suspecting an effect of vascular-derived NO (extracellular NO) on RBC deformability was the study of Starzyk et al. [84] who demonstrated that intravenous infusion of L-NAME (an eNOS inhibitor) in rats caused a reduction of RBC elongation index. Bor-Kucukatay et al. [10] independently reported several eNOS inhibitors (L-NAME and S-methylisothiourea) that reduced NO availability also decreased RBC elongation index, suggesting that basal release of NO actively maintains RBC deformability. Korbut et al. [49] demonstrated that, in the presence of a small number of polymorphonuclear leukocytes, RBC filterability was increased by NO donors, such as sydnonimine or sodium nitroprusside, but was reversed by NOS inhibition. The positive effects of NO donors, including sodium nitroprusside and diethylenetriamine, for increased RBC deformability were also reported [10]. Horn et al. [41], using intra-vital microscopy, tested the effects of NO on RBC velocity in the microcirculation of the chorioallantoic membrane of the avian embryo at day 7 post-fertilization, when all vessels lack smooth muscle cells and therefore blood velocity is dependent more on blood rheology since the vessels do not change diameter. The authors confirmed that inhibition of enzymatic NO synthesis and NO scavenging decreased intracellular RBC NO concentration and avian RBC velocity in vitro. In addition, these authors demonstrated that injection of an eNOS-inhibitor or NO-scavenger into the previously defined microcirculation in situ decreased capillary RBC velocity and deformation, while the diameter of the vessels remained constant. These findings indicate that NO scavenging and inhibition of NO synthesis decrease RBC velocity (i.e., increases RBC transit time) by the active modulation of RBC deformability [41]. Nevertheless, it is important to note that very high NO concentrations, as can be the case in various inflammatory states, may be deleterious for RBC deformability [49,56,84]. Thus, in summary, it appears that extracellular NO influences RBC structural and functional properties in a non-linear, dose-dependent manner [56].
While NO from extracellular sources may have an impact on the deformability of RBC, several works strongly suggest that endogenous RBC NO synthesis may also modulate RBC deformability [47]. Suhr and colleagues [86] demonstrated that exercise-induced increase in shear stress activated RBC NOS (increased RBC NOS phosphorylation at Ser1177) via the PI3-kinase/Akt kinase pathway and this enhanced activity caused a greater production of NO, which was critical to maintain ability of RBC to deform in flow (i.e., RBC elongation index) during exercise. This exercise bout did not change the total RBC NOS content or RBC NOS phosphorylation at threonine 495 – a residue associated with inhibition of NO production. Grau et al. [38] further extend these findings by showing that RBC NOS activation by pharmacological treatment (insulin) lead to increased RBC NO content and improved RBC elongation index through direct S-nitrosylation of cytoskeleton proteins, most likely α- and β-spectrins. In contrast, the use of RBC NOS inhibitors (wortmannin or L-N5-(1-Iminoethyl)-ornithin) resulted in a decrease of RBC NOS Ser1177 phosphorylation, NO content, cytoskeleton proteins S-nitrosylation and RBC elongation index.
There are currently several explanations for the NO-mediated alteration in RBC deformability. As previously outlined, the effects of NO on the vascular smooth muscle are known to be mediated by pathways involving sGC – an enzyme also expressed in RBC [73,74]. It is not surprising to observe that guanylyl cyclase inhibition, thereby limiting the production of the second messenger cGMP, results in decreased RBC elongation index [10]. However, both sodium nitroprusside and diethylenetriamine reversed the effect of guanylyl cyclase inhibition in a dose-dependent manner [10] indicating that the effect of NO on RBC deformability is partially mediated by sGC. NO has also been shown to affect potassium transport across RBC membrane [12]. Bor-Kucukatay et al. [10] have demonstrated that the use of a potassium channel blocker (triethylammonium chloride) reversed the deleterious effects of L-NAME on RBC elongation index [10] suggesting that the inhibition of NO synthesis by this nonspecific NOS inhibitor may lead to deterioration of RBC mechanical properties by increasing the potassium permeability of the RBC membrane. Baskurt et al. [5] also demonstrated that sodium nitroprusside prevented the deterioration of RBC (i.e. sub-hemolytic damage) exposed to high shear stress for 15–120 s. The authors observed a similar protective effect of the triethylammonium chloride suggesting that the beneficial effect of NO on both RBC deformability and fragility is mediated by the inhibition of potassium leakage from RBC [5].
In addition, NO could decrease the risk for hemolysis and increase RBC survival rate through its effects on eryptosis since NO is able to down-regulate caspase 3 activity through S-nitrosylation [14]. More recently, another group demonstrated that the NO donor sodium nitroprusside inhibited the decrease of RBC deformability induced by ionophore A23187-mediated calcium influx in RBC [2]. Increased intra-cellular calcium concentration activates calcium-sensitive K+ (gardos) channels, resulting in potassium-efflux and decreased cell-volume, which in turn increases the stiffness of RBC; however, the presence of sodium nitroprusside abolished this calcium-induced impairment in RBC deformability [2]. Barodka et al. [2] suggested that sodium nitroprusside may have limited calcium influx, thereby inhibiting the activation of gardos channels, and thus, maintaining cell volume and RBC deformability. However, this interpretation is in contrast to the findings discussed previously regarding the importance of calcium influx for RBC NOS activation and intracellular NO accumulation, which positively improves RBC deformability [93]. Nicolay et al. [64] demonstrated that sodium nitroprusside, as well as the NO-donor papanonoate, was able to reverse the phosphatidylserine exposure and cell shrinkage induced by the use of Ca2+ ionophore ionomycin, but not the increase of cytosolic Ca2+ activity. Moreover, papanonoate was able to reverse the decrease of protein nitrosylation and thioredoxin activity induced by ionomycin [64]. These findings collectively suggest a protective role of NO on RBC rheological properties [2] and eryptosis [9] through its effects on cGMP, protein nitrosylation and thioredoxin activity [64].
