

Abstract
The Nr4a subfamily of nuclear receptor comprises three members in mammalian cells: Nur77/Nr4a1, Nurr1/Nr4a2, and Nor1/Nr4a3. Nr4a proteins play key roles in the regulation of glucose homeostasis in peripheral metabolic tissues. However, their biological functions in β-cells remain relatively uncharacterized. Here we sought to investigate the potential role of Nor1 in the regulation of β-cell mass and, in particular, β-cell survival/apoptosis. We used histological analysis to examine the consequences of genetic deletion of either Nur77 and Nor1 on β-cell mass, investigated the expression patterns of Nr4as in human islets and INS cells and performed gain- and loss-of-function experiments to further characterize the role of Nor1 in β-cell apoptosis. Surprisingly, Nor1 knockout mice displayed increased β-cell mass, whereas mice with genetic deletion of Nur77 did not exhibit any significant differences compared with their WT littermates. The increase in β-cell mass in Nor1 knockout mice was accompanied by improved glucose tolerance. A gene expression study performed in both human islets and INS cells revealed that Nor1 expression is significantly increased by pro-inflammatory cytokines and, to a lesser extent, by elevated concentrations of glucose. Nor1 overexpression in both INS and human islet cells caused apoptosis, whereas siRNA-mediated Nor1 knockdown prevented cytokine-induced β-cell death. Finally, Nor1 expression was up-regulated in islets of individuals with type 2 diabetes. Altogether, our results uncover that Nor1 negatively regulates β-cell mass. Nor1 represents a promising molecular target in diabetes treatment to prevent β-cell destruction.
Keywords: β-cell, diabetes, apoptosis, nuclear receptor, cytokine, pancreas, insulin, Nr4a
Introduction
Type 2 diabetes is characterized by progressive deterioration of both β-cell mass and function, resulting in a relative deficit in insulin secretion (1, 2). Autopsy studies have indeed reported significant reductions in β-cell mass in patients with type 2 diabetes (3, 4), an effect that has been largely attributed to an increase in β-cell apoptosis (3, 5). Several environmental factors might contribute to β-cell death in diabetes (5), including elevated circulating levels of long-chain fatty acids, chronic hyperglycemia, and production of pro-inflammatory cytokines (6). Despite recent progress in this field, the instructive signals and the precise intracellular pathways that orchestrate β-cell death remain elusive.
The Nr4a family of orphan nuclear receptors comprises three members in mammalian cells: Nur77/Nr4a1, Nurr1/Nr4a2, and Nor1/Nr4a3 (7). To date, no ligand has been identified for Nr4as (8). Thus, their activity has been suggested to be regulated at the level of expression (consistent with their designation as immediate-early genes) as well as through posttranslational modifications and proteasome-dependent degradation (9–15). Nr4as have been implicated in numerous pathologies such as Parkinson disease, cancer, and inflammatory diseases (16, 17). Importantly, the potential involvement of Nr4as in metabolic diseases has received increasing attention (reviewed in Ref. 16). This stems from several key observations: Nr4as expression varies in animal models of obesity and/or diabetes (18–20), their expression is increased in adipose tissue of obese patients compared with lean subjects (21), Nr4a expression is modulated by physical activity and nutritional interventions (16, 22–24), and Nr4as have been shown to exert tissue-specific functions, resulting in the regulation of glucose homeostasis (18, 20, 25–32).
The biological roles of Nr4as in pancreatic β-cells remain relatively unexplored but have recently attracted interest. A previous publication has suggested that Nr4as expression in the pancreas is restricted to endocrine cells (26). Further, Nur77 was induced by cytotoxic concentrations of free fatty acids to mediate some of the deleterious effects of lipotoxicity in MIN6 cells (26). Importantly, Nur77 and Nor1 were found to regulate different transcriptional targets in MIN6 cells, suggesting nonredundant functions for the two Nr4a members (26). More recently, the homeodomain transcription factor Nkx6.1 has been characterized as a transcriptional regulator of Nur77 and Nor1 (33). The authors went on to show that the proliferative action of Nkx6.1 is blunted in Nur77 knockout animals.
Here we sought to investigate the potential role of Nor1 in pancreatic β-cell mass. We thus examined the cross-sectional β-cell area in animals with genetic deletion of either Nur77 or Nor1. Surprisingly, β-cell mass was significantly increased in Nor1 knockout mice. This prompted us to further characterize the function of Nor1 in β-cells by studying its regulation and potential implication in β-cell death.
Results
Nor1 knockout mice display increased β-cell mass and improved glucose homeostasis
To investigate the potential roles of Nr4a members in the regulation of β-cell mass, we measured the cross-sectional β-cell areas in pancreata harvested from whole-body Nor1 or Nur77 knockout mice compared with WT littermates. Because homozygous deletion of Nurr1 is embryonic lethal, we could not directly compare the consequences of loss of Nurr1. Fig. 1A shows typical immunohistochemistry images for each phenotype. Our morphological study indicated that Nor1 knockout mice have increased β-cell mass compared with their WT littermates (Fig. 1B). Indeed, the β-cell area was ∼2.5-fold greater in Nor1 knockout mice versus control animals. This phenotype was unique to Nor1 because Nur77 knockout islets did not present any significant difference compared with their respective controls. In addition, Nor1 knockout animals consistently displayed a 50% increase in the number of replicating (Ki67-positive) β-cells compared with control animals (Fig. 1C). α-Cell mass remained unchanged in Nor1 knockout mice (Fig. 1D).
Figure 1.
