

Abstract
Key points
Impaired growth during fetal life can reprogramme heart development and increase the risk for long‐term cardiovascular dysfunction.
It is uncertain if the developmental window during which the heart is vulnerable to reprogramming as a result of inadequate nutrition extends into the postnatal period.
We found that adult female mice that had been undernourished only from birth to 3 weeks of age had disproportionately smaller hearts compared to males, with thinner ventricle walls and more mononucleated cardiomyocytes.
In females, but not males, cardiac diastolic function, and heart rate responsiveness to adrenergic stimulation were limited and maximal exercise capacity was compromised.
These data suggest that the developmental window during which the heart is vulnerable to reprogramming by inadequacies in nutrient intake may extend into postnatal life and such individuals could be at increased risk for a cardiac event as a result of strenuous exercise.
Abstract
Adults who experienced undernutrition during critical windows of development are at increased risk for cardiovascular disease. The contribution of cardiac function to this increased disease risk is uncertain. We evaluated the effect of a short episode of postnatal undernutrition on cardiovascular function in mice at the whole animal, organ, and cellular levels. Pups born to control mouse dams were suckled from birth to postnatal day (PN) 21 on dams fed either a control (20% protein) or a low protein (8% protein) isocaloric diet. After PN21 offspring were fed the same control diet until adulthood. At PN70 was measured by treadmill test. At PN80 cardiac function was evaluated by echocardiography and Doppler analysis at rest and following β‐adrenergic stimulation. Isolated cardiomyocyte nucleation and Ca2+ transients (with and without β‐adrenergic stimulation) were measured at PN90. Female mice that were undernourished and then refed (PUN), unlike male mice, had disproportionately smaller hearts and their exercise capacity, cardiac diastolic function, and heart rate responsiveness to adrenergic stimulation were limited. A reduced left ventricular end diastolic volume, impaired early filling, and decreased stored energy at the beginning of diastole contributed to these impairments. Female PUN mice had more mononucleated cardiomyocytes; under resting conditions binucleated cells had a functional profile suggestive of increased basal adrenergic activation. Thus, a brief episode of early postnatal undernutrition in the mouse can produce persistent changes to cardiac structure and function that limit exercise/functional capacity and thereby increase the risk for the development of a wide variety of cardiovascular morbidities.
Keywords: neonatal nutrition, developmental programming, heart, exercise
Key points
Impaired growth during fetal life can reprogramme heart development and increase the risk for long‐term cardiovascular dysfunction.
It is uncertain if the developmental window during which the heart is vulnerable to reprogramming as a result of inadequate nutrition extends into the postnatal period.
We found that adult female mice that had been undernourished only from birth to 3 weeks of age had disproportionately smaller hearts compared to males, with thinner ventricle walls and more mononucleated cardiomyocytes.
In females, but not males, cardiac diastolic function, and heart rate responsiveness to adrenergic stimulation were limited and maximal exercise capacity was compromised.
These data suggest that the developmental window during which the heart is vulnerable to reprogramming by inadequacies in nutrient intake may extend into postnatal life and such individuals could be at increased risk for a cardiac event as a result of strenuous exercise.
Introduction
There is increasing evidence that, in addition to genetic predisposition or lifestyle factors, a suboptimal nutritional environment in early life can modulate developmental processes to produce permanent changes that increase the risk for the development of chronic diseases, including heart disease (Barker, 1999; Osmond & Barker, 2000; Thornburg, 2015). An increased incidence of cardiovascular anomalies that parallel those observed in humans also occurs in a variety of animal models (mouse, rat, sheep, guinea‐pig, and primate) subjected to undernutrition during gestation and/or early postnatal life (Briscoe et al. 2004; Louey et al. 2007; Morrison et al. 2007; Botting et al. 2012; Goyal & Longo, 2013; Zohdi et al. 2014; Kuo et al. 2017). Thus, affected individuals would not only be at increased risk for cardiovascular disease in adulthood, but are likely to have limited functional capacity that restricts their tolerance for exercise and, thereby, exacerbates the development of other chronic morbidities.
By full‐term birth in mammals, structural development of the heart, including the acquisition of most cardiomyocytes, is largely complete although the extent of this is species‐dependent. Postnatal growth is primarily through hypertrophy of existing cardiomyocytes accompanied by varying degrees of cell division, nuclear division, and/or polyploidization depending on species. The capacity of cardiomyocytes for division is retained until the cells undergo terminal differentiation, and is associated with the cells becoming binucleated and/or polyploid (Patterson et al. 2017). In humans, cardiomyocyte numbers increase until 20 years of age by some studies (Mollova et al. 2013), albeit at low rates after the first year of life; the number of mononucleated cells remains relatively constant, whereas the number of polyploid nuclei increases into adulthood (Mollova et al. 2013). In sheep, left ventricular (LV) cardiomyocyte division is largely complete by birth, but persists for a short while in the right ventricle; nuclear division continues in both ventricles for 2–3 weeks (Burrell et al. 2003; Jonker et al. 2015). In rodents, myocyte proliferation continues for a few days postnatally with nuclear division extending into the second week of life (Li et al. 1996; Soonpaa et al. 1996; Ikenishi et al. 2012) coincident with attainment of functional maturity (Li et al. 1996; Soonpaa et al. 1996; Leu et al. 2001; Hamaguchi et al. 2013; Foglia & Poss, 2016). During this terminal phase of cardiomyocyte development when the loss of functionally impaired mononucleated cardiomyocytes also occurs, there is maturation of the sarcoplasmic reticulum and t‐tubule membrane systems (Hamaguchi et al. 2013) which, together with the increase in cell mass, contribute to the attainment of mature cardiac function, especially diastolic function (Chen et al. 2007). Thus, perturbations to the growing organism's environment during the prenatal versus the postnatal period will impact on distinct cellular processes in the heart and, as a result, the long‐term effects incurred may differ.
In humans, the cardiac dysfunction with altered heart size and shape in term fetuses that experience intrauterine growth retardation (IUGR) suggests that some aspects of heart development are vulnerable to the adverse environment they have experienced (Perez‐Cruz et al. 2015). These functional deficits persist into childhood and early adulthood (Crispi et al. 2010; Tennant et al. 2014). Recently, there also has been increasing awareness that premature infants, which comprise 5–18% of births (Blencowe et al. 2012) and where cardiac maturation occurs postnatally, are at a greater risk of early onset heart failure regardless of growth status (Carr et al. 2017). Thus, the extent of cardiac maturation that occurs after birth will depend on the degree of prematurity, which could be substantial, and subject to the postnatal nutritional state. In sheep and rodents, newborns that experienced prenatal growth impairment have fewer cardiomyocytes (Corstius et al. 2005; Stacy et al. 2009; Master et al. 2014) and their capacity to recover appears to depend on the developmental stage at recovery (Bai et al. 1990; Lim et al. 2010; Black et al. 2012; Botting et al. 2012; Vranas et al. 2017). However, the extent to which alterations in cardiac cell size, number and nucleation/ploidy are impacted when a nutritional deficit occurs only after birth, i.e. during the terminal phase of maturation, and whether these are recoverable or contribute to cardiac dysfunction in later life has received less attention.
From the functional perspective, physiological adaptations may compensate for differences in heart size and cardiomyocyte function under conditions of low cardiovascular demand. Thus, consequences of a reduction in cardiac functional capacity are more likely to be manifested acutely when there is an increased demand for blood flow to the peripheral tissues, as occurs with exercise (Powers & Howley, 2015), or chronically when there is increased afterload, as occurs with hypertension. During acute exercise, cardiac output increases through increases in stroke volume and heart rate. These chronotropic, inotropic and lusitropic responses, driven in large part by β‐adrenergic pathways, are dependent on the excitation‐contraction coupling mechanisms mediated by calcium cycling in response to action potentials. Thus, we hypothesized that if developmental anomalies in cardiovascular function incurred by inadequate nutrition during the terminal phase of cardiac maturation are not reversible following nutritional rehabilitation, exercise capacity would be compromised into adulthood even after full nutritional rehabilitation. To test the hypothesis, we assessed cardiac function in vivo and cardiomyocyte structure and function in vitro in nutritionally rehabilitated adult mice that experienced normal intrauterine development, but were undernourished from birth to weaning by suckling them on dams fed a protein‐restricted diet. Global cardiovascular fitness was assessed from performance on a treadmill maximal exercise‐stress test, whereas at the organ and cellular levels, cardiac and cardiomyocyte parameters of function were measured under baseline conditions and following β‐adrenergic stimulation. We established that in females there were effects of postnatal undernutrition on cardiac function, especially diastolic function. These differences incurred adaptive responses to maintain cardiovascular function under basal conditions, but they served to limit the capacity to respond to increases in cardiovascular demand and resulted in the development of exercise intolerance.
Methods
Ethical approval
All experiments were conducted according to the National Research Council's Guide for the Care and Use of Laboratory Animals and approved by the Institutional Animal Care and Use Committee at Baylor College of Medicine.
Experimental design and nutritional model
The study was designed to evaluate the long‐term effects of global postnatal undernutrition during the suckling period following a normal intrauterine growth experience. To induce postnatal undernutrition we used a model that we and others have described previously (Langley‐Evans, 2000; Fiorotto et al. 2014) in which mouse pups born to well‐nourished dams are cross‐fostered at birth and nursed until 21 days of age on dams fed a low protein (LP) diet (Fig. 1). This intervention results in the production of less milk that contains slightly less protein and more fat. Thus, pups principally experience protein‐energy malnutrition with a marginally greater deficit in protein intake (Sampson et al. 1986; Grimble & Mansaray, 1987; Pine et al. 1994; Derrickson & Lowas, 2007; Agnoux et al. 2015).
Figure 1. A schematic representation of the experimental design showing the number of litters generated and mice used for each set of measurements.
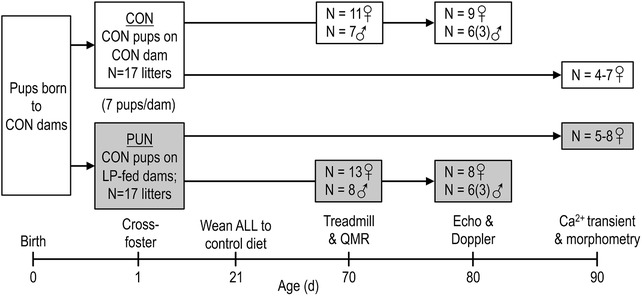
Mice born to well‐nourished control dams were cross‐fostered at 1 day of age to dams fed either a control diet (CON) or an isocaloric low protein diet (LP) to induce postnatal undernutrition (PUN) until weaning. All mice were weaned at 21 day of age to the control diet, thus limiting the period of undernutrition from day 1 to 21 days of age. During adulthood (70–90 days of age) mice were subjected to several tests to assess cardiac function via a maximal test, echocardiography and Doppler measurements, and cardiomyocyte calcium transient analysis.
