

Abstract
Frizzled family receptor 7 (FZD7), a Wnt signaling receptor, is associated with the maintenance of stem cell properties and cancer progression. FZD7 has emerged as a potential therapeutic target because it is capable of transducing both canonical and noncanonical Wnt signals. In this study, we investigated the regulatory pathway downstream of FZD7 and its functional roles. We found that FZD7 expression was crucial to the maintenance of the mesenchymal phenotype, anoikis resistance, and spheroid and tumor formation in ovarian cancer (OC). We identified TWIST1 as the crucial downstream effector of the FZD7 pathway. TWIST1, a basic helix loop helix transcription factor, is known to associate with mesenchymal and cancer stem cell phenotypes. Manipulating TWIST1 expression mimicked the functional consequences observed in the FZD7 model, and overexpression of TWIST1 partially rescued the functional phenotypes abolished by FZD7 knockdown. We further proved that FZD7 regulated TWIST1 expression through epigenetic modifications of H3K4me3 and H3K27ac at the TWIST1 proximal promoter. We also identified that the FZD7‐TWIST1 axis regulates the expression of BCL2, a gene that controls apoptosis. Identification of this FZD7‐TWIST1‐BCL2 pathway reaffirms the mechanism of anoikis resistance in OC. We subsequently showed that the FZD7‐TWIST1 axis can be targeted by using a small molecule inhibitor of porcupine, an enzyme essential for secretion and functional activation of Wnts. In conclusion, our results identified that the FZD7‐TWIST1 axis is important for tumorigenesis and anoikis resistance, and therapeutic inhibition results in cell death in OCs.
Keywords: anoikis, BCL2, FZD7, ovarian carcinoma, tumorigenesis, TWIST1
Abbreviations
- CAM
chorioallantoic membrane
- CSC
cancer stem cells
- ECM
extracellular membrane
- ED
embryonic day
- EMT
epithelial to mesenchymal transition
- EOC
epithelial ovarian cancer
- FACS
florescence‐activated cell sorting
- FZD7
frizzled 7
- FZD
frizzled
- IgG
immunoglobulin G
- Mes
mesenchymal
- OC
ovarian cancer
- PCP
planar cell polarity
- qPCR
quantitative PCR
- shFZD7
short hairpin against FZD7
- shLuci
short hairpin luciferase
- siRNA
small interfering RNA
- TSS
transition start site
1. Introduction
The Wnt signaling pathway comprises of Wnt ligands that are secreted glycoprotein and frizzled (FZD) receptors, which are seven‐pass transmembrane receptors with an extracellular N‐terminal cysteine‐rich domain (Bhanot et al., 1996). Wnt signaling involves two pathways, namely the canonical and noncanonical pathway (Veeman et al., 2003). In the canonical Wnt pathway, the T‐cell factor/lymphoid enhancer factor‐1 and β‐catenin act as a core regulator through nuclear translocation and transcriptional activation of target genes critical for cancer cell stemness and differentiation (Reya and Clevers, 2005; Reya et al., 2003; Yu et al., 2013). The noncanonical Wnt pathway can be further divided into two distinct pathways, Wnt/planar cell polarity (PCP) and Wnt/calcium (Ca2+) pathway. In the Wnt/PCP pathway, binding of Wnt ligands to FZD receptors stimulates the activities of the small GTPases Rho, Rac, and cdc42, which regulates diverse processes, such as cell polarity and migration (Nobes and Hall, 1999; Raftopoulou and Hall, 2004). An aberration in Wnt/PCP pathway has been shown to enhance cell proliferation, survival, abnormal tissue polarity, and cell invasion and cancer metastasis (Katoh, 2005; Ridley, 2004). In the Wnt/Ca2+ pathway, an increase in the level of intracellular Ca2+ is triggered by Wnt‐FZD binding and activation of phospholipase C via G protein. The increase of intracellular Ca2+ also activates protein kinase C. Deregulation of the Wnt/Ca2+ pathway has been implicated to drive cellular motility, cellular proliferation, and epithelial–mesenchymal transition (EMT) during cancer progression (Dissanayake et al., 2007; Wang et al., 2010) .
Frizzled family receptor 7 (FZD7) is one of the FZD receptors that mediates both canonical and noncanonical Wnt signaling (Abu‐Elmagd et al., 2006; Zhang et al., 2013). It is an important regulator of pluripotency resulting in the undifferentiated phenotype (Fernandez et al., 2014; Melchior et al., 2008) in embryonic stem cells and also regulates cellular proliferation in human embryonal carcinoma cells. FZD7 contributes to cell stemness in several normal and cancer cells (Chakrabarti et al., 2014; Mei et al., 2014; Song et al., 2006). Aberrant activation of FZD7 has been found in several types of cancer such as breast (Yang et al., 2011), colorectal (Ueno et al., 2009), hepatocellular carcinoma (Merle et al., 2005), and gastric cancer (Kirikoshi et al., 2001). Recently, it has been shown that FZD7 regulates spheroid proliferation in ovarian cancer stem cells (CSCs) (Condello et al., 2018). Our group previously showed that FZD7 drives in vitro aggressiveness in ovarian cancer (OC) via the noncanonical Wnt/PCP pathway (Asad et al., 2014) while inhibition of FZD receptors through antibody binding reduced tumor‐initiating cell populations in a range of tumor types (Gurney et al., 2012) making the FZD7 pathway very intriguing and warrants further exploration of its regulatory mechanisms and potential for therapeutic targeting.
TWIST1, a transcription factor with a basic helix loop helix domain (Thisse et al., 1988), plays important roles in EMT and cancer metastasis. TWIST1 recruits the nucleosome remodeling and deacetylase complex to upregulate mesenchymal (Mes) markers, to repress epithelial genes, and therefore to induce EMT (Qin et al., 2012). It has been shown to cause apoptosis resistance (Ansieau et al., 2008; Maestro et al., 1999; Valsesia‐Wittmann et al., 2004), chemoresistance (Cheng et al., 2007; Li et al., 2009), enrichment of CSC populations (Battula et al., 2010; Mani et al., 2008; Vesuna et al., 2009), and cell invasiveness (Yang et al., 2004). Several groups have reported the association of TWIST1 with increased tumorigenicity in breast (Yang et al., 2004), prostate (Yuen et al., 2007), and gastric cancers (Feng et al., 2009; Luo et al., 2008). In OC, TWIST1 overexpression correlated with poorer clinical outcomes (Hosono et al., 2007). It has also been linked to both the canonical and noncanonical Wnt pathways. In the canonical pathway, TWIST1 acts as a downstream effector of Wnt3a (Reinhold et al., 2006), Wnt1 (Howe et al., 2003), and β‐catenin (Goodnough et al., 2016), while in the noncanonical pathway, high expression of TWIST1 correlates with the expression of FZD receptor 6 (FZD6), which is associated with increased spheroid‐forming activity and poor survival in neuroblastoma (Cantilena et al., 2011).
Ovarian carcinoma is a highly heterogeneous entity with multiple genetic aberrations and distinct molecular subtypes (Cancer Genome Atlas Research Network, 2011; Tan et al., 2013; Tothill et al., 2008). During trans‐coelomic metastasis of OC, detachment of OC cells from the primary site via an EMT‐like mechanism (Ahmed et al., 2010; Chua et al., 2011; Davidson et al., 2012) allows the dissemination to occur. EMT describes a transition from polarized epithelial cells to motile and invasive Mes cells (Nieto et al., 2016), while anoikis refers to apoptosis induced by lack of proper cell/extracellular membrane (ECM) attachment (Gilmore, 2005). As OC cells tend to shed from primary tumors and suspend in ascites to form secondary lesions throughout the peritoneal cavity, anoikis resistance is considered as one of the most important features of aggressive OC (Cai et al., 2015). Coupling between resistance to anoikis and EMT has been observed in several cancer cell lines (Frisch et al., 2013). Some of the common pathways shared by EMT and anoikis resistance include the PI3K‐Akt pathway (Chiarugi and Giannoni, 2008), Hippo pathway (Cordenonsi et al., 2011), overexpression of Notch3 (Brown et al., 2015), and loss of E‐cadherin (Derksen et al., 2006).
In this study, we use OC as the model system to explore how the FZD7 pathway contributes to the aggressiveness of cancer cells. We found that FZD7 expression was crucial to the maintenance of Mes phenotype, anchorage‐independent growth, and tumorigenesis. We further identified TWIST1 as the downstream effector of FZD7. Manipulating TWIST1 expression mimicked the functional consequences observed in the FZD7 model, while TWIST1 overexpression partially rescued the functional phenotypes abolished by FZD7 knockdown. We subsequently identified the regulation of TWIST1 was by FZD7 through epigenetic modifications of H3K4me3 and H3K27ac at the TWIST1 proximal promoter. In addition, BCL2 expression positively correlated with TWIST1 expression which could be from direct transcriptional regulation. Clinically, the enrichment of FZD7‐TWIST1 axis correlated with poorer survival. We also provided evidence that this axis was amenable to therapeutic targeting by a small molecule porcupine (PORCN) inhibitor, C59.
2. Materials and methods
2.1. Cell culture
Ovarian cancer cell lines OVCA429 and CH1 were grown in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% FBS; OV7 and OV17R were grown in DMEM/F12 plus 10% FBS.
2.2. Generation of stable TWIST1 overexpression and knockdown cell lines
For TWIST1 overexpression, lentiviral plasmids encoding full‐length wide‐type TWIST1 with a pLenti‐GIII‐CMV‐GFP‐2A‐Puro backbone (Applied Biological Materials Inc., Vancouver, BC, Canada) were used. For TWIST1 knockdown, two shRNA clones (#TRCN0000020541 and #TRCN0000020542; Sigma‐Aldrich; subsidiary of Merck KGaA: St. Louis, MO, USA) were selected with pLKO.1‐puro Luciferase shRNA plasmid (#SHC007; Sigma‐Aldrich) as a control. Plasmids were mixed with MISSION® Lentiviral Packaging Mix (#SHP001; Sigma‐Aldrich) before added to a mixture of transfection reagent Fugene 6 (#11814443001; Roche, Basel, Switzerland) and OptiMEM. After 10–15 min’ incubation at room temperature, they were added to 293T cells seeded in the 6‐cm dishes. For infection, virus‐containing supernatants were harvested 48 and 72 h after transfection, filtered, and added to selected cells, together with polybrene (Sigma‐Aldrich). Twenty‐four hours after infection, cells were treated with puromycin at a proper concentration decided by their respective puromycin kill curve.
2.3. BCL2 siRNA Knockdown and Generation of stable TWSIT1‐overexpressing OVCA429
BCL2 small interfering RNA (siRNA; SMART pool ON‐TARGET plus), nontargeting control siRNA (ON‐TARGET plus control pool), and DharmaFECT 4 (# T‐2004‐02) transfection reagents were purchased from Dharmacon (Lafayette, CO, USA). CH1, OV17R, TWIST1 short hairpin against FZD7‐1 (shFZD7‐1), and OVCA429 TWIST1 cells were seeded in 6‐cm dish (Corning, Corning City, NY, USA). BCL2 expression was quantified after 72 h. Plasmid pCMV6‐AC‐tGFP‐TWSIT1 was generated by molecular cloning from pCMV6‐Entry‐TWIST1 (RC202920; OriGene, Rockville, MD, USA). TWSIT1‐overexpressing OVCA429 cells were established by transfection and then sorted into low, intermediate, and high GFP subgroups by florescence‐activated cell sorting (FACS). The high GFP subgroup cells were maintained by G418 (#10131027; Life Technologies, Carlsbad, CA, USA) at 250 μg·mL−1. For negative control, OVCA429 was transfected with pCMV6‐AC‐tGFP empty vector and sorted for GFP‐positive cells every time before the experiment. No stable EV‐OVCA429 survived after G418 selection.
2.4. Reverse transcription and quantitative PCR (RT–qPCR)
mRNA were extracted using an RNeasy mini kit (SAbiosciences, Qiagen, Hilden, Germany) according to manufacturer's protocol and reverse‐transcribed to cDNA using RT2 first‐strand kit (SAbiosciences, Qiagen). The cDNA were mixed with SYBR green master mix (SAbiosciences, Qiagen) and primers for quantitative PCR (qPCR) analysis. Five housekeeping genes ACTB, B2M, GAPDH, HPRT1, and RPL13A were used for normalization. Details of the primers are listed in Tables 1 and 2. For reverse transcription (RT)–qPCR data analysis, the mRNA expression of each gene was normalized to the average of the five housekeeping genes and presented as average fold change 2−∆∆Ct with respect to their control. All qPCR experiments were done using ABI 7900HT (Life Technologies). Cycling conditions were set as 95 °C incubation for 10 min at the first stage, 40 cycles of a two‐step stage 95 °C for 15 s and 60 °C for 1 min, followed by dissociation stage.
Table 1.
Details for commercially available primers (SAbiosciences, Qiagen)
| Target | Cat. No. | Target | Cat. No. |
|---|---|---|---|
| ACTB | PPH00073E | SNAI2 | PPH02475A |
| B2M | PPH01094E | TWIST1 | PPH02132A |
| GAPDH | PPH00150E | ZEB1 | PPH01922A |
| HPRT1 | PPH01018C | ZEB2 | PPH09021A |
| RPL13A | PPH01020B | CD44 | PPH00114A |
| SNAI1 | PPH02459A | BMI1 | PPH577784A |
Table 2.
