

The developed acetylated LysargiNase (Ac-LysargiNase), with superior activity and stability, provides complementary ion types compared with trypsin for MS/MS analysis. Based on the two mirror proteases, we developed a novel de novo sequencing algorithm, pNovoM, which performed with higher efficiency and accuracy compared with other software tools.
Keywords: De novo sequencing, Peptide mass fingerprinting, Mass Spectrometry, Protein engineering, Enzyme catalysis*, Ac-LysargiNase, Mirror proteases, Trypsin
Graphical Abstract

Highlights
The developed Ac-LysargiNase showed higher stability and activity than before.
The merged spectra of the mirror peptides achieved nearly complete ion coverage.
pNovoM obviously increased the efficiency and accuracy of peptide sequencing.
The mirror enzymatic strategy achieved precision de novo sequencing on proteome scales.
Abstract
De novo peptide sequencing for large-scale proteomics remains challenging because of the lack of full coverage of ion series in tandem mass spectra. We developed a mirror protease of trypsin, acetylated LysargiNase (Ac-LysargiNase), with superior activity and stability. The mirror spectrum pairs derived from the Ac-LysargiNase and trypsin treated samples can generate full b and y ion series, which provide mutual complementarity of each other, and allow us to develop a novel algorithm, pNovoM, for de novo sequencing. Using pNovoM to sequence peptides of purified proteins, the accuracy of the sequence was close to 100%. More importantly, from a large-scale yeast proteome sample digested with trypsin and Ac-LysargiNase individually, 48% of all tandem mass spectra formed mirror spectrum pairs, 97% of which contained full coverage of ion series, resulting in precision de novo sequencing of full-length peptides by pNovoM. This enabled pNovoM to successfully sequence 21,249 peptides from 3,753 proteins and interpreted 44–152% more spectra than pNovo+ and PEAKS at a 5% FDR at the spectrum level. Moreover, the mirror protease strategy had an obvious advantage in sequencing long peptides. We believe that the combination of mirror protease strategy and pNovoM will be an effective approach for precision de novo sequencing on both single proteins and proteome samples.
De novo sequencing is an alternative approach to identify unknown proteins, post-translational modifications (PTM)1 and amino acid mutations, which deduces peptide sequences directly from tandem mass spectra (MS2) instead of searching reference databases (1, 2). This technique is applicable to protein expression profiling or sequencing pure proteins if no accurate proteome database is available (3–6).
The challenge in de novo sequencing is mainly because of the poor coverage of ion series after the precursor ions fragmented in MS. This is even more complicated as multiple and redundant ion series are present with the high background noises (7, 8). To improve the coverage of ion series, combination of various fragmentation modes, such as collision-induced dissociation (CID)/electron transfer dissociation (ETD) (9, 10), higher-energy collisional dissociation (HCD)/ETD (11, 12), CID/HCD/ETD (13) and electron-transfer/higher-energy collision dissociation (EThcD) (14, 15), were investigated. Although the combination can improve the coverage of ion series, these technologies depend heavily on additional MS equipment, and the low throughput caused by the slow scan speed of ETD is also a limitation (16). In the meantime, several de novo sequencing algorithms have been developed, such as PEAKS (17), PepNovo (18, 19), NovoHMM (20) and pNovo (11, 21), as well as various machine learning approaches, including Open-pNovo (22), Novor (23), UVnovo (24, 25) and DeepNovo (26). Although these existing methods partially improved the performance of de novo sequencing, the precision and throughput were still far lower than those of database search. Muth et al. (27) reported that less than 40% of the full-length peptide sequences were consistent with the results from database search regardless of which de novo sequencing algorithm was tested.
Heck and Overall groups reported the development of sequencing techniques using mirror proteases, such as Lys-N and Lys-C, LysargiNase and trypsin. These proteases can produce complementary mirror peptides with basic amino acid residues (K/R) at their N- and C-termini, which allowed them to generate a decent coverage of b and y ion series (28–30). However, cleaving at N- and C-termini of lysines alone with Lys-N and Lys-C respectively results in long peptides, which makes it difficult to generate a full coverage of ion series with MS (11). Alternatively, the pair of mirror protease LysargiNase (31, 32) and trypsin, which cleaves at both N- and C-termini of lysines and arginines to generate relatively shorter peptides, is potentially more suitable for powerful de novo sequencing (11).
Here, we developed an Ac-LysargiNase with superior activity and stability. The combination of Ac-LysargiNase and trypsin can dramatically improve the coverage of b and y ions, respectively, which allowed us to develop a novel de novo sequencing algorithm, pNovoM. We used these techniques to sequence the full-length light and heavy chains of purified antibodies and achieved 1% FDR at the amino acid level. We applied the same technique to yeast and Escherichia coli proteome samples, and about 50% of the peptides generated from trypsin and Ac-LysargiNase digested samples are paired mirror peptides, 97% of which have full coverage of b or y ion series in mirror spectrum pairs and hence achieved for the first time high-throughput and high-precision de novo sequencing of a complicated proteome sample.
MATERIALS AND METHODS
Plasmids, E. coli and Yeast Strains and Cell Culture
The plasmids, E. coli and yeast strains used in this study are described in supplemental Table S1. The recombinant E. coli strain XPZ1 was constructed to express pro-LysargiNase (32, 39). The strain was cultured at 37 °C in LB medium (0.5% yeast extract, 1% tryptone and 1% NaCl). The two monoclonal antibodies (PXL1 and PXL2) were expressed in Chinese hamster ovary (CHO) cells. The yeast strain JMP024 was cultured at 30 °C in YPED medium (1% yeast extract, 2% Bacto-peptone and 2% dextrose) and was harvested in the early log phase (A600 = 1.5) (33).
Production of LysargiNase and Ac-LysargiNase
The E. coli XPZ1 strain expressing LysargiNase was induced by 1 mm isopropyl β-d-thiogalactoside (IPTG). The clarified supernatant sample was loaded onto a pre-equilibrated HiTrap Chelating HP column (GE Healthcare, Chicago, IL), and the fraction containing pro-LysargiNase was collected. Then, the pooled fraction was buffer-exchanged and activated at 25 °C overnight by adding 10 mm CaCl2. We acetylated LysargiNase with acetic anhydride as previously described (34). Finally, the MWs of purified LysargiNase and Ac-LysargiNase were measured using ESI-TOF 6210 (Agilent, Santa Clara, CA) and Exactive Plus EMR (Thermo Scientific, San Jose, CA), respectively (35).
Proteolytic Activity Assessment
The purified proteins or proteome samples were used as substrates to assess the proteolytic activity of LysargiNase and Ac-LysargiNase. The purified proteins included BSA, azocasein and antibody, whereas the proteome samples included E. coli and yeast TCLs. The samples were digested using LysargiNase and Ac-LysargiNase and then were analyzed using LC-MS/MS. The proteolytic activity was determined based on the substrate of azocasein according to a previously described method (31, 41). The protease activity was measured and compared using the following formulas:
where df = dilution time, and c = starting concentration.
