

Abstract
Background & Aims:
Development of celiac disease is believed to involve the transglutaminase-dependent response of CD4+ T cells toward deamidated gluten peptides in the intestinal mucosa of individuals with specific HLA-DQ haplotypes. We investigated the antigen presentation process during this mucosal immune response.
Methods:
We generated monoclonal antibodies (mAbs) specific for the peptide–MHC complex HLA-DQ2.5 and the immunodominant gluten epitope DQ2.5-glia-α1a using phage display. We used these mAbs to assess gluten peptide presentation and phenotypes of presenting cells by flow cytometry and ELISPOT in freshly prepared single-cell suspensions from intestinal biopsies from 40 patients with celiac disease (35 untreated and 5 on a gluten-free diet) as well as 18 subjects with confirmed non-inflamed gut mucosa (controls, 12 presumed healthy, 5 undergoing pancreatoduodenectomy, and 1 with potential celiac disease).
Results:
Using the mAbs, we detected peptide–MHC complex on cells from intestinal biopsies from patients with celiac disease who consume gluten, but not from patients on gluten-free diets. We found B cells and plasma cells to be the most abundant cells that present DQ2.5-glia-α1a in the inflamed mucosa. We identified a subset of plasma cells that expresses B-cell receptors specific for gluten peptides or the autoantigen transglutaminase 2. Expression of MHC class II was not restricted to these specific plasma cells in patients with celiac disease but was observed in an average 30% of gut plasma cells from patients and controls.
Conclusions:
A population of plasma cells from intestinal biopsies of patients with celiac disease express MHCII; this is the most abundant cell type presenting the immunodominant gluten peptide DQ2.5-glia-α1a in the tissues from these patients. These results indicate that plasma cells in the gut can function as antigen presenting cells and might promote and maintain intestinal inflammation in patients with celiac disease or other inflammatory disorders.
Keywords: TG2, APC, autoimmunity, immune activation
Graphical Abstract
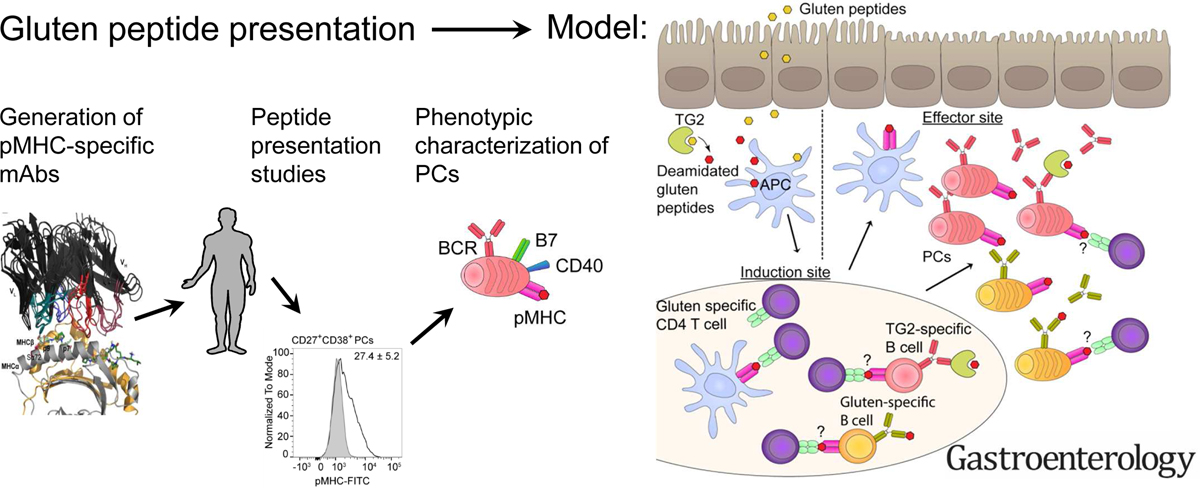
Introduction
Gluten-reactive CD4+ T cells are believed to play a key role in celiac disease1. Genetic risk factors, presence of autoantibodies and T-cell mediated destruction of organ-specific cells are shared features between celiac disease and many other autoimmune disorders1. However, unlike most of these other conditions, the driving antigen of celiac disease is known, namely dietary gluten proteins from wheat, barley and rye.
The strongest genetic risk factor of celiac disease is HLA, with 90% of the patients expressing HLA-DQ2.5 (DQA1*05-DQB1*02), and the vast majority of the remaining patients expressing HLA-DQ8 (DQA1*03-DQB1*03:02) or HLA-DQ2.2 (DQA1*02:01-DQB1*02)2. A consistent finding in HLA-DQ2.5+ celiac disease patients is a CD4+ T-cell response towards the post-translationally modified epitopes DQ2.5-glia-α1a (9mer core region PFPQPELPY) and DQ2.5-glia-α2 (PQPELPYPQ) of α-gliadin, and DQ2.5-glia-ω1 (PFPQPEQPF) and DQ2.5-glia-ω2 (PQPEQPFPW) of ω-gliadin3,4. Transglutaminase 2 (TG2)-mediated deamidation of glutamine residues to glutamate introduces negatively charged anchor residues which make the gluten peptides bind more strongly to HLA-DQ2.5, thus increasing the pMHC half-life critical for T-cell engagement4–6.
Initial activation of mucosal T-cells is thought to take place in gut-draining mesenteric lymph nodes and in gut-associated lymphoid tissue, while re-activation of gut-homing memory/effector T cells primarily occurs in the lamina propria (LP) of the small intestine1,7. Previous studies on the characteristics of antigen presenting cells (APC) in the duodenum have identified the macrophages (Mfs) and dendritic cells (DCs) as the main populations8–10, and a particular CD11c+ population appears to accumulate in active celiac lesions in response to gluten antigen challenge11. When DCs and Mfs were isolated from intestinal biopsies of untreated celiac patients, loaded with peptide in vitro and incubated with gluten-reactive T-cell clones, T-cell activation was indeed observed, the CD11c+ enriched population being most effective8. A hallmark of celiac disease is the presence of terminally differentiated plasma cells in the LP that are secreting antibodies against deamidated gluten peptides and TG212,13. However, a pathogenic role of these plasma cells and the antibodies they secrete have not been established.
To enable specific characterization of the APC subsets involved in gluten peptide presentation, we isolated mAbs with specificity for the DQ2.5-glia-α1a epitope in complex with HLA-DQ2.5 from a naïve, human scFv-phage library. The isolated mAbs discriminate between highly similar pMHC complexes. We further utilized the mAbs to detect both in vitro gluten peptide-loading of APCs, and endogenous gluten peptide presentation by cells isolated from intestinal biopsies from celiac patients. Surprisingly, B cells and plasma cells were the main cell types found to present. Our data thus show that plasma cells, and not DCs and Mfs, is the most abundant cell type that actually presents gluten peptides in the LP of celiac patients. Furthermore, the plasma cells do this by virtue of a hitherto unappreciated ability to express MHCII and present specific antigenic peptides to T cells. This observation may also hold true for other MHCII-associated diseases, and mechanistically explain the beneficial effects seen after B cell depletion therapy even in the absence of pathogenic autoantibodies14,15.