2.3. NO and RBC aggregation
Few works have tested the effects of NO on RBC aggregation properties [11,85]. Bor-Kucukatay et al. [11] demonstrated that incubation of RBC with sodium nitroprusside decreased RBC aggregation while giving L-NAME to rats resulted in a rise of their RBC aggregation. The underlying mechanisms at the origin of these findings are unclear but might involve membrane/cytoskeletal protein nitrosylation or oxidative stress modulation [11].
To our knowledge, there have been no studies designed to investigate the effects of RBC aggregation on endogenous NO production by RBC. One could theorize the effects of RBC aggregation on available surface area for NO diffusion, changes to the unstirred layer surrounding the RBC and most importantly, the effect of the depletion layer on NO scavenging and NO release. This would be particularly important in the venous system and in conditions that would create passive blood flow to an arterial system, i.e., pulmonary arteries receiving passive blood flow in the Fontan type circulation for single ventricle palliation.
Few works have investigated the effects of RBC aggregation on NO production by endothelial cells. It has been established that blood flow and wall shear stress in the vascular system are important determinants of NO synthesis by endothelial cells [20,61,77], whereas decreased blood flow suppressed eNOS expression and flow-mediated dilation responses in small arteries [95]. An elegant study performed by Baskurt et al. [6] on rats submitted to exchange transfusion with aggregating RBC suspensions demonstrated that enhanced RBC aggregation resulted in suppressed expression of endothelial NO synthesizing mechanisms, thereby leading to altered vasomotor tonus. The authors interpreted these findings by proposing that the decreased blood flow associated with increased RBC aggregation ultimately results in reduced wall shear stresses [6]. It should be noted, however, that the effects of RBC aggregation on hemodynamic resistance are not a straightforward relationship, but rather depends on the perfusion pressure and the ability of the vascular system to dilate [101]. Moreover, enhanced RBC aggregation would also tend to promote axial migration of RBC away from blood vessels towards central flow streamlines, resulting in a less-viscous, plasma-rich region near vessel walls [16]. Decreased wall shear stress resulting from this non-uniform radial composition of blood should be expected to influence the NO-related mechanisms in the vascular system [16,36]. In that case, the viscosity of plasma near the vessel wall could be the main modulator of shear stress, eNOS activation and NO production. Yalcin et al. [102] tested the effect of enhanced RBC aggregation, with (use of 500 kDa dextran) or without (use of poloxamer-coated RBC) altering plasma viscosity, on NO production and eNOS activation in perfused human umbilical vein endothelial cells. Perfusion with poloxamer-coated RBC resulted in decreased NO production and reduced phosphorylation of eNOS Ser1177. In contrast, perfusion with RBC suspended in plasma containing dextran resulted in a NO concentration that remained elevated, highlighting the role of plasma viscosity on NO production. Tsai et al. [88] found that increasing plasma viscosity in anemic hamster increased wall shear stress, stimulated eNOS and NO production, and thus promoted vasodilation. These two scenarios (decreased blood flow and a less-viscous region near the vessel wall) are not mutually exclusive and most likely are synergistic in decreasing mechanical forces acting on endothelial cells [6–8]. However, while increased RBC aggregation could decrease eNOS activation and NO production, several findings suggest that enhanced RBC aggregation (for a given hematocrit) promote NO bioavailability because the increased plasma cell-poor layer width may actually provide a spatial buffer against the scavenging effects of hemoglobin-containing RBC [66]. Decreased RBC aggregation and RBC deformability (a situation encountered in sickle cell disease [87]) have been shown to interfere with axial migration and to decrease the plasma cell-poor layer width [104], a scenario expected to decrease NO bioavailability because RBC near the vessel wall would consume NO. Finally, the width of the glycocalyx maintains physical separation of RBC and the vessel wall [96]; it follows then that the glycocalyx also represents a spatial–diffusional barrier for endothelium-derived NO to be scavenged by RBC. Indeed, the decreased glycocalyx volume found in sickle cell patients [94] may contribute to the decreased NO bioavailability previously reported in this population.
3. Future considerations and clinical targets
Nitric oxide is a ubiquitous signaling molecule that is present in multiple tissues. The role of RBC involvement in NO bioactivity is still being determined, but the evidence thus far supports that RBC are integral in contributing to vascular regulation through several mechanisms. SNO–Hb is an exciting mechanism to improve tissue oxygenation, thereby mitigating the RBC storage lesion that has been shown to associate increased blood storage age with adverse clinical vascular outcomes [78]. RBC NOS contributes approximately 50% of the circulating nitrite and regulates systemic blood pressure [99]. Nitrite also decreases platelet aggregation in the circulation and is dependent on the presence of RBC [40]. The ongoing discussion regarding the role of specific nitric oxide species in hypoxic vasodilation is not currently settled, and the relative contribution of the RBC in producing these NO species is still being determined; however, our understanding of the role RBC play in NO equilibrium and vascular regulation has improved significantly in the last decade. Figure 2 demonstrates the potential interaction that is at work in the RBC to generate NO. The individual species contribute to NO in the RBC and may each represent one limb of the NO pool, represented as legs on a stool. Whether these mechanisms act in concert to support the NO pool in the circulation or operate independently without regard for the others remains to be seen; however, the interdependence of each would weave an interesting fabric by which the RBC may act as an NO “recharger” in some situations or an NO “sink” in others. Further studies in the setting of different disease states that include abnormal rheology, hemoglobin subtypes, redox disturbances and blood flow conditions will provide insight into the importance for RBC-derived NO for contributing to systemic NO bioavailability.
Fig. 2.
