

Abstract
Hepatitis B virus X protein (HBx) critically contributes to the development of hepatocellular carcinoma (HCC). However, the mechanisms by which HBx promotes HCC remain unclear. In the present study, using a combination of gene expression profiling and immunohistochemistry, we found higher levels of SH2 domain–containing 5 (SH2D5) in liver tissue from HBV-associated HCC (HBV-HCC) patients than in adjacent nontumor tissues. Moreover, HBV infection elevated SH2D5 levels, and we observed that HBx plays an important role in SH2D5 induction. We also found that HBx triggers SH2D5 expression through the NF-κB and c-Jun kinase pathways. Employing SH2D5 overexpression or knockdown, we further demonstrate that SH2D5 promotes HCC cell proliferation both in vitro and in vivo. While investigating the mechanism of SH2D5-mediated stimulation of HCC cell proliferation, we noted that HBV induces SH2D5 binding to transketolase (TKT), a pentose phosphate pathway enzyme, thereby promoting an interaction between and signal transducer and activator of transcription 3 (STAT3). Furthermore, HBx stimulated STAT3 phosphorylation at Tyr-705 and promoted the activity and downstream signaling pathway of STAT3 via the SH2D5–TKT interaction. Taken together, our results suggest that SH2D5 is an HBV-induced protein capable of binding to TKT, leading to induction of HCC cell proliferation.
Keywords: hepatitis B virus (HBV, Hep B); cancer biology; cell migration; interleukin 6 (IL-6); inflammation; HBV X protein; HBV-associated HCC; IL-6-STAT3 signaling; SH2D5; transketolase
Introduction
Hepatocellular carcinoma (HCC),2 a common aggressive carcinoma of the liver, is the fifth most common cancer in the world and the second leading cause of cancer deaths (1, 2). The 5-year survival rate of HCC patients is less than 10% (3, 4). Chronic hepatitis B virus (HBV) infection has been confirmed as a major risk factor in the development of HCC (5). HBV is a noncytopathic and partially double-stranded DNA virus with four open reading frames (C, P, S, and X) in the genome, resulting in expression of the seven HBV proteins via use of varying in-frame start codons (6, 7). Among these, HBV X protein (HBx) participates in the establishment and development of HCC as a multifunctional regulation factor (8). Studies showed that HBx did not directly bind the target motifs of downstream-regulated genes; however, it exhibited co-transcription factor activity through interaction with nuclear transcriptional factors (9). As a result, many host factors were induced during HBV infection. Although co-transcription factor functions of HBx have been described, it remains obscure as to how HBx alters regulated proteins, post-translational modifications, and signaling networks.
Over the past decade, several studies have demonstrated that the pathogenesis of HCC was closely linked with chronic inflammation and the level of interleukin-6 (IL-6) (10). IL-6 first binds to the IL-6 receptor, initiating signaling cascades that lead to the activation of signal transducer and activator of transcription 3 (STAT3) (11). STAT3 activation has been implicated in abnormal oncogenic processes, including initiation, proliferation, angiogenesis, and progression in HCC (12). To be activated, STAT3 requires phosphorylation for its homodimerization, nuclear translocation, and transcriptional activity (13). Many proteins are involved in the regulation of STAT3 phosphorylation, and transketolase (TKT) is one of most important regulators of STAT3 phosphorylation in HCC (14). TKT is an enzyme in the pentose phosphate pathway, catalyzing the conversion of sedoheptulose 7-phosphate and d-glyceraldehyde 3-phosphate to d-ribose 5-phosphate and d-xylulose 5-phosphate (15). Because of its activity regarding the production of NADPH to counteract oxidative stress, TKT is needed for tumor growth (16). Several studies demonstrated that TKT expression was tightly regulated by oxidative stress sensor pathways in cancers (17). In particular, genetic knockdown or pharmacological inhibition of TKT in ovarian and other cancer cells disturbed redox homeostasis and sensitized cancer cells to existing targeted therapy (18, 19). Consistent with this notion, a study showed that TKT bound to STAT3 and regulated STAT3 phosphorylation in HCC (20). However, little is known about the role of TKT in HBV-associated HCC (HBV-HCC).
Src homology 2 domain–containing protein 5 (SH2D5) is a mammalian-specific, uncharacterized adaptor-like protein that is highly enriched in brain at the transcriptional level (21). In the present study, we demonstrated that SH2D5 played an important role in the proliferation of HCC cells mediated by HBx. We further demonstrate that HBV-induced SH2D5 interacted with TKT, leading to activation of STAT3. In addition, we provide evidence that SH2D5 may have therapeutic benefits in the treatment of HBV-HCC.
Results
SH2D5 expression was elevated in HBV-HCC patients
To determine the molecular mechanisms of HBV-HCC development, we performed microarray analysis to identify genes that may play important roles in HBV-HCC development in cancer tissues (n = 4) and in adjacent nontumor tissues (ANT) (n = 4). We found that 11 gene expression levels were altered in HBV-HCC liver tissues compared with ANT (Table S1). We chose SH2D5 for further study.
We obtained snap-frozen or paraffin-embedded HCC tissues and surrounding ANT tissues from 223 patients. This study involved two independent cohorts of HCC patients. Cohorts 1 and 2 included 121 and 102 HCC patients, respectively. To investigate whether SH2D5 could be an important factor in determining the clinical outcomes of HBV-HCC patients, we analyzed the SH2D5 expression in cohort 1. As determined by real-time PCR assay, SH2D5 expression levels were higher in HBV-HCC liver tissues than in ANT tissues, and patients with high SH2D5 RNA levels had poor overall survival (Fig. 1, A and B). Furthermore, expression levels of IL-6 were also higher in HBV-HCC liver tissues than in their paired ANT tissues (Fig. S1A). Interestingly, high levels of SH2D5 expression were correlated with high levels of IL-6 expression in HBV-HCC liver tissues (Fig. S1B). In cohort 2, SH2D5 protein levels were determined by immunohistochemistry. HBV-HCC patients showed higher levels of SH2D5 staining than did healthy individuals (Fig. 1C). Similarly, patients with high SH2D5 protein levels had poor overall survival (Fig. 1D). Subsequently, we analyzed the correlation between SH2D5 expression and the clinical–virological characteristics of HBV-HCC patients. There were positive correlations between SH2D5 expression and tumor size, distant metastasis, microvascular invasion, malignant differentiation, and tumor-node metastasis (TNM) stage in HBV-HCC patients (Tables S2 and S3). Nevertheless, there were no significant differences between SH2D5 expression and gender, age, HBV DNA levels, alanine aminotransferase levels, and α-fetoprotein levels (Tables S2 and S3). Furthermore, SH2D5 expression levels were measured in several human HCC cell lines with varying metastatic capabilities. Real-time PCR assays showed that SH2D5 expression increased progressively from normal liver cells to low-metastatic HCC cells, and finally to highly-metastatic HCC cells (Fig. 1E). Chang liver is the reference sample for significance. Taken together, these data suggest that SH2D5 contributes to progression of HBV-associated hepatocarcinogenesis.
Figure 1.