Material and Methods
Human material
Duodenal biopsy material was obtained according to approved protocols (Regional Ethics Committee of South-Eastern Norway approval 2010/2720 S-97201), and all subjects gave informed written consent. celiac disease diagnosis was given according to the British Society for Gastroenterology guidelines including clinical history, anti-TG2 serological testing, HLA typing and histological analysis of small intestinal biopsies obtained by esophagogastroduodenoscopy and forceps sampling from the duodenum16. Small intestinal resections (duodenum-proximal jejunum tissue) were obtained from nonpathological small intestine during Whipple procedure (pancreatoduodenectomy) of pancreatic cancer patients who gave informed written consent (approval 2010/2720 S-97201). Only material with confirmed normal histology was included.
Recombinant pMHC
Recombinant pMHCs are detailed in Supplementary Table 2. Recombinant pMHC used for SPR was purified by size exclusion using Superdex 200 after biotinylation.
Selection and rescue of scFv phage libraries and reformatting and expression of clones
HLA-DQ2.5:DQ2.5-glia-α1a-specific binders were isolated from a naïve human scFv library17. Selection and rescue was performed essentially as described18 with the following specifications: pre-blocked phage samples were incubated 1 h with 80 nM biotinylated HLA-DQ2.5:CLIP2 before capture onto Dynabeads MyOne Streptavidin T1 beads for 30 min; unbound phage was transferred to tubes containing 80 nM biotinylated HLA-DQ2.5:DQ2.5-glia-α1a for 1 h before transfer to beads; R4 stringency included 100x less antigen and 20 + 20 washes. Selection alternative 1 was performed as previously described for the OMV selection18; alternative 2 included 16.6 nM non-biotinylated HLA-DQ2.5:DQ2.5-glia-α1a as competitor in solution. Phage rescue (E. coli XL1-Blue and M13K07), PEG/NaCl precipitation, spot titration, reformatting and soluble expression was performed as described18. scFv’s were purified as described19. SDS-PAGE was performed using standard procedures.
Binding analysis by SPR and ELISA
SPR was performed using a Biacore T100 (T200 sensitivity enhanced, GE) essentially as described19. Samples were 3-fold diluted from 2 µM for single-cycle kinetics experiments and specificity assessments, while 0.25 µM was used for half-life comparisons of scFv and hIgG1 variants.
Presence of functionally folded pMHC was verified after running samples by injection of 0.2 µM mAb SPV-L3, binding only correctly folded HLA-DQ2. ELISA was performed as described18 and detailed in Supplementary Table 3.
IgG cloning, eukaryotic protein expression and purification
hIgG1 cloning was performed as described18. Alternatively, synthetic gene fragments encoding VH and VL were ordered together with codon optimized mouse Ig gamma2b or mouse Ig kappa cDNA, respectively, and cloned as BsmI-BamHI fragments. HEK293E cells were co-transfected as described20. mAbs were purified on either HiTrap protein L (GE) or a NIP-coupled column (inhouse prepared) before size exclusion.
Retroviral transduction of A20 murine B cells
A20 B cells expressing HLA-DQ2.5 with the covalently attached DQ2.5-glia-α1a (deamidated=E, native=Q, pL7Q and pY9F variants), DQ2.5-glia-ω1, DQ2.5-glia-α2 as well as HLA-DQ2.5:CLIP2 have been described19. The construct encoding HLA-DQ2.5:DQ2.5-glia-α1a-Sα72I was generated by cloning a BglII/BamHI codon-optimized synthetic DNA fragment (Genscript) encoding the HLA-DQ2.5 α-chain (DQA1*05:01) with the Sα72I mutation into the pMIG-II-eGFP retroviral plasmid (gift from Dario Vignali, University of Pittsburgh, Pittsburgh, USA) already encoding HLA-DQ2.5:DQ2.5-glia-α1a19. HLA-DQ2.2:DQ2.5-glia-α1a was generated by exchange of the HLADQ2.5 α-chain (DQA1*05:01) with a BglII/BamHI codon-optimized synthetic DNA fragment encoding the HLA-DQ2.2 α-chain (DQA1*02:01). Cells were transduced and stained for flow cytometric analysis as described19 (Supplementary Table 4).
Differentiation and flow cytometric detection of peptide-loaded monocyte-derived DCs
Monocyte-derived DCs were prepared from PBMCs from DR3/DQ2-positive blood donors as described21. DCs were matured using 150 ng/ml LPS for 48 h, supplemented with 40 µM deamidated DQ2.5-glia-α1a after 24 h. Flow cytometric staining and analysis was performed as specified in Supplementary Table 4.
Isolation of single-cell suspensions from duodenal biopsies and small intestinal resections and flow cytometry
Single-cell suspensions from duodenal biopsies or from small intestinal resection were prepared as described22 and analyzed by flow cytometry as detailed in Supplementary Table 4.
Sorting of plasma cells, microscopy and ELISPOT
Single-cell suspensions were stained and sorted as described in Supplementary Table 5. Sorted cells (using RPMI1640 without phenol red for microscopy) were spun down, resuspended and incubated at 37°C/5% CO2 over night before imaging live cells in culture using a 40x NA 0.5 objective on an inverted Leica DM IL microscope equipped with a Axiocam MRc camera (Zeiss). ELISPOT was performed essentially as described13, except that cells were incubated in the plates for 3 days instead of 1. TG2, gliadin digest or anti-human Ig antibody were coated at 5 μg/ml, 50 μg/ml, and 5 μg/ml, respectively. Chymotrypsin digested gliadin was generated as described12, with the exception that 20 mM HCl pH 1.7 was used during heating at 95°C for 1h to introduce deamidation. The plate was read by an ImmunoSpot S5 Analyzer (Cellular Technology Limited). BW 364 T-cells was used as negative control19.
