

Abstract
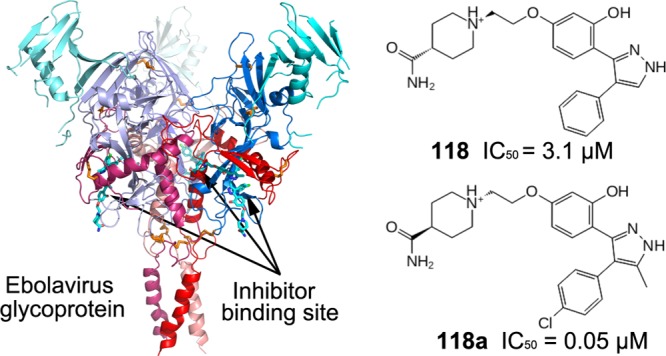
Potent Ebolavirus (EBOV) inhibitors will help to curtail outbreaks such as that which occurred in 2014–16 in West Africa. EBOV has on its surface a single glycoprotein (GP) critical for viral entry and membrane fusion. Recent high-resolution complexes of EBOV GP with a variety of approved drugs revealed that binding to a common cavity prevented fusion of the virus and endosomal membranes, inhibiting virus infection. We performed docking experiments, screening a database of natural compounds to identify those likely to bind at this site. Using both inhibition assays of HIV-1-derived pseudovirus cell entry and structural analyses of the complexes of the compounds with GP, we show here that two of these compounds attach in the common binding cavity, out of eight tested. In both cases, two molecules bind in the cavity. The two compounds are chemically similar, but the tighter binder has an additional chlorine atom that forms good halogen bonds to the protein and achieves an IC50 of 50 nM, making it the most potent GP-binding EBOV inhibitor yet identified, validating our screening approach for the discovery of novel antiviral compounds.
Introduction
Ebola hemorrhagic fever, a deadly disease infecting both human and nonhuman primates, is caused by the highly virulent negative-stranded RNA, membrane-enveloped filovirus—Ebolavirus (EBOV). The 2014–16 West African outbreak claimed over 11 000 lives because suitable therapeutics were not available. The membrane envelope of EBOV is decorated by trimers of glycoprotein (GP), each monomer of which is cleaved by furin into two polypeptides, GP1 and GP2. GP is solely responsible for host cell attachment, endosomal entry, and membrane fusion,1−8 making it an obvious target for therapeutic intervention. A large number of Food and Drug Administration (FDA)-approved drugs have been found to be active against EBOV infection in vitro using either EBOV or pseudotyped virus assays;9−17 however, the precise mechanisms of inhibition remain largely unknown. We have recently demonstrated, using X-ray crystallography, that nine such drugs (Figure S1) interact directly with EBOV GP.18−20 The approved drugs bind in a cavity between the attachment (GP1) and fusion (GP2) subunits, stabilized by predominantly hydrophobic interactions. The cavity lies at the entrance to a large tunnel linking to equivalent tunnels from the other monomers of the trimer at the threefold axis. Residues lining the binding site are highly conserved among filoviruses, with the exception of Marburg viruses (MARVs). The cavity is occupied by residues 192–194 (DFF lid, which immediately follow the putative cathepsin B/L cleavage site) in the apo structure of the GP. Inhibitor binding expels the DFF lid from the cavity, reducing the stability of the protein as judged by its melting temperature. These results suggested that inhibitor binding might trigger the premature release of GP2, preventing fusion between the viral and endosome membranes. Alternatively because inhibitor binding alters the conformation of the cathepsin B/L cleavage site, it might inhibit cleavage, preventing removal of the glycan cap domain, thus blocking the engagement of GP with its receptor NPC1.19,20
The discovery of an inhibitor-binding site on EBOV GP offers opportunities for structure-based drug design against EBOV. Natural compounds have been shown to be effective against different stages of viral infection21,22 and have considerable structural diversity and remain a major source of new drugs. We have therefore performed structure-based in silico screening of a traditional Chinese medicine (TCM) database against EBOV GP to identify novel drug leads. This approach has been combined with thermal shift assays, pseudovirus entry assays, and crystallography to identify and validate potential inhibitors. Our study reveals that although the predictive power of the in silico screening is limited, it still identified two novel compounds (out of eight tested) that display inhibitory activity, as confirmed by pseudovirus entry assays and proof of binding from crystallography. Indeed, one of these compounds appears to be the most potent GP binder yet identified.
Results
Virtual Screening Method Validation Using a Set of Known Binders
Our previous work provides a set of eight drugs known to inhibit EBOV by direct interaction with the GP (Figure S1).18−20 To validate the docking methods, all drugs were subjected to a virtual screening workflow, which we established using the Schrödinger suite (http://www.schrodinger.com/). The IC50 values of the drugs against EBOV10 were converted to pIC50 (−log IC50) values and together with the docking scores Glide XP23,24 and quantum mechanics-polarized ligand docking (QPLD)25 are listed in Table S1. The correlation between the docking scores and pIC50 values is shown in Figure 1. Docking scores of Glide XP show only very weak correlation to experimental pIC50 values (R2 = 0.18), whereas docking scores of QPLD have better correlation to the experimental pIC50 values (R2 = 0.51), although, given the small number of compounds tested, this result is not in itself robust. We think that the relatively poor correlation between the docking scores and the experimental pIC50 values may be attributed to the conformational flexibility of the side chains within the binding pocket of the GP, which cannot be accounted for by the docking program. Nevertheless, this validation method suggests that our virtual screening workflow combining Glide XP docking and QPLD may be able to select binding compounds for the target Ebola GP (although both false positives and negatives would also be expected). As QPLD Emodel scores show some correlation with the experimental IC50, we used these to rank screened compounds for conducting further experiments (QPLD was also found to be useful for the prediction of binding to another viral protein, suggesting general utility26).
Figure 1.

Correlation between the docking score and IC50 for eight known EBOV GP binders. (A) Plot of Glide XP Emodel against pIC50. (B) Plot of QPLD Emodel against pIC50.
Virtual Screening of Novel Natural Compound Inhibitors
To identify novel inhibitors of Ebola GP, we screened the ZINC natural compound library from the TCM database.27 Out of nearly 2.5 million compounds, high-throughput virtual screening (HTVS) selected 416 compounds for subsequent docking calculations. Among these candidates, 88 compounds were selected based on their Glide XP docking and ligand efficiency scores using scores from a known inhibitor (toremifene) as cutoff values. QPLD calculations further reduced the list to 16 compounds, all of which have passed filters for pan assay interference compounds.28 A total of eight of these best-scoring compounds were purchased for in vitro experiments and crystallographic studies based on availability and price (Figure 2 and Table S2).
Figure 2.

Natural compounds selected from the in silico screen results for experimental evaluations. Toremifene is included as a reference. (A) ZINC32540717 (118), 1-{2-[3-hydroxy-4-(4-phenyl-1H-pyrazol-3-yl)phenoxy]ethyl}piperidine-4-carboxamide. (B) ZINC00167626, 5-amino-2-{[3-(trifluoromethyl)phenyl]sulfanyl}benzonitrile. (C) ZINC09410451 (118a), 1-[2-[(4Z)-4-[4-(4-chlorophenyl)-5-methyl-1,2-dihydropyrazol-3-ylidene]-3-oxocyclohexa-1,5-dien-1-yl]oxyethyl]piperidine-4-carboxamide. (D) ZINC00407254, 2-(1-benzofuran-2-yl)-1-(2,4-dihydroxyphenyl)ethan-1-one. (E) ZINC12893941, (2E)-3-(2H-chromen-3-yl)-1-(4-hydroxyphenyl)prop-2-en-1-one. (F) ZINC04772639, (2E)-3-(6-bromo-2,4-dihydro-1,3-benzodioxin-8-yl)-1-(5-fluoro-2-hydroxyphenyl)prop-2-en-1-one. (G) ZINC11865143, 1-(2,4-dihydroxy-6-methylphenyl)-2-(4-methoxyphenyl)ethan-1-one. (H) ZINC00056827, 4-[(4-aminophenyl)disulfanyl]aniline. (I) Toremifene, 2-[4-[(Z)-4-chloro-1,2-diphenylbut-1-enyl]phenoxy]-N,N-dimethylethanamine.
