

Abstract
Exposure to elevated levels of manganese (Mn) causes manganism, a neurological disorder with similar characteristics to those of Parkinson’s disease (PD). Valproic acid (VPA), an antiepileptic, is known to inhibit histone deacetylases and exert neuroprotective effects in many experimental models of neurological disorders. In the present study, we investigated if VPA attenuated Mn-induced dopaminergic neurotoxicity and the possible mechanisms involved in VPA’s neuroprotection, focusing on modulation of astrocytic glutamate transporters (glutamate aspartate transporter, GLAST and glutamate transporter 1, GLT-1) and histone acetylation in H4 astrocyte culture and mouse models. The results showed that VPA increased promoter activity, mRNA/protein levels of GLAST/GLT-1 and glutamate uptake, and reversed Mn-reduced GLAST/GLT-1 in in vitro astrocyte cultures. VPA also attenuated Mn-induced reduction of GLAST and GLT-1 mRNA/protein levels in midbrain and striatal regions of the mouse brain when VPA (200 mg/kg, i.p., daily, 21 d) was administered 30 min prior to Mn exposure (30 mg/kg, intranasal instillation, daily, 21 d). Importantly, VPA attenuated Mn-induced dopaminergic neuronal damage by reversing Mn-induced decrease of tyrosine hydroxylase (TH) mRNA/protein levels in the nigrostriatal regions. VPA also reversed Mn-induced reduction of histone acetylation in astrocytes as well as mouse brain tissue. Taken together, VPA exerts attenuation against Mn-induced decrease of astrocytic glutamate transporters parallel with reversing Mn-induced dopaminergic neurotoxicity and Mn-reduced histone acetylation. Our findings suggest that VPA could serve as a potential neuroprotectant against Mn neurotoxicity as well as other neurodegenerative diseases associated with excitotoxicity and impaired astrocytic glutamate transporters.
Keywords: Valproic acid, manganese, tyrosine hydroxylase, GLT-1, GLAST, neuroprotection
1. Introduction
Manganese (Mn) is an essential element for various metabolic functions, but overexposure to this metal can lead to accumulation in the basal ganglia, such as globus pallidus, striatum and substantia nigra, causing neurotoxic effects [1–3]. Individuals exposed to Mn occupationally and environmentally may develop a neurological disorder referred to as manganism, a syndrome that resembles Parkinson’s disease (PD) [4–6].
Multiple cellular and molecular mechanisms involved in Mn neurotoxicity have been reported, including excitotoxicity [7–10]. Dysregulation of astrocytic glutamate transporters has been documented to modulate Mn-induced excitotoxic neuronal injuries [11–14]. Among five identified glutamate transporters in human, two main glutamate transporters excitatory amino acid transporters (EAAT) 1 or EAAT2, known as glutamate aspartate transporter (GLAST) and glutamate transporter 1 (GLT-1) in rodents, respectively, are predominantly localized in astrocytes and vital for maintaining glutamate homeostasis in the brain [15–17]. It is well established that Mn decreases expression and function of the glutamate transporters EAAT1/GLAST and EAAT2/GLT-1 in astrocytes [18–20] and induces dysregulation of glutamate homeostasis [21]. Interestingly, excitotoxic neuronal injuries are common features in various neurological disorders such as amyotrophic lateral sclerosis (ALS), Alzheimer’s disease (AD), PD and Huntington’s disease (HD) [12, 22].
Although dopaminergic neurodegeneration in the nigrostriatal pathway is a hallmark of PD [23, 24], several studies have also shown that Mn toxicity is also associated with dopaminergic toxicity [25–27]. Since the nigrostriatal pathway is responsible for motor functions, impairment of dopaminergic neurons in the substantia nigra leads to dysregulation of locomotor function and motor coordination [28]. As Mn induces excitatory neurotoxicity in the rat brain [29] and decreases expression of astrocytic glutamate transporters in vitro [30, 31], impairment of astrocytic glutamate transporters may play a critical role in Mn-induced dopaminergic neurotoxicity. Accordingly, pharmacological agents such as valproic acid (VPA) with proven effectiveness to attenuate Mn-induced dysregulation of astrocytic glutamate transporters [31, 32] may protect from Mn-induced nigrostriatal DA neuronal injury.
Given that inhibition of histone deacetylases (HDACs) exerts neuroprotection in several brain disorders [33, 34], HDAC inhibitors (HDACi) including VPA have been investigated as a potential therapeutics for several neurodegenerative diseases [33–35]. VPA is widely used to treat epilepsy, mood disorders, migraines and neuropathic pain [36–39]. The molecular mechanism of VPA as therapeutics is not completely understood, but regulation of gamma-aminobutyric acid (GABA), activation of pro-survival protein kinases and inhibition of HDACs have been suggested [40, 41].
Modulation of astrocytic glutamate transporters appears to be involved in VPA neuroprotection. VPA exerts neuroprotective effects in glaucoma by enhancing GLAST which is a main glutamate transporter in the retina [42]. VPA also exerts protection against malonate toxicity in the rat striatum by enhancing GLT-1 protein levels and glutamate uptake [43]. Thus, it is reasonable to investigate if VPA may exert neuroprotection against Mn neurotoxicity via regulation of GLAST and GLT-1. We have previously shown that HDACs regulate GLAST and GLT-1 promoter activities and their mRNA and protein levels by interacting with several transcription factors in primary astrocytes [31, 44]. Mn increased interaction of HDACs with transcription factor yin yang 1 (YY1) to repress expression of GLAST and GLT-1 [31, 44]. HDAC subtypes of classes I and II have shown to repress GLAST and GLT-1 promoter activities in astrocytes [45, 46]. Among many HDACi, VPA exerted relatively high stimulatory effects on GLAST and GLT-1 promoter activities [31, 44]. HDACi, VPA and sodium butyrate, attenuated Mn-induced behavioral deficits in mice and reduction of astrocytic glutamate transporters in cortex and cerebellum [32]. Thus, the present study investigated if VPA attenuated Mn-induced decrease in tyrosine hydroxylase (TH) expressing dopaminergic neurons, GLT-1 and GLAST in midbrain and striatum. Modulation of histone acetylation by Mn and VPA was also tested in in vitro astrocytes as well as in vivo mouse model to determine if VPA’s protective effects against Mn toxicity involve HDAC inhibition.
2. Materials and methods
2.1. Reagents
Cell culture media and reagents were purchased from Invitrogen (Carlsbad, CA) unless stated otherwise. Luciferase reporter assay kit was obtained from Promega (Madison, WI). Manganese chloride (MnCl2) and valproic acid (VPA) were obtained from Sigma-Aldrich (St. Louis, MO). EAAT1 and EAAT2 antibodies were from Abcam (Cambridge, MA). Antibodies for β-actin, acetylated histone H3 (Ac-histone H3), acetylated histone H4 (Ac-histone H4), TH and GAPDH were from Santa Cruz Biotechnology (Santa Cruz, CA). L-[3H] glutamate was purchased from PerkinElmer (Waltham, MA).
2.2. Astrocyte culture
Human astrocyte H4 cell line (ATCC®HTB-148™) was acquired from the American Type Culture Collection (ATCC, Manassas, VA) and grown in DMEM with 10% fetal bovine serum and 1% penicillin and streptomycin. Cells were plated in 24-well plates for luciferase assay and glutamate uptake assay, and in 6-well plates for mRNA/protein analysis. Cells were maintained at 37°C in a 95% air, 5% CO2 incubator.