Details for other primers
| Forward primers | Reverse primers | |
|---|---|---|
| BCL2 | CTGCACCTGACGCCCTTCACC | CACATGACCCCACCGAACTCAAAGA |
| PTEN | CCAGTGGCACTGTTGTTTCACA | CAGGTAACGGCTGAGGGAGCTC |
| BCLXL | GATCCCCATGGCAGCAGTAAAGCAAG | CCCCATCCCGGAAGAGTTCATTCACT |
| BAX | TGG AGCTGCAGAGGATGATTG | GAAGTTGCCGTCAGAAAACATG |
| ALDH1 | CTGCTGGCGACAATGGAGT | GTCAGCCCAACCTGCACAG |
| CD133 | CAACCCTGAACTGAGGCAGC | TTGATAGCCCTGTTGGACCAG |
2.5. Western blot analysis
Cell lysates were harvested using cold RIPA buffer (#R0278; Sigma‐Aldrich) with protease inhibitor (#539134) and phosphatase (#524625) inhibitor cocktails from Calbiochem, Millipore (Burlington, MA, USA). BCA assay (#23225; Thermo Scientific, Waltham, MA, USA) was performed for protein quantification. After heating, lysates were resolved by standard reducing SDS/PAGE, transferred to poly(vinylidene difluoride) membranes, blocked with 5% BSA for at least 1 h, and incubated overnight at 4 °C with the following antibodies diluted in 1% BSA in TBST: anti‐E‐cadherin (#610182) from BD Biosciences, Franklin Lakes, NJ, USA at 1 : 1000; anti‐TWIST1(#sc‐81417) from Santa Cruz Biotechnology, Dallas, TX, USA at 1 : 200; antivimentin (#M7020) from Agilent Technologies (Dako, Santa Clara, CA, USA) at 1 : 1000; anti‐actin (#A1978) and anti‐GAPDH (#G9545) from Sigma at 1 : 1000. After washing, membranes were then incubated in dark at room temperature for half an hour with Infrared dye‐conjugated secondary antibodies from Li‐COR Biosciences, Lincoln, NE, USA at 1 : 5000 diluted in TBST: IRDye 800CW goat anti‐mouse or anti‐rabbit (#926‐32210, #926‐32211) and IRDye 680LT goat anti‐mouse or anti‐rabbit (#926‐68020, #926‐68021). Blots were scanned using the Odyssey Infrared Imaging System (Li‐COR). Images were transferred to grayscale.
2.6. Immunofluorescence staining
Cells were seeded on 15‐mm glass coverslips (Paul Marienfeld GmbH & Co. KG, Lauda‐Königshofen, Germany) to proper confluent, fixed with 4% paraformaldehyde for 10 min, and permeabilized with 0.05% Triton X for 5 min. The fixed cells were incubated with blocking buffer (3% BSA in PBS) at room temperature for 1 h, with primary antibodies (diluted in 1% BSA in PBS) overnight at 37 °C, and then incubated with secondary antibodies for 1 h at room temperature in the dark. The primary antibodies used include anti‐E‐cadherin (#610182) from BD Transduction Laboratories; antivimentin (#M7020) from Dako; and anti‐β‐Catenin (#8480) from Cell Signaling Technology (Danvers, MA, USA). Secondary antibodies are Alexa Fluor 488‐conjugated anti‐mouse (#A11029) and Alexa Fluor 594‐conjugated anti‐mouse (#A11032) from Invitrogen (Carlsbad, CA, USA). For F‐actin staining, rhodamine phalloidin (#R415; Life Technologies) was used directly after blocking. The stained coverslips were mounted onto glass slides using Vectashield mounting medium with DAPI (#H‐1200) from Vector Laboratories (Burlingame, CA, USA). Images were taken using Nikon (Minato, Tokyo, Japan) A1R confocal microscope. To quantify immunofluorescence (IF) expression of E‐cadherin and vimentin, mean fluorescence intensity was measured by using the automated measurement function in the nikon software.
2.7. Spheroid formation assay
In spheroid formation assay, cells were seeded to ultralow attachment (ULA), flat‐bottom 96‐well plate (#7007; Corning) at 500 cells (OV17R and OVCA429 clones) or 200 cells (CH1 and OV7 clones) in 100 μL growth medium per well. After incubated at 37 °C, 5% CO2 for 10 or 14 days, calcein‐AM (#C3100MP; Life Technologies) was used to stain the living cells and ethidium homodimer‐1 (EthD‐1; #E1169; Life Technologies) was used to stain the dead cells. Images were taken and analyzed using Zeiss (Oberkochen, Germany) Axio Imager M2 imaging system. Each experiment was performed by seeding 10 wells per clone. Three independent replicate experiments were done. Only spheroids above 50 μm in diameter were included for counting. Spheroid counts were calculated by adding the total number of spheroids from 10 wells in each experiment. The data are represented by the average number of total spheroids from three experiments per 5000 (OV17R and OVCA429) and 2000 (CH1 and OV7) cells.
2.8. Anoikis assay (FACS and caspase 3/7 activity)
For FACS, cells were seeded in ULA 10‐cm dish (#3263; Corning) at a density of 500 000 cells per dish. After incubated at 37 °C, 5% CO2 for 48–72 h, cells were collected, trypsinized to get single cell suspension, and stained with propidium iodide (PI) and Annexin V (#V13242; Sigma) for 15 min at room temperature in dark. LSRII FACS analyzer was used to do the FACS with proper gating.
For the caspase 3/7 activity assay, cells were seeded in ULA 96‐well plates (#7007; Corning) at a density of 10 000 (OV17R, OVCA429, and OV7 clones) or 5000 (CH1 clones) cells per well. After 72‐h or 96‐h incubation, 20 μL of CellTiter‐Fluor reagent (for cell viability, #TB371; Promega (Madison, WI, USA)) was added to all the wells and the fluorescence was measured using a Tecan plate reader (Tecan, Männedorf, Switzerland; infinite 200) after 1‐h incubation at 37 °C. One hundred microlitee of caspase‐Glo 3/7 reagent (#TB323; Promega) was then added to all the wells, and the luminescence was measured after 1–2 h incubation at room temperature. The caspase 3/7 activities were divided by the cell viabilities and then normalized to their respective controls.
2.9. Molecular cloning
For TWIST1 promoter amplification, the first pair of PCR primers were forward: GCGTATCCAAGCATTTGGAATTGGGG and reverse CTCTCGAGCGGCGACGCGTGGCCTC. The second round of PCR using primers: forward, CCGGGTACCCTTTCAAGGTCACAATGCGGAGCC and reverse, ATACTCGAGTGGGCGAGAGCTGCAGACTTGG was developed to add RE sites.
KOD hot start kit was used to do the PCR (#71086; Merck Biosciences (Novagene, Darmstadt, Germany)) with a system containing: 1× reaction buffer, 1.5 mm MgSO4: 0.2 mm dNTPs, 0.3 μm primers; 100 ng of Template DNA, 0.02 U·μL−1 KOD Hot Start DNA Polymerase and water with total reaction volume 50 μL. For PCR, step 1, polymerase activation with 95 °C for 2 min; step 2, denature with 95 °C for 20 s; step 3, annealing with lower primer T m (°C) for 10 s; and step 4, extension with 70 °C for 30 s to 1.5 min. Totally 35 cycles of steps 2–4 were followed by a 70 °C, 4 min further extension.
After PCR, the purified PCR products and pGL3 vectors were digested with double enzymes (KpnI and XhoI; New England Biolabs, Ipswich, MA, USA) in 37 °C for 3–4 h. The digested products were then loaded into 0.8% agarose gel for gel extraction. For ligation, digested pGL3 vectors and inserts were incubated at 16 °C overnight together with T4 ligase (#M0202S, NEB) and ligation buffer. After ligation, the whole ligation system was used to do the transformation. Sequences of the inserts were validated by plasmid sequencing.
2.10. Promoter assay
On day 1, the cells were seeded in white 96‐well plates (#655098; Greiner Bio‐One, Kremsmünster, Austria) with 4000 cells per well and incubated at 37 °C for 24 h. On day 2, cells were transfected with pGL3‐TWIST1‐promoter (100 ng per well) together with pCMV‐Renilla (1.5 ng per well) and then incubated at 37 °C for 48 h. On day 4, the medium was removed from the wells and 75 μL Dual‐Glo reagent together with 75 μL fresh medium was added to each well. The plate was then put on the shaker for 30 min in dark for cell lysis, and the firefly luminescence was measured using the Tecan plate reader. After the first read, 75 μL of Dual‐Glo Stop&Glo Reagent was added to each well and incubated on the shaker in dark for another 30 min. The Renilla luminescence was then read in the same plate/well order as the firefly luminescence. For the data presentation, the value of firefly luminescence was divided by Renilla luminescence for the same well and then normalized to their respective control.
2.11. ChIP‐qPCR assays
Cells grown in 10‐cm dishes were cross‐linked by 1% of formaldehyde for 10 min at room temperature followed by a final concentration of 0.125 m glycine for 5 min to stop the cross‐linking. The fixed cells were washed twice with tris‐buffered saline buffer and harvested in 1 mL SDS buffer with additional protease inhibitors. The cell lysates were kept on ice for around 20 min, centrifuged at 300 g, 4 °C for 10 min, reconstituted in IP buffer (0.5 v of SDS buffer and 1 v of Triton dilution buffer), and then sonicated by Bioruptor® sonifier for seven pulses (25″ on 45″ off intervals) to form DNA fragments with 200–500 bp in size. Prior to immunoprecipitation, the chromatin samples were precleared with FBS blocked protein G Sepharose beads for 1 h at 4 °C. The precleared samples were incubated with 5 μg of the following antibodies including goat immunoglobulin G (IgG) control (# sc‐2028; Santa Cruz), anti‐TWIST1 (# sc‐6070; Santa Cruz), rabbit IgG control (# sc‐2027; Santa Cruz), anti‐H3K4me3 (CS‐003‐100; Diagenode SA, Liege, Belgium), anti‐H3K27me3 (#ab6002; Abcam, Cambridge, UK), anti‐H3K27ac (#ab4729; Abcam), anti‐H3K9me3 (#ab8898; Abcam), overnight at 4 °C. The samples were then incubated with preblocked protein G Sepharose beads for 4 h at 4 °C followed by four rounds of washes. The bound DNA was eluted, de‐cross‐linked at 65 °C overnight, and subsequently purified using QIAquick PCR purification kit from Qiagen. The purified samples were subjected to qPCR analysis. For ChIP‐qPCR, the primer pairs are listed in Table 3.
Table 3.
Details of the ChIP‐qPCR primer pairs
| Forward | Reverse | |
|---|---|---|
| TWIST1 promoter | CTCCTCCTCACGTCAGGCCAATG | TGGATGGCCCCGAGGTCCAAA |
| BCL2 promoter region 1 | GCCTCCCAAAGTGCTGAGATT | TTCGCCACCTGCTGGTTGTTT |
| BCL2 promoter region 4 | TTAGGACGGTGGGCCTGAAAG | CCCGAGCGTGGTGTTTACTTT |
| BCL2 promoter region 5 | TGTCGTAAAGCCCTTGATAAACC | TGTCGTAAAGCCCTTGATAAACC |
2.12. Statistical analyses
For all the qPCR experiments, unpaired t‐test was used to analyze the expression of the certain genes in overexpression or knockdown clones compared to controls. For ChIP‐qPCR experiments, unpaired t‐tests were used to determine statistical significance in the enrichment of TWIST1 and the epigenetic markers at the selected DNA regions. All of the t‐tests were performed using graphpad prism® version 6 (GraphPad Software, La Jolla, CA, USA).
To derive the FZD7‐TWIST1 signature, we extracted preprocessed gene expression data from CSIOVDB. The 77 genes whose expressions are most positively correlate the average expression of FZD7 and TWIST1 (Spearman correlation coefficient ρ > 0.3) were selected as an FZD7‐TWIST1 signature. To compute the enrichment score of the signature, a two‐sample Kolmogorov–Smirnov‐based test was used. Kaplan–Meier analyses were performed using graphpad prism® version 5.04 (GraphPad Software). Patients were categorized into low and high groups corresponding to the first (lowest 25%) and last quartiles (highest 25%) of signature enrichment score, respectively. P‐values of survival analyses were computed using log‐rank test.
2.13. CAM xenograft
Fertilized chicken eggs were obtained from a local farm and labeled as embryonic day 0 (ED0). Eggs were wiped mechanically with dry paper towels and incubated at 37–38 °C in 60–62% humidity in a R‐COM MX‐50 incubator. On ED3, 2–3 mL of albumin was removed with 18‐G needle inserted at tip of the egg. Semipermeable adhesive films were applied at the marked upper side of shell to prevent spilling of shell particles onto the chorioallantoic membrane (CAM) while cutting the window. Small holes were made and a window cut of 1 cm2 in the shell using sterile sharp‐pointed surgical scissors. The windows were sealed with semipermeable adhesive films, and the eggs were kept back into the incubator. On ED9, CH1 [short hairpin luciferase (shLuci), shFZD7‐1, shFZD7‐2, and shFZD7‐1 Twist1] and OV‐17R (shLuci, shFZD7‐1, shFZD7‐2, and shFZD7‐1 Twist1) cells at 70% confluency were trypsinized, washed with 1× PBS, and resuspended in 100 μL of Matrigel (Corning, Cat#354234) per well. One million CH1 cells and 2 million OV17R cells were seeded onto the CAM by gently tapping the membrane with an autoclaved glass rod. The windows were again sealed with semipermeable adhesive films before keeping the eggs back in the incubator. On ED15, the tumors were cut out carefully from the CAM and the chick embryos were decapitated. The tumors were washed in 1× PBS in order to remove blood from the tumors. After washing, the tumors were kept and aligned to take images.