Evaluation of the Virtual Screen Results by Thermal Shift Assay and Crystallography
We first performed thermal shift assays to test if the eight selected compounds could perturb the thermal stability of GP. The results show that compounds ZINC32540717 (118) and ZINC09410451 (118a) [derivatives of natural isoflavones29 interact with the dye (SYPRO Orange) and interfere with the fluorescence emission; the remaining six compounds at pH 5.2 and at 500 μM concentration do not alter the melting temperature of the GP (data not shown). Nevertheless, we then carried out crystal soaking experiments at two compound concentrations, 2.5 and 5.0 mg/mL for all compounds. For each compound, eight crystals were soaked, four at each concentration for 5–20 min. Soaked crystals were mounted in loops and frozen in liquid nitrogen for diffraction data collection on beamlines I04-1 and I24 of Diamond Light Source. High multiplicity X-ray data were collected from crystals soaked with compounds 118 and 118a to 2.05 and 2.30 Å, respectively. |Fo – Fc| difference electron density maps phased with rigid-body refined models based on our previously published GP–bepridil structure, excluding the ligand and water molecules,18 indicated that compounds 118 and 118a bind to GP (Figure 4). No binding was observed for the other six compounds for data collected under the same regime.
Figure 4.

Overall structure of EBOV GP and electron density maps. (A) Surface representation of the trimeric EBOV GP (PDB ID 6HRO). GP1, GP2, and the glycan cap domain are colored in blue, red, and cyan, respectively; for clarity, one GP monomer is in bright color and other two in light colors. The bound inhibitor 118a is shown as cyan sticks. (B) Close-up of the inhibitor-binding site. (C,D) Simulated annealing omit |Fo – Fc| electron density maps contoured at 3σ for bound compounds 118 (C) and 118a (D). In both cases, two inhibitor molecules are bound. There is no density for the pyrazole ring (indicated by a red arrow) of 118 molecule I.
Compounds 118 and 118a Inhibit Ebola Pseudovirus Infection
Compounds 118 and 118a were tested for their ability to inhibit EBOV infection in vitro, using HIV-1-derived pseudoviruses expressing the Ebola virus envelope GPs (EBOV pseudoparticle, EBOVpp) as described previously.19 We used TZM-bl cells in the EBOVpp infection assay because TZM-bl cells contain a β-Gal expression cassette with an HIV-1-induced promoter, infected cells can be identified through the hydrolysis of X-gal.30,31 The best-known inhibitor that directly interacts with EBOV GP, toremifene,18−20 was used as a positive control. Multiple concentrations (0.01–25 μM) of 118, 118a, and toremifene were evaluated in the EBOVpp infection assay, and the experiment was done in triplicate (Figures 3 and S2). The results show that all three compounds inhibit EBOVpp infection and fusion (Figure S2) in a dose-dependent manner. The IC50s derived from the experiment are 3.1 ± 0.02, 0.05 ± 0.01, and 0.09 ± 0.08 μM for 118, 118a, and toremifene, respectively. Thus, 118a has the lowest IC50 among the inhibitors known to bind EBOV GP directly.18−20 Although the experiments were not performed on live EBOV, the relative ranking of inhibition constants is likely to be indicative of relative potency against EBOV.
Figure 3.

Compound 118a is a potent inhibitor of EBOVpp infection in live cells. (A–C) Infectivity assays to recover the IC50 and IC80 with different dilutions for compounds 118a (A), 118 (B), and toremifene (C) were performed using a β-Gal assay. The percentage of infection inhibition in a number of cells per condition is plotted against inhibitor concentration. The error bars show the standard error coming from three independent measurements, and the solid lines show a fit using a sigmoidal mathematical model. The IC50 for 118a is 0.05 ± 0.01, 3.1 ± 0.02 μM for 118, and 0.09 ± 0.08 μM for toremifene.
Overall Structures of GP–118 and GP–118a Complexes
The complexes were refined with good R-factors and stereochemistry (Table 1). The resultant electron density maps unambiguously show the binding of the compounds. In both cases, there are two inhibitor molecules bound in each GP-binding pocket (Figure 4). Hereafter, we name the molecule that binds closest to Y517 as molecule I (this is the most interior and presumably the tighter binder) and that closest to M548 as molecule II.
Table 1. Data Collection and Refinement Statistics.
| GP–118 | GP–118a | |
|---|---|---|
| Data Collection | ||
| space group | R32 | |
| Cell Dimensions | ||
| a, b, c (Å) | 114.2, 114.2, 305.4 | 115.1, 115.1, 307.9 |
| α, β, γ (deg) | 90, 90, 120 | 90, 90, 120 |
| resolution (Å) | 57.1–2.05 (2.09–2.05)a | 83.8–2.30 (2.34–2.30) |
| Rmerge | 0.063 (-) | 0.098 (-) |
| I/σI | 21.1 (1.1) | 36.5 (1.4) |
| completeness (%) | 98.4 (90.4) | 100 (99.9) |
| redundancy | 15.8 (6.0) | 91.0 (16.8) |
| CC1/2 | 1.0 (0.44) | 1.0 (0.75) |
| Refinement | ||
| resolution (Å) | 57.1–2.05 | 83.8–2.30 |
| no. of reflections | 32976/1758 | 33711/1696 |
| Rwork/Rfree | 0.191/0.221 | 0.180/0.211 |
| No. Atoms | ||
| protein | 3045 | 3029 |
| ligand/glycan/ion | 213 | 223 |
| water | 165 | 143 |
| Mean B-Factors | ||
| protein | 66 | 92 |
| ligand/glycan/ion | 104 | 134 |
| water | 54 | 77 |
| rms Deviations | ||
| bond lengths (Å) | 0.005 | 0.003 |
| bond angles (deg) | 0.8 | 0.7 |
Values in parentheses are for the highest-resolution shell.
There is no electron density for the pyrazole ring of 118 molecule I, although other groups of the molecule have reasonably well-defined density (Figure 4C). We initially thought that the density might represent an impurity molecule that is very similar to compound 118 produced during its synthesis. However, the NMR spectrum showed no sign of other molecules in the sample. Because the pyrazole ring can potentially undergo hydrolysis, it is conceivable that for some molecules the pyrazole ring of 118 I is hydrolyzed during crystal soaking.32,33
The overall structures of the protein parts of the GP–118 and GP–118a complexes are very similar to each other, as well as to the previously published GP–drug complex structures.18,19 Apart from some local conformation changes around the binding cavity, the binding of different inhibitors does not introduce significant variations in the overall structure of the protein. For example, by superimposition of these complexes using SHP,34 GP–118a overlaps 386 (out of 388), 382, and 381 Cαs of GP–118, GP–toremifene, and the unliganded GP with root-mean-square deviations of 0.38, 0.53, and 0.55 Å, respectively.
