

Abstract
Colorectal cancer (CRC) is the third leading cause of cancer‐related deaths worldwide. Therapies that take advantage of defects in DNA repair pathways have been explored in the context of breast, ovarian, and other tumor types, but not yet systematically in CRC. At present, only immune checkpoint blockade therapies have been FDA approved for use in mismatch repair‐deficient colorectal tumors. Here, we discuss how systematic identification of alterations in DNA repair genes could provide new therapeutic opportunities for CRCs. Analysis of The Cancer Genome Atlas Colon Adenocarcinoma (TCGA‐COAD) and Rectal Adenocarcinoma (TCGA‐READ) PanCancer Atlas datasets identified 141 (out of 528) cases with putative driver mutations in 29 genes associated with DNA damage response and repair, including the mismatch repair and homologous recombination pathways. Genetic defects in these pathways might confer repair‐deficient characteristics, such as genomic instability in the absence of homologous recombination, which can be exploited. For example, inhibitors of poly(ADP)‐ribose polymerase are effectively used to treat cancers that carry mutations in BRCA1 and/or BRCA2 and have shown promising results in CRC preclinical studies. HR deficiency can also occur in cells with no detectable BRCA1/BRCA2 mutations but exhibiting BRCA‐like phenotypes. DNA repair‐targeting therapies, such as ATR and CHK1 inhibitors (which are most effective against cancers carrying ATM mutations), can be used in combination with current genotoxic chemotherapies in CRCs to further improve therapy response. Finally, therapies that target alternative DNA repair mechanisms, such as thiopurines, also have the potential to confer increased sensitivity to current chemotherapy regimens, thus expanding the spectrum of therapy options and potentially improving clinical outcomes for CRC patients.
Keywords: colorectal cancer, genome instability, homologous recombination, microsatellite instability, mismatch repair
Abbreviations
- ATRi
ataxia telangiectasia‐mutated and Rad3‐related inhibitors
- BER
base excision repair
- CHK1i
checkpoint kinase 1 inhibitors
- CRC
colorectal cancer
- DDR
DNA damage response
- FA
Fanconi anemia
- HR
homologous recombination
- MMR
mismatch repair
- MSI
microsatellite instability
- MSS
microsatellite stable
- PARPi
poly(ADP)ribose polymerase inhibitors
Introduction
Colorectal cancer (CRC) is the third most common cancer worldwide and the second leading cause of cancer‐related deaths (Bray et al., 2018). In Europe, CRC accounts for the second highest number of cancer cases and deaths (Malvezzi et al., 2018), and in North America, CRC has the fourth highest rate of incidence and the second highest number of cancer‐related deaths (Jemal et al., 2017). While CRC death rates are slowly declining in the United States and Europe (Jemal et al., 2017; Malvezzi et al., 2018), the five‐year overall survival for patients with metastatic CRC (mCRC) remains poor (approximately 14.0%; NCI 2017). The standard chemotherapeutic regimen for mCRC is 5‐fluorouracil (5‐FU) in combination with either oxaliplatin (FOLFOX) or irinotecan (FOLFIRI) (Cremolini et al., 2015). These chemotherapy agents induce genotoxic damage in tumor cells that is recognized and repaired by DNA repair proteins (Helleday et al., 2008).
In 2012, The Cancer Genome Atlas (TCGA) conducted a comprehensive characterization of CRC tumors, including exome sequences, DNA copy numbers, and RNA expression levels (Network, 2012). Of the cases analyzed, 16% were classified as hypermutated (greater than 12 mutations per 106 bases) and exhibited mutation enrichment in microsatellite regions indicating microsatellite instability (MSI) phenotype. The other 84% of cases were classified as microsatellite stable (MSS) and exhibited a higher frequency of somatic copy number alterations, suggesting chromosomal and subchromosomal defects (Network, 2012). The most frequently identified gene mutations in CRC tumors occur in APC, TP53, and KRAS (Huang et al., 2018; Wolff et al., 2018; Yaeger et al., 2018). Recent analysis of TCGA data identified mutations associated with DNA damage response genes and found that cases in the colon adenocarcinoma (COAD) and rectal adenocarcinoma (READ) datasets carried mutations in several DNA damage response and repair (DDR) genes (Knijnenburg et al., 2018).
Acquisition of mutations is a critical step for tumor development (Hanahan and Weinberg, 2011), and mutations that occur in DNA repair genes impair cells’ ability to restore damaged DNA and can lead to cell death or genome instability (Aguilera and Gomez‐Gonzalez, 2008). Mutations in MMR genes are observed in 2–3% of CRC patients (Lorans et al., 2018), while approximately 10% of CRC patients exhibit hypermethylation of MLH1 (AlDubayan et al., 2018; Pearlman et al., 2017), contributing to a MMR‐deficient (MMRd) phenotype. The remaining CRC patient population can be classified as MMR‐proficient (MMRp). Defects in the MMR pathway are commonly used to classify CRCs, while mutations in HR and FA genes have been historically linked with breast and ovarian cancers (Hoang and Gilks, 2018, Knijnenburg et al., 2018). The TCGA‐COAD and TCGA‐READ PanCancer Atlas cohort (Cerami et al., 2012; Gao et al., 2013; Liu et al., 2018) includes mutational data for 528 patient tumor samples, and analysis of these samples identified 141 cases that carried mutations in at least one of 420 DNA repair genes. The majority of mutations identified were classified as ‘putative passenger’. While these mutations are not currently known to drive carcinogenesis, it is possible that the presence of these mutations will cause the cells to be DNA repair deficient. Using criteria that excluded likely passenger mutations, putative driver mutations were identified in 29 DNA damage response and repair genes (Table 1).
Table 1.
Missense, truncating, and frameshift mutations (putative driver) in DNA damage response and repair genes identified in 528 colorectal cancer cases reported in The Cancer Genome Atlas Colon Adenocarcinoma (COAD) and Rectal Adenocarcinoma (READ) PanCancer Atlas datasets (Liu et al., 2018). Asterisk (*) indicates methylation data acquired from the TCGA‐COADREAD Provisional dataset
| Gene | Cases affected (%) |
|---|---|
| TRP53 | 92 (17.4) |
| ATM | 18 (3.4) |
| BRCA2 | 10 (1.9) |
| TP53BP1 | 9 (1.7) |
| MSH6 | 7 (1.3) |
| ATR | 7 (1.3) |
| MTOR | 5 (0.9) |
| SMARCB1 | 4 (0.8) |
| ATRX | 3 (0.6) |
| BARD1 | 3 (0.6) |
| BLM | 3 (0.6) |
| MSH3 | 3 (0.6) |
| BRIP1 | 2 (0.4) |
| FANCA | 2 (0.4) |
| RAD50 | 2 (0.4) |
| EPC2 | 1 (0.2) |
| ERCC4 | 1 (0.2) |
| MLH1 | 1 (0.2) |
| 37 (10.3)* | |
| MSH2 | 1 (0.2) |
| PMS2 | 1 (0.2) |
| RAD21 | 1 (0.2) |
| RAD21L1 | 1 (0.2) |
| RAD51C | 1 (0.2) |
| SMC1A | 1 (0.2) |
| XRCC2 | 1 (0.2) |
| XRCC3 | 1 (0.2) |
Epigenetic modulation of gene expression can also lead to a repair‐defective phenotype. For example, hypermethylation of the MLH1 promoter has been associated with the MSI phenotype in sporadic endometrial and hereditary nonpolyposis colorectal cancers (Esteller et al., 1998; Niv, 2007; Planck et al., 2003). Epigenetic down‐regulation of MMR genes has also been linked with resistance to alkylating chemotherapy agents in CRC tumor models (Planck et al., 2003), and studies have demonstrated that preventing down‐regulation of MMR genes can restore cellular sensitivity to these agents (Francia et al., 2005). Methylation data were not available for the PanCancer Atlas dataset; however, analysis of the TCGA‐COADREAD Provisional dataset (Cerami et al., 2012; Gao et al., 2013) determined that 10.3% of cases (37 out of 358) exhibited hypermethylation of MLH1. The presence of mutations or hypermethylation of promoter regions in one or more DNA repair genes in a CRC cell may contribute to a DNA repair‐defective phenotype that can be used to classify tumor subtypes and to choose an appropriate therapy regimen.
DNA repair‐defective phenotypes in colorectal cancers
Mismatch repair
The MMR pathway recognizes and removes DNA base pair mismatches that occur due to replication errors (Iyer et al., 2006; Modrich, 2006) (Fig. 1; left panel). First, the mismatch is recognized by MutSα (MSH2/MSH6) and MutLβ (MLH1/PMS1) or MutSβ (MSH2/MSH3) and MutLα (MLH1/PMS2) heterodimer complexes that bind the DNA surrounding the mismatch. Downstream, the exonuclease EXO1 interacts with proliferating cell nuclear antigen (PCNA), initiating DNA resection in a 5′ to 3′ manner. Finally, polymerase δ replicates across the excised region and the DNA is ligated by DNA ligase I. Inactivating mutations in any of these genes decreases recognition of base pair mismatches, leading to increased mutational burden, particularly in microsatellite regions of the genome (Cortes‐Ciriano et al., 2017; Hause et al., 2016; Popat et al., 2005). One of the phenotypes exhibited in MMRd cells is MSI (Zeinalian et al., 2018), and several studies have shown that MSI corresponds with favorable prognosis and better survival (Popat et al., 2005; Thibodeau et al., 1993).
Figure 1.
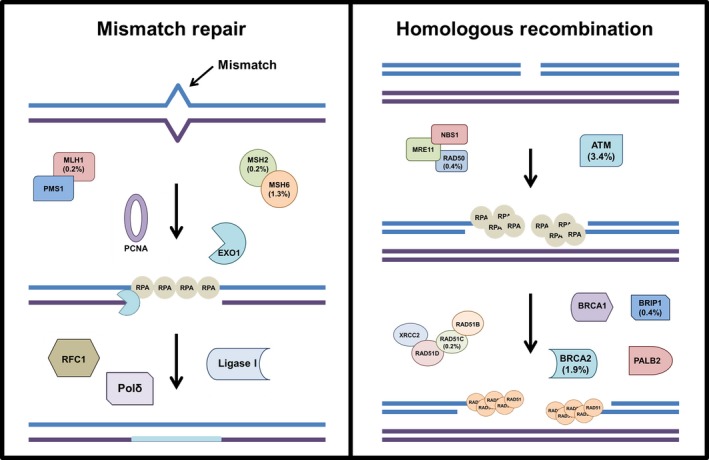
DNA repair pathways with mutated genes highlighted. Left panel: DNA mismatch repair pathway recognizes and removes incorrect DNA base pairs generated during replication. Right panel: homologous recombination proteins recognize and repair DNA double‐strand breaks. Key proteins in each pathway are shown and the percent of cases that carried mutations in these genes are listed (see Table 1 for details).
Microsatellite instability
A phenotype of cells that carry defects in mismatch repair is microsatellite instability (MSI), defined as high mutational burden in sequences along the genome that contain repetitive, short‐tandem sequences containing 1–6 nucleotide units up to 100 times, known as microsatellites (Zeinalian et al., 2018). The National Institutes of Health has defined five biomarkers that contain mono‐ or dinucleotide repeats in specific regions of the genome to be used for clinical determination of MSI status (Boland et al., 1998). PCR amplification of biomarker regions is performed, and the product size in tumor cells is compared with matched normal cells to determine whether mutations are present (Zeinalian et al., 2018). New advents in sequencing technology have allowed for the detection of MSI using whole‐exome sequencing (WES) data from tumor samples and normal tissue isolated from the same patient (Hause et al., 2016). A comprehensive analysis of microsatellite stability in 5930 exome samples demonstrated that these strategies are capable of distinguishing MSI cancer from MSS cancers, and these analyses identified genomic ‘hot spots’ that exhibit higher frequencies of MSI across cancer subtypes (Hause et al., 2016). Immunohistochemistry using antibodies against MLH1, MSH2, MSH6, and PMS2 is another effective technique that is used to assess MMR status (Yuan et al., 2015; Zeinalian et al., 2018).
