

Abstract
The incidence of childhood cancer is increasing and recent evidence suggests an association between childhood cancer and environmental exposure to genotoxins. In the present study, the Big Blue transgenic mouse model was used to determine whether specific periods in early life represent windows of vulnerability to mutation induction by genotoxins in mouse liver. Groups of mice were treated with single doses of 120 mg N-ethyl-N-nitrosourea (ENU)/kg body weight or the vehicle either transplacentally to the 18-day-old fetus or at postnatal days (PNDs) 1,8, 15, 42 or 126; the animals were sacrificed 6 weeks after their treatment. The cII mutation assay was performed to determine the mutant frequencies (MFs) in the livers of the mice. Liver cII MFs for both sexes were dependent on the age at which the animals were treated. Perinatal treatment with ENU (either transplacental treatment to the 18-day-old fetus or i.p. injection at PND 1) induced relatively high MFs. However, ENU treatment at PNDs 8 and 15 resulted in the highest mutation induction. The lowest mutation induction occurred in those animals treated as adults (PND 126). For instance, the cII MF for the PND 8 female group was 646 × 10–6 while the MF for female adults was only 145 × 10–6, a more than 4-fold difference. Molecular analysis of the mutants found that A:T → T:A transversions and A:T → G:C transitions characterized the pattern of mutations induced by ENU in both the neonate and adult mice, while the predominate type of mutation in the controls was G:C → A:T. The results indicate that mouse liver is most sensitive to ENU-induced mutation during infancy. This period correlates well with the age-dependent sensitivity to carcinogenicity in mouse liver, suggesting that mutation is an important rate-limiting factor for age-related carcinogenesis.
Keywords: Ethylnitrosourea, Age, Neonate, Mutagenicity, Transgenic mice, Sensitivity
1. Introduction
The protection of children from environmental toxins is a major challenge to modern society because children can be more vulnerable due to their unique exposures and susceptibilities. Childhood exposure to environmental chemicals may contribute to or exacerbate certain chronic and disabling diseases like cancer [1]. Although the causes of cancer in newborns and infants remain unknown, environmental exposure to genotoxins during the perinatal period and susceptibility to these toxins during early development are strong possibilities [2]. Data from the National Cancer Institute’s Surveillance Epidemiology and End Results program suggest that the annual incidence of childhood cancers is increasing and about 12% of the cancer cases were tumors acquired during infancy [3–5].
The mouse model has been used to evaluate factors modifying the carcinogenic response of various tissues to different types of chemical carcinogens. The age at which animals are exposed is the most effective modulator of carcinogenesis in liver and lung, as well as some other tissues. Infancy appears to be the most susceptible period for carcinogenesis in a great variety of tissues [6]. Exposure of newborn (≤24-h-old) and neonatal (≤3-week-old) mice to only a few doses of any one of a number of genotoxic chemical carcinogens results in tumors approximately one year after the treatment. The primary sites of tumor induction are the liver and the lung. For example, polycyclic aromatic hydrocarbons usually do not induce liver cancer when administered to young adult mice, but do so when given to newborn and neonatal animals [7].
Mutations play an important role in the etiology of cancer. Newborn and neonatal mice are highly sensitive to carcinogens that exert tumorigenicity through a genotoxic mechanism [8–11]. Therefore, it is reasonable to suspect that these animals also possess higher sensitivity to the mutagenicity of genotoxins than do adult animals. However, studies on age-related chemical mutagenicity are rare because mutagenesis studies are often conducted using in vitro tests or in vivo tests in which endogenous genes are utilized. Assays that test mutations in endogenous genes often require testing rapidly dividing tissues like lymphocytes, and may not have the capability to detect the effect of age because cells in these tissues are rapidly dividing regardless of age [12]. The recently developed Big Blue transgenic mutation models represent a novel approach for studying mutant frequencies (MFs) and types of mutations in nearly all tissues, thus permitting the direct comparison of cancer incidence with MF [13–15]. In these systems, the chromosomally-integrated λLIZ/lacI or cII gene is used as the target for mutation. Following recovery of the transgene from the tissue(s) of choice, the target sequence is packaged into phage particles, bacteria are infected, and mutants are identified and quantified. In present study, we treated different age Big Blue transgenic mice with N-ethyl-N-nitrosourea (ENU) using a carcinogenesis protocol to explore the age-related mutagenic sensitivity in liver and its relationship to car-cinogenesis.
2. Materials and methods
2.1. Animals and treatments
The recommendations set forth by our Institutional Animal Care and Use Committee for the handling, maintenance, treatment, and sacrifice of the animals were followed. Pregnant female Big Blue C57BL/6 transgenic mice were purchased from Taconic Laboratories (Germantown, NY). These mice had been bred with non-transgenic C3H male mice by the supplier. The B6C3F1 offspring were separated according to sex, pooled, and then distributed to individual litters during the first week following birth. Six to eight same-sex pups were assigned to each foster mother. The ENU was purchased from Sigma (St. Louis, MO) and administered in dimethylsulfoxide (DMSO). Animals were given a single i.p. injection of 120 mg ENU/kg body weight or vehicle control in a volume of 2 ml/kg body weight at various times during their development. For the prenatal treatment groups, five pregnant mice were treated at 18 days of gestation, and at birth the pups were pooled and assigned randomly to the mothers. For the postnatal treatment groups, the pups were pooled and distributed randomly into five groups at birth. Male and female mice received treatment at postnatal days (PNDs) 1, 8, 15, 42, or 126. For male mice, there were 3 concurrent controls. Control 1 was administered DMSO at PND 8 to serve as a control for the prenatal, PNDs 1, 8 and 15 treatment groups. Controls 2 and 3 were treated with DMSO at PNDs 42 and 126 to serve as concurrent controls for those treatment groups. Since we found no significant difference among the male controls, only one group of female mice was treated with DMSO at PND 8 and served as a control for all of the female treatment groups. The animals were sacrificed six weeks after their treatment. The livers were isolated, frozen quickly by using liquid nitrogen, and stored at –80 °C.
2.2. cII Mutant assay
High-molecular-weight genomic DNA was extracted from the livers using a RecoverEase DNA Isolation Kit (Stratagene, La Jolla, CA) and stored at 4 °C until DNA packaging was performed. The packaging of the phage, plating of the packaged DNA samples, and determination of MF were carried out following Stratagene’s procedure fort he λ select-cII mutation detection system for Big Blue rodents. The shuttle vector containing the cII target gene was rescued from total genomic DNA with phage packaging extract (Transpack; Stratagene). The plating was performed with the Escherichia coli host strain G1250. To determine the total titer of packaged phages, G1250 bacteria were mixed with a 1:3000 dilution of phage, plated on TB1 plates, and incubated overnight at 37 °C (nonselective conditions). For mutant selection, the packaged phages were mixed with G1250, plated on TB1 plates and incubated at 24 °C for about 42 h (conditions for cII–selection). At 24 °C, phages with wild-type cII genes undergo lysogenization and infected bacteria become part of the developing lawn, whereas phages with mutated cII genes undergo lytic growth and give rise to plaques. When incubated at 37 °C, phages with wild-type cII genes also undergo a lytic cycle, resulting in plaque formation. The cII MF was calculated as the ratio of the total number of mutant plaques (determined at 24 °C) to the total number of plaques screened (determined at 37 °C). The packaging was repeated until a minimum of approximately 2 × 105 plaque-forming units (pfus) from each liver sample was screened for mutations.