Two Molecules of 118a and 118 Bind in Each Cavity of GP
The inhibitor-binding site of EBOV GP is located between the N-terminus of GP1 and the stem of the GP2 fusion loop (Figure 4). In the apo structure, the inhibitor-binding cavity is occupied by residues 192–194 (the DFF lid) of GP1, which may function to hold the putative cleavage site35−39 in position for the removal of the glycan cap by the host cathepsin B/L—allowing binding of the receptor NPC1 in the late endosome/lysosome.19,40,41 Binding of an inhibitor in the cavity expels the DFF lid. The inhibitor-binding cavity is also the entrance of a tunnel that connects with the corresponding tunnels in the other monomers of the GP trimer at the threefold axis. The β1−β2 hairpin, β3, β6, and β13 of GP1 and the stem of the fusion loop (β19−β20) and α3 of GP2 contribute residues lining the inhibitor-binding pocket (Figures 4B and S3). The volume occupied by F193 and F194 (the FF volume) in apo GP is important for inhibitor binding and is occupied by all inhibitors whose complex structures with GP are known.18−20 In the cases of benztropine and imipramine, this is achieved by two drug molecules, one molecule occupies part of the volume in front of M548 and the other molecule fills the space in front of Y517. Despite 118a and 118 being the largest inhibitors known to bind GP, once again in each case two inhibitor molecules bind (Figures 4 and S3). Compound 118a has a molecular volume of 382 Å3, and, excluding atoms lying outside the cavity, the two bound molecules alone sample about 640 Å3 of the total ∼1000 Å3 volume of the pocket (in contrast, the previous nine inhibitors, with molecular volumes ranging from 188 Å3 for ibuprofen to 362 Å3 for toremifene, in aggregate occupy 878 Å3).
Interactions between EBOV GP and 118a
Molecule I of compound 118a binds with its chlorophenyl ring deep in the subpocket adjacent to residues V66 and A101, making extensive interactions with the side chains of residues V106, A101, L515, and Y517, as well as main-chain atoms of G67 and G102 (Figure 5A). This subpocket is partially occupied by F194 in apo GP, by a phenyl ring in toremifene, and by the benzodioxol ring of paroxetine and the phenyl ring of bepridil in their complex structures. The chlorine atom makes a strong halogen bond with the carbonyl oxygen of G67 with a bond distance of 3.1 Å, C–Cl–O angle of 167°, and Cl–O–C angle of 116° (a classic halogen bonding interaction of chlorine with the backbone Lewis bases at a glycine residue,42Figure 5C). The V66 side chain in this subpocket rotates to avoid clashes on binding. The methylpyrazole group, apart from the close contacts with the corresponding group of molecule II as discussed below, is positioned to make parallel ring stacking interactions with the side chain of Y517 and hydrophobic interactions with L515 and M548. The hydroxyphenoxy ethyl moiety interacts extensively with the side chains of its flanking residues, R64 and T519. The position and orientation of the phenoxy ring are similar to the phenoxy and benzyl ring of bound toremifene and bepridil, respectively. The piperidine carboxamide group extends fully into the tunnel, exploiting protein interactions not used by other known GP binders. Upon binding, residue D522 refolds toward and makes bifurcated hydrogen bonds (of length 3.0 and 3.2 Å) with the nitrogen group of the piperidine ring. In addition, the carboxamide moiety hydrogen-bonds to the side chains of N61 and R587 (from a neighboring monomer) (Figure 5A).
Figure 5.
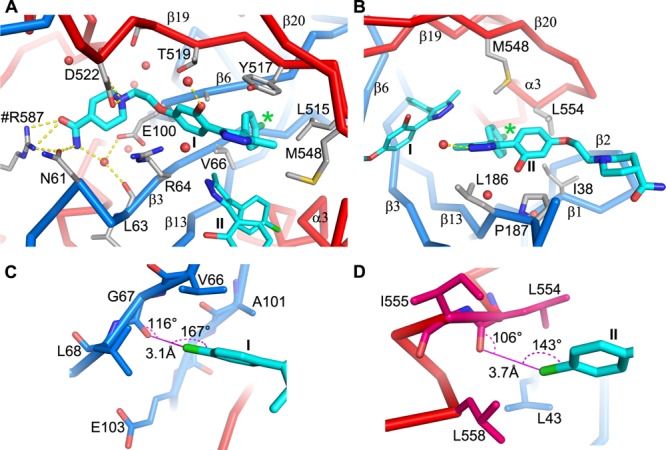
Protein–inhibitor interactions for compound 118a. (A,B) Molecule I (panel A) and molecule II (panel B) in the binding pocket (PDB ID 6HRO). Protein main chains are drawn as thick blue (GP1) and red (GP2) sticks, the inhibitor as cyan sticks, and water molecules as red balls. Protein residues that interact with the inhibitor are shown as gray sticks. Yellow broken sticks represent hydrogen bonds. Residue R587 from a neighboring monomer is labeled with a # prefix. The position of the chlorine atom is indicated by a green *. (C,D) Environment of the chlorine atoms of molecule I (panel C) and molecule II (panel D). Halogen bonds are shown as magenta lines with bond distances and angles labeled. Protein residues are drawn as blue (GP1) and red (GP2) sticks.
Compound 118a molecule II binds the GP with its chlorophenyl group nestled in a subpocket adjacent to α3 interacting with the side chains of I38, L186, M548, and L554 (Figure 5B). This subvolume is occupied by F193 in the apo structure and also by inhibitors in other inhibitor complexes, for example, by a phenyl ring of benztropine molecule A and the isobutoxy group of bepridil. The chlorine atom makes a halogen bond with the carbonyl oxygen of L554 with a bond distance of 3.7 Å, C–Cl–O angle of 143°, and Cl–O–C angle of 106°, although this is less ideal geometry than that seen in molecule I (Figure 5D). The methylpyrazole ring of molecule II is sandwiched between, and extensively contacted by, L186 and the pyrazole ring of molecule I. The methyl group contacts all five nonhydrogen atoms of the pyrazole ring of molecule I, and the interactions are so intimate (separation 3.2–3.8 Å) that the electron density of the two groups is connected even at high contour level (Figure 4D), which may not be favorable for binding. The hydroxyphenoxy group of 118a molecule II makes off-center ring stacking interactions with P187 and contacts the side chain of M548. The hydroxyphenoxy group is also protected from the solvent by the main chain of residues 189–191, the putative cathepsin B/L cleavage site, which becomes partially ordered in the complex. The piperidine carboxamide moiety hangs out of the binding cavity and has weak electron density.
Several water molecules are trapped in the binding cavity. Three have direct interactions with the inhibitor, the first bridges interactions from the carboxamide to the carboxyl group of E100 and carbonyl oxygen of L63, the second hydrogen-bonds to the hydroxyl oxygen of the hydroxylphenoxy, and the third hydrogen-bonds to the pyrazole ring of molecule II.
Interactions between EBOV GP and 118
Compared to 118a, compound 118 has phenyl and pyrazole groups instead of the chlorophenyl and methylpyrazole groups. As noted above, the pyrazole ring of 118 molecule I appears to be hydrolyzed. In addition, the hydroxyl group of the hydroxyphenoxy moiety may be modified because there is extra density connected to this group. The rest of 118 molecule I is bound in a very similar fashion to molecule I of 118a; the hydrogen bond interactions from the piperidine carboxamide group and even the nearby water molecules are conserved (Figure 6A). The phenyl ring is positioned similarly in the subpocket adjacent to V66 and A101; however, lacking the chlorine atom, it does not make any interactions with the main chain of G67 and G102. Molecule II of 118 binds in a similar position to molecule II of 118a (Figure 6B); although it lacks the chlorine atom on the phenyl ring and the methyl group on the pyrazole ring, it is positioned slightly deeper in the cavity and closer to molecule I. The phenyl and pyrazole rings make similar interactions with I38, L186, M548, and L554 and with molecule I to those made by molecule II of 118a. The hydroxyphenoxy moiety makes fewer contacts to P187 and has no interactions with the putative cathepsin B/L cleavage site (residues 190–191), which is disordered.
