

Significance Statement
Antibody-mediated rejection (AMR) in renal allografts, which is usually caused by antibodies (Abs) directed against HLAs, is associated with a poor transplant outcome. However, evidence of AMR in the absence of anti-HLA Abs suggests the presence of non-anti–HLA Abs, presumed to react with other antigens on endothelial cells. The authors describe the clinicopathologic profiles of kidney recipients who experienced acute rejection with microvascular inflammation within 3 months after transplantation in the absence of anti-HLA donor-specific Abs. Using a new endothelial cell crossmatch assay and transcriptomic and proteomic analyses, they discovered that before transplantation, these patients carried unknown anti–endothelial cell Abs in their sera that specifically targeted the glomerular microvascular endothelium. An assessment of these unknown potentially deleterious Abs may provide important diagnostic tools to prevent AMR.
Keywords: acute microvascular rejection, AECAs, acute rejection, antibody mediated rejection
Visual Abstract

Abstract
Background
Although anti-HLA antibodies (Abs) cause most antibody-mediated rejections of renal allografts, non-anti–HLA Abs have also been postulated to contribute. A better understanding of such Abs in rejection is needed.
Methods
We conducted a nationwide study to identify kidney transplant recipients without anti-HLA donor-specific Abs who experienced acute graft dysfunction within 3 months after transplantation and showed evidence of microvascular injury, called acute microvascular rejection (AMVR). We developed a crossmatch assay to assess serum reactivity to human microvascular endothelial cells, and used a combination of transcriptomic and proteomic approaches to identify non-HLA Abs.
Results
We identified a highly selected cohort of 38 patients with early acute AMVR. Biopsy specimens revealed intense microvascular inflammation and the presence of vasculitis (in 60.5%), interstitial hemorrhages (31.6%), or thrombotic microangiopathy (15.8%). Serum samples collected at the time of transplant showed that previously proposed anti–endothelial cell Abs—angiotensin type 1 receptor (AT1R), endothelin-1 type A and natural polyreactive Abs—did not increase significantly among patients with AMVR compared with a control group of stable kidney transplant recipients. However, 26% of the tested AMVR samples were positive for AT1R Abs when a threshold of 10 IU/ml was used. The crossmatch assay identified a common IgG response that was specifically directed against constitutively expressed antigens of microvascular glomerular cells in patients with AMVR. Transcriptomic and proteomic analyses identified new targets of non-HLA Abs, with little redundancy among individuals.
Conclusions
Our findings indicate that preformed IgG Abs targeting non-HLA antigens expressed on glomerular endothelial cells are associated with early AMVR, and that in vitro cell-based assays are needed to improve risk assessments before transplant.
Despite the development of potent immunosuppressive regimens, antibody-mediated rejection (AMR) remains a significant hurdle to long-term organ acceptance. Although histologic findings suggestive of AMR (i.e., microvascular inflammation) usually indicate an anti-HLA–mediated injury, a subset of patients develop these lesions in the absence of detectable anti-HLA donor-specific antibodies (DSAs). The potential involvement of non-HLA antibodies (Abs) is mentioned in the current Banff classification, which requires the presence of “serological evidence of DSAs against HLA or other antigens.”1 However, in the absence of other, clearly defined antigens, the assumption that acute rejections with significant microvascular inflammation (called acute microvascular rejections [AMVRs]) are true AMRs remains hypothetical. In addition, although this issue is of utmost importance for treatment decisions, a clear indication that the observed graft injury is induced by Abs may be difficult to obtain.
These particular types of immune injuries are presumed to be because of Abs that react with non-HLA antigens expressed on endothelial cells (ECs). These Abs might be alloantibodies directed against non-HLA polymorphic antigens that differ between the recipient and donor or autoantibodies that recognize self-antigens after a disruption of self-tolerance.2 The identification and characterization of pathogenic anti–endothelial cell Abs (AECAs) would improve our understanding of the mechanisms involved in AMRs and would enable the development of new tools for patient monitoring. Several hurdles hamper the identification of these AECAs. First, the development of acute renal dysfunction with histologic lesions suggestive of AMR in the absence of anti-HLA DSAs is a relatively rare event. Consequently, previous studies that aimed to identify AECAs often included patients with heterogeneous clinical presentations ranging from hyperacute rejection3–5 to chronic allograft dysfunction,5,6 or patients with a positive EC crossmatch independent of any clinical presentation.7 Second, the identification of deleterious non-HLA Abs is particularly difficult to achieve in long-term patients, as a broad autoantibody response develops over time after transplantation.8,9
We aimed to study a highly selected cohort of patients with a homogeneous clinical and pathologic presentation of AMVR without anti-HLA DSAs during the first 3 months post-transplantation, to overcome these challenges. We reasoned that early AMVR would likely be caused by preformed AECAs, facilitating their identification in pretransplant serum samples. We report here the clinicopathologic description of this cohort and our efforts to identify the pathogenic AECAs.
Methods
Patients
Kidney transplant recipients (KTRs) were identified through a nationwide survey aimed at identifying suspected cases of early AMVRs of renal allografts in the absence of anti-HLA DSAs. Inclusion criteria were a first transplantation or retransplantation, a deceased or living donor, acute dysfunction or delayed graft function occurring within the first 3 months post-transplantation, histologic features of microvascular inflammation with a glomerulitis plus peritubular capillaritis score ≥3 according to the Banff classification, and the absence of historical or current anti-HLA DSA (A/B/Cw/DR/DQ/DP), as assessed using a Luminex single-antigen bead assay (all mean fluorescence intensities <500). All biopsy specimens were centrally reassessed, and the absence of anti-HLA DSAs was also centrally confirmed (see Supplemental Material for details).
For the case-control histologic study (Figure 1), a control group of 20 KTRs with early full-blown AMR who presented with anti-HLA DSAs in the first 3 months was identified. The patients were matched for age, sex, time of transplantation, and immunosuppressive regimen at transplantation.
Figure 1.

Study design and workflow. A nationwide survey identified patients suspected having of early (<3 months post-transplant) microvascular (glomerulitis and peritubular capillaritis score ≥3 according to the Banff classification) rejections of a renal allograft. After centralized Luminex SAFB assay testing and central reading of the biopsy specimens, 38 patients were retained for two parallel substudies. A case-control histologic study (study 1) addressed the histologic characteristics of the 38 AMVRs compared with 20 patients with early acute AMR associated with anti-HLA DSAs. A case-control biologic study (study 2) focused on identifying non-HLA antibodies using several approaches and used pretransplant serum samples from unsensitized KTRs who remained stable during the first year after transplant and were used as controls. Finally, an integrated analysis of transcriptomic and proteomic data were performed to identify antibodies targeting glomerular cell-specific antigens (study 3). For this aim, the differential transcriptomic profiles of microvascular glomerular ECs and macrovascular ECs were combined with the global seroreactivity toward protein arrays of serum samples collected immediately before transplantation from KTRs with AMVR or stable KTRs.
For the case-control biologic study (Figure 1), a second control group of ten highly stable patients (i.e., no rejection during the first year) was identified. Patients in this control group were also matched to patients in the AMVR group for age, sex, time of transplantation, and immunosuppressive regimen at transplantation.
Non-HLA Antibody Detection
Methods for the detection of non-HLA Abs, including anti-MICA, anti-AT1R, anti-ETAR, natural Abs, and Abs against a panel of 62 non-HLA antigens, in patients’ sera are described in detail in the Supplemental Material.
EC Crossmatch
Different types of ECs were incubated with patients’ serum samples, and IgG fixation was detected using flow cytometry. A comparative analysis of the reactivity of the patient’s serum was performed on parallel crossmatches using primary cultures of non-donor-specific arterial ECs and the immortalized human glomerular microvascular EC line CiGEnC (see the Supplemental Material for details).
RNA Sequencing and Protein Array
RNA sequencing (RNAseq) was performed to assess the differences in the transcriptomes between microvascular and macrovascular ECs. Patients’ serum samples were applied to a protein array to assess the seroreactivity of stable KTRs and patients with AMVR (see the Supplemental Material for details).
