

Abstract
Twist1 is a basic helix-loop-helix transcription factor that plays a key role in embryonic development, and its expression is down-regulated in adult cells. However, Twist1 is highly expressed during cancer development, conferring a proliferative, migratory, and invasive phenotype to malignant cells. Twist1 expression can be regulated post-translationally by phosphorylation or ubiquitination events. We report in this study a previously unknown and relevant Twist1 phosphorylation site that controls its stability. To identify candidate phosphorylation sites in Twist1, we first conducted an in silico analysis of the Twist1 protein, which yielded several potential sites. Because most of these sites were predicted to be phosphorylated by protein kinase C (PKC), we overexpressed PKCα in several cell lines and found that it phosphorylates Twist1 on Ser-144. Using a combination of immunoblotting, immunoprecipitation, protein overexpression, and CRISPR/Cas9-mediated PKCα knockout experiments, we observed that PKCα-mediated Twist1 phosphorylation at Ser-144 inhibits Twist1 ubiquitination and consequently stabilizes it. These results provide evidence for a direct association between PKCα and Twist1 and yield critical insights into the PKCα/Twist1 signaling axis that governs cancer aggressiveness.
Keywords: phosphorylation, ovarian cancer, ubiquitylation (ubiquitination), epithelial-mesenchymal transition (EMT), post-transcriptional regulation, embryonic development, NetphosK3.1, PKCalpha, Twist1
Introduction
Twist1 is a highly conserved member of the regulatory basic helix-loop-helix (bHLH)2 family of transcription factors that plays a pivotal role in utero from regulating embryonic and mesodermal development to organogenesis (1, 2). During development, mutations in Twist1 are known to result in the failure of cranial neural tube closure, indicating its role in the proper migration and differentiation of the neural crest (2). Whereas Twist1 is mostly absent in normal adult cells, it is overexpressed in cancer cells, wherein its ability to promote epithelial-to-mesenchymal transition (EMT) provides a prosurvival advantage to cancer cells and confers a migratory and invasive phenotype (3), which is a prerequisite in the establishment of metastatic disease. This is especially true in solid tumors such as breast, prostate, and ovarian cancer (3–6), although the role of Twist1 in the survival and progression of hematological malignancies has also been reported (7). In addition to its role in metastasis formation, the ability of Twist1 to promote EMT has been shown to also confer cancer stem cell properties and promote chemoresistance (3). Thus, it is not surprising that studies have aimed to understand the regulatory pathways that control the levels of Twist1.
The most characterized regulatory modification on Twist1 is its phosphorylation on Ser-68 by the mitogen-activated protein kinases p38 and c-Jun N-terminal kinase (8). The phosphorylation on Ser-68 results in enhanced stability of the Twist1 protein as it inhibits Twist1 ubiquitination and degradation by the proteasome (8). Other known phosphorylation sites on Twist1 are Thr-121 and Ser-123 by protein kinase A (PKA) and Ser-42 by Akt, which result in the regulation of its dimerization activity and anti-apoptotic function, respectively (9, 10).
In addition to phosphorylation, Twist1 is also a target for ubiquitination and subsequent proteasomal degradation (11, 12). It is a known substrate of the ubiquitin ligase F-box and leucine-rich repeat protein 14 (FBXL14), as well as the β-transducin repeat-containing protein (β-TRCP) (11). We have shown in our work in ovarian cancer that epithelial ovarian cancer stem cells constitutively target Twist1 to the ubiquitin-proteasome system for degradation to maintain an epithelial phenotype (12). A similar finding has been reported in a recent study by Lin et al. (13) showing that Twist1 is subjected to degradation mediated by E3 ligases, and deubiquitinase dub3 aids its stabilization.
Protein kinase Cα (PKCα) is a family of phospholipid-dependent serine/threonine kinases that has been shown to promote invasiveness of solid tumors such as breast (14–16), pancreatic (17, 18), and lung (19) cancer. Similar to Twist1, PKCα has been linked to the process of EMT (16, 20). Although it has been previously shown that an indirect PKC/Twist1 axis exists in prostate cancer models (21), any direct interaction between a specific PKCα isoenzyme and Twist1 has not been elucidated. The objective of this study was to determine whether PKCα has a direct regulatory role on Twist1 expression.
We report the identification of Ser-144 as a novel Twist1 phosphorylation site that is a direct target of PKCα. Moreover, we demonstrate that this phosphorylation leads to Twist1 stabilization as it prevents Twist1 ubiquitination.
Results
PKC is predicted to phosphorylate Twist1
Our first objective was to identify novel effectors of Twist1 phosphorylation. Thus, we carried out in silico analysis of the Twist1 protein using NetphosK3.1 (http://www.cbs.dtu.dk/services/NetPhos/) (47),3 which allows kinase-specific predictions of phosphorylation sites on eukaryotic proteins. We identified at least 12 kinases predicted to phosphorylate Twist1: PKC, PKA, CDC2, p38, PKG, CKII, PKB, CDK5, GSK3, RSK, CKI, and DNAPK. Table 1 lists these kinases and their corresponding predicted phosphorylatable amino acid residues. Of the 12 kinases identified, PKC is predicted to have the highest number of possible phosphorylation sites within the Twist1 protein. Eleven of 34 predicted serine/threonine/tyrosine phosphorylation sites on Twist1 protein contain the consensus for PKC (Table 1). As such, we focused our succeeding experiments on the classical PKC isoform, PKCα.
Table 1.
Kinases predicted to phosphorylate Twist1 and their corresponding predicted phosphorylation sites
| Kinase | No. of phosphorylation sites | Phosphorylation sites |
|---|---|---|
| PKC | 11 | Ser-7, Ser-31, Ser-41, Ser-42, Thr-137, Ser-140, Ser-144, Thr-148, Ser-176, Ser-188 |
| PKA | 5 | Ser-41, Ser-42, Ser-78, Ser-123, Ser-174 |
| CDC2 | 5 | Ser-8, Ser-18, Ser-176, Ser-188, Ser-199 |
| p38 MAPK | 4 | Ser-8, Ser-11, Ser-68, Ser-99 |
| PKG | 4 | Ser-42, Ser-78, Ser-123, Ser-144 |
| CKII | 4 | Ser-18, Ser-20, Ser-102, Ser-165 |
| PKB | 3 | Ser-42, Thr-121, Ser-123 |
| CDK5 | 3 | Ser-8, Ser-11, Ser-99 |
| GSK3 | 3 | Ser-11, Ser-68, Ser-99 |
| RSK | 2 | Ser-42, Ser-123 |
| CKI | 2 | Ser-45, Ser-95 |
| DNAPK | 1 | Thr-121 |
PKCα activity and expression directly correlates with Twist1 protein levels
To determine whether PKCα would have an effect on Twist1 expression, we overexpressed a constitutively active form of PKCα (PKCαCAT) in the epithelial ovarian cancer (EOC) cell clone R182, which does not express the Twist1 protein (Fig. S1) (22–24). PKCαCAT is a truncated mutant form of PKCα (45 kDa versus 75 kDa for full-length WT PKCα), which is missing its regulatory domain and hence is constitutively active (25). Transient transfection of PKCαCAT into clone R182 resulted in an increase in PKCαCAT, which peaked at 48 h but was not sustained by 72 h (Fig. 1A). This transient increase in PKCαCAT, however, corresponded with an increase in endogenous Twist1 protein expression as early as 48 h post-transfection and reached statistical significance by 72 h (Fig. 1, A and B). PKCα-induced increase in Twist1 was not due to an increase in Twist1 transcription because we did not observe a change in Twist1 mRNA (Fig. 1C). These results indicate that the observed effect of PKCα on Twist1 is exerted at the protein level.