Figure 6.

Protein–inhibitor interactions for compound 118. (A,B) Molecule I (panel A) and molecule II (panel B) in the binding pocket (PDB ID 6HS4). Protein chains, water molecules, and hydrogen bonds are shown as in Figure 5; compound 118 is drawn as orange sticks.
Structural Changes Introduced by Inhibitor Binding
All 11 inhibitors, of considerable chemical diversity,18−20 bind within the same hydrophobic cavity, with affinity derived from shape complementary enabled by small conformational changes in the protein. Superimposition of inhibitor-bound structures on apo GP shows that while inhibitor binding does not introduce significant main-chain structural changes around the major binding area of the cavity (Figure 7A), there are various side-chain rearrangements, most notably for residues V66, M548, L554, and L558. V66 changes conformation to allow binding of different chemical groups in the subpocket adjacent to it, whereas M548, L554, and L558 changes are associated with different shaped groups occupying the subvolume adjacent to α3. The piperidine carboxamide of 118 or 118a and the dimethylethanamine group of toremifene are positioned to make direct interactions with D522 via either hydrogen bonds or hydrophobic contacts and stabilize the N-terminal end of the fusion loop (residues 522–526) in a different conformation to the apo form (for smaller inhibitors, these residues become disordered and show only weak electron density) (Figure 7B). The structural changes, in turn, lead to ordering of two or three residues contributed from the expression vector pNeosec at the N-terminus of GP1.
Figure 7.
Protein structural changes and binding modes of different inhibitors. (A) Structural differences at the inhibitor-binding site of apo GP and 10 GP–inhibitor complexes. Superimpositions were done using the whole GP; the apo structure is shown as thicker gray sticks and inhibitor bound structures as thinner sticks. (B) Piperidine carboxamide group of 118a (red and cyan, PDB ID 6HRO) and the dimethylethanamine group of toremifene (blue and yellow, PDB ID 5JQ7) introduced structural changes at the N-terminus of the fusion loop compared to the apo GP (gray). The bifurcated hydrogen bonds from D522 to the nitrogen atom of the piperidine ring are shown as broken sticks. (C) Comparison of the binding mode of 118 (orange sticks, PDB ID 6HS4) and 118a (cyan sticks, PDB ID 6HRO) in the cavity. (D–G) Comparison of the binding pose of 118a with toremifene (D), bepridil (E, PDB ID 6F5U), thioridazine (F, PDB ID 6G95), and clomipramine (G, PDB ID 6G9I).
Comparison between the Predicted and Observed Binding Modes
The QPLD docking algorithm docked both 118 and 118a compounds at the site corresponding to molecule I of the crystal structure with the phenyl or chlorophenyl ring in the subpocket adjacent to V66 and A101 (Figure 8 and Tables S3 and S4). However, the pyrazole ring in both cases tilts away from Y517. Because the side chain of D522 points away from the binding site in the structure used for docking, the docking program was unable to predict the hydrogen bond interactions to the piperidine ring that requires a side-chain rotation of 180°. The hydrogen bond between the carboxamide group and N61 was predicted for compound 118. We used the structure of GP observed when toremifene binds for the in silico screening. In this structure, the side chain of L554 partially occupies the subpocket adjacent to α3 where the chlorophenyl ring of 118a molecule II (or phenyl ring of 118) is bound, perhaps explaining why the two compounds were not docked at the site.
Figure 8.
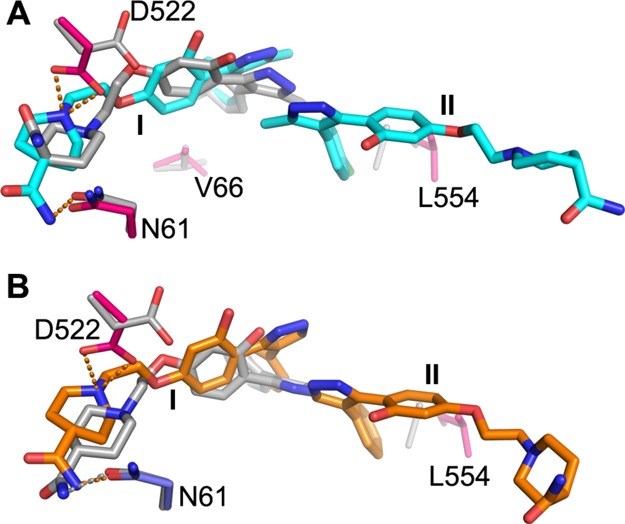
Comparison of the crystal structure and the docked pose. (A) Docked pose of compound 118a is overlaid with the crystal structure of GP–118a. 118a in the crystal structure is shown in cyan and gray for the docked pose; protein side chains that have large conformational differences or hydrogen bond to the inhibitor are shown as blue (GP1) and red (GP2) sticks for the crystal structure and gray sticks for the structure used in docking. (B) Comparison of the docked pose of compound 118 (gray sticks) with the bound mode in the crystal structure (orange sticks). Protein side chains are colored as in (A). The orange broken bonds represent hydrogen bonds.
Discussion and Conclusions
Compound 118a is the best EBOV inhibitor known to directly interact with the viral GP. Its potency is about 2-fold better than toremifene. Two molecules of 118a bind each monomer of GP, whereas only one molecule of toremifene binds each monomer of GP. Molecule I of 118a has stronger electron density and an average B-factor 75% lower than for molecule II and is therefore the major contributor to inhibitory potency. In line with this, molecule I of 118a overlaps well with its predicted binding position and also with bound toremifene and bepridil (Figure 7). By comparing binding modes and potencies, we previously noted that the FF volume and the subvolumes adjacent to V66 and α3 are crucial for binding affinity.18,20 Here, we show the piperidine carboxamide group in molecule I of 118a exploring additional volume inside the tunnel and making hydrogen bonds with the protein. We have previously suggested that substitution of the chlorine atom of toremifene with a benzyl ring to occupy the subvolume adjacent to α3 to mimic the interactions made by clomipramine and thioridazine might improve the potency of toremifene (Figure 7). With knowledge of the GP–118a structure, we suggest that a further modification to toremifene by substituting the dimethylamine group with the piperidine carboxamide group of 118a may greatly increase its potency. Similarly, modifications could also be made to bepridil by replacing the isobutoxy group with a benzyl ring and addition of an oxyethylpiperidine-4-carboxamide group of 118a to its benzyl ring. As more GP complex structures with chemically divergent inhibitors are determined, the knowledge of protein–inhibitor interactions will guide design of potent drugs to combat Ebola virus.
This study reveals that a structure-based high-throughput in silico screen in combination with an inhibitory assay of EBOVpp and crystallography can be an effective way to identify highly potent small-molecule inhibitors effective against EBOV. Because only the top hits of the screen need to be verified experimentally, the method is much more efficient in the requirements of both time and manpower compared to viral or pseudovirus entry assays. Nevertheless, it should be borne in mind that only two of the eight compounds selected by in silico screening showed inhibitory properties. This probably reflects the limited reliability of the scoring functions used and also perhaps the difficulty of predicting binding in the face of extensive side-chain flexibility in the binding cavity. For such flexible targets, we suggest that where a database of potential binding cavity structures is experimentally available, then screening against all possible structures and selecting the best docking score might increase robustness. A fundamental limitation of course will remain—the method can only identify the inhibitors that directly interact with EBOV GP. Despite these caveats, the method used was able to identify two inhibitors of a novel chemical group of EBOV GP inhibitors, one of which represents the most potent known to directly interact with the GP. Given its strong potency, 118a should be tested in vivo using the murine infection model reported by Johansen et al. to determine its protective ability.10
Both inhibitors bind with a unique binding mode, especially their piperidine carboxamide group, exploiting hydrogen bond interactions that have not been seen before, while, in the case of 118a, the presence of strong halide bonding is likely to explain much of the additional potency. The inhibitor-binding cavity of the GP is large and can accommodate various inhibitors with chemically divergent structures so that we believe that features such as the halide bonding might be usefully grafted onto other chemical scaffolds. Such approaches could provide more potent inhibitors to combat EBOV infection; indeed, several compounds have been designed based on these structures and will be made and tested soon.