2.3. Sequence analysis of the cII mutants
cII Mutant plaques were selected at random from different animals and replated at low density to verify the mutant phenotype. Single, well-isolated plaques were selected from these plates and transferred to a microcentrifuge tube containing 100 μl of autoclaved distilled water. The tubes were placed in a thermocycler at 99.9 °C for 5min and centrifuged at 1500 g for 5min immediately after the heating. The cII target DNA was amplified by PCR with primers 5′-AAAAAGGGCATCAAATTAACC-3′ and 5′-CCGAAGTTGAGTATTTTTGCTG-3′. For PCR amplification, 10 μl of the supernatant and 0.03 μl of each primer (148 μM) were added to 10 μl of 2× PCR Master Mix (Promega, Madison, WI). The PCR reaction was carried out with the following cycling parameters: a 3 min denaturation at 95 °C; followed by 30 cycles of 30 s at 95 °C, 1 min at 60 °C, and 1 min at 72 °C; with a final extension of 10 min at 72 °C. The PCR products were purified using PCR purification kits (Qiagen, Chatsworth, CA). The cII mutant DNA was sequenced and analyzedwith a CEQDTCS-Quick Start Kit and a CEQ 8000 Genetic Analysis System (Beckman Coulter, Fullerton, CA) according to the manufacturer’s instructions. The primers for cII mutation sequencing were the same as those used for the PCR.
2.4. Statistical analyses
Statistical analyses of MFs were performed using SigmaStat (SPSS Science, Chicago, IL). All MF data are expressed as the mean ± standard error (SE) from different animals. Statistical significance was determined by one-way analysis of variance (ANOVA) followed by Student–Newman–Keuls test for comparison of multiple treatment groups. Since the variance increased with the magnitude of the MF, the data were log-transformed before conducting the analysis. Mutational spectra were compared using the computer program written by Cariello [16] forthe Monte Carlo analysis developed by Adams and Skopek [17].
3. Results
3.1. Determination of appropriate MF sampling time for Big Blue mice treated transplacentally with ENU
A time–course study was conducted to determine the proper sampling time. This time–course study was designed to determine whether the cII transgene is genetically neutral when animals were treated perinatally. Pregnant female mice were treated with a single dose of ENU by i.p. injection 3 days prior to giving birth (18-day-old fetus). Their male pups were sacrificed at different time points. The MFs in the liver cII gene at 6, 12 and 24 weeks after the treatment were 279 ± 55, 271 ± 25, and 290 ± 41 × 10–6, respectively, while the MF in the concurrent control mice were 45 ± 6, 36 ± 13 and 42 ± 6 × 10–6 (Fig. 1). ENU increased MFs in all the time points over the controls (p <0.001), whereas no significant difference was found among the treated animals sampled at the different times (p ≥ 0.79).
Fig. 1.
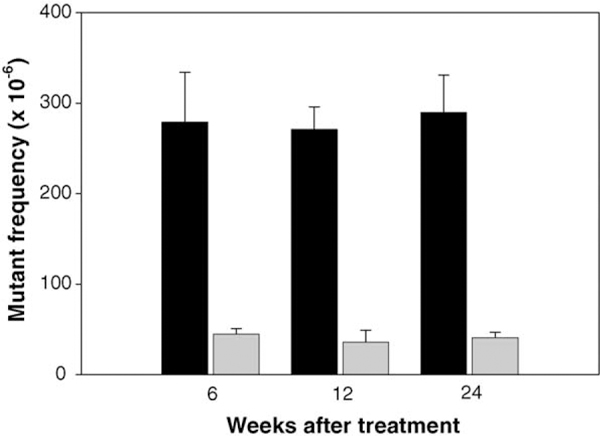
Mutant frequencies in the liver cII gene at different times after dosing male Big Blue mice with ENU transplacentally. The dark bars represent the MFs for the mice treated with ENU and the light bars are the data from the concurrent controls. ENU increased MFs at all of the time points over the controls (p <0.001), whereas no significant difference was found among the treatment groups (p ≥ 0.79).
3.2. Effect of age at treatment on the cII MFs induced by ENU in the liver
Big Blue mice were treated with 120 mg ENU/kg body weight at different ages and sacrificed 6 weeks after the treatment. The livers from the animals of both sexes were assayed for cII gene mutants (Tables 1 and2). Although ENU significantly increased the MFs over the controls at every age (p <0.01 for male and p < 0.001 for female), the level of the MF induction was not the same among the treatment groups. For both sexes, MFs were the highest for mice treated at PNDs 8 and 15 (neonate) and the lowest for mice treated at PND 126 (adult) (p <0.05 for male and p <0.01 for female). ENU also induced higher cII MFs in neonatal mice than at PND 42 (young adult), the difference being significant in males (p<0.05) but not in females (p = 0.18). For the ENU-treated female mice, there were significant differences for the MFs between the neonate and PND 1 (newborn) (p <0.001), as well as between young adult and adult mice (p <0.01). Although the MF for each age group of females was higher than males (Tables 1 and 2), the differences were not significant (p >0.05).
Table 1.