Sample Preparation for LC-MS/MS Analysis
The samples for the LC-MS/MS analysis were either in-gel or in-solution digested, as previously described (36, 37). The samples were reduced using 5 mm DTT at 60 °C for 15 min and were fully alkylated using 15 mm IAA at RT in the darkness for 30 min. For LysargiNase digestion, the samples were solubilized in buffer (20 mm HEPES, 0.05% RapiGest, and 10 mm CaCl2, pH 8.3) with a final ratio of 1:100 (weight ratio of protease to protein) and incubated at 37 °C overnight. The digested peptides were desalted using StageTip (36, 38). The MS analysis was performed using LTQ-Orbitrap Velos (Thermo Electron, San Jose, CA) as previously described (36, 38). The resulting peptides were resolved in buffer A (2% acetonitrile in 0.2% formic acid) and loaded onto an in-house-packed capillary column (75 μm i.d. × 15 cm) with 3-μm C18 beads (Michrom Bioresources, Inc., Auburn, CA). The LC separation was performed with a 60-min linear gradient from 2% to 40% buffer B (98% acetonitrile in 0.2% formic acid) at a flow rate of 0.3 μl/min. The MS1 were acquired in the Orbitrap analyzer with a survey scan (300–1600 m/z) at a resolution of 30,000 at 400 m/z. The automatic gain control (AGC) was set as 1e6 with max injection time (MIT) of 150 ms. The MS2 product ions were detected in Orbitrap with a resolution of 7500 at 400 m/z and fragmented in the HCD mode with normalized collision energy of 40%. The top 10 most intense ions were sequentially detected with a target value of 5e4 and the MIT allowed ion accumulation times were 50 ms. The dynamic exclusion time was set to 30 s. For all full scan with the Orbitrap, a lock-mass ion from ambient air (445.120024 m/z) was used as an internal calibrant.
Mirror-spectra-finding Algorithm of pNovoM
To compare the retention time of the mirror peptides, the samples derived from the digestion of trypsin and Ac-LysargiNase were parallelly analyzed through MS without off-line separation. The retention time was recorded for 3,235 mirror peptide pairs. In addition, 22 of the same peptides identified on both of trypsin and Ac-LysargiNase digested samples were used to calibrate the systematic shift between these two different LC-MS/MS analysis. The retention time differences were always maintained at 0.0 ± 4.0 min throughout the entire experiment (supplemental Table. S14), suggesting that these mirror peptide pairs could not be fragmented into the same MS2 spectrum.
To determine the mirror pairs, all the spectra of the trypsin and Ac-LysargiNase data sets were first de novo sequenced by pNovo+ (11). For each trypsin spectrum, all Ac-LysargiNase spectra whose precursor masses fitted the rules in supplemental Table S2 were found, and then the top one sequence of each Ac-LysargiNase spectrum was matched in the trypsin spectrum. The sensitivities and precisions of the mirror-spectra-finding algorithm with different score thresholds were tested (supplemental Fig. S15). When the matched score was higher than 10, the sensitivities and precisions were both higher than 90% on both the yeast and E. coli data sets. Therefore, two sequences were considered a mirror pair if the matched score was higher than 10.
De Novo Sequencing Algorithm of pNovoM
Given a mirror spectrum pair, some mathematical notations were defined to simplify the following statement. P and Q were defined as the masses of the precursor ions in the trypsin and Ac-LysargiNase spectra, respectively. Pp = [mp, ip] was defined as the p-th peak in the trypsin spectrum with mass mp and intensity ip. Qq = [mq, iq] was defined as the q-th peak in the Ac-LysargiNase spectrum. Each peak Pp in the trypsin spectrum was considered to determine whether there was one peak Qq in the Ac-LysargiNase spectrum whose mass conformed to any one of the following formulas. (Note: in the following formulas, 128, 156, and 1 denote the exact masses of a lysine residue, arginine residue and proton, respectively)
| (Eq. 1) |
| (Eq. 2) |
| (Eq. 3) |
| (Eq. 4) |
| (Eq. 5) |
| (Eq. 6) |
| (Eq. 7) |
| (Eq. 8) |
Pp and Qq were called reliable peaks if their masses conformed to any one of these eight formulas, and all other peaks were called common peaks. The ion types (b or y ion) of Pp and Qq could also be judged according to their relationship shown in supplemental Table S3. All peaks, including the reliable peaks and the common peaks in the trypsin and Ac-LysargiNase spectra, were merged together to construct a spectrum graph. The reliable peaks Pp and Qq were merged into one reliable node ps = [ms, is]. The mass ms depended on the ion type of Pp. If Pp was a b ion, then ms = mp − 1; otherwise, ms = P − mp. The intensity is was the sum of the intensities of the two peaks Pp and Qq. Each common peak was converted to two common nodes using the following method. The common peak Pp in the trypsin spectrum was converted to two nodes pr = [mr, ir] and pt = [mt, it], in which mr = mp − 1, mt = P − mp and ir = it = ip. The common peak Qq in the Ac-LysargiNase spectrum was also converted to two nodes qr = [mr, ir] and qt = [mt, it], in which mr = mq − d − 1, mt = Q – mq − d and ir = it = iq (note that d was 128 if the first amino acid in the Ac-LysargiNase spectrum was lysine and 156 otherwise). A source node and a destination node were added, whose masses were set as zero and P – w, respectively, where w denoted the summed masses of a water molecule and a proton. The weights of the two vertices were both set as zero.
In the first step, only the reliable nodes were considered. For each pair of reliable nodes u and v, if the mass difference (<500 Da) was equal to the mass of several amino acid combinations within the fragment ion tolerance, a directed edge was added from u to v. The weight of the edge was the weight of node v. The longest M paths from the source node to the destination node were computed using the pDAG algorithm (11). The numbers of the longest paths were analyzed by considering the candidate numbers and the sensitivities. As shown in supplemental Fig. S16, the default value of M was set as 2 because the average candidate number from the top two paths divided by that from the top one path was only 3.7, and the sensitivity of the correct peptides when considering the top two paths could be as high as 96.6%.
For each path, all the nodes in this path were used to split the spectrum graph into several subgraphs. For the subgraph in the mass interval [mi, mi+1], all common nodes whose masses were in this mass interval were considered, and the highest intensity nodes were retained. The longest N paths from mi to mi+1 were computed using the pDAG algorithm (11). For each subgraph, T candidate subsequences were retained.
All subsequences created by the subgraphs in one path were connected as one candidate sequence. The number of all candidate sequences was M × TP, where M was the number of whole paths, T was the number of subsequences in one subgraph, and P was the number of subgraphs in one whole path. All candidate sequences were scored against the two spectra using the scoring algorithm in pNovo+ (11), and then the two scores were summed together for the final score.