Statistical Analyses
The results are presented as the means±SD for continuous variables, unless specified otherwise. Frequencies of categorical variables are presented as numbers and percentages. Analyses were performed with GraphPad Prism software (version 5.00; GraphPad Software, San Diego, CA). For statistical comparisons of the clinical data between two groups, we used unpaired, two-tailed t tests and chi-squared test. For statistical comparisons of the in vitro data, we used nonparametric tests. P values <0.05 were considered significant.
A detailed description of the statistical methods used to analyze the protein array and RNAseq data are provided in the Supplemental Material.
Results
Clinicopathologic Description
A nationwide survey identified 51 KTRs (from 21 centers) with suspected early AMVR in the absence of anti-HLA DSAs (DSA-negative AMVR). After a central reassessment of anti-HLA DSAs (A.C.) and a central histologic analysis (M.R. and J.-P.D.V.H.), the final cohort included 38 patients with confirmed early acute DSA-negative AMVR (Figure 1).
Patients were 43.0±14.3 years of age (Table 1). Ten of the 38 patients with AMVR (26.3%) received a second (n=9) or a third (n=1) transplant (Table 1). Nineteen patients with AMVR (50%) presented non-DSA anti-HLA Abs at transplant. Comparing retransplantations with first transplantations within the AMVR group, no difference in the anti-HLA sensitization, neither to class 1 (five out of 28 versus four out of ten; P=0.21) or to class 2 HLA molecules (six out of 28 versus four out of ten; P=0.40), was observed.
Table 1.
Patient demographics
| Variables | AMVR without anti-HLA DSAs, n=38 | AMVR with anti-HLA DSAs, n=20 | P Values |
|---|---|---|---|
| Recipient characteristics | |||
| Men, n (%) | 25 (65.8) | 13 (65.0) | 1.00 |
| Age at transplantation, mean±SD, yr | 43.0±14.3 | 50.4±15.9 | 0.11 |
| Cause of ESRD, n (%) | |||
| GN | 10 (26.3) | 4 (20.0) | 0.75 |
| Diabetes | 6 (15.8) | 5 (25.0) | 0.49 |
| Cystic/hereditary/congenital | 7 (18.4) | 3 (15.0) | 1.00 |
| Secondary GN | 3 (7.9) | 2 (10.0) | 1.00 |
| Hypertension | 2 (5.3) | 0 (0.0) | 0.54 |
| Interstitial nephritis | 3 (7.9) | 2 (10.0) | 1.00 |
| Miscellaneous conditions | 2 (5.4) | 3 (15.0) | 0.33 |
| Uncertain etiology | 5 (13.2) | 1 (5.0) | 0.65 |
| Duration of dialysis before transplantation, mean±SD, yr | 3.9±4.4 | 4.8±4.9 | 0.44 |
| Previous transplantation, n (%) | 10 (26.3) | 3 (15.0) | 0.51 |
| Transplant variables | |||
| Donor age, mean±SD, yr | 50.4±12.6 | 52.3±17.4 | 0.93 |
| Deceased donor, n (%) | 28 (73.7) | 17 (85.0) | 0.51 |
| Male donor, n (%) | 17 (44.7) | 8 (40.0) | 0.79 |
| Cold ischemia time, mean±SD, h | 15.9±10.4 | 20.5±9.7 | 0.13 |
| Preformed anti-HLA Abs with an MFI>500, n (%) | 19 (50.0) | 20 (100.0) | <0.001 |
| Delayed graft function, n (%) | 18 (47.3) | 7 (35.0) | 0.41 |
| Number of post-transplant hemodialysis session, mean±SD | 2.5±4.2 | 2.4±2.9 | 0.39 |
| Immunosuppressive protocol | |||
| Induction therapy, n (%) | 38 (100.0) | 19 (95.0) | 0.34 |
| Basiliximab/thymoglobuline, n (%) | 33 (86.8)/5 (13.2) | 14 (75.0)/5 (25.0) | 0.28 |
| Calcineurin inhibitor-based therapy, n (%) | 37 (97.4) | 20 (100.0) | 1.0 |
| Cyclosporine/tacrolimus, n (%) | 11 (28.9)/26 (68.4) | 3 (15.0)/17 (85.0) | 0.34 |
| Purine synthesis inhibitor, n (%) | 37 (93.9) | 19 (95.0) | 0.35 |
| mTOR inhibitor, n (%) | 0 (0.0) | 1 (5.0) | 0.35 |
| Steroid, n (%) | 37 (97.4) | 20 (100.0) | 1.0 |
| Acute rejection description | |||
| Best serum creatinine level before AMVR, mean±SD, µmol/L | 275±187 | 195±137 | 0.15 |
| Best serum creatinine level before AMVR, mean±SD, d | 15.7±21.4 | 8.5±8.2 | 0.64 |
| AMVR diagnosis, mean±SD, d | 22.0±26.2 | 15.9±13.5 | 0.92 |
| Serum creatinine level at rejection, mean±SD, µmol/L | 417±276 | 298±229 | 0.11 |
| Patients on dialysis at time of rejection | 8 (21.1) | 1 (0.05) | 0.14 |
| Acute rejection treatment | |||
| Steroid, n (%) | 35 (92.1) | 19 (95.0) | 1.00 |
| Thymoglobuline, n (%) | 10 (26.0) | 2 (10.0) | 0.19 |
| Rituximab, n (%) | 12 (31.6) | 10 (50.0) | 0.25 |
| Plasmapheresis, n (%) | 25 (65.8) | 15 (75.0) | 0.56 |
| IGIV, n (%) | 18 (47.4) | 17 (85.0) | 0.01 |
| Follow-up | |||
| Serum creatinine level at 3 mo post-Tx, mean±SD, µmol/L | 161±59 | 129±55 | 0.01 |
| Serum creatinine level at 12 mo post-Tx, mean±SD, µmol/L | 145±53 | 125±41 | 0.08 |
| Mean follow-up, mean±SD, yr | 4.3±3.0 | 3.5±2.7 | 0.25 |
| Serum creatinine level at the last follow-up, mean±SD, µmol/L | 169±97 | 136±76 | 0.23 |
| Proteinuria at the last follow-up, mean±SD, g/g creatininea | 1.27±1.7 (n=20) | 1.0±1.4 (n=18) | 0.44 |
| Patient survival at the last follow-up, n (%) | 37 (97.3) | 18 (90.0) | 0.12 |
| Graft survival at the last follow-up, n (%) | 29 (76.3) | 19 (95.0) | 0.51 |
MFI, mean fluorescence intensity; mTOR, mammalian target of rapamycin; IGIV, IG intravenous; post-Tx, post-transplant.
In patients with a follow-up >1 yr.
AMVR was diagnosed at a mean time of 11.2±1.7 days for the 18 patients still requiring hemodialysis. For the other 20 patients, AMVR was diagnosed on the basis of an increase in the serum creatinine level from 275±187 µmol/L at 15.7±21.4 days to 417±276 µmol/L at 31.8±7.3 days post-transplantation.
The AMVR treatment was heterogeneous. However, rituximab was administered to 31.6% of patients, plasmapheresis to 65.8%, and intravenous immunoglobulins to 47.4%, suggesting that the patients were considered as having AMR.
A comparison of patients with DSA-negative AMVR with matched patients with DSA-positive AMR (Table 1) revealed that patients with DSA-negative AMVR displayed similar graft function at the rejection diagnosis (417±276 µmol/L versus 298±229 µmol/L; P=0.11), more severe graft dysfunction at 3 months (161±59 µmol/L versus 129±55 µmol/L; P=0.01), and similar graft function at the last follow-up (P=0.23). Consistent with severe graft injury, proteinuria was common in both groups, and after a similar follow-up period, the proteinuria in the AMVR cohort was similar to the AMR cohort (1.27±1.7 g/g versus 1.0±1.4 g/g; P=0.44).
The central histologic reading of the patients with DSA-negative AMVR showed severe microvascular inflammation, with a mean glomerulitis plus peritubular capillaritis score of 3.9±0.25 (Figure 2, A and B), and severe endothelial/vascular injury (Figure 2, C–H). Vasculitis was present in 60.5% of patients, and thrombotic microangiopathy and interstitial hemorrhages were observed in 15.8% and 31.6% of patients, respectively (Table 2).