2.3. Luciferase assay
Promoter activities of EAAT1 and EAAT2 were determined by transfecting human H4 cells with pGL3 EAAT1 luciferase plasmid vector containing the EAAT1 promoter sequences from bp +99 to −1973, a generous gift from Dr. Volsky (Columbia University, New York City, NY) [47] and pGL3 EAAT2 luciferase plasmid vector containing the EAAT2 promoter sequences from bp +282 to −954, a generous gift from Dr. Baldwin (University of North Carolina at Chapel Hill). H4 cells were transfected with luciferase plasmid vectors assisted by Lipofectamine 2000 (Thermo Fisher Scientific, Rockford, IL). After transfection overnight, cells were treated with the designated compounds in Opti-MEM media, followed by luciferase assay with the Bright-Glo luciferase kit (Promega) according to the manufacturer’s instructions and the activity was normalized to the protein content as determined with the bicinchoninic acid (BCA) assay (Thermo Fisher Scientific).
2.4. Animal procedures
All animal protocols were reviewed and approved by the Meharry Medical College Institutional Animal Care and Use Committee (Nashville, TN). Adult C57BL/6J wild type (WT) and double transgenic GLT-1-eGFP/GLAST-DsRed mice were used in this study. The C57BL/6J male mice (6–8 weeks old; weight 18–20 g) were purchased from the Jackson Laboratory (Bar Harbor, ME) and housed for one week prior to study. The double transgenic GLT-1-eGFP/GLAST-DsRed reporter male mice on a C57BL/6J background were generated by crossing GLT-1-eGFP homozygous and GLAST-DsRed homozygous mice that were from Dr. Rothstein’s laboratory at John Hopkins University [48, 49]. The double transgenic reporter mice used in this study did not show any signs of abnormalities or variations in GLT-1 or GLAST protein levels compared to its background wild-type C57BL/6J mice [48]. Mice were randomly grouped, housed (4 mice/cage) and maintained on a 12-h light/dark cycle at 22 ± 2°C with food, water and enrichment available ad libitum in the Animal Care Facility of Meharry Medical College (Nashville, TN).
2.5. Treatment of Mn and VPA in mice
Mice were randomly separated into four groups (control, Mn, Mn plus VPA groups, and VPA; 8 mice/group; n=32). Each group contained four C57BL/6J mice and four double transgenic GLT-1-eGFP/GLAST-DsRed mice. Only male mice were used to eliminate any possible influence of female sex hormone estrogens on the experimental outcomes [50, 51]. Mice in the VPA and Mn plus VPA groups were treated with 100 μl of VPA (200 mg/kg, i.p.), whereas control and Mn groups were treated with vehicle saline (0.9% NaCl, i.p.) once daily for 21 consecutive days (Diagram 1) [52, 53]. VPA was dissolved in vehicle saline. After 30 min of VPA or saline, groups of Mn and Mn plus VPA received 2 μl of MnCl2 (30 mg/kg) via intranasal instillation in the left nostril [54] with a slight modification. We also published the same Mn exposure protocols of intranasal instillation [32]. VPA and control groups received 2 μl of distilled water (ddH2O) in the left nostril. For Mn instillation, mice were placed under isoflurane-induced anesthesia for 3 min during pre- and post-instillation periods to sedate and prevent expulsion of Mn from the nostril.
Diagram 1.

Schematic diagram of VPA and Mn treatment in astrocytes and mice Saline was used as a vehicle of VPA. H2O was used as a vehicle of Mn.
2.6. Open-field test
Locomotor activities were assessed on day 21 in Seamless Open field Activity Arenas using Activity Monitor 5 software (Med Associates, Fairfax, VT). Each open-field arena was made of clear Plexiglas, measuring 27.3 cm x 27.3 cm x 20.3 cm, and was covered with a Plexiglas lid containing air holes. Each animal went through an acclimation period (1 trial for 30 min, 1 day before Mn and/or VPA treatment). On day 21, animals (control, Mn, VPA, and Mn plus VPA) commenced open-field testing to monitor total distance traveled. Each mouse was placed in the center of the open-field for observation and the activity measures were recorded for each observed mouse within the 30-min period. Distance traveled for each animal was calculated directly by the Activity Monitor 5 software.
2.7. Rota-rod test
Rota-rod system (Med Associates Inc., Fairfax, VT) was used to measure the time an animal maintains balance on a moving cylinder. Mice were trained for three consecutive days, with one daily session consisting of 3 trials separated by 300 s resting periods. On days 1 and 21, mice treated with designated drugs were placed on the rod and trials were deemed to have started when rod begins start, with speed gradually increasing from 4 rpm to 40 rpm up to 10 min by 0.1 revolutions/s. The fall latency, a time measurement of how fast an experimental subject falls from the rota-rod at various speeds was assessed. If the mouse persisted on the rod for the entire duration, the measurement value was recorded for 650 s. The mean duration for each mouse was used for comparison.
2.8. Immunohistochemistry
EGFP and DsRed fluorescence intensities was analyzed in coronal sections of double transgenic GLT-1-eGFP/GLAST-DsRed mouse brain following consecutive 21 d treatment with saline, Mn, VPA, or Mn plus VPA. From each treatment group (n=2 / group), was used for analysis. These EGFP and DsRed fluorescence intensities represent promoter activities of GLT-1 and GLAST, respectively [48]. In brief, the harvested brain tissues were post-fixed overnight with 4% paraformaldehyde, placed in 30% sucrose in PBS buffer for 24 hours, and frozen immediately. Brain sections were embedded in Tissue-Tek OCT (Sakura), cut in HM525NX cryostat (Thermo Fisher Scientific) at 30 microns, and mounted on SuperFrost Plus slides (VWR, Radnor, PA) and coverslipped. Image analysis of striatal region of the mouse brain was performed with a Ts2R fluorescence microscope (Nikon Instruments, Melville, NY).
Coronal sections from the mouse substantia nigra spanning from the bregma −2.90 to −3.65 mm in 30 μm thickness were prepared for immunohistochemistry to assess TH expression levels as described in our previous study with a few modifications [55]. Two mouse brains and 4 sections per group were used for the TH expression analysis. The slides were incubated with blocking buffer (1X PBS/5% normal serum/0.3% Triton™ X-100) for 1 h at room temperature, washed, and incubated with anti-mouse TH primary antibody (1:250; Santa Cruz Biotechnology) diluted in antibody dilution buffer (1X PBS/1% BSA/0.3% Triton™ X-100) at 4oC. After overnight incubation, tissue sections were incubated with fluorochrome-conjugated secondary antibody Alexa Fluoro 405 (1:500; Santa Cruz Biotechnology) for 1–2 h at room temperature in the dark, washed and coverslipped for analysis of TH expression. Fluorescence intensity of TH expression was assessed in substantia nigra from the same histological location of each sample using a Ts2R fluorescence microscope (Nikon Instruments, Melville, NY), and relative fluorescence intensity was determined as a parameter for TH expression by Image J software (National Institutes of Health (NIH), Bethesda, MD) [56].