Method of Obtaining All Amino Acid Permutations
To effectively determine whether two nodes should be connected by an edge in the spectrum graph, all amino acid permutations whose masses were less than 500 Da were enumerated and inserted into a mass hash table (supplemental Fig. S17). The mass hash table had two arrays of A and B. Each item in array A restored one amino acid permutation whose mass was transformed into an integer by multiplying it by 1000. Each item in array B occupied 8 bytes (64 bits), where the first 4 bytes restored the index in A and the last 4 bytes restored the number of amino acid permutations. As shown in supplemental Fig. S17, three amino acid permutations, “DDE”, “DED” and “EDD” had the same mass of 359,096 mDa, assuming that the mass deviation was 5 mDa. Then, all elements in the mass interval (359,091 to 359,101) (mDa) in B contained the same item: 156 (the index in A) and 3 (the number of amino acid permutations). Thus, given a mass m and mass deviation Δm, all amino acid permutations whose masses were within the mass interval [m−Δm, m+Δm] could be obtained in O(1) time, and the total memory of the mass hash table was only 34.6 MB.
Parameters of MS Analysis for Database Searching and De Novo Sequencing
All raw files were searched using pFind (39, 40, 42) (version 3.1.2) for peptide and protein identification. The MS2 spectra of the yeast and E. coli data sets were searched against the UniProt yeast database (released in 2015–01, 6,726 protein entries) and the UniProt E. coli database (released in 2015–08, 4,319 protein entries), respectively. The MS2 spectra of Huesgen et al. (32, 45) (referred to as HDS) and those of Tsiatsiani et al. (30, 37, 45) (referred to as LDS) were searched against the UniProt human database (released in 2014–10). All three different databases were appended with 286 common contaminant protein sequences. For yeast, E. coli and LDS, both precursor and fragment mass tolerance were set as ±20 ppm and the open search mode was set to automatically discover highly abundant modifications that existed in the corresponding data sets. For HDS, precursor and fragment mass tolerance were set as ±20 ppm and ±0.5 Da, respectively. Carbamidomethylation of cysteine was set as a fixed modification, whereas oxidation of methionine and acetylation of N termini were set as variable modifications. The maximum number of modification sites on one peptide sequence was set as 3. After the database search, peptides were filtered using an FDR of 1% at the spectrum level, whereas proteins were filtered with an FDR of 1% at the protein level (41). When computing the protein FDR, only the peptides that uniquely matched to the corresponding proteins (both target and decoy proteins) were retained. The protein FDR was calculated by the number of decoy proteins divided by the number of target proteins.
For de novo sequencing with the three high-resolution data sets, yeast, E. coli and LDS, the tolerance of precursor ions was ±20 ppm for pNovoM, pNovo+ and PEAKS (v7.5). The tolerance of MS2 ions was ±20 ppm for pNovoM and pNovo+ and ±0.02 Da for PEAKS. Carbamidomethylation of cysteine was considered as the fixed modification, and oxidation of methionine was considered as the variable modification for all three algorithms.
RESULTS
Superior Activity and Stability of the Newly Developed Acetylated-LysargiNase
LysargiNase, which improves protein C-terminal and methylation-site identification, was reported by Huesgen et al. from Overall group in 2015 (32, 33). To further explore the utility of this protease, we inserted a DNA coding sequence which codes pro-LysargiNase protein (342 amino acids) and a 6× Histidine-tag at this protein's N termini into pET28a to generate the expression vector pXPZ1 (supplemental Fig. S1 and supplemental Table S1). After transforming the pXPZ1 into E. coli BL21 (DE3) and induced by IPTG, the high-purity LysargiNase was obtained through purification and activation processes (Fig. 1A). The experimental molecular weight (MW) of the LysargiNase was 29,006.0 Da (Fig. 1B), which was essentially the same as its theoretical average MW (29,005.3 Da), confirming the purified LysargiNase is what we expected.
Fig. 1.

Higher activity and stability of newly developed Ac-LysargiNase. A, SDS-PAGE analysis of the purified wild-type LysargiNase (WT) and its Acetylated form (Ac-LysargiNase). 15% SDS-PAGE was used. B, C, The MWs of LysargiNase and Ac-LysargiNase were determined with mass spectrometry. D, Self-digestion of LysargiNase and Ac-LysargiNase. The arrow indicates a chromatograph peak for an autolyzed peptide KIPVVVH of LysargiNase and Ac-LysargiNase. E, Intensity of the same autolyzed peptide from LysargiNase and Ac-LysargiNase. The digestion time is shown as indicated. F, Comparison of the activity of LysargiNase and Ac-LysargiNase using yeast TCLs as substrate. The ratios of protease to substrate are shown as indicated. The samples with an asterisk below were analyzed using LC-MS/MS. G, Comparison of protein identification and self-digestion with the same yeast TCL digested using LysargiNase and Ac-LysargiNase. The same samples were concurrently analyzed as indicated by the asterisk in panel f.
To improve the proteolytic activity and stability of the LysargiNase, we acetylated LysargiNase (Ac-LysargiNase) according to our previous study (34). The LC-MS/MS analysis showed an envelope of the isotope peaks for molecular weights of the modified LysargiNase. The mass difference between every two adjacent isotope peaks was ∼42 Da (Fig. 1C), which is the mass shift for acetylation, indicating the successful acetylation on LysargiNase. Then, we compared the stability of LysargiNase and Ac-LysargiNase. The results showed that the intensity of autolyzed peptide (KIPVVVH) of LysargiNase was much higher than that of Ac-LysargiNase after 18 h of self-digestion with the same amount of proteases (Fig. 1D–1E). The numbers of peptide-spectrum matches (PSMs) and peptides digested by LysargiNase were 214 and 101, respectively, whereas those digested by Ac-LysargiNase were 129 and 63, respectively (supplemental Fig. S2). These results suggested that the acetylation of LysargiNase effectively protected itself from self-digestion. More importantly, we evaluated the digestion efficiency of Ac-LysargiNase and LysargiNase using a total cell lysate (TCL) of yeast. The activity of Ac-LysargiNase was slightly higher than that of LysargiNase as the residual bands in the Ac-LysargiNase-digested sample were less intense (Fig. 1F). In an LC-MS/MS analysis of these digested samples with a 1:50 protease-to-substrate ratio, slightly more peptides and proteins were identified in the same sample digested by Ac-LysargiNase than those digested by LysargiNase. Consistently, one autolyzed peptide was detected in the LysargiNase data set, but it did not exist in Ac-LysargiNase data set (Fig. 1G). These results further confirmed that acetylation of LysargiNase effectively increased the activity and stability of LysargiNase. Therefore, Ac-LysargiNase was selected as the mirror protease of trypsin in this study.
