

Abstract
Serine protease inhibitors of the Kunitz-bovine pancreatic trypsin inhibitor (BPTI) family are ubiquitous biological regulators of proteolysis. These small proteins are resistant to proteolysis, but can be slowly cleaved within the protease-binding loop by target proteases, thereby compromising their activity. For the human protease mesotrypsin, this cleavage is especially rapid. Here, we aimed to stabilize the Kunitz domain structure against proteolysis through disulfide engineering. Substitution within the Kunitz inhibitor domain of the amyloid precursor protein (APPI) that incorporated a new disulfide bond between residues 17 and 34 reduced proteolysis by mesotrypsin 74-fold. Similar disulfide engineering of tissue factor pathway inhibitor-1 Kunitz domain 1 (KD1TFPI1) and bikunin Kunitz domain 2 (KD2bikunin) likewise stabilized these inhibitors against mesotrypsin proteolysis 17- and 6.6-fold, respectively. Crystal structures of disulfide-engineered APPI and KD1TFPI1 variants in a complex with mesotrypsin at 1.5 and 2.0 Å resolution, respectively, confirmed the formation of well-ordered disulfide bonds positioned to stabilize the binding loop. Long all-atom molecular dynamics simulations of disulfide-engineered Kunitz domains and their complexes with mesotrypsin revealed conformational stabilization of the primed side of the inhibitor-binding loop by the engineered disulfide, along with global suppression of conformational dynamics in the Kunitz domain. Our findings suggest that the Cys-17–Cys-34 disulfide slows proteolysis by dampening conformational fluctuations in the binding loop and minimizing motion at the enzyme–inhibitor interface. The generalizable approach developed here for the stabilization against proteolysis of Kunitz domains, which can serve as important scaffolds for therapeutics, may thus find applications in drug development.
Keywords: protease inhibitor, proteolysis, serine protease, protein engineering, disulfide, protein structure, molecular dynamics, crystal structure
Introduction
Intramolecular disulfide bonds in proteins are covalent cross-links formed between two Cys residues within the macromolecule. The formation of such disulfide bonds, a common post-translational modification of secreted proteins, promotes thermal stability, correct folding, and biological activity (1–3). Disulfide bonds also contribute to maintaining protein integrity, because by stabilizing the protein structure and preventing unfolding, they also reduce the susceptibility of the protein to enzymatic degradation by proteases (4). Consequently, protein disulfide engineering, namely the introduction of non-native disulfide bonds to enhance protein function and robustness, has been explored as a means to improve the stability and half-life of protein therapeutics and to expand the operational range of enzymes for many industrial applications (5).
A functional class of proteins that are naturally highly enriched in disulfide bonds are the canonical inhibitors of serine proteases (6), which belong to a variety of convergently evolved protein families (6, 7). In these proteins, nature has taken full advantage of the stabilizing potential of disulfide linkages to produce molecules that are both highly stable to proteolysis and thermodynamically stable to unfolding processes. Common to these inhibitors is a protease-binding loop of highly characteristic “canonical” backbone conformation (8), which, in most cases, is encompassed within at least one disulfide-bridged loop or knot in the protein structure (9). They inhibit target proteases via the Laskowski mechanism (6, 9) by “posing” as ideal substrates, i.e. by binding extremely tightly with a specific reactive site peptide bond positioned for cleavage by the target enzyme and yet undergoing proteolysis many orders of magnitude more slowly than a typical substrate. An additional feature of the Laskowski mechanism is that cleavage of the reactive site bond is reversible; the cleaved inhibitor, held together by disulfide bonds and other intramolecular interactions, can rebind to the protease and undergo peptide bond resynthesis (6, 9). In vitro, the cleavage-resynthesis competition may eventually achieve thermodynamic equilibrium, and thus the interconvertible inhibitor forms may be considered equivalent thermodynamic inhibitors. However, such equilibration is often much slower than experimental or biological time scales, and the poorer inhibition kinetics of the cleaved inhibitor (9) can compromise its inhibitory activity for practical purposes. Furthermore, cleavage at the reactive site can render the inhibitor susceptible to inactivating cleavages at other sites by the same protease, and in biological settings it may lead to degradation by alternative proteases (10).
A well-studied family of such canonical inhibitors comprises the Kunitz-BPTI4 (MEROPS family I2 (11)) inhibitors, which are highly represented in humans and most other animal species. Kunitz-BPTI inhibitors are compact 58-residue globular protein domains that are highly stabilized by the presence of three native disulfide bonds. Despite the impressive resistance of Kunitz domains to proteolysis, they do undergo slow cleavage by target serine proteases at the reactive site bond, resulting in diminished inhibitory capability. For example, the Kunitz protease inhibitor domain of amyloid precursor protein (APPI) inhibits target proteases, such as trypsin and factor XIa, >100-fold more weakly after its cleavage at the reactive site bond (12). Furthermore, a subset of serine proteases, most notably among them human mesotrypsin, has evolved enhanced capability for proteolysis and inactivation of these inhibitors (12–17). In a previous study that identified structural and dynamic determinants of resistance versus susceptibility of different Kunitz domain inhibitors to mesotrypsin cleavage, we found that proteolysis rates correlate closely with the conformational dynamics of the inhibitors (18). We also found that a variant of APPI, evolved through directed evolution for improved affinity and resistance to mesotrypsin proteolysis, exhibited subtle backbone shifts, resulting in improved intramolecular and intermolecular stabilizing interactions (19), and hence reduced amplitude of conformational fluctuations, with consequent stabilization against proteolysis (18).
Our recent study thus illustrated the important role of substrate dynamics in proteolysis, revealing that even a tightly structured Kunitz domain with three disulfide bonds can undergo extensive acrobatic contortions, involving partial unfolding, as a potentially rate-limiting step in the process of proteolysis (18). On the basis of these observations, we hypothesized that it may be possible to further stabilize the Kunitz domain structure against proteolysis by the introduction of a fourth disulfide bond. In this study, we demonstrate that a novel engineered disulfide bond, placed so as to rigidify the canonical binding loop, can improve both affinity and proteolytic stability toward serine protease targets, such as mesotrypsin. Here, we incorporated an additional disulfide bond into the Kunitz inhibitors, APPI, tissue factor pathway inhibitor-1 Kunitz domain 1 (KD1TFPI1), and bikunin Kunitz domain 2 (KD2bikunin), and found that the mutant proteins exhibited reduced proteolysis by mesotrypsin. By solving the crystal structures of APPI and KD1TFPI1 variants in a complex with mesotrypsin, we demonstrated that the new engineered disulfide bonds are well-structured and positioned appropriately to stabilize the binding loop. In addition, molecular dynamics (MD) simulations based on our crystal structures provided support for a suppressive effect of the engineered disulfide bonds on the dynamics of the binding loops of APPI and KD1TFPI1 variants, thereby explaining their enhanced resistance to proteolysis. This type of disulfide engineering may be used to improve many Kunitz domain scaffolds that are currently used in approved and developmental protein therapeutics targeting serine proteases of pharmacological interest, thereby highlighting the potential value of this approach for biopharmaceutical applications.
Results
Introduction of an additional novel disulfide bond into the APPI Kunitz domain improves stability against proteolysis by mesotrypsin
Based on our previous work in which we showed that the susceptibility to proteolysis of different Kunitz domains depends on the magnitude of their conformational fluctuations (18), we hypothesized here that proteolysis can be intentionally slowed by design, i.e. through the introduction of an additional disulfide bond to reduce conformational mobility. Examination of the structures of APPI and APPIM17G/I18F/F34V, a triple variant previously optimized to enhance affinity and proteolytic stability toward mesotrypsin (19), suggested several possible residues for mutation. We focused initially on residue 34, an important residue within the inhibitor scaffold that lies below the binding loop (Fig. 1A). In the APPIM17G/I18F/F34V variant, the F34V mutation optimizes intramolecular packing, resulting in an H-bond between the Val-34 carbonyl and the Thr-11 side chain and a closer packing arrangement between this side chain and binding loop residue Gly-17 (Fig. 1A) (19). We reasoned that these contacts, which potentially contribute to the increased conformational stability and proteolytic resistance of APPIM17G/I18F/F34V, could be further strengthened by engineering them to be covalent disulfide bonds (Fig. 1, B and C).
Figure 1.
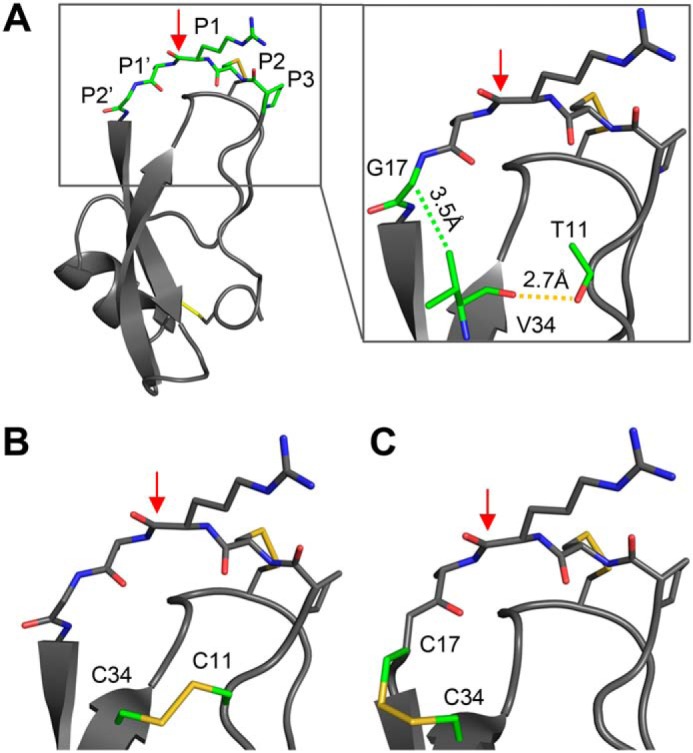
Placement of engineered disulfide bonds in APPI variants. A, structure of APPIM17G/I18F/F34V, with the binding loop colored green (coordinates from PDB code 5C67 (19)). The close-up view of APPIM17G/I18F/F34V shows the H-bond of Val-34 with Thr-11 (dashed yellow line) and the van der Waals interaction of Val-34 with Gly-17 (dashed green line). B, predicted structure of APPIT11C/M17G/I18F/F34C with a disulfide bond between residues 11 and 34. C, predicted structure of APPIM17C/I18F/F34C with a disulfide bond between residues 17 and 34. In each panel, the proteolysis-susceptible Arg-15–Ala-16 bond is indicated by a red arrow.
To test this hypothesis, we used site-directed mutagenesis to generate two new mutant constructs based on APPIM17G/I18F/F34V, one incorporating a pair of Cys residues at positions 11 and 34 (APPIT11C/M17G/I18F/F34C), and the other incorporating a pair of Cys residues at positions 17 and 34 (APPIM17C/I18F/F34C). Notably, the Cα separations of the residue pairs targeted for mutagenesis (5.9 and 5.5 Å for residues 11–34 and 17–34, respectively) lie well within the range typical of natural disulfide bonds (3.8–6.6 Å) (20), suggesting that the engineered disulfide bonds could be accommodated by the APPIM17G/I18F/F34V structure. The soluble recombinant mutant proteins were expressed and purified from Pichia pastoris.