Generation of neoantigens is another phenotype of MMRd cells that has been well characterized in CRC (Germano et al., 2017; Le et al., 2017; Scarpa et al., 2015). The defects in MMR machinery lead to frameshifts and indels that produce novel peptides within a cell. Once translated, these peptides can be exposed to the outside of the cell by the HLA proteins and activate immune surveillance of the tumor cells, thus making MMRd cells more susceptible to immune surveillance (Nakayama, 2014).
Homologous recombination
The HR pathway is essential for preserving genome integrity in the event of DNA double‐strand breaks (DSBs) (Ranjha et al., 2018) (Fig. 1; right panel). If left unrepaired, this type of damage can lead to deletions, frameshifts, chromosome aberrations, and aneuploidy (Jackson and Bartek, 2009). During S phase, a DSB is first recognized by poly(ADP‐ribose) polymerase 1 (PARP1), a protein that scans the genome and detects DSB lesions (Ciccia and Elledge, 2010). PARP1 marks the damage site by attaching ADP‐ribose molecules to chromatin‐bound proteins surrounding the break (Haince et al., 2008). The ADP‐ribose units are essential for recruitment of meiotic recombination 11 (MRE11), RAD50, and Nijmegen breakage syndrome (NBS1) proteins, which form the MRN complex. The exonuclease activity of this complex produces single‐strand DNA (ssDNA) surrounding the break (Dodson et al., 2010; Haince et al., 2008; Huen et al., 2010; You and Bailis, 2010), and localization of MRN triggers ATM‐mediated signaling of downstream repair factors. Following MRN‐mediated resection, the single‐strand binding protein replication protein A (RPA) stabilizes the newly produced ssDNA overhangs (Marechal and Zou, 2015). Recruitment of BRCA1 and BRCA2, along with BRIP1, PALB2, and the RAD51B‐RAD51C‐RAD51D‐XRCC2 (BCDX2) complex, promotes RAD51 binding to the ssDNA overhangs (Candelli et al., 2014; Jensen et al., 2013; Short et al., 2016; Xu et al., 2017). Finally, RAD51 mediates strand invasion of the ssDNA overhang into a homologous DNA region, usually a sister chromatid, enabling repair to be completed (Qi et al., 2015).
Genome instability
In the context of distinct cancers types, such as breast and ovarian, ‘genome instability’ is typically attributed to defects in HR DNA repair genes (Burrell et al., 2013; Chien et al., 2015; Janssen et al., 2011; Vanderstichele et al., 2017). Hanahan and Weinberg classified ‘genome instability’ as an enabling characteristic of cancer and described how defects in DNA repair lead to loss of chromosomes, particularly at the telomere region (Hanahan and Weinberg, 2011). Genome instability is identified by structural alterations that include copy number variations and loss of heterozygosity, often observed in CRC cells (Druliner et al., 2018), and chromosomal rearrangements (Aguilera and Gomez‐Gonzalez, 2008). These elements are characteristic of HR defective cells, and specific genomic signatures have been identified in breast and ovarian cancer cells (Davies et al., 2017; Hillman et al., 2018; Vanderstichele et al., 2017). The detection of genomic rearrangements by whole‐genome sequencing in BRCA1/BRCA2‐deficient samples leads to identification of six distinct mutational signatures that correlated with BRCA status (Davies et al., 2017). Notably, the so‐called BRCA‐ness signature was also identified in cells that did not have detectable BRCA1/BRCA2 mutations, connecting genomic rearrangements with functional HR deficiency, and suggesting that additional molecular alterations might underline BRCA‐like phenotypes (Davies et al., 2017). BRCA‐ness mutational signatures in CRC tumors might be used as predictive biomarkers for HR deficiency regardless of BRCA status.
Telomere defects
In addition to genomic rearrangements, telomere length is a measurement of genome instability (Hackett et al., 2001). HR repair proteins function to protect telomere regions from damage (Claussin and Chang, 2015; Tarsounas et al., 2004), and telomere defects are often observed in genome unstable cells (Venkatesan et al., 2015). A recent study investigating telomere length in CRC determined that KRAS‐mutated cells exhibited extensive telomere shortening compared with control cells. In contrast, cells that carried BRAF mutations or were classified as MSI did not exhibit telomere defects (Balc'h et al., 2017). Another independent study analyzed telomere length in precursor colorectal lesions and observed that telomere shortening was associated with mitochondrial microsatellite instability in the tumor tissue samples and with KRAS and BRAF mutations in the normal tissues (Park et al., 2017). Studies have also demonstrated that KRAS‐mutated CRC cells can become dependent on RAD51‐mediated repair (Kalimutho et al., 2017), a key protein in the HR pathway that is essential for maintaining telomere integrity (Badie et al., 2010; Le et al., 1999; Lu et al., 2014; Signon et al., 2001). Together, these data suggest that telomere shortening is indicative of DNA repair defects and may be a biomarker of early CRC carcinogenesis.
Targeting DNA repair in colorectal cancer
Current medical regimens for CRC patients include combination therapies with oxaliplatin, irinotecan, and 5‐FU (Cremolini et al., 2015). These ‘genotoxic’ drugs directly or indirectly induce DNA damage that is recognized by specific repair pathways (Fig. 2). Oxaliplatin is a platinum‐based compound that can induce cell death through several mechanisms, such as inducing ribosome biogenesis stress (Bruno et al., 2017). The genotoxic activity of this drug is attributed to its ability to bind the N7 of guanine nucleotides in DNA, generating interstrand cross‐links that can inhibit replication during S phase (Ray et al., 2018). Irinotecan, a camptothecin analog, binds to topoisomerase I and DNA, preventing dissociation of topoisomerase I during S phase and ultimately leading to DNA DSBs (Li et al., 2017). These two types of DNA damage are recognized and repaired by the FA/HR pathways (Ceccaldi et al., 2016). 5‐FU is an antimetabolite that inhibits thymidylate synthase, an enzyme involved in nucleotide synthesis, and is thought to inhibit DNA replication thus leading to abasic sites that are repaired by base excision repair (BER) proteins (Huehls et al., 2016). 5‐FU can also be incorporated into DNA, resulting in DNA mismatches that are recognized and repaired by the MMR pathway (Iwaizumi et al., 2011). An alternative nucleotide analog, TAS‐102, has been approved by the FDA as a treatment option for mCRC patients (Marcus et al., 2017). TAS‐102 inhibits nucleoside synthesis in a similar mechanism of action to the standard therapy 5‐FU and is effective in treating patients that are refractory to 5‐FU therapy (Lenz et al., 2015). Recent studies have linked mutations in DNA repair genes, specifically those associated with HR, as predictive markers of the efficacy of TAS‐102 in patients (Suenaga et al., 2017).
Figure 2.
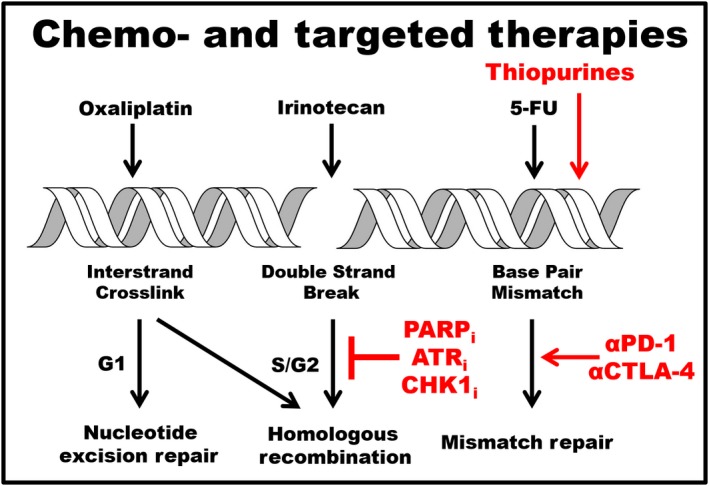
Therapies targeting cancer specific DNA repair defects. Current chemotherapy agents (black) used to treat mCRC. Oxaliplatin induces DNA interstrand cross‐links that are repaired by nucleotide excision repair proteins during G1 and by Fanconi anemia and HR proteins during S phase. Irinotecan (SN38) is a topoisomerase inhibitor that induces single (SSB)‐ and double‐strand breaks (DSBs) that are repaired by HR and BER proteins. 5‐Fluorouracil is an antimetabolite that can lead to DNA base pair mismatches repaired by the MMR pathway. Alternative therapies (red) that can be used in combination with current chemotherapy agents. PARPi and ATRi induce stalled replication forks and DSBs that are lethal in cells carrying mutations in HR genes, and CHKi block cell cycle arrest in the presence of replication stress. Chemosensitivity can be further induced in cells treated with genotoxic agents in combination with targeted therapies. Thiopurines induce DNA base pair mismatches that can lead to increased mutational and neoantigen burdens. Anti‐PD‐1 and CTLA‐4 immunotherapies target MMRd CRC tumors and are most effective against tumors with high neoantigen burdens.
These compounds are most effective in cells with defects in the respective repair pathways, and CRC patients that carry mutations in repair‐associated genes can be predicted to respond well to these types of therapies.
Immune checkpoint blockade
Colorectal cancer tumors that have increased neoantigen production due to MMR deficiency also have higher levels of tumor infiltrating lymphocytes (TILs) and increased programmed death ligand‐1 (PDL‐1) protein expression (Germano et al., 2018; Kim et al., 2016; Llosa et al., 2015). Recent studies have shown that MSI tumor cells are responsive to PD‐1 and CTLA‐4 immune blockade (Germano et al., 2017; Le et al., 2017; Luksza et al., 2017; McGranahan et al., 2016). In one study, patients with higher TILs and PD‐L1 expression responded to checkpoint blockade better compared with patients with lower PD‐L1 expression, suggesting that this phenotype can be used as a predictor of therapy response (Wang et al., 2018). A 2017 Phase II clinical trial of nivolumab, a PD‐L1 immune checkpoint inhibitor, in patients with MMRd and MSI‐H CRC who had received at least three rounds of prior therapy and were no longer responsive to first‐line treatments found that this therapy provided durable response and disease control in these patients. Of the 74 patients enrolled, 31% achieved objective response and 51% achieved disease control (Overman et al., 2018). Based on these data, nivolumab was given FDA approval for the treatment of mCRC with MSI‐H or MMRd tumors (Sarshekeh et al., 2018). In addition to CRC, studies have demonstrated that solid tumors of multiple tissue types exhibiting MSI show durable response to PD‐L1 blockade, and based on this evidence, the FDA approved these agents for use in any cancers that histologically exhibit MSI (Lemery et al., 2017).
The MMRd tumors have increased mutational burden (TMB), a phenotype that can also be observed in a small subset of tumors that are MSS. One study analyzed over 6000 CRC cases, 95% of which were classified as MSS, and observed that 2.9% of MSS tumors exhibited high TMB and, within this subset of tumors, 54% responded to anti‐PD‐L1 immunotherapy (Fabrizio et al., 2018). The results of this study suggest that in MMRd tumors that do not exhibit MSI, TMB can be used as a predictor for therapy response to tumor checkpoint inhibition.
PARP inhibitors
In the presence of directly induced DNA breaks, PARP1 poly(ADP‐ribosyl)ates chromatin surrounding the damage to initiate activity of downstream HR proteins (Haince et al., 2008). Additionally, PARP functions to regulate replication fork progression and maintain fork stability. Chemical inhibition of PARP activity can interfere with either of these activities, leading to replication fork collapse or accelerated fork progression that generates DNA single‐ and double‐strand breaks (D'Andrea, 2018; Maya‐Mendoza et al., 2018). Poly‐(ADP) ribose polymerase inhibitors (PARPi) have been used as anticancer agents since the early 2000s (McCabe et al., 2006), and the first PARPi, olaparib, was approved for BRCA‐mutated ovarian cancer in 2014 (Kim et al., 2015). Approval of these drugs was first given for the treatment of breast and ovarian cancers, and studies found that this therapy regimen was most effective in cells that carry functional defects in DNA DSB pathways, most notably in BRCA‐mutated cells (Cortesi et al., 2018; Ghiringhelli et al., 2016; Lin et al., 2014; Lord and Ashworth, 2017; Mittica et al., 2018; Sunada et al., 2018). Recent studies ascribed this synthetic lethality phenotype to the loss of PARP activity at replication forks, suggesting that PARP inhibition promotes rapid fork progression, leading to increased genome instability that the cell cannot overcome when HR defects are also present (Maya‐Mendoza et al., 2018).