Experimental Section
Data Collection and Ligand Library Preparation
The natural ligand library comprising about 2.5 million compounds was downloaded from the TCM database@Taiwan.27 Prior to screening of the natural ligand library, a drug candidate list with proven inhibitory activity against EBOV was collected.10 Eight compounds with known IC50 values and known complex structures with EBOV GP were used for the evaluation of our in silico workflow. Molecular structures of all drugs were retrieved from the Drug Bank.43 Ligands were prepared for simulation using the ligprep module from the preparation step of the HTVS workflow of Schrödinger suite (http://www.schrodinger.com/).
Docking Structure Preparation
The crystal structure of EBOV GP in complex with toremifene was taken from the Protein Data Bank (PDB ID 5JQ7).19 The protein structure was preprocessed using the protein preparation wizard of Schrödinger/Maestro 11.1. The processed structure was subjected to energy minimization using the OPLS3 force field in the Impact module. The grid box for docking was created in Glide23,24 by picking toremifene as the center and expanding the box size to cover residues of the whole binding pocket. The final grid box dimensions were 44.9 × 15.6 × 8 Å3.
Virtual Screening and Binding Affinity Calculation
The natural ligand library was subjected to three levels of docking using the virtual screening workflow in Glide.23,24 Each molecule was docked in the HTVS mode, from which the top 10% of the compounds were selected for standard precision (SP) docking, followed by refinement in the extra precision (XP) docking. The last step was crucial to reduce false positives returned from SP and better predict binding poses using a more expensive scoring function. The final docking poses and binding affinities of known inhibitors and candidate compounds returned from XP docking were subjected to QPLD calculations.25 This method combines Glide docking with QSite to redock ligands using quantum mechanically derived partial charges on them in the pocket accounting for the polarization effect from the protein.
Reagents
The eight selected compounds used for evaluation of the virtual screen results were purchased from MolPort with a specified purity of >90%. The high degree of purity of compound 118 was further confirmed by NMR analysis and, together with 118a, demonstrated to be the active component by crystallographic structure determination in complex with EBOV GP.
Ebola Pseudovirus Production and Titration
HIV-1-derived pseudoviruses expressing the Ebola virus envelope GPs (EBOV pseudoparticle, EBOVpp) were produced as described previously.19 HEK-293T cells were seeded in T175 flasks one day prior to transfection. Cells were transfected with 2 μg pR8ΔEnv, 2 μg BlaM-Vpr, 1 μg pcREV, and 3 or 2 μg of plexm-EBOV_GP plasmids (containing Zaire EBOV GP residues 1–676 under control of a β-actin/CMV chimeric promoter). After 10 h of transfection, the medium was replaced by fresh Dulbecco’s modified Eagle’s medium (DMEM) with 10% fetal bovine serum (FBS). Virus-containing medium was collected at 48 and 72 h and passed through a 0.45 μm filter to isolate the viral particles which were then concentrated using the Lenti-X concentrator (Clontech). Virus titers were determined by infecting TZM-bl cells (PTA-5659, no mycoplasma contamination) with a serial dilution of concentrated pseudovirus, followed by a β-Gal assay. Because the TZM-bl cells contain a β-Gal expression cassette with an HIV-1-induced promoter, infected cells can be identified through the hydrolysis of X-gal.31
Infectivity Assay
TZM-bl cells were plated 24 h before the assay at 2 × 104 cells per well in black clear-bottomed 96-well plates. On the day of assay, cells were cooled on ice before the addition of EBOVpp. Viral supernatants were added onto the cells with 1 in 10 dilution, and they were centrifuged at 2100g for 30 min at 4 °C. Viral supernatants were removed, and cells were washed with 1× phosphate-buffered saline (PBS). Then, 100 μL of DMEM plus 5% FBS-containing toremifene, a well-characterized ebola fusion inhibitor,19118a and 118 in a concentration range of 100–0.125 μM, or no drug, was added to each well before placing in a 37 °C, CO2 incubator to initiate viral entry. After 48 h, cells were fixed using 2% paraformaldehyde (PFA) for 20 min, followed by a β-Gal assay. Cells were then imaged using a wide-field Olympus microscope equipped with 20× air objective and transmitted light. All cells in each well were measured and tiled using Cellsens software (Olympus). The relative number of infected cells versus the total population of cells was calculated using an automated algorithm (spot tracker) with Icy software (http://icy.bioimageanalysis.org/).
BlaM Assay and Analysis
The β-lactamase assay30,31 was applied to assess EBOVpp fusion. The procedure was similar to that used for the infectivity assay, except that TZM-bl cells were plated at 4 × 104 cells per well, viral supernatants were added at MOI 0.5, and after removal of the virus supernatant and washing, DMEM plus 10% FBS-containing toremifene, 118 or 118a in a concentration range of 12–0.4 μM, or no drug, was added to each well. After 120 min, cells were loaded with 1× CCF2-AM from the LiveBLAzer FRET—B/G Loading Kit (Life Technologies) and incubated at room temperature in the dark for 2 h. After CCF2-AM removal, cells were washed with 1× PBS and fixed with 2% PFA before viewing. Cells were excited using a 405 nm continuous laser (Leica), and the emission spectra between 430 and 560 nm were recorded pixel by pixel (512 × 512) using a Leica SP8 X-SMD microscope with a 20× objective. The ratio of blue emission (440–480 nm, cleaved CCF2-AM) to green (500–540 nm, uncleaved CCF2-AM) was then calculated pixel by pixel using a customized macro34 for ImageJ (http://imagej.nih.gov/ij/) with 25 different observation fields for each condition. A blue/green threshold (fusion threshold) was set using only media. The fusion threshold was calculated recovering the signal (blue/green intensity ratio) coming from individual cells plus 2 × standard deviation from ∼300 cells in each observation field using a custom-made macro with ImageJ.30 This threshold was then applied to all conditions. Cells above the threshold were pseudocolored in red and cells below the threshold were pseudocolored in blue. “Red” cells were then compared with blue cells (nonfusogenic) as an accurate measure of fusion in different conditions.
Protein Expression, Purification, and Crystallization
The production of Zaire EBOV (strain Mayinga-76) recombinant GP extracellular domain has been described previously.18,19 In brief, the construct contains residues 32–312 and 464–632 of the GP with mutations T42A and H613A and a C-terminal tag of a fold on trimerization sequence from the bacteriophage T4 fibritin and 6 histidines. The construct was cloned in the mammalian expression vector pNeosec44 and then transfected into HEK293T cells with polyethylenimine and supplemented with 5 μM kifunensine (Cayman Chemical). The His-tagged protein from dialyzed conditional media was captured with talon beads, treated with endo-β-acetylglucosaminidase F1, and further purified by size exclusion chromatography. The resulting protein has three amino acids (ETG) from the expression vector pNeosec added at the N-terminus. Crystallization of EBOV GP was performed using microcrystal seeding and the sitting-drop vapor diffusion method as described previously.18,19 Crystals were grown in conditions containing 9% (w/v) PEG 6000 and 0.1 M sodium citrate tribasic dihydrate at pH 5.2.