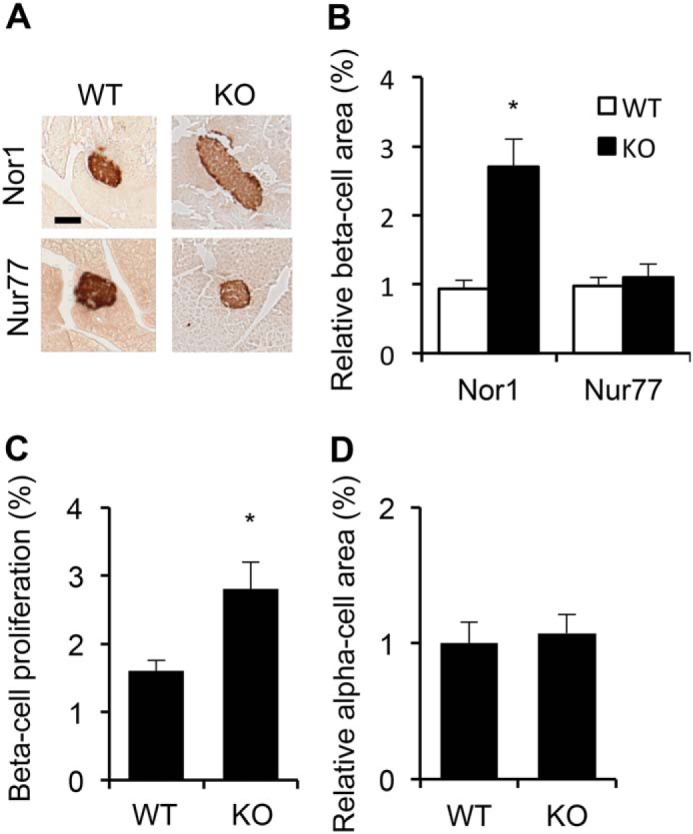
Nor1 knockout mice display increased β-cell mass. A, images show representative insulin immunostaining in WT, Nor1−/− and Nur77−/− pancreata. B, quantification of cross-sectional β-cell areas. Results are represented as -fold-change over WT animals. n = 6 for each genotype. C, β-cell proliferation was evaluated by counting the number of Ki67-positive nuclei. n = 4 for each genotype. D, quantification of cross-sectional α-cell area in WT and Nor1 knockout mice. n = 4 for each genotype. All results are represented as means ± S.E.; *, p < 0.05 versus WT animals. Scale bar = 100 μm.
We next investigated the tissue distribution of Nor1 in the pancreas by immunofluorescence in mice (Fig. 2). Our results indicate that, in WT pancreata, Nor1 is most strongly expressed in islets and co-localizes with both insulin (Fig. 2A) and glucagon (Fig. 2B). The signal for Nor1 was absent in tissue sections from Nor1 knockout mice (Fig. 2D), confirming the specificity of the detected signal.
Figure 2.

Nor1 expression in pancreatic islets. A, double immunofluorescence staining for Nor1 (red) and insulin (green) in WT mice. B, double immunofluorescence staining for Nor1 (red) and glucagon (green) in WT mice. C and D, double immunofluorescence staining for Nor1 and insulin in WT (C) and Nor1−/− animals (D). Scale bars = 100 μm. DAPI, 4′,6-diamidino-2-phenylindole.
We next sought to test whether the increase in β-cell mass in Nor1 knockout animals translated into improved glucose tolerance. We thus measured nonfasting blood glucose and also performed an i.p. glucose tolerance test in Nor1 knockout mice versus controls. Nonfasting blood glucose was significantly lower in Nor1 knockout mice with an average of 4.7 mm compared with 5.5 mm in WT mice (Fig. 3A). Also, mice with genetic deletion of Nor1 displayed improved glucose tolerance, an effect that was not observed in Nur77 knockout mice (Fig. 3, B and C, respectively). Indeed, blood glucose was significantly lower 15, 30, and 45 min following glucose injection in Nor1 knockout versus WT mice. After 120 min, the area under the curve was significantly reduced in Nor1-knockout mice versus controls (Fig. 3D), whereas it was unchanged in Nur77 knockout animals compared with their respective controls. The effect of Nor1 on islet mass and glucose homeostasis could not be accounted for by differences in body weight (Fig. 3E) because this parameter was similar in Nor1 knockout versus control mice. These results are consistent with a hitherto uncharacterized role for Nor1 in the regulation of β-cell mass.
Figure 3.

Invalidation of Nor1 improves glucose tolerance. A, random-fed blood glucose in WT versus Nor1 knockout mice. B and C, glucose tolerance test in Nor1 (B) and Nur77 knockout mice. C, area under the curve for each genotype. D, body weight of 6-month-old WT and Nor1 knockout mice. E, body weight of WT and Nor1 knockout mice. All results are represented as means ± S.E.; n = 10 mice in each group. *, p < 0.05 versus WT animals; AUC, area under the curve.
Nor1 expression is up-regulated by pro-inflammatory cytokines
Although Nor1 has been extensively studied in muscle (34), its regulation and biological functions remain relatively unexplored in β-cells. We therefore studied the expression profile of Nor1 and explored the mechanisms by which the nuclear receptor could exert its effect on β-cell mass.
To investigate the regulation of Nr4a gene expression, we exposed INS cells to either a cytokine mixture comprising IL-1β and IFNγ or elevated concentrations of glucose (glucotoxicity) for various periods of time ranging from 0 to 24 h and measured the expression of all three members of the Nr4a family by qPCR.3 Cytotoxic concentrations of cytokines induced a rapid and transient increase in the expression of Nor1, with a maximum induction of ∼14-fold at 4 h (Fig. 4A). Interestingly, this effect was relatively specific to Nor1. Indeed, Nur77 was increased by a much lesser extent compared with Nor1 (∼2-fold at 1 h), whereas Nurr1 expression remained unaltered. Fig. S1 expresses the results differently to illustrate the relative expression of each Nr4a member before (Fig. S1A) and after cytokine treatment (Fig. S1B). High glucose induced an ∼5-fold increase in Nor1 expression after 8 h (Fig. 4B). Again, this effect of glucose was specific to Nor1 because Nur77 and Nurr1 mRNA levels were not significantly altered. We confirmed the up-regulation of Nor1 by cytokines at the protein level. Exposure of INS cells to cytokines for 6 h caused a 3-fold increase in Nor1 protein levels (Fig. 4C).
Figure 4.

