

SUMMARY
The collection of proteins secreted from a cell—the secretome—is of particular interest in cancer pathophysiology due to its diagnostic potential and role in tumorigenesis. However, cancer secretome studies are often limited to one tissue or cancer type or focus on biomarker prediction without exploring the associated functions. We therefore conducted a pan-cancer analysis of secretome gene expression changes to identify candidate diagnostic biomarkers and to investigate the underlying biological function of these changes. Using transcriptomic data spanning 32 cancer types and 30 healthy tissues, we quantified the relative diagnostic potential of secretome proteins for each cancer. Furthermore, we offer a potential mechanism by which cancer cells relieve secretory pathway stress by decreasing the expression of tissue-specific genes, thereby facilitating the secretion of proteins promoting invasion and proliferation. These results provide a more systematic understanding of the cancer secretome, facilitating its use in diagnostics and its targeting for therapeutic development.
In Brief
Robinson et al. compare secreted protein expression changes across different cancer types and healthy tissues to identify candidate biomarkers likely to be detectable in biological fluids. Functional analyses reveal a pattern whereby cancers decrease the expression of secreted proteins responsible for tissue of origin function in favor of those supporting proliferation and invasion.
Graphical Abstract
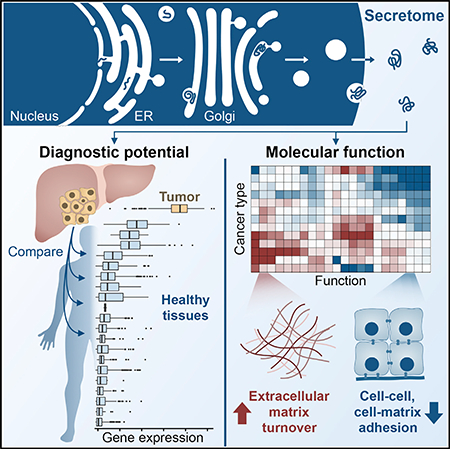
INTRODUCTION
Early diagnosis is a major factor contributing to cancer treatment success (Etzioni et al., 2003; World Health Organization, 2017). As such, there have been extensive efforts to identify with improved accuracy and sensitivity biomarkers that indicate the presence of cancerous cells in a subject (Belczacka et al., 2019; Sawyers, 2008). Recent work has focused on the analysis of markers in biofluids, such as urine, plasma, or cerebrospinal fluid, as they are non-invasive and can be tested with greater frequency than tissue biopsies (Crowley et al., 2013; Diaz and Bardelli, 2014; Webb, 2016). A class of proteins that are of particular interest in this context is the secretome, which is the set of proteins secreted to the extracellular space, as they are generally more abundant in biological fluids than intracellular proteins (Kulasingam and Diamandis, 2008; Stastna and Van Eyk, 2012).
The secretome is considered a valuable reservoir of potential biomarkers for cancer and other diseases (Makridakis and Vlahou, 2010; Xue et al., 2008), and a number of studies have aimed to explore this class of proteins in search of tumor biomarker candidates. For example, Welsh et al., 2003 used Gene Ontology (GO) terms associated with an extracellular location and protein sequence patterns to define the secretome to compare the microarray gene expression profiles of 150 carcinomas spanning 10 tissues of origin to those of 46 healthy tissue samples. Biomarker candidates were validated via comparison with previous studies that had measured increased expression of the gene or protein in cancer tissue or in the serum of cancer patients. Other bioinformatics-based approaches to predict secreted cancer biomarkers include those of Prassas et al. (2012) for colon, lung, pancreatic, and prostate cancers, and Vathipadiekal et al. (2015) for ovarian cancer. These and other, similar investigations demonstrate the validity of using a bioinformatics-based approach to predict proteomic biofluid markers and to identify many new, promising biomarker candidates. However, these studies were generally restricted to a limited number of samples, tissue types, and/or cancer types; were often based on microarray data rather than RNA sequencing (RNA-seq) data; provided only a single set of candidates rather than a complete ranked list; and conducted little or no exploration of the biological functions associated with the proposed biomarkers.
Proteomic approaches have often been used to profile the cancer secretome (Brandi et al., 2018; Geyer et al., 2017; Hanash et al., 2008; Makridakis and Vlahou, 2010; Papaleo et al., 2017; Schaaij-Visser et al., 2013; Xue et al., 2008). These studies generally involve in vitro analyses of cell-line conditioned media or analysis of tumor interstitial fluid (or a more distant fluid such as blood, plasma, urine, or saliva) (Papaleo et al., 2017). For example, Wu et al. (2010) used SDS-PAGE followed by liquid chromatography-tandem mass spectrometry (LC-MS/MS) to analyze the secretome of conditioned media for 23 human cancer cell lines spanning 11 cancer types, which enabled the identification of both cancer-specific and pan-cancer serological biomarker candidates. Four of the candidates were validated experimentally, showing significantly elevated levels in the serum or plasma of liver, lung, or nasopharyngeal carcinoma patients relative to healthy controls. Despite the extensive information gained from these experimental investigations, there still exist a number of challenges that result in high variability and conflicting results among studies. For example, the use of cell lines is not an ideal representation of the in vivo system, culturing conditions can affect cell physiology and protein detection, there is a bias toward high-abundance proteins, protein concentrations span a large dynamic range in plasma, studies differ in sample collection and storage methods, and artifactual proteins are often identified, despite little or no relation to the disease in question (Geyer et al., 2017; Hanash et al., 2008; Kulasingam and Diamandis, 2008; Papaleo et al., 2017).
In the present study, we conducted a systematic analysis of cancer-associated changes in secretome expression to predict candidate biomarkers that could be significantly elevated in the biofluids of individuals with cancer and are therefore more likely to be detectable. We then investigated the patterns and biological functions associated with shifts in secretome expression among different cancer types, focusing on shared “core” secretome behaviors, as well as cancer-specific features. The cancer secretome was explored in the context of tissue-specific genes, revealing a general pattern whereby tumor cells reduce their secretory pathway burden in an effort to relieve endoplasmic reticulum (ER) stress and the associated unfolded protein response (UPR). We expect the resulting ranked lists of biomarkers for each of the 32 different cancer types, in addition to the insight gained from the functional analysis of the cancer secretome and associated modulation of the secretory pathway in cancer cells, to expedite the development of effective diagnostic biomarkers and illuminate potential strategies for improved anti-cancer therapies.
RESULTS
Evaluation of Secretome Biomarker Candidates
To focus on proteins that are intentionally and actively secreted from the cell, we defined the secretome as all proteins possessing an N-terminal signal peptide and annotated as having a subcellular location of “secreted” (UniProt; Bateman et al., 2017). This yielded a set of 1,816 secretome genes for evaluation. In our investigation of cancer-specific secretome changes, we first sought to identify secretome genes whose encoded proteins were most likely to exhibit detectable changes in a biofluid as a result of their altered expression in a tumor. Our analysis pipeline therefore involved the comparison of primary tumor transcriptomes with those of (1) paired-normal tissue, (2) healthy tissue corresponding to the cancer tissue of origin, and (3) all healthy tissues in the human body (Figure 1A). Primary tumor and paired-normal RNA-seq profiles were retrieved for 32 cancer types from The Cancer Genome Atlas (TCGA), whereas healthy tissue profiles were obtained from the Genotype-Tissue Expression (GTEx) database (STAR Methods; Table S1).
Figure 1. Overview of Cancer Secretome Biomarker Consensus Scoring Approach and Results.

(A) Schematic overview of the scoring method. Primary tumor transcriptomic (RNA-seq) profiles were compared to those of (1) paired-normal tissue of the same patients, (2) healthy tissue matching the tumor tissue of origin, and (3) all healthy tissues in the body for which data were available. Genes were ranked according to their relative expression in tumor versus other samples; those with significantly elevated expression in the tumor were ranked highly, and these were combined into a single consensus rank score.
(B) For three representative cancer types, a t-SNE projection illustrates the separation of primary tumor (red), paired-normal tissue (yellow), and healthy tissue of origin (green) samples based on the abundance (log10TPM) of the top 10 consensus-ranked genes for that cancer type. t-SNE plots for the remaining 19 cancer types with at least one of each sample type are presented in Figure S1.
(C) Box and whisker plots showing the expression of CST1 and ANGPTL4 in all healthy tissue samples, as well as in tumor samples of the five cancer types with the highest median expression of the gene. The CST1 and ANGPTL4 genes are representative of multi-cancer and cancer-specific candidate markers, which ranked highly in the third comparison (tumor versus all healthy tissues) in multiple or only one cancer type, respectively.
(D) A heat-scatterplot presenting the results of the three comparisons for the top 10 consensus-ranked secretome biomarker candidates from each cancer type. Only cancer types with sufficient tumor, paired-normal tissue, and healthy tissue of origin samples to conduct all three comparison types are shown. The color of the circles corresponds to the mean log2FC from the first two comparison methods (FC1 and FC2), while circle size is based on the extent to which a gene is expressed at higher levels in the tumor compared to all healthy tissues (quantified as p3). Stars next to gene names indicate those whose encoded proteins are present in the human plasma proteome (Schwenk et al., 2017), in which a filled star represents a canonical (confirmed) protein, an empty star indicates some evidence but the status is uncertain, and proteins with no star are either undetected or not included in the database. Rows and columns were clustered based on Euclidean distance between mean log2FC values.
Generation of a Consensus Score
To integrate information from the three comparisons performed, the results were combined to generate a consensus score for each gene in each cancer type. Top-ranked (high-scoring) genes for each cancer type were those with elevated expression in tumor samples compared to paired-normal tissue, healthy tissue of origin, and all healthy tissues. The complete set of consensus scores for all cancer types, as well as the fold changes (log2FC) and significance values (p values) used to determine the scores, are presented in Table S2.
Transcriptomic data of top-ranked genes were examined to confirm their distinct and elevated expression in tumor versus non-tumor samples. T-distributed stochastic neighbor embedding (t-SNE) was performed on tumor, paired-normal tissue, and healthy tissue transcript per million (TPM) values of the top 10 consensus-ranked genes for each cancer type (Figures 1B and S1). The majority of tumor samples exhibited clear clustering and separation from non-tumor samples, confirming distinct expression profiles between these groups among the highly ranked genes. The t-SNE plots also demonstrate differences between paired-normal tissue and healthy tissue samples, highlighting the importance of including both tumor versus paired-normal tissue and tumor versus healthy tissue comparisons in the consensus rank. Although a difference in data sources (TCGA versus GTEx) could contribute to the observed paired-normal tissue versus healthy tissue separation, a previous analysis of the same two datasets found robust differences even after normalizing for potential batch effects (Aran et al., 2017), thus supporting a biological component.
The elevated expression of top-scoring candidates in tumors compared to all normal tissues is illustrated in Figure 1C for two example genes, cystatin SN (CST1) and angiopoietin-like 4 (ANGPTL4). These genes are representative of two types of biomarker candidates: those with elevated expression in one cancer type (ANGPTL4) and markers with elevated expression in multiple cancer types (CST1). Previous studies have experimentally confirmed significantly elevated protein levels of ANGPTL4 in the serum of patients with renal cell carcinoma (Dong et al., 2017) and of CST1 in the serum and urine of colorectal cancer subjects (Yoneda et al., 2009) relative to non-cancer controls.