Second and third parity FVB (FVB/N; Charles River Laboratories, Wilmington, MA, USA) mouse dams were fed a control diet (20% protein, 7% fat; Research Diets, New Brunswick, NJ, USA) based on AIN93G, or an isocaloric low‐protein (LP) diet (8% protein, 7% fat) throughout gestation and lactation. The diets were identical other than the replacement of casein with an equicaloric amount of the carbohydrate mix (starch, sugar and maltodextrins) (Fiorotto et al. 2014). Mating was timed by introducing males for a 24‐h period. On postnatal day (PN) 1 pups born on the same day to dams fed the control diet were pooled, and then reassigned in litters of seven pups (males and females) to either control (n = 17 litters) or LP dams (n = 17 litters) to generate the two experimental groups: controls (CON), i.e. pups born to and suckled by well‐nourished control dams; postnatal undernutrition (PUN), i.e. pups born to control dams and cross‐fostered to and suckled by LP dams. All offspring were weaned at PN21 to the control diet which they then consumed for the rest of the experiment (Fig. 1).
Mice were housed on wood‐chip bedding in a single room maintained at 23°C with a 12 h light/dark cycle. Body weight was measured weekly. Starting at PN70 mice were subjected to functional measurements detailed below. Exercise testing and in vivo cardiac function measurements were performed on only one pup of each sex from each litter; cardiomyocyte isolation and measurements were performed on a separate female littermate. The number of mice for which measurements were completed is summarized in Fig. 1. The experiments met the recommendations set out by ARRIVE guidelines for executing research with experimental animals. For all measurements the individuals performing the tests and analysing the data were blinded to the animals’ treatment group (PUN vs. CON).
Body composition
At PN70 (adulthood) mice undergoing the treadmill test were weighed and body composition was measured by quantitative magnetic resonance (EchoMRI, Houston, TX, USA). Body fat and lean masses were estimated using the supplied software.
treadmill test
A graded treadmill test with oxygen consumption () and carbon dioxide production () monitoring (Metabolic Modular Treadmill, Columbus Instruments, Columbus, OH, USA) was performed. At PN61, mice were randomly selected from each litter and a 5‐day treadmill acclimation protocol was initiated to familiarize the mice with the test procedure, but insufficient to produce a training effect. The treadmill test was performed at PN70. On testing day mice were weighed and placed in the metabolic treadmill for 5 min, followed by a 10 min warm‐up at 10 m/min at 0 gradient. After the warm‐up the incline was raised to 10 deg and the speed increased 5 m/min every 2 min until voluntary exhaustion when the mouse sat on the shock grid for greater than 15 s or touched the shock grid 5 times in a 30 s period. and were measured every 30 s. was measured at the time point prior to the mouse touching the shock grid to terminate the test. Instantaneous and production rates were derived mathematically by deconvolution of the measured chamber values of and using the first order differential equation that describes the flow characteristics of the chamber (per Columbus Instruments). Maximal work performed was calculated at the point of test termination using the equation (Pederson et al. 2005):
Echocardiography/Doppler blood flow
On PN78 mice were transferred to the DeBakey Heart Center Research Core at Methodist Hospital (Houston, TX, USA) and allowed 2 days to acclimate to minimize potential stress effects before evaluation of cardiovascular function. Echocardiography and Doppler blood flow analyses were performed at PN80 as described previously (Milner et al. 1999; Chintalgattu et al. 2010). Mice were maintained under 1% isoflurane anaesthesia on an ECG/heated board with the limbs taped to the four electrodes. Echocardiography (Vevo 770 ultrasound, with 30 MHz transducer; Visualsonics, Toronto, Canada) measurements were performed in 2D and M‐modes with images taken in the short axis position at the level of the papillary muscles, and used to determine left ventricle (LV) systolic and diastolic dimensions. Mitral and aortic blood flow velocities were measured by Doppler from the apical view (10 MHz pulsed Doppler probe with real time Doppler spectrum analyser; Indus Instruments, Webster, TX, USA). Doppler and echocardiograph recordings were stored for offline analysis; all analyses were done with observer blinded to the animal's treatment group. After obtaining a set of baseline measurements, dobutamine (dobutamine hydrochloride (1.5 μg/g BW); Hospira, Lake Forest, IL, USA) was administered i.p. and the measurements were repeated. The mice recovered for 48 h, after which they were deeply anesthetized (4% isoflurane), the hearts dissected quantitatively, and weighed. Heart weight was normalized for lean mass or body surface area (BSA) calculated using the formula (Cheung et al. 2009):
The parametrized diastolic filling (PDF) parameters and derivatives that describe a spring model of diastolic filling (Kovacs et al. 1987) were computed as previously described (Trial et al. 2017). The E‐wave envelope acquired by Doppler was fitted to a curve that describes a damped harmonic oscillator described by the following formula:
The fit for the E‐wave was accepted when the goodness of fit (r²) was >0.99. Five beats were analysed per mouse. The derived parameters represent ventricular stiffness (k), visco‐elasticity (c), and volumetric load (x 0); h is a time offset and t is time. From these, we computed the stored elastic strain energy preceding valve opening , and the maximum damping slowing filling (cEpeak).
Isolation of cardiomyocytes and Ca2+ transient, and spontaneous spark and wave analysis
At PN90 cardiomyocytes were isolated from 7 CON and 8 PUN female littermates of mice described above (Reynolds et al. 2016). Briefly, under deep anaesthesia with 4% isoflurane, the heart was removed, the aorta cannulated, and the heart was secured to a Langendorff apparatus and perfused retrograde with a zero calcium, collagenase‐containing Tyrode buffer, pH 7.4 (Sigma‐Aldrich, St Louis, MO, USA). Released cardiomyocytes were re‐introduced to calcium through successive centrifugations and resuspensions in Tyrode buffer to a final concentration of 1.8 mM Ca2+. An aliquot of cells was incubated with 10 μM Fura‐4 AM (Sigma‐Aldrich) for Cai 2+ release analysis. Cells measured were selected based on structure (clearly visible sarcomeres using bright field microscopy) and contractile properties (contracted in in response to field stimulation, but were quiescent in the absence of pacing) (n = 20 cells per mouse heart). To record Ca2+ transients, a line scan through the midline of the Fura‐4 AM‐loaded cardiomyocytes was performed with confocal microscopy (LSM 510 META). Transients were recorded at room temperature without field stimulation for 20 s and then paced for 120 s at 1 Hz; transients were recorded for the last 20 s (Fig. 6 A). To assess spontaneous Ca2+ activity, the recording continued for an additional 20 s after pacing had ceased as described elsewhere (Reynolds et al. 2016). To simulate an adrenergic response, cells were incubated for 5 min in Tyrode buffer with 500 nM isoproterenol (isoprenaline; Sigma) (n = 20 cells/mouse heart). Ca2+ transients were analysed using the Peak Analysis module in LabChart software (ADInstruments). For a subset of mice (n = 4 CON; n = 5 PUN), Ca2+ spark frequency was measured after pacing (either at 1 Hz or 2 Hz) had ceased, and analysed using SparkMaster in ImageJ (Picht et al. 2007). Ca2+ wave rates were determined by counting unstimulated increases in internal calcium that propagated in a wave‐like manner after pacing had ceased. A separate aliquot of cells from the same mouse heart was analysed for each measurement condition.
Figure 6. Calcium transient analysis of isolated binucleated cardiomyocytes from CON and PUN females at PN90 before (basal) and after stimulation with isoproterenol.
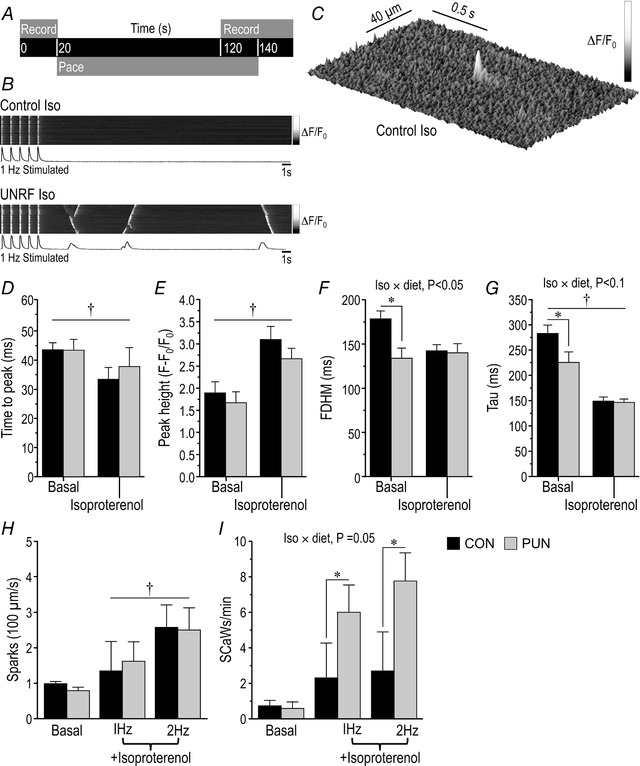
A, pacing and recording protocol; B, representative line scan images of CON and PUN cardiomyocytes in the presence of isoproterenol; C, representative Ca2+ spark in a CON cardiomyocyte in the presence of isoproterenol; D, time from external stimulation to transient peak; E, peak height of transient measured as the relative change in fluorescence intensity; F, full duration of Ca2+peak at half‐maximum (FDHM); G, rate of Ca2+ decay from maximal peak height to baseline as indicated by the decay constant, tau. Values are means ± SEM; n = 7 CON and 8 PUN female mice per dietary group with 20 cells measured per basal and isoproterenol treatment per mouse; diet × isoproterenol P < 0.05–0.1; * P < 0.05 CON vs. PUN; † P < 0.001 baseline vs. +isoproterenol. H, quantification of Ca2+ spark rate (CON, n = 4 mice, 24 cells 1 Hz, 20 cells 1 Hz +isoproterenol, 18 cells 2 Hz, 20 cells 2 Hz +isoproterenol; PUN, n = 5 mice, 20 cells 1 Hz, 22 cells 1 Hz +isoproterenol, 19 cells 2 Hz, 20 cells 2 Hz +isoproterenol; † P = 0.055 for 1 Hz vs. 2 Hz stimulation. I, quantification of spontaneous Ca2+ wave frequencies rate (CON, n = 4 mice, 24 cells 1 Hz, 20 cells 1 Hz +isoproterenol, 18 cells 2 Hz, 20 cells 2 Hz +isoproterenol; PUN, n = 5 mice, 20 cells 1 Hz, 22 cells 1 Hz +isoproterenol, 19 cells 2 Hz, 20 cells 2 Hz +isoproterenol); diet × isoproterenol P < 0.05–0.1; * P < 0.05 CON vs. PUN.
Isolated cardiomyocyte morphology
An aliquot of cardiomyocytes from each mouse also was fixed in 10% zinc‐formalin (Fisher Scientific, Waltham, MA, USA) and stored in phosphate buffered saline with 1 mM 5‐bromo‐5‐nitro‐1,3‐dioxane (Sigma‐Aldrich) to preserve the integrity of the sample during staining and imaging procedures and to optimize measurement of nucleation and cardiomyocyte cross‐sectional area (CSA). Following washing and permeabilization, cells were positively identified by staining with an antibody to myosin heavy chain (MF20; Developmental Studies Hybridoma Bank, Iowa City, IA, USA) followed by an AlexaFluor 647 tagged‐secondary antibody. Nuclei were visualized by staining with Sytox Green (Invitrogen, Waltham, MA, USA). Images were captured with a confocal fluorescence microscope and using Image Pro software (MediaCybernetics, Rockville, MD, USA), cell CSA, and the number of nuclei per cardiomyocytes was assessed; for each mouse 75–100 cells were analysed.