Nor1 is up-regulated by pro-inflammatory cytokines in INS and human islet cells. A and B, mRNA levels of Nor1, Nur77, and Nurr1 were determined by qPCR in INS832/13 cells exposed to cytokines (10 ng/ml IL-1β and 10 ng/ml IFNγ, A) or an elevated concentration of glucose (25 mm, G25, B) for the indicated periods of time (n = 5). Results were normalized to actin and are expressed as -fold change over nonstimulated cells. C, Nor1 protein levels were determined in INS832/13 cells incubated for 6 h in the presence or absence of cytokines (n = 4). A representative image is shown. D–F, the effect of cytokines on Nor1, Nur77, and Nurr1 expression was determined by qPCR in human islets after 8 h of treatment (n = 3). G, dose–response relationship between Nor1 gene expression and cytokine concentrations. INS832/13 cells were exposed to increasing concentrations of IL-1β and IFNγ for 4 h (n = 5). H, the effects of 10 ng/ml IL-1β and IFNγ were tested separately or in combination for 4 h (n = 5). All results are represented as means ± S.E.; *, p < 0.05 versus control; #, p < 0.05 versus IL-1β.
We next sought to confirm the effects of cytokines on Nor1 mRNA expression in human islets. Consistent with our previous results obtained in INS cells, cytokines up-regulated Nor1 expression in human islets (Fig. 4D), albeit to a lesser extent. On the contrary, both Nur77 and Nurr1 mRNA levels remained unchanged (Fig. 4, E and F).
To further characterize the effects of cytokines on Nor1 expression, we tested increasing concentrations of IL-1β and IFNγ and evaluated the respective contribution of each cytokine singly. Nor1 expression was increased in a dose-dependent manner in response to the cytokine mixture in INS cells (Fig. 4G). Also, when both cytokines were used separately, the effect of IL-1β on Nor1 expression was greater than that of IFNγ alone (∼6-fold versus ∼1.5-fold, respectively) (Fig. 4H).
Altogether, these results identify Nor1 as a cytokine-responsive gene in β-cells and thus raise the possibility that Nor1 could exert pro-apoptotic effects in β-cells. Indeed, such a pro-apoptotic action of Nor1 would dovetail with the increased β-cell mass observed in Nor1−/− animals.
Nor1 is a novel mediator of β-cell death
To investigate the potential pro-apoptotic role of Nor1 in β-cells, we sought to perform gain-of-function and loss-of-function studies using Nor1 overexpression and siRNA-mediated knockdown, respectively. Fig. 5A shows that Nor1 protein levels were increased by 3- to 4-fold in INS cells overexpressing Nor1-GFP or Nor1-FLAG compared with control cells. We then transfected INS cells with either Nor1-FLAG or a control vector before incubating them in the presence or absence of cytokines. Nor1 overexpression induced a 2-fold increase in apoptosis (Fig. 5, B and C), and this effect was not additive to that of cytokines. The pro-apoptotic action of Nor1 was confirmed in human islets. Human islet cells were transfected with either Nor1-GFP or a control GFP plasmid. Then we measured apoptosis by counting the number of TUNEL-positive (red) nuclei in the transfected cell population only (green). Ectopic expression of Nor1 increased apoptosis by 5-fold (Fig. 5, D and E).
Figure 5.

Nor1 plays a critical role in INS and islet cell apoptosis. A, Nor1 protein levels were evaluated in nontransfected (NT) INS832/13 cells or 24 h after transfection with a control GFP plasmid, Nor1-GFP, or Nor1-FLAG (n = 5). A representative immunoblots is shown. Quantification results are represented as means ± S.E.; *, p < 0.05 versus control GFP. The arrows indicate the predicted Nor1 bands, with Nor1-GFP migrating at a higher molecular weight. B and C, apoptosis was assessed by TUNEL assay in INS832/13 cells transfected with Nor1 (black columns) or a control vector (white columns) and subsequently incubated in the presence or absence of cytokines for 24 h (n = 3). D and E, apoptosis was measured by TUNEL assay in dispersed human islets 24 h after transfection with Nor1-GFP or GFP alone. The results are represented as the percentages of TUNEL-labeled nuclei in the population of GFP-positive cells (n = 3). DAPI, 4′,6-diamidino-2-phenylindole. F, Nor1, Nur77, and Nurr1 expression was evaluated by qPCR in INS832/13 cells 48 h after transfection with a control siRNA or two different Nor1-specific siRNA sequences (n = 3). G and H, siRNA-mediated knockdown of Nor1 prevented cytokine-induced apoptosis. Apoptosis was determined by TUNEL in INS832/13 cells transfected with control or Nor1-specific siRNAs and incubated in the presence (black columns) or absence (white columns) of cytokines as described above (n = 3). I, the effects of cytokines on islet cell apoptosis was also investigated in intact WT and Nor1-KO mouse islets ex vivo by measuring the fragmentation of DNA using an ELISA assay (n = 5). J and K, siRNA-mediated knockdown of Nor1 prevented cytokine-induced apoptosis in human islet cells. L, Nor1 caused cytochrome c release. Cells were treated as described in B, fractionated into cytoplasmic and mitochondrial fractions, and analyzed by ELISA (n = 4). All results are represented as means ± S.E.; *, p < 0.05 versus control; # p < 0.05 versus PCMV-Nor1-FLAG without cytokines. Scale bars = 50 μm.
To further test the hypothesis that Nor1 represents a novel mediator of cytokine-induced β-cell apoptosis, we performed siRNA-mediated Nor1 knockdown (Fig. 5F). Cytokine-induced β-cell apoptosis was completely abolished by two different siRNA sequences (Fig. 5, G and H), indicating that Nor1 is necessary for the pro-apoptotic action of cytokines in β-cells. We next sought to confirm this result in isolated mouse islets from WT and Nor1 KO mice. Thus, intact islets were cultured ex vivo in the presence or absence of cytokines, and DNA fragmentation was measured by ELISA (Fig. 5I). The results show that Nor1 KO islets were resistant to cytokine-mediated apoptosis. siRNA-mediated knockdown of Nor1 in human islets also prevented cytokine-induced apoptosis like it did in INS cells (Fig. 5, J and K).