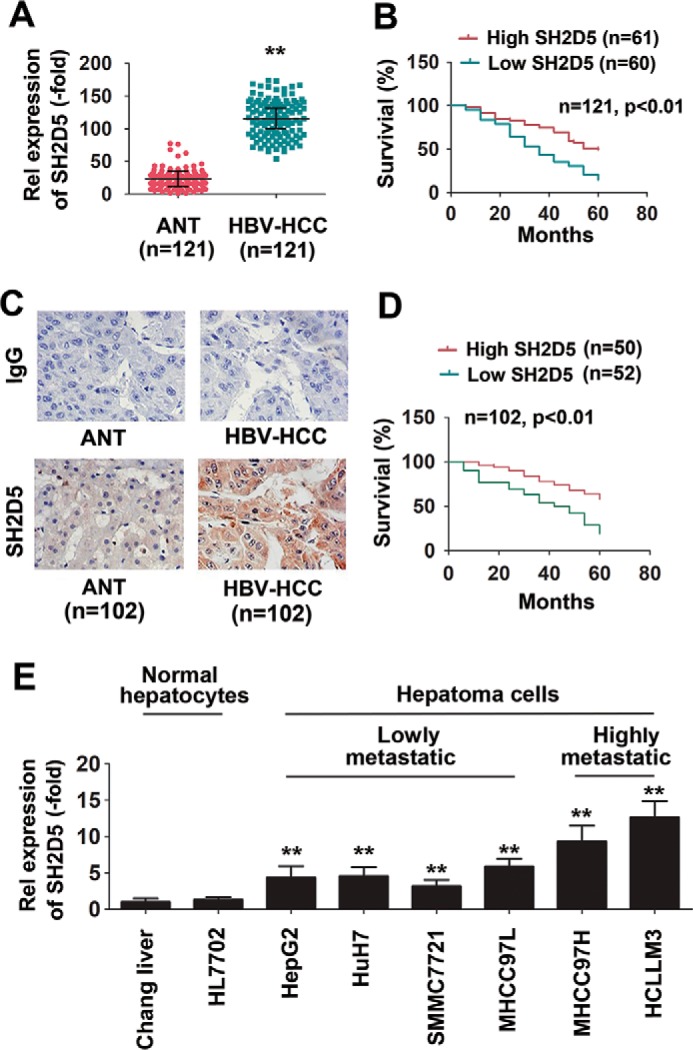
SH2D5 is increased in HCC and is clinically relevant to patient prognosis. A, real-time PCR assays of SH2D5 expression levels in HCC tissues and their corresponding ANT. The lowest value of ANT was designated as 1. SH2D5 data are expressed as fold induction (-fold) relative to the lowest value of healthy individuals. Data represent means ± S.E. B, Kaplan-Meier plots of HCC patients stratified by SH2D5 RNA levels. The median level of SH2D5 expression in each panel was used as the cutoff, with log-rank test for significance. C, immunohistochemical staining of SH2D5 in HCC tissues and their corresponding ANT. IgG was use as isotype control antibody. D, Kaplan-Meier plots of HCC patients stratified by SH2D5 protein levels. The median level of SH2D5 expression in each panel was used as the cutoff, with log-rank test for significance. E, real-time PCR analysis of SH2D5 expression in human normal hepatocytes and HCC cell lines. All experiments were repeated at least three times. (**, p < 0.01).
Transcriptional regulation of SH2D5 by HBV X protein
To test whether expression of SH2D5 was affected by HBV, SH2D5 mRNA and protein expression levels in HepG2 cells were compared with those of HepG2.2.15 HBV (genotype D)-positive cells. Real-time PCR and Western blotting assays showed that SH2D5 mRNA and protein expression levels were higher in HepG2.2.15 cells than in HepG2 cells (Fig. 2A, left panel). To make sure that this was not a cell-specific event, an additional pair of human hepatoma cells, HuH7 and HuH7.37 (HuH7.37 cells contain an integrated HBV genotype B and stably express HBV), were also tested for SH2D5 expression. Consistent with the results obtained with HepG2.2.15 cells, HuH7.37 cells expressed higher levels of SH2D5 mRNA and protein (Fig. 2A, right panel). Next, pHBV-1.3, a 1.3-fold overlength fragment of the HBV genome (genotype A–D) that retains the ability to produce mature HBV virions, was then transfected into HepG2 cells, and SH2D5 mRNA expression levels were detected by real-time PCR assays. pHBV-1.3 (A–D) also stimulated SH2D5 mRNA (Fig. 2B). To investigate which viral protein played a role in SH2D5 regulation, HepG2 cells were co-transfected with each of the seven HBV gene-expressing plasmids and pSH2D5-Luc. Luciferase activity reporter assays indicated that HBx protein stimulated SH2D5 promoter activity, whereas the other HBV proteins had no significant effect (Fig. 2C). Real-time PCR and Western blotting analyses also indicated that HBx increased SH2D5 mRNA and protein expression (Fig. 2D). To further investigate the influence of HBx on SH2D5 expression, Huh7 cells were co-transfected with pHBV-1.2 or pHBV-1.2 (ΔHBx), an HBV mutant that does not express HBx. Real-time PCR and Western blotting analyses indicated that WT HBV activated SH2D5 mRNA and protein expression, whereas the HBV mutant did not induce SH2D5 mRNA or protein expression (Fig. 2E).
Figure 2.

SH2D5 was induced by HBx at the transcriptional level. A, SH2D5 mRNA levels and protein levels in HepG2 and HepG2.2.15 cells (left panel) and Huh7 and Huh7.37 cells (right panel). B, HepG2 cells were transfected with vector control or pHBV-1.3 (genotype A-,-D) for 48 h prior to real-time PCR assays. C, HepG2 cells were co-transfected with pSH2D5-Luc and the indicated plasmids with viral protein-coding sequences for 48 h prior to luciferase activity reporter assays. D, HepG2 cells were transfected with the vector control or pCMV-HBx for 48 h prior to real-time PCR (upper panel) and Western blotting (lower panel) analyses. E, Huh7 cells were transfected with vector control, pHBV-1.2, or pHBV-1.2 (ΔHBx), an HBx-deficient HBV mutant for 48 h. SH2D5 expression was quantified prior to real-time PCR (upper panel) and Western blotting (lower panel) analyses. F, schematic diagram of individual SH2D5 promoter cis-regulatory elements and SH2D5-truncated or site-specific mutants (left), and the results are from luciferase activity assays (right). Huh7 cells were transfected with the indicated plasmids for 48 h prior to luciferase assays. G, experiments were performed similar to those in D, except c-Jun–specific siRNA (siRNA–c-Jun) was used. H, Huh7 cells were transfected with indicated plasmids and treated with or without NF-κB inhibitor Bay11-7082 (left panel) or transfected with vector control, pCMV-HBx, pCMV-NF-κB, or pCMV-NS1 of IAV (right panel) for 48 h, respectively. SH2D5 RNA levels were quantified by real-time PCR assays. I, experiments were performed similar to those in H, except siRNA–c-Jun (left panel) or pCMV–c-Jun (right panel) was used. J, Huh7 cells were transfected with vector control or pCMV-HBx for 48 h. ChIP assays were performed with anti-NF-κB (left panel), anti-c-Jun (right panel), or IgG-conjugated agarose. Promoter sequences in the input DNA and the DNA recovered from antibody-bound chromatin segments were detected using real-time PCR. Enrichment was determined relative to input controls. In the real-time RT-PCR experiments, the control was designated 1. In the Western blotting experiments, intensities of bands were analyzed by ImageJ and normalized to the corresponding controls. All experiments were repeated at least three times. Bar graphs present means ± S.D., n = 3 (**, p < 0.01).
To further investigate the transcriptional regulation of SH2D5, we examined the SH2D5 promoter region for possible consensus cis-elements. Three promoter truncations and two binding-site mutants (NF-κB- and c-Jun–binding sites) were generated from the full-length SH2D5 promoter plasmid. In reporter assays, HBx expression stimulated expression of the WT promoter and the two truncated mutants. However, mutation of either NF-κB and c-Jun abolished HBx-stimulated promoter activity (Fig. 2F). To determine whether SH2D5 expression was NF-κB- and c-Jun-dependent, we chose NF-κB and c-Jun overexpression plasmid, an NF-κB inhibitor (Bay11), and we designed two specific small-interfering RNAs (siRNAs) for c-Jun (siRNA–c-Jun #1 and #2) and tested their efficacy (Fig. 2G). siRNA–c-Jun #2 was selected for the experiments described below.
NF-κB inhibitor decreased HBx-induced SH2D5 mRNA levels, whereas NF-κB overexpression increased HBx-induced SH2D5 levels (Fig. 2H). Influenza A virus (IAV) nonstructural protein-1 (NS1) transfection was included as a negative control. Similar results were obtained by using siRNA–c-Jun or c-Jun overexpression plasmid (Fig. 2I). We further examined the effect of the HBx on the association of NF-κB and c-Jun and SH2D5 promoters. Results from chromatin immunoprecipitation (ChIP) experiments confirmed that HBx increased NF-κB and c-Jun binding to the SH2D5 promoter (Fig. 2J). Together, these results demonstrated that NF-κB and c-Jun signaling pathways were involved in the up-regulation of SH2D5 expression in response to HBx.
SH2D5 promoted proliferation of hepatoma cells in vitro
First, we used cell proliferation assays to determine whether SH2D5 affected cell viability. As shown in Fig. 3A, SH2D5 overexpression increased HBx-regulated proliferation rates of HCC cells. TNFα treatment was included as a positive control for comparison. To further verify the role SH2D5 in HCC cell viability, we designed two siRNAs for SH2D5 (siRNA-SH2D5 #1 and #2) and tested their knockdown efficiency (Fig. 3B). siRNA-SH2D5 #2 was selected for experiments described below. The proliferation rates of HCC cells were inhibited by siRNA-SH2D5 (Fig. 3C). Next, we examined the effect of SH2D5 expression on the migration and invasion of HCC cells. Overexpression of SH2D5 facilitated the HBx-induced migration and invasion of HCC cells (Fig. 3D). Conversely, knockdown of SH2D5 resulted in the inhibition of HBx-induced cell migration and invasion (Fig. 3E). All these results supported the notion that SH2D5 promoted the proliferation of HCC cells.