Statistics
In general, data were analysed with a two‐way ANOVA with diet group (CON and PUN) and sex as main effects (MINITAB, release 14, State College, PA, USA). Interactions were first evaluated and where P ≤ 0.1, the main effects were examined individually; when P ≥ 0.1 for an interaction, it was removed from the model. Post hoc analysis was done using Tukey's method for multiple comparisons. Because pups within a litter are not statistically independent, litter was used as the statistical unit; thus, where the same variable was measured for more than one offspring in the same litter (e.g. body weight, composition, heart weights), data were averaged within the litter for each sex. Exercise testing results were analysed by ANCOVA using resting as covariate. For in vivo and in vitro cardiac function analysis, an ANOVA was used comparing the main effects of diet (CON or PUN), sex (male or female), and β‐adrenergic stimulation (baseline or dobutamine/isoproterenol). Because each mouse was measured both at baseline and upon adrenergic stimulation, mouse was treated as a repeated measure within dietary treatment. For assessment of Ca2+ sparks and waves, baseline activity was used as a covariate. Differences with P ≤ 0.05 were considered significantly different. Values are expressed as least square means ± SE. All tests were performed in MINITAB with an alpha level set a 0.05.
Results
Growth and body composition
By PN4, PUN mice weighed less than CON (P < 0.001) and by PN21 body weight differed by 30% (Fig. 2 A); there was no sex difference over this time (diet × sex, n.s.; sex, n.s.). On refeeding PUN mice underwent some catch‐up growth, and weighed approximately 80% of CON mice at PN70. Sex differences were evident by PN28 (sex, P < 0.05), but there was no sex difference in the extent of recovery. The body weight differences in PUN mice reflected deficits in both fat and lean masses (diet effect for both, P < 0.001; Fig. 2 B); the deficits in fat and lean were approximately 45% and 15%, respectively in male and female PUN mice.
Figure 2. Body weight growth curves and body composition of CON and PUN female and male mice.
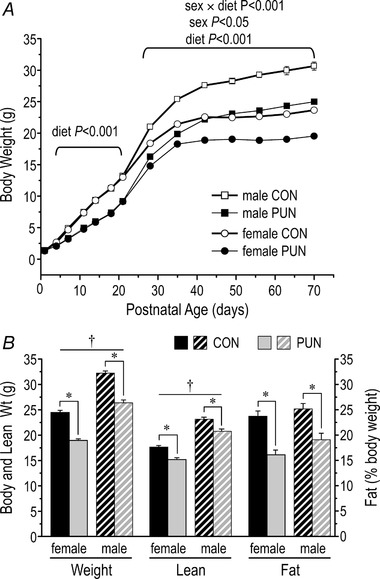
A, average body weight of individual male and female mice from birth to 70 days of age (PN70). Body weight was reduced in PUN mice compared to CON starting at PN4, with no sex difference until PN28, after which males were significantly heavier than females within each dietary group; n = 17 litters/group; values are means ± SE. B, body weight, lean mass, and fat (% of body weight) at PN70 of male and female CON and PUN mice. Values are mean ± SEM. * P < 0.001 for CON vs. PUN; † P < 0.001 for male vs. female; diet × sex, n.s.).
Absolute heart weight was lower in both male and female PUN mice (diet, P < 0.001), with a greater deficit in female mice (sex × diet, P < 0.05; sex, P < 0.001; Fig. 3 A). The difference was proportional to the difference in body weight. When normalized for metabolic (lean) mass or BSA, heart weight was disproportionately small in the PUN females (sex × diet, P < 0.03; diet effect in females, P < 0.001, Fig. 3 B). For PUN males, the smaller hearts were proportional to their smaller lean mass or BSA (diet effect in males, n.s.).
Figure 3. Heart weight of female and male CON and PUN mice at PN80.
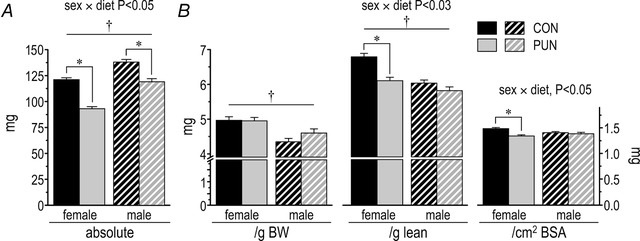
Data are presented in absolute terms (A), or normalized to body weight (BW), lean mass, or body surface area (BSA) (B). Values are mean ± SEM; sex × diet P < 0.05; * P < 0.001 for CON vs. PUN; † P < 0.001 for male vs. female)
Exercise tests
There was a significant effect of baseline O2 consumption on ( over 1 min before the treadmill was started vs. , r = 0.85, P < 0.001). Thus, to assess the effects of the postnatal undernutrition on independently of baseline differences, at baseline was included as a covariate in the statistical analysis. The effects of diet on parameters of exercise capacity measured by the treadmill‐stress test were sex‐dependent. Both total work performed over the entire test (Fig. 4 A) and the maximal work achieved before exhaustion (Fig. 4 B) were approximately 20% less in the PUN than CON females (P < 0.001). These parameters did not differ between PUN and CON male mice. , normalized for basal was still lower in PUN than CON female but not male mice (P < 0.01; Fig. 4 C).
Figure 4. Exercise responses of male and female CON and PUN mice at PN70.
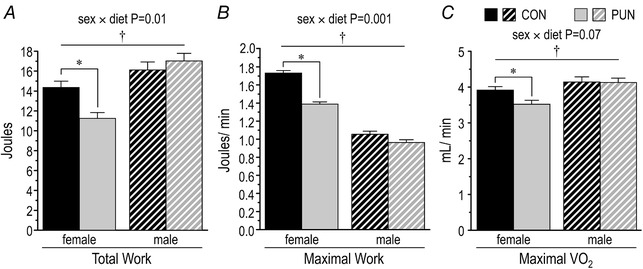
A, total work (joules (J)) performed; B, maximal work (J/min) performed in the last stage of the exercise test; C, maximal oxygen consumption ( , mL/min, corrected for at the start of the test) during the last stage of the exercise protocol. Values are means ± SEM; diet × sex P < 0.001–0.07; * P < 0.001 for CON vs. PUN; † P < 0.05 for male vs. female.
Echocardiography and Doppler blood flow
In vivo cardiac function assessments were performed in female mice and a subset of male mice (Tables 1, 2 and 3). Calculated LV mass of both male and female PUN mice was less than for CON (Table 1), although when corrected for lean mass there was no longer a difference. Posterior and anterior LV walls in diastole were 25% thinner in female (P < 0.05) but not male PUN mice (diet × sex, P = 0.1 and 0.04, respectively; Table 1).
Table 1.
Cardiac parameters obtained by echocardiography at baseline and after dobutamine treatment of CON and PUN female and male mice at PN80
| Female | Male | P | ||||||
|---|---|---|---|---|---|---|---|---|
| CON | PUN | CON | PUN | |||||
| n | 9 | 8 | 6 (3)2 | 6 (3)2 | Diet × Sex1 | Diet | Sex | |
| LV mass (mg) | 67.9 ± 5.2 | 51.9 ± 5.5 | 101.9 ± 6.3 | 84.6 ± 6.3 | n.s. | 0.01 | <0.001 | |
| LV mass (mg/g lean) | 3.80 ± 0.29 | 3.37 ± 0.31 | 4.45 ± 0.36 | 4.16 ± 0.36 | n.s. | n.s. | 0.04 | |
| LV posterior wall (mm) | 0.66 ± 0.05a | 0.48± 0.05b | 0.87 ± 0.06c | 0.87 ± 0.06c | 0.1 | |||
| LV anterior wall (mm) | 0.71 ± 0.05a | 0.53± 0.05b | 0.64 ± 0.05a | 0.68 ± 0.05a | 0.04 | |||
| LV diastole vol (μL) | B | 67.6 ± 2.8 | 59.1 ± 3.0 | 84.3 ± 3.5 | 68.5 ± 3.5 | n.s. | 0.001 | <0.001 |
| +Db | 59.1 ± 2.4 | 50.2 ± 2.5 | 59.4 ± 4.1 | 51.8 ± 4.1 | n.s. | 0.04 | n.s. | |
| Stroke volume (μL) | B | 42.7 ± 1.7 | 36.9 ± 1.8 | 47.7 ± 2.1 | 45.4 ± 2.1 | n.s. | 0.05 | 0.002 |
| +Db | 43.6 ± 1.8 | 37.8 ± 1.9 | 45.1 ± 3.1 | 42.8 ± 3.1 | n.s. | 0.04 | n.s. | |
| FS (%) | B | 37.6± 1.7b | 31.6 ± 1.7ab | 29.5 ± 2.0a | 37.4 ± 2.0b | 0.001 | ||
| +Db | 44.4 ± 2.4 | 42.4 ± 2.6 | 44.1 ± 4.2 | 53.0 ± 4.2 | n.s. | n.s. | n.s. | |
| Cardiac index3 (cm/s) | B | 27.6 ± 1.2 | 25.7 ± 1.2 | 30.4 ± 1.4 | 31.4 ± 1.3 | n.s. | n.s. | 0.01 |
| +Db | 39.2 ± 1.5a | 33.7 ± 1.5b | 49.6 ± 2.4c | 47.9 ± 2.4c | 0.1 | |||
Values are means ± SEM for mice at baseline (B) and when stimulated with 1.5 μg dobutamine/g BW (+Db).
1Where P ≤ 0.1 for diet × sex interaction, post hoc analysis was performed to test for the effect of sex within diet, or diet within sex; significance is indicated by superscripts a, b, and c, such that P < 0.05 when means do not share the same superscript; n.s., not significant.
2Dobutamine injections were successful in only 3 male mice/dietary treatment.
3From mean aortic velocity (by Doppler).
Table 2.