Figure 2.

Representative pathologic characteristics of the early AMVRs. (A) Mean (±SEM) values of the elementary lesions assessed using the Banff classification in the biopsy samples from 38 KTRs at time of AMVR. (B) The glomerulitis and peritubular capillaritis (g+ptc) scores of the 38 individual patients with AMVR. (C) Image of periodic acid–Schiff (PAS) staining showing severe glomerulitis with partial to complete occlusion of glomerular capillaries by infiltrating leukocytes (mononuclear cells and neutrophils cells) (arrows). (D) Image of PAS staining showing severe peritubular capillaritis (ptc3) with more than ten inflammatory cells in dilated capillaries (arrows) associated with diffuse interstitial edema (+). (E) Image of PAS staining showing intimal arteritis v2 with mononuclear cells underneath the endothelium and occlusion of more than 25% of the arterial lumen (⋆) associated with peritubular capillaritis (+) and sparse inflammatory cells within the interstitium. (F) Image of Masson trichrome staining showing severe glomerulitis with complete occlusion of glomerular capillaries by infiltrating leukocytes and EC enlargement. EC enlargement is also present in arterioles (arrows). (G) Image of Masson trichrome staining showing thrombotic microangiopathy characterized by thrombi in the glomerular capillaries (⋆) associated with glomerulitis, peritubular capillaritis, and diffuse interstitial hemorrhage (+). (H) Image of Masson trichrome staining showing a mixed rejection with diffuse interstitial inflammation and tubulitis (arrow), glomerulitis open star, peritubular capillaritis (+), arteriolitis (⋆), and interstitial hemorrhage.
Table 2.
Description of the histologic findings
| Histologic Lesions | AMVR without anti-HLA DSAs, n=38 | AMVR with anti-HLA DSAs, n=20 | P Value |
|---|---|---|---|
| Glomerulitis (g) | |||
| % With a g score >0 | 38 (100.0%) | 18 (90.0%) | 0.11 |
| g score, mean±SD. | 2.1±0.8 | 1.7±0.9 | 0.18 |
| Peritubular capillaritis (ptc) | |||
| % With a ptc score >0 | 36 (94.7%) | 19 (95.0) | 1.0 |
| ptc score, mean±SD | 2.0±0.9 | 1.7±0.7 | 0.66 |
| C4d deposition, C4d | |||
| % With a C4d score >0 | 9 (23.7%) | 3 (15.0%) | 0.52 |
| C4d score, mean±SD | 0.5±1.1 | 0.5±0.8 | 0.98 |
| Interstitial infiltrates (i) | |||
| % With an i score>0 | 21 (55.3%) | 2 (10.0%) | <0.001 |
| i score, mean±SD. | 0.9±1.0 | 0.1±0.3 | 0.003 |
| Tubulitis (t) | |||
| % With a t score >0 | 14 (36.8%) | 14 (70.0%) | 0.03 |
| t score, mean±SD | 1.1±1.1 | 0.5±0.7 | 0.02 |
| TCMR diagnostic criteria, n (%) | 8 (21.1) | 2 (10.0) | 0.18 |
| IA | 3 (8.8) | 2 (10.0) | 0.29 |
| IB | 3 (8.8) | 0 (0) | 0.27 |
| IIA | 0 (0) | 0 (0) | 1.00 |
| IIB | 1 (2.6) | 0 (0) | 1.00 |
| III | 1 (2.6) | 0 (0) | 1.00 |
| Vasculitis (v) | |||
| % With a v score >0 | 23 (60.5%) | 3 (15.0%) | <0.001 |
| v score, mean±SD | 1.3±1.1 | 0.3±0.8 | <0.001 |
| Interstitial hemorrhages, n (%) | 12 (31.6) | 3 (15.0) | 0.22 |
| Thrombotic microangiopathy, n (%) | 6 (15.8) | 0 (0.0) | 0.08 |
| Allograft glomerulopathy (cg) | |||
| % With a cg score >0 | 0 (0.0%) | 0 (0.0%) | 1.00 |
| cg score, mean±SD | 0.0±0.0 | 0.0±0.0 | 1.00 |
| Mesangial expansion (mm) | |||
| % With an mm score >0 | 2 (5.3%) | 0 (0.0%) | 0.54 |
| mm score, mean±SD | 0.1±0.4 | 0.0±0.0 | 0.59 |
| Interstitial fibrosis (ci) | |||
| % With a ci score >0 | 4 (10.5%) | 4 (20.0%) | 0.43 |
| ci score, mean±SD | 0.2±0.7 | 0.3±0.6 | 0.97 |
| Tubular atrophy (ct) | |||
| % With a ct score >0 | 4 (10.5%) | 4 (20.0%) | 0.42 |
| Ct score, mean±SD | 0.2±0.7 | 0.2±0.4 | 0.80 |
| Chronic vascular changes (cv) | |||
| % With a cv score >0 | 16 (42.1%) | 13 (65.0%) | 0.16 |
| cv score, mean±SD | 1.0±1.1 | 0.9±1.1 | 0.87 |
| Arteriolar hyalinosis (ah) | |||
| % With an ah score >0 | 15 (39.5%) | 11 (55.5%) | 0.28 |
| ah score, mean±SD | 0.8±0.9 | 0.8±1.1 | 0.59 |
TCMR, T cell mediated rejection.
Compared with DSA-positive AMR biopsy specimens, DSA-negative AMVR biopsy specimens exhibited more severe endothelial/vascular injury, with significantly more vasculitis lesions (1.3±1.1 versus 0.3±0.8; P<0.001), a greater number of patients with vasculitis lesions (60.5% versus 15%; P=0.001), and numerically more thrombotic microangiopathy (15.8% versus 0%; P=0.08) (Table 2). Compared with patients with AMR, patients in the AMVR group showed significantly more interstitial infiltrates. Overall, T cell–mediated rejection defined according to the Banff classification was not significantly different between the two groups (31.5% versus 10.0%; P=0.18).
Assessment of Known AECAs
The presence of previously proposed AECAs10,11 was assessed in available serum samples collected at the time of transplant (day 0), corresponding to a mean time of 22.0±26.2 days before the AMVR diagnosis, in 23 patients with early AMVR and ten stable KTRs used as controls (Supplemental Table 1).
Anti-MICA Abs were detected in only two patients with AMVR. Titers of angiotensin type 1 receptor (AT1R) and endothelin-1 type A (ETAR) Abs were similar in both groups (Figure 3A). Regarding AT1R Abs, we did not observe any positivity in the AMVR group or in the stable group (Figure 3A), using the threshold of 17 UI/ml proposed by Hönger et al.12. When the positive threshold of 10 UI/ml proposed by Dragun et al.3 was used, six out of 23 patients with AMVR (26%) were positive for AT1R Abs compared with no patients (zero out of ten, 0%) in the stable group (P=0.14, chi-squared test). However, we observed a good correlation between ETAR and AT1R Ab titers, with an r2 >0.8 (P<0.001), suggesting a spread of the Ab response toward more autoreactivity (Figure 3C).
Figure 3.

Assessment of known AECAs. (A) Titers of anti–AT-1R and anti-ETAR antibodies in serum samples collected on the day of transplantation from 23 patients with early AMVR without anti-HLA DSAs and ten nonsensitized KTRs who did not experience any rejection during their first year after transplant and were used as controls. P values were determined using the Mann–Whitney test. (B) Assessments of natural polyreactive antibodies were conducted using flow cytometry to detect reactivity to apoptotic cells or using a dissociation-enhanced lanthanide fluoroimmunoassay (DELFIA) to detect reactivity to malondialdehyde (MDA) in 19 patients with AMVR and eight controls. P values were determined using the Mann–Whitney test. (C) Correlation between anti–AT-1R and anti-ETAR antibody titers at the time of transplantation. (D) Correlation between NAbs reactive to MDA and anti-ETAR antibodies at the time of transplantation. (E) Correlation between NAbs reactive to MDA and anti–AT-1R antibodies at the time of transplantation. (F) Analysis of the seroreactivity of serum samples from ten stable patients and 23 patients with AMVR toward 62 non-HLA antigens using single-antigen flow bead assays. The color of each box indicates the mean fluorescence intensity (MFI) of the reaction of the sample to an individual antigen. The thresholds for defining a positive reaction of the patients with to each individual antigen were calculated on the basis of the mean MFI of the control group of stable patients. Samples with an MFI less than the mean+3 SD were classified as negative and samples with an MFI greater than the mean+3 SD were classified as positive. The number of positive samples is provided on the right and the samples that reached the threshold for positivity are indicated with a cross.