We next evaluated the ability of the disulfide-engineered APPI variants to inhibit mesotrypsin, a step that was designed to confirm whether the mutant proteins retain the correct protein folding and conformational presentation of the binding loop in a manner compatible with protease inhibitory activity. Both engineered disulfide variants did indeed retain inhibitory activity consistent with correct folding, but the potency of the APPIT11C/M17G/I18F/F34C variant for mesotrypsin inhibition was significantly diminished relative to the parental variant APPIM17G/I18F/F34V. The inhibitory kinetics of APPIT11C/M17G/I18F/F34C toward mesotrypsin was determined using a classic competitive inhibition approach, yielding data well-fitted by the competitive inhibition model (Fig. 2, A and B), with an equilibrium inhibition constant (Ki) of 150 nm. Ki for APPIT11C/M17G/I18F/F34C was >1500-fold weaker than that of the parental variant APPIM17G/I18F/F34V but similar to that of wildtype (WT) APPI (Table 1). In crystal structures, APPI residues 11 and 34 do not make extensive contact with mesotrypsin, and their substitution for Cys residues is not expected to introduce any direct steric clash with mesotrypsin that would explain the diminished binding. Instead, our inhibition data may suggest that introduction of the disulfide bond between residues 11 and 34 may subtly alter the shape of the APPI-binding loop and stabilize it in a conformation with a less compatible fit to the enzyme. This explanation is also consistent with the much faster dissociation rate koff measured for APPIT11C/M17G/I18F/F34C relative to other APPI variants (Fig. 2C and Table 1).
Figure 2.

Disulfide bond engineering preserves the mesotrypsin inhibitory capability of APPI variants. Representative examples are shown for determination of kinetic constants for mesotrypsin inhibition by APPI disulfide variants. A and B, mesotrypsin cleavage of the peptide substrate Z-GPR-pNA is inhibited by APPIT11C/M17G/I18F/F34C with a kinetic profile fitting the competitive model of inhibition. Concentrations of APPIT11C/M17G/I18F/F34C are given at the top of each plot; concentration of mesotrypsin was 0.25 nm. A, data were fitted globally to the competitive inhibition equation to obtain the equilibrium inhibition constant Ki. B, Lineweaver-Burk transform of the data shown in A converges on the y axis, as is characteristic of the competitive model of inhibition. C, progress curve is shown for the recovery of mesotrypsin activity due to dissociation from APPIT11C/M17G/I18F/F34C upon 60-fold dilution into assay buffer containing 150 μm Z-GPR-pNA substrate. Experimental values are shown in black, and the fitted curve is shown as a dashed red line. Curve fitting was performed as described under “Experimental procedures” to determine first-order rate constant koff, which describes the transition from complete inhibition to a steady-state rate of partial inhibition. D–F, slow, tight-binding inhibition of mesotrypsin by APPIM17C/I18F/F34C. D, progress curves are shown for the inhibition of mesotrypsin cleavage of the peptide substrate Z-GPR-pNA (150 μm) by APPIM17C/I18F/V34C. Experimental values are shown in black and fitted curves, as dashed red lines, with APPIM17C/I18F/V34C concentrations indicated on the right. Vs and V0 are the steady-state rates in the presence and absence of inhibitor, and Kobs is the observed first-order rate constant, which describes the transition from V0 to Vs from which the kinetic constants were calculated as described under “Experimental procedures.” E, values of the equilibrium inhibition constant Ki were calculated from the steady-state portion of the progress curves from D as described under “Experimental procedures.” F, plot showing the linear dependence of Kobs on APPIM17C/I18F/V34C concentration facilitated the calculation of kon and koff as described under “Experimental procedures.”
Table 1.
Kinetic constants of the reactions between mesotrypsin and different Kunitz type inhibitors
| Inhibitor | Ki | Ki (fold decrease) | kon | koff | kcata | kcat (fold decrease) |
|---|---|---|---|---|---|---|
| m | m−1·s−1 | s−1 | s−1 | |||
| APPIWT | (1.3 ± 0.2) × 10−7b,c | (2.4 ± 0.3) × 106i | (3.1 ± 0.02) × 10−1f | (35.6 ± 2.3) × 10−3c | ||
| APPII18F | (3.3 ± 0.2) × 10−9b,c | (5.9 ± 0.4) × 106i | (19.5 ± 0.7) × 10−3f | (10.4 ± 0.9) × 10−3c | ||
| APPIM17G/I18F/F34V | (9.8 ± 0.1) × 10−11f,g | 34h | (5.0 ± 0.1) × 106g,i | (5.5 ± 0.1) × 10−4g,i | (4.3 ± 0.3) × 10−4c | 24h |
| APPIT11C/M17G/I18F/F34C | (1.5 ± 0.1) × 10−7b | (4.1 ± 0.3) × 106i | (6.0 ± 0.04) × 10−1f | (5.2 ± 0.1) × 10−3 | ||
| APPIM17C/I18F/F34C | (6.1 ± 0.1) × 10−11f | 54h | (15.5 ± 0.2) × 106i | (1.0 ± 0.01) × 10−3i | (1.4 ± 0.02) × 10−4 | 74h |
| KD1TFPI1K15R | (4.2 ± 0.6) × 10−5b | (8.9 ± 0.1) × 10−1 | ||||
| KD1TFPI1K15R/I17C/I34C | (2.0 ± 0.1) × 10−7b | 215h | (5.1 ± 0.1) × 10−2 | 17h | ||
| KD2Bikunin | (5. 1 ± 0.1) × 10−8b | (6.6 ± 0.4) × 10−3 | ||||
| KD2BikuninF17C/P34C | (4. 5 ± 0.1) × 10−8b | 1.13h | (1.0 ± 0.1) × 10−3 | 6.6h |
a kcat was determined by HPLC-based inhibitor cleavage time course.
b Data were determined using Equation 1 (see “Experimental procedures”).
c Results were taken from Ref. 19.
d Data were determined using Equation 2 (see “Experimental procedures”).
e Data were determined using Equation 3 (see “Experimental procedures”).
f Data were determined using Equation 7 (see “Experimental procedures”).
g Results were taken from Ref. 45.
h Fold decrease relative to the parent protein scaffolds APPII18F, KD1TFPI1K15R, or KD2bikunin.
i Data were determined using Equation 8 (see “Experimental procedures”).
In contrast, the APPIM17C/I18F/F34C variant retained the extremely potent inhibitory activity of the parental APPIM17G/I18F/F34V variant toward mesotrypsin and even showed modest improvement. Affinity of this variant toward mesotrypsin exceeded the practical limit of determination using the classical competitive inhibition treatment, and we therefore used an alternative kinetic treatment suitable for the quantification of slow, tight-binding behavior (Fig. 2, D–F). The Ki value of 61 pm so obtained was ∼1.5-fold stronger than that of the parental APPIM17G/I18F/F34V variant measured by the same procedure (Table 1). The improved inhibition relative to APPIM17G/I18F/F34Vappears to be driven by a faster kon (Table 1), suggesting that the presence of the Cys-17–Cys-34 disulfide bond may stabilize the binding loop in a conformation better pre-configured for association with the enzyme. Compared with WT APPI (APPIWT) or with APPII18F, a single mutant that differs from APPIM17C/I18F/F34C only at the two introduced Cys positions, APPIM17C/I18F/F34C showed improved inhibition by >2000-fold or >50-fold, respectively (Table 1).
We then went on to determine whether incorporation of engineered disulfide bonds into the APPIT11C/M17G/I18F/F34C and APPIM17C/I18F/F34C variants had indeed succeeded in conferring improved resistance to proteolysis by mesotrypsin. To test the proteolytic stability of the inhibitors, we measured their rates of cleavage (kcat) by mesotrypsin in an HPLC-based time-course hydrolysis assay (Fig. 3). APPIWT was previously shown to undergo cleavage by mesotrypsin specifically at the Arg-15–Ala-16 reactive site bond (12), and because the disulfide mutant inhibitors bind and inhibit mesotrypsin even more strongly than does APPIWT (Table 1), the same bond is expected to be targeted for cleavage in the mutants. APPIT11C/M17G/I18F/F34C was ∼12-fold more susceptible to mesotrypsin proteolysis than the parental APPIM17G/I18F/F34V variant, although still ∼7-fold more resistant than APPIWT (Table 1). In contrast, the APPIM17C/I18F/F34C variant displayed a mesotrypsin cleavage rate that was 250-fold slower than that of APPIWT, 74-fold slower than that of APPII18F, and 3-fold slower than that of the parental APPIM17G/I18F/F34V variant, which had previously been optimized for resistance to proteolysis using a directed evolution strategy (Table 1) (19). Thus, introduction of a disulfide linkage between residues 17 and 34 of APPI appears to be an effective approach for stabilization against proteolysis by mesotrypsin, while also maintaining and enhancing binding affinity toward mesotrypsin.
Figure 3.
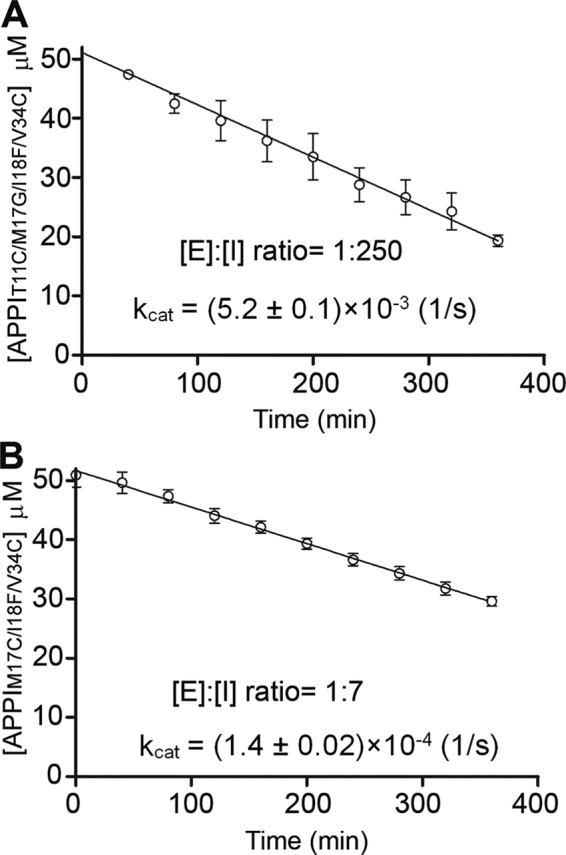
Disulfide bond engineering modulates the stability of APPI variants to proteolysis by mesotrypsin. A, time course showing APPIT11C/M17G/I18F/F34C hydrolysis by mesotrypsin in a reaction containing 50 μm APPIT11C/M17G/I18F/F34C and 200 nm enzyme. Disappearance of intact APPIT11C/M17G/I18F/F34C was quantified by integration of HPLC peaks over the time course. B, time course showing APPIM17C/I18F/F34C hydrolysis by mesotrypsin in a reaction containing 50 μm APPIM17C/I18F/F34C and 7.14 μm enzyme. Disappearance of intact APPIM17C/I18F/F34C was quantified by integration of HPLC peaks over the time course. Note the higher ratio of mesotrypsin to APPIM17C/I18F/F34C required for cleavage of this more proteolytically-resistant variant on a similar time scale. For each variant, kcat was calculated from the observed initial rate of hydrolysis averaged over at least three independent experiments.