Cardiac parameters obtained by Doppler blood flow measurements at baseline and after dobutamine treatment of CON and PUN female and male mice at PN80
| Female | Male | P | ||||||
|---|---|---|---|---|---|---|---|---|
| CON | PUN | CON | PUN | |||||
| n | 9 | 8 | 6 (3)2 | 6 (3)2 | Diet × Sex1 | Diet | Sex | |
| Mitral inflow (diastole) | ||||||||
| Heart rate (beats/min) | B | 405 ± 8a | 435 ± 8b | 428 ± 10b | 417 ± 10b | 0.05 | ||
| +Db | 511 ±17 | 504 ± 18 | 588 ± 30 | 577 ± 30 | n.s. | n.s. | 0.007 | |
| Isovolumic contraction (ms) | B | 9.76 ± 0.48a | 11.92 ± 0.51b | 10.17 ± 0.62a | 10.17 ± 0.62a | 0.09 | ||
| +Db | 7.72 ± 0.28 | 7.92 ± 0.29 | 6.38 ± 0.51 | 7.01 ± 0.51 | n.s. | n.s. | 0.01 | |
| Isovolumic contractioncorr 3 | B | 0.065 ± 0.004a | 0.085 ± 0.004b | 0.071 ± 0.004a | 0.069 ± 0.004a | 0.01 | ||
| +Db | 0.065 ± 0.002 | 0.64 ± 0.003 | 0.062 ± 0.004 | 0.067 ± 0.004 | n.s. | n.s. | n.s. | |
| Isovolumic relaxation (ms) | B | 14.81 ± 0.74a | 17.29 ± 0.79b | 16.98 ± 0.97b | 16.00 ± 0.97b | 0.006 | ||
| +Db | 13.22 ± 0.80 | 15.51 ± 0.76 | 12.61 ± 1.30 | 11.59 ± 1.30 | n.s. | n.s. | 0.05 | |
| Isovolumic relaxationcorr 3 | B | 0.099 ± 0.005a | 0.124 ± 0.006b | 0.121 ± 0.006b | 0.111 ± 0.006b | 0.006 | ||
| +Db | 0.112 ± 0.005a | 0.124 ± 0.005b | 0.123 ± 0.008b | 0.108 ± 0.008b | 0.05 | |||
| Epeak velocity (cm/s) | B | 81.0 ± 2.0a | 66.7 ± 2.1b | 86.3 ± 2.6a | 83.9 ± 2.1a | 0.02 | ||
| +Db | 85.1 ± 1.8 | 79.6 ± 1.9 | 97.9 ± 3.1 | 91.1 ± 3.1 | n.s. | 0.03 | <0.001 | |
| Apeak velocity (cm/s) | B | 52.0 ± 1.6a | 47.7 ± 1.7b | 54.5 ± 2.0a | 55.5 ± 2.0a | 0.1 | ||
| +Db | 66.9 ± 2.4 | 60.3 ± 2.6 | 84.1 ± 4.2 | 79.3 ± 4.2 | n.s. | 0.05 | <0.001 | |
| E/A peak velocity | B | 1.56 ± 0.04 | 1.38 ± 0.05 | 1.64 ± 0.06 | 1.54 ± 0.06 | n.s. | 0.01 | 0.05 |
| +Db | 1.27 ± 0.03 | 1.33 ± 0.03 | 1.17 ± 0.05 | 1.15 ± 0.05 | n.s. | n.s. | 0.004 | |
| Aortic Outflow (systole) | ||||||||
| Peak velocity (cm/s) | B | 111.4 ± 2.7 | 98.5 ± 2.7 | 130.1 ± 3.4 | 124.3 ± 3.1 | n.s. | 0.005 | <0.001 |
| +Db | 133.4 ± 5.8 | 131.1 ± 5.8 | 164.8 ± 9.4 | 159.7 ± 9.4 | n.s. | n.s. | 0.001 | |
| Stroke distance (cm) | B | 3.91 ± 0.11 | 3.47 ± 0.11 | 4.24± 0.13 | 4.18 ± 0.13 | n.s. | 0.05 | <0.001 |
| +Db | 4.35 ± 0.14 | 4.14 ± 0.13 | 5.02 ± 0.25 | 4.36 ± 0.25 | n.s. | 0.05 | 0.04 | |
| Ejection time (ms) | B | 49.7 ± 0.9 | 47.3 ± 1.0 | 44.7 ± 1.1 | 45.90 ± 1.1 | n.s. | n.s. | 0.004 |
| +Db | 46.4 ± 2.1 | 45.3 ± 2.2 | 40.8 ± 3.6 | 38.4 ± 3.6 | n.s. | n.s. | 0.05 | |
| LV MPITEI 4 | B | 0.50 ± 0.03a | 0.60 ± 0.03b | 0.59 ± 0.03b | 0.59 ± 0.03b | 0.07 | ||
| +Db | 0.47 ± 0.02a | 0.53 ± 0.02b | 0.49 ± 0.04ab | 0.48 ± 0.04ab | 0.1 | |||
Values are means ± SEM for mice at baseline (B) and when stimulated with 1.5 μg/g BW of dobutamine (+Db).
1Where P ≤ 0.1 for the diet × sex interaction, post hoc analysis was performed to test for the effect of sex within diet, or diet within sex; the significance is indicated by superscripts a and b, such that P < 0.05 when means do not share the same superscript; n.s., not significant.
2Dobutamine injections were only successful in 3 male mice/dietary treatment group.
3Isovolumic contractioncorr and isovolumic relaxationcorr, values divided by R‐R interval to correct for heart rate;
4MPITEI, calculated as (IVRT+IVCT)/ejection time.
Table 3.
Left ventricular parametrized diastolic filling (PDF) parameters for a spring model of diastolic filling obtained from baseline echocardiographic analysis of CON and PUN female and male mice at PN80
| Female | Male | P | |||||
|---|---|---|---|---|---|---|---|
| CON | PUN | CON | PUN | ||||
| n | 9 | 8 | 6 | 6 | Diet × Sex1 | Diet | Sex |
| x 0 (cm) | 0.021 ± 0.001a | 0.014 ± 0.001b | 0.019 ± 0.002a | 0.020 ± 0.002a | 0.02 | ||
| k (g/s2) | 10247 ± 373a | 11041 ± 373a | 9971 ± 456ab | 9249 ± 456b | 0.08 | ||
| c (g/s) | 194 ± 10 | 145 ± 10 | 173 ± 11 | 157 ± 11 | n.s. | 0.006 | n.s. |
| 1/2kx o 2 (mJ) | 2.14 ± 0.21a | 1.22 ± 0.21b | 1.89 ± 0.25a | 1.80 ± 0.25a | 0.09 | ||
| cEpeak (N) | 157.0 ± 11.2 | 114.5 ± 11.2 | 145.4 ± 13.7 | 129.7 ± 13.7 | n.s. | 0.03 | n.s. |
Values are means ± SEM; PDF Parameters: x 0, load; k, chamber stiffness; c, viscoelasticity; , stored elastic strain energy preceding valve opening; cEpeak, maximum damping slowing filling.
1Where P ≤ 0.1 for the diet × sex interaction, post hoc analysis was performed to test for the effect of sex within diet, or diet within sex; the significance is indicated by superscripts a and b, such that P < 0.05 when means do not share the same superscript; n.s., not significant.
Baseline measurements
LV end‐diastolic and stroke volume were smaller in male and female PUN compared to CON mice. Fractional shortening (FS) was lower in PUN females than CON (P < 0.05), whereas the opposite was true for males (P = 0.04; diet × sex, P = 0.001). Thus, the female PUN hearts compensated for the lower stroke volume to maintain cardiac index (mean aortic velocity by Doppler, which is already corrected for the CSA of the aorta) by maintaining a higher heart rate at baseline (Table 2). PUN males, however, maintained stroke volume and cardiac index by increasing FS without a change in heart rate compared to CON males.
The effect of PUN on baseline diastolic function differed for males and females. In PUN female mice, isovolumic contraction (P < 0.01) and relaxation (P < 0.05) times were longer even after correcting for heart rate (Table 2, P < 0.001 and P < 0.01, respectively), whereas there were no differences between PUN and CON males (diet × sex, P < 0.01–0.09). These differences contributed to a poorer myocardial performance index (MPI) in PUN females, suggesting that overall cardiac function at rest is somewhat impaired compared to CON females (P = 0.003); this was not the case for males (diet × sex, P = 0.07). Peak LV early diastolic filling velocity (Epeak velocity) was decreased by 15% in females (P < 0.001), with the late component (left atrial systole, A) contributing a greater proportion of the LV filling, as indicated by a lower E/A peak ratio compared to CON female mice (Table 2). In males, differences between PUN and CON in LV filling were less than in females, but there was an overall sex effect on the E/A ratio (P < 0.05). Of the systolic function indices at baseline, peak aortic flow velocity and stroke distance were significantly lower in PUN compared to CON mice (Table 2), and the differences were greater for females than males.
To further explore the differences in diastolic function, we used the PDF formalism that models LV filling as a spring (Kovacs et al. 1987). At the start of diastole, energy is stored in the ventricle and aids in LV filling as it unwinds. The initial stored elastic strain energy (1/2kx 0²) was 40% lower in PUN females compared to CON (P < 0.01), with no difference between males (Table 3; diet × sex, P < 0.09). The PDF parameter‐based comparison showed that, compared to CON mice, in female PUN mice there was a 25% decrease in preload (x 0), a trend for a small increase in stiffness (k; P = 0.1), and a 16% decrease in the viscoelasticity parameter (c; P < 0.001). In males, only c was lower in PUN mice. As x 0 represents the spring's displacement, the smaller ventricle in females has a negative impact on diastolic function. The damping needed to stop mitral filling (cEpeak) was also lower in both male and female PUN mice.
Stimulated measurements
Adrenergic stimulation increased heart rate to attain a similar value in PUN and CON, but with a larger change in males (sex, P = 0.003, Table 2). However, because baseline values were higher in the PUN females, both absolute and percentage increases in heart rate were smaller than for CON females (CON, 20 ± 3%; PUN, 13 ± 4%; P = 0.02). In both CON and PUN mice there was little change in stroke volume with dobutamine, which was lower for PUN mice (Table 1). Dobutamine increased FS in all groups, but the change from baseline was greater in males than females (52 ± 8% and 30 ± 4%, respectively, P < 0.001 vs. baseline; sex, P < 0.05, sex × diet, n.s.). The increase in cardiac index with dobutamine was largely due to the change in heart rate and was greater in males than females (62 ± 8% and 33 ± 4%, respectively, P < 0.001 vs. baseline; sex, P < 0.001; sex × diet, n.s.).
Both Epeak and Apeak velocities increased with dobutamine in all groups, but values were lower for PUN than CON hearts and in females compared to males (Table 2). Thus, the E/A peak ratios were similar for PUN and CON in the stimulated state, but lower in males than females. In all groups, peak aortic flow velocity and stroke distance increased (P < 0.001) and ejection time decreased (P = 0.002) with adrenergic stimulation and there was no sex or diet effect on the absolute amount of change. For peak velocity, the difference between PUN and CON mice became less evident, but was still higher in males. The changes in stroke distance and ejection time with dobutamine were equivalent across all groups so that the sex and diet effects present at baseline were retained (Table 2).
Cardiomyocyte morphology
Because exercise capacity and corrected heart weight were similar in male PUN and CON mice, cardiomyocyte measurements were performed only in females. In female CON, 80 ± 5% of cardiomyocytes were binucleated, 3 ± 5% had more than two nuclei, and 16 ± 5% were mononucleated (Fig. 5 A). PUN progeny had approximately 175% more mononucleated cells (a total of 41 ± 3%), and proportionally fewer binucleated cardiomyocytes (Fig. 5 A), but no difference in the proportion of polynucleated cells between groups. Cardiomyocyte CSA was primarily determined by the number of nuclei/cell, with mononucleated cells being smallest, and polynucleated cells largest (Fig. 5 B and C). The CSA of PUN cardiomyocytes tended to be smaller than for CON but attained significance only for mononucleated cells (P < 0.05).
Figure 5. Cardiomyocyte morphometry of female CON and PUN mice at PN90.