2.9. Quantitative real-time PCR
Tissue samples (3 samples/group) were extracted from left midbrain and striatum regions of randomly mixture from C57BL/6J and transgenic mice. We previously confirmed that transgenic mice did not show any difference from the WT in behavioral and GLT-1/GLAST expression in the original characterization [48]. The mRNA levels of GLT-1, GLAST and TH from these regions in each group were analyzed. Briefly, total RNA was extracted from mouse brain tissue using RNeasy Mini Kit (Qiagen, Valencia, CA). The purified RNA was transcribed to cDNA with high-capacity cDNA reverse transcription kit (Applied Biosystems, Foster City, CA). For quantitative real-time PCR (qPCR), the following primers were used: for EAAT1, 5′-ACG GTC ACT GCT GTC ATT G-3′ (forward) and 5′-TGT GAC GAG ACT GGA GAT GA-3′ (reverse); for EAAT2, 5’-ATT CTC GAG CCG CGC CTT CTC AGC CGG CAC-3’ (forward) and 5’-GCC AAG CTT TGG CTT CCC CGA GAG AGC GAT-3’ (reverse); GLT-1, 5′-GCC AAT ACA ACC AAG GCA GTC-3′ (forward) and 5′-TTC ATC CCG TCC TTG AAC TC-3′ (reverse); for GLAST, 5′-GAT CGG AAA CAT GAA GGA GC-3′ (forward) and 5′-CAA GAA GAG GAT GCC CAG AG-3′ (reverse); for TH, 5′-CAC TAT GCC CAC CCC CAG-3′ (forward) and 5′- CGC CGT CCA ATG AAC CTT-3′ (reverse); and for GAPDH, 5′-TCA ACG GGA AGC CCA TCA-3’ (forward) and 5′-CTC GTG GTT CAC ACC CAT CA-3′ (reverse). Total volume of 25 μl for qPCR contained 1μl of cDNA template of each sample 0.4 μM of primers and RT2 SYBR Green qPCR Master Mix (SABiosciences/Qiagen) for each reaction. The PCR was performed in CFX96 real-time detection system (BioRad) with one cycle at 95ºC for 10 min and 40 cycles at 95ºC for 15 sec and 60ºC for 1 min. GAPDH was used to normalize all samples. The web-based RT2 PCR Array Profiler Data Analysis (SABiosciences/Qiagen) was used to analyze the data.
2.10. Preparation of nuclear extracts
After H4 astrocytes were treated with designated compounds, cells were rinsed twice with ice-cold PBS and lysed in hypotonic buffer (20 mM HEPES-KOH, pH 7.4, 10 mM KCl, 2 mM MgCl2) containing 0.5% Nonidet P-40 (NP-40). Cells were carefully collected with a cell scraper and transferred to a microcentrifuge tube. After 15 min on ice, the samples were centrifuged for 5 min at 3,000 rpm at 4°C. The supernatant for cytoplasmic extract was collected, and the pellet (nuclei) was washed with hypotonic buffer without NP-40. To obtain the pure nuclear fractions, the nuclei were incubated in hypertonic buffer (20 mM HEPES-KOH, pH 7.9, 0.4 M NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, and 25% glycerol) on ice for 30 min with periodic vortexing, followed by centrifugation at 20,000 × g for 10 min at 4°C, and the supernatant nucleic extracts were collected and stored at −80°C until used. Brain tissues from midbrain and striatum were processed for nuclear extraction. Briefly, tissues from midbrain and striatum were homogenized with Dounce homogenizer in 1 ml cold PBS lysis buffer (1×PBS, 200 mM PMSF, and 2 mg/kg aprotinin). Homogenate was then transferred to microcentrifuge tube and centrifuged for 5 min at 3,000 rpm at 4°C. Supernatant was removed and nuclei pellet was resuspended in 1 ml extraction lysis buffer (1 M Tris, 5 M NaCl, 1 M MgCl2, 0.5 M EDTA, 12% glycerol, 0.1% NP40, and protease inhibitors). Nuclear pellets in the extraction buffer were sonicated with two 15 sec burst and centrifuged at 20,000 × g for 15 min at 4°C. The supernatant was recovered and used for western blot analysis.
2.11. Western blot analysis
For protein analysis, three mice randomly selected from each group were harvested and brain tissues of left midbrain and striatum were used for protein extraction and further analysis. The protein concentration of homogenized tissue samples [3:1 ratio of radioimmunoprecipitation assay (RIPA) buffer containing protease inhibitors with tissue sample] was determined by the BCA assay. Equal amounts of proteins were run on either 10% SDS-PAGE (GLT-1, GLAST and TH) or 15% SDS-PAGE (Ac-histone H3 and Ac-histone H4) followed by western blot analysis. Primary antibodies for GLT-1 and GLAST (1:5,000 dilution), and those for Ac-histone H3, Ac-histone H4 and TH (1:1,000 dilution), were followed by HRP-conjugated secondary antibody (1:5,000 dilution). All blots were developed using a chemiluminescence detection kit (Pierce, Rockford, IL), followed by blot imaging and quantification with the BioRad ChemDoc Imaging System and Image Lab Software version 5.2.1 (BioRad, Hercules, CA), respectively.
2.12. Glutamate uptake assay
After treated with VPA and Mn for designated time periods, H4 astrocytes were washed twice with prewarmed glutamate uptake buffer (122 mM NaCl, 3.3 mM KCl, 0.4 mM MgSO4, 1.3 mM CaCl2, 1.2 mM KH2PO4, 25 mM HEPES, and 10 mM D-(+)-glucose, pH 7.4). Five μl of prewarmed uptake buffer containing 0.25 μCi/ml L-[3H] glutamate (specific activity, 49.0 Ci/mmol; PerkinElmer Life Sciences) and 100 nM unlabeled glutamate was added to each well. The reactions were terminated by washing three times with ice-cold PBS, immediately followed by cell lysis in 1 ml of 1 N NaOH. An aliquot of 750 μl of each sample was transferred into scintillation vials and neutralized with 75 μl of 10 N HCl. The radioactivity was measured in liquid scintillation counter (LS 6500; Beckman Coulter).
2.13. Statistical Analysis
Data were expressed as a mean ± SEM. Data analyses were carried out with GraphPad software (GraphPad, San Diego, CA). Statistical differences between control and treated groups were determined by Student’s t-test, one-way or two-way analysis of variance (ANOVA) followed by Tukey’s post hoc test with statistical significance set at p<0.05.
3. Results
3.1. VPA reverses Mn-decreased promoter activities of EAAT1 and EAAT2 both in in vitro astrocyte cultures and in vivo mouse brain.
We first determined if VPA increases and attenuates Mn-induced reduction of promoter activities of EAAT1 and EAAT2 in human H4 astrocytes. VPA alone increased EAAT1 and EAAT2 promoter activities in a concentration-dependent manner (Fig. 1A). Concentration of VPA (2 mM) used for the experiments was not toxic to H4 astrocytes, confirmed by cell viability assay (data not shown). Based on these results, VPA 2 mM was used for the rest of the in vitro experiments to determine the effect of VPA to attenuate Mn-induced decrease in expression of EAAT1 and EAAT2. Treatment of astrocytes with VPA (2 mM, 18 h) prior to Mn (250 μM, 6 h) exposure (Diagram 1) reversed Mn-induced decrease in promoter activities of both EAAT1 and EAAT2 (Fig. 1B, p<0.01). We also tested promoter activity in vivo using double transgenic GLT-1-eGFP/GLAST-DsRed reporter mice. Mice were treated with VPA (200 mg/kg, i.p., daily, 21 d) 30 min prior to Mn (30 mg/kg, nostril instillation, daily, 21 d). Coronal sections from the striatal region where Mn is known to accumulate preferentially showed that VPA attenuated Mn-induced reduction promoter activity (Fig. 1C).