Figure 3.

SH2D5 promoted HCC cell proliferation, migration, and invasion in vitro. A, HepG2 cells were transfected with the indicated plasmids for 24 h and treated with or without TNFα (50 ng/ml) for 48 h prior to cell proliferation (upper panel) and Western blotting assays (lower panel). B, Huh7 cells were transfected with siRNA control or siRNA-SH2D5s for 48 h prior to real-time PCR (upper panel) and Western blotting (lower panel) analyses. C, experiments were performed similar to those in A, except the indicated siRNA-SH2D5 #2 was used. D, HepG2 cells were transfected with the indicated plasmids and treated with or without TNFα (50 ng/ml) for 24 h, after culture in a Transwell (upper panel) or invasion (lower panel) system for 24 h. E, experiments were performed similar to those in D, except the indicated siRNA-SH2D5 #2 was used. In the real-time RT-PCR experiments, the control was designated 1. All experiments were repeated at least three times. Bar graphs present means ± S.D., n = 3 (**, p < 0.01).
SH2D5 promoted proliferation of hepatoma cells in vivo
We further investigated the proliferation enhancement effect of SH2D5 in vivo. Results from the colony formation assay indicated that overexpression of SH2D5 increased the HBx-induced colony formation ability of HCC cells. Conversely, knockdown of SH2D5 decreased HBx-regulated HCC cell colony formation (Fig. 4, A and B). We also examined the effect of SH2D5 on the tumorigenicity of HCC cells in vivo. SH2D5 knockdown cells and control cells were subcutaneously injected into nude mice. With no substantial effects on mouse weight, the volume and the weight of the tumors were dramatically lower in the SH2D5 knockdown mice than in control mice (Fig. 4, C–E). Real-time PCR confirmed that expression levels of SH2D5 were lower in tumor tissues of the SH2D5 knockdown group than in the control group (Fig. 4F). Therefore, we concluded that SH2D5 promoted the proliferation of hepatoma cells in vivo.
Figure 4.
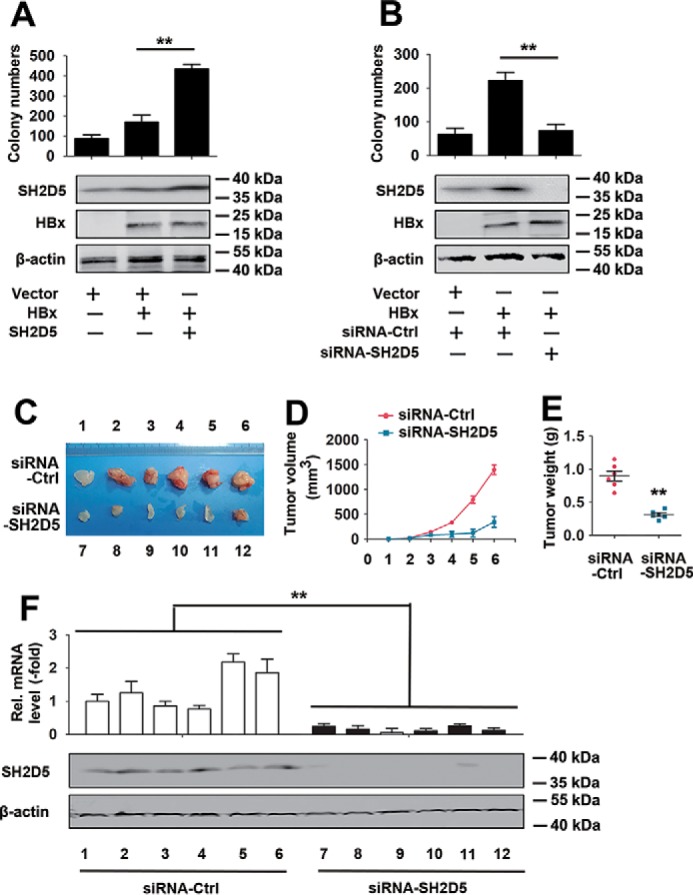
SH2D5 promoted proliferation of hepatoma cells in vivo. A, HepG2 cells were transfected with vector control, pCMV-HBx, or pCMV-SH2D5 and seeded into the soft agar dish. Colonies under the microscope were counted after a 4-week incubation (upper panel). SH2D5 and HBx expressions were quantified by Western blot assays (lower panel). B, experiments were performed similar to those in A, except the indicated siRNA-SH2D5 was used. C, nude mice were sacrificed and photographed after 1 month of subcutaneous injection. D, growth curves of tumors derived from HepG2-X cells transfected with si-SH2D5 or si-Ctrl. E, average weight of tumors. F, relative mRNA and protein levels of SH2D5 in the tumor tissues from mice were detected by real-time PCR (upper panel) and Western blot assays (lower panel). All experiments were repeated at least three times. Bar graphs present means ± S.D., n = 3 (**, p < 0.01).
HBV promotes the interaction of SH2D5 and TKT and the subsequent recruitment of STAT3
To explore the molecular mechanisms by which SH2D5 exerted its tumor-inductive effect on HCC cells, we used immunoprecipitation coupled to MS to identify binding partners for SH2D5. We found that TKT was a potential target of SH2D5 (Fig. 5A). A previous report showed that TKT, a key enzyme in the pentose phosphate pathway, might regulate STAT3 phosphorylation by interacting with STAT3 (20). We suspected that SH2D5 might regulate STAT3 phosphorylation by interacting with TKT during HBV infection. To test this hypothesis, TKT or STAT3 was fused in-frame with the GAL4 DNA-binding domain to create the plasmids pM-TKT and pM-STAT3, respectively. The SH2D5 protein was fused in-frame with the VP16 transactivation domain in the plasmid pVP16-SH2D5. According to the results of a mammalian two-hybrid analysis, TKT, but not STAT3, interacted with SH2D5 in mammalian cells (Fig. 5B). In vivo co-immunoprecipitation and reverse co-immunoprecipitation experiments were performed to further confirm the binding of the SH2D5 to TKT or STAT3. As shown in Fig. 5, C and D, the SH2D5 protein interacted with TKT, but not with STAT3. We further performed endogenous co-immunoprecipitation experiments, and the results indicated that the SH2D5–TKT interaction increased upon transfection with HBV 1.3 plasmid (Fig. 5E). Importantly, overexpression of SH2D5 promoted the HBV-induced TKT–STAT3 interaction, whereas SH2D5 knockdown suppressed the HBV-induced TKT–STAT3 interaction (Fig. 5F). Similarly, exogenous co-immunoprecipitation experiments showed that overexpression of SH2D5 promoted TKT–STAT3 interaction, whereas SH2D5 knockdown suppressed TKT–STAT3 interaction (Fig. 5G). Collectively, these results indicated that HBV-induced SH2D5 interacted with TKT and that association of SH2D5 and TKT in a complex in turn recruited STAT3.
Figure 5.

HBV promotes the interaction of SH2D5 and TKT, and the subsequent recruitment of STAT3. A, HepG2 cells were transfected with vector control or FLAG-SH2D5 for 48 h. Cells were lysed and were immunopurified with anti-FLAG affinity columns and eluted with FLAG peptide. The eluates were resolved by SDS-PAGE and silver-stained. The differential protein bands were retrieved and analyzed by MS. B, determination of SH2D5 and TKT interaction in 293T cells by mammalian two-hybrid analysis. 293T cells were co-transfected with control plasmids, pG5-luc (a luciferase reporter plasmid), pVP16-SH2D5, pM-TKT (left panel), or pM-STAT3 (right panel). Interaction between proteins was monitored by luciferase activity, which was measured 48 h after transfection. C, 293T cells were transfected with FLAG-tagged TKT (FLAG-TKT) and Myc-tagged SH2D5 (Myc-SH2D5). Forty eight hours post-transfection, co-immunoprecipitation (IP) and immunoblot (IB) analyses were performed with the indicated antibodies. D, experiments were performed similar to those in C, except FLAG-STAT3 was used. E, HepG2 cells were transfected with increasing amounts of HBV 1.3 plasmid. Forty eight hours post-transfection, co-immunoprecipitation and immunoblot analyses were performed with the indicated antibodies. F, experiments were performed similar to those in E, except SH2D5 plasmid (left panel) or si-SH2D5 (right panel) was used. G, 293T cells were transfected with the indicated plasmids or siRNAs. Forty eight hours post-transfection, co-immunoprecipitation and immunoblot analyses were performed with the indicated antibodies. All experiments were repeated at least three times. WCL, whole-cell lysates.