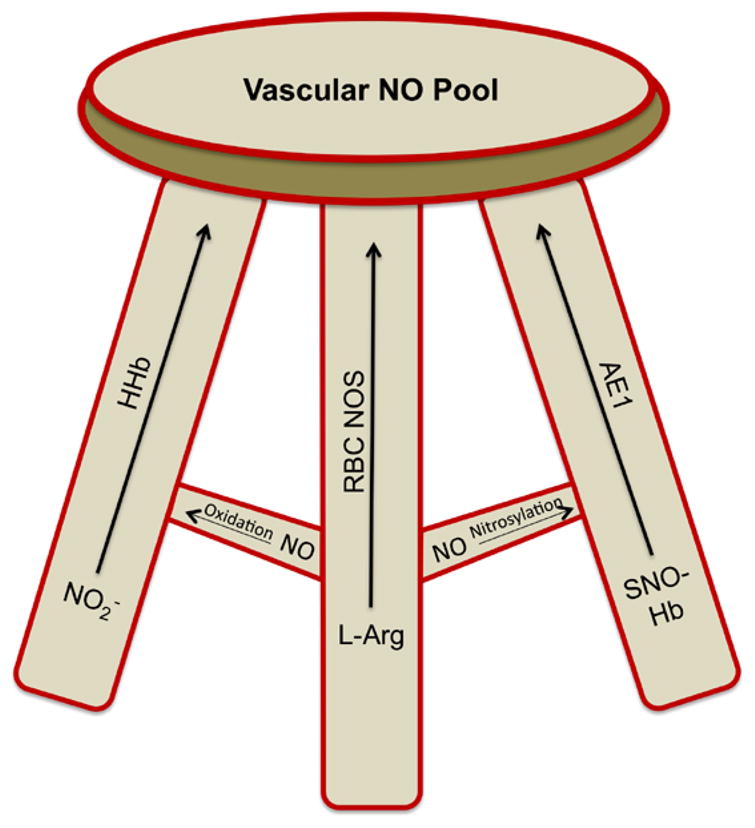
A model that represents the interrelationships between the various nitric oxide (NO) sources in red blood cells (RBC), and their collective contributions to NO bioavailability. Recent findings support that there is a certain amount of redundancy in this system, when one ‘leg’ is impaired (e.g., inhibition of S-nitrosohemoglobin – SNO–Hb – formation in mutant mice), NO bioavailability may be preserved. However, a central role of RBC NO-synthase in providing a basal load of NO that in somehow ‘recharges’ the nitrite/SNO–Hb pool, in addition to a potential extracellular export of NO, is hypothesized. Further explanations are provided in-text. (Colors are visible in the online version of the article; http://dx.doi.org/10.3233/BIR-140653.)
Footnotes
In memory of our inspiring mentor and friend, Prof. Oguz K. Baskurt
References
- 1.Balligand JL, Ungureanu D, Kelly RA, Kobzik L, Pimental D, Michel T, et al. Abnormal contractile function due to induction of nitric oxide synthesis in rat cardiac myocytes follows exposure to activated macrophage-conditioned medium. J Clin Invest. 1993;91:2314–2319. doi: 10.1172/JCI116461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Barodka V, Mohanty JG, Mustafa AK, Santhanam L, Nyhan A, Bhunia AK, et al. Nitroprusside inhibits calcium-induced impairment of red blood cell deformability. Transfusion. 2013;54(2):434–444. doi: 10.1111/trf.12291. [DOI] [PubMed] [Google Scholar]
- 3.Baskurt OK, Meiselman HJ. Blood rheology and hemodynamics. Semin Thromb Hemost. 2003;29:435–450. doi: 10.1055/s-2003-44551. [DOI] [PubMed] [Google Scholar]
- 4.Baskurt OK, Temiz A, Meiselman HJ. Effect of superoxide anions on red blood cell rheologic properties. Free Radic Biol Med. 1998;24:102–110. doi: 10.1016/s0891-5849(97)00169-x. [DOI] [PubMed] [Google Scholar]
- 5.Baskurt OK, Uyuklu M, Meiselman HJ. Protection of erythrocytes from sub-hemolytic mechanical damage by nitric oxide mediated inhibition of potassium leakage. Biorheology. 2004;41:79–89. [PubMed] [Google Scholar]
- 6.Baskurt OK, Yalcin O, Ozdem S, Armstrong JK, Meiselman HJ. Modulation of endothelial nitric oxide synthase expression by red blood cell aggregation. Am J Physiol Heart Circ Physiol. 2004;286:H222–H229. doi: 10.1152/ajpheart.00532.2003. [DOI] [PubMed] [Google Scholar]
- 7.Bishop JJ, Nance PR, Popel AS, Intaglietta M, Johnson PC. Erythrocyte margination and sedimentation in skeletal muscle venules. Am J Physiol Heart Circ Physiol. 2001;281:H951–H958. doi: 10.1152/ajpheart.2001.281.2.H951. [DOI] [PubMed] [Google Scholar]
- 8.Bishop JJ, Popel AS, Intaglietta M, Johnson PC. Effects of erythrocyte aggregation and venous network geometry on red blood cell axial migration. Am J Physiol Heart Circ Physiol. 2001;281:H939–H950. doi: 10.1152/ajpheart.2001.281.2.H939. [DOI] [PubMed] [Google Scholar]
- 9.Bogdanova A, Makhro A, Wang J, Lipp P, Kaestner L. Calcium in red blood cells – a perilous balance. Int J Mol Sci. 2013;14:9848–9872. doi: 10.3390/ijms14059848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Bor-Kucukatay M, Wenby RB, Meiselman HJ, Baskurt OK. Effects of nitric oxide on red blood cell deformability. Am J Physiol Heart Circ Physiol. 2003;284:H1577–H1584. doi: 10.1152/ajpheart.00665.2002. [DOI] [PubMed] [Google Scholar]
- 11.Bor-Kucukatay M, Yalcin O, Gokalp O, Kipmen-Korgun D, Yesilkaya A, Baykal A, et al. Red blood cell rheological alterations in hypertension induced by chronic inhibition of nitric oxide synthesis in rats. Clin Hemorheol Microcirc. 2000;22:267–275. [PubMed] [Google Scholar]
- 12.Caramelo C, Riesco A, Outeirino J, Millas I, Blum G, Monzu B, et al. Effects of nitric oxide on red blood cells: changes in erythrocyte resistance to hypotonic hemolysis and potassium efflux by experimental maneuvers that decrease nitric oxide. Biochemical and Biophysical Research Communications. 1994;199:447–454. doi: 10.1006/bbrc.1994.1249. [DOI] [PubMed] [Google Scholar]
- 13.Chaar V, Tarer V, Etienne-Julan M, Diara JP, Elion J, Romana M. ET-1 and ecNOS gene polymorphisms and susceptibility to acute chest syndrome and painful vaso-occlusive crises in children with sickle cell anemia. Haematologica. 2006;91:1277–1278. [PubMed] [Google Scholar]
- 14.Chowdhury KD, Sen G, Biswas T. Regulatory role of nitric oxide in the reduced survival of erythrocytes in visceral leishmaniasis. Biochim Biophys Acta. 2010;1800:964–976. doi: 10.1016/j.bbagen.2010.05.008. [DOI] [PubMed] [Google Scholar]