Fig. 1. Effects of VPA on Mn-decreased promoter activities of EAAT1 and EAAT2 both in vitro and in vivo.
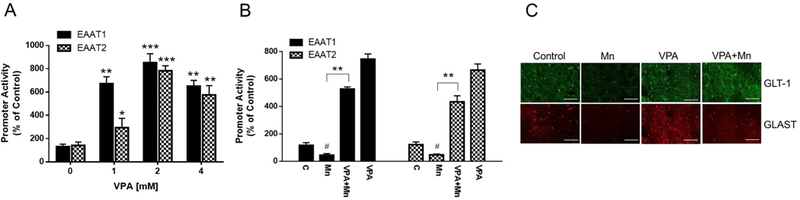
(A) H4 astrocytes were treated with VPA (0, 1, 2, 4 mM) for 24 h and promoter activities of EAAT1 and EAAT2 were measured by luciferase assay as described in the Methods. (B) H4 astrocytes were treated with VPA (2 mM) for 18 h prior to Mn (250 μM) exposure for 6 h. (C) Promoter activities of GLAST and GLT-1 in the mouse brain; DsRed (red) for GLAST and eGFP (green) for GLT-1 in the striatum of GLAST–DsRed/GLT-1–eGFP transgenic mice at 20X magnification. Mice were exposed to 100 μl of VPA (200 mg/kg, i.p. daily) for 30 min prior to Mn exposure (2 μl/30 mg/kg, intranasal instillation, daily) for 21 days as described in the Methods section. Saline (0.9% NaCl, i.p.) and H2O were used as vehicles for VPA and Mn, respectively. #, p < 0.05; *, p < 0.05; **, p < 0.01; ***, p < 0.001 (ANOVA followed by Tukey’s post hoc test; n = 4 for in vitro, n = 2 for in vivo).
3.2. VPA reverses Mn-decreased EAAT1/2 mRNA levels in H4 astrocytes
Next, we tested if these changes in promoter activities were corroborated with mRNA levels of EAAT1 and EAAT2. VPA (2, 4 mM) significantly increased mRNA levels of EAAT1 and EAAT2 (p<0.01) (Fig. 2A). Treatment of astrocytes with VPA prior to Mn exposure reversed Mn-induced reduction in mRNA levels of EAAT1 and EAAT2 (p<0.01) (Fig. 2B).
Fig. 2. Effects of VPA on Mn-decreased mRNA levels of EAAT1 and EAAT2 in astrocytes.

(A) H4 astrocytes were treated with VPA (0, 1, 2, 4 mM), followed by measurement of mRNA levels of EAAT1 and EAAT2 by qPCR. (B) H4 astrocytes were treated with VPA (2 mM) for 18 h prior to Mn (250 μM) for 6 h, followed by mRNA measurement. GAPDH was used for loading controls of mRNA. #, p < 0.05; **, p < 0.01 (ANOVA followed by Tukey’s post hoc test; n = 3).
3.3. VPA reverses Mn-decreased protein levels of EAAT1/2 and glutamate uptake
To determine if VPA modulates Mn effects on EAAT1/2 protein levels, we assessed protein levels of EAAT1 and EAAT2 in H4 astrocytes treated with VPA and Mn. The results showed that VPA alone (2 and 4 mM, 24 h) significantly increased protein levels of EAAT1 (p<0.001) and EAAT2 (p<0.01) (Fig. 3A). Moreover, treatment of astrocytes with VPA (2 mM, 18 h) prior to Mn (250 μM, 6 h) exposure reversed Mn-induced reduction in EAAT1/2 protein levels (p<0.01) (Fig. 3B). Next, we determined if these altered protein levels were corroborated by altered glutamate uptake activity as glutamate uptake is a functional indicator of EAAT1/2 [57]. The results showed that VPA at 2 and 4 mM for 24 h significantly (p<0.001) increased glutamate uptake (Fig. 3C), and reversed Mn-reduced glutamate uptake activity (p<0.01) (Fig. 3D).
Fig. 3. Effects of VPA on Mn-decreased protein levels of EAAT1 and EAAT2, and glutamate uptake in astrocytes.

(A) H4 astrocytes were treated with VPA (0, 1, 2, 4 mM) for 24 h, followed by western blot analysis. (B) Cells were treated with VPA (2 mM) for 18 h prior to Mn (250 μM) exposure for 6 h, followed by western blot. (C) Cells were treated with VPA (0, 1, 2, 4 mM), followed by glutamate uptake assay as described in the Methods section. β-actin was used for loading controls of protein. (D) Cells were treated with VPA (2 mM) prior to Mn (250 μM) exposure, followed by glutamate uptake assay. #, p < 0.05; **, p < 0.01;***, p < 0.001 (ANOVA followed by Tukey’s post hoc test; n = 3 for western blot, n = 4 for glutamate uptake).
3.4. VPA reverses Mn-induced behavioral deficits in mice
We performed the open-field test to determine whether motor function upon treatment with VPA and Mn in C57BL/6J WT and double transgenic mice (as described in the Methods section) was altered. Both WT and transgenic mice showed normal behavior and motor function. Mice treated with Mn (30 mg/kg, intranasal instillation, daily, 21 d) showed decreased total distance traveled (p<0.001) (Fig. 4A) compared to controls. The VPA-treated group (200 mg/kg, i.p., daily, 21 d) exhibited motor activities that were indistinguishable from controls. Treatment of VPA 30 min prior to Mn daily for 21 days attenuated Mn-induced motor deficits (p<0.01). We also performed the rota-rod test to determine if Mn and VPA modulated motor-coordination. As shown in Fig. 4B, Mn significantly decreased motor coordination as measured by the overall latency for falling off the rota-rod (p<0.001). VPA attenuated this Mn-induced reduction (p<0.05), while VPA alone did not alter the overall latency time compared to controls.
Fig 4. Effects of VPA on Mn-induced motor deficits in mice.

(A) Mice (4 mice of C57BL/6J and transgenic were treated with 100 μl of VPA (200 mg/kg, i.p. daily) for 30 min prior to 2 μl of Mn exposure (30 mg/kg, left intranasal instillation, daily) for 21 days. Twenty four h after the last treatment, total distance traveled was measured in open-field system as described in the Methods section. (B) Mice treated with the same paradigm as in (A) were assessed for motor coordination using rota-rod test. Saline (0.9% NaCl, i.p.) and H2O were used as vehicles for VPA and Mn, respectively. ###, p < 0.001; *, p < 0.05; **, p < 0.01 (ANOVA followed by Tukey’s post hoc test; n = 8).
3.5. VPA reverses Mn-decreased mRNA levels of GLT-1 and GLAST in mouse midbrain and striatum
To determine if VPA attenuates Mn-induced impairment of astrocytic glutamate transporters in vivo, we used brain tissues from mice treated with Mn and VPA (as described in the previous sections). Regions of midbrain containing dopaminergic cell bodies in the substantia nigra, and striatum containing dopaminergic nerve terminals were dissected to determine mRNA levels of GLT-1 and GLAST. The results showed that Mn significantly decreased mRNA levels of GLT-1 and GLAST (p<0.001) in midbrain (Fig. 5A and 5B) and striatum (Fig. 5C and 5D). VPA attenuates Mn-induced decrease in mRNA levels of GLT-1 and GLAST in the midbrain (p<0.01) as well as striatal mRNA levels of GLT-1 (p<0.01) and GLAST (p<0.05), while VPA alone was similar to controls.