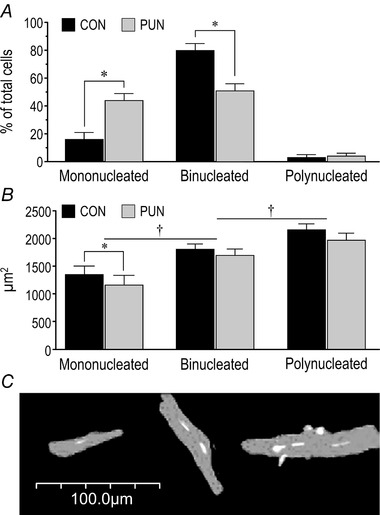
A, relative proportion of mono‐, bi‐ and poly‐nucleated cardiomyocytes; * P < 0.05 for CON vs. PUN. B, cross‐sectional areas of cardiomyocytes according to the number of nuclei/cell. Values are means ± SEM; n = 6 female mice per group with 75 cells measured per mouse; * P < 0.05 for CON vs. PUN; † P < 0.05 between nucleation number. C, representative image of mono‐, bi‐, and poly‐nucleated cardiomyocytes stained for myosin heavy chain (grey) and nuclei (white).
Ca2+ transient analysis
Only binucleated cardiomyocyte met the selection criteria for analysis. Under basal conditions, when paced at 1 Hz, differences in the time from external stimulation to peak maximum (0–100%) (Fig. 6 D) and the peak height of the transient relative to baseline (Fig. 6 E) were not significantly different between CON and PUN. The full duration at half‐maximum (FDHM) of the PUN transient (Fig. 6 F), however, was 25% shorter than that of CON (P < 0.01; diet × stimulation, P = 0.03) and the rate of Ca2+ decay (from maximal peak height to baseline), as indicated by the decay constant, tau (Fig. 6 G), was approximately 20% faster in PUN compared to CON (P = 0.05; diet × stimulation, P = 0.03). The total area under the Ca2+ signal curve, therefore, was lower in PUN cells.
Isoproterenol reduced time to peak by 20% in both CON and PUN mice, (Fig. 6 D; P < 0.005) and increased transient height by 60% (Fig. 6 E; P < 0.001). In CON mice, FDHM decreased (Fig. 6 F; P < 0.001) to values similar to those in the PUN group under basal conditions, whereas in the PUN mice there was no further change. In both groups isoproterenol accelerated Ca2+ reuptake (tau) (P < 0.001) to similar values (Fig. 6 G).
There was no difference in Ca2+ spark frequency between PUN and CON at baseline or when paced at 1 or 2 Hz (Fig. 6 C); an increase in spark frequency was observed at 2 Hz in both groups with isoproterenol (Fig. 6 H; P = 0.055). There was no difference in Ca2+ waves between PUN and CON at baseline. In CON, the frequency did not increase with isoproterenol either at 1 or 2 Hz. For PUN, the Ca2+ waves increased with isoproterenol treatment and values were significantly higher than in CON after pacing at 1 or 2 Hz (Fig. 6 I; P = 0.05).
Discussion
Exposure to suboptimal nutrition during critical stages of development can increase the risk for the development of several chronic diseases including cardiovascular disease (Barker, 1999). Anomalies of the coronary and general vasculature and haemodynamic parameters have been documented extensively, but less is known about the long‐term effects on the heart. In humans, follow‐up studies on the effects of IUGR on heart size or LV mass report positive correlations with birth weight (Hietalampi et al. 2012; Toemen et al. 2016), no difference in children and adolescents (Jiang et al. 2006), or increases in adults (Arnott et al. 2015). Adults born prematurely, and who probably experienced extrauterine growth retardation, have a larger LV mass and potentially pathological LV hypertrophy (Lewandowski et al. 2013 a). In adults born at term who experienced postnatal growth delays, no differences (Tennant et al. 2014) or hypertrophy in older males (Vijayakumar et al. 1995) have been reported. Whether nutritional inadequacies were responsible for the observed anomalies is difficult to ascertain because numerous factors can either modulate the nutrient effects or independently affect the response. These include the stage of development of the growth delay, the age at follow‐up, the presence of insulin resistance, hypertension, and/or obesity, the assessment techniques used, and the method of data normalization. Additionally, the plane of nutrition during rehabilitation can influence the long‐term outcomes (Toemen et al. 2016). Thus, studies with animal models provide a means to isolate the role of individual factors.
Numerous studies in animal models have focused on the long‐term cardiovascular effects of IUGR (reviewed in Kuo et al. 2017). In utero, however, factors in addition to nutrient supply are likely to contribute to the outcomes, such as hypoxia, high catecholamine levels or other stress hormones, and adverse changes to maternal and feto‐placental physiology (Morrison et al. 2007; Bjarnegard et al. 2013; Cohen et al. 2016; Aljunaidy et al. 2017; Limesand & Rozance, 2017). Less is known about the consequences of postnatal undernutrition and growth retardation during the later stages of cardiac maturation, and if deficiencies incurred during this time are recoverable. To address this gap in our knowledge, we studied in mice the effect of a brief episode of exclusively postnatal undernutrition on adult cellular, organ and whole‐body responses using exercise performance as a global measure of cardiovascular fitness following complete nutritional rehabilitation. Our findings are likely to translate to understanding the consequences of undernutrition and growth retardation in the human during the third trimester of gestation or the immediate postnatal period in preterm births when many of the maturational changes correspond to those occurring in the early postnatal period in the mouse.
Heart size
Despite consuming the control diet ad lib. from weaning, a reduction in body weight, lean mass and fat mass of both male and female PUN mice persisted. Although absolute heart weight was smaller for PUN than CON mice in both sexes, in PUN males the deficit was proportionate to their smaller lean mass and BSA, but in females the deficit was proportionately greater and persisted after normalization for lean mass or BSA. The observation of a disproportionate reduction in heart weight has been reported previously in some animal models of undernutrition (Desai et al. 1996, 2005), but not always (Lim et al. 2010; Kuo et al. 2017). The discrepancies probably are attributable to differences in the nutritional models studied (protein vs. global restriction of the dams, altered litter size), the stage of development during which undernutrition and refeeding occurred (intrauterine vs. postnatal), the species (rats, mice, sheep, primates), and sex (Kuo et al. 2018). Results are further confounded by how data are normalized to adjust for differences in metabolic mass. Indeed, as shown by our own data, when there are differences in body composition, normalization of heart size to body weight instead of lean mass or BSA can lead to different conclusions.
The smaller heart weight in PUN mice was manifested in the smaller LV mass derived by echocardiography, although when normalized for lean mass the differences in LV mass were less evident. This discrepancy could be attributed to a smaller effect of postnatal nutrition on the LV mass compared to the other heart chambers, although Bai et al. found this not to be the case (Bai et al. 1990). Alternatively, the geometry of the LV may have been altered so that assumptions inherent in the estimation of LV mass from echocardiography‐derived dimensions would be less accurate for PUN than CON. Indeed, we confirmed that in PUN females the hearts were more spherical compared to CON (data not shown). Similar findings were reported for young adult baboons malnourished during gestation and lactation (Kuo et al. 2017) and in children who were small at birth (Crispi et al. 2010) compared to their counterparts who experienced normal growth. Importantly, however (and consistent with the normalized heart weight) PUN females, but not males, had thinner LV posterior and anterior walls compared to CON.
The smaller hearts of the adult PUN female mice could be attributable to fewer and/or smaller cardiomyocytes. In rats, postnatal undernutrition induced by suckling pups in large litters decreased cardiomyocyte proliferation, resulting in fewer cardiomyocytes at weaning (Hollenberg et al. 1977; Bai et al. 1990). This deficit persisted after nutritional rehabilitation, consistent with the fact that cardiomyocyte proliferation is minimal after PN21. Sheep, in which cardiomyocyte maturation occurs prenatally, developed smaller hearts with fewer cardiomyocytes when there was IUGR and these differences persisted postnatally even with adequate nutrition (Stacy et al. 2009; Vranas et al. 2017). Thus, although we did not measure cell number, it probably was reduced in the PUN hearts and contributed to the smaller hearts.
The average CSA of PUN cardiomyocytes also was smaller than for CON and was accounted for by both a higher proportion of the smaller mononucleated myocytes, and an independent effect of PUN on the CSA of mononucleated cells, as observed previously for rats (Hollenberg et al. 1977). The reciprocal decrease in the proportion of binucleated cardiomyocytes in PUN hearts suggests that nuclear division and karyokinesis is sensitive to inadequacies in postnatal nutrient intake and that it is not entirely recoverable when nutritional rehabilitation is delayed to weaning. In contrast, differences in nucleation were not observed in either newborns of protein malnourished rat dams (Corstius et al. 2005) or after 2 weeks of postnatal undernutrition followed by nutritional rehabilitation (Lim et al. 2010). The latter study differed from ours in the age of nutritional rehabilitation (PN14 vs. PN21, respectively). It suggests that nuclear proliferation in the mouse may extend beyond PN14, but before PN21, thereby enabling deficits incurred before PN14 to be recuperated. This conclusion is supported by recent observations that in the mouse there is a limited, thyroid hormone‐regulated burst in nuclear proliferation at PN14–15 (Naqvi et al. 2014, 2015). Malnutrition during the suckling period, either because the dams are suckling a large litter (Fiorotto & Davis, 1997) or are fed a low‐protein diet (Ramos et al. 1997), results in reduced T3 levels in the pups, which could mitigate the extent of nuclear proliferation at this age. Thus, the higher proportion of mononucleated cardiomyocytes, their smaller size, and a strong likelihood of fewer cells in total, would account for the smaller heart mass and thinner LV wall thickness of adult PUN females.
Sex differences
Our data suggest that the terminal postnatal phase of cardiac maturation and/or its subsequent hypertrophy are influenced by sex. In PUN males, the lighter hearts were proportional to lean body mass or BSA, suggesting that their smaller size was a physiological adaptation to the diminished functional demand. The sex‐specific differences in heart size were corroborated by the interaction between sex and postnatal undernutrition on cardiovascular function. These sex differences implicate a role of sex hormones that are known to affect cardiovascular physiology including the effects on the autonomic nervous system and excitation‐contraction coupling (Parks & Howlett, 2013; Salerni et al. 2015). Studies have demonstrated that a maternal low protein diet during lactation reduces oestrogen and progesterone levels in female rat offspring, but with no effect on testosterone levels in either male or female progeny (Zambrano et al. 2005; Guzman et al. 2006). These alterations in the hypothalamic‐pituitary‐gonadal axis persist into adult life after the restoration of a normal diet. Sex differences have also been documented following IUGR, with most studies demonstrating more severe long‐term consequences for males (Aiken & Ozanne, 2013; Intapad et al. 2014; Dasinger & Alexander, 2016), although in primates effects are seen in both males and females (Kuo et al. 2017). Future studies will need to establish the extent to which the observed responses are consequences of the direct effects of sex hormones on the cardiac structures vs. indirect consequences on the physiological systems that regulate and modulate the cardiac response to growth stimuli.