IgG natural polyreactive antibody (NAb) levels were assessed in serum samples from patients with AMVR and control patients using two separate methods. No difference in IgG NAbs was observed between the two groups with either method (Figure 3B). However, as reported in Figure 3D, the level of IgG NAbs measured using an ELISA was significantly correlated with the level of anti-ETAR Abs, supporting the view of a broad autoimmune component.
Sera were also tested against a panel of 62 non-HLA antigens (Figure 3F). At the time of transplant, 19 out of 23 (83%) patients with AMVR tested positive for at least one of the non-HLA antigens examined, whereas no stable patients reached the positivity threshold. Sixteen of the 62 antigens were positive in at least one patient with AMVR. A total of 45 antigens were found positive, with a maximum of eight patients with AMVR exhibiting positivity for protein kinase Cζ. Overall, no antigen appeared to be a positive target in the majority of patients with AMVR.
EC Crossmatch
Because no AECA candidate explained the majority of AMVR cases, we developed an EC crossmatch assay to assess serum reactivity to human microvascular glomerular ECs.13 Two EC types were used as cellular targets: non-donor-specific primary cultures of human vascular arterial ECs and the CiGEnC line, an established thermosensitive conditionally immortalized cell line that allows cells to differentiate into glomerular microvascular ECs at 37°C with a preserved endothelial phenotype. The CiGEnC phenotype observed after differentiation is shown in Supplemental Figure 1. As ECs express class 1 and class 2 HLA antigens, this analysis was restricted to patients with AMVR, stable KTRs, or healthy volunteers with no circulating anti-HLA Abs to avoid any HLA-dependent cell reactivity. Strikingly, the seroreactivity against glomerular ECs was significantly increased in sera from patients with AMVR (Figure 4A), whereas limited reactivity was observed in healthy volunteers (n=6) or stable KTRs (n=10). Seroreactivity against non-HLA antigens was only due to IgG, as no IgM reactivity was observed (data not shown). This IgG reactivity was present on day 0 (Figure 4B) and persisted to the time of rejection. Serial titration of positive sera revealed high Ab titers (Figure 4C).
Figure 4.


The seroreactivity against glomerular ECs was significantly increased in sera from patients with AMVR. Sera (diluted 1:4) were incubated with ECs. Antibody binding was detected using fluorescently labeled anti-human IgG, and the mean fluorescence intensity (MFI) was measured using flow cytometry. (A) Comparison of the reactivity of sera from healthy volunteers (HVs; n=6) and KTRs with (n=19) or without (n=10) early AMVR and without anti-HLA DSAs toward unstimulated immortalized human glomerular CiGEnCs. The data are presented as a fold increase in the MFI compared with a pool of AB serum samples used as negative control. The P value was calculated using the Kruskal–Wallis test. Asterisks depict significant differences in pairwise group comparisons calculated using Dunn posttest. ***P<0.01; ****P<0.001. (B) Sera (diluted 1:4) collected on the day of transplantation or at rejection from four patients with AMVR without anti-HLA DSAs were incubated with unstimulated microvascular CiGEnCs. Representative histograms showing IgG binding are shown; values indicate the geometric MFIs. (C) Serial dilutions of sera from patient AMVR#11 or a pool of HVs were incubated with renal microvascular CiGEnCs before the detection of antibody binding using anti-human IgG. Data are presented as the geometric MFIs. (D and E) Sera (diluted 1:4) collected on the day of transplantation from 19 patients with AMVR were incubated with (D) renal microvascular CiGEnCs or (E) primary cultures of macrovascular arterial ECs before (unstimulated) or after a 48-hour stimulation with TNF-α and IFN-γ. A pool of AB sera was used as a negative control (CTL). A cut-off of a two-fold increase in the geometric mean value of patients’ sera compared with the negative control was established to define reactive sera. (F) Sera (diluted 1:4) collected on the day of transplantation from two patients with early AMVR or a pool of serum samples from HVs (n=6) were incubated with renal microvascular ECs or epithelial cells. Microvascular ECs were used before or after in vitro differentiation. Representative histograms showing IgG binding are shown, and the values indicate the geometric MFIs.
Crossmatches were also performed in resting ECs and after TNF-α and IFN-γ stimulation to better characterize the seroreactivity of patients with AVMR. The stimulated status of the CiGEnCs after cytokine treatment was controlled by the upregulation of the HLA molecules (Supplemental Figure 2). In healthy controls, even after cell activation, no significant reactivity toward glomerular ECs was observed, compared with 89% positivity in patients with AMVR. Interestingly, the high seroreactivity observed in patients with AMVR did not depend on inflammation (Figure 4D), suggesting that the antigen targets are basally expressed on CiGEnCs and are not regulated by TNF-α or IFN-γ. Moreover, no significant reactivity was observed using primary cultures of human macrovascular ECs as targets, even after cell stimulation (Figure 4E), or using human renal epithelial cells as targets. Finally, patients with AMVR exhibited higher seroreactivity toward fully differentiated glomerular ECs than against undifferentiated ECs (Figure 4F).
On the basis of these results, the targeted antigens are selectively and constitutively expressed on the surface of glomerular ECs.
Integrated cDNA-Protein Array Analyses of Glomerular EC-Specific Immunogenicity
As AMVR seroreactivity specifically targeted glomerular ECs but not macrovascular ECs, we first assessed the differences in the transcriptomic profiles of these two cell types to identify antigens restricted to microvascular ECs (Figure 5A).
Figure 5.

Integrated RNAseq-protein array analysis. (A) Unsupervised principal component analysis of the global seroreactivity profiles of serum samples collected immediately before transplantation from patients with AMVR (n=20) and stable KTRs (n=10). Average fixation signals of the immunogenic antigens were used. Ellipses of confidence (0.95) are presented for each group. (B) Clustering and heat map representations of the transcriptomic data from microvascular and macrovascular ECs. Cell samples (n=3 for microvascular ECs and n=5 for macrovascular ECs) are arranged along the x-axis and differentially expressed genes (n=3427) are arranged along the y-axis. The color of each cell reflects the fold change in the expression of each gene. (C) Heat map representation of the seroreactivity patterns of patients with AMVR and stable KTRs. Sera are arranged along the x-axis, whereas immunogenic antigens are arranged along the y-axis. The color of each cell reflects the normalized average fixation signal of an individual serum to one antigen.
Unsupervised hierarchical clustering of mRNA expression patterns correctly classified the microvascular and macrovascular ECs (Figure 5A), suggesting that microvascular glomerular ECs have a distinct transcriptomic profile. Next, read count normalizations and group comparisons were performed using three independent and complementary methods that identified 3427 differentially expressed transcripts in the two cell types (Supplemental Figure 3), including 2195 genes that were significantly overexpressed in microvascular ECs compared with macrovascular ECs (available at https://www.ebi.ac.uk/fg/annotare; ID: E-MTAB-7003).
We then used a protein array platform to assess the reactivity of serum samples collected immediately before transplantation from 20 patients with early AMVR and ten patients who remained stable over the first year after transplant to approximately 9375 antigens. An evaluation of the average signals for the anti-human IgG revealed values that were within the expected ranges and were consistent across the arrays, indicating the good quality of the samples from both groups. An unsupervised principal component analysis revealed a clear separation of sera from patients with AMVR from sera from stable patients (Figure 5B), suggesting that the global seroreactivity profile of patients with AMVR was different.