HBx regulated the phosphorylation of STAT3 and the expression of downstream genes in the JAK–STAT3 pathway through SH2D5 and TKT
To test the role of SH2D5 and TKT in HBx-regulated STAT3 signaling, we designed two siRNAs for TKT (siRNA-TKT#1 and #2) and tested their knockdown efficiency (Fig. S2). siRNA-TKT #2 was selected for the experiments described below. It has been shown that TKT enhanced STAT3-Y705 phosphorylation but suppressed STAT3-S727 phosphorylation (20). We next examined the role of SH2D5 and TKT in HBx-regulated STAT3 phosphorylation. As shown in Fig. 6, A and B, HBx promoted STAT3-Y705 phosphorylation but reduced STAT3-S727 phosphorylation. Overexpression SH2D5 or TKT enhanced the effect of HBx on the phosphorylation of STAT3-Y705 and STAT3-S727, whereas SH2D5 or TKT knockdown inhibited the effect of HBx on the phosphorylation of STAT3-Y705 and STAT3-S727 (Fig. 6, A and B). To further explore the role of SH2D5 and TKT on the activity of STAT3, we constructed STAT3–Luc that contained the STAT3 response element. Luciferase activity assays indicated that SH2D5 and TKT overexpression promoted HBx-induced STAT3 activation (Fig. 6C). Conversely, SH2D5 or TKT knockdown diminished HBx-induced STAT3 activation (Fig. 6D). The mRNA levels of the downstream genes in the JAK–STAT3 pathway were also examined. Based on real-time PCR results, overexpression of SH2D5 or TKT promoted HBx-induced expression of the downstream genes in the JAK–STAT3 pathway, and conversely, SH2D5 and TKT knockdown inhibited HBx-induced expression of the downstream genes in the JAK–STAT3 pathway (Fig. 6, E and F). Similar results were also obtained in Huh7 cells (Fig. S3). We then constructed an SH2D5 or TKT knockout (SH2D5-KO or TKT-KO) cell line in HepG2 cells using the CRISPR–Cas9 system (Fig. S4A). We found that HBx promoted the proliferation of HCC cells, the activity of STAT3, and the expression of the JAK–STAT3 pathway downstream genes in wildtype (WT) cells, without any effect in SH2D5-KO cells (Fig. S4, B and C). Similar results were also obtained in TKT-KO cells (Fig. S4, D and E).
Figure 6.

Effect of SH2D5 and TKT on HBx regulated phosphorylation and downstream signaling of STAT3. A, HepG2 cells were transfected with the indicated plasmids for 24 h prior to Western blotting analyses. B, experiments were performed similar to those in A, except the indicated siRNA-TKT was used. C, HepG2 cells were transfected with the indicated plasmids for 36 h prior to luciferase activity assays. D, experiments were performed similar to those in C, except the indicated siRNA-TKT was used. E, HepG2 cells transfected with the indicated plasmids for 36 h prior to real-time PCR analyses. F, experiments were performed similar to those in E, except the indicated siRNA-TKT was used. G and H, experiments were performed similar to those in A and B, except without HBx plasmid. I and J, experiments were performed similar to those in C and D, except without HBx plasmid. K and L, experiments were performed similar to those in E and F, except without HBx plasmid. All experiments were repeated at least three times. Bar graphs present means ± S.D., n = 3 (**, p < 0.01; *, p < 0.05).
Because TKT was specifically associated with SH2D5, and both proteins were involved in HBx-regulated STAT3 activation, we examined whether TKT was involved in the regulation of SH2D5-mediated signaling. Results from Western blotting assays showed that TKT overexpression augmented the effect of SH2D5 on the phosphorylation STAT3-Y705 and STAT3-S727 (Fig. 6G). By contrast, TKT knockdown diminished the effect of SH2D5 on the phosphorylation STAT3-Y705 and STAT3-S727 (Fig. 6H). Similarly, reporter assays indicated that overexpression of TKT increased SH2D5-induced STAT3 activation, whereas knockdown of TKT decreased SH2D5-induced STAT3 activation (Fig. 6, I and J). The regulation of SH2D5 on the mRNA levels of the downstream genes in the JAK–STAT3 pathway was also affected by TKT knockdown or overexpression (Fig. 6, K and L). The role of TKT in the SH2D5-mediated signaling was also investigated by using TKT-KO cells. Results indicated that SH2D5 induced the proliferation of HCC cells, the activity of STAT3, and the expression of the JAK–STAT3 pathway downstream genes in WT cells. Conversely, low levels of the proliferation of HCC cells, the activity of STAT3, and the expression of the JAK–STAT3 pathway downstream genes were observed in TKT-KO cells when SH2D5 was overexpressed (Fig. S4, F and G). We next examined the roles of SH2D5–TKT interaction on the viability of HCC cells. Overexpression of SH2D5 or TKT enhanced the effect of HBx on the proliferation rates of HCC cells, whereas SH2D5 or TKT knockdown inhibited the effect of HBx on the proliferation rates of HCC cells (Fig. 7, A and B). Similarly, overexpression of TKT increased SH2D5-induced HCC cell proliferation rates, and conversely, knockdown of TKT decreased SH2D5-induced HCC cell proliferation rates (Fig. 7, C and D).
Figure 7.
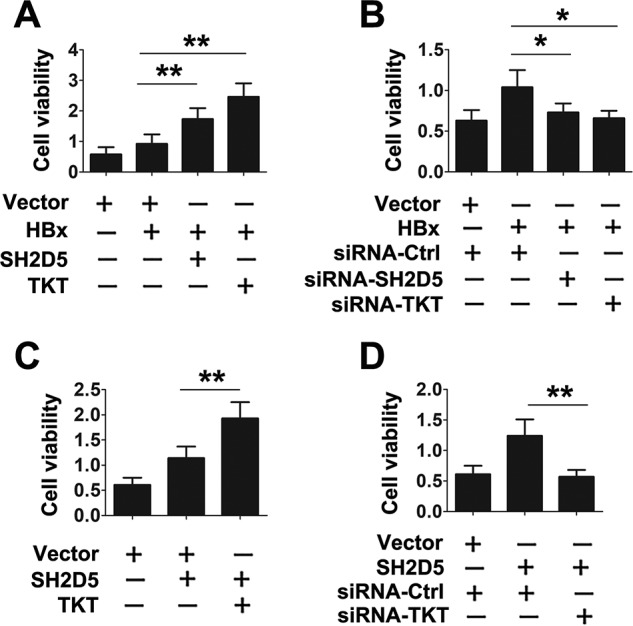
Verification of the interaction of SH2D5 and TKT in HCC cells proliferation. A, HepG2 cells were transfected with vector control, pCMV-HBx, pCMV-TKT, or pCMV-SH2D5 for 3 days prior to cell proliferation assays. B, HepG2 cells were transfected with vector control, pCMV-HBx, or indicated siRNAs for 3 days prior to cell proliferation assays. C, HepG2 cells were transfected with vector control, pCMV-TKT, or pCMV-SH2D5 for 3 days prior to cell proliferation assays. D, HepG2 cells were transfected with vector control, pCMV-SH2D5, siRNA control, or siRNA-TKT for 3 days prior to cell proliferation assays. All experiments were repeated at least three times for consistent results. Bar graphs present means ± S.D., n = 3 (**, p < 0.01; *, p < 0.05).
IL-6 is the most important STAT3 activator that promotes tumor cell proliferation, survival, invasion, and angiogenesis. We also tested the effects of SH2D5 and TKT on the IL-6–STAT3 signaling pathway in HCC cells. Results from luciferase activity assays indicated that SH2D5 or TKT overexpression promoted IL-6–induced STAT3 activation (Fig. 8A). Conversely, SH2D5 and TKT knockdown diminished IL-6–induced STAT3 activation (Fig. 8B). Similarly, overexpression of SH2D5 or TKT promoted IL-6–induced expression of the downstream genes in the JAK–STAT3 pathway. Conversely, SH2D5 and TKT knockdown inhibited IL-6–induced expression of the downstream genes in the JAK–STAT3 pathway (Fig. 8, C and D). Taken together, these findings indicated that SH2D5 and TKT were involved in HBx-regulated activation of STAT3.
Figure 8.

Verification of the role of SH2D5 and TKT in IL-6–STAT3 signaling pathway. A, HepG2 cells transfected with the indicated plasmids for 12 h and treated with or without IL-6 (10 ng/ml) for 12 h prior to luciferase assays. B, HepG2 cells transfected with the indicated siRNAs for 12 h and treated with or without IL-6 (10 ng/ml) for 12 h prior to luciferase assays. C and D, experiments were performed similar to those in A and B, except the downstream genes in the JAK–STAT3 pathway were detected by real-time PCR. All experiments were repeated at least three times for consistent results. Bar graphs present means ± S.D., n = 3 (**, p < 0.01; *, p < 0.05).