Antibody modeling and docking to pMHC
Antibody homology models were generated using RosettaAntibody essentially as described23. Multiple templates of the VL–VH orientation24 were used, resulting in 10 grafted models. During modeling, the CDR H3 was constrained to the kinked conformation with a harmonic potential25. Chothia numbering was used. Models for docking were selected from 2800 Fv models based on low Rosetta energy and good VL-VH orientation. Models from at least three different VL-VH orientation templates were considered for docking onto HLA-DQ2.5:DQ2.5-glia-α1a (PDB ID 1S9V)26). The pMHC structure was “relaxed” in the Rosetta energy function27. The top 10 Fv models and the relaxed pMHC structure were prepared for docking by running the ensemble prepack protocol as described23. The initial orientation was chosen based on the solved TCR:pMHC interaction (4OZI28). Docking using SnugDock29 consisted of an initial spin around the Ab–Ag center-of-mass axis uniformly sampled from 0 to 360°, and additional random perturbations consisting of small translations and rotations, with values sampled from Gaussian distributions centered at 3 Å and 8°, respectively. During docking, CDR H2 and H3 loops were refined by kinematic loop closure and the VL-VH orientation was refined by VL–VH docking, generating 1000 models. The final models were picked based on low Rosetta energy, reasonable orientation relative to the pMHC, and agreement with experimentally observed specificities.
Statistics
For comparisons between two groups unpaired two-tailed t-test was used. Correlation between pair was determined by linear regression using Pearson correlation to give R2 and two-tailed p-values. Asterisks were used as follows (also indicated in figure legends): *P≤.05; **P≤.01; ***P≤.001; ****P≤.0001; ns, not significant. Details on sample size, experimental replicates and statistics are included in the figure legends.
Results
Phage selection of recombinant antibodies highly specific for HLA-DQ2.5 with bound DQ2.5-glia-α1a
To isolate HLA-DQ2.5:DQ2.5-glia-α1a-specific binders, we performed phage selections using a naïve, fully-human scFv-phage library and soluble pMHC molecules17. SPR analysis of binding affinity and specificity of purified scFv clones identified 2 lead clones (termed 106 and 107) differing only in one amino acid that bound specifically to HLA-DQ2.5:DQ2.5-glia-α1a, and not to HLA-DQ2.5 with the control peptides DQ2.5-glia-α2 and CLIP2 (Fig. 1A, Supplementary Fig. 1A-C). The scFv clones bound with a monomeric affinity between 70 and 100 nM (Fig. 1A and Supplementary Table 1).
Figure 1.

Binding properties of the lead HLA-DQ2.5:DQ2.5-glia-αIa-specific binders. (A) Representative SPR single-cycle kinetics sensorgrams of the lead scFv clones 106 and 107 for binding to HLA-DQ2.5:DQ2.5-glia-αIa (n=2–3). KD values were derived by fitting the data to a 1:1 Langmuir model. Steady state affinity evaluations are shown as inset figures. (B-D) The scFv clones and isotype control scFv (anti-NIP) were reformatted to hIgG1 mAbs, expressed and purified before assessment of specificity. (B,C) Competition ELISAs where the hIgG1 mAbs were pre-incubated with titrated amounts of (B) soluble pMHCs, HLA-DQ2.5:DQ2.5-glia-α1a and HLA-DQ2.5:DQ2.5-glia-α2, or the corresponding free peptides, and (C) 33mer α-gliadin before assessment of ability to compete with binding to plate-bound HLA-DQ2.5:DQ2.5-glia-α1a (n=3). (D) Eight different HLA-DQ2.5:gluten peptide complexes and HLA-DQ2.5:CLIP2 were used in ELISA for specificity analysis (n=2). mAb 2.12.E11 specific for the β-chain of HLA-DQ2 was included to control pMHC capture levels. Error bars illustrate mean ± SD of duplicates.
We reformatted and expressed the two scFvs as human IgG1 (hIgG1) mAbs and performed SPR to confirm a gain in functional affinity, resulting in an approximately 160-fold increase in half-life (Supplementary Fig. 1D, E). To confirm DQ2.5-glia-α1a peptide recognition strictly in the context of HLA-DQ2.5, we performed competition ELISA using soluble pMHC and free peptide. Indeed, only soluble HLA-DQ2.5:DQ2.5-glia-α1a, and not peptide alone, competed with the plate-bound complex for binding to the mAbs (Fig. 1B). Of note, DQ2.5-glia-α1a provided as part of a 33mer peptide fragment which binds efficiently to HLA-DQ2.530 was not able to inhibit mAb binding to pMHC (Fig. 1C).
Next, we extended the specificity analysis by including 7 HLA-DQ2.5-gluten-peptide complexes in ELISA. This panel included common epitopes from γ-and ω-gliadin to which celiac patients mount T-cell responses. Neither of the mAbs bound any of the complexes besides HLA-DQ2.5:DQ2.5-glia-α1a (Fig. 1D), not even the pMHC complex of the highly homologous DQ2.5glia-ω1. Collectively, these results show that the mAbs exclusively recognize DQ2.5-glia-α1a bound to HLA-DQ2.5 and are not cross-reactive with HLA-DQ2.5 in complex with the other gluten peptides tested.
Mapping fine-specificity of the candidate mAbs
To validate mAb binding in a cellular context, we utilized murine A20 B cells transduced with HLA-DQ2.5 with covalently linked DQ2.5-glia-α1a or CLIP2 peptides. Both mAbs bound specifically to cells displaying the DQ2.5-glia-α1a epitope, while neither bound CLIP2 (Fig. 2A and Supplementary Fig. 2A).
Figure 2.

Mapping the fine-specificity and the structural basis for specificity. (A,B) Flow cytometric analysis of A20 B cells expressing HLA-DQ2 with covalently bound peptides. (A) Binding to cells expressing HLA-DQ2.5 with DQ2.5-glia-α1a or CLIP2 peptides (n=2). (B) Binding to a panel of cells expressing HLA-DQ2.5:peptide or HLA-DQ2.2:peptide. Data are shown as ratio median fluorescent intensity (MFI) compared to isotype control mAb (n=2). Q indicate native (glutamine) DQ2.5-glia-α1a epitope. Unless specified, all peptides were tested in their deamidated form. (C) Alignment and structural conformation of MHC-bound DQ2.5-glia-α1a (green) and DQ2.5-glia-ω1 (cyan). Residues differing are underlined in the peptide sequences. Structure of DQ2.5-glia-α1a is based on PDB ID 1S9V while the structure of DQ2.5-glia-ω1 is modeled. (D) Overlay of the top three docking models of the Fvs onto HLA-DQ2.5:DQ2.5-gliaα1a26. Peptide residues mutated for fine-specificity analysis and the HLA-DQ2.5/DQ2.2 polymorphism at Sα72 is illustrated. (E) Fv residues from the three models that are within 5 Å of p7, p9 and Sα72 are shown. (D-E) Coloring as follows: VH and VL, black; MHCα, grey; MHCβ, light orange; DQ2.5-glia-α1a, green; CDR-H1 and CDR-H2, deep purple; CDR-H3, red; CDR-L1 and CDR-L2, deep teal; CDR-L3, blue; Sα72, pink; p7 and p9, pale green; H-bonds, yellow dashes.