Liver cII mutant frequencies in ENU-treated and control Big Blue male mice
| Group name | Age at treatment (postnatal day) |
Treatmenta | Mouse ID | Total plaques screened (× 103) |
Mutant plaques |
Mutant frequency (×10–6) |
Average MF ± S.E. (×10–6) |
|---|---|---|---|---|---|---|---|
| Control 1b | 8 | DMSO | C6W1M | 330 | 12 | 36 | 45 ± 6 |
| C6W2M | 284 | 10 | 35 | ||||
| C6W3M | 373 | 24 | 64 | ||||
| C6W4M | 301 | 10 | 33 | ||||
| C6W5M | 317 | 18 | 57 | ||||
| Control 2 | 42 | DMSO | C12W1M | 323 | 9 | 28 | 36 ± 13 |
| C12W2M | 428 | 32 | 75 | ||||
| C12W3M | 853 | 16 | 19 | ||||
| C12W4M | 330 | 8 | 24 | ||||
| Control 3 | 126 | DMSO | C24W1M | 287 | 12 | 42 | 41 ± 6 |
| C24W2M | 274 | 17 | 62 | ||||
| C24W3M | 326 | 13 | 40 | ||||
| C24W4M | 309 | 7 | 23 | ||||
| C24W5M | 201 | 8 | 39 | ||||
| PND –3 | –3c | ENU | EP1M | 252 | 112 | 444 | 279 ± 55* |
| EP2M | 274 | 61 | 223 | ||||
| EP3M | 636 | 149 | 234 | ||||
| EP4M | 313 | 67 | 214 | ||||
| PND 1 | 1 | ENU | E1D1M | 223 | 68 | 305 | 206 ± 50* |
| E1D2M | 202 | 32 | 159 | ||||
| E1D3M | 313 | 32 | 102 | ||||
| E1D4M | 357 | 42 | 118 | ||||
| E1D5M | 244 | 84 | 344 | ||||
| PND 8 | 8 | ENU | E8D1M | 561 | 78 | 139 | 442 ± 87*,# |
| E8D2M | 232 | 112 | 483 | ||||
| E8D3M | 308 | 204 | 662 | ||||
| E8D4M | 190 | 76 | 400 | ||||
| E8D5M | 253 | 133 | 526 | ||||
| PND 15 | 15 | ENU | E15D1M | 466 | 149 | 320 | 416 ± 65*,# |
| E15D2M | 353 | 127 | 360 | ||||
| E15D3M | 382 | 99 | 259 | ||||
| E15D4M | 689 | 160 | 232 | ||||
| E15D5M | 265 | 178 | 672 | ||||
| E15D6M | 250 | 154 | 616 | ||||
| E15D7M | 201 | 91 | 452 | ||||
| PND 42 | 42 | ENU | E6W1M | 245 | 27 | 110 | 158 ± 24* |
| E6W2M | 211 | 45 | 213 | ||||
| E6W3M | 292 | 44 | 151 | ||||
| E6W4M | 445 | 46 | 103 | ||||
| E6W5M | 289 | 61 | 211 | ||||
| PND 126 | 126 | ENU | E18W1M | 193 | 19 | 99 | 136 ± 30* |
| E18W2M | 226 | 26 | 115 | ||||
| E18W3M | 213 | 48 | 225 | ||||
| E18W4M | 519 | 55 | 106 | ||||
All mice were sacrificed 6 weeks after the treatment.
Control 1 serves as the concurrent control for PNDs –3, 1, 8 and 15 treatment groups. Controls 2 and 3 serves as the concurrent controls for PNDs 42 and 126, respectively.
The mice were treated transplacentally at 3 days prior to birth (gestational day 18). Five pregnant female mice were used for the treatment.
There is a significant difference between the treatment group and its concurrent control (p < 0.01).
There is a significant difference between the treatment group and mice treated with ENU at PNDs 42 or 126 (p < 0.05).
Table 2.
Liver cII mutant frequencies in ENU-treated and control Big Blue female mice
| Group name | Age at treatment (postnatal day) |
Treatmenta | Mouse ID | Total plaques screened (× 103) |
Mutant plaques |
Mutant frequency (×10–6) |
Average MF ± S.E. (×10–6) |
|---|---|---|---|---|---|---|---|
| Control | 8 | DMSO | C6W1F | 396 | 15 | 38 | 39 ± 6 |
| C6W2F | 412 | 13 | 32 | ||||
| C6W3F | 269 | 12 | 45 | ||||
| C6W4F | 263 | 6 | 23 | ||||
| C6W5F | 193 | 11 | 57 | ||||
| PND –3 | – 3b | ENU | EP1F | 188 | 60 | 319 | 382 ± 59*,# |
| EP2F | 423 | 110 | 260 | ||||
| EP3F | 328 | 174 | 530 | ||||
| EP4F | 245 | 103 | 420 | ||||
| PND 1 | 1 | ENU | E1D1F | 348 | 50 | 143 | 201 ± 32* |
| E1D2F | 387 | 112 | 289 | ||||
| E1D3F | 202 | 33 | 163 | ||||
| E1D4F | 347 | 72 | 207 | ||||
| PND 8 | 8 | ENU | E8D1F | 386 | 193 | 500 | 646 ± 107*,& |
| E8D2F | 315 | 117 | 371 | ||||
| E8D3F | 308 | 233 | 756 | ||||
| E8D4F | 244 | 241 | 988 | ||||
| E8D5F | 248 | 152 | 616 | ||||
| PND 15 | 15 | ENU | E15D1F | 224 | 139 | 620 | 531 ± 44*,& |
| E15D2F | 257 | 127 | 494 | ||||
| E15D3F | 235 | 108 | 460 | ||||
| E15D4F | 274 | 178 | 650 | ||||
| E15D5F | 291 | 126 | 432 | ||||
| PND 42 | 42 | ENU | E6W1F | 301 | 92 | 306 | 388 ± 76* |
| E6W2F | 326 | 75 | 230 | ||||
| E6W3F | 272 | 157 | 577 | ||||
| E6W4F | 280 | 123 | 439 | ||||
| PND 126 | 126 | ENU | E18W1F | 365 | 62 | 170 | 145 ± 7* |
| E18W2F | 339 | 45 | 133 | ||||
| E18W3F | 275 | 38 | 138 | ||||
| E18W4F | 271 | 39 | 144 | ||||
| E18W5F | 297 | 41 | 138 | ||||
All mice were sacrificed 6 weeks after the treatment.
The mice were treated transplacentally at 3 days prior to birth. Five pregnant female mice were used for the treatment.
There is a significant difference between the treatment group and its concurrent control (p <0.001).
There is a significant difference between the treatment group and mice treated with ENU at PND 126 (p < 0.01).
There is a significant difference between the treatment group and mice treated with ENU at PNDs 1 or 126 (p <0.001).
3.3. Mutational spectra in the liver cII gene from mice treated with ENU as neonates and adults
ENU-induced cII mutations were evaluated by DNA sequence analysis of 90 mutants from 12 male mice treated as neonates (PNDs 8 and 15), and 47 mutants from the 4 adult male mice from the PND 126 ENU treatment group (Table 3). The average MFs were 427 ± 50 for the neonate mice and 136 ± 30 for the adult mice. Mutations that were found more than once in the mutants isolated from a single animal were assumed to be siblings resulting from a single independent mutation. Accordingly, a total of 89 independent mutations from neonate and 41 from adult mice were identified. Table 4 summarizes the types of mutations in the liver cII gene from the ENU-treated neonate and adult male mice, along with the types of spontaneous mutation in the liver cII gene from neonate and adult male mice reported previously [18]. Among the independent mutations from ENU-treated animals, 97% for neonates and 86% for adults were base pair substitutions. A:T → T:A transversion was the major type of mutation (33% in neonate and 29% in adult versus 0% in both the neonate and adult controls). No significant difference was found between the mutation spectra for ENU-treated neonate and adult mice (p =0.17). The overall patterns of mutations in ENU-treated neonate or adult groups, however, were significantly different from those in their control groups (p <0.0001 for both the neonate and adult comparisons). G:C → A:T transition (50% in neonate control and 41% in adult control) and frameshifts (13% in neonate control and 22% adult control) were the predominant types of spontaneous mutations, and both types of mutations were found at lower frequencies in ENU-treated mice.