- 15.Cohen RA, Vanhoutte PM. Endothelium-dependent hyperpolarization. Beyond nitric oxide and cyclic GMP. Circulation. 1995;92:3337–3349. doi: 10.1161/01.cir.92.11.3337. [DOI] [PubMed] [Google Scholar]
- 16.Cokelet GR, Goldsmith HL. Decreased hydrodynamic resistance in the two-phase flow of blood through small vertical tubes at low flow rates. Circ Res. 1991;68:1–17. doi: 10.1161/01.res.68.1.1. [DOI] [PubMed] [Google Scholar]
- 17.Connes P, Bouix D, Py G, Prefaut C, Mercier J, Brun JF, et al. Opposite effects of in vitro lactate on erythrocyte deformability in athletes and untrained subjects. Clin Hemorheol Microcirc. 2004;31:311–318. [PubMed] [Google Scholar]
- 18.Connes P, Caillaud C, Py G, Mercier J, Hue O, Brun JF. Maximal exercise and lactate do not change red blood cell aggregation in well trained athletes. Clin Hemorheol Microcirc. 2007;36:319–326. [PubMed] [Google Scholar]
- 19.Connes P, Simmonds MJ, Brun JF, Baskurt OK. Exercise hemorheology: classical data, recent findings and unresolved issues. Clin Hemorheol Microcirc. 2013;53:187–199. doi: 10.3233/CH-2012-1643. [DOI] [PubMed] [Google Scholar]
- 20.Corson MA, James NL, Latta SE, Nerem RM, Berk BC, Harrison DG. Phosphorylation of endothelial nitric oxide synthase in response to fluid shear stress. Circ Res. 1996;79:984–991. doi: 10.1161/01.res.79.5.984. [DOI] [PubMed] [Google Scholar]
- 21.Cosby K, Partovi KS, Crawford JH, Patel RP, Reiter CD, Martyr S, et al. Nitrite reduction to nitric oxide by deoxyhemoglobin vasodilates the human circulation. Nat Med. 2003;9:1498–1505. doi: 10.1038/nm954. [DOI] [PubMed] [Google Scholar]
- 22.Dejam A, Hunter CJ, Pelletier MM, Hsu LL, Machado RF, Shiva S, et al. Erythrocytes are the major intravascular storage sites of nitrite in human blood. Blood. 2005;106:734–739. doi: 10.1182/blood-2005-02-0567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dimmeler S, Fleming I, Fisslthaler B, Hermann C, Busse R, Zeiher AM. Activation of nitric oxide synthase in endothelial cells by Akt-dependent phosphorylation. Nature. 1999;399:601–605. doi: 10.1038/21224. [DOI] [PubMed] [Google Scholar]
- 24.Doctor A, Platt R, Sheram ML, Eischeid A, McMahon T, Maxey T, et al. Hemoglobin conformation couples erythrocyte S-nitrosothiol content to O2 gradients. Proc Natl Acad Sci USA. 2005;102:5709–5714. doi: 10.1073/pnas.0407490102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ellsworth ML, Ellis CG, Goldman D, Stephenson AH, Dietrich HH, Sprague RS. Erythrocytes: oxygen sensors and modulators of vascular tone. Physiology (Bethesda) 2009;24:107–116. doi: 10.1152/physiol.00038.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Fatini C, Mannini L, Sticchi E, Cecchi E, Bruschettini A, Leprini E, et al. eNOS gene affects red cell deformability: role of T-786C, G894T, and 4a/4b polymorphisms. Clin Appl Thromb Hemost. 2005;11:481–488. doi: 10.1177/107602960501100417. [DOI] [PubMed] [Google Scholar]
- 27.Fatini C, Sofi F, Sticchi E, Gensini F, Gori AM, Fedi S, et al. Influence of endothelial nitric oxide synthase gene polymorphisms (G894T, 4a4b, T-786C) and hyperhomocysteinemia on the predisposition to acute coronary syndromes. Am Heart J. 2004;147:516–521. doi: 10.1016/j.ahj.2003.10.032. [DOI] [PubMed] [Google Scholar]
- 28.Feron O, Saldana F, Michel JB, Michel T. The endothelial nitric-oxide synthase-caveolin regulatory cycle. J Biol Chem. 1998;273:3125–3128. doi: 10.1074/jbc.273.6.3125. [DOI] [PubMed] [Google Scholar]
- 29.Fischer UM, Schindler R, Brixius K, Mehlhorn U, Bloch W. Extracorporeal circulation activates endothelial nitric oxide synthase in erythrocytes. Ann Thorac Surg. 2007;84:2000–2003. doi: 10.1016/j.athoracsur.2007.07.034. [DOI] [PubMed] [Google Scholar]
- 30.Fisslthaler B, Dimmeler S, Hermann C, Busse R, Fleming I. Phosphorylation and activation of the endothelial nitric oxide synthase by fluid shear stress. Acta Physiol Scand. 2000;168:81–88. doi: 10.1046/j.1365-201x.2000.00627.x. [DOI] [PubMed] [Google Scholar]
- 31.Fleming I, Busse R. Molecular mechanisms involved in the regulation of the endothelial nitric oxide synthase. Am J Physiol Regul Integr Comp Physiol. 2003;284:R1–R12. doi: 10.1152/ajpregu.00323.2002. [DOI] [PubMed] [Google Scholar]
- 32.Fulton D, Gratton JP, McCabe TJ, Fontana J, Fujio Y, Walsh K, et al. Regulation of endothelium-derived nitric oxide production by the protein kinase Akt. Nature. 1999;399:597–601. doi: 10.1038/21218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gladwin MT, Crawford JH, Patel RP. The biochemistry of nitric oxide, nitrite, and hemoglobin: role in blood flow regulation. Free Radic Biol Med. 2004;36:707–717. doi: 10.1016/j.freeradbiomed.2003.11.032. [DOI] [PubMed] [Google Scholar]