Top-Scoring Biomarker Candidates
The results for the top-ranked genes across the 20 cancer types included in all three comparisons are illustrated in Figure 1D. There was a marked clustering of high ranks among many cancers for the collagen (COL) and matrix metalloproteinase (MMP) genes. Many members of the MMP family have been detected at significantly elevated levels in the plasma, serum, and/or urine of patients with cancers such as bladder (Eissa et al., 2007), esophageal (Mroczko et al., 2008), colorectal (Dragutinović et al., 2011), prostate (Roy et al., 2008), lung (Izbicka et al., 2012), breast (Patel et al., 2011), and renal (Sarkissian et al., 2008), compared to non-cancer controls. Collagens have similarly been validated as tumor biomarkers. Previous studies have, for example, measured a significant increased abundance of type IV collagens in the plasma of pancreatic cancer subjects (Ohlund et al., 2009), COL10A1 in the serum of colorectal cancer subjects (Solé et al., 2014), COL6A3 in the urine of bladder cancer subjects (Lindén et al., 2012), or degradation products of types I, III, and IV collagens in the serum of ovarian and breast cancer patients (Bager et al., 2015) relative to controls.
Many of the top-scoring, cancer-specific markers have also been experimentally validated as significantly elevated in a biofluid of subjects harboring that particular type of cancer, some of which are currently used in the clinic for diagnosis. For example, four of the top five scoring candidates for liver hepatocellular carcinoma (LIHC) have been experimentally validated as biofluid LIHC biomarkers (ESM1, AFP, GPC3, and MDK); AFP is the most commonly used serological LIHC marker in the clinic (Capurro et al., 2003; Lou et al., 2017; Spangenberg et al., 2006; Yang et al., 2017; Zhu et al., 2013). Likewise, two (ANGPT2 [Gayed et al., 2015] and ANGPTL4 [Dong et al., 2017]) of the top five candidates for kidney renal clear cell carcinoma (KIRC) and the top candidate (LAMC2 [Kosanam et al., 2013]) for pancreatic ductal adenocarcinoma (PAAD) have been measured at significantly higher concentrations in the plasma, serum, or urine of subjects harboring the respective cancer types compared to non-cancer controls.
Extension to an Experimentally Defined Secretome
The biomarker analysis described here included only classically secreted proteins that contain a signal peptide, but many proteins are secreted through unconventional routes and possess similar diagnostic potential (Rabouille, 2017). However, defining a list of unconventionally secreted proteins is non-trivial due to the many secretion routes available (e.g., exosomes, pore-mediated translocation, ATP-driven transport [Rabouille, 2017]), as well as the variation in their protein cargo across different cell types or conditions (Vlassov et al., 2012). We therefore used the Human Cancer Secretome Database (HCSD) (Feizi et al., 2015) to generate a list of all of the proteins (regardless of signal peptide) that had been experimentally detected in the secretome among any of the 35 studies encompassed by the database. This yielded an “experimental secretome” consisting of ~6,500 proteins, ~800 of which were present in our signal peptide-derived secretome. The results and associated consensus ranks for the experimental secretome are presented in Table S3.
Exploration of the “Core” Cancer Secretome Definition of the Core Secretome
Shifts in secretome expression associated with malignant transformation can be used to identify candidate cancer biomarkers; however, based on our global analysis across different cancer types, it is also possible to address the more fundamental question of why cancer cells restructure their secretome profile throughout tumorigenesis. We therefore sought to investigate the biological features underlying the altered secretome expression. Motivated by the large number of multi-cancer candidates in our biomarker analysis, we first explored the core cancer secretome—the subset of the secretome exhibiting strong differential expression across most or all of the cancer types studied. Secretome genes were ranked based on the magnitude and significance of their expression fold changes (tumor versus paired normal) across all cancer types, referred to here as the PF rank (STAR Methods).
Members of the Core Secretome
Upon inspection of the genes populating the top 1% (16 of 1,563 genes) of the pan-cancer PF ranks, two key features were immediately apparent (Figure 2A). First, each gene exhibited an expression change in the same direction across all (or nearly all) of the cancer types, despite ignoring the fold change direction in the rank calculation. This is supportive of an important and defined tumorigenic role for each of the associated encoded proteins, independent of the tissue or cell type from which it originates. Second, 15 of the 16 top-ranked genes exhibited an expression decrease across all or nearly all of the cancer types, suggesting that cancer type-independent shifts in secretome expression tend to be decreases.
Figure 2. Constituents and Functions of the Core Cancer Secretome.

(A) A heat-scatterplot presenting the log2FCs and corresponding significance (false discovery rate [FDR]-adjusted p values) for the 16 genes making up the top 1% of the non-directional core secretome. The color and size of the points correspond to the log2FC and log-transformed p values, respectively, from the DE analysis between tumor and paired-normal samples.
(B) The top 1% of the increased core secretome, obtained in the same manner as the non-directional set in (A), except the fold change direction was incorporated to identify secretome genes exhibiting increased expression across many cancer types.
(C) Gene sets found to be significantly enriched in the decreased (left column), non-directional (center column), or increased core secretome (right column), in which the top 20 most significant sets from each directional class are shown. The intensity of the color in the heatmap indicates the enrichment significance of the gene set. Gene set names are colored according to the Molecular Signatures Database (MSigDB) collection from which they originate: Hallmark, Kyoto Encyclopedia of Genesand Genomes (KEGG), Reactome, GO biological process, and GO molecular function. A non-stacked bar plot to the left of the heatmap shows the sizes (number of genes) of the original gene sets (gray bars) and of the filtered gene sets containing only secretome genes (black bars).
See also Figure S2.
Given the high number of cancer types exhibiting a coordinated expression decrease (or increase) of these core secretome genes, we reasoned that these genes would likely be responsible for important tumor-specific functions. Many of the genes exhibiting decreased expression are putative or established tumor suppressors (e.g., ANGPTL1, C2orf40, CHRDL1, OGN, C7, GREM2) (Hu et al., 2018; Kuo et al., 2013; Li et al., 2015; Pei et al., 2017; Tsubamoto et al., 2016; Ying et al., 2016), are involved in the remodeling of the extracellular matrix (ECM) (e.g., DNASE1L3, CLEC3B, PI16, CCBE1) (Barton et al., 2010; Hawes et al., 2015; Hazell et al., 2016; Obrist et al., 2004), and/or participate in cell-matrix adhesion functions (e.g., MFAP4, DPT, MAMDC2) (Avilés-Vázquez et al., 2017; Pilecki et al., 2016; Yamatoji et al., 2012).
The only top-ranking core secretome gene exhibiting an increased expression was MMP11, which was also one of the MMPs that scored highly as a potential candidate biomarker for many cancer types. In addition to the tumor-specific functions attributed to the MMP family, MMP11 is somewhat unique in that it is secreted in its active form and its ECM substrates differ from those commonly targeted by MMPs (Pei et al., 1994). MMP11 has been reported to enable tumor invasion by inducing de-differentiation of surrounding adipocytes and supporting the accumulation of peritumoral fibroblasts (Andarawewa et al., 2005).
To investigate core secretome genes that exhibited pan-cancer expression increases, the gene-ranking process was repeated, except that the direction of expression fold change was incorporated instead of using the absolute log2FC values. The set of 16 secretome genes with the highest directional PF ranks (top 1%) across the different cancer types exhibited a lower degree of coordination compared to the non-directional set (Figure 2B). Regarding function, the majority of the core increased secretome genes were involved in the structure and composition (e.g., COL1A1, ACAN, ZP3) (Iozzo and Schaefer, 2015; Pickup et al., 2014; Rankin and Dean, 2000) or modification (e.g., metalloprotease MMPs and a disintegrin and metalloproteinase with thrombospondin motifs [ADAM(TS)]) (Egeblad and Werb, 2002) of the ECM. Another function shared by many of the proteins was signaling, either as receptors or effectors. For example, EFNA4, NXPH4, and GPC2 facilitate signaling associated with neuronal and developmental events, which supports essential tumor functions such as angiogenesis, cell adhesion, and motility (Kurosawa et al., 2001; Missler and Südhof, 1998; Wilkinson, 2001). Other proteins with signaling-related functions included CTHRC1 and C1QTNF6, which are involved in vascular remodeling (Park et al., 2013; Takeuchi et al., 2011), and SPP1, which is known to facilitate cell-matrix interactions (Shevde and Samant, 2014). Overall, core secretome shifts contribute to diverse malignant processes, particularly those relating to ECM remodeling, or to a reduction in tumor-suppressive activity.
Enrichment of Functions in the Core Cancer Secretome
Although analysis of the top-ranked core secretome genes offered insight into common functions that were downregulated (or upregulated) across the different cancer types, it excludes information about the remaining 99% of secretome members. We therefore conducted a gene set analysis (GSA) to account for the PF ranks of all of the secretome genes in determining coordinated shifts in secretome function. The GSA was performed using both non-directional and directional PF ranks.
The most significant gene sets associated with the core secretome were related to ECM turnover, cell-matrix adhesion, and signaling processes involving the ECM or immunity and inflammation (Figure 2C). Furthermore, the secretome expression increase associated with the epithelial-mesenchymal transition (EMT) underscores the importance of the cancer secretome in metastatic and invasive processes, regardless of cancer type. Gene sets related to glycosaminoglycan (GAG) binding, specifically heparin, were among the most significant coordinated decreases in secretome expression. As the genes within these sets encode for proteins associated with cell-matrix and basement membrane adhesion, their decreased expression further supports a contribution of the secretome to a more migratory and invasive phenotype.
The Effects of Tumor Purity on Core Secretome Expression Profiles
Tumors are infiltrated to varying degrees by non-cancerous cells, such as stromal or immune cells (Hanahan and Weinberg, 2011). Molecular profiles of bulk tumor samples will therefore contain signatures from these infiltrating cells, which can obscure or be misinterpreted as those originating from tumor cells. To assess whether infiltrating cells were responsible for any of the identified features of the core secretome, we repeated the analyses using only tumor samples with a consensus purity estimate (CPE) (Aran et al., 2015) of at least 80% (Figure S2). The major features remained largely unchanged, supporting their association with the cancerous cells themselves. For example, all 16 genes in the top 1% of the core secretome exhibited a significant expression decrease in most or all of the included cancer types, and 11 of those genes were also present in the top 1% for the original analysis. Functions related to ECM turnover were again enriched among core secretome expression increases, although to a lesser extent when considering only high-purity tumor samples.
Cancer Type-Specific Secretome Expression Profiles
Following the investigation of coordinated pan-cancer secretome shifts, we were interested in evaluating the cancer types individually and determining which processes and functions exhibited strong changes within each type. We therefore conducted a directional and non-directional GSA of the differential expression (DE) analysis results, in which the direction of expression fold changes were included or excluded, respectively (STAR Methods; Varemo et al., 2013).