Thermal Shift Assay
Thermal shift assays were performed using a Mx3005p qPCR machine following exactly the method and protocol described previously.18,19
Crystal Soaking, X-ray Data Collection, and Structure Determination
To obtain GP–inhibitor complexes, the inhibitors were diffused into the GP crystals by soaking. The inhibitors were first dissolved in 100% dimethyl sulfoxide and then diluted with a solution containing 15% (w/v) PEG 6000 and 0.1 M sodium citrate (pH 5.2) to concentrations of 5 and 2.5 mg/mL. Eight crystals (four for each inhibitor concentration) were soaked for each inhibitor in the above solutions for different lengths of time, ranging from 2 to 20 min.
The inhibitor-soaked crystals were mounted in loops and then dipped into cryoprotectants containing 75% inhibitor soaking solution and 25% (v/v) glycerol for a couple of seconds before freezing in liquid nitrogen prior to data collection.
All diffraction data were collected at 100 K with a frame size of 0.1° rotation using PILATUS 6M detectors at Diamond Light Source, UK. GP–118 data were acquired on beamline I24 with a beam size of 50 × 50 μm2 and a wavelength of 0.9686 Å. The exposure time per data frame was 0.01 s with 45% beam transmission.45,46 GP–118a data were collected on beamline I04-1 with a beam size of 60 × 50 μm2 and a wavelength of 0.9282 Å. Data (360°) were collected from every crystal that diffracted.
Diffraction images were indexed, integrated, and scaled with the automated data processing program Xia2 using the 3dii or Dials protocols.47,48 Data from each crystal were initially phased with rigid-body refinement using the GP–bepridil structure (PDB ID 6F5U) by omitting the inhibitor and water molecules. The electron density maps calculated at this stage were checked carefully. Only those data sets that gave high-quality electron density for the soaked inhibitors were used for the later structure refinement. Thus, the final data set for GP–118 to 2.05 Å resolution is from a single crystal, while the GP–118a complex to 2.3 Å is merged from five crystals.
Structure refinement used REFMAC549 or PHENIX,50 and models were rebuilt with COOT.51 Data collection and structure refinement statistics are given in Table 1. Structural comparisons used SHP,34 simulated annealing omit electron density maps were calculated with CNS,52 volumes of the drug-binding cavity and drug molecules were calculated with VOLUMES (Robert Esnouf, unpublished), and figures were prepared with PyMOL.53
Acknowledgments
The authors would like to thank Diamond Light Source for beamtime (proposal MX10627) and the staff of beamlines I04-1 and I24 for assistance with crystal testing and data collection. Y.Z. was supported by the Biostruct-X project (283570) funded by the EU seventh Framework Programme (FP7), J.R. by the Wellcome Trust (101122/Z/13/Z), and D.I.S. and E.E.F. by the UK Medical Research Council (MR/N00065X/1). This is a contribution from the UK Instruct Centre. The Wellcome Trust Centre for Human Genetics is supported by the Wellcome Trust (grant 090532/Z/09/Z). F.S. and S.W.I.S. were supported by the University of Macau (grant MYRG2015-00212-FST), and S.P.-P. acknowledges support by the Nuffield Department of Medicine Leadership Fellowship. C.J.S. and C.L. thank the Welcome Trust and Cancer Research UK for funding.
Glossary
Abbreviations
- EBOV
Ebolavirus
- EBOVpp
Ebolavirus pseudoparticle
- FDA
Food and Drug Administration
- GP
glycoprotein
- MARV
Marburgvirus
- VLP
virus-like particle
Supporting Information Available
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.8b01328.
Chemical structures of drugs whose structures with EBOV GP have been reported previously, inhibition of EBOVpp cell entry, comparison of binding modes of inhibitors that have two molecules bound in the GP cavity; Glide and QPLD docking scores of known EBOV GP binders and top hits of the in silico screening, respectively; and coordinates for docked 118 and 118a (PDF)
SMILES (CSV)
Accession Codes
The coordinates and structure factors have been deposited with the RCSB Protein Data Bank under accession codes 6HS4 and 6HRO for GP–118 and GP–118a, respectively. The authors will release the atomic coordinates and experimental data upon article publication.
Author Contributions
F.S. and Y.Z. contributed equally to this work. Y.Z., J.R., and D.I.S. designed the project. F.S. performed in silico study, guided by S.W.I.S. Y.Z. and J.R. determined the structures. M.I., L.A., and S.P.-P. performed inhibitory assay experiments. S. P.-P. analyzed VLP fusion and infection experiments. C.L. and C.J.S. ascertained structures of the compounds by the NMR spectrum. J.R., Y.Z., and D.I.S. analyzed the results and together with E.E.F. and F.S. wrote the manuscript. All authors read and approved the manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Carette J. E.; Raaben M.; Wong A. C.; Herbert A. S.; Obernosterer G.; Mulherkar N.; Kuehne A. I.; Kranzusch P. J.; Griffin A. M.; Ruthel G.; Cin P. D.; Dye J. M.; Whelan S. P.; Chandran K.; Brummelkamp T. R. Ebola virus entry requires the cholesterol transporter Niemann-Pick C1. Nature 2011, 477, 340–343. 10.1038/nature10348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hacke M.; Bjorkholm P.; Hellwig A.; Himmels P.; de Almodovar C. R.; Brugger B.; Wieland F.; Ernst A. M. Inhibition of Ebola virus glycoprotein-mediated cytotoxicity by targeting its transmembrane domain and cholesterol. Nat. Commun. 2015, 6, 7688. 10.1038/ncomms8688. [DOI] [PubMed] [Google Scholar]
- Nanbo A.; Imai M.; Watanabe S.; Noda T.; Takahashi K.; Neumann G.; Halfmann P.; Kawaoka Y. Ebolavirus is internalized into host cells via macropinocytosis in a viral glycoprotein-dependent manner. PLoS Pathog. 2010, 6, e1001121 10.1371/journal.ppat.1001121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saeed M. F.; Kolokoltsov A. A.; Albrecht T.; Davey R. A. Cellular entry of ebola virus involves uptake by a macropinocytosis-like mechanism and subsequent trafficking through early and late endosomes. PLoS Pathog. 2010, 6, e1001110 10.1371/journal.ppat.1001110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takada A.; Robison C.; Goto H.; Sanchez A.; Murti K. G.; Whitt M. A.; Kawaoka Y. A system for functional analysis of Ebola virus glycoprotein. Proc. Natl. Acad. Sci. U.S.A. 1997, 94, 14764–14769. 10.1073/pnas.94.26.14764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aleksandrowicz P.; Marzi A.; Biedenkopf N.; Beimforde N.; Becker S.; Hoenen T.; Feldmann H.; Schnittler H.-J. Ebola virus enters host cells by macropinocytosis and clathrin-mediated endocytosis. J. Infect. Dis. 2011, 204, S957–S967. 10.1093/infdis/jir326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hunt C. L.; Kolokoltsov A. A.; Davey R. A.; Maury W. The Tyro3 receptor kinase Axl enhances macropinocytosis of Zaire ebolavirus. J. Virol. 2011, 85, 334–347. 