- 34.Gladwin MT, Schechter AN. NO contest: nitrite versus S-nitroso-hemoglobin. Circ Res. 2004;94:851–855. doi: 10.1161/01.RES.0000126697.64381.37. [DOI] [PubMed] [Google Scholar]
- 35.Gladwin MT, Shelhamer JH, Schechter AN, Pease-Fye ME, Waclawiw MA, Panza JA, et al. Role of circulating nitrite and S-nitrosohemoglobin in the regulation of regional blood flow in humans. Proc Natl Acad Sci USA. 2000;97:11482–11487. doi: 10.1073/pnas.97.21.11482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Goldsmith HL, Cokelet GR, Gaehtgens P. Robin Fahraeus: evolution of his concepts in cardiovascular physiology. Am J Physiol. 1989;257:H1005–H1015. doi: 10.1152/ajpheart.1989.257.3.H1005. [DOI] [PubMed] [Google Scholar]
- 37.Granath B, Taylor RR, van Bockxmeer FM, Mamotte CD. Lack of evidence for association between endothelial nitric oxide synthase gene polymorphisms and coronary artery disease in the Australian Caucasian population. J Cardiovasc Risk. 2001;8:235–241. doi: 10.1177/174182670100800408. [DOI] [PubMed] [Google Scholar]
- 38.Grau M, Pauly S, Ali J, Walpurgis K, Thevis M, Bloch W, et al. RBC-NOS-dependent S-nitrosylation of cytoskeletal proteins improves RBC deformability. PLoS One. 2013;8:e56759. doi: 10.1371/journal.pone.0056759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Griffith TM, Edwards DH, Davies RL, Harrison TJ, Evans KT. EDRF coordinates the behaviour of vascular resistance vessels. Nature. 1987;329:442–445. doi: 10.1038/329442a0. [DOI] [PubMed] [Google Scholar]
- 40.Helms C, Kim-Shapiro DB. Hemoglobin-mediated nitric oxide signaling. Free Radic Biol Med. 2013;61C:464–472. doi: 10.1016/j.freeradbiomed.2013.04.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Horn P, Cortese-Krott MM, Keymel S, Kumara I, Burghoff S, Schrader J, et al. Nitric oxide influences red blood cell velocity independently of changes in the vascular tone. Free Radical Research. 2011;45:653–661. doi: 10.3109/10715762.2011.574288. [DOI] [PubMed] [Google Scholar]
- 42.Huang Z, Shiva S, Kim-Shapiro DB, Patel RP, Ringwood LA, Irby CE, et al. Enzymatic function of hemoglobin as a nitrite reductase that produces NO under allosteric control. J Clin Invest. 2005;115:2099–2107. doi: 10.1172/JCI24650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hutcheson IR, Griffith TM. Central role of intracellular calcium stores in acute flow- and agonist-evoked endothelial nitric oxide release. Br J Pharmacol. 1997;122:117–125. doi: 10.1038/sj.bjp.0701340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ignarro LJ, Cirino G, Casini A, Napoli C. Nitric oxide as a signaling molecule in the vascular system: an overview. J Cardiovasc Pharmacol. 1999;34:879–886. doi: 10.1097/00005344-199912000-00016. [DOI] [PubMed] [Google Scholar]
- 45.Isbell TS, Sun CW, Wu LC, Teng X, Vitturi DA, Branch BG, et al. SNO-hemoglobin is not essential for red blood cell-dependent hypoxic vasodilation. Nat Med. 2008;14:773–777. doi: 10.1038/nm1771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Khurana RN, Deng PF, Epstein DL, Vasantha Rao P. The role of protein kinase C in modulation of aqueous humor outflow facility. Exp Eye Res. 2003;76:39–47. doi: 10.1016/s0014-4835(02)00255-5. [DOI] [PubMed] [Google Scholar]
- 47.Kleinbongard P, Schulz R, Rassaf T, Lauer T, Dejam A, Jax T, et al. Red blood cells express a functional endothelial nitric oxide synthase. Blood. 2006;107:2943–2951. doi: 10.1182/blood-2005-10-3992. [DOI] [PubMed] [Google Scholar]
- 48.Kojda G, Cheng YC, Burchfield J, Harrison DG. Dysfunctional regulation of endothelial nitric oxide synthase (eNOS) expression in response to exercise in mice lacking one eNOS gene. Circulation. 2001;103:2839–2844. doi: 10.1161/01.cir.103.23.2839. [DOI] [PubMed] [Google Scholar]
- 49.Korbut R, Gryglewski RJ. Nitric oxide from polymorphonuclear leukocytes modulates red blood cell deformability in vitro. Eur J Pharmacol. 1993;234:17–22. doi: 10.1016/0014-2999(93)90700-r. [DOI] [PubMed] [Google Scholar]
- 50.Kuo L, Davis MJ, Chilian WM. Endothelium-dependent, flow-induced dilation of isolated coronary arterioles. Am J Physiol. 1990;259:H1063–H1070. doi: 10.1152/ajpheart.1990.259.4.H1063. [DOI] [PubMed] [Google Scholar]
- 51.Kuzman D, Znidarcic T, Gros M, Vrhovec S, Svetina S, Zeks B. Effect of pH on red blood cell deformability. Pflugers Arch. 2000;440:R193–R194. [PubMed] [Google Scholar]
- 52.Liu X, Miller MJ, Joshi MS, Sadowska-Krowicka H, Clark DA, Lancaster JR., Jr Diffusion-limited reaction of free nitric oxide with erythrocytes. J Biol Chem. 1998;273:18709–18713. doi: 10.1074/jbc.273.30.18709. [DOI] [PubMed] [Google Scholar]