Discussion
HBV infection is one of the leading causes of HCC worldwide. HBx participates in the establishment and development of HCC as a multifunctional regulation factor. However, the underlying molecular mechanisms remain obscure. In the present study, we identified SH2D5, a previously uncharacterized adaptor-like protein, as a tumor regulator in HBV-HCC.
SH2D5 is a recently evolved, uncharacterized adaptor-like protein, containing an N-terminal PTB domain and a C-terminal SH2-like domain (21). To the best of our knowledge, only one study reported the function of SH2D5. They found that SH2D5 regulated Rac1-GTP levels by binding to the breakpoint cluster region protein (22). In the present study, for the first time, we identified the role of SH2D5 in HBV-HCC. We observed that SH2D5 was frequently increased in HBV-HCC tissues relative to levels in ANT tissues. In addition, clinical evidence showed that SH2D5 overexpression correlated with tumor size, distant metastasis, microvascular invasion, malignant differentiation, and TNM stage. Furthermore, Kaplan-Meier analysis showed that patients with HBV-HCC who had high SH2D5 expression had worse prognoses than did those with low SH2D5 expression. These clinical data strongly suggested that SH2D5 may contribute to the progression of HBV-HCC. Upon investigating the mechanisms behind this event, we found that SH2D5 interacted with TKT, and this association positively regulated the STAT3-mediated signaling pathway. TKT is a ubiquitous enzyme that catalyzes the reversible transfer of two-carbon ketol units between ketose and aldose phosphates, governing carbon flow through the nonoxidative branch of the pentose phosphate pathway (23). A question is raised whether SH2D5 also controlled the glycolysis of HCC cells. When considering this question, studies exploring this question would be of great help in further clarifying the role of SH2D5 in HBV infection.
The role of STAT3 phosphorylation in HCC is intriguing. There is a phosphorylation site on the Tyr-705 residue, functioning in the activation of STAT3 (24). There is another phosphorylation site on the Ser-727 residue that is thought to enhance transcriptional activity of the protein (25, 26). Ser-727 is an equally important but less studied phosphorylation site on STAT3 (25, 26). Similarly, the oncogenic effects of STAT3-Y705 phosphorylation have been confirmed in various cancer types; the role of STAT3-S727 phosphorylation in cancer is not well defined. A previous study showed that TKT regulated STAT3-Y705 and STAT3-S727 phosphorylation in prostate cancer cells. Another recent report suggested that TKT enhanced STAT3-Y705 phosphorylation and inhibited STAT3-S727 phosphorylation in HCC cells (20). However, the role of HBV in TKT-regulated STAT3-Y705 and STAT3-S727 phosphorylation has not been reported. We showed here that HBx increased STAT3-Y705 phosphorylation but decreased STAT3-S727 phosphorylation via SH2D5 and TKT. Further experiments showed that SH2D5 increased STAT3-Y705 phosphorylation but decreased STAT3-S727 phosphorylation by interacting with TKT. Our data also supported previous findings that TKT enhanced STAT3-Y705 phosphorylation and inhibited STAT3-S727 phosphorylation in HCC cells. Furthermore, we identified a previously-unknown mechanism in which HBx regulated STAT3-Y705 and STAT3-S727 phosphorylation by promoting SH2D5 and TKT interaction. However, numerous studies have shown other pathways of STAT3 activation. For example, STAT3 is also activated by IL-22 and TGFα in HCC cells (27). Inhibition of negative regulators SHP1/2 and SOCS proteins by NF-κB- and methylation-dependent mechanisms further augments STAT3 activity (28). STAT3 promotes hepatocellular carcinoma cell survival and proliferation via direct regulation of target genes and via establishment of an epigenetic circuit that amplifies IL-6 signaling through miRNA-induced down-regulation of HNF4a (29). Moreover, sphingosine signaling through SphK1, S1P, S1PR1, and JAK2 contributes to persistent STAT3 activation in tumor cells and cells of the tumor microenvironment (30). Further studies are needed to verify whether SH2D5 participates in those pathways of STAT3 activation.
We propose a working model of the role of the SH2D5–TKT complex in an HBV-induced proliferation of HCC cells (Fig. 9). In this model, HBV infection strongly induces SH2D5 expression through the NF-κB and c-Jun pathways. The enhanced SH2D5 recruits TKT, which in turn recruits and activates STAT3. These complex signals for translocation of STAT3 from the cytoplasm to the nucleus and the subsequent production of downstream genes leads to HCC. Although more studies are needed to understand the intricate regulatory mechanisms of SH2D5 for HBV-HCC, our findings reveal a previously undescribed role for HBx in regulating SH2D5-mediated hepatocarcinogenesis and may provide a potential target for the treatment of HCC in the future.
Figure 9.
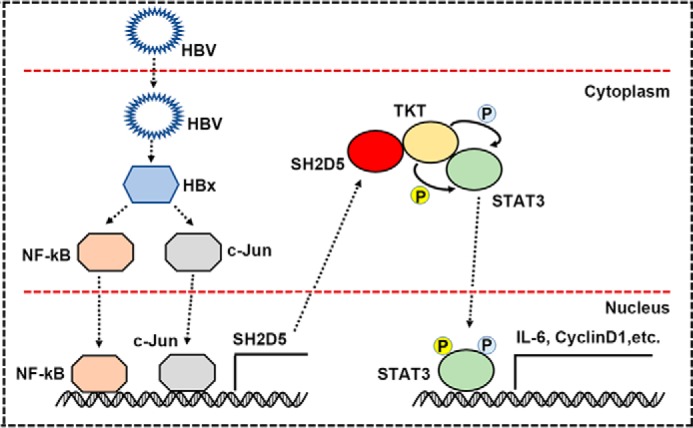
Hypothetical model for SH2D5 induction following HBV infection and the role of SH2D5 in the progression of HBV-HCC.
Experimental procedures
Ethics statement
The collection of human HCC liver tissue was conducted according to the principles of the Declaration of Helsinki and was approved by the Institutional Review Board of Renmin Hospital of Wuhan University in accordance with guidelines for the protection of human subjects. All study participants provided written informed consent for the collection of samples and subsequent analyses.
Samples and cases
HBV-HCC samples and adjacent nontumor tissues (located >3 cm away from the tumor) were collected from 223 patients undergoing surgery at the Renmin Hospital of Wuhan University from February 2012 to February 2018. The preoperative clinical diagnosis of HCC met the diagnostic criteria of the American Association for the Study of Liver Diseases (31). Briefly, the inclusion criteria were as follows: 1) distinctive pathological diagnosis; 2) absence of pre-operative anti-cancer treatment and distant metastases; 3) curative liver resection; and 4) complete clinicopathological and follow-up data. Grading of differentiation status was performed according to the method of Edmondson and Steiner (37). The pTNM classification for HCC was based on the American Joint Committee on Cancer/International Union Against Cancer staging system (2002, 6th Ed.). In addition, 121 paired tumor liver tissues and their peripheral nontumor tissues after surgical resection were cut into two parts. One part was fixed in buffered formalin and embedded in paraffin using standard methods. Immunohistochemistry results were reviewed by two expert pathologists to verify the histological diagnosis. The other part was immediately snap-frozen in liquid nitrogen and stored in liquid nitrogen and subsequently used to detect mRNA levels of SH2D5. The characteristics of patients are described in Tables S2 and S3.
Immunohistochemistry of formalin-fixed paraffin-embedded samples was performed as described previously (7). Briefly, the formalin-fixed paraffin sections were deparaffinized, rehydrated, and pretreated with 3% H2O2 for 20 min. The antibody-binding epitopes of the antigens were retrieved by microwave treatment, and the sections were then preincubated with 10% goat serum to block nonspecific binding. The specimens were incubated with the primary antibodies for 1 h at room temperature, and IgG was used as isotype control. Sections were then incubated with anti-rabbit or anti-mouse secondary antibody and streptavidin-horseradish peroxidase. DAB was used as a chromogen, and hematoxylin was used for counterstaining.
Follow-up data were summarized at the end of December, 2017, with a median follow-up of 60 months. Patients were checked every 2–3 months during the first 2 years and every 3–6 months thereafter. All follow-up examinations were performed by physicians who were unaware of the study. A computed tomography and/or MRI examination was performed every 3–6 months together with a chest radiographic examination. The diagnostic criteria for HCC recurrence were the same as for preoperative criteria. Overall survival was considered the primary end point. Overall survival was calculated from the date of resection to the date of death or the last follow-up.