Fig. 5. Effects of VPA on Mn-decreased mRNA levels of GLT-1 and GLAST in mouse midbrain and striatum.

(A, B) Mice treated with the paradigm described in the Methods were prepared to obtain left midbrain tissues for mRNA analysis of GLT-1 (A) and GLAST (B) by qPCR. (C, D) Mice treated with the same paradigm were prepared to obtain striatal tissues, followed by mRNA analysis of GLT-1 (C) and GLAST (D) by qPCR. GAPDH was used for loading controls of mRNA. ###, p < 0.001; *, p < 0.05; **, p< 0.01 (ANOVA followed by Tukey’s post hoc test; n = 3).
3.6. VPA attenuates Mn-decreased protein levels of GLT-1 and GLAST in mouse brain
We also determined if Mn-induced reduction in mRNA levels of GLT-1 and GLAST was paralleled by changes in protein levels. Notably, Mn decreased protein levels of both GLAST and GLT-1 in midbrain (p<0.001) (Fig. 6A and 6B) as well as in striatum (p<0.001 for GLAST, p<0.01 for GLT-1, respectively, Fig. 6C and 6D). VPA attenuated Mn-induced decrease in GLT-1 and GLAST (p<0.001) protein levels in the midbrain as well as striatum, while the effect of VPA alone was indistinguishable from controls.
Fig. 6. Effects of VPA on Mn-decreased GLT-1 and GLAST protein levels in mouse midbrain and striatum.

(A, B) Mice treated with the paradigm described in the Methods were prepared to obtain left midbrain tissues for protein analysis of GLT-1 (A) and GLAST (B) by western blot. (C, D) Mice treated with the same paradigm were prepared to obtain striatal tissues, followed by protein analysis of GLT-1 (C) and GLAST (D) by western blot. B-actin was used for loading controls of protein. ##, p < 0.01; ###, p < 0.001; ***, p < 0.001 (ANOVA followed by Tukey’s post hoc test; n = 3).
3.7. VPA attenuates Mn-decreased mRNA and protein levels of TH in mouse brain
TH is the rate-limiting enzyme of dopamine biosynthesis in the substantia nigra and serves as a dopaminergic neuronal marker [58–60]. Since motor dysfunction-related diseases such as PD show decreased TH expression in the substantia nigra [23] and VPA has been shown to attenuate Mn-induced reduction in locomotor activity [32], we tested if VPA reversed Mn-induced reduction in motor function was correlated with changes of TH expression in the nigrostriatal pathway. Notably, Mn decreased mRNA and protein levels of TH in midbrain (p<0.01) and striatum (p<0.01 for TH mRNA, p<0.001 for TH protein) compared to controls. VPA attenuated Mn-induced decrease of TH mRNA levels in midbrain (p<0.01) and striatum (p<0.05), as well as TH protein levels in midbrain (p<0.01) and striatum (p<0.001) (Fig. 7A-D). We have also examined TH protein expression by immunohistochemistry in the substantia nigra of the mouse brain. The results showed that Mn significantly decreased the percent of TH expression levels in substantia nigra (p<0.01), while VPA attenuated this Mn-decreased TH expression levels (p<0.01) (Fig. 7E and 7F).
Fig. 7. Effects of VPA on Mn-decreased TH protein/mRNA levels in mouse midbrain and striatum.

(A, B) Midbrain (left) tissues from mice treated with the same paradigm described in the Methods, were analyzed for mRNA levels of TH (A) and protein levels of TH (B). (C, D) Striatal (left) tissues from mice treated with the same paradigm were analyzed for TH mRNA (C) and protein (D) levels. GAPDH and β-actin were used for loading controls of mRNA and protein, respectively. (E, F) Immunohistochemistry was performed to detect TH protein expression in substantia nigra of the mouse brain treated with VPA and Mn using the same paradigm as described in the Methods section. Fluorescence images for TH were captured at 10X magnification (scale bar, 500 μm). Quantification of TH fluorescence intensity of dopaminergic neurons are shown as a percentage of the control. ##, p < 0.01; ###, p < 0.001; *, p < 0.05; **, p < 0.01; ***, p < 0.001 (ANOVA followed by Tukey’s post hoc test; n = 3). Imaging data is representative of 4 samples from two mice per group.
3.8. VPA reverses Mn-decreased acetylation of histone H3 and H4 both in vitro and in vivo
VPA inhibits HDACs which are one of the main classes of histone modifiers. HDACs modulate transcription of variety of genes at the epigenetic level [61, 62]. HDACs have been previously shown to be associated with Mn-induced repression of EAAT1/GLAST and EAAT2/GLT-1 [31, 44]. Accordingly, we tested if VPA attenuation against Mn toxicity is associated with histone acetylation. Mn decreased acetylation of histone H3 and histone H4 (p<0.05) in in vitro astrocyte cultures, while VPA attenuated Mn-decreased acetylation of histone H3 and H4 (p<0.001) (Fig. 8A and 8B). In mouse brain, VPA also attenuated Mn-induced reduction in acetylation of histone H3 and histone H4 (p<0.001) (Fig. 8C and 8D).
Fig. 8. Effects of VPA on Mn-induced reduction in acetylation of histone H3 and histone H4 in vitro and in vivo.

(A, B) H4 astrocyte cells were treated with VPA (2 mM) for 18 h prior to Mn (250 μM) exposure for 6 h, followed by measurement of histone H3 (A) and H4 (B). (C, D) Midbrain (left) tissues from mice treated with the paradigm described in the Methods section were analyzed for protein levels of acetylated histone H3 (C) and H4 (D) by western blot. #, p < 0.05; ###, p < 0.01; ***, p < 0.001 (ANOVA followed by Tukey’s post hoc test; n = 3).
4. Discussion
The findings in the present study demonstrate that VPA reverses Mn-induced decrease in mRNA and protein levels of GLT-1 and GLAST in in vitro astrocytes as well as in the in vivo mouse brain nigrostriatal regions (midbrain and striatum). Furthermore, VPA attenuates Mn-induced reduction of TH expressing dopaminergic neurons in the substantia nigra and striatum, restoring locomotor activity and motor coordination to levels to the control. VPA also reverses Mn-induced reduction in histone acetylation both in vitro astrocyte cultures and in vivo mouse brain.
Mn exposure via intranasal instillation affected nigrostriatal pathway in the mouse brain, corroborating with instilled Mn diffusion into regions of striatum and midbrain [54]. Mn accumulation in these regions, in turn, resulted in impairment in GLT-1 and GLAST function and motor dysfunction accompanied by dopaminergic neurodegeneration. The nigrostriatal pathway is one of the major dopaminergic tracts in the substantia nigra of the midbrain innervating the striatal regions. This tract as a part of the basal ganglia contributes to movement [24, 28]. Depletion of nigrostriatal dopaminergic neurons in PD leads to motor dysfunctions [23], and as evidenced herein, this region is associated with Mn-induced motor function deficits (Fig 4).