3. Results
3.1. FZD7 knockdown cells demonstrate an epithelial trait
Transient silencing of FZD7 by siRNA was shown to induce colony compaction in a subset of OC cells (Asad et al., 2014). We confirmed this phenotypic change by using shRNA‐mediated knockdown of FZD7 (shFZD7) in two cell lines with high FZD7 expression, CH1 and OV17R (Fig. 1C,F). shFZD7 cells demonstrated a more epithelial‐like morphology hallmarked by colony compaction, an increase of linear junctional staining of β‐catenin, loss of stress fibers, and a decrease in vimentin in both CH1 and OV17R cells (Fig. 1A,D). The effect of colony compaction in shFZD7 cells compared to their luciferase control cells was evident by the significantly decreased internuclear distance (Fig. 1B,E) in both cell lines. There was restoration of junctional staining of Pan‐cadherin (Fig. S1A) and E‐cadherin (Fig. S1B) in shFZD7 cells which suggested strengthening of cadherin‐based cell–cell adhesions. qPCR analysis of a panel of related EMT genes in shFZD7 cells showed decreases in vimentin, EMT transcription factors TWIST1 and SNAI2, type I and type V collagen components COL1A2 and COL5A2, and insulin‐like growth factor pathway component IGFBP4 in both CH1 and OV17R cells (Fig. 1C,F), while significant increases in CDH2, CDH3 in CH1 (Fig. S1C) and CDH1, CDH3 and EPCAM in OV‐17R cells were observed (Fig. 1D). The demonstration of colony compaction involving cytoskeleton rearrangement, strengthening of cadherin‐based cell–cell adhesion with increased cell polarity in shFZD7 cells, suggested that FZD7 mediated the maintenance of the Mes trait.
Figure 1.
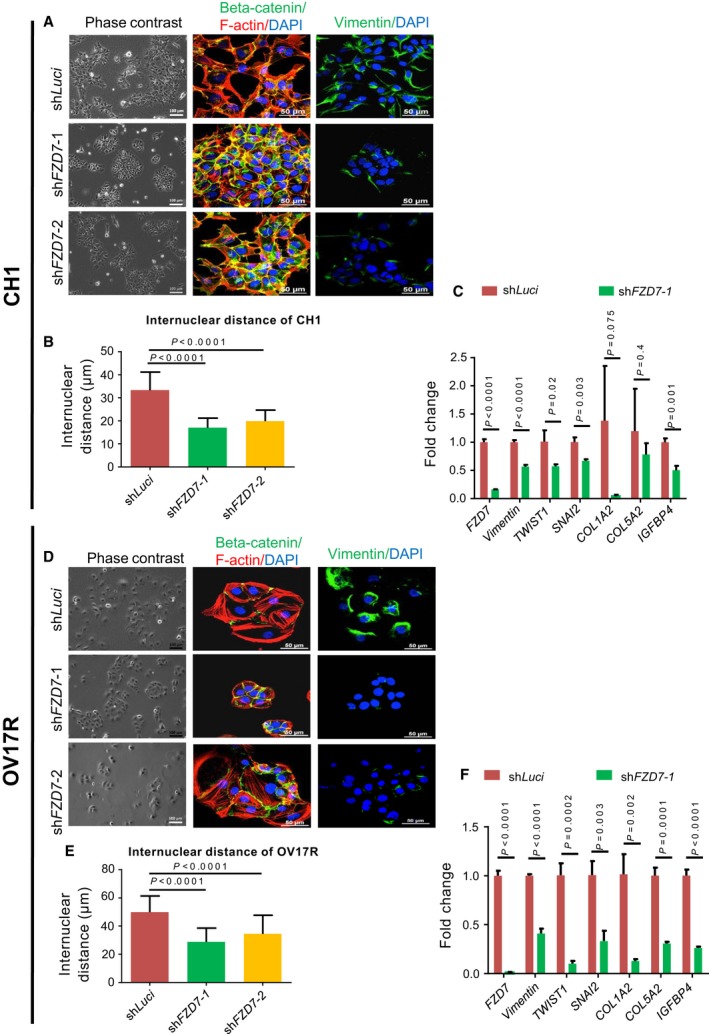
shFZD7 conferred an epithelial‐like phenotype and downregulation of EMT‐related genes. Morphology of (A) CH1‐shLuci, CH1‐shFZD7‐1, and CH1‐shFZD7‐2 clones and (D) OV17R‐shLuci, OV17R‐shFZD7‐1, and OV17R‐shFZD7‐2 clones shown in phase‐contrast images (left panel), IF staining (middle panel) of β‐catenin (green) and F‐actin (red), and vimentin (right panel) together with the nuclear staining of DAPI (blue). Scale bars indicated 100 μm for phase‐contrast images and 50 μm for IF images. Bar charts showing the internuclear distance (y‐axis, μm) of (B) CH1‐shLuci (dark red bars), CH1‐shFZD7‐1 (green bars), and CH1‐shFZD7‐2 (orange bars)clones (x‐axis) and (E) OV17R‐shLuci (dark red bars), CH1‐shFZD7‐1 (green bars), and CH1‐shFZD7‐2 (orange bars) clones. Error bars indicated SEM. Unpaired t‐tests were performed for statistical significance. Bar charts showing the fold change (y‐axis) of FZD7, vimentin, TWIST1,SNAI2,COL1A2,COL5A2, and IGFBP4 mRNA expression (2−∆∆Ct) in (C) CH1‐shLuci (dark red bars), shFZD7‐1(green bars) clones normalized to CH1‐shLuci and in (F) OV17R‐shLuci (dark red bars), shFZD7‐1 (green bars) clones normalized to OV17R‐shLuci. mRNA expression levels were measured by qPCR normalized to a panel of housekeeping genes, ACTB, B2M,GAPDH,RPL13A, and HPRT1. Error bars indicated SEM. Unpaired t‐tests were performed for statistical significance.
3.2. FZD7 is essential for anoikis resistance and tumor formation
Anoikis resistance, the ability to evade cell death in an environment devoid of adhesion to the ECM, is a prerequisite step for cancer metastasis, particularly during the initial phase of trans‐coelomic spread in OC (Gilmore, 2005). Following the acquisition of anoikis resistance, the ability to form spheroids or multicellular aggregates is crucial for the intraperitoneal implantation in OC. We then evaluated anoikis resistance and spheroid‐forming ability of CH1 and OV17R upon FZD7 knockdown. FZD7 knockdown rendered CH1 and OV17R cells more prone to anoikis, evidenced by the significant increase of Annexin V‐positive early apoptotic cells from 15.56% in CH1‐shLuci to 25.33% in CH1‐shFZD7‐1 and 30.73% in CH1‐shFZD7‐2 and from 18.2% in OV17R‐shLuci to 34% in OV17R‐shFZD7‐1 and 31% in OV17R‐shFZD7‐2 (Fig. 2A,F, respectively). We subsequently interrogated these cells for caspase 3/7 activity indicating late apoptotic events and demonstrated an increase in apoptotic activity as evidenced by an increase in caspase 3/7 activity in both CH1 and OV17R shFZD7 clones (except for the CH1 shFZD7‐1 clone) compared to their respective luciferase controls (Fig. 2B,G). Results from the cell viability readouts also show both CH1 and OV17R shFZD7 clones had significantly lower numbers of live cells compared to their shLuci controls (Fig. 2C,H). When cells were seeded in an ULA 96‐well plate at a density of 200 cells per well (Materials and methods), CH1‐shLuci formed much larger spheroids with less EthD‐1‐positive death signal as compared with shFZD7 clones, CH1‐shFZD7‐1 and CH1‐shFZD7‐2 (Fig. 2D). The total number of spheroids (with diameter > 50 μm) formed significantly decreased upon FZD7 knockdown in both cell lines (Fig. 2E,J) with OV17R‐shLuci showed the tendency to form smaller spheroids while the shFZD7 knockdown OV17R clones failed to form any spheroids and displayed higher EthD‐1‐positive signals (Fig. 2I). The tumorigenic efficiency was also affected by the loss of FZD7. By using the CAM xenograft assay (Materials and methods), tumor volume of CH1‐shFZD7‐1 and CH1‐shFZD7‐2 was significantly reduced (Fig. S1E) compared to CH1‐shLuci controls. Our results suggest FZD7 is critical in cell survival and resistance to cell death in an anchorage‐independent environment which is crucial for tumorigenesis.
Figure 2.
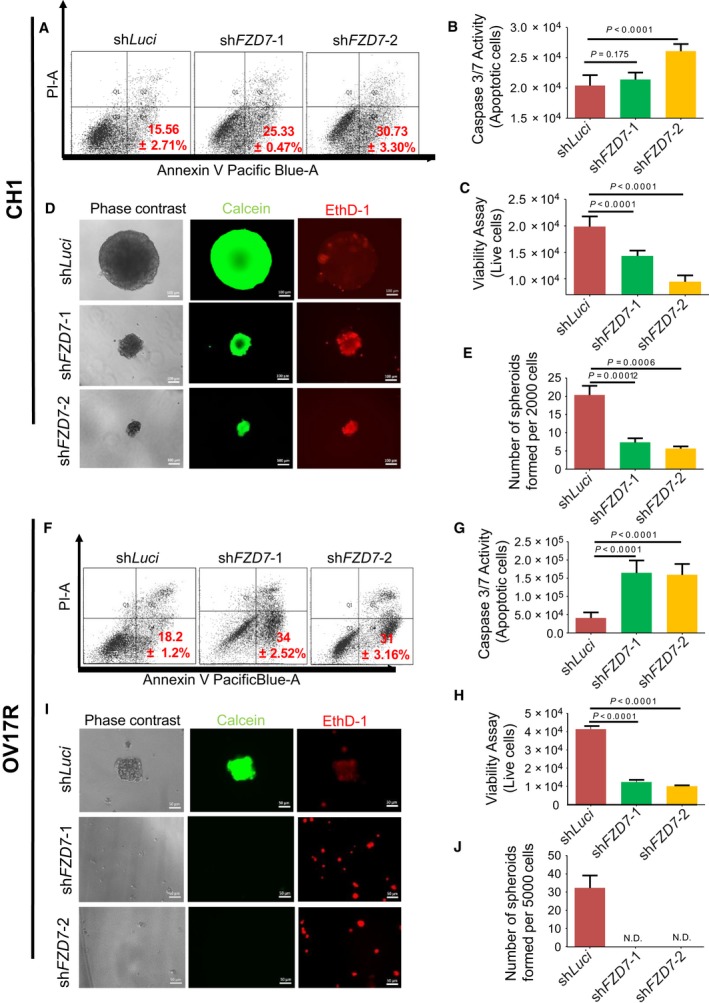
shFZD7 reduced the anoikis resistance and spheroid formation ability of CH1 and OV17R. Percentage of apoptotic (A) CH1‐shLuci, CH1‐shFZD7‐1, and CH1‐shFZD7‐2 and (F) OV17R‐shLuci, OV17R‐shFZD7‐1, and OV17R‐shFZD7‐2 cells after 48 h in suspension, as analyzed by flow cytometry (10 000 events). The percentage of Annexin V+/PI− were considered apoptotic. Values (mean ± SD) in red denoted the percentage of apoptotic cells from three independent experiments. X‐axis: the levels of Annexin V tagged with pacific blue; y‐axis: the levels of PI. Bar charts showing the caspase 3/7 activities—apoptotic cells (y‐axis) for (B) CH1‐shLuci (dark red bars), CH1‐shFZD7‐1 (green bars), and CH1‐shFZD7‐2 (orange bars) cells (x‐axis) and (G) OV17R‐shLuci (dark red bars), OV17R‐shFZD7‐1 (green bars), and OV17R‐shFZD7‐2 (orange bars) cells (x‐axis). Viability assay showing live cells (y‐axis) for (C) CH1‐shLuci (dark red bars), CH1‐shFZD7‐1(green bars), and CH1‐shFZD7‐2 (orange bars) cells (x‐axis) and (H) OV17R‐shLuci (dark red bars), OV17R‐shFZD7‐1 (green bars), and OV17R‐shFZD7‐2 (orange bars) cells (x‐axis). Cells were seeded at a density of 10 000 cells per well in flat‐bottom ULA 96‐well plates for three wells per clone. After 72 h, cell viability was measured by fluorescence reading (400Ex/505Em), and cell death was measured by luminescence readout. (D) CH1‐shLuci, CH1‐shFZD7‐1, and CH1‐shFZD7‐2 cells were seeded at a density of 200 cells per well and (I) OV17R‐shLuci, OV17R‐shFZD7‐1, and OV17R‐shFZD7‐2 cells were seeded at a density of 500 cells per well in flat‐bottom ULA 96‐well plates for 10 wells per clone. After 14 days in culture, phase‐contrast images (left panel), calcein‐AM staining (middle panel) for viable cells, and EthD‐1 staining (right panel) for dead cells were analyzed. Scale bars represented 100 μm (CH1) and 50 μm (OV17R). Bar charts showing numbers of spheroids formed (y‐axis) in (E) CH1 clones (x‐axis) and (J) OV17R clones after 7 days in suspension. Only spheroids with a diameter greater than 50 μm were counted. Error bars indicated SEM. Unpaired t‐tests were performed for statistical significance.