Table 3.
Mutations in the liver cII gene from ENU-treated neonate and adult Big Blue male mice
| Positiona | Mutationb | Amino acid change | Sequence context 5′→ 3′c | Number of mutations (independent) |
|
|---|---|---|---|---|---|
| Neonate | Adult | ||||
| –14 | G → T | N/A | ctaaggaaa | 1 | |
| 1 | A → T | Met → Leu | catATGgtt | 1 | |
| 2 | T → A | Met → Lys | catATGgtt | 2(1) | |
| 2 | T → C | Met → Thr | catATGgtt | 1 | |
| 5 | T → A | Val → Asp | atgGTTcgt | 1 | |
| 25 | G → A | Glu → Lys | aacGAGgct | 1 | |
| 25 | G → T | Glu → Stop | aacGAGgct | 1 | |
| 29 | C → A | Ala → Asp | gagGCTcta | 1 | |
| 32 | T → C | Leu → Pro | gctCTAcga | 1 | |
| 34 | C → T | Arg → Stop | ctaCGAatc | 2 (2) | |
| 35 | G → A | Arg → Gln | ctaCGAatc | 1 | |
| 35 | G → T | Arg → Leu | ctaCGAatc | 1 | |
| 38 | T → A | Ile → Asn | cgaATCgag | 1 | |
| 40 | G → A | Glu → Lys | atcGAGagt | 1 | |
| 40 | G → T | Glu → Stop | atcGAGagt | 3(1) | |
| 41 | A → C | Glu → Ala | atcGAGagt | 1 | |
| 41 | A → T | Glu → Val | atcGAGagt | 1 | |
| 42 | G → T | Glu → Asp | atcGAGagt | 1 | |
| 53 | T → C | Leu → Pro | ttgCTTaac | 2 (2) | |
| 55 | A → T | Asn → Tyr | cttAACaaa | 1 | |
| 58 | A → T | Lys → Stop | aacAAAatc | 2 (2) | |
| 58 | A → C | Lys → Gln | aacAAAatc | 1 | |
| 61 | A → T | Ile → Phe | aaaATCgca | 1 | |
| 62 | T → C | Ile → Thr | gcaATCgca | 1 | |
| 64 | G → A | Ala → Thr | atcGCAatg | 3 (2) | |
| 68 | T → A | Met → Lys | gcaATGctt | 1 | |
| 74 | G → A | Gly → Glu | cttGGAact | 1 | |
| 84 | G → C | Lys → Asn | gagAAGaca | 1 | |
| 85 | A → C | Thr → Pro | aagACAgcg | 1 | |
| 85 | A → G | Thr → Ala | aagACAgcg | 1 | |
| 86 | C → A | Thr → Lys | aagACAgcg | 1 | |
| 89 | C → T | Ala → Val | acaGCGgaa | 1 | |
| 92 | A → G | Glu → Gly | gcgGAAgct | 1 | |
| 94 | G → A | Ala → Thr | gaaGCTgtg | 1 | |
| 98 | T → A | Val → Glu | gctGTGggc | 2 (2) | |
| 99–100 | GG → AT | ValGly → ValCys | gctGTGGGCgtt | 1 | |
| 103 | G → A | Val → Ile | ggcGTTgat | 1 | |
| 103 | G → T | Val → Phe | ggcGTTgat | 1 | |
| 104 | T → C | Val → Ala | ggcGTTgat | 1 | |
| 108 | T → A | Asp → Glu | gttGATaag | 1 | 1 |
| 109 | A → G | Lys → Glu | gatAAGtcg | 1 | |
| 110 | A → C | Lys → Thr | gatAAGtcg | 1 | |
| 112 | T → C | Ser → Pro | aagTCGcag | 2 (2) | |
| 113 | C → A | Ser → Stop | aagTCGcag | 2 (2) | |
| 118 | A → C | Ile → Leu | cagATCagc | 1 | |
| 118 | A → T | Ile → Phe | cagATCagc | 1 | |
| 119 | T → G | Ile → Ser | cagATCagc | 1 | |
| 127 | T → A | Trp → Arg | aggTGGaag | 1 | |
| 129 | G → A | Trp → Stop | aggTGGaag | 1 | |
| 130 | A → G | Lys → Glu | tggAAGagg | 1 | |
| 132 | G → T | Lys → Asn | tggAAGagg | 1 | |
| 133 | A→G | Arg → Gly | aagAGGgac | 1 | |
| 137 | A→G | Asp → Gly | aggGACtgg | 1 | |
| 139 | T→A | Trp → Arg | gacTGGatt | 1 | |
| 141 | G→A | Trp → Stop | gacTGGatt | 3 (3) | |
| 142 | A→T | Ile → Phe | tggATTcca | 2(2) | |
| 143 | T→C | Ile → Thr | tggATTcca | 1 | |
| 145 | C→T | Pro → Ser | attCCAaag | 2 (2) | |
| C→T | Pro → Leu | attCCAaag | 1 | ||
| 152 | T→C | Phe → Ser | aagTTCtca | 1 | |
| 155 | C→A | Ser → Stop | ttcTCAatg | 1 | |
| 161 | T→A | Leu → Gln | atgCTGctt | 1 | |
| 164 | T→C | Leu → Pro | ctgCTTgct | 1 | |
| 169 | G→C | Val → Leu | gctGTTctt | 1 | |
| 170 | T→A | Val → Asp | gctGTTctt | 1 | |
| 170 | T→C | Val → Ala | gctGTTctt | 1 | |
| 176–177 | –A | Frameshift | cttGAAtgg | 1 | |
| 178/185 | +G | Frameshift | gaaTGGGGGGTCgtt | 3 (3) | 7 (4) |
| 178 | T→A | Trp → Arg | gaaTGGggg | 2 (2) | |
| 180 | G→A | Trp → Stop | gaaTGGggg | 1 | |
| 185 | T→A | Val → Asp | gggGTCgtt | 1 | 1 |
| 185 | T→C | Val → Ala | gggGTCgtt | 1 | |
| 191 | A→C | Asp → Als | gttGACgac | 1 | 1 |
| 193 | G→A | Asp → Asn | gacGACgac | 2 (2) | |
| 194 | A→G | Asp → Gly | gacGACgac | 1 | |
| 197 | A→C | Asp → Ala | gacGACatg | 1 | |
| 197 | A→T | Asp → Val | gacGACatg | 1 | |
| 200 | T→A | Met → Lys | gacATGgct | 1 | |
| 203 | C→T | Ala → Val | atgGCTcga | 1 | |
| 209 | T→A | Leu → Stop | cgaTTGgcg | 1 | |
| 212 | C→A | Ala → Glu | ttgGCGcga | 2 (2) | |
| 212 | C→T | Ala → Val | ttgGCGcga | 1 | 1 |
| 214 | C→T | Arg → STOP | gcgCGAcaa | 3 (3) | 1 |
| 221 | T→C | Val → Ala | caaGTTgct | 1 | 1 |
| 221 | T→G | Val → Gly | caaGTTgct | 1 | |
| 230 | T→A | Ile → Asn | gcgATTctc | 2 (2) | |
| 235 | A→C | Thr → Pro | ctcACCaat | 2 (2) | 1 |
| 241 | A→T | Lys → Stop | aatAAAaaa | 1 | 1 |
| 245 | A→C | Lys → Thr | aaaAAAcgc | 1 | |
| 289 | T→A | Phe → Ile | gagTTCtga | 1 | |
| 290 | T→A | Phe → Tyr | gagTTCtga | 1 | 2 (2) |
| 292 | T→A | Stop → Arg | ttcTGAggt | 1 | 1 |
| 294 | A→G | Stop → Trp | ttcTGAggt | 1 | |
| 294 | A→T | Stop → Cys | ttcTGAggt | 2 (2) | |
| Total | 90 (89) | 47 (41) | |||
Abbreviations: –, deletion; +, insertion.