Disulfide bridging between residues 17 and 34 offers a general approach to stabilize Kunitz domain serine protease inhibitors against proteolysis
Several human Kunitz protease inhibitor domains have been studied as promising scaffolds for drug development, yet stability against proteolysis in vivo remains a concern, as it does for therapeutic proteins in general. We hypothesized that incorporation of an engineered disulfide bond between residues 17 and 34 into other Kunitz domain scaffolds of pharmaceutical interest might offer a general method for stabilization against proteolysis. To test this idea, we expressed and purified new disulfide-containing and WT proteins of two additional representative Kunitz domains, namely KD1TFPI1 and KD2bikunin. For KD1TFPI1, a particularly weak inhibitor of mesotrypsin (17), we also introduced a K15R mutation at the P1 primary specificity site in an attempt to strengthen its preexisting affinity for mesotrypsin, as the enzyme is known to prefer Arg over Lys at this position (21). We then evaluated the equilibrium inhibition constants (Ki) for mesotrypsin and rates of proteolysis (kcat) by mesotrypsin, using approaches similar to those employed for the APPI variants above. Consistent with our hypothesis, both Kunitz domains were considerably stabilized against proteolysis by mesotrypsin upon incorporation of the X17C/X34C disulfide mutations (Table 1). Intriguingly, for the weak inhibitor KD1TFPI1K15R, the engineered disulfide also resulted in greatly enhanced Ki of KD1TFPI1K15R/I17C/I34C for mesotrypsin, whereas affinity of KD2bikunin for mesotrypsin was barely affected by the engineered disulfide (Table 1).
Crystal structures of mesotrypsin bound to APPI and TFPI disulfide variants reveal a well-ordered engineered disulfide bond positioned to stabilize the binding loop
To gain insight into the impact of the engineered Cys-17–Cys-34 disulfide bond on the structure and function of the Kunitz domain, we co-crystallized both APPIM17C/I18F/F34C and KD1TFPI1K15R/I17C/I34C with the catalytically inactive mesotrypsin-S195A mutant and solved the crystal structures of these complexes. X-ray data collection and refinement statistics are summarized in Table 2. The mesotrypsin/APPIM17C/I18F/F34C complex crystallized in space group P21 with one copy of the inhibited complex in the asymmetric unit. The structure was refined against data diffracting to 1.5 Å resolution. The structure showed that the inhibitor bound to the enzyme in the expected fashion, with binding loop residues Pro-13–Phe-18 occupying the enzyme S3–S3′ subsites (Fig. 4A). The crystal structure unambiguously confirmed the formation of the engineered disulfide bond between residues Cys-17 and Cys-34; the electron density showed the bond to be well-ordered in a single conformation (Fig. 4A, inset). Superposition of the APPIM17C/I18F/F34C structure on the structure of the parental variant APPIM17G/I18F/F34V (PDB code 5C67 (19)) showed that incorporation of the Cys-17–Cys-34 disulfide bond caused backbone rearrangements to bring these residues into closer proximity (Fig. 4B). Whereas Gly-17 and Val-34 Cα atoms are 5.5 Å apart in the parental variant, this distance was reduced to 4.5 Å in APPIM17C/I18F/F34C. The greater part of this adjustment was accomplished through alterations in the ψ angle of Cys-34 and the φ angle of Tyr-35, resulting in shifts of Cys-34 Cα and Cβ closer to Cys-17 by 0.9 and 1.2 Å, respectively. Cys-17 also underwent adjustment of the backbone angles, resulting in a conformation with greater similarity to that of the native Met-17 residue of APPIWT (data not shown). The net effect of these adjustments reduced the Cβ–Cβ distance between Cys-17 and Cys-34 to 3.7 Å, a value typical of native disulfide bonds (20).
Table 2.
X-ray data collection and refinement statistics
| Structure name | Mesotrypsin/APPIM17C/I18F/F34C | Mesotrypsin/KD1TFPI1K15R/I17C/I34C |
|---|---|---|
| PDB code | 6HAR | 6BX8 |
| Data collection | ||
| Resolution range (Å) | 46.47–1.50 | 60.06–1.98 |
| Space group | P21 | P1 |
| a, b, c (Å) | 34.09, 82.78, 46.56 | 35.14, 87.20, 90.84 |
| α, β, γ (°) | 90.00, 93.63, 90.00 | 94.81, 93.03, 92.46 |
| Total/unique reflections | 78,755/40,015 | 116,262/70,450 |
| R-merge | 0.048 (0.627)a | 0.067 (0.249) |
| R-meas | 0.068 (0.887) | 0.095 (0.352) |
| R-pim | 0.057 (0.771) | 0.0674 (0.249) |
| CC1/2 | 0.997 (0.492) | 0.971 (0.887) |
| Multiplicity | 4.3 (3.4) | 1.7 (1.7) |
| Completeness (%) | 96.8 (87.2) | 93.0 (92.2) |
| Mean I/σ(I) | 7.3 (1.0) | 6.5 (2.5) |
| Refinement | ||
| Reflections used in refinement | 40,008 (3687) | 69,456 (6906) |
| Reflections used for R-free | 2000 (172) | 3375 (333) |
| R-work/R-free | 0.169/0.193 | 0.192/0.221 |
| No. of non-hydrogen atoms | 2437 | 9070 |
| Macromolecules | 2182 | 8229 |
| Ligands | 5 | 20 |
| Solvent | 250 | 821 |
| Protein residues | 278 | 1113 |
| RMSD bonds (Å) | 0.005 | 0.019 |
| RMSD angles (°) | 0.79 | 1.81 |
| Ramachandran favored (%) | 98.54 | 97.72 |
| Ramachandran allowed (%) | 1.46 | 2.28 |
| Ramachandran outliers (%) | 0.00 | 0.00 |
| Average B-factor | 19.93 | 30.91 |
| Macromolecules | 19.10 | 29.84 |
| Ligands | 28.89 | 50.53 |
| Solvent | 27.03 | 41.1 |
a Numbers in parentheses indicate statistics for the highest resolution shell.
Figure 4.

Crystal structures of APPIM17C/I18F/F34C and KD1TFPI1K15R/I17C/I34C complexes with mesotrypsin. A, overall structure of the mesotrypsin/APPIM17C/I18F/F34C complex (PDB code 6HAR). Mesotrypsin (gray) is bound to APPIM17C/I18F/F34C (green); magnification box shows the engineered disulfide bond, with the electron density map contoured at 2σ. Black arrows indicate original positions of Gly-17 and Val-34 Cα carbons; black dotted line shows closer proximity of Cys-17 and Cys-34 Cα carbons in the disulfide mutant. B, superposition of APPIM17C/I18F/F34C (green) with the structure of parental variant APPIM17G/I18F/F34V (blue; PDB code 5C67 chain C) reveals backbone rearrangements to accommodate formation of the engineered disulfide bond. Most notably, shifts are observed in the ψ angle of Cys-34 and the φ angle of Tyr-35, resulting in movement of Cys-34 closer to Cys-17 by ∼1 Å. C, overall structure of the mesotrypsin/KD1TFPI1K15R/I17C/I34C complex (PDB code 6BX8). Mesotrypsin (gray) is bound to KD1TFPI1K15R/I17C/I34C (purple); magnification box shows the engineered disulfide bond, with the electron density map contoured at 2σ. D, superposition of the Kunitz domain chains of the two new structures reveals closely similar positioning and geometry of the engineered disulfide bonds of APPIM17C/I18F/F34C (green) and KD1TFPI1K15R/I17C/I34C (purple). B and D, structures were aligned by Cα of the Kunitz inhibitor chains in PyMOL.
The mesotrypsin/KD1TFPI1K15R/I17C/I34C complex crystallized in space group P1, with four copies of the inhibited complex in the asymmetric unit; this structure was refined against data diffracting to 2.0 Å resolution. This structure represents the first reported structure for KD1TFPI1 bound to a serine protease. As has been found for many protease complexes with other Kunitz inhibitor domains, the inhibitor-binding loop assumed a canonical conformation (6, 8), with residues Pro-13–Met-18 occupying the enzyme S3–S3′ subsites in a substrate-like manner (Fig. 4C). Similarly to the APPIM17C/I18F/F34C structure, the electron density for KD1TFPI1K15R/I17C/I34C unambiguously confirmed the formation of a well-ordered engineered disulfide bond between residues Cys-17 and Cys-34 (Fig. 4C, inset). Superposition of the Kunitz domains from the mesotrypsin/APPIM17C/I18F/F34C and mesotrypsin/KD1TFPI1K15R/I17C/I34C structures revealed that the engineered disulfide bonds had adopted very closely similar conformations in the two inhibitors (Fig. 4D). The highly similar structural impact of Cys-17–Cys-34 disulfide engineering on these two distantly related Kunitz domains (possessing only 36% sequence identity) is consistent with the idea that disulfide bridging between residues 17 and 34 may offer an engineering approach that is generally applicable to Kunitz domains.
Molecular dynamics simulations reveal stabilization of the binding loop conformation by the engineered disulfide bond
In earlier work, we showed that proteolysis rates of different Kunitz domains by mesotrypsin correlate closely with the global conformational dynamics of the inhibitors on the nanosecond–microsecond time scale (18). Based on these prior observations, we hypothesized here that the Cys-17–Cys-34 disulfide bond may stabilize Kunitz domains against proteolysis by reducing the flexibility and dampening the conformational fluctuations of the Kunitz domain fold. To explore this idea, we conducted a series of free all-atom MD simulations, based upon our crystal structures, in which we compared the conformational fluctuations undergone by APPIM17C/I18F/F34C, KD1TFPI1K15R/I17C/I34C, and their complexes with mesotrypsin, versus APPIM17G/I18F/F34V, KD1TFPI1K15R (the parental Kunitz domains lacking the engineered disulfide bond), and their complexes with mesotrypsin. Because we lack experimentally determined crystal structures for KD1TFPI1K15R and for its complex with mesotrypsin, the starting point for these simulations was based on a model derived from in silico back-mutation of the KD1TFPI1K15R/I17C/I34C structure, followed by energy minimization. After energy minimization and equilibration protocols, each Kunitz domain or complex simulation was run for >1000 ns, along with several shorter replicates to assess convergence. Subsequently, we evaluated differences in conformational dynamics between the Kunitz domains and their complexes by aligning MD simulation frames on backbone atoms and comparing root-mean-square deviation (RMSD) plots of backbone atom deviation from average positions for the Kunitz domain residues over the course of the simulations. We also examined per residue root-mean-square fluctuation (RMSF) plots that reveal which residues within the Kunitz domain are most impacted by the introduction of the engineered disulfide bond.