3.3. TWIST1 confers similar functional phenotypes as FZD7 in EMT and anoikis resistance
We next explored the possible downstream effectors of FZD7 related to the Mes trait and anoikis resistance. Upon FZD7 knockdown, the EMT transcription factor TWIST1 was repressed in both CH1 and OV17R (Fig. 1C,F). TWIST1 has been shown to be responsible for maintaining the Mes phenotype and cell survival in suspension in several cancer types (Qin et al., 2012). Thus, we hypothesized that TWIST1 might be a downstream effector of FZD7. We first explored whether depletion of TWIST1 resulted in a similar functional phenotype as depletion of FZD7 in OC. Upon screening a group of relevant OC cell lines, we identified two OC cell lines OVCA429 and OV7 with appreciable low and high TWIST1 protein expression, respectively (data not shown). OVCA429 was transfected with a GFP‐tagged TWIST1 construct for overexpression. Three subpopulations, negative GFP, intermediate GFP, and high GFP, were subsequently sorted according to different GFP levels. These subgroups displayed a spectrum of phenotypic EMT traits with increasing levels of TWIST1 (Fig. 2A). IF stains confirmed the levels of TWIST1 correlated with the morphology as well as the expression of the epithelial marker, E‐cadherin, and the Mes marker, vimentin (Fig. S2B). The high GFP subgroup (TWIST1‐tGFP) in OVCA429 cells displayed actin reorganization with the formation of stress fibers and loss of cell–cell junction with the cytoplasmic relocalization of β‐catenin (Fig. 2B). Conversely, in OV7 cells with high TWIST1 protein expression, loss‐of‐function experiments with knockdown of TWIST1 using two different shRNA sequences, shTWIST1‐1 and shTWIST1‐2, demonstrated colony compaction compared to the dispersed morphology observed in the shLuci control of OV7 cells (Fig. 2C). IF staining of OV7 knockdown clones also demonstrated a slight restoration of E‐cadherin and a decrease in vimentin staining, which is further quantified by measuring the mean fluorescence intensity of E‐cadherin and vimentin (Fig. S2D,E). Concentrated junctional β‐catenin and epithelial‐like cortical actin bundles (Fig. 2C) were also found. The decrease of internuclear distance in OV7 shTWIST1‐1 and OV7 shTWIST1‐2 further indicated the increase of apical polarity (Fig. 2F).
After 48 h in suspension, TWIST1 overexpressed OVCA429 cells (TWIST1‐tGFP) had approximately 10% less apoptotic activity when compared to controls (EV‐OVCA429; Fig. 3A). Adopting the same criteria to look for spheroids with diameters more than 50 μm (Materials and methods), TWIST1‐tGFP‐OVCA429 cells showed spheroid formation while the control EV‐OVCA429 failed to form sizable spheroids but rather numerous smaller aggregates in suspension after 14 days (Fig. 3C,D). The data indicate that TWIST1 overexpression increases spheroid‐forming ability of OC cells. Conversely, we investigated whether the loss of TWIST1 expression would result in an opposite phenotype. We observed that the apoptotic population in suspension cultures increased with the knockdown of TWIST1 in OV7 cells to 19.7% in shTWIST1‐1 and 32.6% in shTWIST1‐2 (Fig. 3E) compared to 13% in shLuci control cells. We also demonstrated that OV7 shTWIST1‐1 and shTWIST1‐2 clones formed smaller and fewer spheroids compared to the shLuci OV7 cells in three independent experiments (Fig. 3G,H). OV7 shTWIST1‐2, which had better TWIST1 knockdown efficiency, showed the least spheroid‐forming ability (Fig. 3G,H). The TWIST1 and E‐cadherin protein levels in these clones were subsequently validated using western blots (Fig. 3B,F). Collectively, these findings indicate that TWIST1 contributes to the Mes phenotype and anoikis resistance similar to FZD7.
Figure 3.
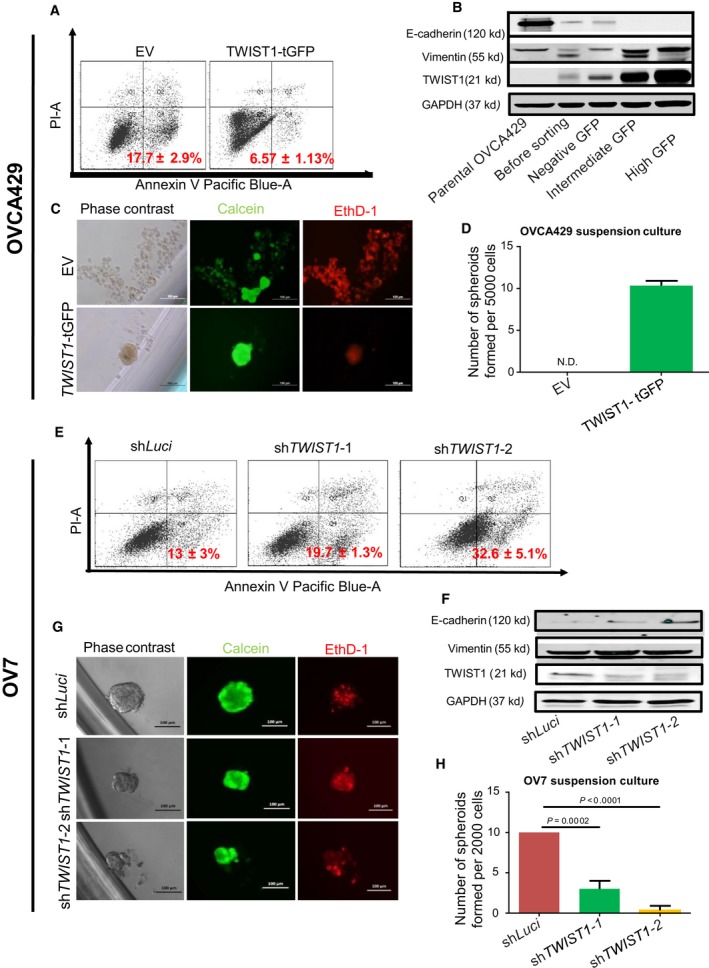
TWIST1 played a crucial role in anoikis resistance in OVCA429 and OV7. Percentage of apoptotic (A) EV‐OVCA429 and TWIST1‐tGFP‐OVCA429 and (E) OV7‐shLuci, OV7‐shTWIST1‐1, and OV7‐shTWIST1‐2 (G) after 48 h in suspension, as analyzed by FACS (10 000 events). The percentage of Annexin V+/PI − were considered apoptotic. Values (mean ± SD) in red denote the percentage of apoptotic cells from three independent experiments. X‐axis: the levels of Annexin V tagged with pacific blue; y‐axis: the levels of PI. (B) Western blots for E‐cadherin, vimentin, TWIST1, and GAPDH in parental OVCA429, TWIST1‐tGFP transfected OVCA429 before FACS sorting, as well as negative GFP, intermediate GFP, and high GFP subgroup OVCA429 after sorting. (C) EV‐ and TWIST1‐tGFP‐OVCA429 cells were seeded at a density of 500 cells per well and (G) OV7 shLuci, shTWIST1‐1, and shTWIST1‐2 cells were seeded at a density of 200 cells per well in flat‐bottom ULA 96‐well plates for 10 wells per clone. After 14 days in culture, phase‐contrast images, calcein‐AM staining for viable cells, and EthD‐1 staining for dead cells were analyzed. Scale bars represented 100 μm. Bar charts showing numbers of spheroids formed by (D) OVCA429 clones TWIST1 tGFP (green bars) or by (H) OV7 clones shLuci (dark red bars), shTWIST1‐1 (green bars) and shTWIST1‐2 (orange bars) after 14 days in suspension. Only spheroids with a diameter more than 50 μm were counted. Error bars indicated SEM. Unpaired t‐tests were performed for statistical significance. (F) Western blots for E‐cadherin, vimentin, TWIST1, and GAPDH in OV7 shLuci, shTWIST1‐1, and shTWIST1‐2 clones.
3.4. TWIST1 is required for FZD7‐driven mesenchymal trait and anoikis resistance
To determine whether TWIST1 is a downstream regulator of FZD7, TWIST1 was overexpressed in two FZD7 knockdown clones, CH1‐shFZD7‐1 and OV17R‐shFZD7‐1, to investigate whether exogenous restoration of TWIST1 expression could reverse the effects of FZD7 knockdown. Morphologically, TWIST1 overexpression in CH1‐shFZD7‐1 resulted in reduced colony compaction, a decrease in cell–cell junctional β‐catenin, and an increase in vimentin staining (Fig. S3A). Similarly, TWIST1 overexpressed OV17R‐shFZD7‐1 cells also showed similar changes, albeit with more marked depletion of E‐cadherin staining (Fig. 3C). In OV17R‐shFZD7‐1 TWIST1 overexpressing cells, the expression of E‐cadherin and vimentin was further quantified by measuring the mean fluorescence intensity (Fig. 3D,E). Increases in the internuclear distance were also observed in both TWIST1 overexpressing CH1‐shFZD7‐1 and OV17R‐shFZD7‐1 clones (Fig. 3B,F). Meanwhile, in CH1‐shFZD7‐1 cells, TWIST1 overexpression rescued the spheroid‐forming ability of FZD7‐depleted CH1 cells (Fig. 4C,D). However, TWIST1 overexpression did not appear to rescue the spheroid‐forming abilities in OV17R‐shFZD7‐1 cells. Notably, we did observe an increase in calcein‐AM‐positive live cells and a decrease in EthD‐1‐positive dead cells in TWIST1‐OV17R‐shFZD7‐1 cells (Fig. 4I). Concordant with the spheroid‐forming nature of TWIST1 overexpressed cells, the population of Annexin V‐positive apoptotic cells decreased by 13% in CH1‐shFZD7‐1 and 7% in OV17R‐shFZD7‐1 upon TWIST1 overexpression (Fig. 4A,G). In addition, there was significant reduction in caspase activity with concomitant increase of live cell signals in the TWIST1 overexpressing CH1‐shFZD7 (Fig. 4E,F) and OV17R‐shFZD7 (Fig. 4J,K) clones. The protein expression of TWIST1, vimentin, and E‐cadherin was further validated using western blots (Fig. 4B,H). The data suggest that TWIST1 rescued the anoikis resistance abolished by FZD7 knockdown and lead to higher cell survival rates in long‐term suspension cultures.
Figure 4.
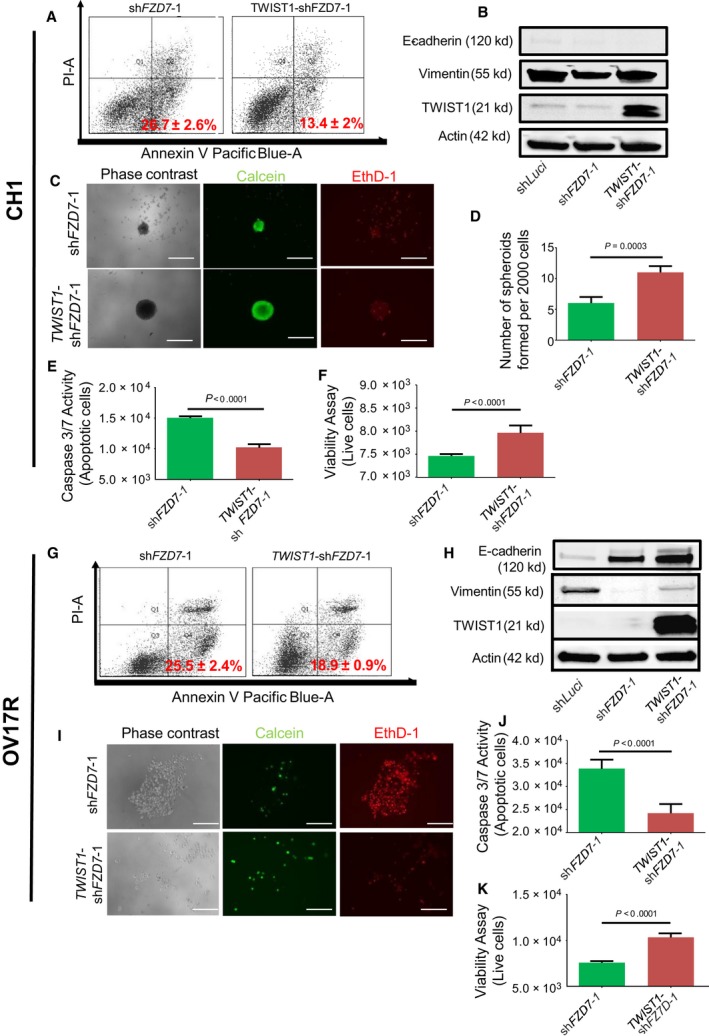
TWIST1 rescued the anoikis resistance in shFZD7 clones. Percentage of apoptotic (A) CH1‐shFZD7‐1 and TWIST1‐CH1‐shFZD7‐1 and (G) OV17R‐shFZD7‐1 and TWIST1‐OV17R‐shFZD7‐1 after 72 h in suspension, as analyzed by FACS (10 000 events). The percentage of Annexin V+/PI − were considered apoptotic. Values (mean ± SD) in red denote the percentage of apoptotic cells from three independent experiments. X‐axis: the levels of Annexin V tagged with pacific blue; y‐axis: the levels of PI. Western blots for E‐cadherin, vimentin, TWIST1, and Actin in (B) CH1‐shFZD7‐1 and TWIST1‐CH1‐shFZD7‐1 clones and in (H) OV17R‐shFZD7‐1 and TWIST1‐OV17R‐shFZD7‐1 clones. (C) CH1‐shFZD7‐1, TWIST1‐CH1‐shFZD7‐1 cells were seeded at a density of 200 cells per well and (I) OV17R‐shFZD7‐1, TWIST1‐OV17R‐shFZD7‐1 cells were seeded at a density of 500 cells per well in flat‐bottom ULA 96‐well plates for 10 wells per clone. After 10 days, phase‐contrast images, calcein‐AM staining for viable cells, and EthD‐1 staining for dead cells were analyzed. Spheroid quantification (D) shows number of spheroids formed per 2000 cells. Scale bars represented 100 μm. Bar charts showing the caspase 3/7 activities—apoptotic cells (y‐axis) for (E) CH1‐shFZD7‐1 (green bars), TWIST1‐CH1‐shFZD7‐1 (dark red bars; x‐axis) and (J) OV17R‐shFZD7‐1 (green bars), TWIST1‐OV17R‐shFZD7‐1 (dark red bars; x‐axis). Viability assay showing live cells (y‐axis) for (F) CH1‐shFZD7‐1 (green bars), TWIST1‐CH1‐shFZD7‐1 (dark red bars; x‐axis) and (K) OV17R‐shFZD7‐1 (green bars), TWIST1‐OV17R‐shFZD7‐1 (dark red bars; x‐axis). Error bars indicated SEM. Unpaired t‐tests were performed for statistical significance.