Position 1 is the first base of the start codon in the cII coding sequence.
Presented in term of sequence change on nontranscribed DNA strand.
Uppercase indicates target codon and target bases are underlined.
Table 4.
Comparison of spontaneous and ENU-induced mutations in the liver cII gene of neonate and adult Big Blue mice
| Type of mutation | Neonate |
Adult |
||
|---|---|---|---|---|
| Control (%)a |
ENU (%) |
Control (%)a |
ENU (%) |
|
| G:C → C:G | 5 | 2 | 9 | 0 |
| G:C→A:T | 50 | 18 | 41 | 27 |
| G:C → T:A | 21 | 12 | 6 | 7 |
| At CpG site | 43 | 21 | 41 | 17 |
| A:T → T:A | 0 | 33 | 0 | 29 |
| A:T → C:G | 9 | 14 | 9 | 5 |
| A:T → G:C | 2 | 18 | 9 | 17 |
| Frameshift | 13 | 3 | 22 | 12 |
| Others | 0 | 0 | 3 | 2 |
| Total mutants screened |
56 | 89 | 32 | 41 |
Control mutations from literature [18]. Control animals were treated with DMSO at approximately 2 μl/g body weight.
4. Discussion
The results for the time-course study are consistent with the liver cII gene being genetically neutral. In mice treated perinatally with ENU, the cII MF seems to reach a maximum at or before 6 weeks after the treatment and the MF remained relatively constant thereafter. Using ENU-treated Muta mice, Douglas et al. [19] showed that the peak MF in the liver lacZ gene was achieved at 5 weeks after treatment and did not significantly change from 5 to 8 weeks. The evidence for the neutrality of the cII gene is also supported by cII MF analysis of the small intestine in Muta mice treated with ENU. The MFs were similar at 10 and 70 days after treatment [20]. Since the cII gene is non-functional in the mouse, mutants in this transgene in vivo would confer no selective growth advantage or disadvantage to the cells. Therefore, mutational data resulting from the cII mutagenesis assay might be a reasonable estimate of mutations occurring throughout the mouse genome and at different periods during the life span of the mouse.
A review of carcinogenicity studies using benzo(a)pyrene, dibenz(a,h)anthracene, diethylnitrosamine (DEN), dimethylbenzanthracene, ENU, 3-methylchloanthrene, urethane and X-rays indicates that juvenile animals are 3- to 10-fold more sensitive to tumor induction than adult animals for a wide range of tissues including liver, lung, kidney, mammary tissue, the hematopoeitic system and nervous tissues [20]. Also, the neonatal mouse model, in which mice are treated at various time-points between PNDs 1 and 15, has been used experimentally since 1959 [22]. While the strain of mouse can greatly influence tumor incidence, in general, this model is very sensitive to carcinogens that operate via a genotoxic mode of action [11]. Mutations are thought to be involved in carcinogenesis because the transition from a normal somatic cell to a cancer cell is due to mutations in protooncogenes, tumor suppressor genes or genes that function in the maintenance of genomic stability [23,24]. Therefore, it is reasonable to hypothesize that the neonatal animals also possess greater sensitivity to the mutagenicity of genetoxic carcinogens than adult animals. To test this hypothesis, we treated Big Blue mice with ENU using a treatment protocol similar to those used in carcinogenesis studies [25,26]. Both the carcinogenicity and mutagenicity studies were conducted in mice with a B6C3F1 background, thus avoiding strain-specific factors that might specially affect mutagenicity or carcinogenicity. ENU was chosen because it is a direct-acting mutagen, which avoids complications from chemical bioactivation. The 18th day of gestation has been identified as a sensitive time point for the carcinogenicity of ENU during the prenatal period [25]. The PNDs 1, 8, 15 and 42 treatment time points were based on a protocol used for tumorigenicity assays [26]. Mice at PNDs 42 and 126 are in the young adult and adult stages of life, respectively.
Our MF data (Tables 1 and 2) show that the age at the time of ENU exposure is an effective modulator of mutation induction. In both male and female mice, prenatal and PND 1 treatments with ENU induced relatively high MFs. ENU treatment at PNDs 8 and 15, however, resulted in the highest mutation induction. The lowest induced MFs occurred in those animals treated as adults (PND 126). The MFs in the PND 8 treatment groups were about 2- to 4-fold higher than those in the PND 126 treatment groups. This age-dependent pattern of mutation induction is comparable to that for tumor induction. A comparison between the mutant frequencies in the liver cII gene and the incidence of liver tumors in the Big Blue or generic B6C3F1 mice treated with 120 mg ENU/kg body weight is given in Table 5. In the carcinogenesis studies [25,26], development of liver tumors was modified significantly by the age at which ENU was given. The tumor incidence was 11% in untreated male animals and increased following ENU treatment to 67% for the males treated as 18-day-old faetuses. The cancer incidence reached a maximum of 94% for the males treated at PND 15 and then dropped to 35% for the mice treated at PND 42. The pattern of tumor responses to ENU in female mice was similar to that observed in males. Females, however, showed a consistently lower incidence of liver tumors than males treated at the same age. This apparent comparability between the mutagenicity and carcinogenicity of ENU is consistent with mutation induction being the rate-limiting factor in the carcinogenicity of ENU. Also the high sensitivity of neonatal mice to mutation induction by ENU indicates that treatments during this period may result in a general increase in the sensitivity of in vivo mutagenesis assays.
Table 5.