In simulations modeling the dynamics of free, unbound APPIM17C/I18F/F34C versus APPIM17G/I18F/F34V, RMSD plots did not reveal striking global differences in dynamics (Fig. 5A), possibly because the parental APPIM17G/I18F/F34V variant had already been engineered with mutations that broadly dampen global dynamics (18). However, RMSF comparisons did show a suppression of conformational dynamics in a few focused regions of APPIM17C/I18F/F34C relative to APPIM17G/I18F/F34V, most notably in residues 15–18 of the binding loop surrounding the G17C mutation (Fig. 5, B and C). Simulations of KD1TFPI1K15R/I17C/I34C versus KD1TFPI1K15R showed a modest suppressive effect of the engineered disulfide bond on the global dynamics, as illustrated by RMSD plots (Fig. 5D), and a more pronounced effect specifically dampening the motion of the apparent hotspot of dynamics surrounding Ile-17 of KD1TFPI1K15R (Fig. 5, E and F).
Figure 5.

Molecular dynamics simulations reveal suppressed dynamics in Kunitz domains with engineered disulfide bonds. A, global dynamics of unbound APPIM17G/I18F/F34V and APPIM17C/I18F/F34C, compared according to backbone atom (NCαCO) average RMSD from MD simulations, show only modest differences in amplitude. B, magnitudes of positional fluctuations per residue of APPI variants over the length of the MD simulation are plotted as Cα average RMSF. C, positional RMSF amplitudes from B are heat-mapped onto the protein structures in the blue-white-red spectrum (scale bar shown). Note the peak of elevated mobility spanning binding loop residues 15–18 of APPIM17G/I18F/F34V that is absent in APPIM17C/I18F/F34C. D, global dynamics of unbound KD1TFPI1K15R versus KD1TFPI1K15R/I17C/I34C, compared according to backbone atom (NCαCO) average RMSD from MD simulations, show lower amplitudes of backbone atom deviation from average positions in the engineered disulfide variant. E, magnitudes of positional fluctuations per residue of KD1TFPI variants over the length of the MD simulation are plotted as Cα average RMSF. F, positional RMSF amplitudes from E are heat-mapped onto the protein structures in the blue-white-red spectrum (scale bar shown). Note that several peaks of elevated mobility in KD1TFPI1K15R, including binding loop residues 16–19, are reduced in KD1TFPI1K15R/I17C/I34C.
In further analyses of these simulations focusing specifically on alignments of the P3–P3′ residues 13–18, we found clear evidence of reduced dynamics throughout the binding loop of APPIM17C/I18F/F34C compared with APPIM17G/I18F/F34V (Fig. 6, A and B). In APPIM17G/I18F/F34V, the Gly-17 backbone fluctuates between different conformations, resulting in a considerable RMSF peak (Fig. 6, B and C). In contrast, this peak was completely eliminated in APPIM17C/I18F/F34C, as the protein backbone in this region assumed a single, more rigid conformation (Fig. 6, B and D). The suppressive effect of the engineered disulfide bond on the dynamics of the binding loop can also be seen in plots of the backbone φ and ψ angles sampled by binding loop residues: Ramachandran plots for residues 17 and 18, in particular, showed tighter clustering for APPIM17C/I18F/F34C compared with APPIM17G/I18F/F34V (Fig. 6, E and F). Similar observations were obtained for the KD1TFPI1 scaffold simulations: whereas the binding loop of KD1TFPI1K15R samples a variety of conformations, mobility throughout the binding loop was markedly suppressed in KD1TFPI1K15R/I17C/I34C (Fig. 7, A–D). Ramachandran plots revealed multiple populations of conformers at each binding loop residue of KD1TFPI1K15R, whereas throughout the simulations of KD1TFPI1K15R/I17C/I34C, each residue remained better stabilized in a single major backbone conformation (Fig. 7, E and F). The stabilization of the engineered disulfide variants with a preconfigured binding loop conformation favorable for binding can help to explain the faster kon and improved Ki values for these inhibitors toward protease targets.
Figure 6.

Molecular dynamics simulations reveal stabilization of APPI-binding loop conformation by the engineered Cys-17–Cys-34 disulfide bond. A, conformational dynamics of the binding loop of APPIM17G/I18F/F34V versus APPIM17C/I18F/F34C show reduced amplitude in the engineered disulfide variant. Positional deviations were evaluated using backbone atom (NCαCO) average RMSD of binding loop residues 13–18 after alignment of frames based only on these residues. B, Cα positional fluctuations per residue (RMSF) plotted for APPI variants show a peak at Gly-17 of APPIM17G/I18F/F34V that is eliminated in APPIM17C/I18F/F34C. C, superposition of binding loop residues 13–18 from representative frames of the unbound APPIM17G/I18F/F34V simulation highlights the ensemble of conformations sampled over the course of the simulation. Note in particular the variety of backbone conformations assumed by Ala-16 and Gly-17. D, superposition of binding loop residues 13–18 from representative frames of the unbound APPIM17C/I18F/F34C simulation illustrates tighter clustering around a single consensus backbone conformation throughout the simulation. E, Ramachandran plots show multiple populations of conformers at binding loop residues in APPIM17G/I18F/F34V, most notably for Gly-17 and Phe-18. F, Ramachandran plots show tighter clustering of the φ and ψ angles around single backbone conformations at binding loop residues Cys-17 and Phe-18 in APPIM17C/I18F/F34C.
Figure 7.

Molecular dynamics simulations reveal stabilization of the KD1TFPI-binding loop conformation by the engineered Cys-17–Cys-34 disulfide bond. A, conformational dynamics of the binding loop of KD1TFPI1K15R versus KD1TFPI1K15R/I17C/I34C show reduced amplitude in the engineered disulfide variant. Positional deviations were evaluated using backbone atom (NCαCO) average RMSD of binding loop residues 13–18 after alignment of frames based only on these residues. B, Cα positional fluctuations per residue (RMSF) plots show a peak at Gly-17 for KD1TFPI1K15R that is eliminated in KD1TFPI1K15R/I17C/I34C. C, superposition of binding loop residues 13–18 from representative frames of the unbound KD1TFPI1K15R simulation reveals a diverse conformational ensemble, especially at residues Arg-15, Ala-16, and Ile-17. D, superposition of binding loop residues 13–18 from representative frames of the unbound KD1TFPI1K15R/I17C/I34Csimulation illustrates tighter clustering around a single consensus backbone conformation throughout the simulation. E, Ramachandran plots show multiple populations of conformers at all binding loop residues in KD1TFPI1K15R. F, Ramachandran plots show much tighter clustering of the φ and ψ angles around single backbone conformations throughout the binding loop residues of KD1TFPI1K15R/I17C/I34C.
All-atom MD simulations of Kunitz domain complexes with mesotrypsin likewise corroborate the role of the engineered disulfide bond in suppressing binding loop dynamics, although, as expected, the magnitude of the effect was somewhat reduced, because contact of the binding loop with mesotrypsin also restricted the conformations of these contact residues. When frames from MD simulations of mesotrypsin/APPI complexes were aligned on the backbone atoms of the Kunitz domain alone, the RMSF peak for residues 16–19, evident in APPIM17G/I18F/F34V, was eliminated in APPIM17C/I18F/F34C (Fig. S1). The region of suppressed dynamics revealed by parallel analyses of mesotrypsin/KD1TFPI complexes was more extensive, spanning binding loop residues 13–19 (Fig. S2). When frames from the simulations of mesotrypsin/Kunitz domain complexes were instead aligned on the backbone atoms of all protein residues, the enzyme chains aligned well, whereas the Kunitz domains displayed substantial displacements in the β-turn and α-helical regions distal to the enzyme interface (Figs. S3 and S4). These displacements resulted from a rocking motion of the Kunitz domain relative to the enzyme, as described in our previous work, where we reported the amplitude of such intermolecular motions to correlate with proteolytic susceptibility (18). Here, we found that introduction of the engineered disulfide bond into APPIM17C/I18F/F34C subtly reduced the displacements of the α-helix compared with those observed in the mesotrypsin/APPIM17G/I18F/F34V complex (Fig. S3); notably, APPIM17G/I18F/F34V had already exhibited considerably suppressed global dynamics relative to APPIWT due to stabilizing mutations (18). The suppressive effect of the engineered disulfide bond on Kunitz domain displacement was far more striking in MD simulations of mesotrypsin/KD1TFPI1K15Rand mesotrypsin/KD1TFPI1K15R/I17C/I34C (Fig. S4). In summary, our MD simulations of both unbound Kunitz domains and complexes with mesotrypsin are consistent with the hypothesis that substantial conformational stabilization, conferred by the engineered Cys-17–Cys-34 disulfide bond, suppresses both local dynamics of the binding loop and global dynamics of the inhibitor within the complex, resulting in increased stabilization against proteolysis.
Discussion
By virtue of their three native disulfide bonds, the folds of Kunitz serine protease inhibitors are inherently highly stabilized against unfolding. Nonetheless, as a consequence of their mechanism of action for inhibition of serine proteases, these inhibitors remain susceptible to slow cleavage of a reactive site bond within the protease-binding loop, which diminishes their efficacy for inhibiting target enzymes (12, 15). Here, we showed that incorporation of an engineered disulfide bond between residues 17 and 34 in the inhibitor-binding loop represents a generalizable strategy for stabilization of Kunitz domain inhibitors against proteolysis by target serine proteases without impairing the inhibitors' inhibitory potency or affinity for the proteases.
A rigorous validation of this idea may be drawn from the improved stability of the disulfide-engineered inhibitors against proteolysis by mesotrypsin, as this enzyme has a remarkable capacity for cleavage and inactivation of human Kunitz domain inhibitors. These properties of mesotrypsin are the result of multiple evolutionary mutations, the most critical being the substitution of Gly-193 by Arg, which clashes sterically with the inhibitors and weakens affinity of the complexes (14, 16). Additional mutations that contribute to mesotrypsin's proteolytic capability are the substitution of Tyr-39 by Ser, Lys-97 by Asp, and Glu-74 by Lys, which alter interactions at the molecular interface within mesotrypsin/inhibitor complexes, eliminating hydrogen bonds and enhancing mobility (15, 16). These mutations are thus responsible for the unusually low affinity of mesotrypsin (relative to typical trypsins) for polypeptide trypsin inhibitors, and they also contribute to the unique ability of mesotrypsin to cleave several canonical trypsin inhibitors as substrates (13, 17, 21).