To gain insight into the molecular mechanisms by which Nor1 causes β-cell apoptosis, we tested the effects of Nor1 on cytochrome c release in β-cells. Indeed, it was suggested previously that Nor1 could translocate to the mitochondria and activate the intrinsic (mitochondrial) pathway of apoptosis in thymocytes (35). Fig. 5L shows that ectopic expression of Nor1 induced cytochrome c release from the mitochondria to the same extent as cytokines. This result suggests that Nor1-mediated β-cell apoptosis involves the intrinsic pathway as well. Future studies should delve deeper into the mechanisms by which Nor1 promotes β-cell death.
We next sought to perform a “gene rescue” experiment in which we performed siRNA-mediated gene knockdown as described above, followed by Nor1 overexpression to re-establish Nor1 expression levels (Fig. 6A). It could be observed that the two siRNA sequences failed to prevent apoptosis when Nor1 was re-expressed to apoptotic levels (Fig. 6B). This experiment limits the possibility that our results were due to off-target effects.
Figure 6.

Nor1 re-expression following siRNA-mediated knockdown causes apoptosis in INS cells. A and B, cells were transfected with Nor1 siRNAs or siControl, followed by overexpression using Nor1-GFP. GFP was used as a control. Results shown are means ± S.E.; *, p < 0.05 versus GFP controls.
Nor1 expression is increased in type 2 diabetes
Because IL-1β is up-regulated in diabetic islets (36), we reasoned that Nor1 could be increased in islets of patients with type 2 diabetes. Fig. 7 shows that Nor1 mRNA levels are increased by ∼12-fold in human islets obtained from patients with type 2 diabetes compared with healthy individuals. This change was specific to Nor1 because the two other Nr4a members, Nur77 and Nurr1, were not significantly altered in type 2 diabetic islets. As a control, we measured the expression of Pdx1, which was significantly decreased by 50% in diabetic islets. We thus postulate that the up-regulation of Nor1 in type 2 diabetic islets could contribute to β-cell demise and may represent a target for therapeutic interventions.
Figure 7.

Nor1 is up-regulated in type 2 diabetic islets. We compared Nor1, Nur77, Nurr1, and Pdx1 mRNA expression in isolated human islets of healthy non-diabetic (ND) or type 2 diabetic (T2D) donors (n = 7). Islets were procured the morning after their isolation and handpicked, and mRNAs were isolated immediately. We only used islet preparations of regular morphology (circularity and size). Results shown are means ± S.E.; *, p < 0.05.
Discussion
The biological roles of Nr4as in metabolic diseases are gathering increasing interest (16, 37). Although their roles in liver and muscle have been studied extensively, their biological actions in pancreatic β-cells remain relatively unexplored.
Here we demonstrate that adult Nor1 knockout mice show increased β-cell mass and that this phenotype is not reiterated by genetic deletion of Nur77. The increase in β-cell mass in Nor1 knockout animals was accompanied by lower blood glucose in the random fed state and improved glucose tolerance during ipGTT. We believe that the effect of Nor1 on β-cell mass involves, at least in part, an increase in pancreatic β-cell replication because we detected an increase in Ki67 staining in Nor1 knockout mice. This distinct phenotype of Nor1 knockout mice is consistent with the unique DNA-binding properties of Nor1, which support distinct biological functions for Nor1 compared with its two Nr4a orthologs. Indeed, Nur77 and Nurr1 can bind the Nur-responsive element as homodimers (38, 39), whereas Nor1 displays a much lower affinity for the Nur-responsive element, resulting in a negligible transcriptional activation (38, 39). In addition, both Nur77 and Nurr1, but not Nor1, can form heterodimers or interact with retinoid X receptors to target DR5 elements (8, 40). Thus, Nor1 displays distinct binding characteristics to DNA regulatory elements and cofactors. This dovetails with a previous report demonstrating that Nur77 and Nor1 regulate distinct sets of genes in β-cells (26).
Because our study employed whole-body Nor1 knockout animals, we cannot rule out the possibility that some of the effects of Nor1 on pancreatic β-cell mass are secondary to changes in other tissues. We believe this is unlikely because our results were reiterated in vitro using gain- and loss-of-function experiments in both INS cells and human islet tissues. Importantly, Floxed-Nor1 mice have not yet been generated. This precludes tissue-specific deletion of Nor1 in β-cells, and whole-body Nor1-KO mice represent the best available model.
Our expression study shows that Nor1 expression is rapidly increased by cytokines. Indeed, cytokine treatments elicited an ∼14-fold and ∼3-fold increase in Nor1 expression in INS cells and human islets, respectively. High glucose also induced Nor1 expression, but its effect was less pronounced and occurred later. Interestingly, high glucose was found to stimulate production of IL-1β in β-cells (36). Because here we demonstrate that IL-1β is a key inducer of Nor1 expression in β-cells, this raises the possibility that glucose induces Nor1 expression via production of pro-inflammatory cytokines. Be that as it may, the up-regulation of Nor1 upon cytokine treatments hinted at a pro-apoptotic role for Nor1 in β-cells, an effect that, over time, could in part explain the increase in β-cell mass observed in Nor1 knockout mice.
Our functional assays using gain and loss of function for Nor1 in INS cells and human islets demonstrated a critical role for Nor1 in β-cell apoptosis. This holds great clinical significance because Butler et al. (3) showed that type 2 diabetic patients present a lower β-cell mass compared with healthy subjects and that this reduction was associated with increased rates of apoptosis. Because we detected an increase in Nor1 expression in type 2 diabetic islets, our study strongly suggests that Nor1 could participate in the β-cell demise that precedes diabetes onset. To unravel a potential mechanism by which Nor1 could promote β-cell apoptosis, we tested its potential effect on the intrinsic pathway of apoptosis. Nor1 was found to provoke cytochrome c release in INS cells, suggesting that Nor1-mediated β-cells apoptosis could involve mitochondrial stress. This is reminiscent of its role in thymocytes, where Nor1 has been demonstrated to relocate to the mitochondria and to associate with Bcl family members to cause apoptosis (35). Our findings, describing Nor1 as a negative regulator of β-cell mass expansion, conflict with those presented in a recent study (33). Indeed, Tessem et al. (33) characterized Nur77 and Nor1 as transcriptional targets of Nkx6.1 in β-cells. They also showed that both Nur77 and Nor1 were necessary for Nkx6.1-mediated β-cell proliferation in rat islets via a mechanism that could implicate transcriptional repression of p21. The causes of this discrepancy are unknown. However, although the authors confirmed their results in Nur77 knockout mice, they did not study Nor1 knockout animals.