Comparison of mutant frequencies in the liver cII gene and incidence of liver tumors from the Big Blue or generic B6C3F1 mice treated with 120 mg ENU/kg body weighta
We sequenced the cII DNA of mutants from neonate and adult mice treated with ENU to observe if there were any differences between neonatal and adult mice in terms of clonal expansion or mutational spectra. High rates of cell proliferation can increase the probability that the initiated clone will expand before cell death [21]. If an initially mutated clone was disproportionately expanded and resulted in a higher MF, the number of sibling mutants in the animal would increase. Such events, however, were rare. The sequencing data gave no evidence for clonal expansion playing a crucial role for the increased MFs found in the neonatal mice. We sequenced approximately 8 mutants from each animal. Eighty-nine of 90 mutations from ENU-treated neonatal mice and 41 of 47 mutations from ENU-treated adults were clearly of independent origin.
ENU reacts with oxygen in DNA, and results in modification of the O6 position of guanine and the O2 and O4 positions of thymine. O6-ethylguanine, which induces G:C → A:T transition, can be efficiently repaired in normal mammalian systems. A:T → T:A transversion induced by O2-ethylthymine andA:T→ G:Ctransitioninducedby O4-ethylthymine become the major types of mutation because O2- and O4-ethylthymine adducts are relatively persistent in normal mammalian cells [27–32]. In this study, the overall pattern of mutations induced by ENU in the neonate and adult mice was very similar (p =0.17). A:T → T:A transversion and A:T → G:C transition were the predominant mutations in both the spectra (33 and 18% in neonate, and 29 and 17% in adult). The percentages of ENU-induced mutation at A:T base pairs in this study (65% in neonates and 51% in adults) are consistent with the ENU-induced mutation spectrum for the liver lacZ gene of adult Muta mouse (56.7% at A:T [19]). Hence, our spectra results suggest that the DNA repair system is similar in neonate and adult mice and may not contribute to the higher MFs induced by ENU in neonates.
The mechanisms for the age-related mutagenesis and carcinogenesis are still unknown although a number of factors have been suggested as determining susceptibility at different stages [33]. Cell proliferation has been regarded as a major factor since mutations are fixed by DNA replication [34,35]. It has been reported that each liver cell in the neonatal mouse divides an average of 3 times, resulting in an 8- to 10-fold increase in liver mass and about a 3-fold increase in the number of cells. Hepatocytes rarely divide in the adult mouse, less than one division every 3 months [36–38]. Genotoxic carcinogens are generally more effective in rapidly dividing tissues because the higher rate of cell division increases the number of cells in S phase. This rapid division makes the cell population more vulnerable to the genotoxic effects of chemicals and leaves less time for DNA repair prior to fixation of the damage as a mutation [39,40]. This reasoning is substantiated by studies using partial hepatectomy, which increases liver cell turnover rate in adult animals. Hepatoecotmyzed mice treated with ENU have enhanced hepatocarcino- genesis [41] and liver MFs [42]. The same principle may also apply to neonatal mice, considering the much faster rate of cell division in neonatal animals. In apparent contradiction with this theory, mice treated with ENU perinatally had lower liver MFs than neonates although fetal and newborn mice have a faster rate of cell proliferation. One explanation for this may be that most of the liver cells are hematopoietic rather than hepatic at these developmental stages and these cells will largely be absent from the liver when MF assays are conducted and/or these hematopoietic cells may respond differently to the ENU insult from hepatocytes [25]. Another possibility is that the level of liver damage in fetal or PND 1 mice is less than the level of damage in neonates. It is also conceivable that DNA repair activity is greater in embryonic cells compared to neonatal cells.
It also seems a paradox that ENU induced significantly higher tumor incidences and lower MFs in male mice than in females at nearly every age (Table 5). For example, the cII liver MFs for mice treated with ENU at PND 15 were 416 × 10–6 in males and 531 × 10–6 in females, whereas the tumor incidences were 94% in males and 67% in females. This pattern of mutagenicity and carcinogenicity has also been found in other chemical-treated male and female mice [43–45]. The functions of sex hormones may be a reason for this inconsistency. The hormonal environment plays an important role in the development of liver tumors. Experimental and clinical data have shown that both estrogens and androgens have important effects in controlling liver cell proliferation and promoting liver tumors [46]. Androgens have been implicated in hepatic carcinogenesis because men develop liver tumors with a much greater frequency and worse prognosis than women. Males tend to have a lower survival rate and an increased recurrence of the disease after treatment. In rodents, carcinogen-induced and spontaneous liver tumors also occur at a higher incidence in males than in females [47,48]. The role of estrogens in tumor incidence, however, is unclear. Although estrogens also have been suggested as cancer promoters in rat liver [49–51], a number of subsequent studies using animal models observed opposite findings. In some studies, estrogens have shown unexpected suppressive effects on spontaneous and chemical-induced liver tumors [52–54].
In summary, the mutagenicity of ENU in the liver cII gene of Big Blue mice was significantly modified by the age at which the animals were exposed. Neonatal Big Blue mice were the most vulnerable to the mutagenicity of ENU while adults displayed much less sensitivity. Although there was a significant difference between the MFs of the neonatal and adult mice, the overall patterns of mutations inducedby ENU in the neonates and adults were very similar. The age-dependent MF responses are comparable to the tumor incidence observed in carcinogenesis studies, suggesting that mutation induction is a major factor for the high susceptibility of neonates to tumor formation by genotoxic carcinogens.
Acknowledgments
This research was supported by an appointment (N. Mei) to the Postgraduate Research Program at the NCTR administered by the Oak Ridge Institute for Science and Education through an interagency agreement between the US Department of Energy and the US Food and Drug Administration.
Abbreviations:
- ENU
N-ethyl-N-nitrosourea
- PNDs
postnatal days
- MF
mutant frequency
- DMSO
dimethylsulfoxide
Footnotes
The views presented in this article do not necessarily reflect those of the Food and Drug Administration.