3.5. TWIST1 is regulated by epigenetic modifications downstream to FZD7
The complex regulation of TWIST1 transcription has been reported to be regulated by stat3 (Cheng et al., 2008; Lo et al., 2007), HIF1α (Yang et al., 2008), and NF‐κB (Sosic et al., 2003) through direct binding at the TWIST1 promoter (Fig. 5A). As such, a dual luciferase reporter assay for TWIST1 promoter activity [−933 bp to +37 bp from transition start site (TSS)] was performed (Fig. 5A) on CH1 and OV17R parental and shFZD7 clones. We did not observe any significant difference in the TWIST1 promoter activity in both CH1 and OV17R cells (Fig. 5B,D) which suggests that the suppression of TWIST1 upon FZD7 knockdown was not through direct repression of TWIST1 promoter activity. As TWIST1 regulation has previously been demonstrated to be mediated by DNA methylation within exon 1 active histone mark H3K4me3 and inactive histone mark H3K9me3 (Sakamoto et al., 2015), we explored whether epigenetic modifications might be an alternative mechanism of TWIST1 regulation by FZD7. Previous ChIP‐sequencing data in OC (Chung et al., 2016) have suggested that histone modification marks were enriched at the TWIST1 exon 1 but not at the distal promoter region. Thus, several histone marks (H3K4me3, H3K27ac, H3K9me3, and H3K27me3) at the TWIST1 proximal promoter and exon 1 regions were interrogated via ChIP‐qPCR. A set of qPCR primers were designed for 8 regions to cover the full length of TWIST1 exon 1. The highest enrichment of the relevant histone marks listed above was observed at the proximal promoter region of TWIST1 (−65 bp to +136 bp from TSS) after normalizing to IgG control (Fig. 5C,E). At this region, significant enrichment of H3K4me3 and H3K27ac was observed in both CH1 and OV17R shLuci cells compared to their respective knockdown clones (shFZD7‐1; Fig. 5C,E). Other histone marks, including H3K9me3 and H3K27me3, demonstrated low‐level enrichment in CH1 shLuci cells but not in OV17R shLuci cells (similar level to the IgG control) throughout the screened regions in the shFZD7 clones (Fig. 5C,E). Collectively, the data would suggest that TWIST1 transcription is epigenetically controlled by active histone marks H3K4me3 and H3K27ac in CH1 and H3K4me3 in OV17R. The depletion of these marks with FZD7 knockdown could be the mechanism which dynamically controls TWIST1 expression.
Figure 5.
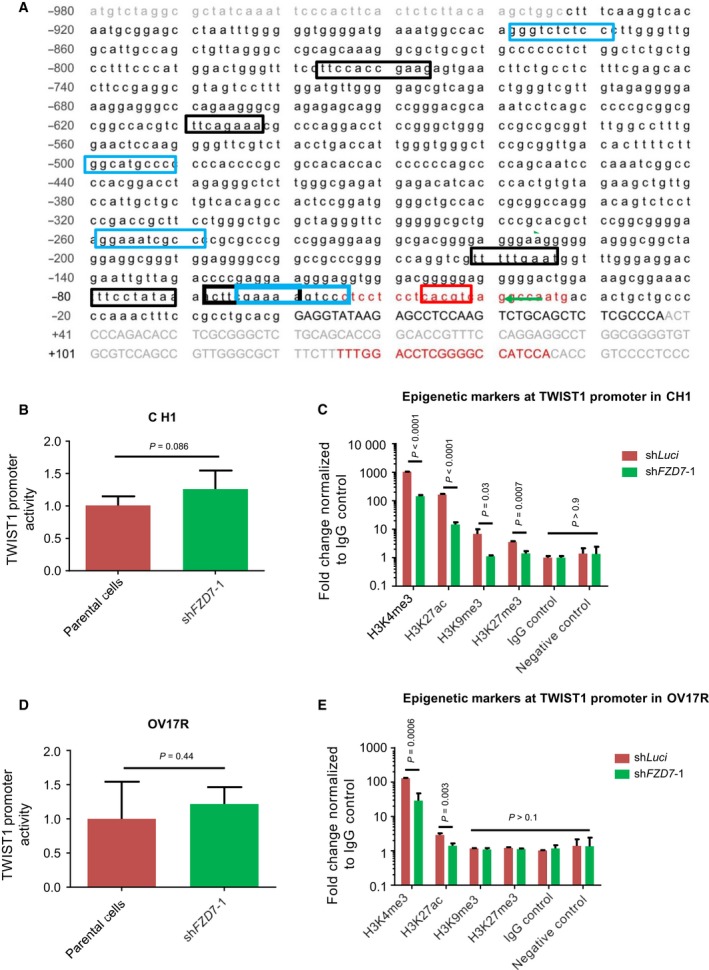
shFZD7 induced epigenetic modifications of TWIST1 promoter. (A) The sequence of the TWIST1 promoter region (black text) from −933 to +37 relative to the TSS with NF‐κB binding sites (blue box), STAT3 binding sites (black box), and an HIF1α binding site (red box). The primer sequences for histone marker ChIP‐qPCR (red text) amplifying the region from −65 to +136 were also shown. Bar charts showing the TWIST1 promoter activities in (B) CH1‐shFZD7‐1 normalized to CH1 and in (D) Ov17R‐shFDZ7‐1 normalized to OV17R. Bar charts showing the enrichments represented by the fold change of percentage of input normalized to IgG control (y‐axis) of H3K4me3, H3K27ac, H3K9me3, H3K27ac, and IgG control (x‐axis) at the TWIST1 promoter in (C) CH1‐shLuci (dark red bars) and CH1‐shFZD7‐1 (green bars) and in (E) OV17R‐shLuci (dark red bars) and OV17R‐shFZD7‐1 (green bars). Unpaired t‐tests were performed for statistical significance.
3.6. BCL2 acts as a downstream effector of the FZD7‐TWIST1 axis
TWIST1 expression has been shown to inversely correlate with the tumor suppressor PTEN (Yin et al., 2010), the intrinsic apoptosis‐related pro‐apoptotic gene BAX (Kwok et al., 2005), and positively correlate with the anti‐apoptotic gene BCL2 (Banerjee et al., 2012). In addition, BCLXL has been shown to contribute to EMT‐related resistance to apoptosis and chemotherapy (Keitel et al., 2014). Therefore, we screened for these genes to explore their involvement in the FZD7‐TWIST1 axis. As expected, BCL2 was upregulated in TWIST1 overexpressed (TWIST1‐tGFP) OVCA429 cells and downregulated in the TWIST1 knockdown clones of OV7 cells (shTWIST1; Fig. 6A,B). When tested in other relevant cell lines, we found that BCL2 was downregulated upon FZD7 knockdown in CH1 and OV17R cells (Fig. 6C,D), and when TWIST1 was overexpressed in these FZD7 knockdown clones, CH1‐ and OV17R‐shFZD7‐1, we observed not only an increase in the expression of BCL2 but also BCL‐XL and BAX (Fig. 6E,F). To confirm whether BCL2 regulates anoikis resistance as a downstream target of FZD7‐TWIST1 axis, siRNA‐mediated knockdown of BCL2 was performed in parental CH1 and OV17R cells, TWIST1 overexpressing OVCA429 and CH1 shFZD7‐1 cells (Fig. S4C). Anoikis assays showed that compared to the negative control siRNA pool siOTP, siBCL2 induced significantly the Annexin V+/PI− population (Fig. 6G). The data indicate highly correlated regulations among FZD7, TWIST1, and BCL2 expressions. While TWIST1 has been shown to suppress BAX in prostate cancer cell lines (Kwok et al., 2005), further work is warranted to explore whether the upregulation of BAX and BCL‐XL in TWIST1 overexpressed CH1‐ and OV17R‐shFZD7 clones is indeed directly induced by TWIST1.
Figure 6.
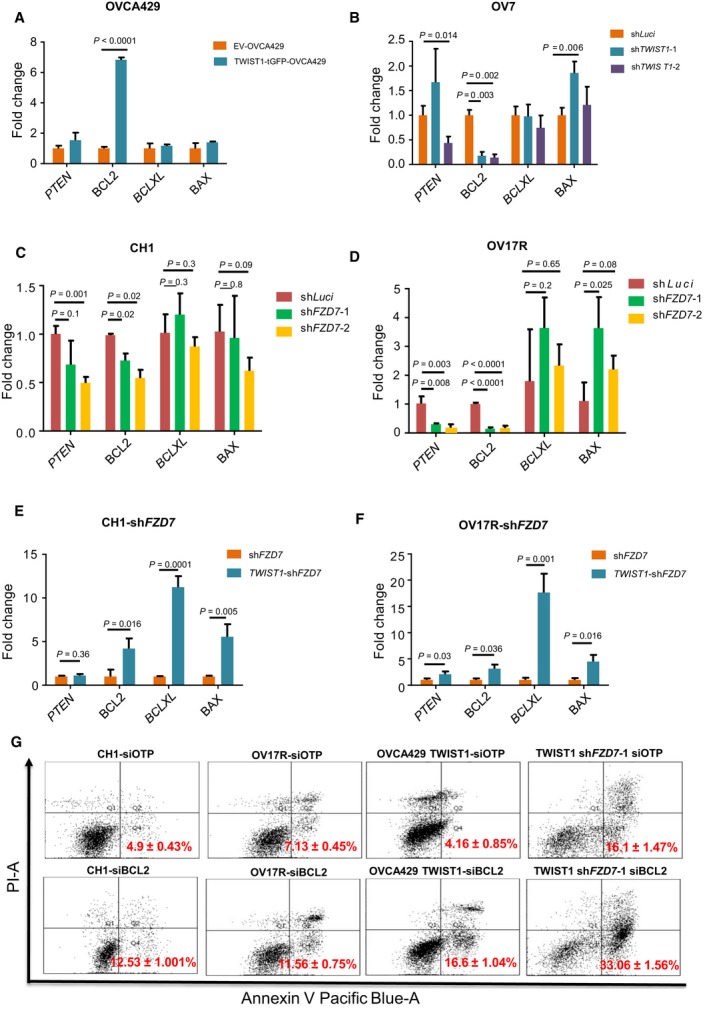
FZD7‐TWIST1 regulates anoikis resistance by activating its downstream effector BCL2. Bar charts showing the fold change (y‐axis) of PTEN,BCL2,BCLXL, and BAX (x‐axis) mRNA expression (2−∆Ct) in (A) TWIST1‐tGFP‐OVCA429 (dark aqua bars) normalized with EV‐OVCA429 (orange bars); (B) OV7‐shTWIST1‐1 (dark aqua bars) and shTWIST1‐2 (purple bars) normalized with OV7‐shLuci (orange bars); (C) CH1‐shFZD7‐1 (green bars), CH1 shFZD7‐2 (orange bars) normalized with CH1shLuci (dark red bars); (D) OV17R‐shFZD7‐1 (green bars), OV17R‐shFZD7‐2 (orange bars) normalized with OV17R‐shLuci (dark red bars); (E) TWIST1‐CH1‐shFZD7 (aqua dark bars) normalized with CH1‐shFZD7 (dark orange bars); (F) TWIST1‐OV17R‐shFZD7 (aqua dark bars) normalized with OV17R‐shFZD7 (dark orange bars). mRNA expression levels are measured by qPCR normalized with a panel of housekeeping genes, ACTB, B2M,GAPDH,RPL13A, and HPRT1. (G) Percentage of apoptotic cells in CH1 (siOTP and siBCL2), OV17R (siOTP and siBCL2), OVCA429 TWIST1 (siOTP and siBCL2) and TWIST1‐CH1‐shFZD7 (siOTP and siBCL2) after 72 h in suspension, as analyzed by FACS (10 000 events). Values (mean ± SD) in red denoted the percentage of apoptotic cells from three independent experiments. X‐axis: the levels of Annexin V tagged with pacific blue; y‐axis: the levels of PI. Error bars indicated SEM. Unpaired t‐tests were performed for statistical significance.