Our results show that introduction of a disulfide linkage between residues 17 and 34 of APPI, i.e. in the APPIM17C/I18F/F34Cvariant, is an effective approach for stabilization against proteolysis by mesotrypsin. In particular, APPIM17C/I18F/F34C displays a mesotrypsin cleavage rate that is 250-fold slower than that of APPIWT, which makes it the most resistant APPI variant yet reported. We found similar disulfide bond placement to likewise stabilize the Kunitz domain inhibitors KD1TFPI1 and KD2bikunin against mesotrypsin proteolysis, demonstrating applicability of the strategy across the Kunitz inhibitor family.
Mesotrypsin is a pancreatic enzyme transcribed from the PRSS3 gene, and several alternative splice isoforms of PRSS3 are transcribed by tumors and other tissues (22). Although active mesotrypsin has not been directly detected in tumors, indirect evidence suggests that PRSS3-encoded trypsinogen 4 may become activated to produce mesotrypsin and contribute to malignancy, driving cellular invasion and migration. The pro-metastatic potential of mesotrypsin has been suggested, for example, in cell culture and xenograft models of pancreatic (23) and prostate cancers (24), where silencing of the PRSS3 gene by RNA interference (RNAi) markedly reduced the invasiveness and metastasis of these cancers both in vitro and in vivo. The enhancement of tumor cell migration and invasion has been linked to the specific proteolytic activity of mesotrypsin, because invasive behavior of prostate cancer cells could be stimulated by exogenously added mesotrypsin (but not a catalytically inactive mutant) and could be blocked by either PRSS3 knockdown or treatment with an inhibitor of mesotrypsin activity (19, 24). These studies suggest that mesotrypsin could be a potential target for therapeutic intervention in cancer, and thus, stabilization of the Kunitz domain against proteolysis by mesotrypsin may be regarded as a step toward the development of potent inhibitors to inhibit the progression and metastasis of various cancers.
A number of previous studies have reported attempts to stabilize proteins for both medical and industrial applications by applying structure-based design for engineering disulfide bonds into the molecules (5, 25). For example, in their recent work, Schultz and co-workers (26) genetically encoded noncanonical amino acids that are capable of forming disulfide bonds that bridge longer distances than the native disulfide bonds. These longer and more flexible linkages made it much easier to crosslink sites within the molecule to produce disulfide-engineered proteins with improved thermal stability vis à vis their WT counterparts (26). Indeed, disulfide engineering has achieved considerable success in a wide range of applications, yet contrary to expectations, many designed disulfide bonds have resulted in decreased stability of the modified protein, probably in cases where the introduction of cysteines disrupts or removes intramolecular interactions that stabilize the native structure (5, 27). These previous studies have most often focused on protein thermodynamic stability and rates of folding and unfolding (as fold stability is an important property for therapeutic proteins and for enzymes engineered for various industrial applications). In contrast, in this study, we focused on the impact of engineered disulfides on a different property that impacts protein activity and longevity in biological settings, namely resistance to proteolytic inactivation and degradation.
Our crystal structures and molecular dynamics simulations support the interpretation that engineered disulfides can slow proteolysis by suppressing conformational dynamics. Most studies of protein dynamics in enzyme catalysis have focused on conformational changes within the enzyme and their impact on substrate/inhibitor binding, progression along the catalytic reaction coordinate, and product release (28–33). However, for the many enzymes that catalyze reactions of protein substrates, we (18) and others (34) have shown that it is necessary to look beyond the enzyme to the protein substrate as well (in this case Kunitz inhibitor domain) if we are to fully appreciate the impact of protein dynamics on enzyme-catalyzed reactions. In our previous work, we found that rates of proteolysis of different Kunitz domains by mesotrypsin correlated closely with the global and local conformational dynamics of the Kunitz domain substrates in the nanosecond to microsecond time regime. Our current MD simulations of APPIM17C/I18F/F34C and KD1TFPI1K15R/I17C/I34C Kunitz domains, both unbound and when complexed with mesotrypsin, confirm and extend these findings and are consistent with the premise that the well-ordered engineered disulfide bond between residues Cys-17 and Cys-34 suppresses both the local dynamics of the binding loop and the global dynamics of the inhibitor within the complex, resulting in increased stabilization against proteolysis.
This novel general method for extending the biological stability and potency of Kunitz domain scaffolds may be of particular importance for the development of new and second-generation pharmaceuticals. An illustrative example is the Food and Drug Administration-approved orphan drug ecallantide (Kalbitor or DX-88); this drug was engineered on the scaffold of KD1TFPI1 to target plasma kallikrein for the treatment of hereditary angioedema (HAE) (35). Treatment with ecallantide compensates for the insufficient quantity of the natural kallikrein inhibitor C1 in HAE patients and is expected to replace conventional therapy with C1 that is isolated from human plasma (36). Another drug that is based on the KD1TFPI1 scaffold is the plasmin inhibitor DX-1000, which is in preclinical development for blocking tumor growth and metastasis in breast cancer (37). Yet another example is a drug based on the KD2bikunin scaffold that targets neutrophil elastase. Known as depelestat (DX-890), this protein is currently in clinical trials for acute respiratory distress syndrome (38) and cystic fibrosis (39).
Although the application of our approach may be successful for biopharmaceuticals and other commercial proteins, we need to bear in mind several limitations. The utility of our approach may be limited to reducing target protease cleavage at the inhibitor reactive site and may not offer protection against other types of proteolytic attack. A second limitation relates to the potential susceptibility of cysteine disulfides to intra- or intermolecular disulfide bond exchange or to several pathways of chemical degradation, thereby rendering some disulfide-containing proteins susceptible to degradation, aggregation, and precipitation (2, 40, 41). In the future, further advantage might be gained by engineering redox-stable disulfide analogs, such as has been explored in the case of sunflower trypsin inhibitor, a very small canonical inhibitor accessible through total synthesis (42, 43). This type of engineering would require incorporation of stable cross-links into proteins through the use of genetically encoded, noncanonical amino acids (26, 44). We predict that such disulfide-engineered proteins with enhanced stability could lead to the development of a new generation of biopharmaceuticals.
Experimental procedures
Protein expression and purification
Kunitz domain inhibitors (including APPI, KD1TFPI1, and KD2bikunin variants) were cloned into a pPIC9K vector, transformed, expressed in P. pastoris (GS115 strain), and purified by nickel-affinity chromatography, followed by size-exclusion chromatography, as described for APPI in our previous work (19). The concentrations of the Kunitz inhibitors were determined by titration with pre-titrated bovine trypsin (Sigma, Poole, UK) and the substrate benzoyl-l-arginine-p-nitroanilide (Sigma), as described previously (14). Mesotrypsin and the catalytically inactive mutant mesotrypsin-S195A were expressed recombinantly as zymogen forms in Escherichia coli, isolated from inclusion bodies, refolded, purified, and proteolytically activated, essentially as described previously (14, 16). Active mesotrypsin was quantified by titration with 4-nitrophenyl 4-guanidinobenzoate (Sigma) (14), and mesotrypsin-S195A was quantified by UV-visible absorbance readings at 280 nm using a calculated extinction coefficient of 41,535 m−1 cm−1.
For the generation of the progress curves described below, the concentration of the chromogenic substrate Z-GPR-pNA (Sigma) was determined in an end-point assay from the change in the absorbance caused by the release of p-nitroaniline (ϵ410 = 8480 m−1 cm−1). All reactions were monitored with a Synergy2 microplate spectrophotometer (BioTek).
Progress curves for human mesotrypsin with APPIM17C/I18F/F34Cwere generated as we have previously described for mesotrypsin with APPIM17G/I18F/F34V (45). Briefly, stock solutions of enzyme, substrate, and APPI proteins were prepared at 40× the desired final concentrations. Assays were performed in 96-well microplates at 37 °C as follows: trypsin buffer (296 μl of 100 mm Tris-HCl, pH 8.0, 1 mm CaCl2), Z-GPR-pNA substrate (8 μl; 6 mm concentration), and APPI (8 μl; concentrations ranging from 0 to 40 nm) were mixed and incubated at 37 °C for 10 min. Reactions were then initiated by dilution of the enzyme (8 μl; 1 nm concentration) into the pre-equilibrated mixture and were followed spectroscopically (from the increase in absorbance at 410 nm) for 1 h.
Progress curves for mesotrypsin with APPIWT, APPII18F, and APPIT11C/M17G/I18F/F34C for independent determination of koff were obtained in 96-well microplates at 37 °C as follows. APPI (10 μm) and mesotrypsin (60 nm) were mixed in a 1:1 ratio and incubated at 37 °C for 1 h. The substrate (Z-GPR-pNA) was prepared in trypsin buffer at a final concentration of 150 μm and incubated at 37 °C for 10 min. Reactions were then initiated by dilution of the enzyme/inhibitor mixture (5 μl) by 60× into the pre-equilibrated substrate (295 μl) and followed spectroscopically.
Progress curves for mesotrypsin with APPIT11C/M17G/I18F/F34C,KD1TFPI1K15R, KD1TFPI1K15R/I17C/I34C, KD2bikunin, and KD2bikuninF17C/P34C for determination of the dissociation constant Ki were generated as we have previously described (19). Briefly, stock solutions of enzyme, substrate, and inhibitors were prepared at 40× the desired final concentrations. Assays were performed in 96-well microplates at 37 °C in the presence of different concentrations of Z-GPR-pNA substrate (50, 100, 150, 200, and 250 μm). The final concentrations of the inhibitors were 0, 50, 100, 200, and 400 nm for APPIT11C/M17G/I18F/F34C; 0, 11, 22, 45, and 85 μm for KD1TFPI1K15R; 0, 0.125, 0.25, 0.5, and 1 μm for KD1TFPI1K15R/I17C/I34C; and 0, 30, 60, 120, and 250 nm for KD2bikunin and KD2bikuninF17C/P34C. Trypsin buffer (296 μl), substrate (8 μl), and APPI (8 μl) were equilibrated together in a 96-well microplate prior to the addition of enzyme (8 μl of 10 nm mesotrypsin). Reactions were followed spectroscopically for 5 min, and initial rates were determined from the increase in absorbance due to the release of p-nitroaniline (ϵ410 = 8480 m−1 cm−1).
Progress curve analysis
The dissociation constants Ki for APPIT11C/M17G/I18F/F34C, KD1TFPI1K15R, KD1TFPI1K15R/I17C/I34C, KD2bikunin, and KD2bikuninF17C/P34C, in complex with mesotrypsin, were determined by fitting their progress curves by multiple regression to Equation 1, the classic competitive inhibition equation (Fig. 2, A and B),
| (Eq. 1) |
where V is the velocity of product formation at the start of the reaction; Km (Michaelis constant) and kcat are the kinetic parameters for substrate hydrolysis; [E]0 is the total concentration of enzyme, and [S]0 and [I]0 are the initial concentrations of substrate and inhibitor, respectively.