Figure 1.

Constitutively active PKCα (PKCαCAT) increases Twist1 protein expression independent of RNA. Human EOC cell clone R182 were transfected with PKCαCAT or empty vector control. A and B, expression of endogenous Twist1 and overexpressed PKCαCAT proteins were determined by Western blotting (A) and quantified by densitometry (B) reported as fold changes compared with empty vector control. *, p = 0.0008. C, effect on mRNA level was analyzed by qPCR, normalized to GAPDH, and reported as fold changes compared with empty vector control 24 h after transfection. ns, not significant (p > 0.05). D, different amounts of plasmid carrying WT Twist1 were transfected in HEK293T cells in the presence or absence of different amounts of plasmid carrying PKCαCAT, and the effect on Twist1 protein determined by Western blotting. Empty vector was used to control for total amount of plasmids transfected. E, HEK293T cells (panels i and ii) and EOC cells clone R182 (panels iii and iv) were transfected with WT Twist1 in the presence of different activation status of PKCα or empty vector control as indicated, and the effect on Twist1 protein was determined by Western blotting analysis, and the resulting bands were quantified by densitometry. CAT, constitutively active; DN, dominant negative; WT, full-length inactive WT. Densitometry readings are reported as fold changes compared with empty vector control. **, p = 0.0017; ****, p = 0.0001. Note that the truncated PKCαCAT (48 kDa) can be distinguished from the full-length DN and WT proteins (80 kDa). F, PKCα was knocked out in OCSC1-F2 ovarian cancer cells using CRISPR/Cas9, and the levels of PKCα and endogenous Twist1 were determined by Western blotting analysis. GAPDH was used as loading control for all Western blots. Each experiment was performed at least three independent times. Representative data are shown.
To further elucidate whether there is a direct correlation between PKCα and Twist1 expression, we determined the ability of PKCαCAT to increase Twist1 expression in HEK293T cells, which express neither Twist1 nor PKCα. As shown in Fig. 1D, the protein levels of Twist1 were significantly higher in cells co-transfected with PKCαCAT compared with the cells transfected with Twist1 alone (Fig. 1D).
To determine whether the observed effect of PKCα on Twist1 expression is secondary to its kinase activity, we compared the effect of constitutively active PKCα (PKCαCAT), dominant-negative PKCα (PKCαDN), and WT inactive PKCα (PKCαWT) on Twist1 protein expression. In both HEK293T cells and EOC cells, a significant increase in Twist1 protein was observed only in the presence of PKCαCAT (Fig. 1E). These results demonstrate that the kinase activity of PKCα is required for its observed effect on the Twist1 protein.
Finally, to conclusively show that PKCα directly regulates Twist1 expression, we used CRISPR/Cas9 to knock out PKCα in OCSC1-F2 ovarian cancer cells, which we previously reported to endogenously express high levels of Twist1 (26). As shown in Fig. 1F, we successfully depleted PKCα in these cells, and notably, the lack of PKCα is associated with decreased Twist1 protein expression. Indeed, OCSC1-F2 cells lacking PKCα have undetectable levels of Twist1 protein. Taken together, these results demonstrate the direct regulatory role of PKCα in Twist1 protein expression.
PKCα phosphorylates Twist1
We next sought to demonstrate that Twist1 is indeed a direct phosphorylation target of PKCα. Thus, we transiently co-transfected PKCαCAT and Twist1 in HEK293T cells and immunoprecipitated (IP) Twist1. Western blotting analysis of the resulting IP complex using anti–phospho-Ser/Thr/Phe antibody demonstrated an increase in the levels of phosphorylated epitopes corresponding to the molecular weight of Twist1 (Fig. 2A). These results show that Twist1 is a substrate of PKCα.
Figure 2.

PKCα induces Twist1 phosphorylation. A, Twist1 was transfected in HEK293T cells in the presence or absence of constitutively active PKCαCAT. Empty vector was used as control. Twist1 was subsequently immunoprecipitated, and the levels of phosphorylated Twist1 were determined by Western blotting using anti–phospho-serine/tyrosine/threonine antibody. GAPDH was used as a loading control for the input. B, in vitro kinase assay was performed as described under “Experimental procedures” with recombinant PKCα in the presence of increasing concentration of recombinant Twist1 as substrate. The ability of PKCα to phosphorylate Twist1 was quantified by the amount of ADP produced and presented as percentages of increase in ADP. *, p = 0.0022; **, p = 0.0001, compared with kinase reaction in the absence of Twist1. The experiments were performed at least three independent times. Representative data are shown.
We then sought to confirm whether Twist1 is a direct substrate of PKCα by performing an in vitro kinase assay. Using human recombinant active PKCα and human recombinant Twist1, we observed a significant increase in ADP levels with increasing concentration of Twist1 in the presence of constant concentration of PKCα concentration (5 ng) (Fig. 2B), suggesting the conversion of ATP to ADP and hence the occurrence of a phosphorylation reaction. These results demonstrate that Twist1 is a direct substrate of PKCα.
PKCα directly interacts with Twist1 protein at the Twist box (WR) domain
We then proceed to demonstrate the binding interaction between PKCα and Twist1 with the objective of identifying the Twist1 domain required for its interface with PKCα. Thus, we transiently expressed PKCαCAT and Twist1 in HEK293T cells and IP PKCα. Western blotting analysis of the obtained IP complex showed the presence of Twist1 (Fig. 3A). We then performed another IP, this time pulling down Twist1. Similarly, we observed the presence of PKCα in the IP complex when Twist1 was pulled down in the IP (Fig. 3A). Together, these results show that PKCα and Twist1 are able to form a complex.
Figure 3.