3.7. FZD7‐TWIST1 axis correlates with poor clinical outcomes
Next, to explore the clinical relevance of the FZD7‐TWIST1 axis, gene expressions for both FZD7 and TWIST1 were examined in several OC cohorts. Firstly, in a Japanese OC cohort (GSE30311), FZD7 and TWIST1 expressions were observed to positively correlate (Fig. 7A, P < 0.0001). Secondly, from CSIOVDB, an in‐house OC microarray database comprising of 3431 OC samples (Tan et al., 2015), we derived a FZD7‐TWIST1 signature consisting of 15 genes with positive Spearman correlation (ρ > +0.3) to the average expression of FZD7 and TWIST1. The FZD7‐TWIST1 signature was significantly enriched in the Mes subtype (Fig. 7B, Kolmogorov–Smirnov test, Mes vs rest P = 0). Significant differences in both overall and disease‐free survival were observed in patients stratified by either the median (Fig. 7C,E) or the highest and lowest quartiles of the FZD7‐TWIST1 signature score (Fig. 7D,F). These data suggest that the FZD7‐TWIST1 axis is clinically relevant in OC.
Figure 7.
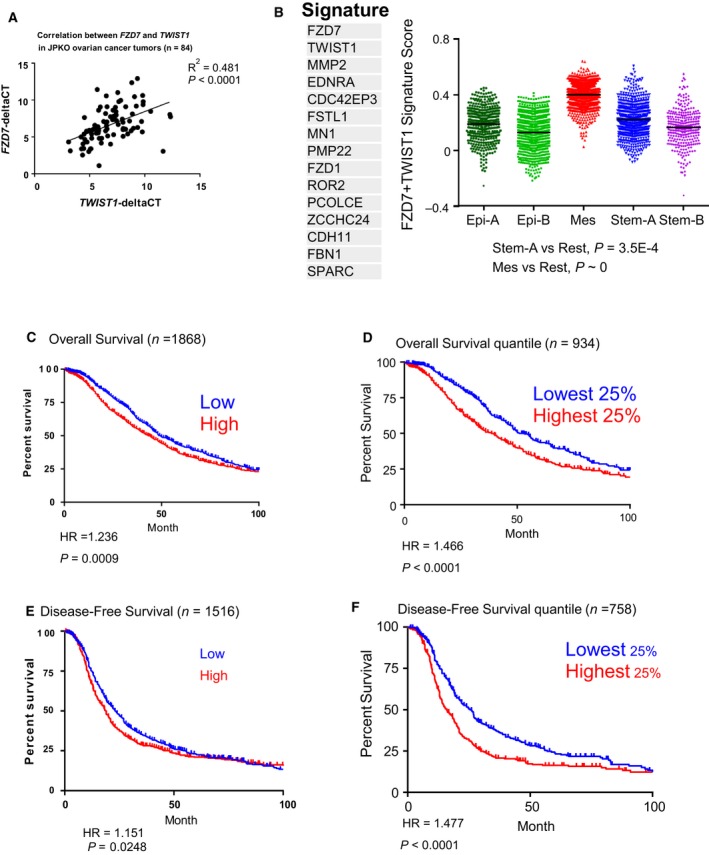
FZD7‐TWIST1 signature predicts poor survival. (A) Scatter plot showing the correlation between FZD7 (y‐axis) and TWIST1 (x‐axis) mRNA expression in a Japanese OC cohort (GSE30311). (B) Distribution of FZD7‐TWIST1 signature score (y‐axis) among the Epi‐A (dark green), Epi‐B (light green), Mes (red), Stem‐A (blue), and Stem‐B (purple) subtypes in CSIOVDB. Kaplan–Meier curves of median overall survival (C), disease‐free survival (E), and the highest and lowest quartiles of the FZD7‐TWIST1 signature score for overall survival (D), disease‐free survivals (F) in OC patients. Stratified by the first and last quartiles of the FZD7‐TWIST1 signature score.
3.8. Targeting the FZD7‐TWIST1 axis with a porcupine inhibitor
We next explored potentially relevant therapy for targeting of the FZD7‐TWIST1 axis. PORCN is an enzyme essential for palmitoylation of Wnts, and this post‐translational modification is necessary for all mammalian Wnts secretion and the binding to their receptors (Madan and Virshup, 2015). Thus, inhibition of PORCN will block the secretion and the receptor binding affinity of all human Wnts. We thus tested whether a small molecule PORCN inhibitor C59 (Proffitt et al., 2013) would block the functional phenotypes of the FZD7‐TWIST1 axis. In CH1‐shLuci, C59 treatment resulted in a decrease in both the number and size of spheroids formed (Fig. 8A,C,D) and an increase in caspase 3/7 activity in suspension culture was observed (Fig. 8E). The level of caspase 3/7 activity was similar in C59‐treated CH1‐shLuci and DMSO‐treated CH1‐shFZD7‐1 (Fig. 8E). Interestingly, C59 treatment did not further enhance caspase 3/7 activity (late apoptotic activity) in CH1‐shFZD7‐1 clone (Fig. 8E). However, early apoptotic activity was observed to increase after C59 treatment as shown by an increase in Annexin V‐positive cells in the CH1 cell line (Fig. 8H). In OV17R, the effect of C59 treatment was limited both in number and in size of the spheroids formed in suspension (Fig. 8B,C,D). In line with this, C59 treatment only induced a slight increase of caspase 3/7 activity in OV17R‐shLuci and to a lesser degree as compared with untreated OV17R‐shFZD7‐1 (Fig. 8F). However, C59 treatment did slightly increase caspase 3/7 activity in OV17R‐shFZD7‐1 (Fig. 8F). We also observed OV17R cells showed marginal increase in Annexin V‐positive cells when treated with C59 (Fig. 8I). In both CH1 and OV17R, TWIST1 and BCL2 expressions were downregulated after C59 treatment (Fig. 8G); thus, the regulation of TWIST1 and BCL2 was, at least partially, Wnt ligand‐dependent. Of note, we observed that the alterations in the FZD7‐TWIST1 axis do not necessarily correlate with the expression changes of the Wnt ligands. The expression of Wnt5a, a known ligand to FZD7 for the noncanonical Wnt pathway, did not show consistent patterns in either shFZD7 clones or in TWIST1 rescue clones (Fig. S4A,B). Collectively, the data indicate that C59 could be utilized to mimic the functional effect of depleting FZD7 in CH1 and to a lesser extent OV17R OC cells.
Figure 8.
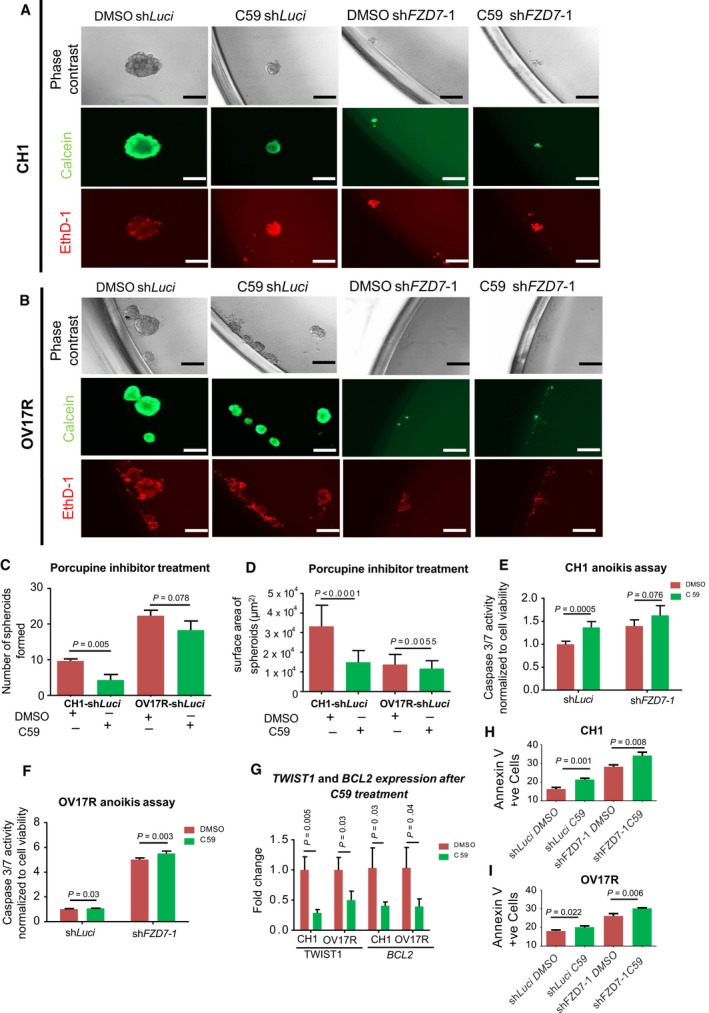
PORCN inhibitor C59 treatment in CH1 and OV17R clones. (A) CH1‐shLuci, CH1‐shFZD7‐1 cells were seeded at a density of 200 cells per well and (B) OV17R‐shLuci, OV17R‐shFZD7‐1 cells were seeded at a density of 500 cells per well in flat‐bottom ULA 96‐well plates for 10 wells per clone. After 10 days in culture with DMSO or C59 treatment, phase‐contrast images, calcein‐AM staining for viable cells, and EthD‐1 staining for dead cells were analyzed. Scale bars represented 200 μm. Bar charts showing (C) the numbers and (D) the surface areas of spheroids formed by CH1‐shLuci and OV17R‐shLuci with DMSO (dark red bars) or C59 (green bars) treatment for 10 days in suspension. Only spheroids with a diameter greater than 50 μm were counted. Bar charts showing caspase 3/7 activities (y‐axis) of DMSO (dark red bars) or C59 (green bars) treated (E) CH1‐Luci, CH1‐shFZD7‐1, and (F) OV17R‐Luci, OV17R‐shFZD7‐1 clones. Cells were seeded at a density of 10 000 cells per well in flat‐bottom ULA 96‐well plates for three wells per clone. After 72 h, cell viability was measured by fluorescence reading (400Ex/505Em) and cell death was measured by luminescence readout. (G) Bar charts showing the fold change of TWIST1 (left) and BCL2 (right) mRNA expression (2−∆Ct; y‐axis) in CH1‐shLuci and OV17R‐shLuci with DMSO (dark red bars) or C59 (green bars) treatment for 3 days. mRNA expression levels were measured by qPCR normalized with a panel of housekeeping genes, ACTB, B2M,GAPDH,RPL13A, and HPRT1. (H) Bar chart showing percentage of Annexin V‐positive cells in CH1‐shLuci, CH1‐shFZD7‐1, (I) OV17R‐shLuci and OV17R shFZD7‐1 after treating the cells with DMSO control (dark red bars) and C59 (green bars). Values (mean ± SD) for Annexin V‐positive cells denoted the percentage of apoptotic cells from three independent experiments. Error bars indicated SEM. Unpaired t‐tests were performed for statistical significance. C59 was used to treat cells at a final concentration of 10 nm. Unpaired t‐tests were performed for statistical significance.
4. Discussion
In this study, we showed that FZD7 and TWIST1 expressions were associated with a Mes‐like phenotype and the expression of EMT‐related genes in OC. We also demonstrated that FZD7 and TWIST1 mediated resistance to cell death and formation of spheroids in suspension. The exogenous expression of TWIST1, at least partially, abrogated the functional effects of FZD7 knockdown. In addition, FZD7 regulated TWIST1 expression via epigenetic modifications, by altering H3K4me3 and H3K27ac enrichments at the TWIST1 promoter. These results indicate the functional importance of the FZD7‐TWIST1 axis in a subgroup of OC in maintaining the Mes‐like phenotype and anoikis resistance. We further demonstrated that patients with a high FZD7‐TWIST1 axis score correlated with poor survival and a small molecular weight inhibitor targeting the Wnt secretion pathway may be a potential therapeutic option.
We have previously shown that siRNA‐mediated FZD7 transient knockdown resulted in colony compaction and a reduction in cell proliferation via casein‐kinase‐1ε mediated noncanonical Wnt/PCP pathway (Asad et al., 2014). Here, we show that shRNA‐mediated stable knockdown of FZD7 not only reversed the Mes phenotype but also diminished anoikis resistance, cell survival, spheroids, and tumor formation. These results not only emphasize the importance of proper cell–matrix attachment to cell survival but also indicate crucial factors contributing to aggressiveness. FZD7 is indeed critical for anchorage‐independent growth and tumorigenesis. We demonstrated, for the first time, the regulation of TWIST1 by FZD7 and the existence of the FZD7‐TWIST1 axis. This proposed FZD7‐TWIST1 axis is more likely to be monodirectional rather than a reciprocal feedback regulation as manipulation of TWIST1 expression did not influence FZD7 expression in our system (data not shown). It has been reported that TWIST1 expression is regulated through multiple mechanisms by DNA methylation, active histone mark H3K4me3, and inactive histone mark H3K9me3 in exon 1 (Sakamoto et al., 2015). In OC, we found that FZD7 regulated TWIST1 expression mainly through affecting the active H3K4me3 mark at the TWIST1 promoter region. This is partially in line with the study in gastric cancer cells, although no increase of H3K9me3 was observed after FZD7 knockdown. In addition, considering that FZD7 mediates mainly β‐catenin‐independent noncanonical Wnt signaling in EOC, the correlation among Wnt signaling, EMT, and anoikis resistance in our model further suggests the importance of targeting EMT‐related factors. ZEB1, a classic EMT transcription driver downstream to the β‐catenin‐dependent canonical Wnt signaling, has been shown to be essential for the tumorigenic potential of mantle cell lymphoma with anti‐apoptotic genes MCL1 and BCL2 acting as its downstream effectors (Sánchez‐Tilló et al., 2014). The role of TWIST1 in our system is similar to what has been described in ZEB1. This suggests the importance of EMT transcription drivers in Wnt signaling mediated cell survival in addition to their classic roles in repressing E‐cadherin expression (Thiery et al., 2009).