Early investigations into the use of PARPi for the treatment of CRC began with the inhibitor ABT‐888, later known as veliparib. Prior studies had demonstrated that ABT‐888 was effective in BRCA‐deficient cells compared with proficient counterparts and that response to PARPi was further increased when combined with platinum‐based genotoxic compounds (Clark et al., 2012). Following on the premise that PARPi will increase sensitivity to genotoxic compounds in cancer cells, the effect of ABT‐888 in combination with irinotecan in CRC cells was investigated (Davidson et al., 2013). This study observed a synergistic response to irinotecan or oxaliplatin in combination with ABT‐888 in CRC cells (Davidson et al., 2013). Another study demonstrated that addition of ABT‐888 increased sensitivity of CRC cells to radiation (Shelton et al., 2013), further supporting the hypothesis that PARPi are a viable option to improve response to current therapy regimens in CRC. A recent phase II open‐label study evaluated the efficacy of the veliparib PARPi in combination with temozolomide in mCRC patients that were refractory to standard therapies. Fifty patients were enrolled in the trial, and 24% exhibited disease control response and 4% showed partial response to the combination therapy (Pishvaian et al., 2018).
Synthetic lethality has been clearly demonstrated in cells that harbor defects in DNA DSB repair pathways, specifically BRCA‐mutated cells (Lord and Ashworth, 2017; McCabe et al., 2006). For this reason, it can be predicted that CRC cells that respond well to PARPi most likely carry defects in DSB repair proteins. One study found that CRC cells carrying inactivating mutations in ATM have increased sensitivity to the PARPi olaparib (Wang et al., 2017). These data correlate well with earlier studies in gastric and lung cancers that found that loss of ATM protein expression increased cellular sensitivity to PARP inhibition (Kubota et al., 2014; Schmitt et al., 2017). However, due to the limited number of preclinical models studied, these results should be confirmed in larger cohorts.
In addition to exploiting DSB repair defects in CRC cells, it has been postulated that cells that exhibit MSI may also be susceptible to PARP inhibition. In 2014, a study demonstrated that loss of MRE11 in MSI CRC cells increased cellular sensitivity to ABT‐888 (Vilar et al., 2011). One proposed explanation is the MSI induces mutations within DNA repair genes, conferring a repair‐deficient phenotype and making the cells more susceptible to the effects of PARP inhibition. This hypothesis has been supported in models of myeloid malignancies (Gaymes et al., 2013). More recently, a phase II clinical trial investigated off‐label of use of PARPi in MSS and MSI CRCs to determine whether microsatellite status was a predictive marker of PARPi response. The results of this study suggested that PARPi alone did not affect patient outcomes regardless of microsatellite status (Leichman et al., 2016). While PARPi have been approved for the treatment of other cancer types that exhibit HR deficiency, PARPi are not currently used for CRC patients.
As discussed above, HR deficiency can also occur in cells with no detectable BRCA1/BRCA2 mutations but showing BRCA‐like phenotypes. Accordingly, it will be of interest to assess whether the BRCA‐ness mutational signatures might be used as predictive biomarkers for sensitivity to PARP inhibitors and oxaliplatin.
DNA repair‐mediated resistance mechanisms to PARP inhibition
The DNA repair‐associated resistance mechanisms to PARP inhibition have been well characterized in breast and ovarian cancers (D'Andrea, 2018), and it is reasonable to predict that similar mechanisms may promote resistance in CRC patients following PARP blockade. One mechanism is re‐activation of HR activity, either through acquired mutations in DNA repair genes or through increased activity of effector proteins that promote HR activity. Acquired mutations have also been described in HR genes that restore the reading frame and expression of the protein following exposure to PARPi (Quigley et al., 2017). Restoration of BRCA1 expression reverses HR‐mediated repair deficiency and allows the cells to repair the damage induced through PARP inhibition (D'Andrea, 2018), and mutations that restore activity of other HR proteins have also been observed in PARPi‐resistant cancer cells. A study of 12 pretreatment and postprogression patient samples observed the acquisition of mutations in the RAD51D and RAD51C genes that restored protein expression and promoted resistance to rucaparib (Kondrashova et al., 2017), and an independent study identified a point mutation in the XRCC2 DNA repair gene that decreased sensitivity of CRC cells to olaparib (Xu et al., 2014). In addition to mutations that restore gene expression, epigenetic regulation of gene expression can predict response and resistance to PARPi. In a study of 12 high‐grade serous ovarian cancer (HGSOC) patient‐derived xenografts and 21 patient samples (ARIEL2 trial), response and resistance to rucaparib were correlated with methylation status of BRCA1. Methylation‐mediated silencing of all BRCA1 copies predicted response to rucaparib, while heterozygous methylation was associated with resistance to therapy (Kondrashova et al., 2018).
Homologous recombination activity can also be increased through regulatory effector proteins, such as the demethylase JMJD1C. These enzymes target MDC1 for demethylation at Lys45 to promote its interaction with RNF8 and its function in the HR signaling cascade (Watanabe et al., 2013). Overexpression of JMJD1C has been detected in colon cancer tissues compared with normal tissues (Chen et al., 2018). Furthermore, depletion of JMJD1C in cells induces cellular resistance to ionizing radiation (IR) and PARPi (Watanabe et al., 2013). These data suggest that resistance mechanisms can arise from regulatory proteins in DNA repair pathways and further show that a comprehensive understanding of repair efficiency is necessary to properly predict therapy response.
A second mechanism of PARPi resistance described in BRCA‐mutated cancers is increased activity of alternative DNA repair pathways. In the absence of BRCA1, activity of nonhomologous end joining (NHEJ), an alternative DNA DSB repair pathway, is increased specifically through loss of p53 binding protein 1 (53BP1) expression (Bouwman et al., 2010; Jaspers et al., 2013). 53BP1 regulates pathway choice in response to DNA DSBs and promotes NHEJ activity through inhibition of BRCA1 recruitment during early DSB repair (Bakr et al., 2016). Somatic loss of 53BP1 expression in BRCA1‐mutated cancers leads to partial restoration of HR‐mediated DSB repair and contributed to resistance to PARPi (Jaspers et al., 2013). A recent study observed that cancer cells carrying mutations that lead to expression of a truncated BRCA1 protein, which maintained the ability to interact with PALB2, still developed PARPi resistance even in the absence of 53BP1. In contrast, loss of the interaction between BRCA1 and PALB2 did not confer PARPi resistance when 53BP1 expression was decreased, suggesting that this protein interaction is required for any measurable HR activity (Nacson et al., 2018).
Increased activity of NHEJ in the absence of BRCA1 can also be attributed to expression of REV7, the noncatalytic subunit of DNA polymerase ζ, which functions to promote translesion synthesis in the presence of DNA damage (Lee et al., 2014). In response to DSBs, REV7 interacts with 53BP1 to prevent DNA‐end resection at the break site. This activity promotes end‐ligation mediated by NHEJ proteins and contributes to DSB repair following PARP inhibition (Xu et al., 2015). REV7 also functions as part of the Shieldin complex that promotes 53BP1 mediated NHEJ in Brca1‐deficient cells (Ghezraoui et al., 2018), a function that could potentially contribute to PARP inhibitor resistance. These activities in the presence of DSBs induced through PARP inhibition provide mechanisms to overcome the damage, leading to resistance to PARP targeting therapies.
A third mechanism of resistance to PARPi, specifically in HR‐deficient cancer cells, is restoration of replication fork stability. One study demonstrated that reduced recruitment of the exonuclease MRE11 in BRCA1‐deficient cells prevented end resection at these sites and promoted fork stability (Ray Chaudhuri et al., 2016). Reduced recruitment of another DNA exonuclease, MUS81, has also been shown to promote stability of replication forks in BRCA2‐deficient cancers that develop resistance to PARP inhibition (Rondinelli et al., 2017). Finally, it has been reported that maintenance of replication forks can be regulated at the transcriptional level. One study observed that BRCA2‐deficient cells treated with PARPi overcome therapeutic pressure by down‐regulating expression of the transcription repressor E2F7. One of the genes under the control of E2F7 is RAD51, and loss of E2F7 expression increases expression of RAD51, enhancing HR activity even in the absence of BRCA2 (Clements et al., 2018).
Resistance to PARP inhibition can also arise from altered activity of PARP and PARP‐associated proteins. One example is loss of poly(ADP‐ribose) glycohydrolase (PARG) that has been shown to be a major resistance mechanism to PARP inhibition in Brca2‐mutated cells (Gogola et al., 2018). Under unperturbed cellular conditions, PARG functions to remove poly(ADP‐ribose) chains generated by PARP1 at the site of DNA damage. PARP inhibitors can either trap PARP1 on the chromatin, leading to stalled replication forks and DSBs, or can inhibit the enzymatic activity and prevent generation of poly(ADP‐ribose) polymers that signal HR‐mediated DSB repair (Dziadkowiec et al., 2016). In the case of the former, PARPi are most effective when PARG is still active in order to remove the poly(ADP‐ribose) polymers and inhibit HR signaling. When PARG activity is lost, the polymers are still present and HR‐mediated repair is still functional. This residual HR activity can counteract the effect of the PARPi and lead to resistance (Gogola et al., 2018).
Together, these studies describe mechanisms of resistance associated with re‐activation of HR activity and provide evidence describing how activity of multiple repair pathways can contribute to resistance to DNA repair targeted therapies.
Alternative DNA repair‐targeting therapies for use in colorectal cancer
The most common chemotherapy agents used for the treatment of CRC induce DNA damage that is recognized and repaired by DNA repair pathways. Inherent defects in these pathways make CRC cells more sensitive to these treatments, and for tumors that do not harbor mutations in DNA repair‐associated genes, these pathways can be targeted to induce a repair‐defective phenotype. The therapies described in the previous section either target or exploit characteristics of DNA repair‐deficient CRC cells. However, there are still other alternative therapies that target DNA repair mechanisms that have not been studied in the context of CRC (Gavande et al., 2016). In this section, we will describe alternative, and potentially novel, treatment regimens that can either take advantage of repair defects in CRC cells or induce repair deficiency in CRC tumors and thus make those cells more responsive to current chemotherapy agents.
ATR inhibitors
Colorectal adenomas exhibit endogenous replication stress (Bartkova et al., 2005), a phenotype that can be exploited through therapies that target replication stress signaling proteins (Halazonetis et al., 2008). One target under investigation is the ataxia telangiectasia and Rad3‐related (ATR) protein. ATR functions at the sites of replication forks and is essential for signaling repair proteins when a cell experiences stress due to DNA damage that blocks replication progression. ATR directly interacts with RPA that coats single‐strand DNA generated during replication, and this ability allows ATR to sense stalled replication forks and corresponding DNA damage (Zou and Elledge, 2003). Additionally, ATR functions in conjunction with ATM in response to IR and is required for promoting accurate repair of DNA DSB damage (Marechal and Zou, 2013). Defects in replication fork protection are correlated with sensitivity to ATR inhibitors (ATRi), and patients who do not exhibit defects in HR but have unstable replication forks may benefit from ATRi therapies (Hill et al., 2018). Furthermore, ATRi is also a viable option to target BRCA‐deficient cancer cells that have acquired resistance to PARPi, by inhibiting the ‘rewired’ HR pathway that is promotes resistance to PARP inhibition (Haynes et al., 2018; Yazinski et al., 2017). For these reasons, ATR is an attractive target to disrupt DNA repair in cancer cells.