- 53.Liu X, Miller MJ, Joshi MS, Thomas DD, Lancaster JR., Jr Accelerated reaction of nitric oxide with O2 within the hydrophobic interior of biological membranes. Proc Natl Acad Sci USA. 1998;95:2175–2179. doi: 10.1073/pnas.95.5.2175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Mannini L, Cecchi E, Fatini C, Marcucci R, Alessandriello Liotta A, Matucci-Cerinic M, et al. Clinical haemorheology and microcirculation. Ann Ist Super Sanita. 2007;43:144–155. [PubMed] [Google Scholar]
- 55.Mannino R, Myers DR, Sakurai Y, Ware WE, Barabino GA, Lam W. Increased erythrocyte rigidity is sufficient to cause endothelial dysfunction in sickle disease. Blood. 2012;120 Abstract 818. [Google Scholar]
- 56.Mesquita R, Picarra B, Saldanha C, Martins e Silva J. Nitric oxide effects on human erythrocytes structural and functional properties – an in vitro study. Clin Hemorheol Microcirc. 2002;27:137–147. [PubMed] [Google Scholar]
- 57.Milner P, Kirkpatrick KA, Ralevic V, Toothill V, Pearson J, Burnstock G. Endothelial cells cultured from human umbilical vein release ATP, substance P and acetylcholine in response to increased flow. Proc Biol Sci. 1990;241:245–248. doi: 10.1098/rspb.1990.0092. [DOI] [PubMed] [Google Scholar]
- 58.Minneci PC, Deans KJ, Shiva S, Zhi H, Banks SM, Kern S, et al. Nitrite reductase activity of hemoglobin as a systemic nitric oxide generator mechanism to detoxify plasma hemoglobin produced during hemolysis. Am J Physiol Heart Circ Physiol. 2008;295:H743–H754. doi: 10.1152/ajpheart.00151.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Moncada S. Nitric oxide: discovery and impact on clinical medicine. J R Soc Med. 1999;92:164–169. doi: 10.1177/014107689909200402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Moncada S, Palmer RM, Higgs EA. Nitric oxide: physiology, pathophysiology, and pharmacology. Pharmacol Rev. 1991;43:109–142. [PubMed] [Google Scholar]
- 61.Nadaud S, Philippe M, Arnal JF, Michel JB, Soubrier F. Sustained increase in aortic endothelial nitric oxide synthase expression in vivo in a model of chronic high blood flow. Circ Res. 1996;79:857–863. doi: 10.1161/01.res.79.4.857. [DOI] [PubMed] [Google Scholar]
- 62.Nagababu E, Ramasamy S, Abernethy DR, Rifkind JM. Active nitric oxide produced in the red cell under hypoxic conditions by deoxyhemoglobin-mediated nitrite reduction. J Biol Chem. 2003;278:46349–46356. doi: 10.1074/jbc.M307572200. [DOI] [PubMed] [Google Scholar]
- 63.Nagassaki S, Herculano RD, Graeff CF, Tanus-Santos JE. eNOS T-786C polymorphism affects atorvastatin-induced changes in erythrocyte membrane fluidity. Eur J Clin Pharmacol. 2009;65:385–392. doi: 10.1007/s00228-008-0602-7. [DOI] [PubMed] [Google Scholar]
- 64.Nicolay JP, Liebig G, Niemoeller OM, Koka S, Ghashghaeinia M, Wieder T, et al. Inhibition of suicidal erythrocyte death by nitric oxide. Pflugers Arch. 2008;456:293–305. doi: 10.1007/s00424-007-0393-1. [DOI] [PubMed] [Google Scholar]
- 65.Nishida K, Harrison DG, Navas JP, Fisher AA, Dockery SP, Uematsu M, et al. Molecular cloning and characterization of the constitutive bovine aortic endothelial cell nitric oxide synthase. J Clin Invest. 1992;90:2092–2096. doi: 10.1172/JCI116092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ong PK, Cho S, Namgung B, Kim S. Effects of cell-free layer formation on NO/O2 bioavailability in small arterioles. Microvascular Research. 2012;83:168–177. doi: 10.1016/j.mvr.2011.11.006. [DOI] [PubMed] [Google Scholar]
- 67.Ozuyaman B, Grau M, Kelm M, Merx MW, Kleinbongard P. RBC NOS: regulatory mechanisms and therapeutic aspects. Trends Mol Med. 2008;14:314–322. doi: 10.1016/j.molmed.2008.05.002. [DOI] [PubMed] [Google Scholar]
- 68.Paniagua OA, Bryant MB, Panza JA. Role of endothelial nitric oxide in shear stress-induced vasodilation of human microvasculature: diminished activity in hypertensive and hypercholesterolemic patients. Circulation. 2001;103:1752–1758. doi: 10.1161/01.cir.103.13.1752. [DOI] [PubMed] [Google Scholar]
- 69.Pantely GA, Swenson LJ, Tamblyn CH, Seaman GV, Anselone CG, Johnson WB, et al. Increased vascular resistance due to a reduction in red cell deformability in the isolated hind limb of swine. Microvascular Research. 1988;35:86–100. doi: 10.1016/0026-2862(88)90052-0. [DOI] [PubMed] [Google Scholar]