SNPs located within the Nor1 locus have been associated with increased insulin secretion and a higher insulinogenic index in diabetic as well as nondiabetic patients (29, 31). At this point, it is unclear whether the improvement in insulin secretion in individuals bearing this mutation results from improved islet function, increased β-cell mass, or both. Further, some controversies reside over the role of Nor1 in insulin secretion. In one study, Nor1 has been suggested to mediate Exendin4-induced potentiation of glucose-induced insulin secretion (29). Conversely, another study characterized Nor1 as a negative regulator of insulin secretion (41).
In conclusion, our work shows that expression of the nuclear receptor Nor1 is up-regulated in type 2 diabetes islets and that Nor1 plays a key role in β-cell apoptosis. We propose that Nor1 represents an interesting molecular target for therapeutic intervention.
Experimental procedures
Animals
All animal studies were conducted in accordance with the Canadian Council on Animal Care Guidelines and Policies, with approval from the Animal Care and Use Committee of Laval University and the University of Alberta. Efforts were made to minimize animal suffering and to reduce the number of animals used. Nur77−/− mice were graciously provided by Dr. Jeff Milbrandt (University of Washington) (42). Nor1−/− mice were from Dr. Yves Labele (St-François d'Assise Hospital, Quebec City, QC, Canada). Nur77−/− mice were maintained on a C57/B6N background. Nor1−/− mice were maintained on a 129S4/SV background. 6-month-old male mice were used in this study. The mice were housed in a temperature-controlled environment on a 12-h light/dark cycle with ad libitum access to food and water. Access to food was removed 12 h prior to glucose challenge. Blood glucose was measured with FreeStyle Lite test strips (Abbott, Alameda, CA). Plasma insulin was measured using a commercial ELISA kit (Millipore, Etobicoke, ON, Canada).
Immunostaining
Pancreata were fixed and embedded in paraffin. 5- to 10-μm sections were deparaffinized and rehydrated before microwave antigen retrieval in citrate buffer. Slides were then blocked in 0.5% BSA and 4% donkey serum in 0.1% PBST (0.1% (v/v) Triton in PBS) for 1 h at room temperature, followed by overnight incubation at 4 °C with mouse anti-Nor1 (Abnova, 1:100, RRID AB_922071), guinea pig anti-insulin (Dako, Burlington, ON, Canada; 1:800, RRID AB_10013624), rabbit anti-Ki67 (Millipore, 1:50, RRID AB_2142366), or rabbit anti-glucagon (Dako, 1:800, RRID AB_1841774) antibodies. The next day, slides were incubated for 1 h at room temperature with anti-mouse Alexa Fluor 568, anti-guinea pig Alexa Fluor 488, anti-rabbit Alexa Fluor 568, or anti-rabbit Alexa Fluor 488 secondary antibodies (1:250, Life Technologies). Slides were mounted with Fluoromount-G® containing 4′,6-diamidino-2-phenylindole (Southern Biotech, Birmingham, AL). Images were analyzed using a spinning disk ERS confocal microscope (PerkinElmer Life Sciences, Guelph, ON, Canada) and Volocity imaging software.
Cell culture
INS832/13 cells (43) (passages 52–78) were grown in RPMI 1640 medium supplemented with 10 mm HEPES, 10% heat-inactivated fetal bovine serum, 2 mm l-glutamine, 1 mm sodium pyruvate, and 50 μm β-mercaptoethanol at 37 °C in a humidified 5% CO2 atmosphere. Cells at 80% confluence were treated in serum-free RPMI with a mixture of cytokines (IL-1β and IFNγ, both at 10 ng/ml) or with 25 mm glucose. All cell culture reagents were purchased from Life Technologies.
Human islets
Human islets were purchased from the Alberta Diabetes Institute Islet Core at the University of Alberta with the assistance of the Human Organ Procurement and Exchange Program and the Trillium Gift of Life Network, who provide donor pancreata for research. Islets are obtained the next morning after their isolation. For mRNA expression in response to cytokines and apoptosis assays, islets from seven different healthy lean donors between 48 and 74 years of age were used. We used islet preparations of similar morphology (including size and circularity) and purity of more than 95%. Islets were cultured in 11 mm glucose DMEM supplemented with 10 mm HEPES, 10% heat-inactivated fetal bovine serum, 2 mm l-glutamine, 1 mm sodium pyruvate, 1% penicillin-streptomycin, and 50 μm β-mercaptoethanol at 37 °C in a humidified 5% CO2 atmosphere. Our study was approved by the Human Research Ethics Board at the University of Alberta. The study abided by the Declaration of Helsinki principles.
qPCR
RNAs were extracted using the RNeasy Mini Kit (Qiagen, Germantown, MD) and reverse-transcribed using the SuperScript First-Strand Synthesis System (Life Technologies). For real-time PCR, we used RT2 SYBR Green ROX qPCR Mastermix (Qiagen) and an ABI 7900HT instrument (Life Technologies). Results were analyzed using SDS software 2.3. Actin was chosen as gene of reference. Results are expressed as -fold change (2(−ΔΔCt)) compared with the indicated control condition.