VPA is well established anti-epileptic, possibly by modulating GABA neurotransmission [36, 38] as well as HDAC inhibition [37, 63]. Notably, several studies have reported that VPA has protective effects in various neurological disease models by multiple mechanisms including anti-oxidative stress and anti-inflammation [42, 43, 53]. VPA has also shown to inhibit oxidative stress, endoplasmic reticulum stress and apoptotic signaling in neurons after spinal cord injury in rats [64]. In addition, VPA protected an ischemic rat model by reducing oxidative stress and inflammation [65]. Moreover, VPA also protected rotenone- and lactacystin-induced dopaminergic neuronal lesion in substantia nigra of rats by modulating α-synuclein and proteasome [66, 67]. These results indicate that VPA-induced neuroprotection may involve multiple mechanisms in addition to modulating glutamate transporter-related excitatory neurotoxicity possibly by HDAC inhibition, thereby reversing Mn-induced excitotoxic dopaminergic neuronal death. However, further investigation is warranted to test if VPA-induced neuroprotection against Mn-induced dopaminergic neurotoxicity is through reversing astrocytic glutamate transporters via HDAC inhibition. For example, valpromide, a derivative of VPA which does not inhibit HDACs [68], can be used to determine if protective effects of VPA against Mn toxicity is via HDAC inhibition mechanism.
Astrocytic glutamate transporters play a critical role in prevention of excitotoxic neuronal injury in the brain, and therefore, VPA attenuation of Mn-reduced astrocytic glutamate transporters GLAST and GLT-1 could be an important mechanism of VPA-induced neuroprotection from Mn neurotoxicity. VPA increases GLAST expression in chick Bergmann glial cells [69]. Our findings also showed that VPA increased the expression and function of GLT-1 and GLAST in human astrocytes as well as the nigrostriatal pathway of the mouse brain. This indicates that treatment with VPA is efficacious in treating neurological disorders by enhancing GLT-1/GLAST expression [70, 71]. As astrocytic glutamate transporters are critically important in regulating excitotoxic neuronal death, it is possible that VPA exerts neuroprotection by modulating GLT-1 and GLAST [31, 72], thus decreasing dopaminergic neurodegeneration within the nigrostriatal pathway.
Our findings suggest that dopaminergic neurodegeneration may coincide with the dysfunction of astrocytic glutamate transporters. This is consistent with previous studies in which decreased GLT-1 protein levels were associated with Mn-induced dopaminergic neurotoxicity [55] and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP)-induced dopaminergic neurodegeneration [73]. VPA attenuates Mn-induced deficits in locomotor activity and motor coordination, corroborating its reversing effects of Mn-reduced GLT-1 and GLAST promoter activities in mouse striatum, suggesting VPA-modulation of GLT-1/GLAST at the transcriptional level.
The reversing effects of VPA in Mn-induced reduction of histone acetylation in vitro and in vivo (Fig. 8) demonstrates its ability to reverse histone acetylation, a step that might be critically involved in its attenuating effects against Mn neurotoxicity. Some HDACi have been shown to attenuate striatal degeneration in the HD animal model [74], indicating that VPA may exert neuroprotection, at least in part, by modulating histone acetylation. VPA has also been shown to modify amino-terminal tails of histone H3 as well as histone H4 by increasing their acetylation [75]. In cultured astrocytes as well as in vivo mouse brain, both histone H3 and H4 have been shown to be modulated by Mn and VPA, suggesting that astrocytes are a potential target for VPA. In astrocytes, VPA inhibits HDACs, concomitantly reversing Mn-induced decrease in expression and function of astrocytic glutamate transporters to attenuate Mn neurotoxicity [36, 76]. Multiple HDACi have also shown to enhance expression of GLT-1 and GLAST in astrocytes [31, 77]. Future studies are required to determine whether HDAC inhibition is a mechanism of VPA-induced attenuation of Mn-reduced astrocytic glutamate transporters (EAAT1/GLAST and EAAT2/GLT-1) and thereby reversing Mn-induced dopaminergic neuronal injury.
In conclusion, we demonstrate that VPA attenuates Mn-induced reduction of astrocytic GLT-1/GLAST expression parallel with dopaminergic neurotoxicity in mice. Our findings suggest that VPA should be considered for the treatment of Mn neurotoxicity as well as other neurodegenerative diseases associated with glutamate dysregulation and ensuing excitotoxicity. Further studies are warranted to better understand the precise molecular targets of VPA.
Acknowledgements
The present study has been supported in part by R01 ES024756 (EL), SC1 CA200519 (DS), R01 ES10563 (MA), R03 ES024849 (MA) and R21 ES025415 (MA).
Reference
- 1.Bouchard M, et al. , Manganese cumulative exposure and symptoms: A follow-up study of alloy workers. Neurotoxicology, 2008. 29(4): p. 577–583. [DOI] [PubMed] [Google Scholar]
- 2.Pal PK, Samii A, and Calne DB, Manganese neurotoxicity: A review of clinical features, imaging and pathology. Neurotoxicology, 1999. 20(2–3): p. 227–238. [PubMed] [Google Scholar]
- 3.Aschner M, Vrana KE, and Zheng W, Manganese uptake and distribution in the central nervous system (CNS). Neurotoxicology, 1999. 20(2–3): p. 173–80. [PubMed] [Google Scholar]
- 4.Calne DB, et al. , MANGANISM AND IDIOPATHIC PARKINSONISM - SIMILARITIES AND DIFFERENCES. Neurology, 1994. 44(9): p. 1583–1586. [DOI] [PubMed] [Google Scholar]
- 5.Rodier J, Manganese poisoning in Moroccan miners. Br J Ind Med, 1955. 12(1): p. 21–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aschner M, et al. , Manganese and its role in Parkinson’s disease: from transport to neuropathology. Neuromolecular Med, 2009. 11(4): p. 252–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dobson AW, Erikson KM, and Aschner M, Manganese neurotoxicity. Ann N Y Acad Sci, 2004. 1012: p. 115–28. [DOI] [PubMed] [Google Scholar]
- 8.Finley JW and Davis CD, Manganese deficiency and toxicity: are high or low dietary amounts of manganese cause for concern? Biofactors, 1999. 10(1): p. 15–24. [DOI] [PubMed] [Google Scholar]
- 9.Gavin CE, Gunter KK, and Gunter TE, Manganese and calcium transport in mitochondria: implications for manganese toxicity. Neurotoxicology, 1999. 20(2–3): p. 445–53. [PubMed] [Google Scholar]
- 10.Liu X, et al. , Manganese-induced neurotoxicity: the role of astroglial-derived nitric oxide in striatal interneuron degeneration. Toxicol Sci, 2006. 91(2): p. 521–31. [DOI] [PubMed] [Google Scholar]
- 11.Lauderback CM, et al. , The glial glutamate transporter, GLT-1, is oxidatively modified by 4-hydroxy-2-nonenal in the Alzheimer’s disease brain: the role of Abeta1–42. J Neurochem, 2001. 78(2): p. 413–6. [DOI] [PubMed] [Google Scholar]