Mirror Protease Strategy for De Novo Peptide Sequencing
Traditional de novo sequencing with tryptic peptides suffers from poor coverage of ion series, especially for N-terminal amino acids with low masses. For example, the order of the two N-terminal amino acids, GL or LG, cannot be determined in the trypsin spectrum because of the loss of y6 (Fig. 2A). In addition, the types of all fragment ions are unknown. However, when an Ac-LysargiNase spectrum was introduced as the mirror of the trypsin spectrum, the b2 ion could be used to determine the order of the amino acid residues as GL instead of LG at the N termini. Furthermore, the types of most ions in both spectra could be deduced. For example, the ion types of y5 in the trypsin spectrum and b3 in the Ac-LysargiNase spectrum could be easily determined according to their masses and the mass of the precursor ion (Online Methods and supplemental Table S3).
Fig. 2.

Mirror protease strategy for de novo sequencing. A, The mirror spectra improved the quality of the matched peptide-spectrum pairs by providing a continuous and complete set of product ions and distinguishable directions of most ions (marked with blue circles, e.g. vertices 2, 3, 4 and 5). Two dotted peaks denote that b1 and y6 were missing in the trypsin spectrum. B, Workflow of the pNovoM algorithm based on the mirror protease strategy for de novo sequencing. Mirror spectrum pairs from trypsin- and Ac-LysargiNase-digested samples were found and sequenced using pNovoM. Then, the two spectra in each pair were integrated into one merged spectrum graph for de novo sequencing.
To apply this feature, a mirror peptide pair was defined as two peptides of the form A1A2 … Al[K/R/-] and [K/R/-]A1A2 … Al from trypsin and Ac-LysargiNase digestion, respectively, in which Ai (1 ≤ i ≤ l) denotes any one of the 20 amino acids, and “-” denotes the absence of amino acids (supplemental Table S2). For example, GLEWVAR/KGLEWVA and GLEWVAR/GLEWVA are two mirror peptide pairs. A mirror spectrum pair was defined as two spectra that are respectively matched with a mirror peptide pair.
Based on these two unique advantages of the mirror protease strategy, including nearly full coverage of ion series and unambiguous ion types (Fig. 2A), we proposed a novel de novo peptide sequencing algorithm, pNovoM (Fig. 2B). Given two data sets generated from trypsin- and Ac-LysargiNase-digested samples, the mirror spectrum pairs can be identified using the masses of their precursor and product ions. Then, the two pieces of the mirror spectrum pair can be integrated into a merged spectrum for de novo sequencing. A detailed description of pNovoM is provided in the Online Methods.
Precision De Novo Sequencing of Purified Proteins Using the Mirror Protease Strategy
To evaluate the performance of pNovoM using the mirror protease strategy, we analyzed two monoclonal antibodies, PXL1 and PXL2 (supplemental Fig. S3). The same amounts of proteins were digested with trypsin and Ac-LysargiNase (Fig. 3A) individually before LC-MS/MS analysis. The specificities of these two proteases were further investigated and the result showed that the percentages of peptides specifically digested by trypsin and Ac-LysargiNase were 98 and 89%, respectively (Fig. 3B). The percentages of peptides with at most one missed cleavage were both as high as 97%.
Fig. 3.

Superior precision de novo sequencing of monoclonal antibody PXL1 based on the mirror protease strategy. A, Workflow of the analysis of two antibody data sets (PXL1 and PXL2) generated from trypsin- and Ac-LysargiNase-digested samples. B, Comparison of the specificity and miss cleavage of the spectra sequenced from trypsin or Ac-LysargiNase-digested antibody PXL1. C, The distribution of ion coverage of trypsin, Ac-LysargiNase and mirror spectra. D, Identification rates of the spectra for each peptide from pNovoM, pNovo+ and PEAKS. The blue numbers indicate the average percentage of the correctly sequenced mirror spectrum pairs, and the blue bar chart denotes the number of mirror spectrum pairs for each mirror peptide pair. E, The peptides sequenced by three algorithms on the heavy and light chains of PXL1. The three different color lines were the results of pNovoM (green), pNovo+ (blue) and PEAKS (red). The asterisks denote incorrect residues. The false amino acids matching was listed as the number showed.
pFind (40, 42) was used to analyze these MS2 data (parameters are shown in Online Methods). A total of 2,621 mirror spectrum pairs were found according to the spectrum pairing rules shown in supplemental Table S2. These 2,621 mirror spectrum pairs corresponded to 59 mirror peptide pairs (supplemental Spectra A), and more importantly, 56 of 59 (95%) mirror peptide pairs exhibit the full (100%) coverage of ion series in their mirror spectrum pairs (Fig. 3C), whereas the corresponding figures for those from trypsin or Ac-LysargiNase alone were only 28 (47%) and 25 (42%), respectively.
All the 2,621 mirror spectrum pairs were further analyzed using pNovoM. The same number of spectra from trypsin-digested sample were analyzed using pNovo+ and PEAKS (v7.5) (Fig. 3A), which represented the traditional de novo sequencing. The success rates were 87%, 54% and 57% using pNovoM, pNovo+ and PEAKS, respectively (Fig. 3D). To further validate the de novo sequencing results, the sequences provided by these algorithms were compared with the actual sequences which are known. As shown in Fig. 3E, pNovoM reported four mismatched amino acid annotations in two peptides of PXL1. The FDR was 1.0% (4/416) at the amino acid level and 6.7% (2/30) at the peptide level. In contrast, the numbers of mismatched amino acid annotations reported by pNovo+ and PEAKS for the same PXL1 were 18 and 19, respectively, which were ∼3- to 4-fold higher than that for pNovoM. The FDRs for amino acids were 4.3% and 4.6%, and the corresponding FDRs for peptides were 23.3% (7/30) for pNovo+ and 20.0% (6/30) for PEAKS. Similar results were achieved using another antibody, PXL2 (supplemental Fig. S4). These results indicate that pNovoM coupled with the mirror protease strategy can provide higher precision sequencing results for purified proteins than pNovo+ and PEAKS coupled with the trypsin protease alone.
To examine the mechanism underlying the high precision of mirror protease strategy and pNovoM, we investigated all 15 mismatched peptides reported by these three algorithms (the asterisks in Fig. 3E). Among them, as many as 13 mismatched peptides (87%) were because of N-terminal ambiguity, which was caused by the missing b ions in the low-mass region of traditional MS2 spectra of tryptic peptides. It was even worse when a significant number of background noise interfered with the correct backbone ions (21, 43, 34). However, based on the mirror spectrum pairs, 11 or 85% of these 13 peptides could be completely sequenced by pNovoM, which demonstrated that the mirror protease strategy provided more precise sequencing results, especially for the N termini of peptides.