In vivo cardiac function
The in vivo assessment of cardiac function identified persistent adverse effects of the postnatal nutritional experience on diastolic function at baseline and was more prominent in females. Several indices of LV diastolic function including Epeak velocity, E/A ratio, IVRT, MPI and PDF parameters indicated that this was impaired and probably contributed to the decrease in exercise tolerance (Little et al. 2000). Consistent with their smaller LV mass, the PUN female mice tended to have a reduced end diastolic volume and, at rest, stroke volume was less than in CON. This response correlates with a lower value of x 0 (volumetric load) in the PUN group, another possible consequence of smaller hearts. Furthermore, the lower 1/2kx 0 value in PUN females, but not males, suggests that LV recoil energy at the start of diastole was reduced. Overall, the characterization of the female PUN hearts using the PDF kinematic model correlates with their impaired diastolic function.
At rest, PUN females functionally compensated for the smaller hearts and achieved a cardiac index appropriate for metabolic mass by maintaining a higher heart rate than CON females. However, the blunted response in heart rate and cardiac index following dobutamine suggests that at rest they were operating at a higher percentage of their maximum cardiac output. We deduce from these results that if the ‘resting’ parameters are a higher proportion of the ‘maximum’ attainable value, then limitations may be more easily reached with exertion. While we do not know if the mice experienced dyspnoea, in humans with diastolic dysfunction, exercise haemodynamics predicts the decreased peak and workload that is achieved (Abudiab et al. 2013). These findings, therefore, also provide a potential mechanism for the reduced and exercise tolerance observed in the PUN females. These observations reproduce findings in children who experienced IUGR in whom altered cardiac morphometry and function have been identified, including a reduced stroke volume accompanied by an increased heart rate to maintain cardiac output at rest (Crispi et al. 2010). Increased resting heart rates have also been documented in some studies of adults who were low birth weight (Phillips & Barker, 1997), although this is not a consistent finding (de Rooij et al. 2016).
Despite the effect of PUN on some parameters, unlike females, male PUN mice did not exhibit the same difference relative to CON in resting heart rate, the cardiac index response to dobutamine, and exercise tolerance. Under basal conditions, the males were able to increase FS to sustain stroke volume, and retained the full chronotropic response to dobutamine stimulation. This contrasts with a report of higher resting heart rate and reduced responsiveness to β‐adrenergic stimulation in adult male offspring of rat dams fed a low protein diet during pregnancy and lactation (Fernandez‐Twinn et al. 2006). The increased resting heart rate was attributed to higher basal epinephrine (noradrenaline) levels, whereas a decreased response to dobutamine was related to a compensatory decrease in cardiac β1‐adrenergic receptor levels in response to chronic persistent adrenergic stimulation or desensitization. Zohdi et al. (2011), however, found a reduced response in cardiac output to adrenergic stimulation in both adult males and females, albeit more severe in females, which they attributed to an increase in LV afterload rather than any change in adrenergic sensitivity. While an increased sympathetic activity at rest (de Oliveira et al. 2012; Brito Alves et al. 2015) and circulating catecholamine levels (Petry et al. 2000; Tenhola et al. 2002) appear to be a common long‐term effect of perinatal nutrition on the autonomic nervous system, the rate and extent of desensitization are likely to vary depending on factors that differ among studies. These factors include the animal model, the timing, duration and severity of the nutritional/growth perturbation with respect to stage of development, and the methodology. These variables are affected by sex hormones and, therefore, would contribute to sex differences in the functional outcomes noted by us and others.
Cardiomyocyte Ca2+ flux
Under basal conditions, binucleated cardiomyocytes from PUN and CON showed similar Ca2+ release kinetics (as indicated by time to maximum peak height and peak height), but the rate of Ca2+ uptake was significantly faster in the PUN group, as demonstrated both in FDHM and tau values. The abbreviated rate of Ca2+ decline in the PUN cardiomyocytes under basal conditions might enable a faster relaxation rate, but is consistent with enhanced diastolic function rather than the impairments we measured in situ. The anticipated response in Ca2+ release with adrenergic stimulation was observed in both CON and PUN cells, i.e. a faster time to peak maximum and an increase in peak height, and the changes were of similar magnitude in both groups. However, the degree of change in the rate of uptake upon stimulation was not as marked in PUN cells as in CON, possibly because they were already accelerated. These data suggest that at the standard slow stimulation rates used to study cardiomyocytes in vitro (relative to in vivo rates), the responsiveness of binucleated PUN cells to adrenergic stimulation probably is not blunted. Moreover, the activity of the mechanisms regulating Ca2+ uptake are already upregulated under baseline conditions and would preclude responses of the same magnitude as CON upon stimulation. The discordance between this and the in vivo LV response, probably reflects the contribution of multiple factors to LV function in the intact heart including the contribution of the mononucleated cell population which is discussed below.
We did not observe effects of postnatal nutrition on the frequency of Ca2+ sparks even at higher pacing, suggesting no differences in Ca2+ leaks between PUN and CON cardiomyocytes. However, by increasing the rate of Ca2+ flux in the presence of isoproterenol, the Ca2+ wave frequency was greater in PUN cardiomyocytes suggesting enhanced propagation of spontaneous Ca2+ spikes. While arrhythmias were not seen with dobutamine during the in vivo studies, the results suggest that PUN mice may have an enhanced risk of incurring arrhythmias.
Limitations
These measurements of Ca2+ kinetics were performed on binucleated cells because small, mononucleated cardiomyocytes often did not meet established prior criteria that identify a ‘healthy’ cell for calcium transient analyses (detailed in Methods). However, previous reports indicate that mononucleated cardiomyocytes have prolonged Ca2+transients at lower amplitudes, as well as desynchronized Ca2+release due to underdeveloped T‐tubular systems and sarcoplasmic reticulum (Chen et al. 2007). The female PUN hearts had at least twice as many mononucleated cells. Thus, measurements performed at the whole organ level would reflect the altered Ca2+ flux of both the binucleated and the greater proportion of mononucleated cells. Moreover, because diastole happens at lower pressure, it may be more sensitive to the heterogeneity in function from the mixture of mononucleated and binucleated myocytes. Given the persistence of the mononucleated cells in PUN hearts, studies to evaluate Ca2+ transients of mononucleated cardiomyocytes and measurements on male cardiomyocytes are warranted. Because the phenotype was observed in females, future studies should explore the role of oestrogens following growth restriction on cardiac function. For logistical reasons, in these studies we did not attempt to control for oestrus, which is known to influence some of the outcomes we assessed. However, because the data were collected from several repetitions of the experiment, differences in oestrus among females would be captured by the variance in the data. Additional time course studies that examine the response to varying doses of β‐adrenergic agonist, ideally without the use of anaesthesia, are necessary to better understand the functional implications of our observations at the whole body level. Lastly, as there is evidence that the right ventricular function is susceptible to reprogramming when growth is compromised during early development (Lewandowski et al. 2013b; Kuo et al. 2017, 2018), in vivo measures of the effect of postnatal undernutrition on right ventricular function and its contribution to exercise capacity merit examination.
In conclusion, we have shown that in the mouse a brief period of suboptimal nutrition in the postnatal period when the heart is undergoing terminal maturation can produce long‐term changes in cardiac structure and function. While there are extensive data that have demonstrated the deleterious consequences of intrauterine insults to the fetus, our results extend the developmental window during which the heart is vulnerable to reprogramming by inadequacies in nutrient intake. In females, this resulted in differences in resting diastolic function. Adaptive mechanisms enabled the PUN heart to meet the needs of the body under sedentary conditions, but in females they restricted the heart's ability to meet the increased demand of strenuous exercise. The cardiac impairment is attributable in part to a disproportionate reduction in cardiac size and structure, and possibly differences in cardiomyocyte nucleation. There were also changes in contractile function at the cellular level, possibly due to programmed changes in gene expression in the cardiomyocyte directly, and/or indirectly via changes to the hypothalamic‐pituitary‐gonadal axis and the autonomic system that regulate cardiac function. With this evidence, it is critical to establish if our results are translatable to humans, as those who experience growth restriction could be at risk of a cardiac event as a result of strenuous exercise. Further studies to understand the molecular mechanisms (genetic, proteomic and epigenetic) responsible for the cardiac impairment are also essential to enable the development of therapeutic countermeasures to reduce the risk of cardiac mortality.
Additional information
Competing interests
The authors have no competing interests.
Author contributions
All mice were produced and reared at the Children's Nutrition Research Center (CNRC); body composition analysis, exercise testing and cardiomyocyte morphometry were performed at the CNRC; in vivo echocardiography and Doppler blood flow analysis were performed at the DeBakey Heart Center Research Core at Methodist Hospital (Houston, TX, USA); cardiomyocyte collection and Ca2+ transients were performed in the Department of Molecular Physiology and Biophysics, Baylor College of Medicine. D.P.F., G.E.T., G.G.R. and M.L.F. were responsible for the conception and design of the experiments; D.P.F., T.O.M., R.F., C.P.H., G.G.R., G.E.T. and M.L.F. collected, analysed and interpreted the data; D.P.F., T.O.M., C.P.H., G.G.R., G.E.T. and M.L.F. prepared and reviewed the manuscript. All authors have approved the final version of the manuscript and agree to be accountable for all aspects of the work. All persons designated as authors qualify for authorship, and all those who qualify for authorship are listed.
Funding
This study was funded by NIH training grant (D.P.F.; T32HL07676), Baylor College of Medicine Cardiovascular Research Institute collaborative grant (M.L.F., G.G.R., G.E.T.), USDA/ARS (M.L.F.; 3092‐51000‐056‐01S).
Acknowledgments
The authors wish to thank Guillermo Medrano, Alejandro Granillo and Thuy Pham for the assistance is echocardiography and Doppler blood flow analysis.
Biography
David P. Ferguson is an Assistant Professor of Kinesiology at Michigan State University. He obtained his doctoral degree in exercise physiology with Dr J. Timothy Lightfoot at Texas A&M University in College Station, Texas, and then did a postdoctoral fellowship with Dr Marta Fiorotto at the USDA/ARS Children's Nutrition Research Center at Baylor College of Medicine in Houston, Texas. Currently, he is the director of the Neonatal Nutrition and Exercise Research Laboratory. The goal of his laboratory is to investigate the mechanistic changes in cardiac function associated with early life growth restriction and propose therapeutic countermeasures to increase functional capacity and decrease mortality rates.