Cell culture
Human HCC cells (HepG2 and Huh7) were described in a previous study (7). Human HCC cells (SMMC7721, MHCC97L, MHCC97H, and HCCLM3) and immortalized liver cell lines (Chang liver and HL-7702) were purchased from the China Center Type Culture Collection. SMMC7721 cells are a low-metastatic HCC cell line. MHCC97L, MHCC97H, and HCCLM3 cells are stepwise metastatic potential cell lines with the same genetic background but with different lung metastatic potentials (32). All cells were cultured in Dulbecco's modified Eagle's medium (Gibco) supplemented with 10% fetal bovine serum (FBS) (Gibco), 100 units/ml penicillin, and 100 μg/ml streptomycin sulfate. The HepG2.2.15 cell line, derived from HepG2 cells and stably expressing HBV (subtype ayw), has been described in a previous study (7). The Huh7.37 cell line, derived from Huh7 cells and also stably expressing HBV (subtype adw), was constructed as previously described (7).
SH2D5-KO and TKT-KO cells were produced using the CRISPR–Cas9 system. Briefly, lentiviruses were harvested from the supernatants of HEK293 cells that were co-transfected with package plasmids pSP and pMD, along with lentiCRISPRv2, containing the target guide sequence of SH2D5 (5′-GGAGTTGCAGGCTTCGGATAA-3′) and TKT (5′-TCACCGTGGAGGACCATTATT-3′). Cells were infected with the lentivirus and screened with puromycin (0.9 μg/ml), and the surviving monocolony was selected. The monocolony was cultured, and SH2D5-KO or TKT-KO protein expression levels were determined. SH2D5-KO or TKT-KO cells were obtained by amplification of the monocolony, which does not express SH2D5 or TKT, and maintained with puromycin (0.9 μg/ml). All cell lines were grown at 37 °C in an aerobic incubator with 5% CO2.
Reagents and constructs
Antibodies (Abs) against human SH2D5 (ab170881), human β-actin (ab8227), human glyceraldehyde-3-phosphate dehydrogenase (ab9484), human c-JUN (ab31419), human NF-κB (ab19870), human TKT (ab112997), HA (ab9110), FLAG (ab49763), and Myc (ab32072) were purchased from Abcam. Abs against human STAT3 (9139), phospho-Stat3 (Tyr-705) (9145), and phospho-Stat3 (Ser-727) (9134) were purchased from Cell Signaling Technology. The siRNAs of SH2D5, TKT, and c-Jun were purchased from Qiagen. TRIzol, Lipofectamine-2000, and Enzyme MIX were purchased from Invitrogen (Basel, Switzerland). Unless specified otherwise, all biochemical reagents were purchased from Sigma.
pHBV1.3 (genotype A, serotype adw2, HE974381), pHBV1.3 (genotype B, serotype adw, JN406371), pHBV1.3 (genotype C, serotype adr, FJ899793), and pHBV1.3 (genotype D, serotype ayw, U95551) were provided by Professor Shi Liu of the College of Life Sciences at Wuhan University, China. The coding regions and promoter of SH2D5 and the coding regions of TKT and STAT3 were created in our laboratory for this study. All constructs were confirmed by DNA sequencing (Sangon Biotech, Shanghai, China).
Tumor formation in nude mice
Ten 6-week-old male BALB/c nude mice were divided into two groups randomly. Each group was composed of five mice that were injected with 2 × 106 control cells (siRNA-Ctrl) or SH2D5 knockdown cells (siRNA–SH2D5). The tumor volume was calculated by measuring the length and width of the tumor (tumor volume = 1/2 length × width2) every week. After 5 weeks, the mice were sacrificed by anesthesia, and the tumors were removed and weighed for further analysis. All animal experiments were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. The protocol was approved by the institutional animal care and use committee of Renmin Hospital of Wuhan University.
Quantitative real-time RT-PCR
Quantitative real-time PCR was described previously (33). Briefly, total RNA was extracted using TRIzol reagent, according to the manufacturer's protocol. cDNA was synthesized with the Prime-Script RT reagent Kit (TaKaRa). Real-time PCR analyses were performed using SYBR Premix Ex Taq (Applied Biosystems). Primers are listed in Table S4. Real-time PCR was performed using an ABI 7900 real-time PCR machine. The relative expression of each gene was calculated and normalized using the 2−ΔΔCt method.
Western blot analysis
Western blotting analyses were described in a previous study (34). Briefly, cells were harvested by low-speed centrifugation and washed with PBS. Cells were lysed in RIPA Buffer (Cell Signaling Technology, 9800), and protein concentrations were determined by using BCA assays (Cell Signaling Technology, 7780). Forty micrograms of each protein sample were separated by 12% SDS-PAGE and transferred to nitrocellulose membranes (Bio-Rad). The membranes were blocked with 1× TBS with Tween 20 (TBST) and 5% (w/v) nonfat milk for 1 h at room temperature. Then, the membranes were incubated with primary antibodies overnight at 4 °C. Subsequently, membranes were incubated with horseradish peroxidase–linked secondary antibodies (Jackson ImmunoResearch) for an additional 1 h. Immunoreactive bands were visualized using an enhanced chemiluminescence system (GE Healthcare).
Microarray experiments
HBV-HCC samples (n = 4) and their corresponding adjacent liver tissues (n = 4) were used for microarray assays, performed by KangChen, Inc. (Shanghai, China). Total RNA from each sample was quantified using the NanoDrop ND-1000, and RNA integrity was assessed using standard denaturing agarose gel electrophoresis. For microarray analysis, the Agilent Array platform was employed. Sample preparation and microarray hybridization were performed based on the manufacturer's standard protocols. Briefly, 1 μg of total RNA from each sample was amplified and transcribed into fluorescent cRNA following the manufacturer's Agilent's Quick Amp labeling protocol (version 5.7, Agilent Technologies). The labeled cRNAs were hybridized onto the Whole Human Genome Oligo Microarray (4 × 44K, Agilent Technologies). After the slides were washed, the arrays were scanned by the Agilent Scanner G2505B.
Histology and immunohistochemistry
Immunohistochemical analysis of SH2D5 expression was performed as described previously (35). Immunohistochemical staining was performed with the Dako Envision Plus System (Dako, Carpinteria, CA) according to the manufacturer's instructions. Briefly, the formalin-fixed paraffin sections were deparaffinized, rehydrated, and pretreated with 3% H2O2 for 20 min. The antibody-binding epitopes of the antigens were retrieved by microwave treatment, and the sections were then preincubated with 10% goat serum to block nonspecific binding. Rabbit anti-SH2D5 (ab170881, Abcam, diluted at 1:100) was used as the primary antibodies. IgG was used as negative control. The specimens were incubated with the primary antibodies for 1 h at room temperature, followed by the addition of biotinylated anti-rabbit secondary antibody and streptavidin–horseradish peroxidase. DAB was used as a chromogen, and hematoxylin was used for counterstaining. Staining was scored by two independent observers on the basis of both staining intensity and the extent of SH2D5 expression across the section. The final score of each sample (negative or positive) was assessed by summarization of the results of the intensity and extent of staining. Intensity of staining was scored as 0 (negative), 1 (weak), or 2 (strong). The extent of staining was based on the percentage of positive tumor cells: 0 (negative), 1 (1–25%), 2 (26–50%), 3 (51–75%), and 4 (76–100%). Therefore, each case was finally considered negative if the final score was 0 to 1 (−) or 2 to 3 (±) and positive if the final score was 4 to 5 (+) or 6 to 7 (++), respectively.
Cell proliferation assay and colony formation assay
The Cell Counting Kit-8 (CCK-8, Dojindo Chemical Laboratory, Kumamoto, Japan) and colony formation assay were conducted to determine the cell proliferation activity.
Approximately 5 × 103 cells per well were transferred to 96-well plates after transfection and were subjected to the CCK-8 assay. After incubation at 37 °C for 1.5 h, the absorbance at 450 nm was measured following the addition of 10 μl of CCK-8 solution. There were five replicates in each group, and three independent experiments were performed.
For the colony formation assay, 500–1000 cells were seeded in 6-well plates overnight. The cells were transfected with siRNA or plasmids for 24 h. The medium was removed and was replaced with fresh medium. When the colonies were visible without a microscope, cell colonies were fixed with 4% paraformaldehyde and were stained using 0.1% crystal violet. The number of colonies was quantified by counting the colonies with more than 50 cells.
Migration and invasion assays
Migration and invasion assays were performed with a modified Boyden chamber (Corning) containing Matrigel-coated (invasion) or no Matrigel-coated (migration) membrane matrix (BD Biosciences). Cells were plated in the upper chamber, and the lower chamber contained medium with 10% FBS and were incubated for 24 h. After fixation with 4% formaldehyde, cells on the lower surface of the membranes were stained with crystal violet and were observed under a microscope at ×200 magnification. The average number of cells was determined from six representative fields.