High Precision De Novo Sequencing of Complex Proteomes Using Mirror Protease Strategy
We applied the mirror protease strategy and pNovoM to TCLs of yeast (Fig. 4), E. coli (supplemental Fig. S5) and human tissue (supplemental Note 3). There were a total of 251,375 spectra sequenced from the tryptic TCL of yeast and 257,478 spectra from the same TCL of yeast digested with Ac-LysargiNase. Totally, 839,244 mirror spectrum pairs from 140,580 (55.9%) trypsin spectra and 131,315 (51.0%) Ac-LysargiNase spectra were found by pNovoM (Fig. 4A). Among them, one trypsin spectrum could be paired with ∼6.0 Ac-LysargiNase spectra and one Ac-LysargiNase spectrum could be paired with ∼6.4 trypsin spectra on average. All the mirror spectrum pairs were analyzed using pNovoM, whereas the trypsin spectra that could not be paired were analyzed via pNovo+ (Fig. 4A). To compare the performance of pNovoM with existing de novo sequencing algorithms such as PEAKS and pNovo+, all the trypsin spectra were analyzed using these algorithms. We used the results from database search engine pFind (40) as benchmark to evaluate the performances of different de novo sequencing algorithms (Online Methods).
Fig. 4.

Proteome-level de novo sequencing of yeast based on the mirror protease strategy. A, Workflow for the de novo sequencing and FDR evaluation at the proteome level. B, FDR curves of pNovoM, pNovo+ and PEAKS at the spectrum level. C, Venn diagram of the spectra sequenced with pNovo+, PEAKS and pNovoM at 5% FDR at the spectrum level. The red numbers in parentheses denote the FDRs of the corresponding parts. D, FDR curves of pNovoM, pNovo+ and PEAKS at the peptide level. E, Venn diagram of the peptides sequenced using pNovo+, PEAKS and pNovoM at 5% FDR at the peptide level.
Compared with the benchmark results from pFind, the results from pNovoM could be divided into three classes: (I) the PSMs sequenced by pNovoM did not exist in the pFind results using 1% FDR threshold at the spectrum level; (II) the PSMs sequenced by pNovoM existed in the pFind results but mismatched; and (III) the PSMs sequenced by pNovoM matched those in the pFind results. All PSMs were sorted in descending order, and the FDR at the spectrum level was estimated as . When estimating the FDR, the class I results were not considered because we could not discriminate between the correct and incorrect results in this part. Only the best PSM for the same peptide was retained when estimating the FDR at the peptide level. The results from pNovo+ and PEAKS on trypsin spectra alone were analyzed using the same approach.
The FDR of the pNovoM results at the spectrum level was 25%, whereas the corresponding FDRs were 41% and 48% for pNovo+ and PEAKS, respectively, suggesting the superiority of pNovoM in analyzing complicated proteome samples (Fig. 4B). Consistently, the distribution curves showed that pNovoM reported significantly more spectra than those of the other two algorithms at any FDR threshold applied. For example, pNovoM interpreted 38,001 spectra at 5% FDR at the spectrum level, which corresponded to 21,249 peptides from 3,753 proteins. The numbers of spectra detected by pNovoM were 44.4% and 151.6% more than those obtained from pNovo+ (26,325) and PEAKS (15,105), respectively (Fig. 4B). We further investigated the overlap of the correct PSMs from all three algorithms. pNovoM covered 98.1% of the PSMs detected by pNovo+ and 93.5% of those detected by PEAKS (Fig. 4C). On the other hand, pNovoM sequenced 11,126 unique spectra at 5% FDR, which was quite close to the total number of 15,105 spectra sequenced by PEAKS, suggesting an advantage of pNovoM for large-scale de novo sequencing. More importantly, although the FDR was set at 5% in every method, the consistently sequenced spectra were more credible because the actual FDR of this part was only 2.7%, which was remarkably lower than the threshold of 5% FDR. In contrast, FDRs of the spectra sequenced uniquely by pNovo+ and PEAKS were both over 20%, whereas that of pNovoM was 5.3%, which was very close to the threshold of 5% FDR (Fig. 4C). Further analysis showed that the high precision of the uniquely sequenced spectra from pNovoM may be because of the high coverage of b ions, which was mainly contributed from Ac-LysargiNase (supplemental Fig. S6). Consistently, the FDR of pNovoM was also significantly lower than those of pNovo+ and PEAKS at the peptide level (Fig. 4D). Although the FDRs of the uniquely sequenced peptide subgroup were slightly higher than those of the corresponding spectra parts, pNovoM still reported the most peptides with the lowest FDR (Fig. 4E). These results emphasize that pNovoM can be applied to proteome-level de novo sequencing. However, neither pNovo+ nor PEAKS can be utilized for this purpose, owing to their significantly higher error rates.
Near-complete Ion Coverage from the Complementary Effects of b and y Ion Series Enables the Precision De Novo Sequencing
Because de novo sequencing strongly depends on the coverage of ion series, we were curious about the ion coverage of the peptide/spectrum from trypsin, Ac-LysargiNase and the mirror pair formed from these two proteases. In the yeast data set, we identified 16,514 mirror peptide pairs via pFind (40), which was used to create the benchmark (supplemental Note 1 containing supplemental Figs. S7 and S8). The average coverage of b and y ions was 40.0% and 91.7%, respectively, in the trypsin-digested sample and 87.5% and 33.3%, respectively, in the Ac-LysargiNase-digested sample (Fig. 5A). After merging the spectra to form the mirror spectrum pairs, the average coverage of both b and y ions was increased to 90.0% and 92.3%, respectively (Fig. 5A). The b ion intensity increased 3.9-fold because of the application of Ac-LysargiNase, which increased b-to-y ion intensity ratio from 0.3 (2−1.6) in the trypsin spectra to 0.7 (2−0.6) in the mirror spectrum pairs (Fig. 5B). Impressively, we found that the percentage of mirror spectrum pairs with complete ion coverage was as high as 97%, but was only ∼50% in either trypsin or Ac-LysargiNase spectra alone (Fig. 5C). These results strongly supported that nearly all mirror spectrum pairs covered full set of b and y ions.
Fig. 5.