Edited by: Laura Bennet & Janna Morrison
References
- Abudiab MM, Redfield MM, Melenovsky V, Olson TP, Kass DA, Johnson BD & Borlaug BA (2013). Cardiac output response to exercise in relation to metabolic demand in heart failure with preserved ejection fraction. Eur J Heart Fail 15, 776–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agnoux MA, Antignac JP, Boquien CY, David A, Desnots E, Ferchaud‐Roucher V, Darmaun D, Parnet P & Alexandre‐Gouabau MC (2015). Perinatal protein restriction affects milk free amino acid and fatty acid profile in lactating rats: potential role on pup growth and metabolic status. J Nutr Biochem 26, 784–795. [DOI] [PubMed] [Google Scholar]
- Aiken CE & Ozanne SE (2013). Sex differences in developmental programming models. Reproduction 145, R1–13. [DOI] [PubMed] [Google Scholar]
- Aljunaidy MM, Morton JS, Cooke CM & Davidge ST (2017). Prenatal hypoxia and placental oxidative stress: linkages to developmental origins of cardiovascular disease. Am J Physiol Regul Integr Comp Physiol 313, R395–R399. [DOI] [PubMed] [Google Scholar]
- Arnott C, Skilton MR, Ruohonen S, Juonala M, Viikari JS, Kahonen M, Lehtimaki T, Laitinen T, Celermajer DS & Raitakari OT (2015). Subtle increases in heart size persist into adulthood in growth restricted babies: the Cardiovascular Risk in Young Finns Study. Open Heart 2, e000265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai SL, Campbell SE, Moore JA, Morales MC & Gerdes AM (1990). Influence of age, growth, and sex on cardiac myocyte size and number in rats. Anat Rec 226, 207–212. [DOI] [PubMed] [Google Scholar]
- Barker DJ (1999). Fetal origins of cardiovascular disease. Ann Med 31 (Suppl. 1), 3–6. [PubMed] [Google Scholar]
- Bjarnegard N, Morsing E, Cinthio M, Lanne T & Brodszki J (2013). Cardiovascular function in adulthood following intrauterine growth restriction with abnormal fetal blood flow. Ultrasound Obstet Gynecol 41, 177–184. [DOI] [PubMed] [Google Scholar]
- Black MJ, Siebel AL, Gezmish O, Moritz KM & Wlodek ME (2012). Normal lactational environment restores cardiomyocyte number after uteroplacental insufficiency: implications for the preterm neonate. Am J Physiol Regul Integr Comp Physiol 302, R1101–R1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blencowe H, Cousens S, Oestergaard MZ, Chou D, Moller AB, Narwal R, Adler A, Vera GC, Rohde S, Say L & Lawn JE (2012). National, regional, and worldwide estimates of preterm birth rates in the year 2010 with time trends since 1990 for selected countries: a systematic analysis and implications. Lancet 379, 2162–2172. [DOI] [PubMed] [Google Scholar]
- Botting KJ, Wang KC, Padhee M, McMillen IC, Summers‐Pearce B, Rattanatray L, Cutri N, Posterino GS, Brooks DA & Morrison JL (2012). Early origins of heart disease: low birth weight and determinants of cardiomyocyte endowment. Clin Exp Pharmacol Physiol 39, 814–823. [DOI] [PubMed] [Google Scholar]
- Briscoe TA, Rehn AE, Dieni S, Duncan JR, Wlodek ME, Owens JA & Rees SM (2004). Cardiovascular and renal disease in the adolescent guinea pig after chronic placental insufficiency. Am J Obstet Gynecol 191, 847–855. [DOI] [PubMed] [Google Scholar]
- Brito Alves JL, Nogueira VO, Cavalcanti Neto MP, Leopoldino AM, Curti C, Colombari DS, Colombari E, Wanderley AG, Leandro CG, Zoccal DB & Costa‐Silva JH (2015). Maternal protein restriction increases respiratory and sympathetic activities and sensitizes peripheral chemoreflex in male rat offspring. J Nutr 145, 907–914. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrell JH, Boyn AM, Kumarasamy V, Hsieh A, Head SI & Lumbers ER (2003). Growth and maturation of cardiac myocytes in fetal sheep in the second half of gestation. Anat Rec A Discov Mol Cell Evol Biol 274, 952–961. [DOI] [PubMed] [Google Scholar]
- Carr H, Cnattingius S, Granath F, Ludvigsson JF & Edstedt Bonamy AK (2017). Preterm birth and risk of heart failure up to early adulthood. J Am Coll Cardiol 69, 2634–2642. [DOI] [PubMed] [Google Scholar]
- Chen X, Wilson RM, Kubo H, Berretta RM, Harris DM, Zhang X, Jaleel N, MacDonnell SM, Bearzi C, Tillmanns J, Trofimova I, Hosoda T, Mosna F, Cribbs L, Leri A, Kajstura J, Anversa P & Houser SR (2007). Adolescent feline heart contains a population of small, proliferative ventricular myocytes with immature physiological properties. Circ Res 100, 536–544. [DOI] [PubMed] [Google Scholar]
- Cheung MC, Spalding PB, Gutierrez JC, Balkan W, Namias N, Koniaris LG & Zimmers TA (2009). Body surface area prediction in normal, hypermuscular, and obese mice. J Surg Res 153, 326–331. [DOI] [PubMed] [Google Scholar]
- Chintalgattu V, Ai D, Langley RR, Zhang J, Bankson JA, Shih TL, Reddy AK, Coombes KR, Daher IN, Pati S, Patel SS, Pocius JS, Taffet GE, Buja LM, Entman ML & Khakoo AY (2010). Cardiomyocyte PDGFR‐beta signaling is an essential component of the mouse cardiac response to load‐induced stress. J Clin Invest 120, 472–484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen E, Wong FY, Horne RS & Yiallourou SR (2016). Intrauterine growth restriction: impact on cardiovascular development and function throughout infancy. Pediatr Res 79, 821–830. [DOI] [PubMed] [Google Scholar]
- Corstius HB, Zimanyi MA, Maka N, Herath T, Thomas W, van der Laarse A, Wreford NG & Black MJ (2005). Effect of intrauterine growth restriction on the number of cardiomyocytes in rat hearts. Pediatr Res 57, 796–800. [DOI] [PubMed] [Google Scholar]
- Crispi F, Bijnens B, Figueras F, Bartrons J, Eixarch E, Le Noble F, Ahmed A & Gratacos E (2010). Fetal growth restriction results in remodeled and less efficient hearts in children. Circulation 121, 2427–2436. [DOI] [PubMed] [Google Scholar]
- Dasinger JH & Alexander BT (2016). Gender differences in developmental programming of cardiovascular diseases. Clin Sci (Lond) 130, 337–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Oliveira JC, Grassiolli S, Gravena C & de Mathias PC (2012). Early postnatal low‐protein nutrition, metabolic programming and the autonomic nervous system in adult life. Nutr Metab (Lond) 9, 80–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Rooij SR, Jones A, Phillips DI, Osmond C, Karemaker JM, Roseboom TJ & Painter RC (2016). Prenatal undernutrition and autonomic function in adulthood. Psychosom Med 78, 991–997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derrickson EM & Lowas SR (2007). The effect of dietary protein levels on milk protein levels and postnatal growth in laboratory mice (Mus musculus) . J Mammol 88, 1475–1481. [Google Scholar]
- Desai M, Crowther NJ, Lucas A & Hales CN (1996). Organ‐selective growth in the offspring of protein‐restricted mothers. Br J Nutr 76, 591–603. [DOI] [PubMed] [Google Scholar]
- Desai M, Gayle D, Babu J & Ross MG (2005). Permanent reduction in heart and kidney organ growth in offspring of undernourished rat dams. Am J Obstet Gynecol 193, 1224–1232. [DOI] [PubMed] [Google Scholar]
- Fernandez‐Twinn DS, Ekizoglou S, Wayman A, Petry CJ & Ozanne SE (2006). Maternal low‐protein diet programs cardiac beta‐adrenergic response and signaling in 3‐mo‐old male offspring. Am J Physiol Regul Integr Comp Physiol 291, R429–R436. [DOI] [PubMed] [Google Scholar]
- Fiorotto ML & Davis TA (1997). Food intake alters muscle protein gain with little effect on Na+‐K+‐ATPase and myosin isoforms in suckled rats. Am J Physiol Regul Integr Comp Physiol 272, R1461–R1471. [DOI] [PubMed] [Google Scholar]
- Fiorotto ML, Davis TA, Sosa HA, Villegas‐Montoya C, Estrada I & Fleischmann R (2014). Ribosome abundance regulates the recovery of skeletal muscle protein mass upon recuperation from postnatal undernutrition in mice. J Physiol 592, 5269–5286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foglia MJ & Poss KD (2016). Building and re‐building the heart by cardiomyocyte proliferation. Development 143, 729–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goyal R & Longo LD (2013). Maternal protein deprivation: sexually dimorphic programming of hypertension in the mouse. Hypertens Res 36, 29–35. [DOI] [PubMed] [Google Scholar]
- Grimble RF & Mansaray YK (1987). Effects in rats of dietary protein inadequacy on lactose production, milk volume and components of the lactose synthetase complex (EC 2.4.1.22). Ann Nutr Metab 31, 179–184. [DOI] [PubMed] [Google Scholar]
- Guzman C, Cabrera R, Cardenas M, Larrea F, Nathanielsz PW & Zambrano E (2006). Protein restriction during fetal and neonatal development in the rat alters reproductive function and accelerates reproductive ageing in female progeny. J Physiol 572, 97–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamaguchi S, Kawakami Y, Honda Y, Nemoto K, Sano A, Namekata I & Tanaka H (2013). Developmental changes in excitation‐contraction mechanisms of the mouse ventricular myocardium as revealed by functional and confocal imaging analyses. J Pharmacol Sci 123, 167–175. [DOI] [PubMed] [Google Scholar]
- Hietalampi H, Pahkala K, Jokinen E, Ronnemaa T, Viikari JS, Niinikoski H, Heinonen OJ, Salo P, Simell O & Raitakari OT (2012). Left ventricular mass and geometry in adolescence: early childhood determinants. Hypertension 60, 1266–1272. [DOI] [PubMed] [Google Scholar]
- Hollenberg M, Honbo N & Samorodin AJ (1977). Cardiac cellular responses to altered nutrition in the neonatal rat. Am J Physiol Heart Circ Physiol 233, H356–H360. [DOI] [PubMed] [Google Scholar]
- Ikenishi A, Okayama H, Iwamoto N, Yoshitome S, Tane S, Nakamura K, Obayashi T, Hayashi T & Takeuchi T (2012). Cell cycle regulation in mouse heart during embryonic and postnatal stages. Dev Growth Differ 54, 731–738. [DOI] [PubMed] [Google Scholar]
- Intapad S, Ojeda NB, Dasinger JH & Alexander BT (2014). Sex differences in the developmental origins of cardiovascular disease. Physiology (Bethesda) 29, 122–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang B, Godfrey KM, Martyn CN & Gale CR (2006). Birth weight and cardiac structure in children. Pediatrics 117, e257–e261. [DOI] [PubMed] [Google Scholar]
- Jonker SS, Louey S, Giraud GD, Thornburg KL & Faber JJ (2015). Timing of cardiomyocyte growth, maturation, and attrition in perinatal sheep. FASEB J 29, 4346–4357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kovacs SJ, Barzilai B & Perez JE (1987). Evaluation of diastolic function with Doppler echocardiography: the PDF formalism. Am J Physiol Heart Circ Physiol 252, H178–H187. [DOI] [PubMed] [Google Scholar]