To further map fine-specificity, we screened for binding against a panel of HLA-DQ2.5:peptide or HLA-DQ2.2:peptide expressing A20 B cells (Supplementary Fig. 2B). Covalent tethering of the peptides to MHC largely eliminates effects of differences in peptide off-rates, enabling comparative assessment of binding. None of the mAbs bound the HLA-DQ2.5:DQ2.5glia-ω1 or the HLA-DQ2.2:DQ2.5-glia-α1a complexes (Fig. 2B, Supplementary Fig. 2C-E). As DQ2.5-glia-α1a and DQ2.5-glia-ω1 differ at the p7 and p9 positions (Fig. 2C), we constructed pL7Q and pY9F variants of DQ2.5-glia-α1a to resemble DQ2.5-glia-ω1 in these positions. While both mAbs bound the pL7Q variant, albeit not as strongly as DQ2.5-glia-α1a, neither bound the pY9F variant (Fig. 2B). Of the polymorphic residues that differ between HLA-DQ2.5 and HLADQ2.2, the α72 residue (S in HLA-DQ2.5 and I in HLA-DQ2.2) is the only residue located so that it could potentially make direct interactions with the mAbs (Fig. 2D). As expected, neither of the mAbs bound the Sα72I variant (Fig. 2B). As seen with soluble HLA molecules, we did not observe binding to DQ2.5-glia-α2 (Fig. 2B). Additionally, the native, non-deamidated DQ2.5-glia-α1a (DQ2.5-glia-α1a-Q) was not recognized.
In an effort to visualize how the mAbs bind pMHC, we built Fv homology models using the V region sequence of mAb 107 and docked the models to the crystal structure of HLA-DQ2.5:DQ2.5-glia-α1a26. The three selected docking models were highly similar and positioned the Fv in a TCR-like diagonal manner across the pMHC (Fig. 2D). In all three models, the CDR-H3 was positioned close to p7, and although no direct interactions were identified, the pL7Q substitution could indirectly be sensed, possibly causing the small reduction in MFI observed (Fig. 2B,E). Similarly, both CDR-L1 and CDR-L3 are in close proximity to p9 (Fig. 2E). In the model, three residues are close enough to interact with the p9Y, and one of these residues potentially forms a H-bond with Sα72 of the MHC, thus, giving a molecular explanation to the lost binding (Fig. 2E). In summary, fine-specificity analysis using single mutant pMHC variants revealed that the mAbs discriminate between highly similar pMHC complexes.
Detection of cell-surface HLA-DQ2.5:DQ2.5-glia-α1a complexes
As all efforts to characterize specificity and affinity of the antibodies were conducted using recombinant HLA-DQ2.5 with peptide covalently tethered with a linker to the N-terminus of DQβ, either soluble or cell-bound, we next examined if the mAbs could bind HLA-DQ2.5+ cells loaded with soluble peptide. For this purpose, we isolated monocytes using PBMCs from a healthy HLA-DQ2.5+ donor and in vitro differentiated to monocyte-derived DCs and loaded the cells with peptide. Using mAb 106, we specifically detected cells presenting DQ2.5-glia-α1a (Supplementary Fig. 3A).
B cells and CD19+ plasma cells are the most abundant DQ2.5-glia-α1a presenting cell subsets in the intestinal mucosa of celiac disease patients
Encouraged by the ability of mAb 106 to specifically stain cells exogenously loaded with DQ2.5glia-α1a peptide, we generated single-cell suspensions of intestinal biopsies from HLA-DQ2.5+ untreated celiac patients, and co-stained the freshly isolated cells with mouse IgG2b (mIgG2b) versions of mAb 106 together with antibodies specific for other APC surface markers (Fig. 3A and Supplementary Fig. 3B-D). Unexpectedly, we observed binding of mAb 106 almost exclusively to plasma cells (large, viable, CD19+/−CD27+CD38+) and B cells (smaller, viable, CD19+CD38-), whereas very few CD11c+CD14-DCs and CD11c+CD14+ or CD11c-CD14+ Mfs stained positive (Fig. 3A). Analysis of three patients in parallel showed an average of 27.4% and 35.4% mAb 106 positive plasma cells and B cells, respectively. Importantly, pre-blocking of FcγRs did not affect staining (Supplementary Fig. 3D). We also assessed the abundance of these cells in the LP of celiac patients. Cells of the B cell compartment dominated in numbers, while the DCs and the Mf subpopulations were found in low numbers (Fig. 3B). Notably, plasma cells greatly outnumber the B cells31.
Figure 3.

Specific detection of gluten peptide presentation in context of HLA-DQ2.5 and the abundance of the cell populations in the LP. (A) Single-cell suspensions of intestinal biopsies obtained from patients undergoing gastroduodenoscopy were stained with a panel of antibodies to phenotypically characterize DQ2.5-glia-α1a presenting cells along with mIgG2b 106. Bound mAb 106 was detected using a FITC-conjugated secondary anti-mouse IgG2b Ab. Samples from three HLA-DQ2.5+ untreated celiac disease (UCD) patients with Marsh 3B/C were run in parallel and a representative example is shown. The mean percent of mAb 106 positive cells from the three patients compared to no primary antibody is shown ± SD. (B) The abundance of the cell populations identified in panel A was determined relative to percentage of live cells in samples from HLA-DQ2.5+ UCD patients (Marsh 3A/B/C, n=6). Each data point represents an individual subject. Error bars illustrate mean ± SD. (A,B) PCs, plasma cells.
We then compared the level of peptide presentation by B cells and plasma cells as detected by mAb 106 or 107 staining of both untreated and treated celiac patients (i.e. on gluten-free diet) and matched with non-celiac healthy controls. Notably, these mAbs stained cells similarly (data not shown). Small intestinal plasma cells can be separated into 3 major subsets with distinct longevity based on CD19 and CD45 expression; CD19+CD45+ plasma cells which are dynamically exchanged, and CD19-CD45+ plasma cells and CD19-CD45-plasma cells which are long-lived subsets and exhibit little and no replacement, respectively (we refer to these subsets as CD19+, CD45+, and CD45-plasma cells, respectively; Fig. 4A and Supplementary Fig. 4A)22. Of these plasma cell subsets, we found the CD19+ plasma cells to display the highest level of HLA-DQ2.5:DQ2.5-glia-α1a complexes, followed by the CD45+ and the CD45-plasma cells, with an average of 19%, 11% and 7% positive cells among HLA-DQ2.5+ untreated celiac patients, respectively (Fig. 4B). An average of 16% of the B cells were positive (Fig. 4B). Further analysis of patients revealed that all plasma cell subsets and the B cells of treated celiac patients stained negative, comparable to both HLA-DQ2.5+ and HLA-DQ2.5-healthy controls (Fig. 4C,D and Supplementary Fig. 4B). Additionally, both HLA-DQ8+ and HLA-DQ2.2+ celiac patients with active disease were negative. Possibly, there was a higher number of positive plasma cells from patients with high Marsh scores (Fig. 4E). In summary, we found plasma cells and B cells to be the main cell types presenting DQ2.5-glia-α1a on HLA-DQ2.5, with the highest level of staining in the CD19+ plasma cell subset.