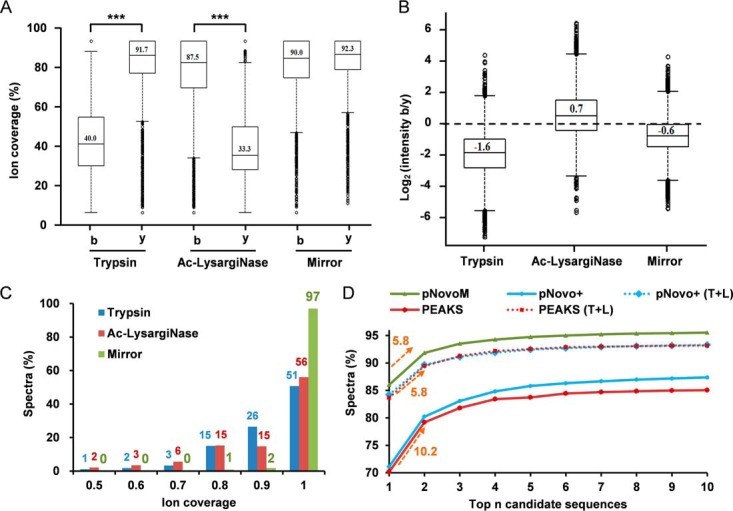
Mirror spectrum pairs achieved near-complete ion coverage. A, Percentages of matched b and y ions in trypsin, Ac-LysargiNase and mirror spectra. B, The distributions of the intensities of the matched b and y ions in trypsin, Ac-LysargiNase and mirror spectra. C, The distributions of ion coverage in trypsin, Ac-LysargiNase and mirror spectra. D, In all 16,514 mirror spectrum pairs, the cumulative curves of the number of correctly matched spectra from top one to top ten candidates of pNovoM, pNovo+, pNovo+ (T+L), PEAKS and PEAKS (T+L). (T+L) denotes that both trypsin and Ac-LysargiNase spectra were considered. Numbers below the arrows indicate the increase from the top one sensitivity to the top two sensitivity.
The 16,514 mirror spectrum pairs were then sequenced using pNovoM, whereas the trypsin spectra alone were sequenced using pNovo+ and PEAKS. For each algorithm, the top ten matched candidate peptide sequences of each spectrum were retained. The database search results were considered as the ground truth for evaluating the sensitivity of these three de novo algorithms. As shown in Fig. 5D, the sensitivity of the top one candidates was only ∼70% for either PEAKS or pNovo+, whereas the sensitivity of pNovoM was as high as 86.0%. Moreover, the difference between the top one sensitivity and the top two sensitivity was 10.2% for both pNovo+ and PEAKS, but was only 5.8% for pNovoM, suggesting that pNovoM is more powerful to distinguish the top two candidates. This is a unique advantage of pNovoM compared with traditional approaches.
We have also tested pNovo+ and PEAKS using the same data set of mirror spectrum pairs. Both the trypsin and Ac-LysargiNase spectra were analyzed with pNovo+/PEAKS as well. Each mirror spectrum pair was assigned two sets of candidate sequences (one from a trypsin spectrum and the other from an Ac-LysargiNase spectrum). The mirror spectrum pair was considered correctly interpreted when either one best peptide from the two sets matched to the database search results, then the sensitivities of both pNovo+ and PEAKS increased to ∼84%. However, even with this optimistic estimation, the sensitivity of either pNovo+ or PEAKS was still ∼2.5% lower than that of pNovoM (Fig. 5D). This result further demonstrated that both the mirror spectrum pairs and the pNovoM algorithm contributed to the precision de novo sequencing at the proteome level. The data set generated from the TCL of E. coli provided a similar result, which supported this conclusion as well (supplemental Fig. S9).
Mirror Protease Strategy for Precision De Novo Sequencing on Long Peptides
Previous studies have reported that the ion coverage negatively correlated with the length of peptides, which indicates a challenge to sequence long peptides (11, 44, 45). We investigated the impact of peptide length on de novo sequencing. The consistency analysis of all correct peptides sequenced using pNovoM, pNovo+ and PEAKS was performed in yeast (Fig. 6A) and E. coli (supplemental Fig. S10A). As shown in Fig. 6A, there were 10,009 peptides sequenced by all three approaches, which represented 70.5% of all peptides sequenced by pNovoM. We investigated the length distributions of the 10,009 consistently sequenced peptides and the 1,930 uniquely sequenced peptides of pNovoM (Figs. 6B and supplemental S10B). Clearly, the length distribution of the uniquely sequenced peptides was skewed to the right side compared with that of the consistently sequenced peptides. The result showed that 15.2% of the peptides uniquely sequenced by pNovoM have more than 18 amino acids. However, only 3.2% of the consistently sequenced peptides have more than 18 amino acids. This result suggested that pNovoM showed unique advantage in sequencing long peptides over the other two algorithms. To further reveal the mechanism of mirror spectrum pairs on de novo sequencing of long peptides, we also studied the ion position and the matched ion ratio of the identified peptides. As shown in Fig. 6C, if the trypsin spectra alone were considered, the b ions near the N termini of peptides and the y ions near the C-termini of peptides (ions with low masses) were easy to be detected, whereas ions in the central part of long peptides (ions with high masses) were more likely to be missed. This resulted in that only short peptides can be sequenced if only trypsin alone was used.
Fig. 6.

Mirror protease strategy sequenced more long peptides and increased the ion coverage, especially for the N termini of peptides. A, Venn diagram of the correct peptides sequenced using pNovoM, pNovo+ and PEAKS. B, The distributions of the length of the peptides consistently sequenced using all three algorithms and uniquely sequenced using pNovoM. C, Relationship between the ion position and the matched ion ratio of the mirror peptide pairs (length ≤ 30). D, The distributions of the N- and C-terminal-most three-product ions for the 1,930 peptides uniquely sequenced using pNovoM. The blue numbers denote the percentage of the spectra without any ions at the corresponding ion position.
A previous study showed that b1 ions could easily be missed in HCD spectra of tryptic peptides (46). Therefore, the order of the first two N-terminal amino acids could not be determined if the yn-1 ions (n was the length of the peptide) were not detected. We also investigated the match between the ions of the N- (leftmost) and C-terminal (rightmost) three amino acids of the 1,930 uniquely sequenced peptides using pNovoM (Fig. 6D). Consistently, there were ∼79.1% of spectra in which both b1 and yn-1 ions missed in our tryptic data sets (38). However, the missing rate of b1 and yn-1 ions dropped to 7.6% if the mirror protease strategy was adopted (Fig. 6D). These results strongly indicated that the Ac-LysargiNase spectra could provide enough complementary ions to enable the precision de novo sequencing.
DISCUSSION
Precision de novo peptide sequencing is hindered by poor coverage of b and/or y ion series. In this study, we developed Ac-LysargiNase, which showed higher activity and stability than the commonly used LysargiNase. Ac-LysargiNase provides not only the better coverage and stronger signal of b ions compared with tryptic peptides, but also can work with trypsin to create a complementary ion series which can nearly cover all amino acid residues of sequenced peptides. The types of these ion series generated independently from Ac-LysargiNase and trypsin can be examined by each other. More importantly, the mirror spectrum pairs provide more N-terminal ions to discriminate N termini amino acid residues of peptides. Based on these characteristics, a novel de novo peptide sequencing algorithm, pNovoM, was designed to merge the mirror spectrum pairs, which made de novo sequencing possible. This new technology can be applied not only to purified proteins but also to complicated proteome samples.