Early investigations into ATRi were performed in breast and ovarian cancer cells. Several studies have identified a synthetic lethality with ATRi and ATM or p53 deficiency (Reaper et al., 2011; Toledo et al., 2011), and this effect is further increased when cells are also treated with genotoxic agents (Reaper et al., 2011; Shi et al., 2018). One study reported that the ATRi NU6027 sensitized cells to cisplatin in wild‐type p53 and functional MMR expressing cells, while mutant p53 cells with functional MMR were most sensitive to temozolomide in combination with NU6027 (Peasland et al., 2011). In addition, ATR inhibition was synthetic lethal in combination with PARPi or in cells that had defective HR (through loss of XRCC1) (Peasland et al., 2011; Sultana et al., 2013). Interestingly, one study reported that inhibition of ATR in BRCA1‐depleted cells further sensitized the cells to damage induced by cisplatin and veliparib, suggesting that ATR inhibition functions independently of BRCA status (Huntoon et al., 2013). More recent studies have shown that pancreatic ductal adenocarcinoma (PDAC) and various gastrointestinal cancer cells that exhibit loss of ATM were more sensitive to ATRi (Min et al., 2017; Perkhofer et al., 2017). One study demonstrated that the ATR inhibitor AZD6738 induces a synthetic lethal phenotype in ATM‐deficient, but not ATM‐proficient, gastric cancer cells, and in vivo tumor growth of ATM‐deficient gastric cell xenografts was effectively controlled by treatment with AZD6738 compared with control (Min et al., 2017).
Recently, DNA DSB repair has been implicated in regulating the expression of PD‐L1 in cancer cells (Sato et al., 2017), and the effect of ATR inhibition on PD‐L1 expression, and consequently on immune surveillance of tumor cells, has been investigated (Sun et al., 2018; Vendetti et al., 2018). A siRNA‐mediated screen of DSB repair genes found that loss of BRCA2 enhanced expression of PD‐L1, specifically in response to DSBs induced by IR or PARPi. Furthermore, loss of genes associated with the error‐prone NHEJ pathway, such as Ku80, substantially enhanced PD‐L1 expression in response to IR (Sato et al., 2017). It was observed that treatment with IR and cisplatin significantly increased expression of PD‐L1 and that this effect was abrogated when cells were also treated with pharmacological inhibitors of ATR. Additionally, decreased PD‐L1 expression in the presence of ATRi led to increased immune surveillance of tumor cells, and controlled tumor growth. These data suggest that ATRi would be effective in MMRp cells that have increased PD‐L1 expression as a mechanism of overcoming immune evasion and re‐activating the immunogenicity of these tumor cells (Sun et al., 2018).
Together, these data suggest that ATR inhibition is most effective when combined with genotoxic agents and support the hypothesis that DNA repair‐defective CRC cells may also experience synthetic lethality when treated with ATRi. It would be interesting to test ATRi in MMRd preclinical models of mCRCs that are able to evade immune surveillance despite high levels of neoantigens.
CHK1 Inhibitors
Another key player in the DNA damage response signaling cascade is checkpoint kinase 1 (CHK1) that directly interacts with ATR in the presence of replication stress during S phase and promotes replication fork stabilization (Chen and Poon, 2008). Following the same principle as ATRi, inhibitors targeting CHK1 (CHK1i) have been developed to inhibit replication stress signaling in cancer cells that already exhibit DNA repair defects.
Prexasertib is one CHK1i that has been thoroughly investigated in patients with squamous cell carcinomas (Hong et al., 2018), non‐small‐cell lung carcinomas (Sen et al., 2017), and high‐grade serous ovarian carcinomas (Brill et al., 2017; Lee et al., 2018), often in combination with other therapies. Early studies demonstrated that prexasertib (LY2606368) induced replication catastrophe and DNA damage while concomitantly disabling cell cycle checkpoints, ultimately leading to apoptosis. This effect was observed in vitro and in vivo in models of acute lymphoblastic leukemia and squamous cell carcinoma (Ghelli Luserna Di Rora et al., 2016; King et al., 2015).
In the context of CRC, one study investigated the effect of CHK1 inhibition on CRC stem cells (Manic et al., 2018). The authors observed that treatment with prexasertib, both in vitro and in vivo, inhibited replication and disabled cell cycle checkpoints, causing the cells to enter mitosis prematurely, ultimately leading to apoptosis. Interestingly, this effect was observed in cells that harbored KRAS mutations, a subset of CRCs particularly difficult to target and treat (Manic et al., 2018). An independent study also observed efficacy of CHK1 inhibition in KRAS‐mutated lung and colon adenocarcinoma cells, especially when combined with MK2 inhibitors (Dietlein et al., 2015). MK2 functions in a pathway parallel to CHK1 and is responsible for maintaining cell cycle checkpoints in response to stress (Reinhardt et al., 2010). KRAS‐mutated cells exhibit intrinsic genotoxic stress that leads to constant activation of CHK1 and MK2, and inhibition of these proteins induced mitotic catastrophe in vitro, in murine cancer models, and in patient‐derived cells (Dietlein et al., 2015). CHK1 inhibitors might be tested in KRAS‐mutated CRC, a subset that currently has limited therapy options.
Thiopurines
Thiopurines are a class of nucleotide analogs that have been used successfully for the treatment of childhood leukemias (Karran, 2006). Thiopurines are incorporated into DNA during replication, leading to DNA base pair mismatches that are removed by MMR proteins (Coulthard and Hogarth, 2005; Karran, 2006; Munshi et al., 2014). The effect of the thiopurine analog 6‐thioguanine (6TG) on cell growth has been tested in CRC cell models. MMRd CRC cell lines are resistant to 6TG compared with MMRp counterparts (Carethers et al., 1996; Yan et al., 2003). 6TG induces reactive oxygen species (Brem and Karran, 2012) can trigger activity of BER proteins, suggesting that BER‐deficient cells may have increased cellular sensitivity to thiopurines. Further studies have also demonstrated that HR and FA proteins function to repair thiopurine induced damage (Brem and Karran, 2012) and that cells deficient for HR proteins, such as RAD51D, have increased sensitivity to thiopurines (Rajesh et al., 2011). These data suggest that this class of compounds might be effective in CRC tumors that are MMRp but carry mutations in genes associated with HR and BER.
Clinical trials of DNA repair inhibitors in colorectal cancer
Targeting DNA repair in CRC has the potential to further increase the efficacy of current therapies, and, as described above, there have been multiple preclinical studies investigating DNA repair‐targeting therapies in CRC. None of these therapies have been approved by the FDA for use in CRC patients; however, several trials are ongoing (Table 2). To date, six clinical trials investigating the efficacy of PARPi in CRC are ongoing or have been completed. Of the five completed trials, only NCT00912743 has reported results (Leichman et al., 2016). In this study, the efficacy of olaparib was investigated in 33 CRC patients stratified by microsatellite status. Thirteen MSI‐H and 20 non‐MSI‐H patients were enrolled and treated with olaparib 400 mg twice a day. The median of progression‐free survival (PFS) was 61 days for the MSI‐H cohort and 55 for the non‐MSI‐H cohort. Overall survival (OS) was reported to be 248 days for the MSI‐H cohort and 209.5 days for the non‐MSI‐H. There was no statistical significance in the median PFS or OS for non‐MSI‐H cohort. The results of this study suggest that olaparib alone is ineffective in CRC patients regardless of microsatellite status, and the authors recommend that further studies investigate the use of olaparib in combination with DNA damaging agents for this patient cohort (Leichman et al., 2016). Importantly, this study was conducted in CRC patients that were not enriched for HR deficiency status.
Table 2.
Clinical trials of PARP, ATR, and CHK1 inhibitors reported to the U.S. National Library of Medicine. (https://clinicaltrials.gov/ct2/home)
| Trial identifier | Therapy | Disease(s) | Status |
|---|---|---|---|
| Clinical trials in colorectal cancer | |||
| NCT00912743 | Olaparib | Chemorefractory metastatic colorectal cancer |
Completed Results Available (Leichman et al., 2016) |
| NCT02484404 |
Olaparib Cediranib MEDI4736 |
Ovarian, triple negative breast, lung, prostate, colorectal cancers | Recruiting |
| NCT02305758 |
FOLFIRI Bevacizumab Veliparib |
Untreated metastatic colorectal cancer | Completed (Gorbunova et al., 2018) |
| NCT01051596 | Temozolomide ABT‐888 | Colorectal cancer | Completed (Pishvaian et al., 2018) |
| NCT01589419 |
Veliparib Capecitabine Radiation |
Locally advanced rectal cancer | Completed (Czito et al., 2017) |
| NCT02033551 |
Veliparib Carboplatin Paclitaxel FOLFIRI |
Metastatic and chemorefractory Breast cancer, ovarian cancer, colon cancer, lung cancer, gastric cancer, solid tumors | Completed (Berlin et al., 2018) |
| Clinical trials in other cancers | |||
| NCT03669601 |
AZD6738 Gemcitabine |
Cancer | Not yet recruiting |
| NCT03682289 |
AZD6738 Olaparib |
Clear cell renal cell carcinoma, Pancreatic ductal adenocarcinoma, Renal cell carcinoma cancers | Not yet recruiting |
| NCT03428607 |
AZD6738 Olaparib |
SCLC | Not yet recruiting |
| NCT02630199 |
AZD6738 Paclitaxel |
Refractory cancers | Recruiting |
| NCT03462342 |
AZD6738 Olaparib |
High‐grade serous carcinoma | Recruiting |
| NCT02223923 | AZD6738 | Solid tumor refractory to conventional therapy | Suspended |
| NCT01955668 | AZD6738 | Chronic lymphocytic leukemia, prolymphocytic leukemia, B‐cell leukemia | Completed |
| NCT02264678 |
AZD6738 Carboplatin Olaparib MEDI4736 |
Advanced solid malignancies—H&N SCC, ATM Pro/Def NSCLC, gastric and breast cancer | Recruiting |
| NCT03328273 | AZD6738 Acalabrutinib | Chronic lymphocytic leukemia | Recruiting |
| NCT03330847 |
AZD6738 Olaparib |
Metastatic triple negative breast cancer | Recruiting |
| NCT03022409 |
AZD6738 Olaparib |
Head and neck squamous cell carcinoma | Recruiting |
| NCT02576444 | AZD6738 | Cancer | Recruiting |
| NCT03527147 | AZD6738 | NHL, DLBCL, non‐Hodgkin's lymphoma, diffuse large B‐cell lymphoma | Recruiting |
| NCT03334617 | AZD6738 | Non‐small‐cell lung cancer | Recruiting |
| NCT02937818 |
AZD6738 Olaparib |
Platinum refractory extensive‐stage small cell lung carcinoma | Recruiting |
| NCT02203513 | LY2606368 | Breast, ovarian, prostate | Recruiting (Lee et al., 2018) |
| NCT01870596 | SCH900776 | Acute myeloid leukemia | Completed |
| NCT03495323 |
LY3300054 Prexasertib |
Cancer | Recruiting |
| NCT02808650 | Prexasertib | Childhood solid neoplasm, Recurrent central nervous system neoplasm, recurrent malignant solid neoplasm, refractory central nervous system neoplasm, refractory malignant solid neoplasm | Recruiting |
| NCT02797964 | SRA737 | Advanced solid tumors or non‐Hodgkin's lymphoma | Recruiting |
| NCT02797977 |
SRA737 Gemcitabine Cisplatin |
Advanced solid tumors | Recruiting |
| NCT02873975 | LY2606368 | Advanced cancers | Recruiting |
| NCT03057145 |
LY2606368 Olaparib |
Solid tumor | Recruiting |
| NCT01115790 | Prexasertib | Advanced cancer, squamous cell carcinoma, carcinoma, squamous cell of head and neck, lung squamous cell carcinoma, anal squamous cell carcinoma, carcinoma, non‐small‐cell lung | Completed |
| NCT01139775 |
LY2603618 Pemetrexed Cisplatin |
Non‐small‐cell lung cancer | Completed |
| NCT02735980 | Prexasertib | Small cell lung cancer | Completed |
| NCT02514603 | Prexasertib | Neoplasm | Completed |
| NCT03735446 |
Prexasertib Mitoxantrone Etoposide Cytarabine |
Acute myeloid leukemia, Myelodysplastic syndromes | Not yet recruiting |
| NCT03377556 | Talazoparib | Squamous cell lung cancer | Recruiting |
Multiple clinical trials investigating the efficacy of the ATRi AZD6738 have been initiated, and most of these include gastrointestinal malignancies other than CRC. NCT01955668 is the only trial that has been completed thus far, and the results of this study have not been reported in full. Currently, the majority of clinical trials for AZD6738 focus on assessing the efficacy and safety of the drug in patients. As yet, there have not been any trials initiated to further elucidate sensitivity of specific cancer subtypes, such as ATM‐deficient tumors, in the clinical setting. In addition to analyzing the efficacy of AZD6738 alone, several studies are investigating the efficacy of this drug in combination with the PARPi olaparib, particularly in patients who were refractory to primary therapies. Once completed, these studies have the potential to describe novel therapy regimens can be used to treat CRC patients.