In the directional GSA (Figure 3A), cancer types generally exhibited expression increases associated with ECM components and metalloprotease activity; however, cholangiocarcinoma (CHOL) and head and neck squamous cell carcinoma (HNSC) accounted for the most significant increases, whereas prostate adenocarcinoma (PRAD), bladder urothelial carcinoma (BLCA), uterine corpus endometrial carcinoma (UCEC), and the kidney cancers displayed no coordinated change or even a modest decrease in expression. For these latter cancers, the non-directional GSA results (Figure 3B) revealed significant expression changes associated with these processes, but it was a mix of increases and decreases rather than a coordinated shift in one direction. Conversely, expression decreases related to adhesion and GAG binding were observed across many cancer types, with the most significant decreases occurring in kidney chromophobe (KICH), BLCA, and UCEC. Again, when ignoring the direction of expression change, virtually all of the cancers exhibited significant shifts in the secretome related to these functions. These results suggest that different cancer types are shifting their secretome expression in accordance with a common set of molecular functions, but the extent and direction of these changes are often tuned specifically to the tissue of origin.
Figure 3. Gene Set Analysis of the Cancer Secretome.

Heatmaps illustrate the (A) directional and (B) non-directional GSA results for secretome genes based on the tumor versus paired-normal fold changes and significance in 17 different cancer types. Only the GO molecular function gene set collection (MSigDB) was evaluated, and sets with <10 genes were excluded. In (A), the distinct directional gene set p values are calculated for coordinated increases (padj,dist-dir-up) and decreases (padj,dist-dir-down) in expression. The more significant (lower value) of the two directional p values for each gene set is shown in the heatmap as a log10-transformed value. The value is also “signed,” meaning that gene sets with a more significant decrease than increase (padj,dist-dir-down < padj,dist-dir-up) are made negative; otherwise, they are positive. Only gene sets with a padj,dist-dir ≤ 0.01 (in either direction) in at least one cancer type are shown. A non-stacked bar plot to the left of the heatmap shows the sizes of the original gene sets (gray bars) and of the filtered gene sets containing only secretome genes (black bars). *The ammonium ion binding gene set was identical to the quaternary ammonium group binding set after removing non-secretome genes; thus, the latter set is not shown. **The chemokine activity gene set was identical to the chemokine receptor binding gene set after removing non-secretome genes; thus, the latter set is not shown. See also Figure S2.
When repeating the analysis with high-purity tumor samples (Figure S2), much of the enrichment of secretome expression increases in ECM-related functions were reduced or absent, indicating a potential contribution of non-tumor cells to this behavior. However, significant coordinated expression decreases among genes associated with adhesion and GAG binding were observed to an even greater extent when using high-purity tumor samples, suggesting a more tumor-specific behavior.
Another feature of interest was the significant decrease in expression associated with several gene sets that was unique to CHOL and LIHC. Even when ignoring the directionality of expression change, only CHOL and LIHC exhibited significant changes in these sets (Figure 3B). These sets included genes associated with normal liver function, including binding or activity related to lipids, alcohols, sterols, and lipoproteins. Thus, it appeared that CHOL and LIHC, which both originate from the liver, were decreasing the expression of their healthy, tissue-specific secretome components in favor of those related to malignant and invasive processes.
Decreased Expression of Genes Specific to Tumor Tissue of Origin
Given that liver-derived cancers CHOL and LIHC exhibited significant and coordinated decreases in the expression of the secretome components specific to liver function, we investigated expression changes in the context of tissue specificity across all cancer types. In addition, to obtain a more comprehensive picture of the secretory pathway clientele, we expanded the analysis to include any protein possessing a signal peptide, not only those that are destined for secretion (e.g., membrane proteins). This corresponded to a set of 3,491 signal-peptide genes, referred to hereafter as SP genes.
Tissue-specificity data from the Human Protein Atlas (HPA) (Uhlen et al., 2015) was used to define the set of SP genes associated with each tissue (STAR Methods; Table S4). The DE analysis (tumor versus paired normal) results for each cancer type were then evaluated in the context of the tissue-specific gene sets to determine whether any of the cancer types exhibited significant expression changes in the subset of SP genes that are typically associated with a particular healthy tissue. As in the previous analyses, directionality of fold change was also taken into account to determine whether there were significantly coordinated expression increases or decreases.
In addition to the liver-derived cancers, the trend of a decrease in tissue-specific SP gene expression generally held true among the other cancers (Figures 4 and S3), all of which exhibited either a significant coordinated decrease or no significant change in the genes specific to their respective tissue of origin. Furthermore, the same behavior was observed even when including only high-purity tumor samples (Figure S4A).
Figure 4. Tissue-Specific Expression Changes in SP Genes.

The heatmap shows the significance and direction of coordinated expression changes in SP genes classified as specific to various tissue types. Cancer and tissue types are organized such that entries along the diagonal represent cancer types paired with their tissue of origin and are outlined in a solid box if there is a significant (padj < 0.05) coordinated expression decrease among the tissue-specific SP genes for that cancer type or in a dotted box otherwise. The log-transformed p values of cancer types sharing the same tissue of origin were averaged to facilitate this organization. The complete results for each individual tissue and cancer type are presented in Figure S3. The number of tissue-specific SP genes for each tissue type are indicated in the bar plot to the left of the heatmap. The distribution of tissue-specific SP gene expression changes across different cancer types is presented for two representative tissue types: prostate and liver. The log2FC values for each set of genes are represented by boxplots, with the individual gene values shown as gray points whose sizes indicate the significance (p value) of their FC. See also Figure S4.
Consistent with the GSA results, LIHC and CHOL exhibited a significant coordinated decreased expression of liver-specific genes. None of the 176 liver-specific SP genes were significantly (padj < 0.05) increased in either LIHC or CHOL relative to paired-normal tissue, whereas 156 (89%) and 174 (99%) of these genes exhibited a significant decrease in expression for LIHC and CHOL, respectively. These genes encoded functions such as lipid and cholesterol transport and metabolism (apolipoproteins), the complement system, coagulation, and protease inhibition (serpins). Similar strong, coordinated decreases in the expression of tissue-specific SP genes were observed in breast, colorectal, and lung cancers, in which only three or fewer genes in each set (<6%) were significantly increased in expression, while the majority were significantly decreased. The four cancer types that did not show a significant coordinated decreased expression in SP genes specific to their corresponding tissue of origin were BLCA, esophageal carcinoma (ESCA), PRAD, and UCEC. However, ESCA, PRAD, and UCEC did exhibit a significant decrease in the expression of genes specific to a tissue near their tissues of origin (stomach, seminal vesicle, and ovary, respectively) (Figure S3), suggesting a similar phenomenon. The data cannot distinguish between tumor cells that have actively decreased their tissue-specific gene expression and those that originated from more stem-like cells from the start; however, the end state is the same in that (most) cancer types exhibit a lower expression of tissue-specific SP genes in tumor cells than in the corresponding normal tissue.
Evaluation of Secretory Pathway Stress Signatures
The common decrease in the expression of tissue-specific SP genes across many different cancer types suggests a general pattern in which tumor cells are relieving the burden on an already strained (Ma and Hendershot, 2004) secretory system. By limiting the production and secretion of tissue-specific components, tumor cells may be able to dedicate more resources to processing proteins that contribute to cell proliferation and other malignant processes. To investigate further, we evaluated the tumor versus paired normal DE data for signs of increased stress or burden on the secretory pathway.
Activation of the UPR
Disruption of the secretory pathway results in the accumulation of misfolded proteins, which in turn activates a series of adaptive processes collectively known as the UPR to restore ER homeostasis (Ron and Walter, 2007). Coordinated expression increases in UPR-associated genes would therefore be indicative of cells undergoing secretory pathway stress and UPR activation. For each cancer type, we evaluated the enrichment of expression changes in genes affiliated with the UPR (all affiliated genes, not only secreted or SP genes). The results revealed a significant coordinated increase in UPR-related gene expression in nearly all cancer types (Figures 5 and S5A), consistent with previous reports regarding the prevalence of UPR activation among many cancers (Dejeans et al., 2014; Ma and Hendershot, 2004). CHOL and papillary thyroid carcinoma (THCA), however, exhibited a negligible coordinated expression increase in UPR-associated genes. The same results were observed when considering only high-purity tumor samples (Figure S4B), although CHOL was excluded due to the absence of purity scores for this cancer type.
Figure 5. Coordinated Expression Increases Associated with the UPR.

Shown are the log-transformed directional p values representing the significance of coordinated expression changes in genes associated with the UPR, defined as those included in the unfolded protein response gene set in the Hallmark gene set collection from MSigDB. Bars are colored blue if there is a significant (padj < 0.05) expression increase among the genes for that cancer type; if not, they are colored yellow. See also Figures S5 and S6.
Given that CHOL and THCA were among the cancer types exhibiting a strong coordinated expression decrease in tissue-specific SP genes (Figure 4), the data are supportive of the observed pattern whereby tumor cells alleviate secretory pathway stress by reducing the expression of SP genes specific to sustaining the function of their tissue of origin. Likewise, cancer types with an insignificant decrease in the expression of their respective tissue-specific SP genes (BLCA, ESCA, PRAD, and UCEC) exhibited coordinated expression increases associated with ER stress and the UPR (Figures 5 and S5A).
Estimation of Secretory Burden
Proteins traversing the secretory pathway undergo a number of maturation processes such as folding and post-translational modifications (PTMs). Larger proteins with a greater number of PTMs will require more cellular resources than shorter, less-modified proteins, and thus may impart a greater burden on the secretory pathway (Feizi et al., 2017; Gutierrez et al., 2018). We reasoned that a shift in expression toward lower-cost proteins may constitute another potential strategy to alleviate secretory pathway stress in tumor cells. To quantify this cost, we formulated a secretory burden (SB) score for each SP gene i as a function of its encoded protein length L (i.e., number of amino acids) and number of disulfide (NDS) and glycosylation (Ngly) sites:
| (Equation 1) |
where each property is normalized by the median (med) value among all of the SP genes.
For each cancer type, the Spearman correlation between gene SB scores and expression fold changes was calculated (Figures 6 and S5B). Although the correlation coefficients were low, the trend was consistent with our observations regarding UPR activation and decreased expression of tissue-specific SP genes, which is best illustrated by the two extremes, BLCA and CHOL. BLCA yielded the strongest negative correlation between SB score and log2FC, suggesting that expression increases tend to be associated with low-burden SP genes, whereas the opposite was true for CHOL. Given that BLCA showed evidence of UPR activation and exhibited the least significant expression decrease in tissue-specific SP genes, it suggests that the inability of BLCA cells to relieve secretory pathway stress via reduction in tissue-specific SP gene expression may constrain their ability to process proteins with a high secretory burden. Conversely, CHOL exhibited the strongest expression decrease in tissue-specific SP genes and showed little evidence of UPR activation, which is indicative of lower secretory pathway stress, thus relaxing the constraint on which proteins the secretory pathway can accommodate. Cancer types mirroring the trend of BLCA included ESCA, PRAD, and UCEC, whereas THCA followed that of CHOL.