- Kuo AH, Li C, Huber HF, Nathanielsz PW & Clarke GD (2018). Ageing changes in biventricular cardiac function in male and female baboons (Papio spp.). J Physiol 596, 5083–5098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuo AH, Li C, Li J, Huber HF, Nathanielsz PW & Clarke GD (2017). Cardiac remodelling in a baboon model of intrauterine growth restriction mimics accelerated ageing. J Physiol 595, 1093–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langley‐Evans SC (2000). Critical differences between two low protein diet protocols in the programming of hypertension in the rat. Int J Food Sci Nutr 51, 11–17. [DOI] [PubMed] [Google Scholar]
- Leu M, Ehler E & Perriard JC (2001). Characterisation of postnatal growth of the murine heart. Anat Embryol (Berl) 204, 217–224. [DOI] [PubMed] [Google Scholar]
- Lewandowski AJ, Augustine D, Lamata P, Davis EF, Lazdam M, Francis J, McCormick K, Wilkinson AR, Singhal A, Lucas A, Smith NP, Neubauer S & Leeson P (2013. a). Preterm heart in adult life: cardiovascular magnetic resonance reveals distinct differences in left ventricular mass, geometry, and function. Circulation 127, 197–206. [DOI] [PubMed] [Google Scholar]
- Lewandowski AJ, Bradlow WM, Augustine D, Davis EF, Francis J, Singhal A, Lucas A, Neubauer S, McCormick K & Leeson P (2013b). Right ventricular systolic dysfunction in young adults born preterm. Circulation 128, 713–720. [DOI] [PubMed] [Google Scholar]
- Li F, Wang X, Capasso JM & Gerdes AM (1996). Rapid transition of cardiac myocytes from hyperplasia to hypertrophy during postnatal development. J Mol Cell Cardiol 28, 1737–1746. [DOI] [PubMed] [Google Scholar]
- Lim K, Zimanyi MA & Black MJ (2010). Effect of maternal protein restriction during pregnancy and lactation on the number of cardiomyocytes in the postproliferative weanling rat heart. Anat Rec (Hoboken) 293, 431–437. [DOI] [PubMed] [Google Scholar]
- Limesand SW & Rozance PJ (2017). Fetal adaptations in insulin secretion result from high catecholamines during placental insufficiency. J Physiol 595, 5103–5113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Little WC, Kitzman DW & Cheng CP (2000). Diastolic dysfunction as a cause of exercise intolerance. Heart Fail Rev 5, 301–306. [DOI] [PubMed] [Google Scholar]
- Louey S, Jonker SS, Giraud GD & Thornburg KL (2007). Placental insufficiency decreases cell cycle activity and terminal maturation in fetal sheep cardiomyocytes. J Physiol 580, 639–648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Master JS, Zimanyi MA, Yin KV, Moritz KM, Gallo LA, Tran M, Wlodek ME & Black MJ (2014). Transgenerational left ventricular hypertrophy and hypertension in offspring after uteroplacental insufficiency in male rats. Clin Exp Pharmacol Physiol 41, 884–890. [DOI] [PubMed] [Google Scholar]
- Milner DJ, Taffet GE, Wang X, Pham T, Tamura T, Hartley C, Gerdes AM & Capetanaki Y (1999). The absence of desmin leads to cardiomyocyte hypertrophy and cardiac dilation with compromised systolic function. J Mol Cell Cardiol 31, 2063–2076. [DOI] [PubMed] [Google Scholar]
- Mollova M, Bersell K, Walsh S, Savla J, Das LT, Park SY, Silberstein LE, Dos Remedios CG, Graham D, Colan S & Kuhn B (2013). Cardiomyocyte proliferation contributes to heart growth in young humans. Proc Natl Acad Sci U S A 110, 1446–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morrison JL, Botting KJ, Dyer JL, Williams SJ, Thornburg KL & McMillen IC (2007). Restriction of placental function alters heart development in the sheep fetus. Am J Physiol Regul Integr Comp Physiol 293, R306–R313. [DOI] [PubMed] [Google Scholar]
- Naqvi N, Li M, Calvert JW, Tejada T, Lambert JP, Wu J, Kesteven SH, Holman SR, Matsuda T, Lovelock JD, Howard WW, Iismaa SE, Chan AY, Crawford BH, Wagner MB, Martin DI, Lefer DJ, Graham RM & Husain A (2014). A proliferative burst during preadolescence establishes the final cardiomyocyte number. Cell 157, 795–807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naqvi N, Singh R, Iismaa SE, Li M, Calvert JW, Martin DI, Harvey RP, Graham RM & Husain A (2015). Cardiomyocytes replicate and their numbers increase in young hearts. Cell 163, 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osmond C & Barker DJ (2000). Fetal, infant, and childhood growth are predictors of coronary heart disease, diabetes, and hypertension in adult men and women. Environ Health Perspect 108 (Suppl. 3), 545–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks RJ & Howlett SE (2013). Sex differences in mechanisms of cardiac excitation‐contraction coupling. Pflugers Arch 465, 747–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patterson M, Barske L, Van Handel B, Rau CD, Gan P, Sharma A, Parikh S, Denholtz M, Huang Y, Yamaguchi Y, Shen H, Allayee H, Crump JG, Force TI, Lien CL, Makita T, Lusis AJ, Kumar SR & Sucov HM (2017). Frequency of mononuclear diploid cardiomyocytes underlies natural variation in heart regeneration. Nat Genet 49, 1346–1353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pederson BA, Cope CR, Schroeder JM, Smith MW, Irimia JM, Thurberg BL, DePaoli‐Roach AA & Roach PJ (2005). Exercise capacity of mice genetically lacking muscle glycogen synthase: in mice, muscle glycogen is not essential for exercise. J Biol Chem 280, 17260–17265. [DOI] [PubMed] [Google Scholar]
- Perez‐Cruz M, Cruz‐Lemini M, Fernandez MT, Parra JA, Bartrons J, Gomez‐Roig MD, Crispi F & Gratacos E (2015). Fetal cardiac function in late‐onset intrauterine growth restriction vs. small‐for‐gestational age, as defined by estimated fetal weight, cerebroplacental ratio and uterine artery Doppler. Ultrasound Obstet Gynecol 46, 465–471. [DOI] [PubMed] [Google Scholar]
- Petry CJ, Dorling MW, Wang CL, Pawlak DB & Ozanne SE (2000). Catecholamine levels and receptor expression in low protein rat offspring. Diabet Med 17, 848–853. [DOI] [PubMed] [Google Scholar]
- Picht E, Zima AV, Blatter LA & Bers DM (2007). SparkMaster: automated calcium spark analysis with ImageJ. Am J Physiol Cell Physiol 293, C1073–C1081. [DOI] [PubMed] [Google Scholar]
- Pine AP, Jessop NS & Oldham JD (1994). Maternal protein reserves and their influence on lactational performance in rats. 3. The effects of dietary protein restriction and stage of lactation on milk composition. Br J Nutr 72, 815–830. [DOI] [PubMed] [Google Scholar]
- Phillips DI & Barker DJ (1997). Association between low birthweight and high resting pulse in adult life: is the sympathetic nervous system involved in programming the insulin resistance syndrome? Diabet Med 14, 673–677. [DOI] [PubMed] [Google Scholar]
- Powers SK & Howley ET (2015). Exercise Physiology: Theory and Application to Fitness and Performance McGraw Hill, New York. [Google Scholar]
- Ramos CF, Lima AP, Teixeira CV, Brito PD & Moura EG (1997). Thyroid function in post‐weaning rats whose dams were fed a low‐protein diet during suckling. Braz J Med Biol Res 30, 133–137. [DOI] [PubMed] [Google Scholar]
- Reynolds JO, Quick AP, Wang Q, Beavers DL, Philippen LE, Showell J, Barreto‐Torres G, Thuerauf DJ, Doroudgar S, Glembotski CC & Wehrens XH (2016). Junctophilin‐2 gene therapy rescues heart failure by normalizing RyR2‐mediated Ca2+ release. Int J Cardiol 225, 371–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salerni S, Di Francescomarino S, Cadeddu C, Acquistapace F, Maffei S & Gallina S (2015). The different role of sex hormones on female cardiovascular physiology and function: not only oestrogens. Eur J Clin Invest 45, 634–645. [DOI] [PubMed] [Google Scholar]
- Sampson DA, Hunsaker HA & Jansen GR (1986). Dietary protein quality, protein quantity and food intake: effects on lactation and on protein synthesis and tissue composition in mammary tissue and liver in rats. J Nutr 116, 365–375. [DOI] [PubMed] [Google Scholar]
- Soonpaa MH, Kim KK, Pajak L, Franklin M & Field LJ (1996). Cardiomyocyte DNA synthesis and binucleation during murine development. Am J Physiol Heart Circ Physiol 271, H2183–H2189. [DOI] [PubMed] [Google Scholar]
- Stacy V, De Matteo R, Brew N, Sozo F, Probyn ME, Harding R & Black MJ (2009). The influence of naturally occurring differences in birthweight on ventricular cardiomyocyte number in sheep. Anat Rec (Hoboken) 292, 29–37. [DOI] [PubMed] [Google Scholar]
- Tenhola S, Martikainen A, Rahiala E, Parviainen M, Halonen P & Voutilainen R (2002). Increased adrenocortical and adrenomedullary hormonal activity in 12‐year‐old children born small for gestational age. J Pediatr 141, 477–482. [DOI] [PubMed] [Google Scholar]
- Tennant IA, Barnett AT, Thompson DS, Kips J, Boyne MS, Chung EE, Chung AP, Osmond C, Hanson MA, Gluckman PD, Segers P, Cruickshank JK & Forrester TE (2014). Impaired cardiovascular structure and function in adult survivors of severe acute malnutrition. Hypertension 64, 664–671. [DOI] [PubMed] [Google Scholar]
- Thornburg KL (2015). The programming of cardiovascular disease. J Dev Orig Health Dis 6, 366–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toemen L, de Jonge LL, Gishti O, Osch‐Gevers L, Taal HR, Steegers EA, Hofman A, Helbing WA & Jaddoe VW (2016). Longitudinal growth during fetal life and infancy and cardiovascular outcomes at school‐age. J Hypertens 34, 1396–1406. [DOI] [PubMed] [Google Scholar]
- Trial J, Heredia CP, Taffet GE, Entman ML & Cieslik KA (2017). Dissecting the role of myeloid and mesenchymal fibroblasts in age‐dependent cardiac fibrosis. Basic Res Cardiol 112, 34–0623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vijayakumar M, Fall CH, Osmond C & Barker DJ (1995). Birth weight, weight at one year, and left ventricular mass in adult life. Br Heart J 73, 363–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vranas S, Heinemann GK, Liu H, De Blasio MJ, Owens JA, Gatford KL & Black MJ (2017). Small size at birth predicts decreased cardiomyocyte number in the adult ovine heart. J Dev Orig Health Dis 8, 618–625. [DOI] [PubMed] [Google Scholar]
- Zambrano E, Rodriguez‐Gonzalez GL, Guzman C, Garcia‐Becerra R, Boeck L, Diaz L, Menjivar M, Larrea F & Nathanielsz PW (2005). A maternal low protein diet during pregnancy and lactation in the rat impairs male reproductive development. J Physiol 563, 275–284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zohdi V, Jane BM & Pearson JT (2011). Elevated vascular resistance and afterload reduce the cardiac output response to dobutamine in early growth‐restricted rats in adulthood. Br J Nutr 106, 1374–1382. [DOI] [PubMed] [Google Scholar]
- Zohdi V, Lim K, Pearson JT & Black MJ (2014). Developmental programming of cardiovascular disease following intrauterine growth restriction: findings utilising a rat model of maternal protein restriction. Nutrients 7, 119–152. [DOI] [PMC free article] [PubMed] [Google Scholar]