The WR domain of Twist1 is required for interaction with PKCα. A, HEK239T cell were co-transfected with Twist1–c-Myc and HA-PKCαCAT and whole-cell lysates were subjected to immunoprecipitation with either anti-HA antibody to capture PKCαCAT or anti-c-Myc antibody to capture Twist1. The resulting IP complexes were immunoblotted for PKCα and Twist1. B, schematic illustration of Twist1 protein deletions used to identify the domain required for binding to PKCα. C and D, Twist1 N-terminal deletion mutants with FLAG tag (C) and C-terminal deletions with Myc tag (D) were co-transfected with PKCαCAT in HEK293T cells. Whole-cell lysates were used to immunoprecipitate the corresponding tag on Twist1, and IP complexes were probed to detect presence of PKCα. IgG was used as control for all IPs. The experiments were performed at least three independent times. Representative data are shown.
To determine which domain of Twist1 is required for this interaction with PKCα, we created deletion mutants in either the N or C terminus of Twist1 (Fig. 3B). We then co-transfected these mutants individually with PKCαCAT in HEK293T cells and IP Twist1. N terminus deletions at Δ51–82 and Δ23–36 of Twist1 did not affect the formation of its complex with PKCα, as shown by the persistent presence of PKCα in the IP complex (Fig. 3C). Likewise, deletion of the C-terminal bHLH domain of Twist1 (ΔHLH; Δ109–164) did not affect PKCα and Twist1 complex formation (Fig. 3D). In contrast, deletion of the C-terminal WR (Δ182–202) domain of Twist1 abrogated the formation of PKCα and Twist1 complex as demonstrated by the absence of PKCα in the Twist1 IP complex in cells co-transfected with PKCαCAT and ΔWR Twist1 (Fig. 3D). These results demonstrate the requirement for Twist1 WR domain in its interaction with PKCα.
Phosphorylation of Twist1 protein at serine 144 prevents its degradation by inhibiting ubiquitination
Having demonstrated that PKCα is able to bind and phosphorylate Twist1, our next objective was to identify the specific amino acid residue on Twist1 that is phosphorylated by PKCα. As shown in Table 1, there are 11 predicted PKCα phosphorylation sites on Twist1. To determine the most likely amino acid, which is targeted by PKCα and, in addition, may be responsible for Twist1 stabilization, we determined which of these 11 phosphorylation sites are near predicted ubiquitination sites. Using UbPred server (http://ubpred.org),3 we identified 10 possible ubiquitination sites on Twist1, and four of these ten sites are adjacent or in close proximity to predicted phosphorylation sites. These ubiquitination sites are Lys-133, Lys-142, Lys-145, and Lys-150, which are adjacent to predicted phosphorylation sites Thr-137, Ser-140, Ser-144, and Thr-148, respectively (Fig. 4A). Fig. 4B shows the location of these phosphorylation sites on a 3D structure of Twist1. This structure is based on computational models that have been previously described (27). We then focused on Thr-137, Ser-140, Ser-144, and Thr-148 and determined which of these sites can control Twist1 stability.
Figure 4.

Phosphorylation on Ser-144 of Twist1 promotes its stability. A, schematic representation of predicted PKCα phosphorylation sites on Twist1 adjacent to predicted Twist1 ubiquitination sites. B, location of phosphorylation sites included in this study (Thr-137, Ser-140, Ser-144, and Thr-148) on Twist1 3D structure (green) shown dimerized with another bHLH protein (gray). Note that the phosphorylation sites (yellow stars) are within the loop and accessible on the exterior of the dimer. WT Twist1 or phospho-mutants were transfected in HEK293T cells (C, panels i and ii) or EOC cells clone R182 (D, panels I and ii) as indicated; the levels of Twist1 protein were determined by Western blotting analysis; and the resulting bands were quantified by densitometry. Densitometry readings are reported as fold changes compared with empty vector control. *, p = 0.0126; **, p = 0.0021. E, WT Twist1 (panel i), phospho-deficient on 144 (144A, panel ii), or phosho-mimic on 144 (144D, panels iii) were transfected in HEK293T cells and treated with cyclohexamide (CHX, 20 μg/ml). The levels of Twist1 protein were determined by Western blotting, and the resulting bands were quantified by densitometry; densitometry readings are reported as fold changes compared with empty vector control. **, p = 0.0096. GAPDH was used as loading control.
Therefore, we introduced mutations into a plasmid-borne Twist1 gene at each of these four sites. Alanine substitution creates a nonpolar side chain unable to be modified by kinases, and we hereafter refer to this type of change as “phospho-deficient.” Meanwhile, aspartic acid is negatively charged at cellular pH, mimicking the chemical and charge character of a phosphate group, appearing to the cell as if the residue is permanently phosphorylated. We refer to this as a “phosphomimetic” mutation. Thus, we created four phospho-deficient (T137A, S140A, S144A, and T148A) and four phosphomimetic mutants (T137D, S140D, S144D, and T148D). Each of these constructs were transfected into two different cell models, HEK293T and EOC cells, and the level of Twist1 protein expressed was then compared with WT Twist1. There is a notable reduction in Twist1 protein expression when a phospho-deficient residue is introduced at position 144 (S144A) (Fig. 4, C and D). This reduction is more evident when transfected in clone R182, which has an active mechanism to promote Twist1 degradation (12). In contrast, there are no changes in the level of Twist1 protein expression shown with other phospho-deficient constructs, i.e. T137A, S140A, and T148A. Interestingly, introduction of a phosphomimetic residue at position 144 (S144D) is shown to rescue and increase the protein expression level of Twist1 over that of WT in both cell models (Fig. 4, C and D). Equivalent mutations at other sites did not affect the expression of Twist1 protein. These results demonstrate that the availability of Twist1 HLH region, particularly position Ser-144, for phosphorylation is important for increased Twist1 expression. Intriguingly, although the phospho-deficient mutant T148A was expressed in lower abundance compared with WT Twist1, the T148D mutant also showed reduced protein expression levels in both cells (Fig. 4, C and D). These data suggest that mutation in Thr-148 affects the expression of Twist1 but not necessarily due to its phosphorylation status.
To further investigate the importance of Ser-144 phosphorylation in Twist1 protein stability, we determined its effect on Twist1 protein turnover. Thus, we transfected HEK293T with either Twist1 WT, Twist1 S144A, or Twist1 S144D followed by treatment with the protein synthesis inhibitor cycloheximide. Addition of the inhibitor would halt further production of Twist1 protein, allowing us to follow the rate of degradation of the existing pool. Our data showed that the expression of Twist1 WT was substantially reduced after 3 h and totally undetectable after 6 h of cycloheximide treatment (Fig. 4E, panel i). On the other hand, Twist1 S144D expression is almost unaffected at 3 h, and 30% of the protein was still expressed after 6 h (Fig. 4E, panels ii and iv), suggesting that phosphorylation on Ser-144 has a positive impact on protein stability. Taken together, these results further highlight the role of phosphorylation at Ser-144 in Twist1 protein stability.