10.1128/jvi.01278-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulherkar N.; Raaben M.; de la Torre J. C.; Whelan S. P.; Chandran K. The Ebola virus glycoprotein mediates entry via a non-classical dynamin-dependent macropinocytic pathway. Virology 2011, 419, 72–83. 10.1016/j.virol.2011.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansen L. M.; Brannan J. M.; Delos S. E.; Shoemaker C. J.; Stossel A.; Lear C.; Hoffstrom B. G.; Dewald L. E.; Schornberg K. L.; Scully C.; Lehar J.; Hensley L. E.; White J. M.; Olinger G. G. FDA-approved selective estrogen receptor modulators inhibit Ebola virus infection. Sci. Transl. Med. 2013, 5, 190ra79. 10.1126/scitranslmed.3005471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johansen L. M.; DeWald L. E.; Shoemaker C. J.; Hoffstrom B. G.; Lear-Rooney C. M.; Stossel A.; Nelson E.; Delos S. E.; Simmons J. A.; Grenier J. M.; Pierce L. T.; Pajouhesh H.; Lehár J.; Hensley L. E.; Glass P. J.; White J. M.; Olinger G. G. A screen of approved drugs and molecular probes identifies therapeutics with anti-Ebola virus activity. Sci. Transl. Med. 2015, 7, 290ra89. 10.1126/scitranslmed.aaa5597. [DOI] [PubMed] [Google Scholar]
- Kouznetsova J.; Sun W.; Martinez-Romero C.; Tawa G.; Shinn P.; Chen C. Z.; Schimmer A.; Sanderson P.; McKew J. C.; Zheng W.; Garcia-Sastre A. Identification of 53 compounds that block Ebola virus-like particle entry via a repurposing screen of approved drugs. Emerging Microbes Infect. 2014, 3, 1. 10.1038/emi.2014.88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yermolina M. V.; Wang J.; Caffrey M.; Rong L. L.; Wardrop D. J. Discovery, synthesis, and biological evaluation of a novel group of selective inhibitors of filoviral entry. J. Med. Chem. 2011, 54, 765–781. 10.1021/jm1008715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards M. R.; Pietzsch C.; Vausselin T.; Shaw M. L.; Bukreyev A.; Basler C. F. High-throughput minigenome system for identifying small-molecule inhibitors of Ebola virus replication. ACS Infect. Dis. 2015, 1, 380–387. 10.1021/acsinfecdis.5b00053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anantpadma M.; Kouznetsova J.; Wang H.; Huang R.; Kolokoltsov A.; Guha R.; Lindstrom A. R.; Shtanko O.; Simeonov A.; Maloney D. J.; Maury W.; LaCount D. J.; Jadhav A.; Davey R. A. Large-scale screening and identification of novel Ebola virus and Marburg virus entry inhibitors. Antimicrob. Agents Chemother. 2016, 60, 4471–4481. 10.1128/aac.00543-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Basu A.; Li B.; Mills D. M.; Panchal R. G.; Cardinale S. C.; Butler M. M.; Peet N. P.; Majgier-Baranowska H.; Williams J. D.; Patel I.; Moir D. T.; Bavari S.; Ray R.; Farzan M. R.; Rong L.; Bowlin T. L. Identification of a small-molecule entry inhibitor for filoviruses. J. Virol. 2011, 85, 3106–3119. 10.1128/jvi.01456-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng H.; Schafer A.; Soloveva V.; Gharaibeh D.; Kenny T.; Retterer C.; Zamani R.; Bavari S.; Peet N. P.; Rong L. Identification of a coumarin-based antihistamine-like small molecule as an anti-filoviral entry inhibitor. Antiviral Res. 2017, 145, 24–32. 10.1016/j.antiviral.2017.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao J.; Rijal P.; Schimanski L.; Tharkeshwar A. K.; Wright E.; Annaert W.; Townsend A. Characterization of an influenza virus pseudotyped with Ebolavirus glycoprotein. J. Virol. 2018, 92, 1–18. 10.1128/JVI.00941-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ren J.; Zhao Y.; Fry E. E.; Stuart D. I. Target identification and mode of action of four chemically divergent drugs against Ebolavirus infection. J. Med. Chem. 2018, 61, 724–733. 10.1021/acs.jmedchem.7b01249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y.; Ren J.; Harlos K.; Jones D. M.; Zeltina A.; Bowden T. A.; Padilla-Parra S.; Fry E. E.; Stuart D. I. Toremifene interacts with and destabilizes the Ebola virus glycoprotein. Nature 2016, 535, 169–172. 10.1038/nature18615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y.; Ren J.; Fry E. E.; Xiao J.; Townsend A. R.; Stuart D. I. Structures of Ebola virus glycoprotein complexes with tricyclic antidepressant and antipsychotic drugs. J. Med. Chem. 2018, 61, 4938–4945. 10.1021/acs.jmedchem.8b00350. [DOI] [PubMed] [Google Scholar]
- Biedenkopf N.; Lange-Grünweller K.; Schulte F. W.; Weisser A.; Müller C.; Becker D.; Becker S.; Hartmann R. K.; Grünweller A. The natural compound silvestrol is a potent inhibitor of Ebola virus replication. Antiviral Res. 2017, 137, 76–81. 10.1016/j.antiviral.2016.11.011. [DOI] [PubMed] [Google Scholar]
- Rebensburg S.; Helfer M.; Schneider M.; Koppensteiner H.; Eberle J.; Schindler M.; Gurtler L.; Brack-Werner R. Potent in vitro antiviral activity of Cistus incanus extract against HIV and Filoviruses targets viral envelope proteins. Sci. Rep. 2016, 6, 20394. 10.1038/srep20394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Friesner R. A.; Banks J. L.; Murphy R. B.; Halgren T. A.; Klicic J. J.; Mainz D. T.; Repasky M. P.; Knoll E. H.; Shelley M.; Perry J. K.; Shaw D. E.; Francis P.; Shenkin P. S. Glide: a new approach for rapid, accurate docking and scoring. 1. Method and assessment of docking accuracy. J. Med. Chem. 2004, 47, 1739–1749. 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- Halgren T. A.; Murphy R. B.; Friesner R. A.; Beard H. S.; Frye L. L.; Pollard W. T.; Banks J. L. Glide: a new approach for rapid, accurate docking and scoring. 2. Enrichment factors in database screening. J. Med. Chem. 2004, 47, 1750–1759. 10.1021/jm030644s. [DOI] [PubMed] [Google Scholar]
- Cho A. E.; Guallar V.; Berne B. J.; Friesner R. Importance of accurate charges in molecular docking: quantum mechanical/molecular mechanical (QM/MM) approach. J. Comput. Chem. 2005, 26, 915–931. 10.1002/jcc.20222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Colibus L.; Wang X.; Spyrou J. A. B.; Kelly J.; Ren J.; Grimes J.; Puerstinger G.; Stonehouse N.; Walter T. S.; Hu Z.; Wang J.; Li X.; Peng W.; Rowlands D. J.; Fry E. E.; Rao Z.; Stuart D. I. More-powerful virus inhibitors from structure-based analysis of HEV71 capsid-binding molecules. Nat. Struct. Mol. Biol. 2014, 21, 282–288. 10.1038/nsmb.2769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C. Y.-C. TCM Database@Taiwan: the world’s largest traditional Chinese medicine database for drug screening in silico. PLoS One 2011, 6, e15939 10.1371/journal.pone.0015939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baell J. B.; Holloway G. A. New substructure filters for removal of pan assay interference compounds (PAINS) from screening libraries and for their exclusion in bioassays. J. Med. Chem. 2010, 53, 2719–2740. 10.1021/jm901137j. [DOI] [PubMed] [Google Scholar]
- Bondarenko S. P. Synthesis of 7-beta-(N,N-dialkylamino)ethoxy derivatives of natural isoflavones and 4-aryl-3-[2-hydroxy-4-beta-(N,N-dialkylamino) ethoxy]phenylpyrazoles based on them. Chem. Nat. Compd. 2013, 49, 36–40. 10.1007/s10600-013-0500-9. [DOI] [Google Scholar]