Values of the equilibrium inhibition constant (Ki) of mesotrypsin with APPIM17C/I18F/F34C were calculated by using Equation 2 from the steady-state portions of the progress curves (Fig. 2, D and E), as described previously (45). Equation 2 describes an equilibrium state of reversible competitive inhibition with slow, tight-binding behavior,
| (Eq. 2) |
where Vs and V0 are the steady-state rates in the presence and absence of inhibitor (Fig. 2, D and E).
Association (kon) and dissociation (koff) constants for slow inhibition of mesotrypsin by APPIM17C/I18F/F34C were obtained using Equations 3–6 (46). Data from the generated curves were first globally fitted by multiple regression to Equation 3, with the integrated rate equation describing slow binding inhibition,
| (Eq. 3) |
where Kobs is the observed first-order rate constant that describes the transition from V0 to Vs (Fig. 2D), and [P] is the concentration of product formed at any time, t.
Slow, tight-binding inhibition can be described by two alternative general mechanisms (46). In brief, one mechanism is a two-step process involving the formation/accumulation of an initial inhibitor–enzyme complex, followed by slow kinetics to form a tighter complex. In this mechanism, kon and koff are both first-order kinetic constants that are characterized by a nonlinear relationship between Kobs and the inhibitor concentration. In contrast, the second mechanism is a direct, single-step process in which the final complex is formed slowly. In this mechanism, kon and koff are second- and first-order kinetic constants, respectively, that are characterized by a linear dependence between Kobs and the inhibitor concentration [I], as shown in Equation 4. For the inhibition of mesotrypsin by APPIM17C/I18F/F34C, a plot of Kobs versus inhibitor concentration displayed a linear dependence, consistent with the single-step mechanism (Fig. 2F).
| (Eq. 4) |
kon and koff were calculated from the linear curve generated by Equation 4 and by using the following relationships shown in Equations 5 and 6.
| (Eq. 5) |
| (Eq. 6) |
Because the inhibition kinetics of mesotrypsin with APPIWT, APPII18F, and APPIT11C/M17G/I18F/F34C were relatively fast, estimation of koff from Equation 4 would be inaccurate. Therefore, koff was calculated independently of the inhibitor concentration by using a multiple regression curve fit to Equation 7 (Fig. 2C) (46).
| (Eq. 7) |
The calculated association rate constant was then obtained from their measured Ki by using Equation 8.
| (Eq. 8) |
Calculations were performed using Km values of 24.7 ± 1.3 μm for mesotrypsin as determined from at least three Michaelis-Menten kinetic experiments that were previously performed in our laboratory (19). All calculations were performed using Prism (GraphPad Software, San Diego). Results (means ±S.D.) were obtained from at least three independent experiments.
The cleavage of intact inhibitors (including APPI, KD1TFPI1, and KD2bikunin variants) in time course incubations with catalytically active mesotrypsin was monitored by HPLC. Mesotrypsin was incubated at 37 °C with the inhibitors in trypsin buffer (100 mm Tris-HCl, pH 8.0, 1 mm CaCl2) for APPIT11C/M17G/I18F/F34C and APPIM17C/I18F/F34C, the reaction was performed for 6 h in the presence of 50 μm inhibitor using 0.2 and 7.14 μm enzyme, respectively; for KD1TFPI1K15R/I17C/I34C,KD2bikunin, and KD2bikuninF17C/P34C, the reaction was performed for 4 h in the presence of 50 μm inhibitor using 0.033, 0.25, and 1 μm enzyme, respectively; for KD1TFPI1K15R the reaction was performed for 1 h in the presence of 300 μm inhibitor using 0.05 μm enzyme; inhibitor and enzyme concentrations are given in Table S1. For HPLC analysis, aliquots of 30 μl were withdrawn at periodic intervals (Table S1), added to a solution containing 80 μl of 6 m urea and 2 mm DTT, incubated for 10 min at 37 °C, quenched by acidification to pH 1, and then frozen at −20 °C until analyzed. Samples were resolved on a 50 × 2.0-mm Jupiter 4 μ 90-Å C12 column (Phenomenex) with a gradient of 0–100% acetonitrile in 0.1% trifluoroacetic acid (TFA) at a flow rate of 0.6 ml/min over 50 min. Intact inhibitors were quantified by peak integration of absorbance traces monitored at 210 nm. Initial rates were obtained by linear regression using a minimum of seven data points within the initial linear phase of the reaction. Hydrolysis rates reported for each inhibitor represent the average of three independent experiments.
Protein crystallization, data collection, structure determination, and refinement
For crystallization of the mesotrypsin-S195A/APPIM17C/I18F/F34Ccomplex, purified mesotrypsin was mixed with APPIM17C/I18F/F34Cin 1:1 molar ratio and subjected to crystallization trials using the sitting drop vapor diffusion method. Initial screening of crystallization conditions was performed with the index screening kit of Hampton Research at 293 K. Each drop contained a mixture of 0.3 μl of crystallization solution and 0.3 μl of mesotrypsin-S195A/APPIM17C/I18F/F34C complex. Crystals grew after 9 days in a drop containing 0.1 m MES, pH 6.8, and 16% polyethylene glycol 20,000. The crystals were harvested, cryoprotected, and flash-cooled in liquid nitrogen prior to data collection. X-ray diffraction data were collected at 100 K on the beamline ID30B of the European Synchrotron Radiation Facility (ESRF, Grenoble, France) to a maximum resolution of 1.5 Å. The crystals belong to the P21 space group, with unit cell dimensions of a 34.09, b 82.78, and c 46.56 and contain one molecule of mesotrypsin and one molecule of APPI in the asymmetric unit. The X-ray data for mesotrypsin-S195A/APPIM17C/I18F/F34C crystals was processed, merged, and scaled using program package XDS (47). Data collection statistics are given in Table 2. Phase acquisitions and structure determination were performed by molecular replacement using Phaser (48) from the CCP4 Program Suite (49). Protein Data Bank (PDB) code 5C67 was used as the search model. Refinement was performed using Phenix.refine (50), and alternating rounds of model building and manual corrections were performed by COOT (51). The coordinates and structure factors have been submitted to the Worldwide Protein Data Bank (PDB) under the accession code 6HAR.
For crystallization of the mesotrypsin-S195A/KD1TFPI1K15R/I17C/I34C complex, KD1TFPI1K15R/I17C/I34C was mixed with mesotrypsin-S195A in an equimolar ratio at a total protein concentration of 4 mg/ml, mixed 1:1 (v/v) with reservoir solution, and crystallized via the hanging drop method over a reservoir containing 0.2 m lithium sulfate, 0.1 m BisTris-HCl, pH 4.6, and 25% v/v PEG 3350 at room temperature. Crystals were harvested, cryoprotected, and flash-cooled in liquid nitrogen. Single wavelength (0.97741 Å) native X-ray diffraction data were collected at 100 K on the Advanced Light Source beamline 5.0.1, Lawrence Berkeley National Laboratory. The structure was solved from a single crystal that diffracted to 1.98 Å resolution. The X-ray data were processed with xia2 (52) using DIALS (53) for indexing, refinement, and integration with POINTLESS (54) and AIMLESS (55) for scaling and merging. R-free flags were assigned to a random 5% of reflections, and this test set was maintained throughout all subsequent stages of structure solution and refinement. The structure was phased with molecular replacement with CCP4 Molrep (56), using as the search model the structure of the mesotrypsin-S195A chain from PDB code 3P95 (57). When electron density maps for the molecular replacement solution showed the presence of continuous positive density peaks protruding from the mesotrypsin-binding site, Molrep was used again to place the inhibitor chain, using PDB code 4BQD (human KD1TFPI (58)) as the search model. The model was refined using an iterative process of Phenix.refine (maximum likelihood) (59) and manual alterations in COOT (51). Molprobity (60) and wwPDB (61) validation servers were used to help guide later refinement and manual structure improvements. The coordinates and structure factors for the mesotrypsin-S195A/KD1TFPI1K15R/I17C/I34C complex structure have been submitted to the Worldwide Protein Data Bank under the accession code 6BX8. Structure figures were generated using PyMOL (Schrodinger, LLC).
Molecular dynamics simulations and analyses
Initial structural models for MD of mesotrypsin/Kunitz domain complexes were generated from the following crystallographic structures: (a) mesotrypsin/APPIM17G/I18F/F34V (PDB code 5C67 (19)); (b) mesotrypsin/APPIM17C/I18F/F34C (PDB ID 6HAR; present study); and (c) mesotrypsin-S195A/KD1TFPI1K15R/I17C/I34C (PDB code 6BX8; present study). Additionally, a model for the mesotrypsin/KD1TFPI1K15R complex was generated via in silico back-mutation of the mesotrypsin/KD1TFPI1K15R/I17C/I34C structure, followed by energy minimization. Initial structural models for MD of the same four Kunitz inhibitor domains in the unbound state were generated by extracting the coordinates only for the inhibitor chains from the models of mesotrypsin complexes described above. All models were further energy-minimized, validated, and prepared for MD, as described previously (18). Well-known MD methodologies and capabilities were used (18, 62–65). Free (unbiased) all atom MD simulations were run in explicit solvents using methods that we have described in detail previously (18, 66–68). Models were solvated with TIP3P water molecules, salt ions (Na+, Cl−), and counter-ions (neutralizing the enzyme) resembling a biological environment to a depth of 15–18 Å from the edge of the molecule forming a simulation box, which was subjected to a particle-mesh Ewald algorithm and periodic boundary conditions for complete electrostatic treatment (69). Simulations were run using NAnoscale Molecular Dynamics 2 engine (NAMD2) and AMBER force-field parameters, which were exported to Yasara for very long runs on a Linux Beowulf cluster. We compared NAMD2 pre-equilibration using CHARMM 27 with CMAP correction force field parameters to check for any inconsistencies in the structure prior to production run simulations. Simulations were carried out using the particle mesh Ewald technique with repeating boundary conditions with a 9 Å nonbonded cutoff, using SHAKE with a 2-fs time step. A constant temperature of 310 K was maintained using the Berendsen weak-coupling algorithm with a time constant of 1.0 ps. Following equilibration cycles of minimization and heating under restrained and unrestrained MD, equilibration was determined from a flattening of RMSD over time. Production runs were subsequently carried out with constant pressure boundary conditions (relaxation time of 1.0 ps) for >1000 ns. Our protocols for equilibration and production runs are described in greater detail elsewhere (18, 66, 67). Following simulations, individual frames were superposed back to the origin to remove rotation and translation effects.
Author contributions
I. C., M. C., A. S., B. S., and S. L. data curation; I. C., M. C., A. S., B. S., T. R. C., E. S. R., and N. P. formal analysis; I. C., M. C., A. S., T. R. C., E. S. R., and N. P. validation; I. C., M. C., B. S., A. H., T. R. C., E. S. R., and N. P. investigation; I. C., M. C., B. S., T. R. C., and N. P. methodology; I. C., E. S. R., and N. P. writing-original draft; I. C., M. C., A. S., S. L., T. R. C., E. S. R., and N. P. writing-review and editing; E. S. R. and N. P. conceptualization; B. S., A. H., T. R. C., and E. S. R. resources; E. S. R. and N. P. supervision; E. S. R. and N. P. funding acquisition; E. S. R. and N. P. visualization.