- 12.Sheldon AL and Robinson MB, The role of glutamate transporters in neurodegenerative diseases and potential opportunities for intervention. Neurochemistry International, 2007. 51(6–7): p. 333–355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tessler S, et al. , Expression of the glutamate transporters in human temporal lobe epilepsy. Neuroscience, 1999. 88(4): p. 1083–91. [DOI] [PubMed] [Google Scholar]
- 14.Trotti D, et al. , Amyotrophic lateral sclerosis-linked glutamate transporter mutant has impaired glutamate clearance capacity. J Biol Chem, 2001. 276(1): p. 576–82. [DOI] [PubMed] [Google Scholar]
- 15.Danbolt NC, Glutamate uptake. Prog Neurobiol, 2001. 65(1): p. 1–105. [DOI] [PubMed] [Google Scholar]
- 16.Bjornsen LP, et al. , The GLT-1 (EAAT2; slc1a2) glutamate transporter is essential for glutamate homeostasis in the neocortex of the mouse. J Neurochem, 2014. 128(5): p. 641–9. [DOI] [PubMed] [Google Scholar]
- 17.Haugeto O, et al. , Brain glutamate transporter proteins form homomultimers. J Biol Chem, 1996. 271(44): p. 27715–22. [DOI] [PubMed] [Google Scholar]
- 18.Mutkus L, et al. , The in vitro uptake of glutamate in GLAST and GLT-1 transfected mutant CHO-K1 cells is inhibited by manganese. Biol Trace Elem Res, 2005. 107(3): p. 221–30. [DOI] [PubMed] [Google Scholar]
- 19.Sidoryk-Wegrzynowicz M, et al. , Manganese disrupts astrocyte glutamine transporter expression and function. J Neurochem, 2009. 110(3): p. 822–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sidoryk-Wegrzynowicz M and Aschner M, Role of astrocytes in manganese mediated neurotoxicity. BMC Pharmacology & Toxicology, 2013. 14: p. 23–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Erikson KM, et al. , Duration of airborne-manganese exposure in rhesus monkeys is associated with brain regional changes in biomarkers of neurotoxicity. Neurotoxicology, 2008. 29(3): p. 377–85. [DOI] [PubMed] [Google Scholar]
- 22.Lin CLG, et al. , Glutamate transporter EAAT2: a new target for the treatment of neurodegenerative diseases. Future Medicinal Chemistry, 2012. 4(13): p. 1689–1700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dauer W and Przedborski S, Parkinson’s Disease: Mechanisms and Models. Neuron, 2003. 39(6): p. 889–909. [DOI] [PubMed] [Google Scholar]
- 24.Schultz W, Getting Formal with Dopamine and Reward. Neuron, 2002. 36(2): p. 241–263. [DOI] [PubMed] [Google Scholar]
- 25.Stanwood GD, et al. , Manganese exposure is cytotoxic and alters dopaminergic and GABAergic neurons within the basal ganglia. J Neurochem, 2009. 110(1): p. 378–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhao F, et al. , Manganese Induces Dopaminergic Neurodegeneration via Microglial Activation in a Rat Model of Manganism. Toxicological Sciences, 2009. 107(1): p. 156–164. [DOI] [PubMed] [Google Scholar]
- 27.Liu M, et al. , Effect of microglia activation on dopaminergic neuronal injury induced by manganese, and its possible mechanism. Neurotox Res, 2009. 16(1): p. 42–9. [DOI] [PubMed] [Google Scholar]
- 28.Robbins TW and Everitt BJ, Functions of dopamine in the dorsal and ventral striatum. Seminars in Neuroscience, 1992. 4(2): p. 119–127. [Google Scholar]
- 29.Brouillet EP, et al. , Manganese injection into the rat striatum produces excitotoxic lesions by impairing energy metabolism. Exp Neurol, 1993. 120(1): p. 89–94. [DOI] [PubMed] [Google Scholar]
- 30.Erikson K and Aschner M, Manganese causes differential regulation of glutamate transporter (GLAST) taurine transporter and metallothionein in cultured rat astrocytes. Neurotoxicology, 2002. 23(4–5): p. 595–602. [DOI] [PubMed] [Google Scholar]
- 31.Karki P, et al. , Yin Yang 1 is a repressor of glutamate transporter EAAT2, and it mediates manganese-induced decrease of EAAT2 expression in astrocytes. Mol Cell Biol, 2014. 34(7): p. 1280–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Johnson J Jr., et al. , Valproate and sodium butyrate attenuate manganese-decreased locomotor activity and astrocytic glutamate transporters expression in mice. Neurotoxicology, 2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dokmanovic M, Clarke C, and Marks PA, Histone deacetylase inhibitors: Overview and perspectives. Molecular Cancer Research, 2007. 5(10): p. 981–989. [DOI] [PubMed] [Google Scholar]
- 34.Zhang H, et al. , Histone deacetylases function as novel potential therapeutic targets for cancer. Hepatology Research, 2017. 47(2): p. 149–159. [DOI] [PubMed] [Google Scholar]
- 35.Karki P, et al. , Role of transcription factor yin yang 1 in manganese-induced reduction of astrocytic glutamate transporters: Putative mechanism for manganese-induced neurotoxicity. Neurochem Int, 2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Johannessen CU, Mechanisms of action of valproate: a commentatory. Neurochemistry International, 2000. 37(2–3): p. 103–110. [DOI] [PubMed] [Google Scholar]
- 37.Ghodke-Puranik Y, et al. , Valproic acid pathway: pharmacokinetics and pharmacodynamics. Pharmacogenet Genomics, 2013. 23(4): p. 236–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Johannessen CU and Johannessen SI, Valproate: past, present, and future. CNS Drug Rev, 2003. 9(2): p. 199–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Shelton CE and Connelly JF, Valproic acid: a migraine prophylaxis alternative. Ann Pharmacother, 1996. 30(7–8): p. 865–6. [DOI] [PubMed] [Google Scholar]
- 40.Wu X, et al. , Histone deacetylase inhibition leads to neuroprotection through regulation on glial function. Molecular Neurodegeneration, 2013. 8(Suppl 1): p. P49–P49. [Google Scholar]
- 41.Wu X, et al. , Histone deacetylase inhibitors up-regulate astrocyte GDNF and BDNF gene transcription and protect dopaminergic neurons. Int J Neuropsychopharmacol, 2008. 11(8): p. 1123–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kimura A, et al. , Valproic acid prevents retinal degeneration in a murine model of normal tension glaucoma. Neurosci Lett, 2015. 588: p. 108–13. [DOI] [PubMed] [Google Scholar]
- 43.Morland C, et al. , Valproate is neuroprotective against malonate toxicity in rat striatum: an association with augmentation of high-affinity glutamate uptake. J Cereb Blood Flow Metab, 2004. 24(11): p. 1226–34. [DOI] [PubMed] [Google Scholar]
- 44.Karki P, et al. , Transcriptional Regulation of the Astrocytic Excitatory Amino Acid Transporter 1 (EAAT1) via NF-kappa B and Yin Yang 1 (YY1). Journal of Biological Chemistry, 2015. 290(39): p. 23725–23737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Itoh M, et al. , Trichostatin A enhances glutamate transporter GLT-1 mRNA levels in C6 glioma cells via neurosteroid-mediated cell differentiation. J Mol Neurosci, 2013. 49(1): p. 21–7. [DOI] [PubMed] [Google Scholar]