Figure 6. Correlation between Protein Secretory Burden and Gene Expression Fold Change.

Cancer types with a significantly (p < 0.05) negative correlation are colored blue, significantly positive cancers are colored red, and those with an insignificant correlation are colored gray. See also Figures S5 and S6.
To further explore the PTM burden, we investigated the expression changes in genes associated with different PTMs: N- and O-linked glycosylation, and protein disulfide bond oxidation and reduction (Figure S5C). Nearly half of the studied cancer types exhibited a significant coordinated expression increase in genes associated with glycosylation and/or disulfide bond formation, suggesting an additional effort to reduce secretory stress. The opposite behavior was observed for CHOL, which exhibited significant expression decreases associated with disulfide redox and N-linked glycosylation. All of the cancer types that did not show a coordinated expression increase associated with these PTMs were those exhibiting a significant decrease in their tissue-specific secretome, providing additional support for this relief strategy.
Additional Contributors to the UPR
Although HNSC and rectum adenocarcinoma (READ) exhibited a coordinated expression decrease in tissue-specific SP genes, as well as a positive correlation between SB score and gene expression fold change, these cancer types still show evidence of an activated UPR, unlike CHOL and THCA. Because the UPR can be triggered by sources of stress other than an overburdened secretory pathway (e.g., genome instability, hypoxia, nutrient deprivation) (Corazzari et al., 2017), it is possible that one or more of these alternative sources are contributing to UPR activation in HNSC and READ cells, despite their modified secretory profile. We therefore compared genome instability among the different cancer types using mutation profiles from TCGA whole-exome sequencing datasets. HNSC and READ samples exhibited similar mutation burdens (median of 134 and 127 somatic mutations per sample, respectively), which were >2-fold greater than CHOL (63 median mutations per sample) and >10-fold greater than THCA (12 mutations per sample) (all p < 10−6, one-sided Wilcoxon rank-sum test) (Figure S6). These results support the possibility that other sources of stress beyond those directly involving the ER and secretory pathway could be responsible for elevated UPR activation in HNSC and READ.
DISCUSSION
The secretome is regarded as an attractive reservoir of disease biomarkers, as its extracellular nature offers the potential to evaluate physiological status through easily accessible biofluids (Kulasingam and Diamandis, 2008; Schaaij-Visser et al., 2013; Stastna and Van Eyk, 2012). Furthermore, there are many protein biomarkers in use for the diagnosis or monitoring of different cancer types based on their abundance in serum, plasma, or urine, such as PSA, CA-125, CA19–9, and NuMA for prostate, ovarian, pancreatic, and bladder cancer, respectively (Füzéry et al., 2013).
Beyond its potential as a reservoir of biomarker candidates, the cancer secretome is known to play a crucial role in tumor development and invasion. We sought to evaluate cancer-associated shifts in secretome expression with regard to the function of the encoded proteins. The majority of shared pan-cancer changes in secretome expression were decreases and included proteins associated with functions such as cell-cell and cell-matrix adhesion, tumor suppressors with anti-proliferative or anti-migratory activities, and immune response. These proteins harbor potential therapeutic opportunities, either by targeting the factors driving their expression decrease or through direct use of the tumor suppressor as a therapeutic peptide (Bonin-Debs et al., 2004; Guo et al., 2014; Oricchio et al., 2011). For example, ANGPTL1, which was among the top 1% core decreased secretome proteins, has been demonstrated to suppress cell migration, invasion, angiogenesis, metastasis, and/or therapy resistance in hepatocellular carcinoma (Chen et al., 2016; Yan et al., 2017), colorectal cancer (Chen et al., 2017), and lung and breast cancers (Kuo et al., 2013).
The trend of expression decreases among the secretome was also observed in the cancer-specific analyses, in which liver-related cancers (LIHC and CHOL) exhibited a particularly strong decrease in the expression of liver-specific SP genes. This reduced expression of tissue-specific genes in hepatocellular carcinoma has been explored previously; the extent of expression decrease was shown to negatively correlate with tumor grade or degree of dedifferentiation (Ge et al., 2005; Uhlen et al., 2017). We investigated this further, focusing on the subset of proteins targeted to the secretory pathway and spanning many different cancer types. Using tissue-specific gene classification from the HPA, this phenomenon of a significant decrease in expression of SP genes specific to the tissue of origin of the cancer was found to hold across the majority of examined cancer types.
Since UPR activation (Urra et al., 2016) and increased expression of secretory pathway machinery (Dejeans et al., 2014) are common in many cancers, our results suggest a common pattern by which tumor cells modify their secretory profile to alleviate ER stress by reducing the production of tissue-specific components in favor of tumorigenic factors. Consistent with this hypothesis, CHOL and THCA, which exhibited among the strongest decreases in their tissue-specific SP genes, were associated with the weakest UPR activation and displayed no bias toward the increased expression of low-burden (shorter and with fewer PTMs) SP genes. Conversely, the few cancer types with an insignificant decrease in their tissue-specific SP gene expression (BLCA, ESCA, PRAD, and UCEC) exhibited increased expression associated with the UPR and displayed an apparent bias in expression toward lower-burden SP genes.
Given that different tissues exhibit fine-tuned expression of their secretory machinery to accommodate their unique secretome profile (Feizi et al., 2017), it is reasonable to expect that a malignant cell could quickly overload this system and induce ER stress upon increasing the production of tumorigenic components without an accompanying decrease in other SP genes. A number of anti-cancer therapies that activate the UPR are under development or approved for clinical use, demonstrating the importance of this system in cancer treatment (Hetz et al., 2013). Although many cancers are known to leverage an activated UPR for its cytoprotective and restorative effects, UPR-targeted therapies function by driving the response further to a pro-apoptotic regime. We reasoned that the strong decrease in tissue-specific SP gene expression observed in CHOL or THCA cells, coupled with the insignificant coordinated expression increase in UPR-associated genes, could indicate a heightened sensitivity of these cancers toward this form of stress. In support of this hypothesis, treatment of CHOL cells in vitro and in a subcutaneous transplantation mouse model with bortezomib, which activates the UPR via proteasome inhibition, was shown to inhibit proliferation and induce apoptosis (Vaeteewoottacharn et al., 2013). Furthermore, bortezomib has been found to induce apoptosis in THCA cell lines with half-maximal inhibitory concentration (IC50) values lower than those of other cancer types (e.g., glioma, colon, renal, ovarian, prostate) (Mitsiades et al., 2006), whereas bortezomib treatment of BLCA cell line 253JB-V did not result in significant apoptosis and could not inhibit 253JB-V tumor growth in mice unless combined with another therapy (gemcitabine) (Kamat et al., 2004).
Overall, the functional diversity and close involvement of the secretome in a number of critical tumorigenic and metastatic processes highlights the importance of this group of proteins in cancer pathophysiology and presents a strong case for its targeting in anti-cancer therapeutic development. In addition, the ranked list of secretome biomarker candidates for each of the 32 different cancer types is expected to help facilitate the development of more accurate, less invasive diagnostic methods.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources should be directed to and will be fulfilled by the Lead Contact, Jens Nielsen (nielsenj@chalmers.se).
METHOD DETAILS
Definition of the secretome and SP genes
The list of proteins comprising the classically secreted secretome was obtained via UniProt (uniprot.org) (Bateman et al., 2017). Beginning with the entire human proteome (UP000005640), proteins were filtered to include those labeled as “UniProtKB/Swiss-Prot (reviewed),” with a subcellular location of “Secreted,” and PTM/Processing of “Signal peptide,” yielding 1,838 unique UniProt entries. The associated Entrez gene IDs and gene names were mapped to Ensembl IDs (GRCh38.p12), where those that did not map were excluded, and duplicated entries were removed, resulting in a secretome of 1,816 unique genes when analyzing TCGA data. For analyses also involving GTEx samples, genes absent from the that dataset were excluded, yielding a secretome comprised of 1,810 genes.
SP (signal peptide) genes were defined and generated in the same way as the secretome, except without the requirement for a subcellular location of “Secreted.” This resulted in a set of 3,491 SP genes, of which 3,111 had associated differential expression data (TCGA primary tumor versus paired normal).
The experimentally-derived secretome
Many proteins are secreted despite not having a signal peptide. To account for these unconventionally secreted proteins, we defined an “experimentally-derived” secretome consisting of proteins that had been detected within the extracellular environment in any one of the 35 secretome studies included in the Human Cancer Secretome Database (HCSD). We first retrieved the label-free proteomic data from HCSD, and extracted a list of all proteins that had been detected in at least one of the studies. For the label-based studies, proteins were retrieved if they had been measured to decrease or increase in concentration among any of the studies, as both cases imply detection. These lists were combined and mapped to the set of genes present in TCGA RNA-Seq data, resulting in a secretome consisting of 6,543 genes.
Retrieval of human plasma proteome data
Given the RNA-based nature of the analysis, we sought to enrich the results through the integration of protein-level data. We therefore retrieved a list of proteins that have been experimentally detected in plasma, which is a result of the Human Plasma Proteome Project (HPPP) (Schwenk et al., 2017). This protein evidence information was integrated with the consensus score results summarized in Figure 1D and Table S2.
The human plasma proteome was retrieved from PeptideAtlas (Farrah et al., 2013) (htpp://www.peptideatlas.org/hupo/c-hppp/). Only entries with a neXtProt protein evidence (PE) level of 1 (evidence at the protein level) were considered. This yielded four sets of proteins with categories of “canonical,” “uncertain,” “redundant,” or “not observed” (see Tables S2 or S3 for category definitions). Non-unique protein entries were combined, where the category of greater evidence was used if multiple categories were assigned to the same entry. Genes in the present study that did not have a corresponding entry in the plasma proteome dataset were categorized as “NA.”
Transcriptomic data retrieval
RNA-Seq data (FPKM and raw gene counts) were retrieved from TCGA on May 4, 2017 using the TCGAbiolinks (Colaprico et al., 2016) package in R (Gentleman et al., 2004; R Development Core Team, 2018), for all 33 cancer types available at that time. One cancer type, acute myeloid leukemia (LAML), did not have any associated primary tumor RNA-Seq data, and was thus excluded from all analyses, resulting in a total of 32 cancer types. GTEx RNA-Seq data (V7, TPM and raw gene counts) were retrieved directly from the site (http://www.gtexportal.org/home/datasets) on October 18, 2017.
Primary tumor and paired-normal transcriptomic (RNA-Seq) data were retrieved for 32 cancer types from TCGA, for a total of 9,760 primary tumor and 730 paired-normal samples, where both sample types were available for 697 patients. Healthy tissue RNA-Seq data was retrieved from the GTEx database, for a total of 11,688 samples spanning 714 donors and 30 tissue/organ types (or 53 subtissue types).
Mutation burden quantification
Mutation annotation files (MAFs) derived from whole-exome sequencing data were retrieved for all available cancer types from TCGA using the TCGAbiolinks R package. The total number of somatic mutation events (insertion, deletion, or single nucleotide polymorphism) for each primary tumor sample were summed to yield a total mutation burden for each sample.