Supplementary Material
Acknowledgments
The structural studies of the mesotrypsin-S195A/APPIM17C/I18F/F34C complex were performed on beamline ID30-B at the European Synchrotron Radiation Facility (ESRF), Grenoble, France. The structural studies of the mesotrypsin-S195A/KD1TFPI1K15R/I17C/I34C complex were performed on beamline 5.0.1 of the Berkeley Center for Structural Biology at the Advanced Light Source, Lawrence Berkeley National Laboratory. We are grateful to the beamline scientists for providing assistance in using these beamlines. X-ray diffraction data were measured at the Berkeley Center for Structural Biology at the Advanced Light Source. The Berkeley Center for Structural Biology is supported in part by the Howard Hughes Medical Institute. The Advanced Light Source is a Department of Energy Office of Science User Facility under Contract No. DE-AC02-05CH11231. The Pilatus detector on 5.0.1 was funded under National Institutes of Health Grant S10OD021832. The ALS-ENABLE beamlines are supported in part by the National Institutes of Health, NIGMS Grant P30 GM124169. X-ray diffraction data were also measured at beamline ID30-B at the European Synchrotron Radiation Facility (ESRF), Grenoble, France.
This work was supported by the European Research Council “Ideas Program” ERC-2013-StG Grant 336041 (to N. P.), a Prostate Cancer Foundation grant (to N. P.), an Israel Science Foundation (ISF) grant (to N. P.), DKFZ-MOST Grant GR2495 (to N. P.), National Institutes of Health Grant R01CA154387 (to E. S. R.), and a United States–Israel Binational Science Foundation grant (to N. P. and E. S. R.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Figs. S1–S4 and Table S1.
The atomic coordinates and structure factors (codes 6HAR and 6BX8) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- BPTI
- bovine pancreatic trypsin inhibitor
- APPI
- amyloid precursor protein Kunitz protease inhibitor domain
- KD1TFPI1
- tissue factor pathway inhibitor-1 Kunitz domain 1
- KD2bikunin
- bikunin Kunitz domain 2
- MD
- molecular dynamics
- RMSD
- root-mean-square deviation
- RMSF
- residue root-mean-square fluctuation
- BisTris
- 2-[bis(2-hydroxyethyl)amino]-2-(hydroxymethyl)propane-1,3-diol
- pNA
- p-nitroaniline
- Z
- benzyloxycarbonyl
- BOC
- t-butoxycarbonyl
- AMC
- aminomethylcoumarin
- HAE
- hereditary angioedema.
References
- 1. Thornton J. M. (1981) Disulphide bridges in globular proteins. J. Mol. Biol. 151, 261–287 10.1016/0022-2836(81)90515-5 [DOI] [PubMed] [Google Scholar]
- 2. Zhang L., Chou C. P., and Moo-Young M. (2011) Disulfide bond formation and its impact on the biological activity and stability of recombinant therapeutic proteins produced by Escherichia coli expression system. Biotechnol. Adv. 29, 923–929 10.1016/j.biotechadv.2011.07.013 [DOI] [PubMed] [Google Scholar]
- 3. Qin M., Wang W., and Thirumalai D. (2015) Protein folding guides disulfide bond formation. Proc. Natl. Acad. Sci. U.S.A. 112, 11241–11246 10.1073/pnas.1503909112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Goldberg A. L. (2003) Protein degradation and protection against misfolded or damaged proteins. Nature 426, 895–899 10.1038/nature02263 [DOI] [PubMed] [Google Scholar]
- 5. Dombkowski A. A., Sultana K. Z., and Craig D. B. (2014) Protein disulfide engineering. FEBS Lett. 588, 206–212 10.1016/j.febslet.2013.11.024 [DOI] [PubMed] [Google Scholar]
- 6. Krowarsch D., Cierpicki T., Jelen F., and Otlewski J. (2003) Canonical protein inhibitors of serine proteases. Cell. Mol. Life Sci. 60, 2427–2444 10.1007/s00018-003-3120-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rawlings N. D., Tolle D. P., and Barrett A. J. (2004) Evolutionary families of peptidase inhibitors. Biochem. J. 378, 705–716 10.1042/bj20031825 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Bode W., and Huber R. (1992) Natural protein proteinase inhibitors and their interaction with proteinases. Eur. J. Biochem. 204, 433–451 10.1111/j.1432-1033.1992.tb16654.x [DOI] [PubMed] [Google Scholar]
- 9. Laskowski M. Jr., and Kato I. (1980) Protein inhibitors of proteinases. Annu. Rev. Biochem. 49, 593–626 10.1146/annurev.bi.49.070180.003113 [DOI] [PubMed] [Google Scholar]
- 10. Schneider S. L., Stasiuk L., and Laskowski M. (1973) Sequence of tryptic cleavages in porcine pancreatic secretory inhibitor II. J. Biol. Chem. 248, 7207–7214 [PubMed] [Google Scholar]
- 11. Rawlings N. D., Barrett A. J., and Finn R. (2016) Twenty years of the MEROPS database of proteolytic enzymes, their substrates and inhibitors. Nucleic Acids Res. 44, D343–D350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Salameh M. A., Robinson J. L., Navaneetham D., Sinha D., Madden B. J., Walsh P. N., and Radisky E. S. (2010) The amyloid precursor protein/protease nexin 2 Kunitz inhibitor domain is a highly specific substrate of mesotrypsin. J. Biol. Chem. 285, 1939–1949 10.1074/jbc.M109.057216 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Szmola R., Kukor Z., and Sahin-Tóth M. (2003) Human mesotrypsin is a unique digestive protease specialized for the degradation of trypsin inhibitors. J. Biol. Chem. 278, 48580–48589 10.1074/jbc.M310301200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Salameh M. A., Soares A. S., Hockla A., and Radisky E. S. (2008) Structural basis for accelerated cleavage of bovine pancreatic trypsin inhibitor (BPTI) by human mesotrypsin. J. Biol. Chem. 283, 4115–4123 10.1074/jbc.M708268200 [DOI] [PubMed] [Google Scholar]
- 15. Salameh M. A., Soares A. S., Alloy A., and Radisky E. S. (2012) Presence versus absence of hydrogen bond donor Tyr-39 influences interactions of cationic trypsin and mesotrypsin with protein protease inhibitors. Protein Sci. 21, 1103–1112 10.1002/pro.2097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Alloy A. P., Kayode O., Wang R., Hockla A., Soares A. S., and Radisky E. S. (2015) Mesotrypsin has evolved four unique residues to cleave trypsin inhibitors as substrates. J. Biol. Chem. 290, 21523–21535 10.1074/jbc.M115.662429 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Pendlebury D., Wang R., Henin R. D., Hockla A., Soares A. S., Madden B. J., Kazanov M. D., and Radisky E. S. (2014) Sequence and conformational specificity in substrate recognition: several human Kunitz protease inhibitor domains are specific substrates of mesotrypsin. J. Biol. Chem. 289, 32783–32797 10.1074/jbc.M114.609560 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Kayode O., Wang R., Pendlebury D. F., Cohen I., Henin R. D., Hockla A., Soares A. S., Papo N., Caulfield T. R., and Radisky E. S. (2016) An acrobatic substrate metamorphosis reveals a requirement for substrate conformational dynamics in trypsin proteolysis. J. Biol. Chem. 291, 26304–26319 10.1074/jbc.M116.758417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Cohen I., Kayode O., Hockla A., Sankaran B., Radisky D. C., Radisky E. S., and Papo N. (2016) Combinatorial protein engineering of proteolytically resistant mesotrypsin inhibitors as candidates for cancer therapy. Biochem. J. 473, 1329–1341 10.1042/BJ20151410 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Dani V. S., Ramakrishnan C., and Varadarajan R. (2003) MODIP revisited: re-evaluation and refinement of an automated procedure for modeling of disulfide bonds in proteins. Protein Eng. 16, 187–193 10.1093/proeng/gzg024 [DOI] [PubMed] [Google Scholar]
- 21. Salameh M. A., Soares A. S., Navaneetham D., Sinha D., Walsh P. N., and Radisky E. S. (2010) Determinants of affinity and proteolytic stability in interactions of Kunitz family protease inhibitors with mesotrypsin. J. Biol. Chem. 285, 36884–36896 10.1074/jbc.M110.171348 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Salameh M. A., and Radisky E. S. (2013) Biochemical and structural insights into mesotrypsin: an unusual human trypsin. Int. J. Biochem. Mol. Biol. 4, 129–139 [PMC free article] [PubMed] [Google Scholar]
- 23. Jiang G., Cao F., Ren G., Gao D., Bhakta V., Zhang Y., Cao H., Dong Z., Zang W., Zhang S., Wong H. H., Hiley C., Crnogorac-Jurcevic T., Lemoine N. R., and Wang Y. (2010) PRSS3 promotes tumour growth and metastasis of human pancreatic cancer. Gut 59, 1535–1544 10.1136/gut.2009.200105 [DOI] [PubMed] [Google Scholar]
- 24. Hockla A., Miller E., Salameh M. A., Copland J. A., Radisky D. C., and Radisky E. S. (2012) PRSS3/Mesotrypsin is a therapeutic target for metastatic prostate cancer. Mol. Cancer Res. 10, 1555–1566 10.1158/1541-7786.MCR-12-0314 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Craig D. B., and Dombkowski A. A. (2013) Disulfide by design 2.0: a web-based tool for disulfide engineering in proteins. BMC Bioinformatics 14, 346 10.1186/1471-2105-14-346 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Liu T., Wang Y., Luo X., Li J., Reed S. A., Xiao H., Young T. S., and Schultz P. G. (2016) Enhancing protein stability with extended disulfide bonds. Proc. Natl. Acad. Sci. U.S.A. 113, 5910–5915 10.1073/pnas.1605363113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Matsumura M., Becktel W. J., Levitt M., and Matthews B. W. (1989) Stabilization of phage T4 lysozyme by engineered disulfide bonds. Proc. Natl. Acad. Sci. U.S.A. 86, 6562–6566 10.1073/pnas.86.17.6562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Bhabha G., Lee J., Ekiert D. C., Gam J., Wilson I. A., Dyson H. J., Benkovic S. J., and Wright P. E. (2011) A dynamic knockout reveals that conformational fluctuations influence the chemical step of enzyme catalysis. Science 332, 234–238 10.1126/science.1198542 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Boehr D. D., McElheny D., Dyson H. J., and Wright P. E. (2006) The dynamic energy landscape of dihydrofolate reductase catalysis. Science 313, 1638–1642 10.1126/science.1130258 [DOI] [PubMed] [Google Scholar]