Figure 4.

Plasma cells and B cells of gut biopsies present the DQ2.5-glia-α1a peptide. Detection of DQ2.5-glia-α1a presentation among plasma cells and B cells in single-cells suspension prepared from intestinal biopsies from either untreated celiac disease (UCD) or treated celiac disease (TCD) patients or healthy controls. mIgG2b mAb 106 or 107 were used for detection and percent positive cells was determined relative to use of secondary antibody alone. (A) Representative flow cytometric gating strategy to identify plasma cells and B cells from singlecell suspensions. (B) Percentage of specific HLA-DQ2.5:DQ2.5-glia-α1a detection among plasma cell subsets and B cells in HLA-DQ2.5+ UCD patients (n=18) compared to controls (grouped healthy and non-HLA-DQ2.5+ celiac patients, n=15). Two-tailed P-values are shown (unpaired ttest). (C) Stratification of the control patients among the CD19+ plasma cells from panel B. Ctrl HLA-DQ2.5+ (n=5), Ctrl HLA-DQ2.5-(n=5), UCD HLA-DQ2.5+ (n=18), TCD HLA-DQ2.5+ (n=3), UCD HLA-DQ8+ (n=1), and UCD HLA-DQ2.2+ (n=1). (D) Representative histograms showing pMHCspecific staining within patient groups as indicated. (E) The HLA-DQ2.5+ UCD patients (n=18) were stratified according to Marsh score as indicated. (B-E) Non-celiac ctrl patients did not have mucosal alterations. Each data point represents an individual subject. Red bars indicate mean percentage. PCs, plasma cells.
DQ2.5-glia-α1a presenting plasma cells express IgA BCR specific for TG2 and gliadin
Ab-secreting cells producing IgA antibodies towards TG2 and gliadin are a hallmark of celiac disease12,13. We first determined the BCR isotype of the plasma cells and found the vast majority to express IgA BCR and the rest to be mainly of IgM isotype (Fig. 5A and Supplementary Fig. 5A), in line with previous descriptions of the intestinal plasma cell compartment31. There was no difference between the pMHC+ and pMHC-plasma cell populations. We next assessed whether TG2-specific plasma cells were among the cells detected by the pMHC-specific mAb. From three untreated celiac patients, we sorted four populations of plasma cells; bulk plasma cells, bulk TG2+ plasma cells, pMHC+TG2+ plasma cells and pMHC+TG2-plasma cells by use of mAb 107 and TG2-antigen multimers. The cells of all four populations were microscopically plasma cells, being large lymphocytes with eccentric nuclei (Fig. 5B). Further, TG2-specific ELISPOT using the sorted plasma cells verified that the cells secrete IgA anti-TG2 antibodies (Supplementary Fig. 5B). To perform a further assessment of antigen reactivity of the presenting plasma cells, we then sorted pMHC+ and pMHC-plasma cell populations and performed TG2 and gliadin ELISPOT (Fig. 5C and Supplementary Fig. 5C), which showed that both groups contained plasma cells secreting TG2-specific antibodies, whereas plasma cells secreting gliadin-specific antibodies could only be detected in the pMHC+ group. Among bulk plasma cells, spots were visible only at high cell numbers (Supplementary Fig. 5C). This is in line with the observation that approximately 10% of the plasma cells in the inflamed mucosa of celiac patients are TG2-specific13. We also evaluated if the pMHC-specific staining level among the CD19+ plasma cells identified in Fig. 4 would reflect the serum IgA anti-TG2 titer of the patients. We did not find any correlation (Fig. 5D), in line with a previously reported lack of correlation between the frequencies of TG2-specific plasma cells and serum Ab titers in the intestinal mucosa32.
Figure 5.

DQ2.5-glia-α1a presenting plasma cells express TG2-and gliadin-specific IgA. (A) Expression profile of surface IgA and IgM BCR on CD38+ plasma cells staining as pMHC+ or pMHC-(HLA-DQ2.5+ UCD patients with Marsh 3B/C, n=3). Error bars illustrate mean ± SD. (B) Representative micrographs of FACS-sorted plasma cell subsets as indicated (HLA-DQ2.5+ UCD patients with positive serum anti-TG2 IgA titers and with Marsh 3B/C, n=3). Two individual cells within each group are shown. (C) TG2 and gliadin ELISPOT on FACS sorted pMHC+ or pMHC-CD38+ plasma cell subsets as indicated (HLA-DQ2.5+ UCD patients with Marsh 3B/C, n=2). TG2-and gliadin-specific IgA antibodies were detected using AP-conjugated anti-IgA Ab. (D) Percentage of HLA-DQ2.5:DQ2.5-glia-α1a detection within CD19+ plasma cells does not correlate with serum anti-TG2 IgA titer (n=18). Each data point represents an individual subject and the Marsh score is indicated. The dotted line indicates upper and lower thresholds for antiTG2 IgA detection (7 and 120, respectively). The line is derived from linear fitting; R2 and twotailed P-values of IgA anti-TG2 antibody value (Pearson correlation); ns=not significant. (B,C) PCs, plasma cells.
Gut plasma cells express MHCII and co-stimulatory molecules
The specific detection of gluten peptide presentation on HLA-DQ2.5 implies MHCII expression on human intestinal plasma cells. It is unclear to which extent human gut plasma cells have the capacity to express MHCII due to Blimp1-mediated silencing22,33,34. Thus, to verify class II expression, we performed flow cytometric staining using single-cell suspensions from celiac patients, showing that the CD19+, CD45+ and CD45-plasma cells indeed express MHCII, albeit at a substantially lower level than DCs and macrophages or B cells (Fig. 6A,B and Supplementary Fig. 6A,B). We detected MHCII expression on approximately 20–30% of the gut plasma cells, and this was not restricted to the TG2-specific plasma cells (Fig. 6C,D). Along with the observed stronger pMHC-specific staining within the CD19+ plasma cells (Fig. 4B-D), this subset also showed the highest frequency of TG2-specific plasma cells. Additionally, this subset was the only one where we detected double positive MHCII and TG2-specific plasma cells (Fig. 6D).
Figure 6.