Western blotting
Cells were lysed in a buffer containing 50 mm Tris (pH 8.0), 150 mm NaCl, 1% Triton X-100, and protease inhibitors (cOmpleteMini, Roche Applied Sciences). Protein concentrations were determined by BCA protein assay. Equal amounts of heat-denaturated proteins from each treatment group were run on Novex gels (Life Technologies) and transferred onto nitrocellulose membranes. After blocking, membranes were incubated overnight with a mouse anti-Nor1 antibody (1:1000). The next day, membranes were incubated with horseradish peroxidase–linked secondary antibodies and exposed to ECL reagents. Band density was determined using the ChemiDoc MP Imaging System and Image Lab software 4.1 (Bio-Rad, Mississauga, ON, Canada). Membranes were stripped and incubated with an anti-Actin antibody (Santa Cruz Biotechnology, Dallas, TX; RRID AB_626630) to confirm equal loading. Blots were developed on film, and band density was determined using ImageJ software.
Plasmid transfection
pCMV-Nor1-GFP and pCMV-Nor1-FLAG were obtained from Origene (Rockville, MD). pCMV-mCherry was a kind gift from Dr. Peter Light (University of Alberta, Edmonton, AB, Canada). INS832/13 cells were transfected via nucleofection (Lonza, Allendale, NJ). Human islet cells were dispersed using 0.05% trypsin–EDTA and transfected the following day using Lipofectamine 2000 (Life Technologies) according to manufacturer's protocol. Cells were assayed 24 h post-transfection.
siRNA-mediated knockdown
Nor1-specific siRNAs were from Life Technologies. The sequences used were RSS329251 (hereafter called siNor1-A) and RSS367608 (siNor1-B). siNor1 and control siRNAs were transfected using Lipofectamine RNAiMax following the manufacturer's protocol.
Apoptosis
Apoptosis was assessed by TUNEL assay using either TMR Red (human islets) or fluorescein green (INS cells) in situ cell death detection kits (Roche Applied Science), following manufacturer's protocol. Images were captured at ×40 magnification using a Zeiss COLIBRI inverted fluorescence microscope. At least 2000 cells were analyzed for each experimental condition in at least three independent experiments. In human islet studies, the results are presented as the percentage of apoptotic cells (red nuclei) in the transfected population only (green, GFP-positive cells). For the determination of apoptosis in WT versus Nor1-KO islets, 40 intact islets of similar size were incubated in the presence or absence of cytokines for 24 h in 12-well plates. Histone-associated DNA fragments were quantified using the Cell Death Detection ELISA Plus Kit (Roche, Laval, QC, Canada) according to the manufacturer's protocol. This kit allows specific determination of mono- and oligonucleosomes generated by apoptotic cleavage of DNA.
Cytochrome c release
Cells were fractionated into nuclear, cytosolic, and mitochondrial fractions using a cell fractionation kit (Abcam, Toronto, ON, Canada). Cytochrome c release from the mitochondria to the cytoplasm was subsequently evaluated using a commercial ELISA kit (Abcam) and an EnVision 2104 Multilabel reader (PerkinElmer Life Sciences).
Statistical analysis
Data are presented as means ± S.E. Statistical analyses were performed with GraphPad Prism® using analysis of variance methods followed by Bonferroni's post test. p < 0.05 was considered statistically significant.
Author contributions
A.-F. C. and J. B. conceptualization; A.-F. C., C. R., and J. B. formal analysis; A.-F. C., N. D., B. S. V., C. R., and J. B. investigation; A.-F. C., C. R., and J. B. methodology; A.-F. C., C. R., and J. B. writing-original draft; A.-F. C., C. R., and J. B. writing-review and editing; C. R. and J. B. resources; C. R. and J. B. supervision; C. R. and J. B. project administration; J. B. validation.
Supplementary Material
Acknowledgments
We thank Renée Paradis (Laval), Joanie Baillargeon (Laval), and Ying Wayne Wang (University of Alberta) for technical support.
This work was supported by the Alberta Diabetes Foundation, the Canadian Institutes of Health Research, and the Natural Sciences and Engineering Research Council of Canada. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Fig. S1.
- qPCR
- quantitative PCR.
References
- 1. Saltiel A. R. (2001) New perspectives into the molecular pathogenesis and treatment of type 2 diabetes. Cell 104, 517–529 10.1016/S0092-8674(01)00239-2 [DOI] [PubMed] [Google Scholar]
- 2. Meier J. J., and Bonadonna R. C. (2013) Role of reduced β-cell mass versus impaired β-cell function in the pathogenesis of type 2 diabetes. Diabetes Care 36, S113–S119 10.2337/dcS13-2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Butler A. E., Janson J., Bonner-Weir S., Ritzel R., Rizza R. A., and Butler P. C. (2003) β-Cell deficit and increased β-cell apoptosis in humans with type 2 diabetes. Diabetes 52, 102–110 10.2337/diabetes.52.1.102 [DOI] [PubMed] [Google Scholar]
- 4. Meier J. J., Breuer T. G., Bonadonna R. C., Tannapfel A., Uhl W., Schmidt W. E., Schrader H., and Menge B. A. (2012) Pancreatic diabetes manifests when β cell area declines by approximately 65% in humans. Diabetologia 55, 1346–1354 10.1007/s00125-012-2466-8 [DOI] [PubMed] [Google Scholar]
- 5. Rhodes C. J. (2005) Type 2 diabetes: a matter of β-cell life and death? Science 307, 380–384 10.1126/science.1104345 [DOI] [PubMed] [Google Scholar]