Analysis of high-purity tumor samples
Consensus purity estimate (CPE) scores for TCGA primary solid tumor samples were obtained from a previous study (Aran et al., 2015), which calculated and combined purity scores using four different methods (ESTIMATE (Yoshihara et al., 2013), ABSOLUTE (Carter et al., 2012), LUMP and IHC (Aran et al., 2015)). All tumor samples with a CPE of less than 80% (0.80), or those that did not have a score available, were removed from the high-purity analysis. Cancer types that did not have any scores available (CHOL, ESCA, PAAD, PCPG, STAD), or had 3 or fewer tumor-normal sample pairs after removing low-purity tumor samples (BLCA, HNSC) were also excluded.
Consensus biomarker score
The consensus biomarker score was generated by combining the results from three types of sample comparison: (1) tumor versus paired normal, (2) tumor versus healthy tissue of origin, and (3) tumor versus all healthy tissues.
Comparison 1: primary tumor versus paired-normal tissue
The first comparison leveraged the paired nature of TCGA samples, meaning the tumor and normal tissue sample originated from the same patient. This enabled an estimation of gene expression changes that were specific to malignant transformation, rather than those arising from variation among patients or tissues of origin. TCGA data were filtered to only keep patients with paired samples; i.e., those with both a primary tumor and normal tissue sample. Furthermore, only cancer types with at least three patients after filtering were included, resulting in a final count of 693 patients spanning 20 cancer types. For each cancer type, a differential expression analysis was performed, comparing primary tumor with paired normal tissue, using the patient ID as a blocking factor.
Comparison 2: primary tumor versus healthy matched tissue
The second comparison was conducted in recognition of the fact that paired-normal samples are not always representative of normal healthy tissue, as nearby tumor cells are known to perturb cellular function (Aran et al., 2017; Huang et al., 2016). Therefore, primary tumor TCGA samples were compared to GTEx healthy tissue samples (of the same tissue-of-origin) from non-cancer patients. For this analysis, all 9,760 primary tumor samples were used, not just those with a corresponding paired-normal tissue sample. A differential expression analysis was performed for each cancer type, comparing primary tumor samples with those of the corresponding healthy tissue from GTEx.
Comparison 3: primary tumor versus all healthy tissues
The final comparison sought to identify genes with relatively low expression throughout all tissues in the body compared to their expression in a tumor. We hypothesized that tumor-derived expression changes in such genes would be more detectable in a biofluid than genes expressed at similar or higher levels in many healthy tissues, as the latter could impart a “dilution” effect on the tumor-associated signal of interest. For this analysis, we were more interested in transcript abundance rather than fold-changes between two conditions. Therefore, normalized gene counts (FPKM) were retrieved from TCGA for all tumor and paired normal tissue samples and converted to transcripts per million (TPM). TPM gene counts were also retrieved from the GTEx database for all measured tissues. The complete set of healthy tissues was obtained by combining healthy tissue samples from GTEx with paired normal samples from TCGA (Table S1).
For each gene in a given cancer type, the TPM values among all TCGA primary tumor samples for that cancer type were compared to the TPM values for that gene across all normal samples for a particular tissue type, using a right-tailed Wilcoxon rank-sum test (i.e., the null-hypothesis being that the tumor counts are not sampled from a distribution with a higher median than that of the normal tissue counts). This yielded a significance (p value) for each gene for a tissue type, where a low p value corresponded to genes with higher TPM values in primary tumor tissues than in the normal tissue. The comparison was repeated for all of the healthy tissue types, to obtain a p value for each tissue. The test was performed with each healthy tissue individually rather than pooling all of the normal samples together, as the pooled test would be biased by variations in the number of samples for different tissues. Each of the p values obtained from the different tissues types were then combined (geometric mean) into a single p-like score (ranging from 0–1). The entire process was repeated for each of the different cancer types, yielding a single score for each gene and each cancer type.
Consensus score formulation
For the first two comparisons (DE analyses), genes were ranked by their combined fold-change and significance (FDR-adjusted p value). Fold-changes were ranked directly, with higher ranks assigned to genes with greater positive log2FC (tumor/normal), and vice versa. Prior to ranking p values, the associated FC direction was incorporated to generate directional p values (pdir) for each gene i (analogous to the approach described in (Väremo et al., 2013)):
| (2) |
where sign(FC) is the sign of the corresponding log2(fold-change). In this manner, genes with low p values and a positive FC receive a pdir near zero, whereas genes with low p values but a negative FC have a pdir close to one. Genes associated with a high p value will therefore have a pdir near 0.5, regardless of FC direction. These pdir values were then ranked such that higher ranks were assigned to genes with lower pdir values. Finally, the p-like scores generated from the third comparison (tumor versus all tissues) were ranked directly, where low p-scores (high significance) were ranked highly, and vice versa.
The consensus rank score was calculated by combining the gene ranks from each of the three comparative analyses, as illustrated in Figure 1A. Specifically, the FC and pdir ranks from the first comparison were averaged, and this mean rank was averaged with the mean of the FC and pdir ranks from the second comparison. The resulting combined rank was averaged with the rank of p-like scores from the third comparison to yield the overall consensus rank score, enabling the prediction of candidate biomarkers for each cancer type. The effective weight ratios from the three comparisons (tumor versus paired normal, tumor versus healthy tissue-of-origin, and tumor versus all healthy tissues) in the consensus score were therefore 1:1:2, respectively. The ratios were assigned as such because the score was designed to place equal weight on expression differences of tumor versus tissue-of-origin, and of tumor versus all tissues. Since comparisons 1 and 2 both quantify tumor versus tissue-of-origin differences, they were each assigned half the weight of comparison 3, which quantified tumor versus all tissue differences. Moreover, since the information from the first two comparisons is likely to exhibit more redundancy (paired normal tissue and healthy tissue-of-origin are relatively similar in their expression profiles compared to other tissue types), they were weighted less than comparison 3.
Cancer types lacking paired-normal or healthy tissue data
Among the 32 TCGA cancer types with available primary tumor samples, 12 lacked sufficient paired-normal tissue data from TCGA to be included in the first comparison, and 6 types could not be appropriately matched to one of the tissue types defined in GTEx (e.g., SARC, “sarcoma”), and thus could not be included in the second comparison. However, the genes were still scored based on the results from the remaining comparisons that could be performed. Although there is less confidence associated with the scores for these particular cancer types, potential biomarkers could still be identified. For example, the top-scoring candidate for ovarian cancer (OV) was WFDC2 (also known as HE4), which is an established OV protein biomarker in both urine and serum (Hellström et al., 2003, 2010), and the next top 6 candidates included FOLR1, KLK6, KLK7, and MSLN, all of which have been experimentally confirmed as biofluid diagnostic markers of OV (Badgwell et al., 2007; Diamandis et al., 2003; Leung et al., 2013; Tamir et al., 2014).
Core secretome definition and analysis
To focus on changes in secretome expression associated specifically with malignant progression rather than inter-individual and inter-tissue variation, the analysis was conducted using paired tumor-normal samples from TCGA. Furthermore, cancer types with only a few sample pairs (CESC, PAAD, and PCPG; each had only 2 or 3 pairs) were excluded, yielding a final dataset spanning 17 cancer types, 683 patients, and 1,563 secretome genes (very low-count or non-detected genes were excluded).
To identify the subset of secretome genes with substantial paired normal versus primary tumor expression changes across many cancer types, a rank-based metric was used. The rationale of implementing a relative metric rather than directly using the fold-change and significance values from the DE analyses was that their ranges, especially those of the p values, vary widely across cancer types due to differences in the number of samples for each. We therefore ranked the genes within each cancer type by p value, and by absolute log2(fold-change) value, then averaged the two ranks to yield a combined “PF-rank.” To identify the genes that exhibited the greatest and most significant changes across all included cancer types (regardless of fold-change direction), the PF-ranks for each cancer were averaged to yield a pan-cancer PF-rank.
Directional PF-ranks were also generated, where the direction of expression fold-change was incorporated instead of using the absolute log2FC values. In addition, the associated p values were converted to directional p values (pdir, Equation 2, Method Details), such that the lowest ranks were assigned to genes exhibiting a significant decrease in expression across many cancers, and the highest to those with a significant increase in expression.
Definition of tissue-specific genes
Gene tissue specificity data was retrieved from the HPA, which has compiled a list of genes for each tissue that are classified as tissue enriched, group enriched, or tissue enhanced, based on their expression in that tissue compared to others (Uhlén et al., 2015). Given the relatively small number of tissue-enriched genes for many tissues, especially when removing all non-SP genes, we defined tissue-specific gene sets for each individual tissue as the combination of all its tissue-enriched, group-enriched, and tissue-enhanced genes (Table S4).
Estimation of UPR activation
Activation of the UPR was estimated using a GSA. In this analysis, the full gene sets were used; i.e., they were not filtered to remove non-secretome genes. The “Hallmark,” “Canonical pathways,” and “GO gene sets” libraries from MSigDB were queried for any set containing the phrase “endoplasmic reticulum stress” or “unfolded protein response,” and sets with the term “negative regulation” were excluded. This yielded 11 gene sets related to UPR and/or ER stress, which are shown in Figures S4 and S5.
Glycosylation and disulfide bond redox
Expression changes related to glycosylation and disulfide bond oxidation/reduction processes were evaluated by conducting a GSA, using the “GO bioprocess: glycosylation” and “GO molecular function: protein disulfide oxidoreductase activity” gene sets from MSigDB, respectively. To add resolution to the analysis of glycosylation activity, two subsets of the glycosylation gene set, “protein N-linked glycosylation” and “protein O-linked glycosylation,” were also evaluated for coordinated changes in gene expression. These gene sets were used in their complete form, and were not filtered (e.g., by removing non-secretome genes).
Secretory burden (SB) score
The SB score was calculated for each gene based on its associated protein length (number of amino acids), number of disulfide sites, and number of glycosylation sites, as described in Equation 1. Data for each of these terms was retrieved from UniProt, where the number of glycosylation sites was the sum of all N-, C-, O-, and S-linked glycosylation sites.
QUANTIFICATION AND STATISTICAL ANALYSIS
Differential expression (DE) analysis
All differential expression analyses reported in the study were conducted using the edgeR package in R (Robinson et al., 2010), with the raw gene count (integer) values as input. For the DE analysis comparing primary tumor expression to that of paired normal tissues, the patient ID number was included as an additional field in the design matrix. When comparing primary tumor gene counts from TCGA to those of healthy tissues from GTEx, only the sample type was considered (tumor or normal). Counts were normalized using the EdgeR calcNormFactors function, which scales library sizes using the trimmed mean of M-values (TMM) between each pair of samples (Robinson and Oshlack, 2010). For each DE analysis, low-count genes were removed beforehand; i.e., only genes with at least 10 counts in at least half of the samples were retained. Furthermore, DE analyses were only performed if there were at least 3 samples in each of the 2 conditions to be compared.