References
- [1].Landrigan PJ, Garg A, Chronic effects of toxic environmental exposures on children’s health, J. Toxicol. Clin. Toxicol. 40 2002. 449–456. [DOI] [PubMed] [Google Scholar]
- [2].Vasilatou-Kosmidis H, Cancer in neonates and infants, Med. Pediatr. Oncol. 41 (2003) 7–9. [DOI] [PubMed] [Google Scholar]
- [3].Gurney JG, Davis S, Severson RK, Fang JY, Ross JA, Robison LL, Trends in cancer incidence among children in the U.S., Cancer 78 (1996) 532–541. [DOI] [PubMed] [Google Scholar]
- [4].Gurney JG, Ross JA, Wall DA, Bleyer WA, Severson RK, Robison LL, Infant cancer in the U.S.: histology-specific incidence and trends, 1973 to 1992, J Pediatr. Hematol. Oncol. 19(1997) 428–432. [DOI] [PubMed] [Google Scholar]
- [5].Kenney LB, Miller BA, Ries LA, Nicholson HS, Byrne J, Reaman GH, Increased incidence of cancer in infants in the U.S.: 1980–1990, Cancer 82 (1998) 1396–1400. [DOI] [PubMed] [Google Scholar]
- [6].Vesselinovitch SD, Koka M, Mihailovich N, V Rao K, Carcinogenicity of diethylnitrosamine in newborn, infant, and adult mice, J. Cancer Res. Clin. Oncol. 108 (1984) 60–65. [DOI] [PubMed] [Google Scholar]
- [7].IARC, Perinatal and multigeneration carcinogenesis, IARC Scientific Publications, Lyon, 1989, pp. 1–436. [PubMed] [Google Scholar]
- [8].Dooley KL, Stavenuiter JF, Westra JG, Kadlubar FF, Comparative carcinogenicity of the food pyrolysis product, 2- amino-5-phenylpyridine, and the known human carcinogen, 4- aminobiphenyl, in the neonatal B6C3F1 mouse, Cancer Lett. 41 (1988) 99–103. [DOI] [PubMed] [Google Scholar]
- [9].Fu PP, Von Tungeln LS, Zhan DJ, Bucci T, Potent tumorigenicity of 7-chlorobenz[a]anthracene and 7- bromobenz[a]anthracene in the neonatal B6C3F1 male mouse, Cancer Lett. 101 (1996) 37–42. [DOI] [PubMed] [Google Scholar]
- [10].Flammang TJ, Tungeln LS, Kadlubar FF, Fu PP, Neonatal mouse assay fortumorigenicity: alternative to the chronicrodent bioassay, Regul. Toxicol. Pharmacol. 26 (1997) 230–240. [DOI] [PubMed] [Google Scholar]
- [11].McClain RM, Keller D, Casciano D, Fu P, MacDonald J, Popp J, Sagartz J, Neonatal mouse model: review of methods and results, Toxicol. Pathol. 29 (Suppl.) (2001) 128–137. [DOI] [PubMed] [Google Scholar]
- [12].Dass SB, Heflich RH, Casciano DA, Mutational response at the splenic T-lymphocyte hprt locus in mice treated as neonates: contrasting effects of the carcinogens N-ethyl-N-nitrosourea, dimethylnitrosamine, and 2-amino-1-methyl- 6-phenylimidazo[4,5-b]pyridine, Environ. Mol. Mutagen. 31 (1998) 243–247. [PubMed] [Google Scholar]
- [13].Kohler SW, Provost GS, Fieck A, Kretz PL, Bullock WO, Putman DL, Sorge JA, Short JM, Analysis of spontaneous and induced mutations in transgenic mice using a lambda ZAP/lacI shuttle vector, Environ. Mol. Mutagen. 18 (1991) 316–321. [DOI] [PubMed] [Google Scholar]
- [14].Kohler SW, Provost GS, Fieck A, Kretz PL, Bullock WO, Sorge JA, Putman DL, Short JM, Spectra of spontaneous and mutagen-induced mutations in the lacI gene in transgenic mice, Proc. Natl. Acad. Sci. U.S.A. 88 (1991) 7958–7962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Provost GS, Kretz PL, Hamner RT, Matthews CD, Rogers BJ, Lundberg KS, Dycaico JM Short, Transgenic systems for in vivo mutation analysis, Mutat. Res. 288 (1993) 133–149. [DOI] [PubMed] [Google Scholar]
- [16].Cariello NF, Software for the analysis of mutations at the human hprt gene, Mutat. Res. 312 (1994) 173–185. [DOI] [PubMed] [Google Scholar]
- [17].Adams WT, Skopek TR, Statistical test for the comparison of samples from mutational spectra, J. Mol. Biol. 194 (1987) 391–396. [DOI] [PubMed] [Google Scholar]
- [18].Chen T, Mittelstaedt R, Heflich R, Moore M, Parsons B, Mutant frequency and spectra of mutations in the liver lacI and cII genes of lambda/lacI transgenic mice treated as neonates with 4-aminobiphenyl, Environ. Mol. Mutagen. 39 (Suppl. 33) (2002) 18.11813292 [Google Scholar]
- [19].Douglas GR, Jiao J, Gingerich JD, Soper LM, Gossen JA, Temporal and molecular characteristics of lacZ mutations in somatic tissues oftransgenic mice, Environ. Mol. Mutagen. 28 (1996) 317–324. [DOI] [PubMed] [Google Scholar]
- [20].Swiger RR, Cosentino L, Shima N, Bielas JH, Cruz-Munoz W, Heddle JA, The cII locus in the MutaMouse system, Environ. Mol. Mutagen. 34 (1999) 201–207. [PubMed] [Google Scholar]
- [21].Ginsberg GL, Assessing cancer risks from short-term exposures in children, Risk Anal. 23 (2003) 19–34. [DOI] [PubMed] [Google Scholar]
- [22].Pietra G, Spencer K, Shubik P, Response of newly born mice to a chemical carcinogen, Nature 183 (1959) 1689. [DOI] [PubMed] [Google Scholar]
- [23].Hennings H, Glick AB, Greenhalgh DA, Morgan DL, Strickland JE, Tennenbaum T, Yuspa SH, Critical aspects of initiation, promotion, and progression in multistage epidermal carcinogenesis, Proc. Soc. Exp. Biol. Med. 202 (1993) 1–8. [DOI] [PubMed] [Google Scholar]
- [24].Loeb LA, A mutator phenotype in cancer, Cancer Res. 61 (2001) 3230–3239. [PubMed] [Google Scholar]
- [25].Vesselinovitch SD, Rao KV, Mihailovich N, Neoplastic response of mouse tissues during perinatal age periods and its significance in chemical carcinogenesis, Natl. Cancer Inst. Monogr. (1979) 239–250. [PubMed] [Google Scholar]
- [26].Vesselinovitch SD, Rao KV, Mihailovich N, Rice JM, Lombard LS, Development of broad spectrum of tumors by ethylnitrosourea in mice and the modifying role of age, sex, and strain, Cancer Res. 34 (1974) 2530–2538. [PubMed] [Google Scholar]
- [27].Bronstein SM, Skopek TR, Swenberg JA, Efficient repair of O6-ethylguanine, but not O4-ethylthymine or O2-ethylthymine, is dependent upon O6-alkylguanine-DNA alkyltransferase and nucleotide excision repair activities in human cells, Cancer Res. 52 (1992) 2008–2011. [PubMed] [Google Scholar]
- [28].Jansen JG, Mohn GR, Vrieling H, van Teijlingen CM, Lohman PH, van Zeeland AA, Molecular analysis of hprt gene mutations in skin fibroblasts of rats exposed in vivo to N-methyl- N-nitrosourea or N-ethyl-N-nitrosourea, Cancer Res. 54 (1994) 2478–2485. [PubMed] [Google Scholar]