- 6. Prentki M., and Nolan C. J. (2006) Islet β cell failure in type 2 diabetes. J. Clin. Invest. 116, 1802–1812 10.1172/JCI29103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nuclear Receptors Nomenclature Committee (1999) A unified nomenclature system for the nuclear receptor superfamily. Cell 97, 161–163 10.1016/S0092-8674(00)80726-6 [DOI] [PubMed] [Google Scholar]
- 8. Kurakula K., Koenis D. S., van Tiel C. M., and de Vries C. J. (2014) NR4A nuclear receptors are orphans but not lonesome. Biochim. Biophys. Acta 1843, 2543–2555 10.1016/j.bbamcr.2014.06.010 [DOI] [PubMed] [Google Scholar]
- 9. Lammi J., and Aarnisalo P. (2008) FGF-8 stimulates the expression of NR4A orphan nuclear receptors in osteoblasts. Mol. Cell Endocrinol. 295, 87–93 10.1016/j.mce.2008.08.023 [DOI] [PubMed] [Google Scholar]
- 10. Arredondo C., Orellana M., Vecchiola A., Pereira L. A., Galdames L., and Andrés M. E. (2013) PIASγ enhanced SUMO-2 modification of Nurr1 activation-function-1 domain limits Nurr1 transcriptional synergy. PLoS ONE 8, e55035 10.1371/journal.pone.0055035 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Galleguillos D., Vecchiola A., Fuentealba J. A., Ojeda V., Alvarez K., Gómez A., and Andrés M. E. (2004) PIASγ represses the transcriptional activation induced by the nuclear receptor Nurr1. J. Biol. Chem. 279, 2005–2011 10.1074/jbc.M308113200 [DOI] [PubMed] [Google Scholar]
- 12. Han Y. H., Cao X., Lin B., Lin F., Kolluri S. K., Stebbins J., Reed J. C., Dawson M. I., and Zhang X. K. (2006) Regulation of Nur77 nuclear export by c-Jun N-terminal kinase and Akt. Oncogene 25, 2974–2986 10.1038/sj.onc.1209358 [DOI] [PubMed] [Google Scholar]
- 13. Kovalovsky D., Refojo D., Liberman A. C., Hochbaum D., Pereda M. P., Coso O. A., Stalla G. K., Holsboer F., and Arzt E. (2002) Activation and induction of NUR77/NURR1 in corticotrophs by CRH/cAMP: involvement of calcium, protein kinase A, and MAPK pathways. Mol. Endocrinol. 16, 1638–1651 10.1210/mend.16.7.0863 [DOI] [PubMed] [Google Scholar]
- 14. Volakakis N., Malewicz M., Kadkhodai B., Perlmann T., and Benoit G. (2006) Characterization of the Nurr1 ligand-binding domain co-activator interaction surface. J. Mol. Endocrinol. 37, 317–326 10.1677/jme.1.02106 [DOI] [PubMed] [Google Scholar]
- 15. Wingate A. D., Campbell D. G., Peggie M., and Arthur J. S. (2006) Nur77 is phosphorylated in cells by RSK in response to mitogenic stimulation. Biochem. J. 393, 715–724 10.1042/BJ20050967 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Close A. F., Rouillard C., and Buteau J. (2013) NR4A orphan nuclear receptors in glucose homeostasis: a minireview. Diabetes Metab. 39, 478–484 10.1016/j.diabet.2013.07.005 [DOI] [PubMed] [Google Scholar]
- 17. Lévesque D., RC (2009) In The Handbook of Neuropsychiatric Biomarkers, Endophenotypes and Genes (Ritsner M. S., and Susser E., eds.) pp. 195–210, Springer, Amsterdam, Netherlands [Google Scholar]
- 18. Fu Y., Luo L., Luo N., Zhu X., and Garvey W. T. (2007) NR4A orphan nuclear receptors modulate insulin action and the glucose transport system: potential role in insulin resistance. J. Biol. Chem. 282, 31525–31533 10.1074/jbc.M701132200 [DOI] [PubMed] [Google Scholar]
- 19. Kanzleiter T., Wilks D., Preston E., Ye J., Frangioudakis G., and Cooney G. J. (2009) Regulation of the nuclear hormone receptor nur77 in muscle: influence of exercise-activated pathways in vitro and obesity in vivo. Biochim. Biophys. Acta 1792, 777–782 10.1016/j.bbadis.2009.05.002 [DOI] [PubMed] [Google Scholar]
- 20. Pei L., Waki H., Vaitheesvaran B., Wilpitz D. C., Kurland I. J., and Tontonoz P. (2006) NR4A orphan nuclear receptors are transcriptional regulators of hepatic glucose metabolism. Nat. Med. 12, 1048–1055 10.1038/nm1471 [DOI] [PubMed] [Google Scholar]
- 21. Veum V. L., Dankel S. N., Gjerde J., Nielsen H. J., Solsvik M. H., Haugen C., Christensen B. J., Hoang T., Fadnes D. J., Busch C., Vage V., Sagen J. V., and Mellgren G. (2012) The nuclear receptors NUR77, NURR1 and NOR1 in obesity and during fat loss. Int. J. Obes. (Lond.) 36, 1195–1202 10.1038/ijo.2011.240 [DOI] [PubMed] [Google Scholar]
- 22. Catoire M., Mensink M., Boekschoten M. V., Hangelbroek R., Müller M., Schrauwen P., and Kersten S. (2012) Pronounced effects of acute endurance exercise on gene expression in resting and exercising human skeletal muscle. PLoS ONE 7, e51066 10.1371/journal.pone.0051066 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Kawasaki E., Hokari F., Sasaki M., Sakai A., Koshinaka K., and Kawanaka K. (2009) Role of local muscle contractile activity in the exercise-induced increase in NR4A receptor mRNA expression. J. Appl. Physiol. 106, 1826–1831 10.1152/japplphysiol.90923.2008 [DOI] [PubMed] [Google Scholar]
- 24. Mahoney D. J., Parise G., Melov S., Safdar A., and Tarnopolsky M. A. (2005) Analysis of global mRNA expression in human skeletal muscle during recovery from endurance exercise. FASEB J. 19, 1498–1500 10.1096/fj.04-3149fje [DOI] [PubMed] [Google Scholar]
- 25. Berriel Diaz M., Lemke U., and Herzig S. (2006) Discovering orphans' sweet secret: NR4A receptors and hepatic glucose production. Cell Metab. 4, 339–340 10.1016/j.cmet.2006.10.005 [DOI] [PubMed] [Google Scholar]