After normalization, individual antigens from protein arrays were ranked according to the frequency of reactivity of AMVR sera compared with control sera. Antigen-specific responses must have been more prevalent in the sera from patients with AMVR than in sera from the stable patients to be considered an antigen of interest, thus possibly representing shared immunogenic events targeting microvascular ECs. Compared with sera from stable patients, sera from patients with AMVR preferentially reacted with 136 out of 9375 antigens (unadjusted P<0.05; Supplemental Table 2), but substantial variability was observed among individuals, as illustrated in Figure 5C.
We next performed an integrated analysis (Figure 1) combining the serologic responses of the patients with AMVR and stable KTRs to the microvascular EC-specific mRNA expression profiles, with the aim of identifying non-HLA Abs in patients with AMVR that target proteins specifically expressed by glomerular microvascular ECs. This strategy allowed us to identify a list of 857 matches of immunogenic antigens and overexpressed genes in microvascular ECs (Figure 1).
Because seroreactivity was highly variable among patients with AMVR, we rank-ordered the 857 potential targets using a previously described method8 that calculates a global score for each candidate by including the frequency of seroreactivity in patients with AMVR compared with stable patients and the relative strength of the reactivity. Thus, numerous unidentified AECAs are present in patients with AMVR, but not in stable patients (Table 3).
Table 3.
Top 20 immunogenic antigens in patients with AMVR of 857 candidate antigens overexpressed in microvascular ECs
| Gene ENS | Symbol | Δ Expression in Micro-ECs versus Macro-ECs | Protein Locus | Description | Frequency in Stable Patients, % | Frequency in Patients with AMVR, % | Intensity in Stable Patientsa | Intensity in Patients with AMVRa | Overall Scoreb | P Value |
|---|---|---|---|---|---|---|---|---|---|---|
| ENSG00000162078 | ZG16B | 148.5 | NM_145252.1 | Zymogen granule protein 16B | 33.33 | 90.91 | 2.11 | 2.92 | 87.2 | <0.001 |
| ENSG00000163431 | LMOD1 | 144.8 | BC001755.1 | Leiomodin 1 | 25.00 | 68.18 | 1.04 | 1.99 | 60.4 | 0.01 |
| ENSG00000107779 | BMPR1A | 1782.7 | NM_004329 | Bone morphogenetic protein receptor, type IA | 16.67 | 72.73 | 0.87 | 1.03 | 57.4 | 0.001 |
| ENSG00000197971 | MBP | 4251.9 | NM_001025100.1 | Myelin basic protein | 25.00 | 63.64 | 1.11 | 2.16 | 56.4 | 0.03 |
| ENSG00000169188 | APEX2 | 23.7 | NM_014481.2 | APEX nuclease 2 | 25.00 | 77.27 | 0.93 | 1.09 | 55.0 | 0.01 |
| ENSG00000106789 | CORO2A | 433 | NM_052820.1 | Coronin, actin binding protein, 2A | 33.33 | 63.64 | 1.01 | 2.34 | 51.1 | 0.18 |
| ENSG00000183287 | CCBE1 | 15,174.7 | BC046645.1 | Collagen and calcium binding EGF domains 1 | 8.33 | 45.46 | 0.60 | 1.59 | 46.0 | 0.06 |
| ENSG00000145242 | EPHA5 | 1609.2 | PV3359 | EPH receptor A5 | 25.00 | 63.64 | 0.90 | 1.23 | 44.0 | 0.08 |
| ENSG00000106829 | TLE4 | 1281.9 | BC059405.1 | Transducin-like enhancer of split 4 | 33.33 | 68.18 | 2.05 | 1.99 | 43.5 | 0.05 |
| ENSG00000142459 | EVI5L | 1270.2 | NM_145245.1 | Ecotropic viral integration site 5-like | 25.00 | 59.09 | 0.84 | 1.33 | 41.4 | 0.12 |
| ENSG00000107679 | PLEKHA1 | 2978.9 | NM_001001974.1 | Pleckstrin homology domain containing, family A1 | 16.67 | 45.46 | 0.69 | 1.71 | 39.7 | 0.06 |
| ENSG00000198959 | TGM2 | 28,594.6 | BC003551.1 | Transglutaminase 2 | 8.33 | 45.46 | 0.68 | 0.96 | 37.6 | 0.06 |
| ENSG00000082805 | ERC1 | 2673.4 | PV3626 | ELKS/RAB6-interacting/CAST family member 1 | 8.33 | 31.82 | 0.39 | 2.31 | 36.0 | 0.07 |
| ENSG00000198081 | ZBTB14 | 285.7 | NM_003409.2 | Zinc finger and BTB domain containing 14 | 33.33 | 63.64 | 0.94 | 1.25 | 36.0 | 0.18 |
| ENSG00000128872 | TMOD2 | 560.4 | BC036184.1 | Tropomodulin 2 | 16.6 | 50.00 | 0.85 | 1.09 | 35.6 | 0.04 |
| ENSG00000168175 | MAPK1IP1L | 3083.3 | NM_144578.1 | Mitogen-activated protein kinase 1 interacting protein 1-like | 8.33 | 40.91 | 0.60 | 1.12 | 35.5 | 0.02 |
| ENSG00000112561 | TFEB | 171.1 | NM_007162.1 | Transcription factor EB | 8.33 | 40.91 | 0.62 | 0.99 | 33.7 | 0.02 |
| ENSG00000123836 | PFKFB2 | 596.2 | NM_006212.1 | 6-Phosphofructo-2-kinase/fructose-2,6-biphosphatase 2 | 41.67 | 68.18 | 1.09 | 1.40 | 33.4 | 0.12 |
| ENSG00000106123 | EPHB6 | 119.4 | NM_004445.1 | EPH receptor B6 | 8.33 | 36.36 | 0.69 | 1.14 | 30.6 | 0.15 |
| ENSG00000240694 | PNMA2 | 684.4 | XM_376764.2 | Paraneoplastic ma antigen 2 | 8.33 | 36.36 | 0.65 | 1.10 | 30.3 | 0.04 |
ENS, Ensembl Gene ID.
Intensity represents the average ratio of observed reactivity exceeding the cut-off for sera from stable patients and patients with AMVR.
The score was calculated using the equation 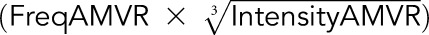 −
− described by Gnjatic et al.7
described by Gnjatic et al.7
Finally, four genes identified using our integrated RNAseq-protein array analysis were selected from Table 3 and validated at the mRNA and protein levels in microvascular ECs. As shown in Supplemental Figure 4, the four genes bone morphogenetic protein receptor type 1A (BMPR1A), ephrin type-B receptor 6 (EPHB6), leiomodin-1 (LMOD1), and myelin basic protein (MBP) are expressed in the endothelial crossmatch target cells.
Discussion
The concept that AMR may arise in the absence of anti-HLA DSA is universally accepted.14 This particular type of rejection is still improperly diagnosed, primarily because of the unknown specificity of the non-HLA Abs associated with its manifestation. Its clinical course and effect on the transplant outcome are also largely unknown. In an effort to better understand this complication, we studied a cohort of highly selected KTRs who experienced an AMR that was likely triggered by non-HLA DSAs.
In addition to circulating Abs, C4d deposition in peritubular capillaries is considered the best surrogate of antibody-induced injury, even if this marker is not always detected in patients with conventional AMR15,16 or in the context of suspected AECA-related AMR.4,17 In the absence of a consensus definition, we restricted our inclusion criteria to patients with significant microvascular inflammation. In addition, we selected KTRs experiencing acute rejection within the first 3 months after transplantation, resulting presumably from preformed Abs. These criteria allowed us to identify patients with a homogeneous clinical and pathologic presentation. In addition to a severe clinical phenotype, the histologic assessment revealed a dramatic involvement of the vascular wall with an unusual frequency of vasculitis lesions, thrombotic microangiopathy, and interstitial hemorrhages. Long-term follow-up of these patients revealed allograft dysfunction and glomerular proteinuria that were also consistent with an antibody-mediated immune injury.