Multiple clinical trials with CHK1 inhibitors are ongoing. One trial (NCT02203513) has reported results from a cohort of BRCA wild‐type high‐grade serous ovarian cancer patients treated with the CHK1 inhibitor prexasertib (Lee et al., 2018). In this cohort, 80% of the patients were platinum‐resistant or refractory at the start of the trial. Sixteen patients (out of 28) exhibited partial response during the treatment time and one patient died during the study due to tumor progression (Lee et al., 2018). Preclinical data that led to another ongoing trial (NCT02555644) investigated the efficacy of CHK1 inhibitors in combination with EGFR targeted therapies and/or radiotherapy (Zeng et al., 2017). In this study, prexasertib combined with EGFR targeting therapies significantly decreased cell proliferation and delayed tumor growth in both HPV‐positive and HPV‐negative head and neck squamous cell carcinoma mouse models (Zeng et al., 2017). These promising results provided rationale to test CHK1 inhibitors in combination with both genotoxic and nongenotoxic therapy regimens for cancer treatment.
Concluding remarks
Standard clinical testing of CRC includes identifying mutations in oncogenes such as KRAS and BRAF, as well as characterization of the microsatellite status. Currently, the status of DNA repair genes is not investigated in CRCs. However, MSS CRCs carry a higher proportion of mutations in HR genes, and defects in this pathway have been associated with genomic instability. Whole‐genome sequencing analysis of breast cancer samples can identify tumors that exhibit genomic rearrangements due to functional deficiencies in homologous recombination even BRCA wild‐type cells, and these characteristics can be used to predict therapy response to PARP inhibitors. We propose that, in addition to genetic screening for mutations in known DNA repair genes, identification of gene alterations and genomic rearrangements indicative of a repair‐defective phenotype should be performed systematically in CRC patients. Characterizations based on functional repair deficiency, rather than analyses based primarily on genetic alterations, are likely to better predict therapy response to inhibitors of DNA repair pathways in CRC patient cohorts.
Author contributions
NMR wrote the manuscript. LN performed bioinformatics analysis of The Cancer Genome Atlas datasets. FDN and AB contributed to the manuscript preparation.
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
This work was supported by European Community's Seventh Framework Programme under grant agreement no. 602901 MErCuRIC (A.B.); H2020 grant agreement no. 635342‐2 MoTriColor (A.B.); AIRC IG n. 17707 (F.D.N.); AIRC 2010 Special Program Molecular Clinical Oncology 5 per mille, Project n. 9970 Extension program (A.B.); AIRC IG n. 16788 (A.B.); Fondazione Piemontese per la Ricerca sul Cancro‐ONLUS 5 per mille 2011, 2014, and 2015 Ministero della Salute (A.B. and F.D.N.); AIRC Special Program 5 per mille metastases project n. 21091 (A.B. and F.D.N); Fondo per la Ricerca Locale (ex 60%), Università di Torino, 2017 (F.D.N). Progetto NET‐2011‐02352137 Ministero della Salute (A.B. and F.D.N). N.M.R. is supported by an AIRC ‘Molini Bongiovanni’ 3‐year fellowship. The results shown here are in whole or in part based upon data generated by the TCGA Research Network: http://cancergenome.nih.gov/.
References
- Aguilera A and Gomez‐Gonzalez B (2008) Genome instability: a mechanistic view of its causes and consequences. Nat Rev Genet 9, 204–217. [DOI] [PubMed] [Google Scholar]
- AlDubayan SH, Giannakis M, Moore ND, Han GC, Reardon B, Hamada T, Mu XJ, Nishihara R, Qian Z, Liu L et al (2018) Inherited DNA‐repair defects in colorectal cancer. Am J Hum Genet 102, 401–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Badie S, Escandell JM, Bouwman P, Carlos AR, Thanasoula M, Gallardo MM, Suram A, Jaco I, Benitez J, Herbig U et al (2010) BRCA2 acts as a RAD51 loader to facilitate telomere replication and capping. Nat Struct Mol Biol 17, 1461–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakr A, Kocher S, Volquardsen J, Reimer R, Borgmann K, Dikomey E, Rothkamm K and Mansour WY (2016) Functional crosstalk between DNA damage response proteins 53BP1 and BRCA1 regulates double strand break repair choice. Radiother Oncol 119, 276–281. [DOI] [PubMed] [Google Scholar]
- Balc'h EL, Grandin N, Demattei MV, Guyetant S, Tallet A, Pages JC, Ouaissi M, Lecomte T, Charbonneau M (2017) Measurement of telomere length in colorectal cancers for improved molecular diagnosis. Int J Mol Sci 18, 1871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartkova J, Horejsi Z, Koed K, Kramer A, Tort F, Zieger K, Guldberg P, Sehested M, Nesland JM, Lukas C et al (2005) DNA damage response as a candidate anti‐cancer barrier in early human tumorigenesis. Nature 434, 864–870. [DOI] [PubMed] [Google Scholar]
- Berlin J, Ramanathan RK, Strickler JH, Subramaniam DS, Marshall J, Kang YK, Hetman R, Dudley MW, Zeng J, Nickner C et al (2018) A phase 1 dose‐escalation study of veliparib with bimonthly FOLFIRI in patients with advanced solid tumours. Br J Cancer 118, 938–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boland CR, Thibodeau SN, Hamilton SR, Sidransky D, Eshleman JR, Burt RW, Meltzer SJ, Rodriguez‐Bigas MA, Fodde R, Ranzani GN et al (1998) A National Cancer Institute Workshop on Microsatellite Instability for cancer detection and familial predisposition: development of international criteria for the determination of microsatellite instability in colorectal cancer. Cancer Res 58, 5248–5257. [PubMed] [Google Scholar]
- Bouwman P, Aly A, Escandell JM, Pieterse M, Bartkova J, van der Gulden H, Hiddingh S, Thanasoula M, Kulkarni A, Yang Q et al (2010) 53BP1 loss rescues BRCA1 deficiency and is associated with triple‐negative and BRCA‐mutated breast cancers. Nat Struct Mol Biol 17, 688–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA and Jemal A (2018) Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 68, 394–424. [DOI] [PubMed] [Google Scholar]
- Brem R and Karran P (2012) Oxidation‐mediated DNA cross‐linking contributes to the toxicity of 6‐thioguanine in human cells. Cancer Res 72, 4787–4795. [DOI] [PubMed] [Google Scholar]
- Brill E, Yokoyama T, Nair J, Yu M, Ahn YR and Lee JM (2017) Prexasertib, a cell cycle checkpoint kinases 1 and 2 inhibitor, increases in vitro toxicity of PARP inhibition by preventing Rad51 foci formation in BRCA wild type high‐grade serous ovarian cancer. Oncotarget 8, 111026–111040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno PM, Liu Y, Park GY, Murai J, Koch CE, Eisen TJ, Pritchard JR, Pommier Y, Lippard SJ and Hemann MT (2017) A subset of platinum‐containing chemotherapeutic agents kills cells by inducing ribosome biogenesis stress. Nat Med 23, 461–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burrell RA, McClelland SE, Endesfelder D, Groth P, Weller MC, Shaikh N, Domingo E, Kanu N, Dewhurst SM, Gronroos E et al (2013) Replication stress links structural and numerical cancer chromosomal instability. Nature 494, 492–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Candelli A, Holthausen JT, Depken M, Brouwer I, Franker MA, Marchetti M, Heller I, Bernard S, Garcin EB, Modesti M et al (2014) Visualization and quantification of nascent RAD51 filament formation at single‐monomer resolution. Proc Natl Acad Sci USA 111, 15090–15095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carethers JM, Hawn MT, Chauhan DP, Luce MC, Marra G, Koi M and Boland CR (1996) Competency in mismatch repair prohibits clonal expansion of cancer cells treated with N‐methyl‐N’‐nitro‐N‐nitrosoguanidine. J Clin Invest 98, 199–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceccaldi R, Sarangi P and D'Andrea AD (2016) The Fanconi anaemia pathway: new players and new functions. Nat Rev Mol Cell Biol 17, 337–349. [DOI] [PubMed] [Google Scholar]
- Cerami E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E et al (2012) The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2, 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Aihemaiti M, Zhang X, Qu H, Sun QL, He QS and Yu WB (2018) Downregulation of histone demethylase JMJD1C inhibits colorectal cancer metastasis through targeting ATF2. Am J Cancer Res 8, 852–865. [PMC free article] [PubMed] [Google Scholar]
- Chen Y and Poon RY (2008) The multiple checkpoint functions of CHK1 and CHK2 in maintenance of genome stability. Front Biosci 13, 5016–5029. [DOI] [PubMed] [Google Scholar]
- Chien J, Sicotte H, Fan JB, Humphray S, Cunningham JM, Kalli KR, Oberg AL, Hart SN, Li Y, Davila JI et al (2015) TP53 mutations, tetraploidy and homologous recombination repair defects in early stage high‐grade serous ovarian cancer. Nucleic Acids Res 43, 6945–6958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ciccia A and Elledge SJ (2010) The DNA damage response: making it safe to play with knives. Mol Cell 40, 179–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark CC, Weitzel JN and O'Connor TR (2012) Enhancement of synthetic lethality via combinations of ABT‐888, a PARP inhibitor, and carboplatin in vitro and in vivo using BRCA1 and BRCA2 isogenic models. Mol Cancer Ther 11, 1948–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Claussin C and Chang M (2015) The many facets of homologous recombination at telomeres. Microb Cell 2, 308–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clements KE, Thakar T, Nicolae CM, Liang X, Wang HG and Moldovan GL (2018) Loss of E2F7 confers resistance to poly‐ADP‐ribose polymerase (PARP) inhibitors in BRCA2‐deficient cells. Nucleic Acids Res 46, 8898–8907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortes‐Ciriano I, Lee S, Park WY, Kim TM and Park PJ (2017) A molecular portrait of microsatellite instability across multiple cancers. Nat Commun 8, 15180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortesi L, Toss A, Cucinotto I (2018) Parp Inhibitors for the treatment of ovarian cancer. Curr Cancer Drug Targets 18, 877–893. [DOI] [PubMed] [Google Scholar]
- Coulthard S and Hogarth L (2005) The thiopurines: an update. Invest New Drugs 23, 523–532. [DOI] [PubMed] [Google Scholar]