The Wnt pathway is known to regulate apoptosis through the canonical pathway (Chen et al., 2001). Among the cell death‐related genes screened in this study, we observed that BCL2 expression consistently correlated with TWIST1 expression. BCL2 has also been shown to correlate with activation of Wnt pathway in several studies (Fani et al., 2016; Wang et al., 2012; Yang et al., 2015; Zhang et al., 2015). Interestingly, knockdown of WNT5A, a known ligand for FZD7, decreases the expression of BCL2 (Zhang et al., 2015). There have been studies to suggest that Wnt signaling is pro‐apoptotic and activation of the canonical Wnt signaling by conditional expression of an active form of β‐catenin would downregulate BCL2 and elevate caspase 3/9 activity leading to a loss of mitochondrial membrane potential resulting in increase in apoptosis in hematopoietic stem/progenitor cells (Ming et al., 2012). Thus, the correlation of Wnt signaling and apoptosis might be context dependent and this should be taken into consideration when we evaluate the role of targeting Wnt signaling.
The treatment with PORCN inhibitor, C59, revealed that CH1 was more sensitive than OV17R in functional blockade of anoikis resistance. However, in both CH1 and OV17R cells, C59 treatment significantly decreased the expression of TWIST1 and BCL2 (Fig. 8G). This suggests that the PORCN inhibitor could still induce downstream transcriptional changes similar to knocking down of FZD7 in both CH1 and OV17R cells. However, OV17R may be intrinsically resistant to C59 or to PORCN inhibitors in general. If the first hypothesis is true, C59 resistance can be overcome by using an alternative best‐in‐class PORCN inhibitor. Further testing of other PORCN inhibitors in development is warranted for confirmation. DMSO‐OV17R‐shFZD7‐1 cells were more prone to anoikis as compared with C59‐OV17R‐shLuci. This suggests that if the later was true, this could result from either the presence of crosstalk between the Wnt pathway with other more dominant pathways, or from the Wnt ligand‐independent role of FZD7 in regulating anoikis resistance. The presence of crosstalk signals between FZD7‐TWIST1 axis and other pathways that may contribute to anoikis resistance is intriguing and requires further investigation. For example, Hedgehog signaling, which is aberrantly activated in several types of cancers, linked Wnt signaling pathways with cell survival genes BCL2 and EMT driver genes SNAI1/2, ZEB1/2 through epigenetic or genetic alterations during carcinogenesis (Katoh and Katoh, 2009). The Wnt ligand‐independent role of FZD7 has not been reported previously.
5. Conclusions
In conclusion, we demonstrate that the FZD7‐TWIST1 axis is involved in the maintenance of Mes phenotype, resistance to anoikis and tumor formation through epigenetic modifications. We also identified BCL‐2 is as a downstream effector of this axis. Furthermore, we provided in vitro evidence that PORCN inhibitors can be used to inhibit this axis to suppress aggressiveness of OC cells that are dependent on FZD7‐TWIST1 axis.
Author contributions
RYH, MT, MA, and VH involved in conception and design. MA, MT, MKW, and RYH involved in development of methodology.MA, MT, VH, KT, and JY involved in acquisition of data. MA, MT, VH, RYH, and TTZ involved in analysis and interpretation of data. MA, MT, VH, and RYH involved in writing, review, and/or revision of manuscript. RH, CS, and JPT involved in study supervision.
Supporting information
Fig. S1. Knockdown of FZD7 results an increase in cadherin based cell‐cell adhesion at cell junction.
Fig. S2. TWIST1 overexpression shows similar phenotype change as compared to FZD7 with an increase of Mes marker and loss of epithelial marker.
Fig. S3. TWIST1 regulates epithelial Mes transition.
Fig. S4. TWIST1 and Wnt5a expression in shLuc and shFZD7 cells.
Acknowledgements
This work is supported by the National Research Foundation Singapore and the Singapore Ministry of Education under its Research Centres of Excellence initiative to Cancer Science Institute of Singapore to JPT and RYH, National Medical Research Council (NMRC) Clinician Scientist‐Individual Research Grant (CS‐IRG) (NMRC/CIRG/1449/2016) to RYH, and National University Cancer Institute Singapore (NCIS) Yong Siew Yoon (YSY) Research Grant through donations from the Yong Loo Lin Trust to JPT and RYH. CS was supported by fellowships and grants from the National Health and Medical Research Council (NHMRC Australia) Project grant 1081178, the Cancer Council Victoria (Sir Edward Dunlop Fellowship in Cancer Research), and the Victorian Cancer Agency (Clinical Fellowships CRF 10‐20, CRF16014).
Ming Tan, Mohammad Asad and Valerie Heong are equally contributing authors.
References
- Abu‐Elmagd M, Garcia‐Morales C and Wheeler GN (2006) Frizzled 7 mediates canonical Wnt signaling in neural crest induction. Dev Biol 298, 285–298. [DOI] [PubMed] [Google Scholar]
- Ahmed N, Abubaker K, Findlay J and Quinn M (2010) Epithelial mesenchymal transition and cancer stem cell‐like phenotypes facilitate chemoresistance in recurrent ovarian cancer. Curr Cancer Drug Targets 10, 268–278. [DOI] [PubMed] [Google Scholar]
- Ansieau S, Bastid J, Doreau A, Morel AP, Bouchet BP, Thomas C, Fauvet F, Puisieux I, Doglioni C, Piccinin S et al (2008) Induction of EMT by twist proteins as a collateral effect of tumour‐promoting inactivation of premature senescence. Cancer Cell 14, 79–89. [DOI] [PubMed] [Google Scholar]
- Asad M, Wong MK, Tan TZ, Choolani M, Low J, Mori S, Virshup D, Thiery JP and Huang RY (2014) FZD7 drives in vitro aggressiveness in Stem‐A subtype of ovarian cancer via regulation of non‐canonical Wnt/PCP pathway. Cell Death Dis 5, e1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee A, Qian P, Wu ZS, Ren X, Steiner M, Bougen NM, Liu S, Liu DX, Zhu T and Lobie PE (2012) Artemin stimulates radio‐ and chemo‐resistance by promoting TWIST1‐BCL‐2‐dependent cancer stem cell‐like behavior in mammary carcinoma cells. J Biol Chem 287, 42502–42515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battula VL, Evans KW, Hollier BG, Shi Y, Marini FC, Ayyanan A, Wang RY, Brisken C, Guerra R, Andreeff M et al (2010) Epithelial‐mesenchymal transition‐derived cells exhibit multilineage differentiation potential similar to mesenchymal stem cells. Stem Cells 28, 1435–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhanot P, Brink M, Samos CH, Hsieh JC, Wang Y, Macke JP, Andrew D, Nathans J and Nusse R (1996) A new member of the frizzled family from Drosophila functions as a Wingless receptor. Nature 382, 225–230. [DOI] [PubMed] [Google Scholar]
- Brown CW, Brodsky AS and Freiman RN (2015) Notch3 overexpression promotes anoikis resistance in epithelial ovarian cancer via upregulation of COL4A2. Mol Cancer Res 13, 78–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai Q, Yan L and Xu Y (2015) Anoikis resistance is a critical feature of highly aggressive ovarian cancer cells. Oncogene 34, 3315–3324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network (2011) Integrated genomic analyses of ovarian carcinoma. Nature 474, 609–615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cantilena S, Pastorino F, Pezzolo A, Chayka O, Pistoia V, Ponzoni M, Sala A (2011) Frizzled receptor 6 marks rare, highly tumourigenic stem‐like cells in mouse and human neuroblastomas. Oncotarget 2, 976–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarti R, Wei Y, Hwang J, Hang X, Andres Blanco M, Choudhury A, Tiede B, Romano RA, DeCoste C, Mercatali L et al (2014) DeltaNp63 promotes stem cell activity in mammary gland development and basal‐like breast cancer by enhancing Fzd7 expression and Wnt signalling. Nat Cell Biol 16, 1004–1015, 1001–1013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Guttridge DC, You Z, Zhang Z, Fribley A, Mayo MW, Kitajewski J and Wang CY (2001) Wnt‐1 signaling inhibits apoptosis by activating beta‐catenin/T cell factor‐mediated transcription. J Cell Biol 152, 87–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng GZ, Chan J, Wang Q, Zhang W, Sun CD and Wang LH (2007) Twist transcriptionally up‐regulates AKT2 in breast cancer cells leading to increased migration, invasion, and resistance to paclitaxel. Cancer Res 67, 1979–1987. [DOI] [PubMed] [Google Scholar]
- Cheng GZ, Zhang WZ, Sun M, Wang Q, Coppola D, Mansour M, Xu LM, Costanzo C, Cheng JQ and Wang LH (2008) Twist is transcriptionally induced by activation of STAT3 and mediates STAT3 oncogenic function. J Biol Chem 283, 14665–14673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiarugi P and Giannoni E (2008) Anoikis: a necessary death program for anchorage‐dependent cells. Biochem Pharmacol 76, 1352–1364. [DOI] [PubMed] [Google Scholar]
- Chua KN, Poon KL, Lim J, Sim WJ, Huang RY and Thiery JP (2011) Target cell movement in tumour and cardiovascular diseases based on the epithelial‐mesenchymal transition concept. Adv Drug Deliv Rev 63, 558–567. [DOI] [PubMed] [Google Scholar]
- Chung VY, Tan TZ, Tan M, Wong MK, Kuay KT, Yang Z, Ye J, Muller J, Koh CM, Guccione E et al (2016) GRHL2‐miR‐200‐ZEB1 maintains the epithelial status of ovarian cancer through transcriptional regulation and histone modification. Sci Rep 6, 19943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Condello S, Sima L, Ivan C, Cardenas H, Schiltz G, Mishra RK and Matei D (2018) Tissue transglutaminase regulates interactions between ovarian cancer stem cells and the tumor niche. Cancer Res 78, 2990–3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordenonsi M, Zanconato F, Azzolin L, Forcato M, Rosato A, Frasson C, Inui M, Montagner M, Parenti AR, Poletti A et al (2011) The Hippo transducer TAZ confers cancer stem cell‐related traits on breast cancer cells. Cell 147, 759–772. [DOI] [PubMed] [Google Scholar]
- Davidson B, Trope CG and Reich R (2012) Epithelial‐mesenchymal transition in ovarian carcinoma. Front Oncol 2, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derksen PW, Liu X, Saridin F, van der Gulden H, Zevenhoven J, Evers B, van Beijnum JR, Griffioen AW, Vink J, Krimpenfort P et al (2006) Somatic inactivation of E‐cadherin and p53 in mice leads to metastatic lobular mammary carcinoma through induction of anoikis resistance and angiogenesis. Cancer Cell 10, 437–449. [DOI] [PubMed] [Google Scholar]
- Dissanayake SK, Wade M, Johnson CE, O'Connell MP, Leotlela PD, French AD, Shah KV, Hewitt KJ, Rosenthal DT, Indig FE et al (2007) The Wnt5A/protein kinase C pathway mediates motility in melanoma cells via the inhibition of metastasis suppressors and initiation of an epithelial to mesenchymal transition. J Biol Chem 282, 17259–17271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fani S, Dehghan F, Karimian H, Mun Lo K, Ebrahimi Nigjeh S, Swee Keong Y, Soori R, May Chow K, Kamalidehghan B, Mohd Ali H et al (2016) Monobenzyltin complex C1 induces apoptosis in MCF‐7 breast cancer cells through the intrinsic signaling pathway and through the targeting of MCF‐7‐derived breast cancer stem cells via the WNT/beta‐catenin signaling pathway. PLoS ONE 11, e0160836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feng MY, Wang K, Song HT, Yu HW, Qin Y, Shi QT and Geng JS (2009) Metastasis‐induction and apoptosis‐protection by TWIST in gastric cancer cells. Clin Exp Metastasis 26, 1013–1023. [DOI] [PubMed] [Google Scholar]
- Fernandez A, Huggins IJ, Perna L, Brafman D, Lu D, Yao S, Gaasterland T, Carson DA and Willert K (2014) The WNT receptor FZD7 is required for maintenance of the pluripotent state in human embryonic stem cells. Proc Natl Acad Sci USA 111, 1409–1414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frisch SM, Schaller M and Cieply B (2013) Mechanisms that link the oncogenic epithelial‐mesenchymal transition to suppression of anoikis. J Cell Sci 126, 21–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilmore AP (2005) Anoikis. Cell Death Differ 12, 1473–1477. [DOI] [PubMed] [Google Scholar]