- Jones D. M.; Padilla-Parra S. Imaging real-time HIV-1 virion fusion with FRET-based biosensors. Sci. Rep. 2015, 5, 13449. 10.1038/srep13449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones D. M.; Padilla-Parra S. The beta-lactamase assay: harnessing a FRET biosensor to analyse viral fusion mechanisms. Sensors 2016, 16, 950. 10.3390/s16070950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sidique S.; Shiryaev S. A.; Ratnikov B. I.; Herath A.; Su Y.; Strongin A. Y.; Cosford N. D. P. Structure-activity relationship and improved hydrolytic stability of pyrazole derivatives that are allosteric inhibitors of West Nile Virus NS2B-NS3 proteinase. Bioorg. Med. Chem. Lett. 2009, 19, 5773–5777. 10.1016/j.bmcl.2009.07.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trukhacheva L. A.; Levina V. I.; Grigor’ev N. B.; Arzamastsev A. P.; Dalinger I. L.; Vatsadze I. A.; Popova G. P.; Shevelev S. A.; Granik V. G. Kinetics of hydrolysis of five-membered C-nitroheterocycles: pyrazole, imidazole, 1,2,4-triazole, and isoxazole derivatives. Russ. Chem. Bull. 2005, 54, 2813–2819. 10.1007/s11172-006-0195-1. [DOI] [Google Scholar]
- Stuart D. I.; Levine M.; Muirhead H.; Stammers D. K. Crystal structure of cat muscle pyruvate kinase at a resolution of 2.6 A. J. Mol. Biol. 1979, 134, 109–142. 10.1016/0022-2836(79)90416-9. [DOI] [PubMed] [Google Scholar]
- Chandran K.; Sullivan N. J.; Felbor U.; Whelan S. P.; Cunningham J. M. Endosomal proteolysis of the Ebola virus glycoprotein is necessary for infection. Science 2005, 308, 1643–1645. 10.1126/science.1110656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flyak A. I.; Shen X.; Murin C. D.; Turner H. L.; David J. A.; Fusco M. L.; Lampley R.; Kose N.; Ilinykh P. A.; Kuzmina N.; Branchizio A.; King H.; Brown L.; Bryan C.; Davidson E.; Doranz B. J.; Slaughter J. C.; Sapparapu G.; Klages C.; Ksiazek T. G.; Saphire E. O.; Ward A. B.; Bukreyev A.; Crowe J. E. Jr. Cross-reactive and potent neutralizing antibody responses in human survivors of natural Ebolavirus infection. Cell 2016, 164, 392–405. 10.1016/j.cell.2015.12.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dube D.; Brecher M. B.; Delos S. E.; Rose S. C.; Park E. W.; Schornberg K. L.; Kuhn J. H.; White J. M. The primed ebolavirus glycoprotein (19-kilodalton GP1,2): sequence and residues critical for host cell binding. J. Virol. 2009, 83, 2883–2891. 10.1128/jvi.01956-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hood C. L.; Abraham J.; Boyington J. C.; Leung K.; Kwong P. D.; Nabel G. J. Biochemical and structural characterization of cathepsin L-processed Ebola virus glycoprotein: implications for viral entry and immunogenicity. J. Virol. 2010, 84, 2972–2982. 10.1128/jvi.02151-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schornberg K.; Matsuyama S.; Kabsch K.; Delos S.; Bouton A.; White J. Role of endosomal cathepsins in entry mediated by the Ebola virus glycoprotein. J. Virol. 2006, 80, 4174–4178. 10.1128/jvi.80.8.4174-4178.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang H.; Shi Y.; Song J.; Qi J.; Lu G.; Yan J.; Gao G. F. Ebola Viral Glycoprotein Bound to Its Endosomal Receptor Niemann-Pick C1. Cell 2016, 164, 258–268. 10.1016/j.cell.2015.12.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y.; Ren J.; Harlos K.; Stuart D. I. Structure of glycosylated NPC1 luminal domain C reveals insights into NPC2 and Ebola virus interactions. FEBS Lett. 2016, 590, 605–612. 10.1002/1873-3468.12089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirimulla S.; Bailey J. B.; Vegesna R.; Narayan M. Halogen interactions in protein-ligand complexes: implications of halogen bonding for rational drug design. J. Chem. Inf. Model. 2013, 53, 2781–2791. 10.1021/ci400257k. [DOI] [PubMed] [Google Scholar]
- Law V.; Knox C.; Djoumbou Y.; Jewison T.; Guo A. C.; Liu Y.; Maciejewski A.; Arndt D.; Wilson M.; Neveu V.; Tang A.; Gabriel G.; Ly C.; Adamjee S.; Dame Z. T.; Han B.; Zhou Y.; Wishart D. S. DrugBank 4.0: shedding new light on drug metabolism. Nucleic Acids Res. 2014, 42, D1091–D1097. 10.1093/nar/gkt1068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y.; Ren J.; Padilla-Parra S.; Fry E. E.; Stuart D. I. Lysosome sorting of beta-glucocerebrosidase by LIMP-2 is targeted by the mannose 6-phosphate receptor. Nat. Commun. 2014, 5, 4321. 10.1038/ncomms5321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen R. L.; Axford D.; Nettleship J. E.; Owens R. J.; Robinson J. I.; Morgan A. W.; Doré A. S.; Lebon G.; Tate C. G.; Fry E. E.; Ren J.; Stuart D. I.; Evans G. Outrunning free radicals in room-temperature macromolecular crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2012, 68, 810–818. 10.1107/s0907444912012553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Owen R. L.; Paterson N.; Axford D.; Aishima J.; Schulze-Briese C.; Ren J.; Fry E. E.; Stuart D. I.; Evans G. Exploiting fast detectors to enter a new dimension in room-temperature crystallography. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2014, 70, 1248–1256. 10.1107/s1399004714005379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winter G.; Lobley C. M. C.; Prince S. M. Decision making in xia2. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2013, 69, 1260–1273. 10.1107/s0907444913015308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterman D. G.; Winter G.; Gildea R. J.; Parkhurst J. M.; Brewster A. S.; Sauter N. K.; Evans G. Diffraction-geometry refinement in the DIALS framework. Acta Crystallogr., Sect. D: Struct. Biol. 2016, 72, 558–575. 10.1107/s2059798316002187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murshudov G. N.; Skubák P.; Lebedev A. A.; Pannu N. S.; Steiner R. A.; Nicholls R. A.; Winn M. D.; Long F.; Vagin A. A. REFMAC5 for the refinement of macromolecular crystal structures. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2011, 67, 355–367. 10.1107/s0907444911001314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janowski P. A.; Moriarty N. W.; Kelley B. P.; Case D. A.; York D. M.; Adams P. D.; Warren G. L. Improved ligand geometries in crystallographic refinement using AFITT in PHENIX. Acta Crystallogr., Sect. D: Struct. Biol. 2016, 72, 1062–1072. 10.1107/s2059798316012225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P.; Cowtan K. Coot: model-building tools for molecular graphics. Acta Crystallogr., Sect. D: Biol. Crystallogr. 2004, 60, 2126–2132. 10.1107/s0907444904019158. [DOI] [PubMed] [Google Scholar]
- Brunger A. T. Version 1.2 of the Crystallography and NMR system. Nat. Protoc. 2007, 2, 2728–2733. 10.1038/nprot.2007.406. [DOI] [PubMed] [Google Scholar]
- DeLano W. L.; Lam J. W.. PyMOL: A communications tool for computational models. Abstracts of Papers of the American Chemical Society, 2005; Vol. 230, pp U1371–U1372.
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.