Finally, we sought to demonstrate that the phosphorylation at Ser-144 directly affects Twist1 ubiquitination. Using HEK293T cells, we co-transfected HA-tagged ubiquitin with either WT Twist1 or Twist1 S144D and IP Twist1. We observed less ubiquitin binding to S144D compared with WT Twist1 (Fig. 5A). This can be seen as a reduction in the ubiquitin smear corresponding to a ladder of high-molecular-weight species in samples with Twist1 S144D. Taken together, these results demonstrate that phosphorylation at Ser-144 is able to inhibit the ubiquitination of Twist1 leading to its stability.
Figure 5.

PKCα-induced Ser-144 phosphorylation on Twist1 inhibits Twist1 ubiquitination and promotes stability. A, WT Twist1 or 144D phosphomimetic mutant were co-transfected in HEK293T cells with HA-tagged ubiquitin. Twist1 was subsequently immunoprecipitated, and the levels of ubiquitinated Twist1 were determined by Western blotting for HA. Ubiquitinated Twist1 appears as a smear on the HA blot. Note less ubiquitination in Ser-144 mutant compared with WT Twist1. Whole cell lysates were used as input control and GAPDH as loading control. B, phospho-deficient 144A, or phosphomimetic 144D were co-transfected in HEK293T cells with constitutively active PKCαCAT or empty vector to control for total amount of plasmids transfected. The levels of Twist1 protein detected by Western blotting. C, quantified by densitometry reported as fold changes compared with 144A. *, p = 0.04. D, WT Twist1 or 144D phosphomimetic mutant were co-transfected in HEK293T cells with PKCαCAT. Twist1 was subsequently immunoprecipitated, and phosphorylated Twist1 was detected by Western blotting using anti–phospho-(Ser/Thr)Phe antibody. Whole cell lysates were used as input control. The experiments were performed at least three independent times. Representative data are shown.
Ser-144 is one of the sites of PKCα-induced Twist1 phosphorylation
Our final objective is to demonstrate that Ser-144 on Twist1 is a PKCα substrate. Thus, Twist1 WT, Twist1 S144A, and Twist1 S144D were transfected in the presence or absence of PKCαCAT in HEK293T cells. We observed that Twist1 S144D protein expression was higher than Twist1 S144A. More importantly, we observed that the presence of PKCαCAT did not further increase the expression of Twist1 S144D nor Twist1 S144A (Fig. 5, B and C). These results demonstrate that the availability of Ser-144 as a phosphorylation substrate for PKCα is critical for the stabilization of Twist1. To further validate that the amino acid Ser-144 is a PKCα substrate, Twist1 was IP from HEK293T cells co-transfected with either Twist1 WT or Twist1 S144D in the presence of PKCαCAT. The IP complexes were then immunoblotted with anti–phospho-Ser/Thr/Phe antibody to determine the level of phosphorylation of Twist1 protein. The co-expression of Twist1 S144D with PKCαCAT did not result in Twist1 phosphorylation to the extent observed when Twist1 WT is co-expressed with PKCαCAT (Fig. 5D). These results strongly demonstrate the importance of amino acid Ser-144 within Twist1 protein as a substrate for PKCα and for its stabilization.
Discussion
In this study, we identified a novel Twist1 phosphorylation site that promotes its stability. Phosphorylation on Ser-144 of Twist1 can prevent its ubiquitination and proteasomal degradation. In addition, we identified PKCα as the primary kinase that can generate this modification. To the best of our knowledge, this is the first demonstration that Twist1 is a direct target of PKCα and that phosphorylation on Ser-144 is critical for Twist1 protein stability (Fig. 6).
Figure 6.
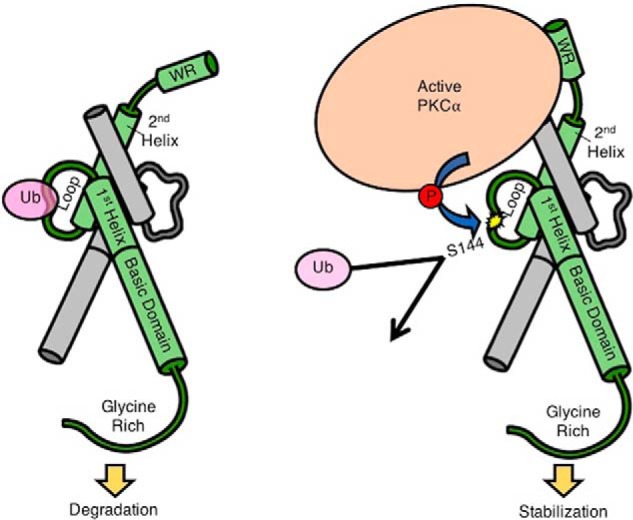
Proposed model of PKCα-dependent Twist1 stabilization. A, Twist1 is a target for ubiquitination around the loop region. Twist1 3D structure is shown in green dimerized with another bHLH protein shown in gray. B, active PKCα binds to Twist1 via Twist1's WR domain and phosphorylates Twist1 on Ser-144 located in the loop region. This phosphorylation precludes ubiquitination, leading to Twist1 stabilization.
Twist1 is a transcription factor critical for a myriad of cellular processes, the most studied of which is cellular migration (28, 29). It is this function that makes it an essential effector during the process of development and embryogenesis. The loss of expression or inactivation of Twist1 leads to Saethre–Chotzen syndrome, a developmental disorder characterized by premature cranial fusion and syndactyly (30, 31). Interestingly, it is this role in cellular migration that makes Twist1 also indispensable during the process of cancer metastasis.
Twist1 is often reactivated in many cancers, wherein it enables the EMT program to confer motility and invasiveness to cancer cells (32). In addition to metastasis formation, Twist1 overexpression in tumors has been linked to chemoresistance, which together with metastasis represents a major clinical challenge. As such, the regulation of Twist1 has been an active and ongoing field of research not only in developmental biology but in cancer as well.
Twist1 has been shown to be regulated both at the transcriptional and post-translational level (6, 33). Transcriptionally, Twist1 mRNA expression can be promoted by several signaling pathways such as NF-κB (34, 35), STAT3 (36), and Wnt (37), as well as the classical EMT inducer TGFβ (38). Post-translationally, Twist1 has been shown to be a target for phosphorylation, acetylation, and ubiquitination (8, 10, 39).
In utero, the expression of Twist1 is tightly regulated not only at the mRNA level but also at the protein level. In mice, although there is a significant expression of Twist1 mRNA, there was no detectable Twist1 protein in the paraxial mesoderm until embryonic day 8.25 (40). We observed similar findings in our in vitro models of EOC, where we found very low to no detectable Twist1 protein expression in the presence of a significant amount of Twist1 mRNA (12). Further studies demonstrated that in EOC, Twist1 protein is constitutively ubiquitinated and targeted for proteasomal degradation.