- 70.Pavlovic D, Hall AR, Kennington EJ, Aughton K, Boguslavskyi A, Fuller W, et al. Nitric oxide regulates cardiac intracellular Na(+) and Ca(2)(+) by modulating Na/K ATPase via PKCepsilon and phospholemman-dependent mechanism. J Mol Cell Cardiol. 2013;61:164–171. doi: 10.1016/j.yjmcc.2013.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Pawloski JR, Hess DT, Stamler JS. Export by red blood cells of nitric oxide bioactivity. Nature. 2001;409:622–626. doi: 10.1038/35054560. [DOI] [PubMed] [Google Scholar]
- 72.Pawloski JR, Hess DT, Stamler JS. Impaired vasodilation by red blood cells in sickle cell disease. Proc Natl Acad Sci USA. 2005;102:2531–2536. doi: 10.1073/pnas.0409876102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Petrov V, Amery A, Lijnen P. Role of cyclic GMP in atrial-natriuretic-peptide stimulation of erythrocyte Na+/H+ exchange. Eur J Biochem. 1994;221:195–199. doi: 10.1111/j.1432-1033.1994.tb18729.x. [DOI] [PubMed] [Google Scholar]
- 74.Petrov V, Lijnen P. Regulation of human erythrocyte Na+/H+ exchange by soluble and particulate guanylate cyclase. Am J Physiol. 1996;271:C1556–C1564. doi: 10.1152/ajpcell.1996.271.5.C1556. [DOI] [PubMed] [Google Scholar]
- 75.Pohl U, Holtz J, Busse R, Bassenge E. Crucial role of endothelium in the vasodilator response to increased flow in vivo. Hypertension. 1986;8:37–44. doi: 10.1161/01.hyp.8.1.37. [DOI] [PubMed] [Google Scholar]
- 76.Pryor WA, Lemercier JN, Zhang H, Uppu RM, Squadrito GL. The catalytic role of carbon dioxide in the decomposition of peroxynitrite. Free Radic Biol Med. 1997;23:331–338. doi: 10.1016/s0891-5849(97)00121-4. [DOI] [PubMed] [Google Scholar]
- 77.Ranjan V, Xiao Z, Diamond SL. Constitutive NOS expression in cultured endothelial cells is elevated by fluid shear stress. Am J Physiol. 1995;269:H550–H555. doi: 10.1152/ajpheart.1995.269.2.H550. [DOI] [PubMed] [Google Scholar]
- 78.Reynolds JD, Bennett KM, Cina AJ, Diesen DL, Henderson MB, Matto F, et al. S-nitrosylation therapy to improve oxygen delivery of banked blood. Proc Natl Acad Sci USA. 2013;110:11529–11534. doi: 10.1073/pnas.1306489110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Rossi GP, Cesari M, Zanchetta M, Colonna S, Maiolino G, Pedon L, et al. The T-786C endothelial nitric oxide synthase genotype is a novel risk factor for coronary artery disease in Caucasian patients of the GENICA study. J Am Coll Cardiol. 2003;41:930–937. doi: 10.1016/s0735-1097(02)03012-7. [DOI] [PubMed] [Google Scholar]
- 80.Sharan K, Surrey S, Ballas S, Borowski M, Devoto M, Wang KF, et al. Association of T-786C eNOS gene polymorphism with increased susceptibility to acute chest syndrome in females with sickle cell disease. Br J Haematol. 2004;124:240–243. doi: 10.1046/j.1365-2141.2003.04762.x. [DOI] [PubMed] [Google Scholar]
- 81.Simmonds MJ, Connes P, Sabapathy S. Exercise-induced blood lactate increase does not change red blood cell deformability in cyclists. PLoS One. 2013;8:e71219. doi: 10.1371/journal.pone.0071219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Simmonds MJ, Meiselman HJ, Marshall-Gradisnik SM, Pyne M, Kakanis M, Keane J, et al. Assessment of oxidant susceptibility of red blood cells in various species based on cell deformability. Biorheology. 2011;48:293–304. doi: 10.3233/BIR-2012-0599. [DOI] [PubMed] [Google Scholar]
- 83.Singel DJ, Stamler JS. Chemical physiology of blood flow regulation by red blood cells: the role of nitric oxide and S-nitrosohemoglobin. Annu Rev Physiol. 2005;67:99–145. doi: 10.1146/annurev.physiol.67.060603.090918. [DOI] [PubMed] [Google Scholar]
- 84.Starzyk D, Korbut R, Gryglewski RJ. The role of nitric oxide in regulation of deformability of red blood cells in acute phase of endotoxaemia in rats. J Physiol Pharmacol. 1997;48:731–735. [PubMed] [Google Scholar]
- 85.Starzyk D, Korbut R, Gryglewski RJ. Effects of nitric oxide and prostacyclin on deformability and aggregability of red blood cells of rats ex vivo and in vitro. J Physiol Pharmacol. 1999;50:629–637. [PubMed] [Google Scholar]
- 86.Suhr F, Brenig J, Muller R, Behrens H, Bloch W, Grau M. Moderate exercise promotes human RBC-NOS activity, NO production and deformability through Akt kinase pathway. PLoS One. 2012;7:e45982. doi: 10.1371/journal.pone.0045982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tripette J, Alexy T, Hardy-Dessources MD, Mougenel D, Beltan E, Chalabi T, et al. Red blood cell aggregation, aggregate strength and oxygen transport potential of blood are abnormal in both homozygous sickle cell anemia and sickle-hemoglobin C disease. Haematologica. 2009;94:1060–1065. doi: 10.3324/haematol.2008.005371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Tsai AG, Acero C, Nance PR, Cabrales P, Frangos JA, Buerk DG, et al. Elevated plasma viscosity in extreme hemodilution increases perivascular nitric oxide concentration and microvascular perfusion. Am J Physiol Heart Circ Physiol. 2005;288:H1730–H1739. doi: 10.1152/ajpheart.00998.2004. [DOI] [PubMed] [Google Scholar]