- [29].Mittelstaedt RA, Smith BA, Heflich RH, Analysis of in vivo mutation induced by N-ethyl-N-nitrosourea in the hprt gene of rat lymphocytes, Environ. Mol. Mutagen. 26 (1995) 261–269. [DOI] [PubMed] [Google Scholar]
- [30].Walker VE, Gorelick NJ, Andrews JL, Craft TR, de¬Boer JG, Glickman BW, Skopek TR, Frequency and spectrum of ethylnitrosourea-induced mutation at the hprt and lacI loci in splenic lymphocytes of exposed lacI transgenic mice, Cancer Res. 56 (1996) 4654–4661. [PubMed] [Google Scholar]
- [31].Jansen JG, Vrieling H, van Teijlingen CM, Mohn GR, Tates AD, van Zeeland AA, Marked differences in the role of O6- alkylguanine in hprt mutagenesis in T-lymphocytes of rats exposed in vivo to ethylmethanesulfonate, N-(2-hydroxyethyl)-N- nitrosourea, or N-ethyl-N-nitrosourea, Cancer Res. 55 (1995) 1875–1882. [PubMed] [Google Scholar]
- [32].Liem LK, Lim A, Li BF, Specificities of human, rat and E. coli O6 -methylguanine-DNA methyltransferases towards the repair of O6-methyl and O6-ethylguanine in DNA, Nucleic Acids Res. 22 (1994) 1613–1619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Anderson LM, Diwan BA, Fear NT, Roman E, Critical windows of exposure for children’s health: cancer in human epidemiological studies and neoplasms in experimental animal models, Environ. Health Perspect. 108 (Suppl. 3) (2000) 573–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Stuart GR, Oda Y, de Boer JG, Glickman BW, Mutation frequency and specificity with age in liver, bladder and brain of lacI transgenic mice, Genetics 154 (2000) 1291–1300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Stuart GR, Glickman BW, Through a glass, darkly: reflections of mutation from lacI transgenic mice, Genetics 155 (2000) 1359–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Schultze B, Kellerer AM, Grossmann C, Maurer W, Growth fraction and cycle duration of hepatocytes in the three-week-old rat, Cell Tissue Kinet. 11 (1978) 241–249. [DOI] [PubMed] [Google Scholar]
- [37].Cameron IL, Cell proliferation and renewal in the mammalian body, in: Cameron IL, Thrasher JD (Eds.), Cellular and Molecular Renewal in the Mammalian Body, Academic Press, New York, 1971, pp. 45–85. [Google Scholar]
- [38].Buetow DH, Cell numbers vs. age in mammalian tissues and organs, in: Cristofalo HV (Ed.), CRC Handbook of Cell Biology of Aging, CRC Press, Boca Raton, FL, 1985, pp. 1–115. [Google Scholar]
- [39].Bertram JS, Heidelberger C, Cell cycle dependency of oncogenic transformation induced by N-methyl-N’-nitro-N- nitrosoquanidine in culture, Cancer Res. 34 (1974) 526–537. [PubMed] [Google Scholar]
- [40].Rabes HM, Muller L, Hartmann A, Kerler R, Schuster C, Cell cycle-dependent initiation of adenosine triphosphatasedeficient populations in adult rat liver by a single dose of N- methyl-N-nitrosourea, Cancer Res. 46 (1986) 645–650. [PubMed] [Google Scholar]
- [41].Vesselinovitch SD, Itze L, Mihailovich N, Rao KV, Modifying role of partial hepatectomy and gonadectomy in ethylnitrosourea-induced hepatocarcinogenesis, Cancer Res. 40 (1980) 1538–1542. [PubMed] [Google Scholar]
- [42].Shane BS, Wood VJ, Cunningham ML, Tindall KT, Effect of partial hepatectomy on the mutant freuency of lacI in Big Blue mice treated with N-ethyl-N-nitrosourea, Environ. Mol. Mutagen. 25 (1995) 47. [Google Scholar]
- [43].Suter W, Ahiabor R, Blanco B, Locher F, Mantovani F, Robinson M, Sreenan G, Staedtler F, Swingler T, Vignutelli A, Perentes E, Evaluation of the in vivo genotoxic potential of three carcinogenic aromatic amines using the Big Blue transgenic mouse mutation assay, Environ. Mol. Mutagen. 28 (1996) 354–362. [DOI] [PubMed] [Google Scholar]
- [44].Suter W, Staedtler F, Poetter-Locher F, Swingler T, Wilson L, 4-Chloro-o-phenylenediamine: a 26-week oral (in feed) mutagenicity study in Big Blue mice, Mutat. Res. 414 (1998) 149–156. [DOI] [PubMed] [Google Scholar]
- [45].Hoel DG, Haseman JK, Hogan MD, Huff J, Mc-Connell EE, The impact of toxicity on carcinogenicity studies: implications for risk assessment, Carcinogenesis 9 (1988) 2045–2052. [DOI] [PubMed] [Google Scholar]
- [46].De Maria N, Manno M, Villa E, Sex hormones and liver cancer, Mol. Cell Endocrinol. 193 (2002) 59–63. [DOI] [PubMed] [Google Scholar]
- [47].Johnson PJ, Krasner N, Portmann B, Eddleston AL, Williams R, Hepatocellular carcinoma in Great Britain: influence of age, sex, HBsAg status, and aetiology of underlying cirrhosis, Gut 19 (1978) 1022–1026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Nagasue N, Ogawa Y, Yukaya H, Ohta N, Ito A, Serum levels of estrogens and testosterone in cirrhotic men with and without hepatocellular carcinoma, Gastroenterology 88 (1985) 768–772. [DOI] [PubMed] [Google Scholar]
- [49].Taper HS, The effect of estradiol-17-phenylpropionate and estradiol benzoate on N-nitrosomorpholine-induced liver carcinogenesis in ovariectomized female rats, Cancer 42 (1978) 462–467. [DOI] [PubMed] [Google Scholar]
- [50].Yager JD Jr., R. Yager, Oral contraceptive steroids as promoters of hepatocarcinogenesis in female Sprague–Dawley rats, Cancer Res. 40 (1980) 3680–3685. [PubMed] [Google Scholar]
- [51].Cameron RG, Imaida K, Tsuda H, Ito N, Promotive effects of steroids and bile acids on hepatocarcinogenesis initiated by diethylnitrosamine, Cancer Res. 42 (1982) 2426–2428. [PubMed] [Google Scholar]
- [52].Shimizu I, Yasuda M, Mizobuchi Y, Ma YR, Liu F, Shiba M, Horie T, Ito S, Suppressive effect of oestradiol on chemical hepatocarcinogenesis in rats, Gut 42 (1998) 112–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Mishkin SY, Farber E, Ho RK, Mulay S, Mishkin S, Evidence for the hormone dependency ofhepatic hyperplastic nodules: inhibition of malignant transformation after exogenous 17 beta-estradiol and tamoxifen, Hepatology 3 (1983) 308–316. [DOI] [PubMed] [Google Scholar]
- [54].Yager JD, Zurlo J, Sewall CH, Lucier GW, He H, Growth stimulation followed by growth inhibition in livers of female rats treated with ethinyl estradiol, Carcinogenesis 15 (1994) 2117–2123. [DOI] [PubMed] [Google Scholar]