- 26. Briand O., Helleboid-Chapman A., Ploton M., Hennuyer N., Carpentier R., Pattou F., Vandewalle B., Moerman E., Gmyr V., Kerr-Conte J., Eeckhoute J., Staels B., and Lefebvre P. (2012) The nuclear orphan receptor Nur77 is a lipotoxicity sensor regulating glucose-induced insulin secretion in pancreatic β-cells. Mol. Endocrinol. 26, 399–413 10.1210/me.2011-1317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Chao L. C., Wroblewski K., Ilkayeva O. R., Stevens R. D., Bain J., Meyer G. A., Schenk S., Martinez L., Vergnes L., Narkar V. A., Drew B. G., Hong C., Boyadjian R., Hevener A. L., Evans R. M., et al. (2012) Skeletal muscle Nur77 expression enhances oxidative metabolism and substrate utilization. J. Lipid Res. 53, 2610–2619 10.1194/jlr.M029355 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chao L. C., Zhang Z., Pei L., Saito T., Tontonoz P., and Pilch P. F. (2007) Nur77 coordinately regulates expression of genes linked to glucose metabolism in skeletal muscle. Mol. Endocrinol. 21, 2152–2163 10.1210/me.2007-0169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ordelheide A. M., Gerst F., Rothfuss O., Heni M., Haas C., Thielker I., Herzberg-Schäfer S., Böhm A., Machicao F., Ullrich S., Stefan N., Fritsche A., Häring H. U., and Staiger H. (2013) Nor-1, a novel incretin-responsive regulator of insulin genes and insulin secretion. Mol. Metab. 2, 243–255 10.1016/j.molmet.2013.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Pearen M. A., Eriksson N. A., Fitzsimmons R. L., Goode J. M., Martel N., Andrikopoulos S., and Muscat G. E. (2012) The nuclear receptor, Nor-1, markedly increases type II oxidative muscle fibers and resistance to fatigue. Mol. Endocrinol. 26, 372–384 10.1210/me.2011-1274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Weyrich P., Staiger H., Stancáková A., Schäfer S. A., Kirchhoff K., Ullrich S., Ranta F., Gallwitz B., Stefan N., Machicao F., Kuusisto J., Laakso M., Fritsche A., and Häring H. U. (2009) Common polymorphisms within the NR4A3 locus, encoding the orphan nuclear receptor Nor-1, are associated with enhanced β-cell function in non-diabetic subjects. BMC Med. Genet. 10, 77 10.1186/1471-2350-10-77 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nonogaki K., Kaji T., Ohba Y., Sumii M., Wakameda M., and Tamari T. (2009) Serotonin 5-HT2C receptor-independent expression of hypothalamic NOR1, a novel modulator of food intake and energy balance, in mice. Biochem. Biophys. Res. Commun. 386, 311–315 10.1016/j.bbrc.2009.06.023 [DOI] [PubMed] [Google Scholar]
- 33. Tessem J. S., Moss L. G., Chao L. C., Arlotto M., Lu D., Jensen M. V., Stephens S. B., Tontonoz P., Hohmeier H. E., and Newgard C. B. (2014) Nkx6.1 regulates islet β-cell proliferation via Nr4a1 and Nr4a3 nuclear receptors. Proc. Natl. Acad. Sci. U.S.A. 111, 5242–5247 10.1073/pnas.1320953111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Pearen M. A., Goode J. M., Fitzsimmons R. L., Eriksson N. A., Thomas G. P., Cowin G. J., Wang S. C., Tuong Z. K., and Muscat G. E. (2013) Transgenic muscle-specific Nor-1 expression regulates multiple pathways that effect adiposity, metabolism, and endurance. Mol. Endocrinol. 27, 1897–1917 10.1210/me.2013-1205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Thompson J., Burger M. L., Whang H., and Winoto A. (2010) Protein kinase C regulates mitochondrial targeting of Nur77 and its family member Nor-1 in thymocytes undergoing apoptosis. Eur. J. Immunol. 40, 2041–2049 10.1002/eji.200940231 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Maedler K., Sergeev P., Ris F., Oberholzer J., Joller-Jemelka H. I., Spinas G. A., Kaiser N., Halban P. A., and Donath M. Y. (2002) Glucose-induced β cell production of IL-1β contributes to glucotoxicity in human pancreatic islets. J. Clin. Invest. 110, 851–860 10.1172/JCI200215318 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Pearen M. A., and Muscat G. E. (2010) Minireview: Nuclear hormone receptor 4A signaling: implications for metabolic disease. Mol. Endocrinol. 24, 1891–1903 10.1210/me.2010-0015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Wilson T. E., Fahrner T. J., and Milbrandt J. (1993) The orphan receptors NGFI-B and steroidogenic factor 1 establish monomer binding as a third paradigm of nuclear receptor-DNA interaction. Mol. Cell Biol. 13, 5794–5804 10.1128/MCB.13.9.5794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Maira M., Martens C., Philips A., and Drouin J. (1999) Heterodimerization between members of the Nur subfamily of orphan nuclear receptors as a novel mechanism for gene activation. Mol. Cell Biol. 19, 7549–7557 10.1128/MCB.19.11.7549 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gissendanner C. R., Kelley K., Nguyen T. Q., Hoener M. C., Sluder A. E., and Maina C. V. (2008) The Caenorhabditis elegans NR4A nuclear receptor is required for spermatheca morphogenesis. Dev. Biol. 313, 767–786 10.1016/j.ydbio.2007.11.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Gao W., Fu Y., Yu C., Wang S., Zhang Y., Zong C., Xu T., Liu Y., Li X., and Wang X. (2014) Elevation of NR4A3 expression and its possible role in modulating insulin expression in the pancreatic β cell. PLoS ONE 9, e91462 10.1371/journal.pone.0091462 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Lee S. L., Wesselschmidt R. L., Linette G. P., Kanagawa O., Russell J. H., and Milbrandt J. (1995) Unimpaired thymic and peripheral T cell death in mice lacking the nuclear receptor NGFI-B (Nur77). Science 269, 532–535 10.1126/science.7624775 [DOI] [PubMed] [Google Scholar]
- 43. Hohmeier H. E., Mulder H., Chen G., Henkel-Rieger R., Prentki M., and Newgard C. B. (2000) Isolation of INS-1-derived cell lines with robust ATP-sensitive K+ channel-dependent and -independent glucose-stimulated insulin secretion. Diabetes 49, 424–430 10.2337/diabetes.49.3.424 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.