Numerous AECAs have been reported in the past decade.10 Unlike other targets, AT1R and ETAR are well established effectors in autoimmune diseases affecting the macro- and microvasculature. AT1R agonistic Abs have been associated with preeclampsia, malignant/refractory hypertension, and primary aldosteronism.18 ETAR agonistic Abs have been associated with systemic sclerosis and SLE associated with pulmonary hypertension.18 Importantly, adoptive transfer experiments19 and pharmacologic inhibition in animal models20 supported their pathogenic effects. In transplantation, the seminal work by Dragun et al.3 reported not only the association of AT1R Abs with refractory vascular rejection, but also their potential pathogenic effects, demonstrated by the transfer of AT1R Abs to a rat model of kidney transplantation. Recent studies also suggest the association of AT1R with histologic features of AMR in indication renal allograft biopsy specimens.21 In this study, we focused on anti-AT1R,3 anti-ETAR22,23 and NAbs.24,25 Although none of these candidates clearly identified our patients with AMVR compared with stable KTRs, the more surprising result was that they were all correlated with each other. Indeed, we identified a strong correlation (r2=0.82) between anti-AT1R and anti-ETAR Abs, a finding that was also reported previously in the context of heart22 and renal transplantation.11 This observation supports the hypothesis that a broad autoimmune response may occur in some patients. Consistent with this hypothesis, Butte et al.26 previously identified an autoantibody signature in patients with renal insufficiency compared with controls, thus suggesting that end-stage renal damage may release proteins that are not otherwise recognized as self-antigens, leading to an adaptive humoral response. In addition, a longitudinal analysis of the Ab responses of pretransplantation and post-transplantation sera using a protein array revealed a significant enrichment of the Ab response against kidney compartments, again suggesting that chronic organ damage induces a broad autoantibody response.8 Interestingly, 26% of our patients with AMVR occurred in retransplanted patients. Thus, AECAs might develop during a previous transplantation. Consistent with this hypothesis, using protein arrays, Li et al.27 revealed that, in addition to HLA sensitization, kidney transplantation is associated with an enrichment of a specific antibody response against different kidney compartments, suggesting that non-anti–HLA Abs might develop in transplant recipients. Further studies are needed to determine whether this autoimmune response observed in patients with ESRD and transplant recipients is due to the release of self-antigens by the damaged organ or to a systemic B cell deregulation. In this regard, our observation that the global Ab response before transplantation clearly distinguished sera from patients with AMVR from sera from stable patients supports the hypothesis of systemic B cell deregulation. More recently, an association between endothelial crossmatch positivity and AT1R Abs has also been reported.28 However, in view of the increased autoimmunity observed in some patients, this association does not prove causation. Indeed, the findings reported by Dinavahi et al.29 and Porcheray et al.9 that autoimmune profiles induced by transplantation are unique to each individual patient also suggest that this response potentially results from systemic B cell deregulation rather than a response to potential cryptic epitopes unmasked during chronic renal injury.
Surprisingly, our assessment of AT1R Abs revealed the paucity of highly positive sera (>17 IU/ml) for AT1R Abs compared with other published studies of renal transplantation. The small sample size and the highly selected cohort are certainly possible explanations for the lack of patients with high levels of AT1R Abs. Nevertheless, the cut-off for AT1R Ab positivity remains controversial and several studies used a threshold of 10 IU/ml,30 whereas others used 15 IU/ml6 or even 17 IU/ml.12 Using a threshold of 10 IU/ml, Giral et al.30 reported that 47% of patients displayed AT1R Ab positivity before transplant, whereas Taniguchi et al.6 reported that 17% of patients displayed AT1R Ab positivity before transplant, using a threshold of 15 IU/ml. In our study, using a threshold of 10 IU/ml, six out of the 23 evaluated patients with AMVR (26%) would have been considered positive for AT1R Abs, supporting the potential role of AT1R Abs in AMVR occurrence.
We also evaluated the seroreactivity to a panel of 62 non-HLA antigens using two single-antigen flow bead assays. Although no antigen appeared to be a positive target in the majority of patients with AMVR, eight out of 23 patients with AMVR presented Abs against protein kinase Cζ, which has been previously associated with acute rejection and graft loss after kidney transplantation.31 The assessment of the seroreactivity to the 62 non-HLA antigens (Figure 3F) facilitated the identification of at least one antigen in 19 out of 23 patients with AMVR, whereas none of the control patients exhibited a positive result, revealing the relatively good discriminative capacity of this approach. However, this experiment also confirmed the broad and variable reactivity among individuals. Considering the highly variable serum reactivity to non-HLA endothelial antigens, our observation that IgG reactivity toward glomerular ECs predicts non-HLA Ab–induced AMRs shares some similarities with historical analyses showing an increased rejection risk associated with high panel reactive antibody values, before the use of sensitive bead assays to more specifically identify anti-HLA DSAs. In this respect, if the 62 non-HLA antigen panel used in this study were complemented with new candidate antigens, we may be able to improve our understanding of the underlying mechanisms and identify the culprits.
Although this “candidate gene” approach did not identify irrefutable candidates, our crossmatch assay identified preformed IgGs targeting antigens that are constitutively expressed on glomerular ECs in a compartment-specific fashion. No response or a minimal response to macrovascular cells, epithelial cells, and smooth muscle cells was detected (data not shown). This reactivity was highly specific to patients with AMVR, thus supporting our primary hypothesis that patients with AMVR are true AMRs. Notably, an increase in IgG binding was not observed between the day of transplantation and the day of rejection (Figure 4B), suggesting that non-HLA AECAs are preformed Abs. Figure 4B even shows a small reduction in the IgG binding when comparing “day 0” and “at rejection,” suggesting that circulating Abs may bind to the graft microvessels after transplantation, similar to anti-HLA DSAs.32
We observed a greater heterogeneity in the results of crossmatches that used microvascular ECs compared with macrovascular ECs. The heterogeneous results observed with the microvascular CiGEnCs are consistent with our results suggesting that each patient has peculiar AECAs and may be due to various titers of the AECAs. The relative homogeneity observed when using the macrovascular ECs as targets suggests that the KTRs have no Abs targeting the macrovascular endothelium. Although our crossmatch analysis did not detect IgG binding to macrovascular ECs isolated from unused pieces of artery taken from organ donors before kidney transplantation, we cannot exclude the presence, in kidney recipients, of other AECAs targeting antigens that are not strictly specific to the microvascular endothelium. However, the specific response to microvascular renal ECs may provide some clues regarding the still unexplained observation that auto-Abs targeting ECs have no pathogenic consequences in nontransplanted patients.33 In addition, their specificity toward the microvascular endothelium of the graft organ may explain why their pathogenicity is confined to the graft. This confinement to the graft of the pathogenic consequences of AECAs may suggest that the pathogenicity of AECAs first requires an initiating injury (i.e., ischemia/reperfusion injury, anti-HLA–mediated alloimmune injury, etc.). For example, our previous study assessing the role of natural Abs suggested some additive effect between anti-HLA DSA and natural Abs,34 a finding that was also observed for anti-HLA DSA and AT1R Abs,6,21,28 thus suggesting that non-anti–HLA autoantibodies have the potential to amplify microcirculation injury caused by alloantibodies in antibody-mediated transplant rejection.33,35
We used non-donor-specific ECs in our crossmatch assays. However, previously identified non-anti–HLA AECAs with suspected deleterious effects on the renal allograft, such as AT1R or ETAR or natural Abs, which are considered autoantibodies, have not be confirmed to be donor-specific allo-Abs.33
Our transcriptomic analysis revealed substantial differences in transcriptomic profiles between macrovascular and microvascular ECs, suggesting that the endothelial crossmatch performance will be highly dependent on the EC that is used as the target. The cell-based assays that have been developed to date have used various ECs (HUVECs,36 primary cultures of macrovascular arterial ECs,37 and circulating endothelial progenitors38), but have never used the ECs that are actually the target cells during the pathologic process (i.e., the renal microvascular ECs). To the best of our knowledge, our EC crossmatch assay is the first to use the target cells of the pathogenic Abs in KTRs. Of course, renal microvascular ECs cannot realistically be derived from every donor. Therefore, the availability of the CiGEnCs may facilitate the development of cell-based assays with a good capability of detecting non-anti–HLA AECAs.