- 30. Torbeev V. Y., Raghuraman H., Hamelberg D., Tonelli M., Westler W. M., Perozo E., and Kent S. B. (2011) Protein conformational dynamics in the mechanism of HIV-1 protease catalysis. Proc. Natl. Acad. Sci. U.S.A. 108, 20982–20987 10.1073/pnas.1111202108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Watt E. D., Shimada H., Kovrigin E. L., and Loria J. P. (2007) The mechanism of rate-limiting motions in enzyme function. Proc. Natl. Acad. Sci. U.S.A. 104, 11981–11986 10.1073/pnas.0702551104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Whittier S. K., Hengge A. C., and Loria J. P. (2013) Conformational motions regulate phosphoryl transfer in related protein tyrosine phosphatases. Science 341, 899–903 10.1126/science.1241735 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Wolf-Watz M., Thai V., Henzler-Wildman K., Hadjipavlou G., Eisenmesser E. Z., and Kern D. (2004) Linkage between dynamics and catalysis in a thermophilic-mesophilic enzyme pair. Nat. Struct. Mol. Biol. 11, 945–949 10.1038/nsmb821 [DOI] [PubMed] [Google Scholar]
- 34. Guerin M. E., Stirnemann G., and Giganti D. (2018) Conformational entropy of a single peptide controlled under force governs protease recognition and catalysis. Proc. Natl. Acad. Sci. U.S.A. 115, 11525–11530 10.1073/pnas.1803872115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Lehmann A. (2008) Ecallantide (DX-88), a plasma kallikrein inhibitor for the treatment of hereditary angioedema and the prevention of blood loss in on-pump cardiothoracic surgery. Expert Opin. Biol. Ther. 8, 1187–1199 10.1517/14712598.8.8.1187 [DOI] [PubMed] [Google Scholar]
- 36. Longhurst H. (2017) Optimum use of acute treatments for hereditary angioedema: evidence-based expert consensus. Front. Med. 4, 245 10.3389/fmed.2017.00245 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Devy L., Rabbani S. A., Stochl M., Ruskowski M., Mackie I., Naa L., Toews M., van Gool R., Chen J., Ley A., Ladner R. C., Dransfield D. T., and Henderikx P. (2007) PEGylated DX-1000: pharmacokinetics and antineoplastic activity of a specific plasmin inhibitor. Neoplasia 9, 927–937 10.1593/neo.07544 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dubois M., Becher F., Herbet A., and Ezan E. (2007) Immuno-mass spectrometry assay of EPI-HNE4, a recombinant protein inhibitor of human elastase. Rapid Commun. Mass Spectrom. 21, 352–358 10.1002/rcm.2844 [DOI] [PubMed] [Google Scholar]
- 39. Attucci S., Gauthier A., Korkmaz B., Delépine P., Martino M. F., Saudubray F., Diot P., and Gauthier F. (2006) EPI-hNE4, a proteolysis-resistant inhibitor of human neutrophil elastase and potential anti-inflammatory drug for treating cystic fibrosis. J. Pharmacol. Exp. Ther. 318, 803–809 10.1124/jpet.106.103440 [DOI] [PubMed] [Google Scholar]
- 40. Trivedi M., Laurence J. S., Williams T. D., Middaugh C. R., and Siahaan T. J. (2012) Improving the stability of the EC1 domain of E-cadherin by thiol alkylation of the cysteine residue. Int. J. Pharm. 431, 16–25 10.1016/j.ijpharm.2012.03.051 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Trivedi M. V., Laurence J. S., and Siahaan T. J. (2009) The role of thiols and disulfides on protein stability. Curr. Protein Pept. Sci. 10, 614–625 10.2174/138920309789630534 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Jiang S., Li P., Lee S.-L., Lin C. Y., Long Y.-Q., Johnson M. D., Dickson R. B., and Roller P. P. (2007) Design and synthesis of redox stable analogues of sunflower trypsin inhibitors (SFTI-1) on solid support, potent inhibitors of matriptase. Organic Lett. 9, 9–12 10.1021/ol0621497 [DOI] [PubMed] [Google Scholar]
- 43. Li P., Jiang S., Lee S.-L., Lin C. Y., Johnson M. D., Dickson R. B., Michejda C. J., and Roller P. P. (2007) Design and synthesis of novel and potent inhibitors of the type II transmembrane serine protease, matriptase, based upon the sunflower trypsin inhibitor-1. J. Med. Chem. 50, 5976–5983 10.1021/jm0704898 [DOI] [PubMed] [Google Scholar]
- 44. Moore E. J., Zorine D., Hansen W. A., Khare S. D., and Fasan R. (2017) Enzyme stabilization via computationally guided protein stapling. Proc. Natl. Acad. Sci. U.S.A. 114, 12472–12477 10.1073/pnas.1708907114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Cohen I., Naftaly S., Ben-Zeev E., Hockla A., Radisky E. S., and Papo N. (2018) Pre-equilibrium competitive library screening for tuning inhibitor association rate and specificity toward serine proteases. Biochem. J. 475, 1335–1352 10.1042/BCJ20180070 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Stein R. (2011) Kinetics of Enzyme Action: Essential Principles for Drug Hunters, pp. 115–140, John Wiley & Sons, Hoboken, NJ [Google Scholar]
- 47. Kabsch W. (2010) XDS. Acta Crystallogr. D Biol. Crystallogr. 66, 125–132 10.1107/S0907444909047337 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J Appl. Crystallogr. 40, 658–674 10.1107/S0021889807021206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 10.1107/S0907444910045749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 10.1107/S0907444904019158 [DOI] [PubMed] [Google Scholar]
- 52. Winter G. (2010) xia2: an expert system for macromolecular crystallography data reduction. J. Appl. Crystallogr. 43, 186–190 10.1107/S0021889809045701 [DOI] [Google Scholar]
- 53. Winter G., Waterman D. G., Parkhurst J. M., Brewster A. S., Gildea R. J., Gerstel M., Fuentes-Montero L., Vollmar M., Michels-Clark T., Young I. D., Sauter N. K., and Evans G. (2018) DIALS: implementation and evaluation of a new integration package. Acta Crystallogr. D Biol. Crystallogr. 74, 85–97 10.1107/S2059798317017235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Evans P. (2006) Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 62, 72–82 10.1107/S0907444905036693 [DOI] [PubMed] [Google Scholar]
- 55. Evans P. R., and Murshudov G. N. (2013) How good are my data and what is the resolution? Acta Crystallogr. D Biol. Crystallogr. 69, 1204–1214 10.1107/S0907444913000061 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Vagin A., and Teplyakov A. (2010) Molecular replacement with MOLREP. Acta Crystallogr. D Biol. Crystallogr. 66, 22–25 10.1107/S0907444909042589 [DOI] [PubMed] [Google Scholar]
- 57. Salameh M. A., Soares A. S., Hockla A., Radisky D. C., and Radisky E. S. (2011) The P(2)′ residue is a key determinant of mesotrypsin specificity: engineering a high-affinity inhibitor with anticancer activity. Biochem. J. 440, 95–105 10.1042/BJ20110788 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Dockal M., Hartmann R., Fries M., Thomassen M. C., Heinzmann A., Ehrlich H., Rosing J., Osterkamp F., Polakowski T., Reineke U., Griessner A., Brandstetter H., and Scheiflinger F. (2014) Small peptides blocking inhibition of factor Xa and tissue factor-factor VIIa by tissue factor pathway inhibitor (TFPI). J. Biol. Chem. 289, 1732–1741 10.1074/jbc.M113.533836 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Afonine P. V., Grosse-Kunstleve R. W., Echols N., Headd J. J., Moriarty N. W., Mustyakimov M., Terwilliger T. C., Urzhumtsev A., Zwart P. H., and Adams P. D. (2012) Towards automated crystallographic structure refinement with phenix.refine. Acta Crystallogr. D Biol. Crystallogr. 68, 352–367 10.1107/S0907444912001308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Chen V. B., Arendall W. B. 3rd., Headd J. J., Keedy D. A., Immormino R. M., Kapral G. J., Murray L. W., Richardson J. S., and Richardson D. C. (2010) MolProbity: all-atom structure validation for macromolecular crystallography. Acta Crystallogr. D. Biol. Crystallogr. 66, 12–21 10.1107/S0907444909042073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Read R. J., Adams P. D., Arendall W. B. 3rd., Brunger A. T., Emsley P., Joosten R. P., Kleywegt G. J., Krissinel E. B., Lütteke T., Otwinowski Z., Perrakis A., Richardson J. S., Sheffler W. H., Smith J. L., Tickle I. J., et al. (2011) A new generation of crystallographic validation tools for the Protein Data Bank. Structure 19, 1395–1412 10.1016/j.str.2011.08.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Caulfield T. R., Devkota B., and Rollins G. C. (2011) Examinations of tRNA range of motion using simulations of cryo-EM microscopy and X-ray data. J. Biophys. 2011, 219515 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Caulfield T. R., Fiesel F. C., Moussaud-Lamodière E. L., Dourado D. F., Flores S. C., and Springer W. (2014) Phosphorylation by PINK1 releases the UBL domain and initializes the conformational opening of the E3 ubiquitin ligase Parkin. PLoS Comput. Biol. 10, e1003935 10.1371/journal.pcbi.1003935 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Puschmann A., Fiesel F. C., Caulfield T. R., Hudec R., Ando M., Truban D., Hou X., Ogaki K., Heckman M. G., James E. D., Swanberg M., Jimenez-Ferrer I., Hansson O., Opala G., Siuda J., et al. (2017) Heterozygous PINK1 p.G411S increases risk of Parkinson's disease via a dominant-negative mechanism. Brain 140, 98–117 10.1093/brain/aww261 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Zhang Y. J., Caulfield T., Xu Y. F., Gendron T. F., Hubbard J., Stetler C., Sasaguri H., Whitelaw E. C., Cai S., Lee W. C., and Petrucelli L. (2013) The dual functions of the extreme N terminus of TDP-43 in regulating its biological activity and inclusion formation. Hum. Mol. Genet. 22, 3112–3122 10.1093/hmg/ddt166 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Caulfield T., and Devkota B. (2012) Motion of transfer RNA from the A/T state into the A-site using docking and simulations. Proteins 80, 2489–2500 10.1002/prot.24131 [DOI] [PubMed] [Google Scholar]
- 67. Caulfield T., and Medina-Franco J. L. (2011) Molecular dynamics simulations of human DNA methyltransferase 3B with selective inhibitor nanomycin A. J. Struct. Biol. 176, 185–191 10.1016/j.jsb.2011.07.015 [DOI] [PubMed] [Google Scholar]
- 68. Caulfield T. R. (2011) Inter-ring rotation of apolipoprotein A-I protein monomers for the double-belt model using biased molecular dynamics. J. Mol. Graph. Model. 29, 1006–1014 10.1016/j.jmgm.2011.04.005 [DOI] [PubMed] [Google Scholar]
- 69. Jorgensen W. L., Chandrasekhar J., Madura J. D., Impey R. W., and Klein M. L. (1983) Comparison of simple potential functions for simulating liquid water. J. Chem. Phys. 79, 926–935 10.1063/1.445869 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.