- Cremolini C, Schirripa M, Antoniotti C, Moretto R, Salvatore L, Masi G, Falcone A and Loupakis F (2015) First‐line chemotherapy for mCRC‐a review and evidence‐based algorithm. Nat Rev Clin Oncol 12, 607–619. [DOI] [PubMed] [Google Scholar]
- Czito BG, Deming DA, Jameson GS, Mulcahy MF, Vaghefi H, Dudley MW, Holen KD, DeLuca A, Mittapalli RK, Munasinghe W et al (2017) Safety and tolerability of veliparib combined with capecitabine plus radiotherapy in patients with locally advanced rectal cancer: a phase 1b study. Lancet Gastroenterol Hepatol 2, 418–426. [DOI] [PubMed] [Google Scholar]
- D'Andrea AD (2018) Mechanisms of PARP inhibitor sensitivity and resistance. DNA Repair (Amst) 71, 172–176. [DOI] [PubMed] [Google Scholar]
- Davidson D, Wang Y, Aloyz R and Panasci L (2013) The PARP inhibitor ABT‐888 synergizes irinotecan treatment of colon cancer cell lines. Invest New Drugs 31, 461–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies H, Glodzik D, Morganella S, Yates LR, Staaf J, Zou X, Ramakrishna M, Martin S, Boyault S, Sieuwerts AM et al (2017) HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat Med 23, 517–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dietlein F, Kalb B, Jokic M, Noll EM, Strong A, Tharun L, Ozretic L, Kunstlinger H, Kambartel K, Randerath WJ et al (2015) A Synergistic Interaction between Chk1‐ and MK2 Inhibitors in KRAS‐Mutant Cancer. Cell 162, 146–159. [DOI] [PubMed] [Google Scholar]
- Dodson GE, Limbo O, Nieto D and Russell P (2010) Phosphorylation‐regulated binding of Ctp1 to Nbs1 is critical for repair of DNA double‐strand breaks. Cell Cycle 9, 1516–1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Druliner BR, Ruan X, Sicotte H, O'Brien D, Liu H, Kocher JA and Boardman L (2018) Early genetic aberrations in patients with sporadic colorectal cancer. Mol Carcinog 57, 114–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dziadkowiec KN, Gasiorowska E, Nowak‐Markwitz E and Jankowska A (2016) PARP inhibitors: review of mechanisms of action and BRCA1/2 mutation targeting. Prz Menopauzalny 15, 215–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esteller M, Levine R, Baylin SB, Ellenson LH and Herman JG (1998) MLH1 promoter hypermethylation is associated with the microsatellite instability phenotype in sporadic endometrial carcinomas. Oncogene 17, 2413–2417. [DOI] [PubMed] [Google Scholar]
- Fabrizio DA, George TJ Jr, Dunne RF, Frampton G, Sun J, Gowen K, Kennedy M, Greenbowe J, Schrock AB, Hezel AF et al (2018) Beyond microsatellite testing: assessment of tumor mutational burden identifies subsets of colorectal cancer who may respond to immune checkpoint inhibition. J Gastrointest Oncol 9, 610–617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Francia G, Green SK, Bocci G, Man S, Ernmenegger U, Ebos JML, Weinerman A, Shaked Y and Kerbel RS (2005) Down‐regulation of DNA mismatch repair proteins in human and murine tumor spheroids: implications for multicellular resistance to alkylating agents. Mol Cancer Therap 4, 1484–1494. [DOI] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E et al (2013) Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci Signal 6, pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gavande NS, VanderVere‐Carozza PS, Hinshaw HD, Jalal SI, Sears CR, Pawelczak KS and Turchi JJ (2016) DNA repair targeted therapy: The past or future of cancer treatment? Pharmacol Ther 160, 65–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaymes TJ, Mohamedali AM, Patterson M, Matto N, Smith A, Kulasekararaj A, Chelliah R, Curtin N, Farzaneh F, Shall S et al (2013) Microsatellite instability induced mutations in DNA repair genes CtIP and MRE11 confer hypersensitivity to poly (ADP‐ribose) polymerase inhibitors in myeloid malignancies. Haematologica 98, 1397–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Germano G, Amirouchene‐Angelozzi N, Rospo G and Bardelli A (2018) The clinical impact of the genomic landscape of mismatch repair‐deficient cancers. Cancer Discov 8, 1518–1528. [DOI] [PubMed] [Google Scholar]
- Germano G, Lamba S, Rospo G, Barault L, Magri A, Maione F, Russo M, Crisafulli G, Bartolini A, Lerda G et al (2017) Inactivation of DNA repair triggers neoantigen generation and impairs tumour growth. Nature 552, 116–120. [DOI] [PubMed] [Google Scholar]
- Ghelli Luserna Di Rora A, Iacobucci I, Imbrogno E, Papayannidis C, Derenzini E, Ferrari A, Guadagnuolo V, Robustelli V, Parisi S, Sartor C et al (2016) Prexasertib, a Chk1/Chk2 inhibitor, increases the effectiveness of conventional therapy in B‐/T‐ cell progenitor acute lymphoblastic leukemia. Oncotarget 7, 53377–53391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghezraoui H, Oliveira C, Becker JR, Bilham K, Moralli D, Anzilotti C, Fischer R, Deobagkar‐Lele M, Sanchiz‐Calvo M, Fueyo‐Marcos E et al (2018) 53BP1 cooperation with the REV7‐shieldin complex underpins DNA structure‐specific NHEJ. Nature 560, 122–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghiringhelli F, Richard C, Chevrier S, Vegran F and Boidot R (2016) Efficiency of olaparib in colorectal cancer patients with an alteration of the homologous repair protein. World J Gastroenterol 22, 10680–10686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gogola E, Duarte AA, de Ruiter JR, Wiegant WW, Schmid JA, de Bruijn R, James DI, Guerrero Llobet S, Vis DJ, Annunziato S et al (2018) Selective loss of PARG restores PARylation and counteracts PARP inhibitor‐mediated synthetic lethality. Cancer Cell 33, 1078–1093.e1012. [DOI] [PubMed] [Google Scholar]
- Gorbunova V, Beck JT, Hofheinz RD, Garcia‐Alfonso P, Nechaeva M, Cubillo Gracian A, Mangel L, Elez Fernandez E, Deming DA, Ramanathan RK et al (2018) A phase 2 randomised study of veliparib plus FOLFIRI+/‐bevacizumab versus placebo plus FOLFIRI+/‐bevacizumab in metastatic colorectal cancer. Br J Cancer 120, 183–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hackett JA, Feldser DM and Greider CW (2001) Telomere dysfunction increases mutation rate and genomic instability. Cell 106, 275–286. [DOI] [PubMed] [Google Scholar]
- Haince JF, McDonald D, Rodrigue A, Dery U, Masson JY, Hendzel MJ and Poirier GG (2008) PARP1‐dependent kinetics of recruitment of MRE11 and NBS1 proteins to multiple DNA damage sites. J Biol Chem 283, 1197–1208. [DOI] [PubMed] [Google Scholar]
- Halazonetis TD, Gorgoulis VG and Bartek J (2008) An oncogene‐induced DNA damage model for cancer development. Science 319, 1352–1355. [DOI] [PubMed] [Google Scholar]
- Hanahan D and Weinberg RA (2011) Hallmarks of cancer: the next generation. Cell 144, 646–674. [DOI] [PubMed] [Google Scholar]
- Hause RJ, Pritchard CC, Shendure J and Salipante SJ (2016) Classification and characterization of microsatellite instability across 18 cancer types. Nat Med 22, 1342–1350. [DOI] [PubMed] [Google Scholar]
- Haynes B, Murai J and Lee JM (2018) Restored replication fork stabilization, a mechanism of PARP inhibitor resistance, can be overcome by cell cycle checkpoint inhibition. Cancer Treat Rev 71, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Helleday T, Petermann E, Lundin C, Hodgson B and Sharma RA (2008) DNA repair pathways as targets for cancer therapy. Nat Rev Cancer 8, 193–204. [DOI] [PubMed] [Google Scholar]
- Hill SJ, Decker B, Roberts EA, Horowitz NS, Muto MG, Worley MJ, Feltmate CM, Nucci MR, Swisher EM, Nguyen H et al (2018) Prediction of DNA repair inhibitor response in short term patient‐derived ovarian cancer organoids. Cancer Discov 8, 1404–1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillman RT, Chisholm GB, Lu KH, Futreal PA (2018) Genomic rearrangement signatures and clinical outcomes in high‐grade serous ovarian cancer. J Natl Cancer Inst 110, 265–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoang LN and Gilks BC (2018) Hereditary breast and ovarian cancer syndrome: moving beyond BRCA1 and BRCA2. Adv Anat Pathol 25, 85–95. [DOI] [PubMed] [Google Scholar]
- Hong DS, Moore K, Patel M, Grant SC, Burris HA 3rd, William WN Jr, Jones S, Meric‐Bernstam F, Infante J, Golden L et al (2018) Evaluation of Prexasertib, a checkpoint kinase 1 inhibitor, in a phase Ib study of patients with squamous cell carcinoma. Clin Cancer Res 24, 3263–3272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang D, Sun W, Zhou Y, Li P, Chen F, Chen H, Xia D, Xu E, Lai M, Wu Y et al (2018) Mutations of key driver genes in colorectal cancer progression and metastasis. Cancer Metastasis Rev 37, 173–187. [DOI] [PubMed] [Google Scholar]
- Huehls AM, Huntoon CJ, Joshi PM, Baehr CA, Wagner JM, Wang X, Lee MY and Karnitz LM (2016) Genomically incorporated 5‐fluorouracil that escapes UNG‐initiated base excision repair blocks DNA replication and activates homologous recombination. Mol Pharmacol 89, 53–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huen MS, Sy SM and Chen J (2010) BRCA1 and its toolbox for the maintenance of genome integrity. Nat Rev Mol Cell Biol 11, 138–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huntoon CJ, Flatten KS, Wahner Hendrickson AE, Huehls AM, Sutor SL, Kaufmann SH and Karnitz LM (2013) ATR inhibition broadly sensitizes ovarian cancer cells to chemotherapy independent of BRCA status. Cancer Res 73, 3683–3691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwaizumi M, Tseng‐Rogenski S and Carethers JM (2011) DNA mismatch repair proficiency executing 5‐fluorouracil cytotoxicity in colorectal cancer cells. Cancer Biol Ther 12, 756–764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer RR, Pluciennik A, Burdett V and Modrich PL (2006) DNA mismatch repair: functions and mechanisms. Chem Rev 106, 302–323. [DOI] [PubMed] [Google Scholar]
- Jackson SP and Bartek J (2009) The DNA‐damage response in human biology and disease. Nature 461, 1071–1078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janssen A, van der Burg M, Szuhai K, Kops GJ and Medema RH (2011) Chromosome segregation errors as a cause of DNA damage and structural chromosome aberrations. Science 333, 1895–1898. [DOI] [PubMed] [Google Scholar]