Gut plasma cells express MHCII and costimulatory molecules. (A-D) Single-cell suspensions from five HLA-DQ2.5+ UCD patients all with positive serum anti-TG2 IgA antibody levels and with Marsh score 3A/B/C were assessed for MHCII expression and TG2 specificity (n=5). (A-E) PCs, plasma cells. (A) Percentage MHCII expression among various cell subsets as indicated compared to isotype control mAb. (B) Representative histograms showing MHCII-specific staining within cell subsets as indicated. The bulk CD38+ plasma cell population was used to represent the isotype staining for all three plasma cell subsets. (C) Representative overview of MHC and anti-TG2 BCR staining among the plasma cell subsets. Percent positive cells within quadrant are indicated. (D) Distributions of cells stained as single MHCII+, single TG2+ or double MHCII+TG2+ within the plasma cells subsets as seen in (C). Background staining (Bg) was determined based on isotype control mAb for the MHCII staining and without TG2 multimer for the anti-TG2 BCR staining. (E) Percentage MHCII expression on plasma cell subsets from non-inflamed intestinal samples (n=6). Cells were obtained either from patients undergoing Whipple procedure (Whipple, n=4) or a treated celiac patient (TCD, n=1) or a potential celiac patient (potential celiac, n=1, non-inflamed duodenum, but symptoms of celiac disease). All subjects were evaluated to have normal histology of the duodenum. (F) Representative staining for costimulatory markers CD40, CD80 and CD86 on plasma cells from UCD or TCD patients (n=4, 3 HLA-DQ2.5+ UCD patients with Marsh 3A/B and 1 TCD with Marsh 0). Red bars indicate mean percentage. Error bars indicate mean ± SD. Each data point represents an individual subject; two-tailed P-values from unpaired t-test.
To investigate if MHCII expression is a general feature of gut plasma cells, and not restricted to the inflamed mucosa of celiac patients, we evaluated the MHCII expression on plasma cells from non-inflamed intestinal mucosa. We either used biopsies from a treated and a potential (Marsh 0 of the duodenum, but symptoms of celiac disease) celiac disease patient or small intestinal resections from patients undergoing Whipple procedure (pancreatoduodenectomy) due to pancreatic cancer. Indeed, we detected MHCII expression to a similar extent as in the celiac disease patient samples (Fig. 6E and Supplementary Fig. 6B). Finally, we assessed gut biopsies for the presence of costimulatory molecules important for T cell interaction. In line with previous observations35, we found that the DCs and Mfs expressed CD40, CD80 and CD86 only in small subpopulations (Supplementary Fig. 7A), and at lower levels than plasma cells. Plasma cells of both untreated and treated celiacs expressed high levels of CD40 (Figure 6F). Interestingly, there appeared to be a difference in the CD80 and CD86 profile between celiacs and subjects with non-inflamed tissue. Whereas plasma cells from non-inflamed tissue showed either no or a clear dichotomous CD80 expression pattern, but no CD86 expression, celiacs expressed no apparent CD80, and medium to low levels of CD86 (Fig. 6F and Supplementary Fig. 7B,C). Taken together, these results show that plasma cells of the gut express MHCII and costimulatory molecules, which suggest that plasma cells may serve as important APCs in the intestinal mucosa.
Discussion
In this study, we report the generation of mAbs highly specific for HLA-DQ2.5 in complex with one of the immunodominant T-cell epitopes in celiac disease, DQ2.5-glia-α1a. By utilizing these mAbs, we identified B cells and plasma cells as the most abundant cells presenting gluten peptides in the inflamed intestine of celiac patients.
This finding was unexpected as the intestinal APC compartment is dominated by macrophages, with smaller populations of DCs, naïve and memory B cells8,9,36. Previous experiments with in vitro gluten peptide-loaded cells isolated from biopsies pointed to a CD11c+ population as particularly efficient APCs8. However, no activation was detected without in vitro peptide loading, which is in line with our data where we did not detect specific pMHC staining of this cell population, despite high MHCII expression. Furthermore, although the plasma cells were clearly positive for MHCII, the expression levels are much lower on plasma cells than on DCs, which may explain why they have been missed in previous studies. Although the affinities of mAbs 106 and 107 are in line with those reported for other TCR-like mAbs isolated from naïve libraries37,38, it might be too low to detect rare cells. Thus, our lack of detection of macrophages and DC as specific APCs, might in part be explained by low cell density in the intestine8,9, and possibly by their migration to the mesenteric lymph node after antigen uptake39. In vivo loaded DCs might well be migratory9 and thus not present high levels of peptide in the duodenal material assessed.
B cells and plasma cells expressing Ig specific for gliadin and TG2 are a characteristic of celiac disease12,13. The dominant B-cell lineage in the LP is plasma cells, which are found in high numbers, constituting 25–35% of the total mononuclear cell population, whereas there is only a minor population of memory B cells and very few naïve B cells36. In celiac disease, approximately 1% and 10% of the plasma cells are specific for gliadin or TG2, respectively12,13. The role of these cells and the antibodies they produce in disease development have so far not been appreciated, as the plasma cells have not been considered APCs and a pathogenic role for their secreted antibodies has yet to be shown. Our findings strongly point to plasma cell involvement as APCs for gluten-reactive CD4+ T cells. Comparing the 3 major subsets of small intestinal plasma cells, we found the CD19+ subset to present DQ2.5-glia-α1a most efficiently. This subset is highly dynamic and undergoes constant renewal, whereas the CD45+ and CD45-plasma cells are long-lived, in particular the CD45-plasma cells, where we detected little peptide presentation22. The finding that the TG2-specific plasma cells are predominantly CD19+, and thus short-lived, parallels the observation that serum anti-TG2 IgA titers drops upon gluten-free diet40.
Despite the fact that intestinal IgA+ plasma cells appear to have a functional BCR, and thus Ag capture ability, they are thought to lack APC properties due to Blimp1-mediated transcriptional silencing of the MHCII loci33,41. However, we show that intestinal plasma cells express MHCII, as well as the costimulatory CD40, CD80 and CD86 molecules, which together may give them ability to activate T cells. The presence of IgA+ DQ2.5-glia-α1a presenting cells among TG2-specific plasma cells strengthens the so-called hapten-carrier hypothesis as a mechanism to explain how TG2 specific B cells get help from gluten reactive T cells42,43. The phenotype of the CD19+CD38-B cells we identified has been thoroughly investigated22,36. This population was found to constitute mostly memory B cells (CD27+IgD-IgA+) with a minor population of naïvemature B cells (CD27-IgD+IgM+) most likely representing a variable contribution from isolated lymphoid follicles and blood.