In an effort to identify the culprits, we profiled the global IgG Ab responses in patients AMVR and compared them with controls, using protein arrays. The two main conclusions of this “antibodyome-wide” approach were that the global antibodyome correctly classified patients with AMVR, but no single specific Ab explained the disease, although several Abs that emerged from our combined analysis of transcriptomic and proteomic data have been already reported in the context of autoimmune diseases (Supplemental Material). The protein array we used in this study was not customized to contain endothelia-specific antigens and/or the whole spectrum of glomerular antigenic molecules, which may have led to false negative or false positive results. Nevertheless, our combined transcriptomic and proteomic approach identified new potential targets, and the expression of several of these targets in the glomerular ECs was validated (Supplemental Figure 4), thus confirming the rationale of our approach. Finally, patients suffering from non-HLA Ab-induced AMRs exhibit profound alterations of their seroreactivity, but with little redundancy, and some of their Abs are able to bind to glomerular cells. Altogether, our observations complement the aforementioned literature and suggest that an attempt to identify a common Ab that may explain the entire spectrum of disease may not succeed.
If a cell-based assay designed to detect AECAs in patient serum is an appealing strategy to circumvent the large individual variability in AECA specificities, it remains a challenging technique for several reasons, such as the variability in cell quality and surface antigen expression, the need for cell culture expertise, and the inability to test high PRA sera. The risk of cell variability is limited by the use of a well phenotyped cell line as opposed to primary cultures of ECs or purified circulating endothelial progenitors. Our robust endothelial phenotyping approach facilitates a longitudinal assessment of the stability of the cell line over time. Finally, AECA detection must be feasible even in highly sensitized patients. As a human cell line, CiGEnCs express class 1 and class 2 HLA molecules that may lead to a positive EC crossmatch because of the presence of anti-HLA Abs in sensitized recipients. The CRISPR/Cas9 technology will be used to delete HLA molecules from the CiGEnC line and establish an endothelial crossmatch that could be useful as a screening test for AECA assessment, even in highly sensitized patients. Finally, the observed binding of AECAs to the CiGEnCs may enable the more precise identification of the antigenic targets, thus facilitating the refinement of the existing solid phase assays for AECA identification. Expectedly, the development of a cell-based assay using a single cell line would not account for the potential donor genetic heterogeneity and would be more suitable for assessment of autoantibodies that recognize public antigens after a disruption of self-tolerance, as opposed to alloantibodies targeting non-HLA polymorphic antigens. A test that could address the tremendous interindividual variability in terms of autoantibody response could constitute a relevant companion test that could screen for the presence of circulating AECA in conjunction with the “candidate gene” approach.
In conclusion, we addressed the challenging problem of AMR in the absence of anti-HLA Abs in an original way by identifying a highly selected cohort of patients who likely suffered from this unusual and difficult to diagnose entity. Previously identified non-HLA Abs failed to differentiate patients with AMVR from stable patients, but an innovative EC crossmatch identified a universal IgG reactivity to microvascular glomerular ECs. An in-depth integrated analysis of transcriptomic and proteomic data revealed a large Ab response mediated by deregulation with little redundancy among individuals. On the basis of our results, in vitro cell-based assays are needed to assess the presence of EC Abs with a potential deleterious effect after transplantation.
Disclosures
None.
Supplementary Material
Acknowledgments
We thank the following colleagues who were willing to include patients who did not completely satisfy our inclusion criteria: D. Ducloux, Y. Lemeur, C. Hurault de Ligny, C. Mousson, P. Zaoui, J.P. Rerolle, V. Moal, G. Mourad, V. Esnault, H. Francois, M. Delahousse, and A. Thierry. We also thank C. Bole for her assistance with the RNA sequencing experiments, Dr. Jarhow Lee from One Lambda Inc. for providing the non-HLA antigen panel used for serum testing, and Dr. Claire Tinel for editing the manuscript.
M.D. and D.A. designed the study, analyzed the data, created the figures, and drafted the paper. S.C.S. provided the cells and reviewed and approved the manuscript. B.L. and N.C. analyzed the data and created the figures. C.B., O.A., S.P., A.C., and S.B.S. performed the experiments. B.C. analyzed the data and created the figures. M.R. and J.-P.D.V.-H. read the renal biopsy specimens. E.Z. analyzed the data and revised the paper. C.L. and J.-L.T. revised the paper. P.G., M.G., O.T., N.A., A.H., M.H., M.M., C.M., S.C., N.K., J.S., P.F.W., C.G., M.L., V.V., J.R., P.M., D.B., and A.L.M. participated in selecting the cohort of patients with AMVR and collected serum samples from the patients with AMVR.
This research was supported by funding from the LabEx Transplantex (grant ANR-11-LABX-0070_TRANSPLANTEX), the French Agence de la Biomédecine, the Centaure Foundation, the Day Solvay Foundation, and the Emmanuel Boussard Foundation.
Footnotes
Published online ahead of print. Publication date available at www.jasn.org.
Supplemental Material
This article contains the following supplemental material online at http://jasn.asnjournals.org/lookup/suppl/doi:10.1681/ASN.2018080868/-/DCSupplemental.
Supplemental Table 1. Baseline characteristics of 38 patients with early AMVR in the absence of anti-HLA DSAs and 10 KTRs who remained stable during the first year after transplant.
Supplemental Table 2. Antigens that are more immunogenic in patients with AMVR than in stable KTRs (P<0.05).
Supplemental Figure 1. CiGEnCs acquire an endothelial phenotype after 7 days of culture at 37°C.
Supplemental Figure 2. Cytokine stimulation increases HLA expression in CiGEnCs.
Supplemental Figure 3. Venn diagram illustrating the number of differentially expressed genes between microvascular ECs and macrovascular ECs, according to three statistical methods.
Supplemental Figure 4. Validation of antigen expression in micro- and macrovascular ECs.
References
- 1. doi: 10.1097/TP.0000000000002366. Roufosse C, Simmonds N, Clahsen-Van Groningen M, Haas M, Henriksen KJ, Horsfield C, et al.: A 2018 Reference Guide to the Banff Classification of Renal Allograft Pathology. Transplantation 102: 1795–1814, 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Reindl-Schwaighofer R, Heinzel A, Signorini L, Thaunat O, Oberbauer R: Mechanisms underlying human genetic diversity: Consequence for antigraft antibody responses. Transpl Int 31: 239–250, 2018 [DOI] [PubMed] [Google Scholar]
- 3.Dragun D, Müller DN, Bräsen JH, Fritsche L, Nieminen-Kelhä M, Dechend R, et al.: Angiotensin II type 1-receptor activating antibodies in renal-allograft rejection. N Engl J Med 352: 558–569, 2005 [DOI] [PubMed] [Google Scholar]
- 4.Jackson AM, Kuperman MB, Montgomery RA: Multiple hyperacute rejections in the absence of detectable complement activation in a patient with endothelial cell reactive antibody. Am J Transplant 12: 1643–1649, 2012 [DOI] [PubMed] [Google Scholar]
- 5.Zou Y, Stastny P, Süsal C, Döhler B, Opelz G: Antibodies against MICA antigens and kidney-transplant rejection. N Engl J Med 357: 1293–1300, 2007 [DOI] [PubMed] [Google Scholar]
- 6.Taniguchi M, Rebellato LM, Cai J, Hopfield J, Briley KP, Haisch CE, et al.: Higher risk of kidney graft failure in the presence of anti-angiotensin II type-1 receptor antibodies. Am J Transplant 13: 2577–2589, 2013 [DOI] [PubMed] [Google Scholar]