In this study, we determined possible mechanisms that can inhibit this constitutive ubiquitination and focused on phosphorylation as a likely post-translational modification that can prevent it. As mentioned above, Twist1 has been previously reported to be phosphorylated at Ser-42, Ser-68, Thr-121, and Ser-123 (9, 10). Phosphorylation at these sites has been demonstrated to affect the dimerization of Twist1, its transcriptional activity, and stability (8, 9). Our results revealed Ser-144 as a novel phosphorylation site on Twist1 protein that can reduce its ubiquitination and consequently lead to its stabilization. Interestingly, previous studies have shown that a missense mutation in the 144 position (S144R) prevents Twist1/E12 heterodimer from binding to DNA promoter regions and, as a result, impairs Twist1 transcriptional activity during development (41). Taken together with these findings, our results further highlight the importance of the Ser-144 position in Twist1 function.
Our data show that active PKCα is able to induce Ser-144 phosphorylation on Twist1 and that the WR region of Twist1 is required for this interaction. Similar to Twist1, PKCα is known to regulate various cellular processes, some of which interestingly have opposing effects. PKCα has been shown to induce proliferation in human glioma U87 cells as well as in MCF-7 human breast cancer cells but promote cell cycle arrest in MCF-10 human mammary epithelial cells and even apoptosis in human gastric cancer cells (16, 42, 43). Also similar to Twist1 is the role of PKCα in cell migration and tumor progression (44). Previous studies using prostate cancer models have shown that a PKC/Twist1 signaling axis exists and contributes to a castration-resistant phenotype (21). In that study, knockdown of PKCβ or PKCϵ was shown to inhibit the expression of Twist1 in the protein level. The PKC/Twist1 signaling pathway involves NF-κB–dependent up-regulation of Twist1 secondary to PKC activation (45). Our results demonstrate that PKCα is able to directly form a complex with and phosphorylate Twist1, consequently inhibiting its ubiquitination and its targeting to the proteasomal degradation system. These results offer further insights into PKC/Twist1 signaling axis and, given their roles in both cancer aggressiveness and chemoresistance, open new venues to the development of novel therapeutic modalities targeted to these factors.
Experimental procedures
Reagents, antibodies, and plasmids
Plasmids
pHACE-PKC-α-CAT (PKCαCAT) (Addgene, catalog no. 21234), pHACE-PKC-α-DN (PKCαDN) (Addgene, catalog no. 21235) and pHACE-PKC-α-WT (PKCαWT) (Addgene, catalog no. 21232) plasmids were gifts from Bernard Weinstein (25). Twist1 plasmids were constructed in pCDNA4 backbone. lentiCRISPRv2 (Addgene, catalog no. 52961) plasmid was a gift from Feng Zhang (46). pVSVg (Addgene, catalog no. 8454) and psPAX2 (Addgene, catalog no. 12260) plasmids were gifts from Bob Weinberg and Didier Trono, respectively.
Antibodies
anti-GAPDH (1:10000, G8795; Sigma), anti-Twist1 (1:200, CS-81417; Santa Cruz, Dallas, TX), anti-PKCα (1:200, CS-8393; Santa Cruz), anti-HA probe (1:200, CS-7392; Santa Cruz), anti–c-Myc tag (1:1000, catalog no. A190-105A; Bethyl Laboratories Inc., Montgomery, TX), phospho-Ser/Thr/Phe (1:1000, catalog no. 9631; Cell Signaling Technology, Danvers, MA), peroxidase-conjugated anti-rabbit IgG (1:10000), and peroxidase-conjugated anti-mouse IgG (1:10000) (Dako, Glostrup, Denmark). For Western blotting detections of IP test, TrueBlot Ultra anti-mouse and anti-rabbit IgG HRP (1:1000, catalog no. 18-8817-31 and 18-8816-31; Rockland Immunochemicals Inc., Limerick, PA) were used.
Cell culture and culture conditions
Human epithelial ovarian cancer cells (R182) used in these studies were isolated from ascites and grown as previously described (22). The isolation and characterization of the EOC cells has been previously reported (23, 24). All patients signed consent forms, and the use of patient samples was approved under Yale University's Human Investigations Committee (approval no. 10425). All studies described abide by the Declaration of Helsinki principles.
EOC cells were maintained at 60–80% confluence in RPMI 1640 medium (Gibco, catalog no. 2340-021) supplemented with 10% FBS (Gemini Bio-products, Woodland, CA; catalog no. 100-106). The confluence of the culture is critical to prevent differentiation. The primary cultures are maintained at low number of passages. We are extremely careful to maintain low passage number for the cells to prevent changes associated with long-term cultures. HEK 293T cells were maintained in Dulbecco's modified Eagle's medium (Gibco, catalog no. 119065-092) supplemented with 10% FBS.
Plasmid transfection
HEK293T cells were seeded at 5 × 105 cells/well in a 6-well plate or 3 × 106 cells/100-mm dish and incubated overnight. The media were then changed to fresh RPMI 1640 medium supplemented with 10% FBS at 4 h prior to transfection. A total of 2 μg of plasmids were mixed with 6 μg of PEI in 100 μl of OPTIMEM medium (Gibco) or for 100-mm dish a total of 20 μg of plasmids were mixed with 60 μg of PEI in 1 ml of OPTIMEM medium and incubated for 15 min at room temperature. The plasmid/PEI mix was then added in a dropwise manner. 24–72 h following transfection, cells were collected and washed with PBS for RNA purification and protein sample extraction. EOC cells were seeded at 7 × 105 cells/100-mm dish and then transfected and harvested as described for 293T cells.
Knockout of PKCα using CRISPR/Cas9
PKCα was knocked out in OCSC1-F2 ovarian cancer cells using CRISPR/Cas9. The guide RNA for PKCα was designed using the CRISPR design tool from the Zhang Laboratory at MIT. This guide was introduced to lentiCRISPRv2 plasmid. 10 μg of the resulting plasmid was then co-transfected with 8 μg of packaging plasmid (pVSVg) and 4 μg of envelope plasmid (psPAX2) in the presence of 60 μg of PEI on HEK293T cells in a 100-mm dish. The packaged plasmids were collected and transduced into the OCSC1-F2 cells. The cells were then subjected to puromycin selection.
Protein preparation and immunoprecipitation
Whole-cell protein extraction was performed using cell lysis buffer (1% Triton X-100, 0.05% SDS, 100 mm Na2PO4, and 150 mm NaCl) supplemented with protease inhibitor mixture (Roche) and PMSF. Cell pellets were lysed with 50 μl of cell lysis buffer mixture on ice for 15 min followed by centrifugation at 16,000 × g for 15 min at 4 °C to remove cell debris. The whole cell lysate was quantified using Pierce BCA reagent (catalog no. 23223, Thermo Scientific).
For IP, 500 μg of whole cell protein lysate were incubated with 1.5 μg of primary antibody at 4 °C overnight. The IP product was then incubated with protein A–Sepharose beads for 3 h, followed by three washes with KLB buffer (20 mm Tris HCl, pH 8.0, 137 mm NaCl, 10% glycerol, 1% Nonidet P-40, 2 mm EDTA). The product was then isolated for Western blotting detection.