Gene set analysis
To quantify the extent to which different groups of genes were enriched in a given metric (e.g., p values from a DE analysis), a gene set analysis (GSA) was performed. This type of analysis was applied in a number of situations throughout the study, and followed the same procedure (described below), unless stated otherwise. The following gene set collections were retrieved from the Molecular Signatures Database (MSigBD (Subramanian et al., 2005)): hallmark (H) (Liberzon et al., 2015), KEGG (C2 CP:KEGG), Reactome (C2 CP:REACTOME), GO biological process (C5 BP), and GO molecular function (C5 MF).
Gene set collections were filtered to remove all non-secretome genes from each set prior to analysis, unless otherwise stated. We note that this filtration can cause the name of a gene set to become less representative if a substantial portion of genes in the set are removed. In this way, the significance of a gene set does not necessarily represent an enrichment in its named function/pathway, but instead represents an enrichment in the set of secretome genes that are associated with that function/pathway. In addition, gene sets containing more than 400 genes (before filtering) were also removed, as these sets tended to have a very low fraction of secretome genes, and were generally uninformative. Finally, to avoid statistical problems with very small gene sets, those with less than 20 genes after filtration were excluded from the analysis, unless otherwise noted.
A Wilcoxon rank-sum test statistic was calculated from the DE analysis p values of genes in a given set, and compared to those of 100,000 randomly shuffled gene sets of the same size. The significance (p value) of a gene set was calculated as:
| (3) |
where Nrand ≥ set is the number of randomly shuffled gene sets with a test statistic greater than or equal to that of the original gene set, and Nperms is the number of random permutations (100,000 in this study). Gene set p values calculated in this manner correspond to “non-directional” p values (pnon-dir), as they do not take into account the direction (increase or decrease) of the fold-change from the DE analysis, only the significance.
“Distinct directional” gene set p values (pdist-dir-up and pdist-dir-down) Väremo et al., 2013) were obtained in the same manner, except the p values from the DE analysis were first converted to directional p values (Equation 2) before calculating the associated Wilcoxon test statistic. The resulting gene set pdist-dir-up values quantify coordinated expression increases in a gene set, where a low pdist-dir-up indicates a get set that is enriched in genes with significant expression increases. Coordinated expression decreases are quantified simply as pdist-dir-down = 1 – pdist-dir-up, where low pdist-dir-down values indicate an enrichment of genes with expression decreases.
Adjustment of p values
All adjusted p values (padj) reported in the study were adjusted to control for the false discovery rate (FDR) using the Benjamini-Hochberg procedure. Statistical significance in this study was defined as padj < 0.05.
Supplementary Material
KEY RESOURCES TABLE.
| REAGENT or RESOURCE Software and Algorithms | SOURCE | IDENTIFIER |
|---|---|---|
| The R Project for Statistical Computing | R Development Core Team, 2018 | https://www.R-project.org/ |
| Bioconductor | Gentleman et al., 2004 | https://www.bioconductor.org/ |
| EdgeR | Robinson et al., 2010 | R Bioconductor |
| TCGAbiolinks | Colaprico et al., 2016 | R Bioconductor |
| MATLAB R2017b | The MathWorks, Inc. | https://ch.mathworks.com/products/matlab.html |
| MSigDB database | Subramanian et al., 2005 | http://software.broadinstitute.org/gsea/msigdb |
Highlights.
Secreted proteins with elevated expression in tumors comprise potential biomarkers
Secretome expression changes common to many cancer types tend to be decreases
Common expression increases include functions such as extracellular matrix turnover
Tumors appear to reduce their tissue-specific secretome to relieve secretory stress
ACKNOWLEDGMENTS
The authors thank Daniel Cook for valuable discussions. Research reported in this publication was supported by funding from the Knut and Alice Wallenberg Foundation and the National Cancer Institute of the NIH under award number F32CA220848. The results shown here are in part based upon data generated by TCGA Research Network (https://cancergenome.nih.gov/). The GTEx Project was supported by the Common Fund of the Office of the Director of the NIH, and by NCI, NHGRI, NHLBI, NIDA, NIMH, and NINDS. The GTEx data used for the analyses described in this article were obtained from the GTEx Portal on October 18, 2017.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental Information can be found with this article online at https://doi.org/10.1016/j.celrep.2019.02.025.
REFERENCES
- Andarawewa KL, Motrescu ER, Chenard MP, Gansmuller A, Stoll I, Tomasetto C, and Rio MC (2005). Stromelysin-3 is a potent negative regulator of adipogenesis participating to cancer cell-adipocyte interaction/cross-talk at the tumor invasive front. Cancer Res. 65, 10862–10871. [DOI] [PubMed] [Google Scholar]
- Aran D, Sirota M, and Butte AJ (2015). Systematic pan-canceranalysis of tumour purity. Nat. Commun 6, 8971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aran D, Camarda R, Odegaard J, Paik H, Oskotsky B, Krings G, Goga A, Sirota M, and Butte AJ (2017). Comprehensive analysis of normal adjacent to tumor transcriptomes. Nat. Commun 8, 1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avilés-Vázquez S, Chávez-Gonzalez A, Hidalgo-Miranda A, Moreno-Lorenzana D, Arriaga-Pizano L, Sandoval-Esquivel MA, Ayala-Sánchez M, Aguilar R, Alfaro-Ruiz L, and Mayani H (2017). Global gene expression profiles of hematopoietic stem and progenitor cells from patients with chronic myeloid leukemia: the effect of in vitro culture with or without imatinib. Cancer Med. 6, 2942–2956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badgwell D, Lu Z, Cole L, Fritsche H, Atkinson EN, Somers E, Allard J, Moore RG, Lu KH, and Bast RC Jr. (2007). Urinary mesothelin provides greater sensitivity for early stage ovarian cancer than serum mesothelin, urinary hCG free beta subunit and urinary hCG beta core fragment. Gynecol. Oncol 106, 490–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bager CL, Willumsen N, Leeming DJ, Smith V, Karsdal MA, Dornan D, and Bay-Jensen AC (2015). Collagen degradation products measured in serum can separate ovarian and breast cancer patients from healthy controls: a preliminary study. Cancer Biomark. 15, 783–788. [DOI] [PubMed] [Google Scholar]
- Barton CA, Gloss BS, Qu W, Statham AL, Hacker NF, Sutherland RL, Clark SJ, and O’Brien PM (2010). Collagen and calcium-binding EGF domains 1 is frequently inactivated in ovarian cancer by aberrant promoter hypermethylation and modulates cell migration and survival. Br. J. Cancer 102, 87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman A, Martin MJ, O’Donovan C, Magrane M, Alpi E, Antunes R, Bely B, Bingley M, Bonilla C, Britto R, et al. ; The UniProt Consortium (2017). UniProt: the universal protein knowledgebase. Nucleic Acids Res 45 (D1), D158–D169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Belczacka I, Latosinska A, Metzger J, Marx D, Vlahou A, Mischak H, and Frantzi M (2019). Proteomics biomarkers for solid tumors: current status and future prospects. Mass Spectrom. Rev 38, 49–78. [DOI] [PubMed] [Google Scholar]
- Bonin-Debs AL, Boche I, Gille H, and Brinkmann U (2004). Development of secreted proteins as biotherapeutic agents. Expert Opin. Biol. Ther 4, 551–558. [DOI] [PubMed] [Google Scholar]
- Brandi J, Manfredi M, Speziali G, Gosetti F, Marengo E, and Cecconi D (2018). Proteomic approaches to decipher cancer cell secretome. Semin. Cell Dev. Biol 78, 93–101. [DOI] [PubMed] [Google Scholar]
- Capurro M, Wanless IR, Sherman M, Deboer G, Shi W, Miyoshi E, and Filmus J (2003). Glypican-3: a novel serum and histochemical marker for hepatocellular carcinoma. Gastroenterology 125, 89–97. [DOI] [PubMed] [Google Scholar]
- Carter SL, Cibulskis K, Helman E, McKenna A, Shen H, Zack T, Laird PW, Onofrio RC, Winckler W, Weir BA, et al. (2012). Absolute quantification of somatic DNA alterations in human cancer. Nat. Biotechnol 30, 413–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HA, Kuo TC, Tseng CF, Ma JT, Yang ST, Yen CJ, Yang CY, Sung SY, and Su JL (2016). Angiopoietin-like protein 1 antagonizes MET receptor activity to repress sorafenib resistance and cancer stemness in hepatocellular carcinoma. Hepatology 64, 1637–1651. [DOI] [PubMed] [Google Scholar]
- Chen H, Xiao Q, Hu Y, Chen L, Jiang K, Tang Y, Tan Y, Hu W, Wang Z, He J, et al. (2017). ANGPTL1 attenuates colorectal cancer metastasis by up-regulating microRNA-138. J. Exp. Clin. Cancer Res 36, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Colaprico A, Silva TC, Olsen C, Garofano L, Cava C, Garolini D, Sabedot TS, Malta TM, Pagnotta SM, Castiglioni I, et al. (2016). TCGAbiolinks: an R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res. 44, e71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corazzari M, Gagliardi M, Fimia GM, and Piacentini M (2017). Endoplasmic Reticulum Stress, Unfolded Protein Response, and Cancer Cell Fate. Front. Oncol 7, 78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crowley E, Di Nicolantonio F, Loupakis F, and Bardelli A (2013). Liquid biopsy: monitoring cancer-genetics in the blood. Nat. Rev. Clin. Oncol 10, 472–484. [DOI] [PubMed] [Google Scholar]
- Dejeans N, Manié S, Hetz C, Bard F, Hupp T, Agostinis P, Samali A, and Chevet E (2014). Addicted to secrete - novel concepts and targets in cancer therapy. Trends Mol. Med 20, 242–250. [DOI] [PubMed] [Google Scholar]
- Diamandis EP, Scorilas A, Fracchioli S, Van Gramberen M, De Bruijn H, Henrik A, Soosaipillai A, Grass L, Yousef GM, Stenman UH, et al. (2003). Human kallikrein 6 (hK6): a new potential serum biomarker for diagnosis and prognosis of ovarian carcinoma. J. Clin. Oncol 21, 1035–1043. [DOI] [PubMed] [Google Scholar]
- Diaz LA Jr., and Bardelli A (2014). Liquid biopsies: genotyping circulating tumor DNA. J. Clin. Oncol 32, 579–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dong D, Jia L, Zhou Y, Ren L, Li J, and Zhang J (2017). Serum level of ANGPTL4 as a potential biomarker in renal cell carcinoma. Urol. Oncol 35, 279–285. [DOI] [PubMed] [Google Scholar]