Transfection and luciferase reporter gene assays
Cells were seeded on 24-well dishes and were transfected using Lipofectamine 3000 (Invitrogen) for 24 h, after which they were serum-starved for an additional 24 h prior to harvest. A Renilla luciferase reporter vector pRL-TK was used as internal control. Luciferase assays were performed with a dual-specific luciferase assay kit (Promega, Madison, WI). Firefly luciferase activities were normalized to Renilla luciferase activities.
Chromatin immunoprecipitation (ChIP)
Formaldehyde was added to the culture medium to a final concentration of 1%. The cells were then washed twice with PBS, scraped, and lysed in lysis buffer (1% SDS, 10 mm Tris-HCl, pH 8.0, 10% protease inhibitor mixture, 50 mg/ml each of aprotinin and leupeptin) for 10 min at 4 °C. The lysates were sonicated on ice, and the debris was removed by centrifugation at 12,000 rpm for 15 min at 4 °C. One-fourth of the supernatant was used as the DNA input control. The remaining sample was diluted 10-fold with dilution buffer (0.01% SDS, 1% Triton X-100, 1 mm EDTA, 10 mm Tris-HCl, pH 8.0, and 150 mm NaCl) followed by incubation with antibodies overnight at 4 °C. Immunoprecipitated complexes were collected using protein A/G-Sepharose. The pellets were washed for four times with dialysis buffer containing 2 mm EDTA and 50 mm Tris-HCl, pH 8.0. After washing, the precipitates were incubated with elution buffer (1% SDS and 0.1 m NaHCO3) at room temperature. Supernatants were transferred to clean tubes, and RNase A was added to destroy the bound RNA in the sample. Samples were incubated at 65 °C for 5 h to reverse the formaldehyde cross-links, and DNA was precipitated with ethanol and extracted twice with phenol/chloroform. Finally, pellets were resuspended in TE buffer and subjected to PCR amplification. Primers used this study are listed in Table S5.
Mammalian two-hybrid analysis
293T cells were transfected with the luciferase reporter plasmid pG5-luc (Promega), test plasmids, negative control plasmids, and positive control plasmids. Forty eight hours post-transfection, cells were harvested, and luciferase activities were assayed using the luciferase reporter assay system (Promega), according to the manufacturer's recommendations.
Co-immunoprecipitation
Co-immunoprecipitation analyses were performed as described previously (36). Briefly, after treatment, cells were collected and lysed using lysis buffer (20 mm Tris, pH 7, 0.5% (v/v) Nonidet P-40, 25 mm NaCl, 3 mm EDTA, 3 mm EGTA, 2 mm DTT, 0.5 mm phenylmethyl sulfonyl fluoride, 20 mm β-glycerol phosphate, 1 mm sodium vanadate, and 1 mg/ml leupeptin). Lysates were mixed and were precipitated with Abs or IgG mixed with protein G-agarose beads by overnight incubation at 4 °C. Beads were washed three to five times with lysis buffer, and bound proteins were separated by SDS-PAGE with subsequent immunoblotting analysis.
Statistical analysis
The data presented in this study were obtained from three independent reproducible experiments. The results were presented as the means ± S.D. Student's t test was performed for statistical comparisons between two groups. One-way analysis of variance was used to compare three or more groups. Kaplan-Meier analysis was used for the survival analyses. A p value < 0.05 was considered significant and was indicated with an asterisk.
Author contributions
Y. Z. and A. C. conceptualization; Y. Z. and A. C. data curation; Y. Z., C. Z., and J. W. validation; Y. Z., P. M., and Y. S. investigation; Y. Z. and B. Z. writing-original draft; P. M. and S. X. project administration; C. Z. resources; C. Z. visualization; Y. S. formal analysis; Y. S., A. C., and J. W. methodology; S. X. and J. W. software; B. Z. supervision; B. Z. writing-review and editing.
Supplementary Material
This work was supported by Quality Test and Operation with the Anesthesia Center of Experimental Animal of Hubei Province Research Grant 2060403, Key Program of National Natural Science Foundation of China Research Grant 81703035, the National Natural Science Foundation of China Research Grants 81600794 and 31571168, and National Key R&D Program of China Research Grant 2016YFC1303800. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S4 and Tables S1–S5.
- HCC
- hepatocellular carcinoma
- HBV
- hepatitis B virus
- HBx
- hepatitis B virus X protein
- PPP
- pentose phosphate pathway
- STAT3
- signal transducer and activator of transcription 3
- TKT
- transketolase
- SH2D5
- Src homology 2 domain–containing protein 5
- IL-6
- interleukin-6
- ANT
- adjacent nontumor tissue
- TNM
- tumor node metastasis
- Ab
- antibody
- FBS
- fetal bovine serum
- IAV
- influenza A virus
- DAB
- 3,3′-diaminobenzidine.
References
- 1. Chen S., Dong Z., Yang P., Wang X., Jin G., Yu H., Chen L., Li L., Tang L., Bai S., Yan H., Shen F., Cong W., Wen W., and Wang H. (2017) Hepatitis B virus X protein stimulates high mobility group box 1 secretion and enhances hepatocellular carcinoma metastasis. Cancer Lett. 394, 22–32 10.1016/j.canlet.2017.02.011 [DOI] [PubMed] [Google Scholar]
- 2. Hsieh A., Kim H. S., Lim S. O., Yu D. Y., and Jung G. (2011) Hepatitis B viral X protein interacts with tumor suppressor adenomatous polyposis coli to activate Wnt/β-catenin signaling. Cancer Lett. 300, 162–172 10.1016/j.canlet.2010.09.018 [DOI] [PubMed] [Google Scholar]
- 3. Torre L. A., Bray F., Siegel R. L., Ferlay J., Lortet-Tieulent J., and Jemal A. (2015) Global cancer statistics, 2012. CA Cancer J. Clin. 65, 87–108 10.3322/caac.21262 [DOI] [PubMed] [Google Scholar]
- 4. Yang P., Li Q. J., Feng Y., Zhang Y., Markowitz G. J., Ning S., Deng Y., Zhao J., Jiang S., Yuan Y., Wang H. Y., Cheng S. Q., Xie D., and Wang X. F. (2012) TGF-β-miR-34a-CCL22 signaling-induced Treg cell recruitment promotes venous metastases of HBV-positive hepatocellular carcinoma. Cancer Cell 22, 291–303 10.1016/j.ccr.2012.07.023 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Liu D., Cui L., Wang Y., Yang G., He J., Hao R., Fan C., Qu M., Liu Z., Wang M., Chen L., Li H., and Guo D. (2016) Hepatitis B e antigen and its precursors promote the progress of hepatocellular carcinoma by interacting with NUMB and decreasing p53 activity. Hepatology 64, 390–404 10.1002/hep.28594 [DOI] [PubMed] [Google Scholar]
- 6. Liu S., Hao Q., Peng N., Yue X., Wang Y., Chen Y., Wu J., and Zhu Y. (2012) Major vault protein: a virus-induced host factor against viral replication through the induction of type-I interferon. Hepatology 56, 57–66 10.1002/hep.25642 [DOI] [PubMed] [Google Scholar]
- 7. Liu S., Peng N., Xie J., Hao Q., Zhang M., Zhang Y., Xia Z., Xu G., Zhao F., Wang Q., Han T., and Zhu Y. (2015) Human hepatitis B virus surface and e antigens inhibit major vault protein signaling in interferon induction pathways. J. Hepatol. 62, 1015–1023 10.1016/j.jhep.2014.11.035 [DOI] [PubMed] [Google Scholar]
- 8. Neuveut C., Wei Y., and Buendia M. A. (2010) Mechanisms of HBV-related hepatocarcinogenesis. J. Hepatol. 52, 594–604 10.1016/j.jhep.2009.10.033 [DOI] [PubMed] [Google Scholar]
- 9. Xia L., Huang W., Tian D., Zhu H., Zhang Y., Hu H., Fan D., Nie Y., and Wu K. (2012) Upregulated FoxM1 expression induced by hepatitis B virus X protein promotes tumor metastasis and indicates poor prognosis in hepatitis B virus-related hepatocellular carcinoma. J. Hepatol. 57, 600–612 10.1016/j.jhep.2012.04.020 [DOI] [PubMed] [Google Scholar]