SDS-PAGE and Western blots
20 μg of each protein lysate were electrophoresed on a 12% SDS-polyacrylamide gel. The proteins were then transferred onto polyvinylidene difluoride membranes (EMD Millipore). After blocking in PBS with 0.05% Tween 20 (PBS-Tween) and 5% milk, the membranes were probed with primary antibody in 2% milk PBS-Tween at 4 °C overnight. The membranes were then washed with PBS-Tween three times followed by a secondary antibody in 2% milk PBS-Tween for 2 h at room temperature. Immunoreactivity was detected by the ECL system (Bio-Rad).
In vitro kinase assay
In vitro kinase assay was performed using ADP-GloTM kinase assay (Promega Corp., Madison, WI) according to the manufacturer's instructions. Briefly, the kinase reaction consisted of 5 ng of recombinant human active full-length PKCα protein (Millipore, catalog no. 14-484), 200 or 300 ng recombinant human Twist1 protein (Abcam, catalog no. ab132349), PKC lipid activator (0.1 mg/ml phosphatidylserine, 0.01 mg/ml diacylglycerol, 4 mm MOPS, pH 7.2, 5 mm β-glycerol phosphate, 0.2 mm sodium orthovanadate, 0.2 mm DTT, 0.2 mm CaCl2), 20 mm HEPES, pH 7.4, 20 mm MgCl2, 0.1 mg/ml BSA, 0.1 mm CaCl2, and 100 μm ATP. The kinase reaction was performed at 37 °C for 1 h. The luminescence signal was measured using a desktop TD20/20 luminometer (Turner Biosystems, Sunnyvale, CA). Percentage of increase in ADP conversion from ATP, suggestive of the occurrence of a kinase reaction was calculated as follows, wherein LS is luminescence signal: % increase in ADP = (LSPKC+Twist-blank) − (LSPKCalone-blank)/(LSPKC+Twist-blank).
RNA quantification
For measuring Twist gene expression level, the cells were transfected as described in the plasmid transfection method section. Following the transfection, the cells were collected and washed with PBS. RNA was isolated using Qiagen RNasey Mini Plus kit (catalog no. 74106, Qiagen) according the manufacturer's instruction with addition of on-column DNase digestion. cDNA was synthesized using iScript cDNA kit (Bio-Rad). qPCR was performed on CFX96TM PCR detection system (Bio-Rad) using SYBR Green Supermix (Bio-Rad). The primers used for detection of Twist gene expression are: forward primer, 5′-GTCATGGCCAACGTGCGGGA-3′, and reverse primer, 5′-GCCGCCAGCTTGAGGGTCTG-3′. Twist gene expression was normalized to reference gene GAPDH. The primers used for detection of GAPDH expression are: forward primer, 5′-CTCTGCTCCTTCCTGTTCGAC-3′, and reverse primer, 5′-ACGACCAAATCCGTTGACTC-3′. Relative expression was calculated using the comparative ΔΔCT method. The quantifications were performed in triplicate.
Statistical analysis
The data are expressed as means ± standard deviation. Statistical significance (p < 0.05) was determined using either two tailed unpaired t tests or one-way analysis of variance using GraphPad Prism7.
Author contributions
R. T., C. M. R., A. B. A., C. C., Y. Y.-H., S. S., and M. P. data curation; R. T., C. M. R., A. B. A., and G. M. formal analysis; R. T. validation; R. T. methodology; R. T., C. M. R., and A. B. A. writing-original draft; R. T., C. M. R., A. B. A., and G. M. writing-review and editing; A. B. A., G. Y., C. A. G., and G. M. conceptualization; G. M. supervision; G. M. funding acquisition; G. M. project administration.
Supplementary Material
This work was supported in part by NCI, National Institutes of Health Grant RO1CA199004 (to G. M., A. B. A., and R. T.) and funds from the Sands Family Foundation (to G. M. and A. B. A.), the Discovery to Cure Program (to G. M.), and the Debra Levine Endowment (to G. M.). The authors declare that they have no conflicts of interest with the contents of this article. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
This article contains Fig. S1.
Please note that the JBC is not responsible for the long-term archiving and maintenance of this site or any other third party hosted site.
- bHLH
- basic helix-loop-helix
- EMT
- epithelial-to-mesenchymal transition
- PKA
- protein kinase A
- PKC
- protein kinase C
- EOC
- epithelial ovarian cancer
- IP
- immunoprecipitated
- FBS
- fetal bovine serum
- PEI
- polyethyleneimine.
References
- 1. Rose C. S. P., and Malcolm S. (1997) A TWIST in development. Trends Genet. 13, 384–387 [DOI] [PubMed] [Google Scholar]
- 2. O'Rourke M. P., and Tam P. P. (2002) Twist functions in mouse development. Int. J. Dev. Biol. 46, 401–413 [PubMed] [Google Scholar]
- 3. Nuti S. V., Mor G., Li P., and Yin G. (2014) TWIST and ovarian cancer stem cells: implications for chemoresistance and metastasis. Oncotarget 5, 7260–7271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Xu Y., Qin L., Sun T., Wu H., He T., Yang Z., Mo Q., Liao L., and Xu J. (2017) Twist1 promotes breast cancer invasion and metastasis by silencing Foxa1 expression. Oncogene 36, 1157–1166 10.1038/onc.2016.286 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Croset M., Goehrig D., Frackowiak A., Bonnelye E., Ansieau S., Puisieux A., and Clézardin P. (2014) TWIST1 expression in breast cancer cells facilitates bone metastasis formation. J. Bone Miner. Res. 29, 1886–1899 10.1002/jbmr.2215 [DOI] [PubMed] [Google Scholar]