- Dragutinović VV, Radonjić NV, Petronijević ND, Tatić SB, Dimitrijević IB, Radovanović NS, and Krivokapić ZV (2011). Matrix metalloproteinase-2 (MMP-2) and −9 (MMP-9) in preoperative serum as independent prognostic markers in patients with colorectal cancer. Mol. Cell. Biochem 355, 173–178. [DOI] [PubMed] [Google Scholar]
- Egeblad M, and Werb Z (2002). New functions for the matrix metalloproteinases in cancer progression. Nat. Rev. Cancer 2, 161–174. [DOI] [PubMed] [Google Scholar]
- Eissa S, Ali-Labib R, Swellam M, Bassiony M, Tash F, and El-Zayat TM (2007). Noninvasive diagnosis of bladder cancer by detection of matrix metalloproteinases (MMP-2 and MMP-9) and their inhibitor (TIMP-2) in urine. Eur. Urol 52, 1388–1396. [DOI] [PubMed] [Google Scholar]
- Etzioni R, Urban N, Ramsey S, McIntosh M, Schwartz S, Reid B, Radich J, Anderson G, and Hartwell L (2003). The case for early detection. Nat. Rev. Cancer 3, 243–252. [DOI] [PubMed] [Google Scholar]
- Farrah T, Deutsch EW, Hoopmann MR, Hallows JL, Sun Z, Huang CY, and Moritz RL (2013). The state of the human proteome in 2012 as viewed through PeptideAtlas. J. Proteome Res 12, 162–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feizi A, Banaei-Esfahani A, and Nielsen J (2015). HCSD: the human cancer secretome database. Database (Oxford) 2015, bav051. [DOI] [PMC free article] [PubMed]
- Feizi A, Gatto F, Uhlen M, and Nielsen J (2017). Human protein secretory pathway genes are expressed in a tissue-specific pattern to match processing demands of the secretome. NPJ Syst. Biol. Appl 3, 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Füzéry AK, Levin J, Chan MM, and Chan DW (2013).Translation of proteomic biomarkers into FDA approved cancer diagnostics: issues and challenges. Clin. Proteomics 10, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gayed BA, Gillen J, Christie A, Peña-Llopis S, Xie XJ, Yan J, Karam JA, Raj G, Sagalowsky AI, Lotan Y, et al. (2015). Prospective evaluation of plasma levels of ANGPT2, TuM2PK, and VEGF in patients with renal cell carcinoma. BMC Urol. 15, 24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge X, Yamamoto S, Tsutsumi S, Midorikawa Y, Ihara S, Wang SM, and Aburatani H (2005). Interpreting expression profiles of cancers by genomewide survey of breadth of expression in normal tissues. Genomics 86, 127–141. [DOI] [PubMed] [Google Scholar]
- Gentleman RC, Carey VJ, Bates DM, Bolstad B, Dettling M, Dudoit S, Ellis B, Gautier L, Ge Y, Gentry J, et al. (2004). Bioconductor: open software development for computational biology and bioinformatics. Genome Biol. 5, R80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geyer PE, Holdt LM, Teupser D, and Mann M (2017). Revisiting biomarker discovery by plasma proteomics. Mol. Syst. Biol 13, 942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo XE, Ngo B, Modrek AS, and Lee WH (2014). Targeting tumor suppressor networks for cancer therapeutics. Curr. Drug Targets 15, 2–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutierrez JM, Feizi A, Li S, Kallehauge TB, Hefzi H, Grav LM, Ley D, Hizal DB, Betenbaugh MJ, Voldborg B, et al. (2018). Genome-scale reconstructions of the mammalian secretory pathway predict metabolic costs and limitations of protein secretion. bioRxiv. 10.1101/351387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanahan D, and Weinberg RA (2011). Hallmarks of cancer: the next generation. Cell 144, 646–674. [DOI] [PubMed] [Google Scholar]
- Hanash SM, Pitteri SJ, and Faca VM (2008). Mining the plasma proteome for cancer biomarkers. Nature 452, 571–579. [DOI] [PubMed] [Google Scholar]
- Hawes MC, Wen F, and Elquza E (2015). Extracellular DNA: A Bridge to Cancer. Cancer Res. 75, 4260–264. [DOI] [PubMed] [Google Scholar]
- Hazell GG, Peachey AM, Teasdale JE, Sala-Newby GB, Angelini GD, Newby AC, and White SJ (2016). PI16 is a shear stress and inflammationregulated inhibitor of MMP2. Sci. Rep 6, 39553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hellström I, Raycraft J, Hayden-Ledbetter M, Ledbetter JA, Schummer M, McIntosh M, Drescher C, Urban N, and Hellström KE (2003).The HE4 (WFDC2) protein is a biomarker for ovarian carcinoma. Cancer Res. 63, 3695–3700. [PubMed] [Google Scholar]
- Hellström I, Heagerty PJ, Swisher EM, Liu P, Jaffar J, Agnew K, and Hellstrom KE (2010). Detection of the HE4 protein in urine as a biomarker for ovarian neoplasms. Cancer Lett. 296, 43–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz C, Chevet E, and Harding HP (2013). Targeting the unfolded protein response in disease. Nat. Rev. Drug Discov 12, 703–719. [DOI] [PubMed] [Google Scholar]
- Hu X, Li YQ, Li QG, Ma YL, Peng JJ, and Cai SJ (2018). Osteoglycin (OGN) reverses epithelial to mesenchymal transition and invasiveness in colorectal cancer via EGFR/Akt pathway. J. Exp. Clin. Cancer Res 37, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang X, Stern DF, and Zhao H (2016). Transcriptional Profiles from Paired Normal Samples Offer Complementary Information on Cancer Patient Survival-Evidence from TCGA Pan-Cancer Data. Sci. Rep 6, 20567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iozzo RV, and Schaefer L (2015). Proteoglycan form and function: a comprehensive nomenclature of proteoglycans. Matrix Biol 42, 11–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izbicka E, Streeper RT, Michalek JE, Louden CL, Diaz A 3rd, and Campos DR (2012). Plasma biomarkers distinguish non-small cell lung cancer from asthma and differ in men and women. Cancer Genomics Proteomics 9, 27–35. [PubMed] [Google Scholar]
- Kamat AM, Karashima T, Davis DW, Lashinger L, Bar-Eli M, Millikan R, Shen Y, Dinney CP, and McConkey DJ (2004). The proteasome inhibitor bortezomib synergizes with gemcitabine to block the growth of human 253JB-V bladder tumors in vivo. Mol. Cancer Ther 3, 279–290. [PubMed] [Google Scholar]
- Kosanam H, Prassas I, Chrystoja CC, Soleas I, Chan A, Dimitromanolakis A, Blasutig IM, Rückert F, Gruetzmann R, Pilarsky C, et al. (2013). Laminin, gamma 2 (LAMC2): a promising new putative pancreatic cancer biomarker identified by proteomic analysis of pancreatic adenocarcinoma tissues. Mol. Cell. Proteomics 12, 2820–2832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulasingam V, and Diamandis EP (2008). Strategies for discovering novel cancer biomarkers through utilization of emerging technologies. Nat. Clin. Pract. Oncol 5, 588–599. [DOI] [PubMed] [Google Scholar]
- Kuo TC, Tan CT, Chang YW, Hong CC, Lee WJ, Chen MW, Jeng YM, Chiou J, Yu P, Chen PS, et al. (2013). Angiopoietin-like protein 1 suppresses SLUG to inhibit cancer cell motility. J. Clin. Invest 123, 1082–1095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurosawa N, Chen GY, Kadomatsu K, Ikematsu S, Sakuma S, and Muramatsu T (2001). Glypican-2 binds to midkine: the role of glypican-2 in neuronal cell adhesion and neurite outgrowth. Glycoconj. J 18, 499–507. [DOI] [PubMed] [Google Scholar]
- Leung F, Dimitromanolakis A, Kobayashi H, Diamandis EP, and Kulasingam V (2013). Folate-receptor 1 (FOLR1) protein is elevated in the serum of ovarian cancer patients. Clin. Biochem 46, 1462–1468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Li L, Wang W, Yang Y, Zhou Y, and Lu S (2015). Soluble purified recombinant C2ORF40 protein inhibits esophageal cancer cell proliferation by inducing cell cycle G-ι phase block. Oncol. Lett 10, 1593–1596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A, Birger C, Thorvaldsdóttir H, Ghandi M, Mesirov JP, and Tamayo P (2015). The Molecular Signatures Database (MSigDB) hallmark gene set collection. Cell Syst 1, 417–425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindén M, Lind SB, Mayrhofer C, Segersten U, Wester K, Lyutvinskiy Y, Zubarev R, Malmström PU, and Pettersson U (2012). Proteomic analysis of urinary biomarker candidates for nonmuscle invasive bladder cancer. Proteomics 12, 135–144. [DOI] [PubMed] [Google Scholar]
- Lou J, Zhang L, Lv S, Zhang C, and Jiang S (2017). Biomarkers for Hepatocellular Carcinoma. Biomark. Cancer 9, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma Y, and Hendershot LM (2004). The role of the unfolded protein response in tumour development: friend or foe? Nat. Rev. Cancer 4, 966–977. [DOI] [PubMed] [Google Scholar]
- Makridakis M, and Vlahou A (2010). Secretome proteomics for discovery of cancer biomarkers. J. Proteomics 73, 2291–2305. [DOI] [PubMed] [Google Scholar]
- Missler M, and Südhof TC (1998). Neurexophilins form a conserved family of neuropeptide-like glycoproteins. J. Neurosci 18, 3630–3638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mitsiades CS, McMillin D, Kotoula V, Poulaki V, McMullan C, Negri J, Fanourakis G, Tseleni-Balafouta S, Ain KB, and Mitsiades N (2006). Antitumor effects of the proteasome inhibitor bortezomib in medullary and anaplastic thyroid carcinoma cells in vitro. J. Clin. Endocrinol. Metab 91, 4013–4021. [DOI] [PubMed] [Google Scholar]
- Mroczko B, Kozłowski M, Groblewska M, Łukaszewicz M, Nikliński J, Jelski W, Laudański J, Chyczewski L, and Szmitkowski M (2008). The diagnostic value of the measurement of matrix metalloproteinase 9 (MMP-9), squamous cell cancer antigen (SCC) and carcinoembryonic antigen (CEA) in the sera of esophageal cancer patients. Clin. Chim. Acta 389, 61–66. [DOI] [PubMed] [Google Scholar]
- Obrist P, Spizzo G, Ensinger C, Fong D, Brunhuber T, Schäfer G, Varga M, Margreiter R, Amberger A, Gastl G, and Christiansen M (2004). Aberrant tetranectin expression in human breast carcinomas as a predictor of survival. J. Clin. Pathol 57, 417–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohlund D, Lundin C, Ardnor B, Oman M, Naredi P, and Sund M (2009). Type IV collagen is a tumour stroma-derived biomarker for pancreas cancer. Br. J. Cancer 101, 91–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oricchio E, Nanjangud G, Wolfe AL, Schatz JH, Mavrakis KJ, Jiang M, Liu X, Bruno J, Heguy A, Olshen AB, et al. (2011). The Eph-receptor A7 is a soluble tumor suppressor for follicular lymphoma. Cell 147, 554–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papaleo E, Gromova I, and Gromov P (2017). Gaining insights into cancer biology through exploration of the cancer secretome using proteomic and bioinformatic tools. Expert Rev. Proteomics 14, 1021–1035. [DOI] [PubMed] [Google Scholar]