- 7.Zitzner JR, Shah S, Jie C, Wegner W, Tambur AR, Friedewald JJ: A prospective study evaluating the role of donor-specific anti-endothelial crossmatch (XM-ONE assay) in predicting living donor kidney transplant outcome. Hum Immunol 74: 1431–1436, 2013 [DOI] [PubMed] [Google Scholar]
- 8.Gnjatic S, Wheeler C, Ebner M, Ritter E, Murray A, Altorki NK, et al.: Seromic analysis of antibody responses in non-small cell lung cancer patients and healthy donors using conformational protein arrays. J Immunol Methods 341: 50–58, 2009 [DOI] [PubMed] [Google Scholar]
- 9.Porcheray F, DeVito J, Yeap BY, Xue L, Dargon I, Paine R, et al.: Chronic humoral rejection of human kidney allografts associates with broad autoantibody responses. Transplantation 89: 1239–1246, 2010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Delville M, Charreau B, Rabant M, Legendre C, Anglicheau D: Pathogenesis of non-HLA antibodies in solid organ transplantation: Where do we stand? Hum Immunol 77: 1055–1062, 2016 [DOI] [PubMed] [Google Scholar]
- 11.Gareau AJ, Wiebe C, Pochinco D, Gibson IW, Ho J, Rush DN, et al.: Pre-transplant AT1R antibodies correlate with early allograft rejection. Transpl Immunol 46: 29–35, 2018 [DOI] [PubMed] [Google Scholar]
- 12.Hönger G, Cardinal H, Dieudé M, Buser A, Hösli I, Dragun D, et al.: Human pregnancy and generation of anti-angiotensin receptor and anti-perlecan antibodies. Transpl Int 27: 467–474, 2014 [DOI] [PubMed] [Google Scholar]
- 13.Satchell SC, Tasman CH, Singh A, Ni L, Geelen J, von Ruhland CJ, et al.: Conditionally immortalized human glomerular endothelial cells expressing fenestrations in response to VEGF. Kidney Int 69: 1633–1640, 2006 [DOI] [PubMed] [Google Scholar]
- 14.Loupy A, Haas M, Solez K, Racusen L, Glotz D, Seron D, et al.: The Banff 2015 kidney meeting report: Current challenges in rejection classification and prospects for adopting molecular pathology. Am J Transplant 17: 28–41, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Haas M: C4d-negative antibody-mediated rejection in renal allografts: Evidence for its existence and effect on graft survival. Clin Nephrol 75: 271–278, 2011 [DOI] [PubMed] [Google Scholar]
- 16.Hönger G, Wahrmann M, Amico P, Hopfer H, Böhmig GA, Schaub S: C4d-fixing capability of low-level donor-specific HLA antibodies is not predictive for early antibody-mediated rejection. Transplantation 89: 1471–1475, 2010 [DOI] [PubMed] [Google Scholar]
- 17.Dragun D, Catar R, Kusch A, Heidecke H, Philippe A: Non-HLA-antibodies targeting Angiotensin type 1 receptor and antibody mediated rejection. Hum Immunol 73: 1282–1286, 2012 [DOI] [PubMed] [Google Scholar]
- 18.Liu C, Kellems RE, Xia Y: Inflammation, autoimmunity, and hypertension: The essential role of tissue transglutaminase. Am J Hypertens 30: 756–764, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhou CC, Zhang Y, Irani RA, Zhang H, Mi T, Popek EJ, et al.: Angiotensin receptor agonistic autoantibodies induce pre-eclampsia in pregnant mice. Nat Med 14: 855–862, 2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Li H, Kem DC, Zhang L, Huang B, Liles C, Benbrook A, et al.: Novel retro-inverso peptide inhibitor reverses angiotensin receptor autoantibody-induced hypertension in the rabbit. Hypertension 65: 793–799, 2015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Fichtner A, Süsal C, Schröder C, Höcker B, Rieger S, Waldherr R, et al.: Association of angiotensin II type 1 receptor antibodies with graft histology, function and survival in paediatric renal transplant recipients. Nephrol Dial Transplant 33: 1065–1072, 2018 [DOI] [PubMed] [Google Scholar]
- 22.Hiemann NE, Meyer R, Wellnhofer E, Schoenemann C, Heidecke H, Lachmann N, et al.: Non-HLA antibodies targeting vascular receptors enhance alloimmune response and microvasculopathy after heart transplantation. Transplantation 94: 919–924, 2012 [DOI] [PubMed] [Google Scholar]
- 23.Banasik M, Boratyńska M, Kościelska-Kasprzak K, Krajewska M, Mazanowska O, Kamińska D, et al.: The impact of non-HLA antibodies directed against endothelin-1 type A receptors (ETAR) on early renal transplant outcomes. Transpl Immunol 30: 24–29, 2014 [DOI] [PubMed] [Google Scholar]
- 24.Gao B, Rong C, Porcheray F, Moore C, Girouard TC, Saidman SL, et al.: Evidence to support a contribution of polyreactive antibodies to HLA serum reactivity. Transplantation 100: 217–226, 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.See SB, Clerkin KJ, Kennel PJ, Zhang F, Weber MP, Rogers KJ, et al.: Ventricular assist device elicits serum natural IgG that correlates with the development of primary graft dysfunction following heart transplantation. J Heart Lung Transplant 36: 862–870, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Butte AJ, Sigdel TK, Wadia PP, Miklos DB, Sarwal MM: Protein microarrays discover angiotensinogen and PRKRIP1 as novel targets for autoantibodies in chronic renal disease. Mol Cell Proteomics 10: M110.000497, 2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li L, Wadia P, Chen R, Kambham N, Naesens M, Sigdel TK, et al.: Identifying compartment-specific non-HLA targets after renal transplantation by integrating transcriptome and “antibodyome” measures. Proc Natl Acad Sci U S A 106: 4148–4153, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Philogene MC, Bagnasco S, Kraus ES, Montgomery RA, Dragun D, Leffell MS, et al.: Anti-angiotensin II type 1 receptor and anti-endothelial cell antibodies: A cross-sectional analysis of pathological findings in allograft biopsies. Transplantation 101: 608–615, 2017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dinavahi R, George A, Tretin A, Akalin E, Ames S, Bromberg JS, et al.: Antibodies reactive to non-HLA antigens in transplant glomerulopathy. J Am Soc Nephrol 22: 1168–1178, 2011 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Giral M, Foucher Y, Dufay A, Van Huyen JP, Renaudin K, Moreau A, et al.: Pretransplant sensitization against angiotensin II type 1 receptor is a risk factor for acute rejection and graft loss. Am J Transplant 13: 2567–2576, 2013 [DOI] [PubMed] [Google Scholar]
- 31.Sutherland SM, Li L, Sigdel TK, Wadia PP, Miklos DB, Butte AJ, et al.: Protein microarrays identify antibodies to protein kinase Czeta that are associated with a greater risk of allograft loss in pediatric renal transplant recipients. Kidney Int 76: 1277–1283, 2009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Bachelet T, Couzi L, Lepreux S, Legeret M, Pariscoat G, Guidicelli G, et al.: Kidney intragraft donor-specific antibodies as determinant of antibody-mediated lesions and poor graft outcome. Am J Transplant 13: 2855–2864, 2013 [DOI] [PubMed] [Google Scholar]
- 33.Halloran PF: Transplantation: Autoantibodies-epiphenomena or biological clues. Nat Rev Nephrol 9: 705–706, 2013 [DOI] [PubMed] [Google Scholar]
- 34.See SB, Aubert O, Loupy A, Veras Y, Lebreton X, Gao B, et al.: Post-transplant natural antibodies associate with kidney allograft injury and reduced long-term survival. J Am Soc Nephrol 29: 1761–1770, 2018 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Porcheray F, Fraser JW, Gao B, McColl A, DeVito J, Dargon I, et al.: Polyreactive antibodies developing amidst humoral rejection of human kidney grafts bind apoptotic cells and activate complement. Am J Transplant 13: 2590–2600, 2013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sun Q, Cheng Z, Cheng D, Chen J, Ji S, Wen J, et al.: De novo development of circulating anti-endothelial cell antibodies rather than pre-existing antibodies is associated with post-transplant allograft rejection. Kidney Int 79: 655–662, 2011 [DOI] [PubMed] [Google Scholar]
- 37.Le Bas-Bernardet S, Hourmant M, Coupel S, Bignon JD, Soulillou JP, Charreau B: Non-HLA-type endothelial cell reactive alloantibodies in pre-transplant sera of kidney recipients trigger apoptosis. Am J Transplant 3: 167–177, 2003 [DOI] [PubMed] [Google Scholar]
- 38.Breimer ME, Rydberg L, Jackson AM, Lucas DP, Zachary AA, Melancon JK, et al.: Multicenter evaluation of a novel endothelial cell crossmatch test in kidney transplantation. Transplantation 87: 549–556, 2009 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.