- Jaspers JE, Kersbergen A, Boon U, Sol W, van Deemter L, Zander SA, Drost R, Wientjens E, Ji J, Aly A et al (2013) Loss of 53BP1 causes PARP inhibitor resistance in Brca1‐mutated mouse mammary tumors. Cancer Discov 3, 68–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jemal A, Ward EM, Johnson CJ, Cronin KA, Ma J, Ryerson B, Mariotto A, Lake AJ, Wilson R, Sherman RL et al (2017) Annual report to the Nation on the status of cancer, 1975–2014, featuring survival. J Natl Cancer Inst 109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen RB, Ozes A, Kim T, Estep A and Kowalczykowski SC (2013) BRCA2 is epistatic to the RAD51 paralogs in response to DNA damage. DNA Repair (Amst) 12, 306–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalimutho M, Bain AL, Mukherjee B, Nag P, Nanayakkara DM, Harten SK, Harris JL, Subramanian GN, Sinha D, Shirasawa S et al (2017) Enhanced dependency of KRAS‐mutant colorectal cancer cells on RAD51‐dependent homologous recombination repair identified from genetic interactions in Saccharomyces cerevisiae . Mol Oncol 11, 470–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karran P (2006) Thiopurines, DNA damage, DNA repair and therapy‐related cancer. Br Med Bull 79–80, 153–170. [DOI] [PubMed] [Google Scholar]
- Kim G, Ison G, McKee AE, Zhang H, Tang S, Gwise T, Sridhara R, Lee E, Tzou A, Philip R et al (2015) FDA approval summary: Olaparib monotherapy in patients with deleterious germline BRCA‐mutated advanced ovarian cancer treated with three or more lines of chemotherapy. Clin Cancer Res 21, 4257–4261. [DOI] [PubMed] [Google Scholar]
- Kim JH, Park HE, Cho NY, Lee HS and Kang GH (2016) Characterisation of PD‐L1‐positive subsets of microsatellite‐unstable colorectal cancers. Br J Cancer 115, 490–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King C, Diaz HB, McNeely S, Barnard D, Dempsey J, Blosser W, Beckmann R, Barda D and Marshall MS (2015) LY2606368 causes replication catastrophe and antitumor effects through CHK1‐dependent mechanisms. Mol Cancer Ther 14, 2004–2013. [DOI] [PubMed] [Google Scholar]
- Knijnenburg TA, Wang L, Zimmermann MT, Chambwe N, Gao GF, Cherniack AD, Fan H, Shen H, Way GP, Greene CS et al (2018) Genomic and molecular landscape of DNA damage repair deficiency across the cancer genome atlas. Cell Rep 23, 239–254.e236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondrashova O, Nguyen M, Shield‐Artin K, Tinker AV, Teng NNH, Harrell MI, Kuiper MJ, Ho GY, Barker H, Jasin M et al (2017) Secondary somatic mutations restoring RAD51C and RAD51D associated with acquired resistance to the PARP inhibitor Rucaparib in high‐grade ovarian carcinoma. Cancer Discov 7, 984–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondrashova O, Topp M, Nesic K, Lieschke E, Ho GY, Harrell MI, Zapparoli GV, Hadley A, Holian R, Boehm E et al (2018) Methylation of all BRCA1 copies predicts response to the PARP inhibitor rucaparib in ovarian carcinoma. Nat Commun 9, 3970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubota E, Williamson CT, Ye R, Elegbede A, Peterson L, Lees‐Miller SP and Bebb DG (2014) Low ATM protein expression and depletion of p53 correlates with olaparib sensitivity in gastric cancer cell lines. Cell Cycle 13, 2129–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, Lu S, Kemberling H, Wilt C, Luber BS et al (2017) Mismatch repair deficiency predicts response of solid tumors to PD‐1 blockade. Science 357, 409–413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le S, Moore JK, Haber JE and Greider CW (1999) RAD50 and RAD51 define two pathways that collaborate to maintain telomeres in the absence of telomerase. Genetics 152, 143–152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee YS, Gregory MT and Yang W (2014) Human Pol zeta purified with accessory subunits is active in translesion DNA synthesis and complements Pol eta in cisplatin bypass. Proc Natl Acad Sci USA 111, 2954–2959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JM, Nair J, Zimmer A, Lipkowitz S, Annunziata CM, Merino MJ, Swisher EM, Harrell MI, Trepel JB, Lee MJ et al (2018) Prexasertib, a cell cycle checkpoint kinase 1 and 2 inhibitor, in BRCA wild‐type recurrent high‐grade serous ovarian cancer: a first‐in‐class proof‐of‐concept phase 2 study. Lancet Oncol 19, 207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leichman L, Groshen S, O'Neil BH, Messersmith W, Berlin J, Chan E, Leichman CG, Cohen SJ, Cohen D, Lenz HJ et al (2016) Phase II study of Olaparib (AZD‐2281) after standard systemic therapies for disseminated colorectal cancer. Oncologist 21, 172–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemery S, Keegan P and Pazdur R (2017) First FDA approval agnostic of cancer site – when a biomarker defines the indication. N Engl J Med 377, 1409–1412. [DOI] [PubMed] [Google Scholar]
- Lenz HJ, Stintzing S and Loupakis F (2015) TAS‐102, a novel antitumor agent: a review of the mechanism of action. Cancer Treat Rev 41, 777–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Jiang T, Li Q and Ling X (2017) Camptothecin (CPT) and its derivatives are known to target topoisomerase I (Top1) as their mechanism of action: did we miss something in CPT analogue molecular targets for treating human disease such as cancer? Am J Cancer Res 7, 2350–2394. [PMC free article] [PubMed] [Google Scholar]
- Lin ZP, Ratner ES, Whicker ME, Lee Y and Sartorelli AC (2014) Triapine disrupts CtIP‐mediated homologous recombination repair and sensitizes ovarian cancer cells to PARP and topoisomerase inhibitors. Mol Cancer Res 12, 381–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J, Lichtenberg T, Hoadley KA, Poisson LM, Lazar AJ, Cherniack AD, Kovatich AJ, Benz CC, Levine DA, Lee AV et al (2018) An integrated TCGA pan‐cancer clinical data resource to drive high‐quality survival outcome analytics. Cell 173, 400–416.e411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llosa NJ, Cruise M, Tam A, Wicks EC, Hechenbleikner EM, Taube JM, Blosser RL, Fan H, Wang H, Luber BS et al (2015) The vigorous immune microenvironment of microsatellite instable colon cancer is balanced by multiple counter‐inhibitory checkpoints. Cancer Discov 5, 43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorans M, Dow E, Macrae FA, Winship IM, Buchanan DD (2018) Update on hereditary colorectal cancer: improving the clinical utility of multigene panel testing. Clin Colorectal Cancer 17, e293–e305. [DOI] [PubMed] [Google Scholar]
- Lord CJ and Ashworth A (2017) PARP inhibitors: synthetic lethality in the clinic. Science 355, 1152–1158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu R, Pal J, Buon L, Nanjappa P, Shi J, Fulciniti M, Tai YT, Guo L, Yu M, Gryaznov S et al (2014) Targeting homologous recombination and telomerase in Barrett's adenocarcinoma: impact on telomere maintenance, genomic instability and tumor growth. Oncogene 33, 1495–1505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luksza M, Riaz N, Makarov V, Balachandran VP, Hellmann MD, Solovyov A, Rizvi NA, Merghoub T, Levine AJ, Chan TA et al (2017) A neoantigen fitness model predicts tumour response to checkpoint blockade immunotherapy. Nature 551, 517–520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malvezzi M, Carioli G, Bertuccio P, Boffetta P, Levi F, La Vecchia C and Negri E (2018) European cancer mortality predictions for the year 2018 with focus on colorectal cancer. Ann Oncol 29, 1016–1022. [DOI] [PubMed] [Google Scholar]
- Manic G, Signore M, Sistigu A, Russo G, Corradi F, Siteni S, Musella M, Vitale S, De Angelis ML, Pallocca M et al (2018) CHK1‐targeted therapy to deplete DNA replication‐stressed, p53‐deficient, hyperdiploid colorectal cancer stem cells. Gut 67, 903–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcus L, Lemery SJ, Khasar S, Wearne E, Helms WS, Yuan W, He K, Cao X, Yu J, Zhao H et al (2017) FDA approval summary: TAS‐102. Clin Cancer Res 23, 2924–2927. [DOI] [PubMed] [Google Scholar]
- Marechal A, Zou L (2013) DNA damage sensing by the ATM and ATR kinases. Cold Spring Harb Perspect Biol 5, a012716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marechal A and Zou L (2015) RPA‐coated single‐stranded DNA as a platform for post‐translational modifications in the DNA damage response. Cell Res 25, 9–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maya‐Mendoza A, Moudry P, Merchut‐Maya JM, Lee M, Strauss R and Bartek J (2018) High speed of fork progression induces DNA replication stress and genomic instability. Nature 559, 279–284. [DOI] [PubMed] [Google Scholar]
- McCabe N, Turner NC, Lord CJ, Kluzek K, Bialkowska A, Swift S, Giavara S, O'Connor MJ, Tutt AN, Zdzienicka MZ et al (2006) Deficiency in the repair of DNA damage by homologous recombination and sensitivity to poly(ADP‐ribose) polymerase inhibition. Cancer Res 66, 8109–8115. [DOI] [PubMed] [Google Scholar]
- McGranahan N, Furness AJ, Rosenthal R, Ramskov S, Lyngaa R, Saini SK, Jamal‐Hanjani M, Wilson GA, Birkbak NJ, Hiley CT et al (2016) Clonal neoantigens elicit T cell immunoreactivity and sensitivity to immune checkpoint blockade. Science 351, 1463–1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Min A, Im SA, Jang H, Kim S, Lee M, Kim DK, Yang Y, Kim HJ, Lee KH, Kim JW et al (2017) AZD6738, A novel oral inhibitor of ATR, induces synthetic lethality with ATM deficiency in gastric cancer cells. Mol Cancer Ther 16, 566–577. [DOI] [PubMed] [Google Scholar]
- Mittica G, Ghisoni E, Giannone G, Genta S, Aglietta M, Sapino A, Valabrega G (2018) PARP inhibitors in ovarian cancer. Recent Pat Anticancer Drug Discov 13, 392–410. [DOI] [PubMed] [Google Scholar]
- Modrich P (2006) Mechanisms in eukaryotic mismatch repair. J Biol Chem 281, 30305–30309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munshi PN, Lubin M and Bertino JR (2014) 6‐thioguanine: a drug with unrealized potential for cancer therapy. Oncologist 19, 760–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nacson J, Krais JJ, Bernhardy AJ, Clausen E, Feng W, Wang Y, Nicolas E, Cai KQ, Tricarico R, Hua X et al (2018) BRCA1 mutation‐specific responses to 53BP1 loss‐induced homologous recombination and PARP inhibitor resistance. Cell Rep 24, 3513–3527.e3517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama M (2014) Antigen presentation by MHC‐dressed cells. Front Immunol 5, 672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- NCI (2017) Surveillance, Epidemiology, and End Results (SEER) Program Populations (1969‐2016). Retrieved April 1, 2018, from www.seer.cancer.gov/popdata.