- Goodnough LH, Dinuoscio GJ and Atit RP (2016) Twist1 contributes to cranial bone initiation and dermal condensation by maintaining Wnt signaling responsiveness. Dev Dyn 245, 144–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurney A, Axelrod F, Bond CJ, Cain J, Chartier C, Donigan L, Fischer M, Chaudhari A, Ji M, Kapoun AM et al (2012) Wnt pathway inhibition via the targeting of Frizzled receptors results in decreased growth and tumorigenicity of human tumours. Proc Natl Acad Sci USA 109, 11717–11722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hosono S, Kajiyama H, Terauchi M, Shibata K, Ino K, Nawa A and Kikkawa F (2007) Expression of Twist increases the risk for recurrence and for poor survival in epithelial ovarian carcinoma patients. Br J Cancer 96, 314–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Howe LR, Watanabe O, Leonard J and Brown AM (2003) Twist is up‐regulated in response to Wnt1 and inhibits mouse mammary cell differentiation. Cancer Res 63, 1906–1913. [PubMed] [Google Scholar]
- Katoh M (2005) WNT/PCP signaling pathway and human cancer (review). Oncol Rep 14, 1583–1588. [PubMed] [Google Scholar]
- Katoh Y and Katoh M (2009) Hedgehog target genes: mechanisms of carcinogenesis induced by aberrant hedgehog signaling activation. Curr Mol Med 9, 873–886. [DOI] [PubMed] [Google Scholar]
- Keitel U, Scheel A, Thomale J, Halpape R, Kaulfuß S, Scheel C and Dobbelstein M (2014) Bcl‐xL mediates therapeutic resistance of a mesenchymal breast cancer cell subpopulation. Oncotarget 5, 11778–11791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirikoshi H, Sekihara H and Katoh M (2001) Up‐regulation of Frizzled‐7 (FZD7) in human gastric cancer. Int J Oncol 19, 111–115. [PubMed] [Google Scholar]
- Kwok WK, Ling MT, Lee TW, Lau TC, Zhou C, Zhang X, Chua CW, Chan KW, Chan FL, Glackin C et al (2005) Up‐regulation of TWIST in prostate cancer and its implication as a therapeutic target. Cancer Res 65, 5153–5162. [DOI] [PubMed] [Google Scholar]
- Li QQ, Xu JD, Wang WJ, Cao XX, Chen Q, Tang F, Chen ZQ, Liu XP and Xu ZD (2009) Twist1‐mediated adriamycin‐induced epithelial‐mesenchymal transition relates to multidrug resistance and invasive potential in breast cancer cells. Clin Cancer Res 15, 2657–2665. [DOI] [PubMed] [Google Scholar]
- Lo HW, Hsu SC, Xia W, Cao X, Shih JY, Wei Y, Abbruzzese JL, Hortobagyi GN and Hung MC (2007) Epidermal growth factor receptor cooperates with signal transducer and activator of transcription 3 to induce epithelial‐mesenchymal transition in cancer cells via up‐regulation of TWIST gene expression. Cancer Res 67, 9066–9076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo GQ, Li JH, Wen JF, Zhou YH, Hu YB and Zhou JH (2008) Effect and mechanism of the Twist gene on invasion and metastasis of gastric carcinoma cells. World J Gastroenterol 14, 2487–2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madan B and Virshup DM (2015) Targeting Wnts at the source–new mechanisms, new biomarkers, new drugs. Mol Cancer Ther 14, 1087–1094. [DOI] [PubMed] [Google Scholar]
- Maestro R, Dei Tos AP, Hamamori Y, Krasnokutsky S, Sartorelli V, Kedes L, Doglioni C, Beach DH and Hannon GJ (1999) Twist is a potential oncogene that inhibits apoptosis. Genes Dev 13, 2207–2217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani SA, Guo W, Liao MJ, Eaton EN, Ayyanan A, Zhou AY, Brooks M, Reinhard F, Zhang CC, Shipitsin M et al (2008) The epithelial‐mesenchymal transition generates cells with properties of stem cells. Cell 133, 704–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mei H, Nakatsu MN, Baclagon ER and Deng SX (2014) Frizzled 7 maintains the undifferentiated state of human limbal stem/progenitor cells. Stem Cells 32, 938–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melchior K, Weiss J, Zaehres H, Kim YM, Lutzko C, Roosta N, Hescheler J and Müschen M (2008) The WNT receptor FZD7 contributes to self‐renewal signaling of human embryonic stem cells. Biol Chem 389, 897–903. [DOI] [PubMed] [Google Scholar]
- Merle P, Kim M, Herrmann M, Gupte A, Lefrançois L, Califano S, Trépo C, Tanaka S, Vitvitski L, de la Monte S et al (2005) Oncogenic role of the frizzled‐7/beta‐catenin pathway in hepatocellular carcinoma. J Hepatol 43, 854–862. [DOI] [PubMed] [Google Scholar]
- Ming M, Wang S, Wu W, Senyuk V, Le Beau MM, Nucifora G and Qian Z (2012) Activation of Wnt/beta‐catenin protein signaling induces mitochondria‐mediated apoptosis in hematopoietic progenitor cells. J Biol Chem 287, 22683–22690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nieto MA, Huang RY, Jackson RA and Thiery JP (2016) Emt: 2016. Cell 166, 21–45. [DOI] [PubMed] [Google Scholar]
- Nobes CD and Hall A (1999) Rho GTPases control polarity, protrusion, and adhesion during cell movement. J Cell Biol 144, 1235–1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Proffitt KD, Madan B, Ke Z, Pendharkar V, Ding L, Lee MA, Hannoush RN and Virshup DM (2013) Pharmacological inhibition of the Wnt acyltransferase PORCN prevents growth of WNT‐driven mammary cancer. Cancer Res 73, 502–507. [DOI] [PubMed] [Google Scholar]
- Qin Q, Xu Y, He T, Qin C and Xu J (2012) Normal and disease‐related biological functions of Twist1 and underlying molecular mechanisms. Cell Res 22, 90–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raftopoulou M and Hall A (2004) Cell migration: Rho GTPases lead the way. Dev Biol 265, 23–32. [DOI] [PubMed] [Google Scholar]
- Reinhold MI, Kapadia RM, Liao Z and Naski MC (2006) The Wnt‐inducible transcription factor Twist1 inhibits chondrogenesis. J Biol Chem 281, 1381–1388. [DOI] [PubMed] [Google Scholar]
- Reya T and Clevers H (2005) Wnt signalling in stem cells and cancer. Nature 434, 843–850. [DOI] [PubMed] [Google Scholar]
- Reya T, Duncan AW, Ailles L, Domen J, Scherer DC, Willert K, Hintz L, Nusse R and Weissman IL (2003) A role for Wnt signalling in self‐renewal of haematopoietic stem cells. Nature 423, 409–414. [DOI] [PubMed] [Google Scholar]
- Ridley AJ (2004) Rho proteins and cancer. Breast Cancer Res Treat 84, 13–19. [DOI] [PubMed] [Google Scholar]
- Sakamoto A, Akiyama Y, Shimada S, Zhu WG, Yuasa Y and Tanaka S (2015) DNA Methylation in the exon 1 region and complex regulation of twist1 expression in gastric cancer cells. PLoS ONE 10, e0145630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez‐Tilló E, Fanlo L, Siles L, Montes‐Moreno S, Moros A, Chiva‐Blanch G, Estruch R, Martinez A, Colomer D, Győrffy B et al (2014) The EMT activator ZEB1 promotes tumour growth and determines differential response to chemotherapy in mantle cell lymphoma. Cell Death Differ 21, 247–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song L, Webb NE, Song Y and Tuan RS (2006) Identification and functional analysis of candidate genes regulating mesenchymal stem cell self‐renewal and multipotency. Stem Cells 24, 1707–1718. [DOI] [PubMed] [Google Scholar]
- Sosic D, Richardson JA, Yu K, Ornitz DM and Olson EN (2003) Twist regulates cytokine gene expression through a negative feedback loop that represses NF‐kappaB activity. Cell 112, 169–180. [DOI] [PubMed] [Google Scholar]
- Tan TZ, Miow QH, Huang RY, Wong MK, Ye J, Lau JA, Wu MC, Bin Abdul Hadi LH, Soong R, Choolani M et al (2013) Functional genomics identifies five distinct molecular subtypes with clinical relevance and pathways for growth control in epithelial ovarian cancer. EMBO Mol Med 5, 983–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan TZ, Yang H, Ye J, Low J, Choolani M, Tan DS, Thiery JP and Huang RY (2015) CSIOVDB: a microarray gene expression database of epithelial ovarian cancer subtype. Oncotarget 6, 43843–43852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiery JP, Acloque H, Huang RY and Nieto MA (2009) Epithelial‐mesenchymal transitions in development and disease. Cell 139, 871–890. [DOI] [PubMed] [Google Scholar]
- Thisse B, Stoetzel C, Gorostizathisse C and Perrinschmitt F (1988) Sequence of the twist gene and nuclear‐localization of its protein in endomesodermal cells of early drosophila embryos. EMBO J 7, 2175–2183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tothill RW, Tinker AV, George J, Brown R, Fox SB, Lade S, Johnson DS, Trivett MK, Etemadmoghadam D, Locandro B et al, Australian Ovarian Cancer Study Group , Gertig D, DeFazio A, Bowtell DD (2008) Novel molecular subtypes of serous and endometrioid ovarian cancer linked to clinical outcome. Clin Cancer Res 14, 5198–5208. [DOI] [PubMed] [Google Scholar]
- Ueno K, Hazama S, Mitomori S, Nishioka M, Suehiro Y, Hirata H, Oka M, Imai K, Dahiya R and Hinoda Y (2009) Down‐regulation of frizzled‐7 expression decreases survival, invasion and metastatic capabilities of colon cancer cells. Br J Cancer 101, 1374–1381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valsesia‐Wittmann S, Magdeleine M, Dupasquier S, Garin E, Jallas AC, Combaret V, Krause A, Leissner P and Puisieux A (2004) Oncogenic cooperation between H‐Twist and N‐Myc overrides failsafe programs in cancer cells. Cancer Cell 6, 625–630. [DOI] [PubMed] [Google Scholar]
- Veeman MT, Axelrod JD and Moon RT (2003) A second canon. Functions and mechanisms of beta‐catenin‐independent Wnt signaling. Dev Cell 5, 367–377. [DOI] [PubMed] [Google Scholar]
- Vesuna F, Lisok A, Kimble B and Raman V (2009) Twist modulates breast cancer stem cells by transcriptional regulation of CD24 expression. Neoplasia 11, 1318–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H, Fan L, Xia X, Rao Y, Ma Q, Yang J, Lu Y, Wang C, Ma D and Huang X (2012) Silencing Wnt2B by siRNA interference inhibits metastasis and enhances chemotherapy sensitivity in ovarian cancer. Int J Gynecol Cancer 22, 755–761. [DOI] [PubMed] [Google Scholar]
- Wang Q, Symes AJ, Kane CA, Freeman A, Nariculam J, Munson P, Thrasivoulou C, Masters JR and Ahmed A (2010) A novel role for Wnt/Ca2+ signaling in actin cytoskeleton remodeling and cell motility in prostate cancer. PLoS ONE 5, e10456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Mani SA, Donaher JL, Ramaswamy S, Itzykson RA, Come C, Savagner P, Gitelman I, Richardson A and Weinberg RA (2004) Twist, a master regulator of morphogenesis, plays an essential role in tumour metastasis. Cell 117, 927–939. [DOI] [PubMed] [Google Scholar]
- Yang J, Wei D, Wang W, Shen B, Xu S and Cao Y (2015) TRAF4 enhances oral squamous cell carcinoma cell growth, invasion and migration by Wnt‐beta‐catenin signaling pathway. Int J Clin Exp Pathol 8, 11837–11846. [PMC free article] [PubMed] [Google Scholar]
- Yang MH, Wu MZ, Chiou SH, Chen PM, Chang SY, Liu CJ, Teng SC and Wu KJ (2008) Direct regulation of TWIST by HIF‐1alpha promotes metastasis. Nat Cell Biol 10, 295–305. [DOI] [PubMed] [Google Scholar]
- Yang L, Wu X, Wang Y, Zhang K, Wu J, Yuan YC, Deng X, Chen L, Kim CC, Lau S et al (2011) FZD7 has a critical role in cell proliferation in triple negative breast cancer. Oncogene 30, 4437–4446. [DOI] [PubMed] [Google Scholar]
- Yin G, Chen R, Alvero AB, Fu HH, Holmberg J, Glackin C, Rutherford T and Mor G (2010) TWISTing stemness, inflammation and proliferation of epithelial ovarian cancer cells through MIR199A2/214. Oncogene 29, 3545–3553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu Q, Liu L, Duan Y, Wang Y, Xuan X, Zhou L and Liu W (2013) Wnt/beta‐catenin signaling regulates neuronal differentiation of mesenchymal stem cells. Biochem Biophys Res Commun 439, 297–302. [DOI] [PubMed] [Google Scholar]
- Yuen HF, Chua CW, Chan YP, Wong YC, Wang X and Chan KW (2007) Significance of TWIST and E‐cadherin expression in the metastatic progression of prostatic cancer. Histopathology 50, 648–658. [DOI] [PubMed] [Google Scholar]
- Zhang Z, Rankin SA and Zorn AM (2013) Different thresholds of Wnt‐Frizzled 7 signaling coordinate proliferation, morphogenesis and fate of endoderm progenitor cells. Dev Biol 378, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Tu C, Zhang D, Zheng Y, Peng Z, Feng Y, Xiao S and Li Z (2015) Wnt/beta‐catenin and Wnt5a/Ca pathways regulate proliferation and apoptosis of keratinocytes in psoriasis lesions. Cell Physiol Biochem 36, 1890–1902. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Knockdown of FZD7 results an increase in cadherin based cell‐cell adhesion at cell junction.
Fig. S2. TWIST1 overexpression shows similar phenotype change as compared to FZD7 with an increase of Mes marker and loss of epithelial marker.
Fig. S3. TWIST1 regulates epithelial Mes transition.
Fig. S4. TWIST1 and Wnt5a expression in shLuc and shFZD7 cells.