Superior activity and stability of LysargiNase are the keys to generate paired mirror peptides for de novo sequencing in this study. As the original LysargiNase could cleave dimethylated but not acetylated lysine residues (31, 32), we developed the acetylated LysargiNase (Ac-LysargiNase) to avoid autolysis. As expected, the acetylation significantly improved the stability and activity of original LysargiNase. This result was further validated by analyzing the data sets generated by Huesgen et al. (32, 39) and Tsiatsiani et al. (30, 39) (supplemental Note 2 containing supplemental Figs. S18, S19, and S20). There were fewer identified peptides from the data sets digested by LysargiNase than those digested by Ac-LysargiNase if the peptides identified in the corresponding data sets digested by trypsin were used as standard.
Different collision modes can also provide more fragment ions in MS2, which may play a similar role to the mirror protease strategy in de novo sequencing. We studied the ion coverage of trypsin or LysargiNase spectra produced with HCD or ETD by Tsiatsiani et al. (30, 39). In the combination of b ions and c ions from HCD and ETD spectra of tryptic peptides (supplemental Fig. S11A), the coverage of N-terminal ions was only 60%, whereas that of the C-terminal ions was close to 100%. This data suggested that the combination of y and z ions in trypsin HCD and ETD spectra could potentially be applied in the near future in de novo sequencing though the two independent series of ions cannot examine each other as mirror proteases do. Although there is only about 70% of c ion coverage in the ETD spectra of LysargiNase digested peptides (supplemental Fig. S11B), there are almost no z ions in the same spectra, which was also previously confirmed by Lys-N spectra in ETD reported by Taouatas et al. (28). More specifically, the N-terminal ion coverage was only 60% in LysargiNase spectra and the C-terminal ion coverage was only 29% in trypsin spectra, suggesting that the HCD/ETD could not generate a good coverage of N- or C-terminal ions in either trypsin or LysargiNase spectra. However, in our mirror protease data sets, both the N- and C-terminal ion coverage was greater than 90%, indicating that the mirror protease strategy could provide nearly full coverage of N- and C-terminal ions compared with the HCD/ETD strategy.
To further investigate the impact of different collision dissociation and mirror protease strategy on de novo sequencing, we also analyzed the public data sets that generated by HCD and ETD on digested TCL of Jurkat T lymphoma cells with trypsin and LysargiNase (30, 39). The parameters described by Tsiatsiani were used for the database search (30). In that data set, each peptide corresponded to one of the four spectra, including trypsin-HCD, trypsin-ETD, LysargiNase-HCD and LysargiNase-ETD (supplemental Fig. S12A–S12C). The sensitivity of the best match (top one) was only 73.6% if only trypsin-HCD spectra were considered as in the traditional de novo sequencing mode (supplemental Fig. S12D). Comparing the HCD/ETD strategy (trypsin-HCD/trypsin-ETD) with the mirror protease strategy (trypsin-HCD/LysargiNase-HCD) developed here, the sensitivities of these two strategies were 90.0% and 87.6%, respectively, suggesting the competitive advantage of these two strategies. If all four spectra were included, the sensitivities of the top one and top three candidates were 95.6% and 99.1%, suggesting that the HCD/ETD and the mirror protease strategies were highly complementary. The advantage of combinations of HCD/ETD and mirror proteases in de novo sequencing remains to be studied in the near future to achieve even better performance than the database search approach.
De Novo Sequencing Is Also a Powerful Approach for PTM Studies, e.g. Phosphorylation with Large Scale MS/MS Data
By using the mirror protease strategy, we have found that pNovoM still outperformed pNovo+ and PEAKS on phosphorylation spectra, which proved that the mirror protease strategy and pNovoM software have more advantages on such post-translational modified data (supplemental Note 3). Another application of de novo sequencing in large scale proteomics is to precisely identify single amino acid polymorphism. In the yeast data set, 57 peptides with single amino acid mutations were sequenced by the mirror spectrum pairs, which is significantly higher than 38 found by the trypsin spectra alone (supplemental Spectra B and C). These peptide mutants were manually validated using high-quality PSMs. The amino acid mutation loci, the titles of the corresponding spectra and the protein accession numbers were included in supplemental Tables S4 and S5. The score distributions demonstrated that these mutant peptides sequenced by the mirror spectrum pairs were more accurate than those sequenced by the trypsin spectra alone (supplemental Fig. S13). These results revealed that the mirror protease strategy will be a powerful tool in the detection of mutations and single amino acid polymorphisms in the near future.
It is worth mentioning that the workload doubles to generate the mirror spectra for de novo sequencing. It is also important that the current mirror proteases of Ac-LysargiNase and trypsin only provide sequence information on peptide level. The continuous exploration of other paired mirror proteases that provides the linkers between the sequenced peptides may be necessary for sequencing the whole protein in de novo sequencing technique.
DATA AVAILABILITY
All raw data from the three samples mentioned in this study have been deposited to the ProteomeXChange Consortium via the PRIDE partner repository with the accession numbers PXD008688, PXD008690 and PXD011562. The database search results of all datasets investigated in this study are shown in supplemental Tables S6–S13. pNovoM was developed in the C/C++ language, and a standalone executable file can be accessed at http://pfind.ict.ac.cn/software/pNovoM/index.html.
Supplementary Material
Acknowledgments
We thank Mr. Chen Deng and Ms. Wen-Jing Zhou for their technical support.
Footnotes
* This work was funded by the National Key R&D Program of China (2016YFA0501300, 2017YFC0906600, 2017YFA0505002 and 2015CB910700), the National Natural Science Foundation of China (31870824, 91839302, 31670834 & 31700723), Youth Innovation Promotion Association CAS (No. 2014091), National Megaprojects for Key Infectious Diseases (2018ZX10302302001), the National Natural Science Foundation of Beijing (Grant No. 5152008), Beijing Training Project for The Leading Talents in S&T (Z161100004916024), the Foundation of State Key Laboratory of Proteomics (SKLP-Y201501 & SKLP-K201705), the Innovation Foundation of Medicine (2017CXJJ19, BWS17J032, 16CXZ027 & BWS14J052) and the CAS Interdisciplinary Innovation Team (Y604061000).
 This article contains supplemental material.
This article contains supplemental material.
We declare no competing financial interests.
1 The abbreviations used are:
- Ac-LysargiNase
- acetylated lysargiNase
- PTM
- post-translational modification
- FDR
- false discovery rate
- PSM
- peptide-spectrum match
- LC-MS
- liquid chromatography mass spectrometry
- SDS-PAGE
- SDS-polyacrylamide gel electrophoresis
- ETD
- electron transfer dissociation
- EThcD
- electron-transfer/higher-energy collision dissociation
- CID
- collision-induced dissociation
- HCD
- higher-energy collision dissociation.