- 10. Liu Y., Li P. K., Li C., and Lin J. (2010) Inhibition of STAT3 signaling blocks the anti-apoptotic activity of IL-6 in human liver cancer cells. J. Biol. Chem. 285, 27429–27439 10.1074/jbc.M110.142752 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Stein B., and Yang M. X. (1995) Repression of the interleukin-6 promoter by estrogen receptor is mediated by NF-κB and C/EBPβ. Mol. Cell. Biol. 15, 4971–4979 10.1128/MCB.15.9.4971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Hirano T., Ishihara K., and Hibi M. (2000) Roles of STAT3 in mediating the cell growth, differentiation and survival signals relayed through the IL-6 family of cytokine receptors. Oncogene 19, 2548–2556 10.1038/sj.onc.1203551 [DOI] [PubMed] [Google Scholar]
- 13. Kitamura H., Ohno Y., Toyoshima Y., Ohtake J., Homma S., Kawamura H., Takahashi N., and Taketomi A. (2017) Interleukin-6/STAT3 signaling as a promising target to improve the efficacy of cancer immunotherapy. Cancer Sci. 108, 1947–1952 10.1111/cas.13332 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Xu I. M., Lai R. K., Lin S. H., Tse A. P., Chiu D. K., Koh H. Y., Law C. T., Wong C. M., Cai Z., Wong C. C., and Ng I. O. (2016) Transketolase counteracts oxidative stress to drive cancer development. Proc. Natl. Acad. Sci. U.S.A. 113, E725–E734 10.1073/pnas.1508779113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Yang H., Wu X. L., Wu K. H., Zhang R., Ju L. L., Ji Y., Zhang Y. W., Xue S. L., Zhang Y. X., Yang Y. F., and Yu M. M. (2016) MicroRNA-497 regulates cisplatin chemosensitivity of cervical cancer by targeting transketolase. Am. J. Cancer Res. 6, 2690–2699 [PMC free article] [PubMed] [Google Scholar]
- 16. Xu X., Zur Hausen A., Coy J. F., and Löchelt M. (2009) Transketolase-like protein 1 (TKTL1) is required for rapid cell growth and full viability of human tumor cells. Int. J. Cancer 124, 1330–1337 10.1002/ijc.24078 [DOI] [PubMed] [Google Scholar]
- 17. Diaz-Moralli S., Tarrado-Castellarnau M., Alenda C., Castells A., and Cascante M. (2011) Transketolase-like 1 expression is modulated during colorectal cancer progression and metastasis formation. PLoS ONE 6, e25323 10.1371/journal.pone.0025323 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Krockenberger M., Engel J. B., Schmidt M., Kohrenhagen N., Häusler S. F., Dombrowski Y., Kapp M., Dietl J., and Honig A. (2010) Expression of transketolase-like 1 protein (TKTL1) in human endometrial cancer. Anticancer Res. 30, 1653–1659 [PubMed] [Google Scholar]
- 19. Schultz H., Kähler D., Branscheid D., Vollmer E., Zabel P., and Goldmann T. (2008) TKTL1 is overexpressed in a large portion of non-small cell lung cancer specimens. Diagn. Pathol. 3, 35 10.1186/1746-1596-3-35 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang J., Li Z., Liu L., Wang Q., Li S., Chen D., Hu Z., Yu T., Ding J., Li J., Yao M., Huang S., Zhao Y., and He X. (2018) Long noncoding RNA TSLNC8 is a tumor suppressor that inactivates the interleukin-6/STAT3 signaling pathway. Hepatology 67, 171–187 10.1002/hep.29405 [DOI] [PubMed] [Google Scholar]
- 21. Liu B. A., and Nash P. D. (2012) Evolution of SH2 domains and phosphotyrosine signalling networks. Philos. Trans. R. Soc. Lond. B Biol. Sci. 367, 2556–2573 10.1098/rstb.2012.0107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Gray E. J., Petsalaki E., James D. A., Bagshaw R. D., Stacey M. M., Rocks O., Gingras A. C., and Pawson T. (2014) Src homology 2 domain–containing protein 5 (SH2D5) binds the breakpoint cluster region protein, BCR, and regulates levels of Rac1-GTP. J. Biol. Chem. 289, 35397–35408 10.1074/jbc.M114.615112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Schenk G., Duggleby R. G., and Nixon P. F. (1998) Properties and functions of the thiamin diphosphate dependent enzyme transketolase. Int. J. Biochem. Cell Biol. 30, 1297–1318 10.1016/S1357-2725(98)00095-8 [DOI] [PubMed] [Google Scholar]
- 24. Bowman T., Garcia R., Turkson J., and Jove R. (2000) STATs in oncogenesis. Oncogene 19, 2474–2488 10.1038/sj.onc.1203527 [DOI] [PubMed] [Google Scholar]
- 25. Sansone P., and Bromberg J. (2012) Targeting the interleukin-6/Jak/stat pathway in human malignancies. J. Clin. Oncol. 30, 1005–1014 10.1200/JCO.2010.31.8907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Wen Z., Zhong Z., and Darnell J. E. Jr. (1995) Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell 82, 241–250 10.1016/0092-8674(95)90311-9 [DOI] [PubMed] [Google Scholar]
- 27. Ki S. H., Park O., Zheng M., Morales-Ibanez O., Kolls J. K., Bataller R., and Gao B. (2010) Interleukin-22 treatment ameliorates alcoholic liver injury in a murine model of chronic-binge ethanol feeding: role of signal transducer and activator of transcription 3. Hepatology 52, 1291–1300 10.1002/hep.23837 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Svinka J., Mikulits W., and Eferl R. (2014) STAT3 in hepatocellular carcinoma: new perspectives. Hepat. Oncol. 1, 107–120 10.2217/hep.13.7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Hatziapostolou M., Polytarchou C., Aggelidou E., Drakaki A., Poultsides G. A., Jaeger S. A., Ogata H., Karin M., Struhl K., Hadzopoulou-Cladaras M., and Iliopoulos D. (2011) An HNF4α-miRNA inflammatory feedback circuit regulates hepatocellular oncogenesis. Cell 147, 1233–1247 10.1016/j.cell.2011.10.043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Liang J., Nagahashi M., Kim E. Y., Harikumar K. B., Yamada A., Huang W. C., Hait N. C., Allegood J. C., Price M. M., Avni D., Takabe K., Kordula T., Milstien S., and Spiegel S. (2013) Sphingosine-1-phosphate links persistent STAT3 activation, chronic intestinal inflammation, and development of colitis-associated cancer. Cancer Cell 23, 107–120 10.1016/j.ccr.2012.11.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Bruix J., Sherman M., and Practice Guidelines Committee, American Association for the Study of Liver Diseases (2005) Management of hepatocellular carcinoma. Hepatology 42, 1208–1236 10.1002/hep.20933 [DOI] [PubMed] [Google Scholar]
- 32. Li Y., Tang Z. Y., and Hou J. X. (2011) Hepatocellular carcinoma: insight from animal models. Nat. Rev. Gastroenterol. Hepatol. 9, 32–43 [DOI] [PubMed] [Google Scholar]
- 33. Jing M., Wang J., Zhu S., Ao F., Wang L., Han T., Yue X., Zhu Y., Ye L., and Liu S. (2016) Development of a more efficient hepatitis B virus vaccine by targeting hepatitis B virus preS to dendritic cells. Vaccine 34, 516–522 10.1016/j.vaccine.2015.11.069 [DOI] [PubMed] [Google Scholar]
- 34. Gui S., Chen X., Zhang M., Zhao F., Wan Y., Wang L., Xu G., Zhou L., Yue X., Zhu Y., and Liu S. (2015) Mir-302c mediates influenza A virus-induced IFNβ expression by targeting NF-κB inducing kinase. FEBS Lett. 589, 4112–4118 10.1016/j.febslet.2015.11.011 [DOI] [PubMed] [Google Scholar]
- 35. Peng N., Liu S., Xia Z., Ren S., Feng J., Jing M., Gao X., Wiemer E. A., and Zhu Y. (2016) Inducible major vault protein plays a pivotal role in double-stranded RNA- or virus-induced proinflammatory response. J. Immunol. 196, 2753–2766 10.4049/jimmunol.1501481 [DOI] [PubMed] [Google Scholar]
- 36. Chen X., Zhou L., Peng N., Yu H., Li M., Cao Z., Lin Y., Wang X., Li Q., Wang J., She Y., Zhu C., Lu M., Zhu Y., and Liu S. (2017) MicroRNA-302a suppresses influenza A virus-stimulated interferon regulatory factor-5 expression and cytokine storm induction. J. Biol. Chem. 292, 21291–21303 10.1074/jbc.M117.805937 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Edmondson H. A., and Steiner P. E. (1954) Primary carcinoma of the liver: a study of 100 cases among 48,900 necropsies. Cancer 7, 462–503 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.