- Network CGA (2012) Comprehensive molecular characterization of human colon and rectal cancer. Nature 487, 330–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niv Y (2007) Microsatellite instability and MLH1 promoter hypermethylation in colorectal cancer. World J Gastroenterol 13, 1767–1769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overman MJ, Lonardi S, Wong KYM, Lenz HJ, Gelsomino F, Aglietta M, Morse MA, Van Cutsem E, McDermott R, Hill A et al (2018) Durable clinical benefit with Nivolumab Plus Ipilimumab in DNA mismatch repair‐deficient/microsatellite instability‐high metastatic colorectal cancer. J Clin Oncol 36, 773–779. [DOI] [PubMed] [Google Scholar]
- Park WJ, Bae SU, Heo YR, Jung SJ and Lee JH (2017) Telomere shortening in non‐tumorous and tumor mucosa is independently related to colorectal carcinogenesis in precancerous lesions. Int J Mol Epidemiol Genet 8, 53–58. [PMC free article] [PubMed] [Google Scholar]
- Pearlman R, Frankel WL, Swanson B, Zhao W, Yilmaz A, Miller K, Bacher J, Bigley C, Nelsen L, Goodfellow PJ et al (2017) Prevalence and spectrum of germline cancer susceptibility gene mutations among patients with early‐onset colorectal cancer. JAMA Oncol 3, 464–471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peasland A, Wang LZ, Rowling E, Kyle S, Chen T, Hopkins A, Cliby WA, Sarkaria J, Beale G, Edmondson RJ et al (2011) Identification and evaluation of a potent novel ATR inhibitor, NU6027, in breast and ovarian cancer cell lines. Br J Cancer 105, 372–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perkhofer L, Schmitt A, Romero Carrasco MC, Ihle M, Hampp S, Ruess DA, Hessmann E, Russell R, Lechel A, Azoitei N et al (2017) ATM deficiency generating genomic instability sensitizes pancreatic ductal adenocarcinoma cells to therapy‐induced DNA damage. Cancer Res 77, 5576–5590. [DOI] [PubMed] [Google Scholar]
- Pishvaian MJ, Slack RS, Jiang W, He AR, Hwang JJ, Hankin A, Dorsch‐Vogel K, Kukadiya D, Weiner LM, Marshall JL et al (2018) A phase 2 study of the PARP inhibitor veliparib plus temozolomide in patients with heavily pretreated metastatic colorectal cancer. Cancer 124, 2337–2346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Planck M, Ericson K, Piotrowska Z, Halvarsson B, Rambech E and Nilbert M (2003) Microsatellite instability and expression of MLH1 and MSH2 in carcinomas of the small intestine. Cancer 97, 1551–1557. [DOI] [PubMed] [Google Scholar]
- Popat S, Hubner R and Houlston RS (2005) Systematic review of microsatellite instability and colorectal cancer prognosis. J Clin Oncol 23, 609–618. [DOI] [PubMed] [Google Scholar]
- Qi Z, Redding S, Lee JY, Gibb B, Kwon Y, Niu H, Gaines WA, Sung P and Greene EC (2015) DNA sequence alignment by microhomology sampling during homologous recombination. Cell 160, 856–869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quigley D, Alumkal JJ, Wyatt AW, Kothari V, Foye A, Lloyd P, Aggarwal R, Kim W, Lu E, Schwartzman J et al (2017) Analysis of circulating cell‐free DNA identifies multiclonal heterogeneity of BRCA2 reversion mutations associated with resistance to PARP inhibitors. Cancer Discov 7, 999–1005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajesh P, Litvinchuk AV, Pittman DL and Wyatt MD (2011) The homologous recombination protein RAD51D mediates the processing of 6‐thioguanine lesions downstream of mismatch repair. Mol Cancer Res 9, 206–214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranjha L, Howard SM, Cejka P (2018) Main steps in DNA double‐strand break repair: an introduction to homologous recombination and related processes. Chromosoma 127, 187–214. [DOI] [PubMed] [Google Scholar]
- Ray Chaudhuri A, Callen E, Ding X, Gogola E, Duarte AA, Lee JE, Wong N, Lafarga V, Calvo JA, Panzarino NJ et al (2016) Replication fork stability confers chemoresistance in BRCA‐deficient cells. Nature 535, 382–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray B, Gupta B, Mehrotra R (2018) Binding of platinum derivative, oxaliplatin to deoxyribonucleic acid: structural insight into antitumor action. J Biomol Struct Dyn, 1–24. [DOI] [PubMed] [Google Scholar]
- Reaper PM, Griffiths MR, Long JM, Charrier JD, Maccormick S, Charlton PA, Golec JM and Pollard JR (2011) Selective killing of ATM‐ or p53‐deficient cancer cells through inhibition of ATR. Nat Chem Biol 7, 428–430. [DOI] [PubMed] [Google Scholar]
- Reinhardt HC, Hasskamp P, Schmedding I, Morandell S, van Vugt MA, Wang X, Linding R, Ong SE, Weaver D, Carr SA et al (2010) DNA damage activates a spatially distinct late cytoplasmic cell‐cycle checkpoint network controlled by MK2‐mediated RNA stabilization. Mol Cell 40, 34–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rondinelli B, Gogola E, Yucel H, Duarte AA, van de Ven M, van der Sluijs R, Konstantinopoulos PA, Jonkers J, Ceccaldi R, Rottenberg S et al (2017) EZH2 promotes degradation of stalled replication forks by recruiting MUS81 through histone H3 trimethylation. Nat Cell Biol 19, 1371–1378. [DOI] [PubMed] [Google Scholar]
- Sarshekeh AM, Overman MJ and Kopetz S (2018) Nivolumab in the treatment of microsatellite instability high metastatic colorectal cancer. Future Oncol 14, 1869–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato H, Niimi A, Yasuhara T, Permata TBM, Hagiwara Y, Isono M, Nuryadi E, Sekine R, Oike T, Kakoti S et al (2017) DNA double‐strand break repair pathway regulates PD‐L1 expression in cancer cells. Nat Commun 8, 1751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarpa M, Ruffolo C, Canal F, Basato S, Erroi F, Fiorot A, Dall'Agnese L, Pozza A, Porzionato A, Castagliuolo I et al (2015) Mismatch repair gene defects in sporadic colorectal cancer enhance immune surveillance. Oncotarget 6, 43472–43482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitt A, Knittel G, Welcker D, Yang TP, George J, Nowak M, Leeser U, Buttner R, Perner S, Peifer M et al (2017) ATM deficiency is associated with sensitivity to PARP1‐ and ATR inhibitors in lung adenocarcinoma. Cancer Res 77, 3040–3056. [DOI] [PubMed] [Google Scholar]
- Sen T, Tong P, Stewart CA, Cristea S, Valliani A, Shames DS, Redwood AB, Fan YH, Li L, Glisson BS et al (2017) CHK1 inhibition in small‐cell lung cancer produces single‐agent activity in biomarker‐defined disease subsets and combination activity with Cisplatin or Olaparib. Cancer Res 77, 3870–3884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelton JW, Waxweiler TV, Landry J, Gao H, Xu Y, Wang L, El‐Rayes B and Shu HK (2013) In vitro and in vivo enhancement of chemoradiation using the oral PARP inhibitor ABT‐888 in colorectal cancer cells. Int J Radiat Oncol Biol Phys 86, 469–476. [DOI] [PubMed] [Google Scholar]
- Shi Q, Shen LY, Dong B, Fu H, Kang XZ, Yang YB, Dai L, Yan WP, Xiong HC, Liang Z et al (2018) The identification of the ATR inhibitor VE‐822 as a therapeutic strategy for enhancing cisplatin chemosensitivity in esophageal squamous cell carcinoma. Cancer Lett 432, 56–68. [DOI] [PubMed] [Google Scholar]
- Short JM, Liu Y, Chen S, Soni N, Madhusudhan MS, Shivji MK and Venkitaraman AR (2016) High‐resolution structure of the presynaptic RAD51 filament on single‐stranded DNA by electron cryo‐microscopy. Nucleic Acids Res 44, 9017–9030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Signon L, Malkova A, Naylor ML, Klein H and Haber JE (2001) Genetic requirements for RAD51‐ and RAD54‐independent break‐induced replication repair of a chromosomal double‐strand break. Mol Cell Biol 21, 2048–2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suenaga M, Schirripa M, Cao S, Zhang W, Yang D, Murgioni S, Rossini D, Marmorino F, Mennitto A, Ning Y et al (2017) Genetic variants of DNA repair‐related genes predict efficacy of TAS‐102 in patients with refractory metastatic colorectal cancer. Ann Oncol 28, 1015–1022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sultana R, Abdel‐Fatah T, Perry C, Moseley P, Albarakti N, Mohan V, Seedhouse C, Chan S and Madhusudan S (2013) Ataxia telangiectasia mutated and Rad3 related (ATR) protein kinase inhibition is synthetically lethal in XRCC1 deficient ovarian cancer cells. PLoS ONE 8, e57098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun LL, Yang RY, Li CW, Chen MK, Shao B, Hsu JM, Chan LC, Yang Y, Hsu JL, Lai YJ et al (2018) Inhibition of ATR downregulates PD‐L1 and sensitizes tumor cells to T cell‐mediated killing. Am J Cancer Res 8, 1307–1316. [PMC free article] [PubMed] [Google Scholar]
- Sunada S, Nakanishi A and Miki Y (2018) Crosstalk of DNA double‐strand break repair pathways in poly(ADP‐ribose) polymerase inhibitor treatment of breast cancer susceptibility gene 1/2‐mutated cancer. Cancer Sci 109, 893–899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarsounas M, Munoz P, Claas A, Smiraldo PG, Pittman DL, Blasco MA and West SC (2004) Telomere maintenance requires the RAD51D recombination/repair protein. Cell 117, 337–347. [DOI] [PubMed] [Google Scholar]
- Thibodeau SN, Bren G and Schaid D (1993) Microsatellite instability in cancer of the proximal colon. Science 260, 816–819. [DOI] [PubMed] [Google Scholar]
- Toledo LI, Murga M, Zur R, Soria R, Rodriguez A, Martinez S, Oyarzabal J, Pastor J, Bischoff JR and Fernandez‐Capetillo O (2011) A cell‐based screen identifies ATR inhibitors with synthetic lethal properties for cancer‐associated mutations. Nat Struct Mol Biol 18, 721–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vanderstichele A, Busschaert P, Olbrecht S, Lambrechts D and Vergote I (2017) Genomic signatures as predictive biomarkers of homologous recombination deficiency in ovarian cancer. Eur J Cancer 86, 5–14. [DOI] [PubMed] [Google Scholar]
- Vendetti FP, Karukonda P, Clump DA, Teo T, Lalonde R, Nugent K, Ballew M, Kiesel BF, Beumer JH, Sarkar SN et al (2018) ATR kinase inhibitor AZD6738 potentiates CD8+ T cell‐dependent antitumor activity following radiation. J Clin Invest 128, 3926–3940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkatesan S, Natarajan AT and Hande MP (2015) Chromosomal instability–mechanisms and consequences. Mutat Res, Genet Toxicol Environ Mutagen 793, 176–184. [DOI] [PubMed] [Google Scholar]
- Vilar E, Bartnik CM, Stenzel SL, Raskin L, Ahn J, Moreno V, Mukherjee B, Iniesta MD, Morgan MA, Rennert G et al (2011) MRE11 deficiency increases sensitivity to poly(ADP‐ribose) polymerase inhibition in microsatellite unstable colorectal cancers. Cancer Res 71, 2632–2642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Gong J, Tu TY, Lee PP and Fakih M (2018) Immune profiling of microsatellite instability‐high and polymerase epsilon (POLE)‐mutated metastatic colorectal tumors identifies predictors of response to anti‐PD‐1 therapy. J Gastrointest Oncol 9, 404–415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Jette N, Moussienko D, Bebb DG and Lees‐Miller SP (2017) ATM‐deficient colorectal cancer cells are sensitive to the PARP inhibitor Olaparib. Transl Oncol 10, 190–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe S, Watanabe K, Akimov V, Bartkova J, Blagoev B, Lukas J and Bartek J (2013) JMJD1C demethylates MDC1 to regulate the RNF8 and BRCA1‐mediated chromatin response to DNA breaks. Nat Struct Mol Biol 20, 1425–1433. [DOI] [PubMed] [Google Scholar]
- Wolff RK, Hoffman MD, Wolff EC, Herrick JS, Sakoda LC, Samowitz WS, Slattery ML (2018) Mutation analysis of adenomas and carcinomas of the colon: early and late drivers. Genes Chromosom Cancer 57, 366–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu G, Chapman JR, Brandsma I, Yuan J, Mistrik M, Bouwman P, Bartkova J, Gogola E, Warmerdam D, Barazas M et al (2015) REV7 counteracts DNA double‐strand break resection and affects PARP inhibition. Nature 521, 541–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu K, Song X, Chen Z, Qin C and He Y (2014) XRCC2 rs3218536 polymorphism decreases the sensitivity of colorectal cancer cells to poly(ADP‐ribose) polymerase 1 inhibitor. Oncol Lett 8, 1222–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu J, Zhao L, Xu Y, Zhao W, Sung P and Wang HW (2017) Cryo‐EM structures of human RAD51 recombinase filaments during catalysis of DNA‐strand exchange. Nat Struct Mol Biol 24, 40–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yaeger R, Chatila WK, Lipsyc MD, Hechtman JF, Cercek A, Sanchez‐Vega F, Jayakumaran G, Middha S, Zehir A, Donoghue MTA et al (2018) Clinical sequencing defines the genomic landscape of metastatic colorectal cancer. Cancer Cell 33, 125–136.e123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan T, Berry SE, Desai AB and Kinsella TJ (2003) DNA mismatch repair (MMR) mediates 6‐thioguanine genotoxicity by introducing single‐strand breaks to signal a G2‐M arrest in MMR‐proficient RKO cells. Clin Cancer Res 9, 2327–2334. [PubMed] [Google Scholar]
- Yazinski SA, Comaills V, Buisson R, Genois MM, Nguyen HD, Ho CK, Todorova Kwan T, Morris R, Lauffer S, Nussenzweig A et al (2017) ATR inhibition disrupts rewired homologous recombination and fork protection pathways in PARP inhibitor‐resistant BRCA‐deficient cancer cells. Genes Dev 31, 318–332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- You Z and Bailis JM (2010) DNA damage and decisions: CtIP coordinates DNA repair and cell cycle checkpoints. Trends Cell Biol 20, 402–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan L, Chi Y, Chen W, Chen X, Wei P, Sheng W, Zhou X and Shi D (2015) Immunohistochemistry and microsatellite instability analysis in molecular subtyping of colorectal carcinoma based on mismatch repair competency. Int J Clin Exp Med 8, 20988–21000. [PMC free article] [PubMed] [Google Scholar]
- Zeinalian M, Hashemzadeh‐Chaleshtori M, Salehi R and Emami MH (2018) Clinical aspects of microsatellite instability testing in colorectal cancer. Adv Biomed Res 7, 28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng L, Beggs RR, Cooper TS, Weaver AN and Yang ES (2017) Combining Chk1/2 inhibition with Cetuximab and radiation enhances in vitro and in vivo cytotoxicity in head and neck squamous cell carcinoma. Mol Cancer Ther 16, 591–600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou L and Elledge SJ (2003) Sensing DNA damage through ATRIP recognition of RPA‐ssDNA complexes. Science 300, 1542–1548. [DOI] [PubMed] [Google Scholar]