The ability of plasma cells to act as APCs is controversial. Whereas murine plasma cells have been shown to process antigen and activate T cells44, this has proven much more difficult to verify for human plasma cells. However, human bone marrow and splenic plasma cells have been shown to express MHCII45. To firmly establish that human gut plasma cells activate gluten-specific T cells, we will pursue functional testing in T-cell assays.
We have only assessed the specific pMHC expression on plasma cells and B cells at severe inflammation, and thus cannot exclude that DCs and the B-cell compartment serve different functions as potential APCs at different physiological sites, as well as at different stages of gut inflammation. Down these lines, it is interesting that the CD80 versus CD86 expression levels appears to differ on plasma cells at homeostasis and during inflammation (Fig. 6F vs Supplementary Fig. 7B).
In summary, we describe the isolation and characterization of two mAbs with specificity for one of the immunodominant epitopes in celiac disease, namely HLA-DQ2.5 in complex with DQ2.5-glia-α1a. This particular epitope is found in the convergent 33mer gluten derived peptide to which the majority of all HLA-DQ2.5+ celiac patients respond3,30,46. Using the mAbs to detect the presence of the target pMHC complex on cells isolated from intestinal lesions of celiac disease patients, they bound to plasma cells and B cells, and not to macrophages and DCs. Although life-long gluten-free diet is a safe treatment for many celiac patients, there is also an unmet medical need for novel treatments. Our data identify plasma cells of the gut as the most prevalent gluten peptide presenting cell in the gut mucosa of celiac patients, which indicates that gut plasma cells may well function as an APC, and thus constituting a potential therapeutic target in celiac disease.
A potential role of plasma cells as APCs and the establishment of the importance of B cells in presentation of gluten peptide may also offer instructive clues for our understanding other T-cell driven autoimmune diseases. Lastly, we found the ability of the plasma cells to express MHCII was not restricted to the inflamed mucosa of celiac patients, which has important implications for our understanding of mucosal adaptive immunity.
Supplementary Material
Acknowledgments
Grant support: This work received funding from the South-Eastern Norway Regional Health Authority (grant 2012046) and by the Research Council of Norway through its Centers of Excellence funding scheme, project number 179573/V40. JRJ and JJG are supported by the U.S. National Institutes of Health grant R01-GM078221. Part of the computations were performed on resources provided by UNINETT Sigma2 -the National Infrastructure for High Performance Computing and Data Storage in Norway. We would like to thank Sivaganesh Sathiaruby, Linn Margrethe Eggesbø and Anna Bujko for excellent technical assistance, Espen Bækkevold for technical advice, Kjetil Taskén (Biotechnology Center of Oslo) for access to the Biacore T100 instrument, The Flow Cytometry Core Facility (FCCF) at Oslo University Hospital for access to equipment and assistance with sorting and the Department of Immunology at Oslo University Hospital-Rikshospitalet for HLA typing.
Abbreviations:
- APC
antigen presenting cell
- BCR
B-cell receptor
- DC
dendritic cell
- LP
lamina propria
- mAb
monoclonal antibody
- Mf
macrophage
- MHCII
MHC class II
- pMHC
peptide-MHC
- TG2
transglutaminase 2
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflict of interest: LSH, RF, IS, LMS and GÅL are holders of a patent application on the mAbs against gluten-pMHC complexes. The remaining of the authors discloses no conflicts of interest.
Author names in bold designate shared co-first authorship.
References
- 1.Sollid LM, Jabri B. Triggers and drivers of autoimmunity: lessons from coeliac disease. Nat Rev Immunol 2013;13:294–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Abadie V, Sollid LM, Barreiro LB, et al. Integration of genetic and immunological insights into a model of celiac disease pathogenesis. Annu Rev Immunol 2011;29:493–525. [DOI] [PubMed] [Google Scholar]
- 3.Tye-Din JA, Stewart JA, Dromey JA, Beissbarth T, et al. Comprehensive, quantitative mapping of T cell epitopes in gluten in celiac disease. Sci Transl Med 2010;2:41ra51. [DOI] [PubMed] [Google Scholar]
- 4.Arentz-Hansen H, Korner R, Molberg O, et al. The intestinal T cell response to alphagliadin in adult celiac disease is focused on a single deamidated glutamine targeted by tissue transglutaminase. J Exp Med 2000;191:603–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Xia J, Sollid LM, Khosla C. Equilibrium and kinetic analysis of the unusual binding behavior of a highly immunogenic gluten peptide to HLA-DQ2. Biochemistry 2005;44:4442–9. [DOI] [PubMed] [Google Scholar]
- 6.Fallang LE, Bergseng E, Hotta K, et al. Differences in the risk of celiac disease associated with HLA-DQ2.5 or HLA-DQ2.2 are related to sustained gluten antigen presentation. Nat Immunol 2009;10:1096–101. [DOI] [PubMed] [Google Scholar]
- 7.Mowat AM, Agace WW. Regional specialization within the intestinal immune system. Nat Rev Immunol 2014;14:667–85. [DOI] [PubMed] [Google Scholar]
- 8.Raki M, Tollefsen S, Molberg Ø, et al. A unique dendritic cell subset accumulates in the celiac lesion and efficiently activates gluten-reactive T cells. Gastroenterology 2006;131:428–38. [DOI] [PubMed] [Google Scholar]
- 9.Beitnes AC, Raki M, Lundin KE, et al. Density of CD163+ CD11c+ dendritic cells increases and CD103+ dendritic cells decreases in the coeliac lesion. Scand J Immunol 2011;74:186–94. [DOI] [PubMed] [Google Scholar]
- 10.Bujko A, Atlasy N, Landsverk OJB, et al. Transcriptional and functional profiling defines human small intestinal macrophage subsets. J Exp Med 2018;215:441–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Beitnes AC, Raki M, Brottveit M, et al. Rapid accumulation of CD14+CD11c+ dendritic cells in gut mucosa of celiac disease after in vivo gluten challenge. PLoS One 2012;7:e33556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Steinsbø Ø, Henry Dunand CJ, Huang M, et al. Restricted VH/VL usage and limited mutations in gluten-specific IgA of coeliac disease lesion plasma cells. Nat Commun 2014;5:4041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Di Niro R, Mesin L, Zheng NY, et al. High abundance of plasma cells secreting transglutaminase 2-specific IgA autoantibodies with limited somatic hypermutation in celiac disease intestinal lesions. Nat Med 2012;18:441–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chen D, Ireland SJ, Davis LS, et al. Autoreactive CD19+CD20-Plasma Cells Contribute to Disease Severity of Experimental Autoimmune Encephalomyelitis. J Immunol 2016;196:1541–9. [DOI] [PubMed] [Google Scholar]