- 89.Tsai EJ, Kass DA. Cyclic GMP signaling in cardiovascular pathophysiology and therapeutics. Pharmacol Ther. 2009;122:216–238. doi: 10.1016/j.pharmthera.2009.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Ulker P, Gunduz F, Meiselman HJ, Baskurt OK. Nitric oxide generated by red blood cells following exposure to shear stress dilates isolated small mesenteric arteries under hypoxic conditions. Clin Hemorheol Microcirc. 2013;54:357–369. doi: 10.3233/CH-2012-1618. [DOI] [PubMed] [Google Scholar]
- 91.Ulker P, Meiselman HJ, Baskurt OK. Nitric oxide generation in red blood cells induced by mechanical stress. Clin Hemorheol Microcirc. 2010;45:169–175. doi: 10.3233/CH-2010-1293. [DOI] [PubMed] [Google Scholar]
- 92.Ulker P, Sati L, Celik-Ozenci C, Meiselman HJ, Baskurt OK. Mechanical stimulation of nitric oxide synthesizing mechanisms in erythrocytes. Biorheology. 2009;46:121–132. doi: 10.3233/BIR-2009-0532. [DOI] [PubMed] [Google Scholar]
- 93.Ulker P, Yaras N, Yalcin O, Celik-Ozenci C, Johnson PC, Meiselman HJ, et al. Shear stress activation of nitric oxide synthase and increased nitric oxide levels in human red blood cells. Nitric Oxide: Biology and Chemistry/Official Journal of the Nitric Oxide Society. 2011;24:184–191. doi: 10.1016/j.niox.2011.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.van Beers EJ, Nieuwdorp M, Duits AJ, Evers LM, Schnog JJ, Biemond BJ. Sickle cell patients are characterized by a reduced glycocalyx volume. Haematologica. 2008;93:307–308. doi: 10.3324/haematol.12027. [DOI] [PubMed] [Google Scholar]
- 95.Varin R, Mulder P, Richard V, Tamion F, Devaux C, Henry JP, et al. Exercise improves flow-mediated vasodilatation of skeletal muscle arteries in rats with chronic heart failure. Role of nitric oxide, prostanoids, and oxidant stress. Circulation. 1999;99:2951–2957. doi: 10.1161/01.cir.99.22.2951. [DOI] [PubMed] [Google Scholar]
- 96.Vink H, Duling BR. Identification of distinct luminal domains for macromolecules, erythrocytes, and leukocytes within mammalian capillaries. Circ Res. 1996;79:581–589. doi: 10.1161/01.res.79.3.581. [DOI] [PubMed] [Google Scholar]
- 97.Wennmalm A, Benthin G, Petersson AS. Dependence of the metabolism of nitric oxide (NO) in healthy human whole blood on the oxygenation of its red cell haemoglobin. Br J Pharmacol. 1992;106:507–508. doi: 10.1111/j.1476-5381.1992.tb14365.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Wilcox CS, Deng X, Doll AH, Snellen H, Welch WJ. Nitric oxide mediates renal vasodilation during erythropoietin-induced polycythemia. Kidney Int. 1993;44:430–435. doi: 10.1038/ki.1993.261. [DOI] [PubMed] [Google Scholar]
- 99.Wood KC, Cortese-Krott MM, Kovacic JC, Noguchi A, Liu VB, Wang X, et al. Circulating blood endothelial nitric oxide synthase contributes to the regulation of systemic blood pressure and nitrite homeostasis. Arterioscler Thromb Vasc Biol. 2013;33:1861–1871. doi: 10.1161/ATVBAHA.112.301068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Xiao Z, Zhang Z, Ranjan V, Diamond SL. Shear stress induction of the endothelial nitric oxide synthase gene is calcium-dependent but not calcium-activated. J Cell Physiol. 1997;171:205–211. doi: 10.1002/(SICI)1097-4652(199705)171:2<205::AID-JCP11>3.0.CO;2-C. [DOI] [PubMed] [Google Scholar]
- 101.Yalcin O, Meiselman HJ, Armstrong JK, Baskurt OK. Effect of enhanced red blood cell aggregation on blood flow resistance in an isolated-perfused guinea pig heart preparation. Biorheology. 2005;42:511–520. [PubMed] [Google Scholar]
- 102.Yalcin O, Ulker P, Yavuzer U, Meiselman HJ, Baskurt OK. Nitric oxide generation by endothelial cells exposed to shear stress in glass tubes perfused with red blood cell suspensions: role of aggregation. Am J Physiol Heart Circ Physiol. 2008;294:H2098–H2105. doi: 10.1152/ajpheart.00015.2008. [DOI] [PubMed] [Google Scholar]
- 103.Yalcin O, Uyuklu M, Armstrong JK, Meiselman HJ, Baskurt OK. Graded alterations of RBC aggregation influence in vivo blood flow resistance. Am J Physiol Heart Circ Physiol. 2004;287:H2644–H2650. doi: 10.1152/ajpheart.00534.2004. [DOI] [PubMed] [Google Scholar]
- 104.Zhang J, Johnson PC, Popel AS. Effects of erythrocyte deformability and aggregation on the cell free layer and apparent viscosity of microscopic blood flows. Microvascular Research. 2009;77:265–272. doi: 10.1016/j.mvr.2009.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]