- Park EH, Kim S, Jo JY, Kim SJ, Hwang Y, Kim JM, Song SY, Lee DK, and Koh SS (2013). Collagen triple helix repeat containing-1 promotes pancreatic cancer progression by regulating migration and adhesion of tumor cells. Carcinogenesis 34, 694–702. [DOI] [PubMed] [Google Scholar]
- Patel S, Sumitra G, Koner BC, and Saxena A (2011). Role of serum matrix metalloproteinase-2 and −9 to predict breast cancer progression. Clin. Biochem 44, 869–872. [DOI] [PubMed] [Google Scholar]
- Pei D, Majmudar G, and Weiss SJ (1994). Hydrolytic inactivation of a breast carcinoma cell-derived serpin by human stromelysin-3. J. Biol. Chem 269, 25849–25855. [PubMed] [Google Scholar]
- Pei YF, Zhang YJ, Lei Y, Wu DW, Ma TH, and Liu XQ (2017). Hypermethylation of the CHRDL1 promoter induces proliferation and metastasis by activating Akt and Erk in gastric cancer. Oncotarget 8, 23155–23166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pickup MW, Mouw JK, and Weaver VM (2014). The extracellular matrix modulates the hallmarks of cancer. EMBO Rep. 15, 1243–1253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pilecki B, Holm AT, Schlosser A, Moeller JB, Wohl AP, Zuk AV, Heumüller SE, Wallis R, Moestrup SK, Sengle G, et al. (2016). Characterization of Microfibrillar-associated Protein 4 (MFAP4) as a Tropoelastin- and Fibrillin-binding Protein Involved in Elastic Fiber Formation. J. Biol. Chem 291, 1103–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prassas I, Chrystoja CC, Makawita S, and Diamandis EP (2012). Bioinformatic identification of proteins with tissue-specific expression for biomarker discovery. BMC Med. 10, 39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- R Development Core Team (2018). R: A language and environment for statistical computing (R Foundation for Statistical Computing; ). [Google Scholar]
- Rabouille C (2017). Pathways of Unconventional Protein Secretion. Trends Cell Biol. 27, 230–240. [DOI] [PubMed] [Google Scholar]
- Rankin T, and Dean J (2000). The zona pellucida: using molecular genetics to study the mammalian egg coat. Rev. Reprod 5, 114–121. [DOI] [PubMed] [Google Scholar]
- Robinson MD, and Oshlack A (2010). A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 11, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson MD, McCarthy DJ, and Smyth GK (2010). edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron D, and Walter P (2007). Signal integration in the endoplasmic reticulum unfolded protein response. Nat. Rev. Mol. Cell Biol 8, 519–529. [DOI] [PubMed] [Google Scholar]
- Roy R, Louis G, Loughlin KR, Wiederschain D, Kilroy SM, Lamb CC, Zurakowski D, and Moses MA (2008). Tumor-specific urinary matrix metalloproteinase fingerprinting: identification of high molecular weight urinary matrix metalloproteinase species. Clin. Cancer Res 14, 6610–6617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarkissian G, Fergelot P, Lamy PJ, Patard JJ, Culine S, Jouin P, Rioux-Leclercq N, and Darbouret B (2008). Identification of pro-MMP-7 as a serum marker for renal cell carcinoma by use of proteomic analysis. Clin. Chem 54, 574–581. [DOI] [PubMed] [Google Scholar]
- Sawyers CL (2008). The cancer biomarker problem. Nature 452, 548–552. [DOI] [PubMed] [Google Scholar]
- Schaaij-Visser TBM, de Wit M, Lam SW, and Jiménez CR (2013). The cancer secretome, current status and opportunities in the lung, breast and colorectal cancer context. Biochim. Biophys. Acta 1834, 2242–2258. [DOI] [PubMed] [Google Scholar]
- Schwenk JM, Omenn GS, Sun Z, Campbell DS, Baker MS, Overall CM, Aebersold R, Moritz RL, and Deutsch EW (2017). The Human Plasma Proteome Draft of 2017: Building on the Human Plasma PeptideAtlas from Mass Spectrometry and Complementary Assays. J. Proteome Res 16, 4299–4310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shevde LA, and Samant RS (2014). Role of osteopontin in the pathophysiology of cancer. Matrix Biol. 37, 131–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solé X, Crous-Bou M, Cordero D, Olivares D, Guineó E, Sanz-Pamplona R, Rodriguez-Moranta F, Sanjuan X, de Oca J, Salazar R, and Moreno V (2014). Discovery and validation of new potential biomarkers for early detection of colon cancer. PLoS One 9, e106748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spangenberg HC, Thimme R, and Blum HE (2006). Serum markers of hepatocellular carcinoma. Semin. Liver Dis 26, 385–390. [DOI] [PubMed] [Google Scholar]
- Stastna M, and Van Eyk JE (2012). Secreted proteins as a fundamental source for biomarker discovery. Proteomics 12, 722–735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, and Mesirov JP (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. USA 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takeuchi T, Adachi Y, and Nagayama T (2011). Expression of a secretory protein C1qTNF6, a C1qTNF family member, in hepatocellular carcinoma. Anal. Cell. Pathol. (Amst.) 34, 113–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tamir A, Jag U, Sarojini S, Schindewolf C, Tanaka T, Gharbaran R, Patel H, Sood A, Hu W, Patwa R, et al. (2014). Kallikrein family proteases KLK6 and KLK7 are potential early detection and diagnostic biomarkers for serous and papillary serous ovarian cancer subtypes. J. Ovarian Res 7, 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsubamoto H, Sakata K, Sakane R, Inoue K, Shibahara H, Hao H, and Hirota S (2016). Gremlin 2 is Repressed in Invasive Endometrial Cancer and Inhibits Cell Growth In Vitro. Anticancer Res. 36, 199–203. [PubMed] [Google Scholar]
- Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu Å, Sivertsson A, Kampf C, Sjöstedt E, Asplund A, et al. (2015). Proteomics. Tissue-based map of the human proteome. Science 347, 1260419. [DOI] [PubMed] [Google Scholar]
- Uhlen M, Zhang C, Lee S, Sjöstedt E, Fagerberg L, Bidkhori G, Benfeitas R, Arif M, Liu Z, Edfors F, et al. (2017). A pathology atlas of the human cancer transcriptome. Science 357, 660. [DOI] [PubMed] [Google Scholar]
- Urra H, Dufey E, Avril T, Chevet E, and Hetz C (2016). Endoplasmic Reticulum Stress and the Hallmarks of Cancer. Trends Cancer 2, 252–262. [DOI] [PubMed] [Google Scholar]
- Vaeteewoottacharn K, Kariya R, Matsuda K, Taura M, Wongkham C, Wongkham S, and Okada S (2013). Perturbation of proteasome function by bortezomib leading to ER stress-induced apoptotic cell death in cholangiocarcinoma. J. Cancer Res. Clin. Oncol 139, 1551–1562. [DOI] [PubMed] [Google Scholar]
- Väremo L, Nielsen J, and Nookaew I (2013). Enriching the gene set analysis of genome-wide data by incorporating directionality of gene expression and combining statistical hypotheses and methods. Nucleic Acids Res. 41, 4378–4391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vathipadiekal V, Wang V, Wei W, Waldron L, Drapkin R, Gillette M, Skates S, and Birrer M (2015). Creation of a Human Secretome: A Novel Composite Library of Human Secreted Proteins: Validation Using Ovarian Cancer Gene Expression Data and a Virtual Secretome Array. Clin. Cancer Res 21, 4960–4969. [DOI] [PubMed] [Google Scholar]
- Vlassov AV, Magdaleno S, Setterquist R, and Conrad R (2012). Exosomes: current knowledge of their composition, biological functions, and diagnostic and therapeutic potentials. Biochim. Biophys. Acta 1820, 940–948. [DOI] [PubMed] [Google Scholar]
- Webb S (2016). The cancer bloodhounds. Nat. Biotechnol 34, 1090–1094. [DOI] [PubMed] [Google Scholar]
- Welsh JB, Sapinoso LM, Kern SG, Brown DA, Liu T, Bauskin AR, Ward RL, Hawkins NJ, Quinn DI, Russell PJ, et al. (2003). Large-scale delineation of secreted protein biomarkers overexpressed in cancer tissue and serum. Proc. Natl. Acad. Sci. USA 100, 3410–3415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkinson DG (2001). Multiple roles of EPH receptors and ephrins in neural development. Nat. Rev. Neurosci 2, 155–164. [DOI] [PubMed] [Google Scholar]
- World Health Organization (2017). Guide to Cancer. Early Diagnosis (World Health Organization; ). [Google Scholar]
- Wu CC, Hsu CW, Chen CD, Yu CJ, Chang KP, Tai DI, Liu HP, Su WH, Chang YS, and Yu JS (2010). Candidate serological biomarkers for cancer identified from the secretomes of 23 cancer cell lines and the human protein atlas. Mol. Cell. Proteomics 9, 1100–1117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xue H, Lu B, and Lai M (2008). The cancer secretome: a reservoir of biomarkers. J. Transl. Med 6, 52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamatoji M, Kasamatsu A, Kouzu Y, Koike H, Sakamoto Y, Ogawara K, Shiiba M, Tanzawa H, and Uzawa K (2012). Dermatopontin: a potential predictor for metastasis of human oral cancer. Int. J. Cancer 130, 2903–2911. [DOI] [PubMed] [Google Scholar]
- Yan Q, Jiang L, Liu M, Yu D, Zhang Y, Li Y, Fang S, Li Y, Zhu YH, Yuan YF, and Guan XY (2017). ANGPTL1 Interacts with Integrin α1β1 to Suppress HCC Angiogenesis and Metastasis by Inhibiting JAK2/STAT3 Signaling. Cancer Res. 77, 5831–5845. [DOI] [PubMed] [Google Scholar]
- Yang WE, Hsieh MJ, Lin CW, Kuo CY, Yang SF, Chuang CY, and Chen MK (2017). Plasma Levels of Endothelial Cell-Specific Molecule-1 as a Potential Biomarker of Oral Cancer Progression. Int. J. Med. Sci 14, 1094–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ying L, Zhang F, Pan X, Chen K, Zhang N, Jin J, Wu J, Feng J, Yu H, Jin H, and Su D (2016). Complement component 7 (C7), a potential tumor suppressor, is correlated with tumor progression and prognosis. Oncotarget 7, 86536–86546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoneda K, Iida H, Endo H, Hosono K, Akiyama T, Takahashi H, Inamori M, Abe Y, Yoneda M, Fujita K, et al. (2009). Identification of Cystatin SN as a novel tumor marker for colorectal cancer. Int. J. Oncol 35, 33–40. [PubMed] [Google Scholar]
- Yoshihara K, Shahmoradgoli M, Martínez E, Vegesna R, Kim H, Torres-Garcia W, Treviño V, Shen H, Laird PW, Levine DA, et al. (2013). Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun 4, 2612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu WW, Guo JJ, Guo L, Jia HL, Zhu M, Zhang JB, Loffredo CA, Forgues M, Huang H, Xing XJ, et al. (2013). Evaluation of midkine as a diagnostic serum biomarker in hepatocellular carcinoma. Clin. Cancer Res 19, 3944–3954. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.