- 6. Gajula R. P., Chettiar S. T., Williams R. D., Nugent K., Kato Y., Wang H., Malek R., Taparra K., Cades J., Annadanam A., Yoon A. R., Fertig E., Firulli B. A., Mazzacurati L., Burns T. F., Firulli A. B., An S. S., and Tran P. T. (2015) Structure–function studies of the bHLH phosphorylation domain of TWIST1 in prostate cancer cells. Neoplasia 17, 16–31 10.1016/j.neo.2014.10.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wang N., Guo D., Zhao Y. Y., Dong C. Y., Liu X. Y., Yang B. X., Wang S. W., Wang L., Liu Q. G., Ren Q., Lin Y. M., and Ma X. T. (2015) TWIST-1 promotes cell growth, drug resistance and progenitor clonogenic capacities in myeloid leukemia and is a novel poor prognostic factor in acute myeloid leukemia. Oncotarget 6, 20977–20992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hong J., Zhou J., Fu J., He T., Qin J., Wang L., Liao L., and Xu J. (2011) Phosphorylation of serine 68 of Twist1 by MAPKs stabilizes Twist1 protein and promotes breast cancer cell invasiveness. Cancer Res. 71, 3980–3990 10.1158/1538-7445.AM2011-3980,10.1158/0008-5472.CAN-10-2914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Firulli B. A., Krawchuk D., Centonze V. E., Vargesson N., Virshup D. M., Conway S. J., Cserjesi P., Laufer E., and Firulli A. B. (2005) Altered Twist1 and Hand2 dimerization is associated with Saethre–Chotzen syndrome and limb abnormalities. Nat. Genet. 37, 373–381 10.1038/ng1525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Vichalkovski A., Gresko E., Hess D., Restuccia D. F., and Hemmings B. A. (2010) PKB/AKT phosphorylation of the transcription factor Twist-1 at Ser42 inhibits p53 activity in response to DNA damage. Oncogene 29, 3554–3565 10.1038/onc.2010.115 [DOI] [PubMed] [Google Scholar]
- 11. Zhong J., Ogura K., Wang Z., and Inuzuka H. (2013) Degradation of the transcription factor Twist, an oncoprotein that promotes cancer metastasis. Discov. Med. 15, 7–15 [PMC free article] [PubMed] [Google Scholar]
- 12. Yin G., Alvero A. B., Craveiro V., Holmberg J. C., Fu H.-H., Montagna M. K., Yang Y., Chefetz-Menaker I., Nuti S., Rossi M., Silasi D.-A., Rutherford T., and Mor G. (2013) Constitutive proteasomal degradation of TWIST-1 in epithelial ovarian cancer stem cells impacys differentiation and metastatic potential. Oncogene 32, 39–49 10.1038/onc.2012.33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lin Y., Wang Y., Shi Q., Yu Q., Liu C., Feng J., Deng J., Evers B. M., Zhou B. P., and Wu Y. (2017) Stabilization of the transcription factors slug and twist by the deubiquitinase dub3 is a key requirement for tumor metastasis. Oncotarget 8, 75127–75140 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Lahn M., Köhler G., Sundell K., Su C., Li S., Paterson B. M., and Bumol T. F. (2004) Protein kinase Cα expression in breast and ovarian cancer. Oncology 67, 1–10 10.1159/000080279 [DOI] [PubMed] [Google Scholar]
- 15. Tan M., Li P., Sun M., Yin G., and Yu D. (2006) Upregulation and activation of PKCα by ErbB2 through Src promotes breast cancer cell invasion that can be blocked by combined treatment with PKCα and Src inhibitors. Oncogene 25, 3286–3295 10.1038/sj.onc.1209361 [DOI] [PubMed] [Google Scholar]
- 16. Tam W. L., Lu H., Buikhuisen J., Soh B. S., Lim E., Reinhardt F., Wu Z. J., Krall J. A., Bierie B., Guo W., Chen X., Liu X. S., Brown M., Lim B., and Weinberg R. A. (2013) Protein kinase Cα is a central signaling node and therapeutic target for breast cancer stem cells. Cancer Cell 24, 347–364 10.1016/j.ccr.2013.08.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Chen Y., Yu G., Yu D., and Zhu M. (2010) PKCα-induced drug resistance in pancreatic cancer cells is associated with transforming growth factor-β1. J. Exp. Clin. Cancer Res. 29, 104–104 10.1186/1756-9966-29-104 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Chow J. Y., Dong H., Quach K. T., Van Nguyen P. N., Chen K., and Carethers J. M. (2008) TGF-β mediates PTEN suppression and cell motility through calcium-dependent PKC-α activation in pancreatic cancer cells. Am. J. Physiol. Gastrointest. Liver Physiol. 294, G899–G905 10.1152/ajpgi.00411.2007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Abera M. B., and Kazanietz M. G. (2015) Protein Kinase Cα mediates erlotinib resistance in lung cancer cells. Mol. Pharmacol. 87, 832–841 10.1124/mol.115.097725 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Garg R., Benedetti L. G., Abera M. B., Wang H., Abba M., and Kazanietz M. G. (2014) Protein kinase C and cancer: what we know and what we do not. Oncogene 33, 5225–5237 10.1038/onc.2013.524 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Shiota M., Yokomizo A., Takeuchi A., Imada K., Kashiwagi E., Song Y., Inokuchi J., Tatsugami K., Uchiumi T., and Naito S. (2014) Inhibition of protein kinase C/Twist1 signaling augments anticancer effects of androgen deprivation and enzalutamide in prostate cancer. Clin. Cancer Res. 20, 951–961 10.1158/1078-0432.CCR-13-1809 [DOI] [PubMed] [Google Scholar]
- 22. Kamsteeg M., Rutherford T., Sapi E., Hanczaruk B., Shahabi S., Flick M., Brown D., and Mor G. (2003) Phenoxodiol–an isoflavone analog–induces apoptosis in chemoresistant ovarian cancer cells. Oncogene 22, 2611–2620 10.1038/sj.onc.1206422 [DOI] [PubMed] [Google Scholar]
- 23. Chen R., Alvero A. B., Silasi D.-A., Kelly M. G., Fest S., Visintin I., Leiser A., Schwartz P. E., Rutherford T., and Mor G. (2008) Regulation of IKKβ by miR-199a affects NF-κB activity in ovarian cancer cells. Oncogene 27, 4712–4723 10.1038/onc.2008.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Alvero A. B., Chen R., Fu H.-H., Montagna M., Schwartz P. E., Rutherford T., Silasi D.-A., Steffensen K. D., Waldstrom M., Visintin I., and Mor G. (2009) Molecular phenotyping of human ovarian cancer stem cells unravels the mechanisms for repair and chemoresistance. Cell Cycle 8, 158–166 10.4161/cc.8.1.7533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Soh J.-W., and Weinstein I. B. (2003) Roles of specific isoforms of protein kinase C in the transcription control of cyclin D1 and related genes. J. Biol. Chem. 278, 34709–34716 10.1074/jbc.M302016200 [DOI] [PubMed] [Google Scholar]