- 46.Aguirre G, et al. , Valproate-dependent transcriptional regulation of GLAST/EAAT1 expression: involvement of Ying-Yang 1. Neurochem Int, 2008. 52(7): p. 1322–31. [DOI] [PubMed] [Google Scholar]
- 47.Kim SY, et al. , Transcriptional regulation of human excitatory amino acid transporter 1 (EAAT1): cloning of the EAAT1 promoter and characterization of its basal and inducible activity in human astrocytes. J Neurochem, 2003. 87(6): p. 1485–98. [DOI] [PubMed] [Google Scholar]
- 48.Regan MR, et al. , Variations in promoter activity reveal a differential expression and physiology of glutamate transporters by glia in the developing and mature CNS. J Neurosci, 2007. 27(25): p. 6607–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tawfik VL, et al. , Propentofylline-Induced Astrocyte Modulation Leads to Alterations in Glial Glutamate Promoter Activation Following Spinal Nerve Transection. Neuroscience, 2008. 152(4): p. 1086–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dhandapani KM and Brann DW, Role of astrocytes in estrogen-mediated neuroprotection. Experimental Gerontology, 2007. 42(1–2): p. 70–75. [DOI] [PubMed] [Google Scholar]
- 51.Lee ESY, et al. , Estrogen and tamoxifen reverse manganese-induced glutamate transporter impairment in astrocytes. Journal of Neurochemistry, 2009. 110(2): p. 530–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Loscher W, Rundfeldt C, and Honack D, Pharmacological characterization of phenytoin-resistant amygdala-kindled rats, a new model of drug-resistant partial epilepsy. Epilepsy Res, 1993. 15(3): p. 207–19. [DOI] [PubMed] [Google Scholar]
- 53.Zaky A, et al. , Valproic acid potentiates curcumin-mediated neuroprotection in lipopolysaccharide induced rats. Frontiers in Cellular Neuroscience, 2014. 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Kim J, et al. , Iron-responsive olfactory uptake of manganese improves motor function deficits associated with iron deficiency. PLoS One, 2012. 7(3): p. e33533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Pajarillo E, et al. , 17β-estradiol and tamoxifen protect mice from manganese-induced dopaminergic neurotoxicity. NeuroToxicology. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.McCloy RA, et al. , Partial inhibition of Cdk1 in G 2 phase overrides the SAC and decouples mitotic events. Cell Cycle, 2014. 13(9): p. 1400–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Danbolt NC, et al. , Properties and localization of glutamate transporters. Prog Brain Res, 1998. 116: p. 23–43. [DOI] [PubMed] [Google Scholar]
- 58.Kaufman S, Coenzymes and hydroxylases: ascorbate and dopamine-beta-hydroxylase; tetrahydropteridines and phenylalanine and tyrosine hydroxylases. Pharmacol Rev, 1966. 18(1): p. 61–9. [PubMed] [Google Scholar]
- 59.Kaufman S, Regulatory properties of tyrosine hydroxylase. Adv Biochem Psychopharmacol, 1975. 13: p. 127–36. [PubMed] [Google Scholar]
- 60.Kaufman S, Tyrosine hydroxylase. Adv Enzymol Relat Areas Mol Biol, 1995. 70: p. 103–220. [DOI] [PubMed] [Google Scholar]
- 61.Khochbin S and Wolffe AP, The origin and utility of histone deacetylases. FEBS Lett, 1997. 419(2–3): p. 157–60. [DOI] [PubMed] [Google Scholar]
- 62.Khochbin S, et al. , Functional significance of histone deacetylase diversity. Curr Opin Genet Dev, 2001. 11(2): p. 162–6. [DOI] [PubMed] [Google Scholar]
- 63.Phiel CJ, et al. , Histone deacetylase is a direct target of valproic acid, a potent anticonvulsant, mood stabilizer, and teratogen. J Biol Chem, 2001. 276(39): p. 36734–41. [DOI] [PubMed] [Google Scholar]
- 64.Lee JY, et al. , Valproic Acid Protects Motor Neuron Death by Inhibiting Oxidative Stress and Endoplasmic Reticulum Stress-Mediated Cytochrome C Release after Spinal Cord Injury. Journal of Neurotrauma, 2014. 31(6): p. 582–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Suda S, et al. , Valproic acid attenuates ischemia-reperfusion injury in the rat brain through inhibition of oxidative stress and inflammation. Eur J Pharmacol, 2013. 707(1–3): p. 26–31. [DOI] [PubMed] [Google Scholar]
- 66.Monti B, et al. , Valproic acid is neuroprotective in the rotenone rat model of Parkinson’s disease: involvement of alpha-synuclein. Neurotox Res, 2010. 17(2): p. 130–41. [DOI] [PubMed] [Google Scholar]
- 67.Harrison IF, Anis HK, and Dexter DT, Associated degeneration of ventral tegmental area dopaminergic neurons in the rat nigrostriatal lactacystin model of parkinsonism and their neuroprotection by valproate. Neurosci Lett, 2016. 614: p. 16–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Eyal S, et al. , Histone deacetylases inhibition and tumor cells cytotoxicity by CNS-active VPA constitutional isomers and derivatives. Biochem Pharmacol, 2005. 69(10): p. 1501–8. [DOI] [PubMed] [Google Scholar]
- 69.Rosas S, et al. , Glutamate-dependent transcriptional regulation of GLAST/EAAT1: a role for YY1. J Neurochem, 2007. 101(4): p. 1134–44. [DOI] [PubMed] [Google Scholar]
- 70.Yoo YE and Ko CP, Treatment with trichostatin A initiated after disease onset delays disease progression and increases survival in a mouse model of amyotrophic lateral sclerosis. Exp Neurol, 2011. 231(1): p. 147–59. [DOI] [PubMed] [Google Scholar]
- 71.Cui SS, et al. , Suberoylanilide hydroxamic acid prevents downregulation of spinal glutamate transporter-1 and attenuates spinal nerve ligation-induced neuropathic pain behavior. Neuroreport, 2016. 27(6): p. 427–34. [DOI] [PubMed] [Google Scholar]
- 72.Karki P, et al. , Genetic Dys-regulation of Astrocytic Glutamate Transporter EAAT2 and its Implications in Neurological Disorders and Manganese Toxicity. Neurochem Res, 2015. 40(2): p. 380–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hsu CY, et al. , Ceftriaxone prevents and reverses behavioral and neuronal deficits in an MPTP-induced animal model of Parkinson’s disease dementia. Neuropharmacology, 2015. 91: p. 43–56. [DOI] [PubMed] [Google Scholar]
- 74.Ferrante RJ, et al. , Histone deacetylase inhibition by sodium butyrate chemotherapy ameliorates the neurodegenerative phenotype in Huntington’s disease mice. Journal of Neuroscience, 2003. 23(28): p. 9418–9427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Wang Z, et al. , Chronic valproate treatment enhances postischemic angiogenesis and promotes functional recovery in a rat model of ischemic stroke. Stroke, 2012. 43(9): p. 2430–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Chen PS, et al. , Valproate protects dopaminergic neurons in midbrain neuron/glia cultures by stimulating the release of neurotrophic factors from astrocytes. Mol Psychiatry, 2006. 11(12): p. 1116–25. [DOI] [PubMed] [Google Scholar]
- 77.Karki P, et al. , Transcriptional Regulation of the Astrocytic Excitatory Amino Acid Transporter 1 (EAAT1) via NF-kappaB and Yin Yang 1 (YY1). J Biol Chem, 2015. 290(39): p. 23725–37. [DOI] [PMC free article] [PubMed] [Google Scholar]