- 15.Li R, Patterson KR, Bar-Or A. Reassessing B cell contributions in multiple sclerosis. Nat Immunol 2018. [DOI] [PubMed]
- 16.Ludvigsson JF, Bai JC, Biagi F, et al. Diagnosis and management of adult coeliac disease: guidelines from the British Society of Gastroenterology. Gut 2014;63:1210–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Løset GA, Lobersli I, Kavlie A, et al. Construction, evaluation and refinement of a large human antibody phage library based on the IgD and IgM variable gene repertoire. J Immunol Methods 2005;299:47–62. [DOI] [PubMed] [Google Scholar]
- 18.Høydahl LS, Nilssen NR, Gunnarsen KS, et al. Multivalent pIX phage display selects for distinct and improved antibody properties. Sci Rep 2016;6:39066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gunnarsen KS, Høydahl LS, Risnes LF, et al. A TCRalpha framework-centered codon shapes a biased T cell repertoire through direct MHC and CDR3beta interactions. JCI Insight 2017;2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Berntzen G, Lunde E, Flobakk M, et al. Prolonged and increased expression of soluble Fc receptors, IgG and a TCR-Ig fusion protein by transiently transfected adherent 293E cells. J Immunol Methods 2005;298:93–104. [DOI] [PubMed] [Google Scholar]
- 21.Qiao SW, Bergseng E, Molberg Ø, et al. Antigen presentation to celiac lesion-derived T cells of a 33-mer gliadin peptide naturally formed by gastrointestinal digestion. J Immunol 2004;173:1757–62. [DOI] [PubMed] [Google Scholar]
- 22.Landsverk OJ, Snir O, Casado RB, et al. Antibody-secreting plasma cells persist for decades in human intestine. J Exp Med 2017;214:309–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Weitzner BD, Jeliazkov JR, Lyskov S, et al. Modeling and docking of antibody structures with Rosetta. Nat Protoc 2017;12:401–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marze NA, Lyskov S, Gray JJ. Improved prediction of antibody VL-VH orientation. Protein Eng Des Sel 2016;29:409–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weitzner BD, Gray JJ. Accurate Structure Prediction of CDR H3 Loops Enabled by a Novel Structure-Based C-Terminal Constraint. J Immunol 2017;198:505–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim CY, Quarsten H, Bergseng E, et al. Structural basis for HLA-DQ2-mediated presentation of gluten epitopes in celiac disease. Proc Natl Acad Sci U S A 2004;101:4175–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Conway P, Tyka MD, DiMaio F, et al. Relaxation of backbone bond geometry improves protein energy landscape modeling. Protein Sci 2014;23:47–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Petersen J, Montserrat V, Mujico JR, et al. T-cell receptor recognition of HLA-DQ2-gliadin complexes associated with celiac disease. Nat Struct Mol Biol 2014;21:480–8. [DOI] [PubMed] [Google Scholar]
- 29.Sircar A, Gray JJ. SnugDock: paratope structural optimization during antibody-antigen docking compensates for errors in antibody homology models. PLoS Comput Biol 2010;6:e1000644. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Shan L, Molberg Ø, Parrot I, et al. Structural basis for gluten intolerance in celiac sprue. Science 2002;297:2275–9. [DOI] [PubMed] [Google Scholar]
- 31.Brandtzaeg P, Johansen FE. Mucosal B cells: phenotypic characteristics, transcriptional regulation, and homing properties. Immunol Rev 2005;206:32–63. [DOI] [PubMed] [Google Scholar]
- 32.Di Niro R, Snir O, Kaukinen K, et al. Responsive population dynamics and wide seeding into the duodenal lamina propria of transglutaminase-2-specific plasma cells in celiac disease. Mucosal Immunol 2016;9:254–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pinto D, Montani E, Bolli M, et al. A functional BCR in human IgA and IgM plasma cells. Blood 2013;121:4110–4. [DOI] [PubMed] [Google Scholar]
- 34.Piskurich JF, Lin KI, Lin Y, et al. BLIMP-I mediates extinction of major histocompatibility class II transactivator expression in plasma cells. Nat Immunol 2000;1:526–32. [DOI] [PubMed] [Google Scholar]
- 35.Chirdo FG, Millington OR, Beacock-Sharp H, et al. Immunomodulatory dendritic cells in intestinal lamina propria. Eur J Immunol 2005;35:1831–40. [DOI] [PubMed] [Google Scholar]
- 36.Farstad IN, Carlsen H, Morton HC, et al. Immunoglobulin A cell distribution in the human small intestine: phenotypic and functional characteristics. Immunology 2000;101:354–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chames P, Hufton SE, Coulie PG, et al. Direct selection of a human antibody fragment directed against the tumor T-cell epitope HLA-A1-MAGE-A1 from a nonimmunized phage-Fab library. Proc Natl Acad Sci U S A 2000;97:7969–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Dahan R, Gebe JA, Preisinger A, et al. Antigen-specific immunomodulation for type 1 diabetes by novel recombinant antibodies directed against diabetes-associates autoreactive T cell epitope. J Autoimmun 2013;47:83–93. [DOI] [PubMed] [Google Scholar]
- 39.Du Pre MF, Kozijn AE, van Berkel LA, et al. Tolerance to ingested deamidated gliadin in mice is maintained by splenic, type 1 regulatory T cells. Gastroenterology 2011;141:61020, 620 e1–2. [DOI] [PubMed] [Google Scholar]
- 40.Sulkanen S, Halttunen T, Laurila K, et al. Tissue transglutaminase autoantibody enzymelinked immunosorbent assay in detecting celiac disease. Gastroenterology 1998;115:1322–8. [DOI] [PubMed] [Google Scholar]
- 41.Tellier J, Shi W, Minnich M, et al. Blimp-1 controls plasma cell function through the regulation of immunoglobulin secretion and the unfolded protein response. Nat Immunol 2016;17:323–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sollid LM, Molberg Ø, McAdam S, et al. Autoantibodies in coeliac disease: tissue transglutaminase--guilt by association? Gut 1997;41:851–2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Iversen R, Fleur du Pre M, Di Niro R, et al. Igs as Substrates for Transglutaminase 2: Implications for Autoantibody Production in Celiac Disease. J Immunol 2015;195:5159–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Pelletier N, McHeyzer-Williams LJ, Wong KA, et al. Plasma cells negatively regulate the follicular helper T cell program. Nat Immunol 2010;11:1110–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Ellyard JI, Avery DT, Phan TG, et al. Antigen-selected, immunoglobulin-secreting cells persist in human spleen and bone marrow. Blood 2004;103:3805–12. [DOI] [PubMed] [Google Scholar]
- 46.Bodd M, Ráki M, Bergseng E, et al. Direct cloning and tetramer staining to measure the frequency of intestinal gluten-reactive T cells in celiac disease. Eur J Immunol 2013;43:2605–2612. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.