- 26. Craveiro V., Yang-Hartwich Y., Holmberg J. C., Joo W. D., Sumi N. J., Pizzonia J., Griffin B., Gill S. K., Silasi D.-A., Azodi M., Rutherford T., Alvero A. B., and Mor G. (2013) Phenotypic modifications in ovarian cancer stem cells following Paclitaxel treatment. Cancer Med. 2, 751–762 10.1002/cam4.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Maia A. M., da Silva J. H., Mencalha A. L., Caffarena E. R., and Abdelhay E. (2012) Computational modeling of the bHLH domain of the transcription factor TWIST1 and R118C, S144R and K145E mutants. BMC Bioinformatics 13, 184–184 10.1186/1471-2105-13-184 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lee M. P., and Yutzey K. E. (2011) Twist1 directly regulates genes that promote cell proliferation and migration in developing heart valves. PLoS One 6, e29758 10.1371/journal.pone.0029758 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Matsuo N., Shiraha H., Fujikawa T., Takaoka N., Ueda N., Tanaka S., Nishina S., Nakanishi Y., Uemura M., Takaki A., Nakamura S., Kobayashi Y., Nouso K., Yagi T., and Yamamoto K. (2009) Twist expression promotes migration and invasion in hepatocellular carcinoma. BMC Cancer 9, 240 10.1186/1471-2407-9-240 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Howard T. D., Paznekas W. A., Green E. D., Chiang L. C., Ma N., Ortiz de Luna R. I., Garcia Delgado C., Gonzalez-Ramos M., Kline A. D., and Jabs E. W. (1997) Mutations in TWIST, a basic helix-loop-helix transcription factor, in Saethre–Chotzen syndrome. Nat. Genet. 15, 36–41 10.1038/ng0197-36 [DOI] [PubMed] [Google Scholar]
- 31. Johnson D., Horsley S. W., Moloney D. M., Oldridge M., Twigg S. R., Walsh S., Barrow M., Njølstad P. R., Kunz J., Ashworth G. J., Wall S. A., Kearney L., and Wilkie A. O. (1998) A comprehensive screen for TWIST mutations in patients with craniosynostosis identifies a new microdeletion syndrome of chromosome band 7p21.1. Am. J. Hum. Genet. 63, 1282–1293 10.1086/302122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Yang J., Mani S. A., Donaher J. L., Ramaswamy S., Itzykson R. A., Come C., Savagner P., Gitelman I., Richardson A., and Weinberg R. A. (2004) Twist, a master regulator of morphogenesis, plays an essential role in tumor metastasis. Cell 117, 927–939 10.1016/j.cell.2004.06.006 [DOI] [PubMed] [Google Scholar]
- 33. Asanoma K., Liu G., Yamane T., Miyanari Y., Takao T., Yagi H., Ohgami T., Ichinoe A., Sonoda K., Wake N., and Kato K. (2015) Regulation of the mechanism of TWIST1 transcription by BHLHE40 and BHLHE41 in cancer cells. Mol. Cell. Biol. 35, 4096–4109 10.1128/MCB.00678-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Šošić D., Richardson J. A., Yu K., Ornitz D. M., and Olson E. N. (2003) Twist regulates cytokine gene expression through a negative feedback loop that represses NF-κB activity. Cell 112, 169–180 10.1016/S0092-8674(03)00002-3 [DOI] [PubMed] [Google Scholar]
- 35. Kasinathan N. K., Subramaniya B., and Sivasithamparam N. D. (2018) NF-κB/twist mediated regulation of colonic inflammation by lupeol in abating dextran sodium sulfate induced colitis in mice. J. Funct. Foods 41, 240–249 10.1016/j.jff.2017.12.048 [DOI] [Google Scholar]
- 36. Cheng G. Z., Zhang W. Z., Sun M., Wang Q., Coppola D., Mansour M., Xu L. M., Costanzo C., Cheng J. Q., and Wang L.-H. (2008) Twist is transcriptionally induced by activation of STAT3 and mediates STAT3 oncogenic function. J. Biol. Chem. 283, 14665–14673 10.1074/jbc.M707429200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Howe L. R., Watanabe O., Leonard J., and Brown A. M. (2003) Twist is up-regulated in response to Wnt1 and inhibits mouse mammary cell differentiation. Cancer Res. 63, 1906–1913 [PubMed] [Google Scholar]
- 38. Dong Y.-F., Soung do Y., Chang Y., Enomoto-Iwamoto M., Paris M., O'Keefe R. J., Schwarz E. M., and Drissi H. (2007) Transforming growth factor-β and Wnt signals regulate chondrocyte differentiation through Twist1 in a stage-specific manner. Mol. Endocrinol. 21, 2805–2820 10.1210/me.2007-0199 [DOI] [PubMed] [Google Scholar]
- 39. Shi J., Wang Y., Zeng L., Wu Y., Deng J., Zhang Q., Lin Y., Li J., Kang T., Tao M., Rusinova E., Zhang G., Wang C., Zhu H., Yao J., Zeng Y.-X., Evers B. M., Zhou M.-M., and Zhou B. P. (2014) Disrupting the interaction of BRD4 with di-acetylated Twist suppresses tumorigenesis in basal-like breast cancer. Cancer Cell 25, 210–225 10.1016/j.ccr.2014.01.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Gitelman I. (1997) Twist protein in mouse embryogenesis. Dev. Biol. 189, 205–214 10.1006/dbio.1997.8614 [DOI] [PubMed] [Google Scholar]
- 41. El Ghouzzi V., Legeai-Mallet L., Aresta S., Benoist C., Munnich A., de Gunzburg J., and Bonaventure J. (2000) Saethre–Chotzen mutations cause TWIST protein degradation or impaired nuclear location. Hum. Mol. Genet. 9, 813–819 10.1093/hmg/9.5.813 [DOI] [PubMed] [Google Scholar]
- 42. Mandil R., Ashkenazi E., Blass M., Kronfeld I., Kazimirsky G., Rosenthal G., Umansky F., Lorenzo P. S., Blumberg P. M., and Brodie C. (2001) Protein kinase Cα and protein kinase Cδ play opposite roles in the proliferation and apoptosis of glioma cells. Cancer Res. 61, 4612–4619 [PubMed] [Google Scholar]
- 43. Sun X.-G., and Rotenberg S. A. (1999) Overexpression of protein kinase Cα in MCF-10A human breast cells engenders dramatic alterations in morphology, proliferation, and motility. Cell Growth Differ. 10, 343–352 [PubMed] [Google Scholar]
- 44. Ways D. K., Kukoly C. A., deVente J., Hooker J. L., Bryant W. O., Posekany K. J., Fletcher D. J., Cook P. P., and Parker P. J. (1995) MCF-7 breast cancer cells transfected with protein kinase C-α exhibit altered expression of other protein kinase C isoforms and display a more aggressive neoplastic phenotype. J. Clin. Invest. 95, 1906–1915 10.1172/JCI117872 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Shiota M., Yokomizo A., Takeuchi A., Kashiwagi E., Dejima T., Inokuchi J., Tatsugami K., Uchiumi T., and Eto M. (2017) Protein kinase C regulates Twist1 expression via NF-κB in prostate cancer. Endocrine-related Cancer 24, 171–180 10.1530/ERC-16-0384 [DOI] [PubMed] [Google Scholar]
- 46. Sanjana N. E., Shalem O., and Zhang F. (2014) Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784 10.1038/nmeth.3047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Blom N., Sicheritz-Pontén T., Gupta R., Gammeltoft S., and Brunak S. (2004) Prediction of post-translational glycosylation and phosphorylation of proteins from the amino acid sequence. Proteomics 4, 1633–1649 10.1002/pmic.200300771 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.