REFERENCES
- 1. Ma B., and Johnson R. (2012) De novo sequencing and homology searching. Mol. Cell. Proteomics 11, O111 014902 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Seidler J., Zinn N., Boehm M. E., and Lehmann W. D. (2010) De novo sequencing of peptides by MS/MS. Proteomics 10, 634–649 [DOI] [PubMed] [Google Scholar]
- 3. Bandeira N., Pham V., Pevzner P., Arnott D., and Lill J. R. (2008) Automated de novo protein sequencing of monoclonal antibodies. Nat. Biotechnol. 26, 1336–1338 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Cheung W. C., Beausoleil S. A., Zhang X., Sato S., Schieferl S. M., Wieler J. S., Beaudet J. G., Ramenani R. K., Popova L., Comb M. J., Rush J., Polakiewicz R. D. (2012) A proteomics approach for the identification and cloning of monoclonal antibodies from serum. Nat. Biotechnol. 30, 447–452 [DOI] [PubMed] [Google Scholar]
- 5. Boutz D. R., Horton A. P., Wine Y., Lavinder J. J., Georgiou G., Marcotte E. M. (2017) Proteomic identification of monoclonal antibodies from serum. Anal Chem 86, 4758–4766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Miyashita M., Kitanaka A., Yakio M., Yamazaki Y., Nakagawa Y., Miyagawa H. (2017) Complete de novo sequencing of antimicrobial peptides in the venom of the scorpion Isometrus maculatus. Toxicon 139, 1–12 [DOI] [PubMed] [Google Scholar]
- 7. Pevtsov S., Fedulova I., Mirzaei H., Buck C., and Zhang X. (2006) Performance evaluation of existing de novo sequencing algorithms. J. Proteome Res. 5, 3018–3028 [DOI] [PubMed] [Google Scholar]
- 8. Bringans S., Kendrick T. S., Lui J., and Lipscombe R. (2008) A comparative study of the accuracy of several de novo sequencing software packages for datasets derived by matrix-assisted laser desorption/ionisation and electrospray. Rapid Commun. Mass Sp. 22, 3450–3454 [DOI] [PubMed] [Google Scholar]
- 9. Horn D. M., Zubarev R. A., and McLafferty F. W. (2000) Automated de novo sequencing of proteins by tandem high-resolution mass spectrometry. Proc. Natl. Acad. Sci. U.S.A. 97, 10313–10317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Bertsch A., Leinenbach A., Pervukhin A., Lubeck M., Hartmer R., Baessmann C., Elnakady Y. A., Müller R., Böcker S., Huber C. G., Kohlbacher O. (2009) De novo peptide sequencing by tandem MS using complementary CID and electron transfer dissociation. Electrophoresis 30, 3736–3747 [DOI] [PubMed] [Google Scholar]
- 11. Chi H., et al. (2013) pNovo+: de novo peptide sequencing using complementary HCD and ETD tandem mass spectra. Journal of proteome research 12, 615–625 [DOI] [PubMed] [Google Scholar]
- 12. Yan Y., Kusalik A. J., and Wu F. X. (2015) A Framework of De Novo Peptide Sequencing for Multiple Tandem Mass Spectra. Ieee T Nanobiosci 14, 478–484 [DOI] [PubMed] [Google Scholar]
- 13. Guthals A., Clauser K. R., Frank A. M., and Bandeira N. (2013) Sequencing-grade de novo analysis of MS/MS triplets (CID/HCD/ETD) from overlapping peptides. J Proteome Res 12, 2846–2857 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Frese C. K., et al. (2012) Toward Full Peptide Sequence Coverage by Dual Fragmentation Combining Electron-Transfer and Higher-Energy Collision Dissociation Tandem Mass Spectrometry. Analytical chemistry 84, 9668–9673 [DOI] [PubMed] [Google Scholar]
- 15. Mommen G. P. M., et al. (2014) Expanding the detectable HLA peptide repertoire using electron-transfer/higher-energy collision dissociation (EThcD). PNatl Acad Sci USA 111, 4507–4512 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Sun R. X., et al. (2010) Improved peptide identification for proteomic analysis based on comprehensive characterization of electron transfer dissociation spectra. JProteome Res 9, 6354–6367 [DOI] [PubMed] [Google Scholar]
- 17. Ma B., et al. (2003) PEAKS: powerful software for peptide de novo sequencing by tandem mass spectrometry. Rapid communications in mass spectrometry : RCM 17, 2337–2342 [DOI] [PubMed] [Google Scholar]
- 18. Frank A., and Pevzner P. (2005) PepNovo: de novo peptide sequencing via probabilistic network modeling. Anal Chem 77, 964–973 [DOI] [PubMed] [Google Scholar]
- 19. Frank A. M., Savitski M. M., Nielsen M. L., Zubarev R. A., and Pevzner P. A. (2007) De novo peptide sequencing and identification with precision mass spectrometry. J Proteome Res 6, 114–123 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Fischer B., et al. (2005) NovoHMM: a hidden Markov model for de novo peptide sequencing. Anal Chem 77, 7265–7273 [DOI] [PubMed] [Google Scholar]
- 21. Chi H., et al. (2010) pNovo: de novo peptide sequencing and identification using HCD spectra. Journal of proteome research 9, 2713–2724 [DOI] [PubMed] [Google Scholar]
- 22. Yang H., et al. (2017) Open-pNovo: De Novo Peptide Sequencing with Thousands of Protein Modifications. JProteome Res 16, 645–654 [DOI] [PubMed] [Google Scholar]
- 23. Ma B. (2015) Novor: real-time peptide de novo sequencing software. Journal of the American Society for Mass Spectrometry 26, 1885–1894 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Robotham S. A., et al. (2016) UVnovo: A de Novo Sequencing Algorithm Using Single Series of Fragment Ions via Chromophore Tagging and 351 nm Ultraviolet Photodissociation Mass Spectrometry. Anal Chem 88, 3990–3997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Horton A. P., et al. (2017) Comprehensive de Novo Peptide Sequencing from MS/MSPairs Generated through Complementary Collision Induced Dissociation and 351 nm Ultraviolet Photodissociation. Anal Chem 89, 3747–3753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tran N. H., Zhang X., Xin L., Shan B., and Li M. (2017) De novo peptide sequencing by deep learning. Proc. Natl. Acad. Sci. U.S.A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Muth T., and Renard B. Y. (2017) Evaluating de novo sequencing in proteomics: already an accurate alternative to database-driven peptide identification? Briefings in bioinformatics [DOI] [PubMed] [Google Scholar]
- 28. Taouatas N., Drugan M. M., Heck A. J., and Mohammed S. (2008) Straightforward ladder sequencing of peptides using a Lys-N metalloendopeptidase. Nature methods 5, 405–407 [DOI] [PubMed] [Google Scholar]
- 29. Boersema P. J., et al. (2009) Straightforward and de novo peptide sequencing by MALDI-MS/MS using a Lys-N metalloendopeptidase. Molecular & cellular proteomics : MCP 8, 650–660 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tsiatsiani L., et al. (2017) Opposite Electron-Transfer Dissociation and Higher-Energy Collisional Dissociation Fragmentation Characteristics of Proteolytic K/R(X)n and (X)nK/RPeptides Provide Benefits for Peptide Sequencing in Proteomics and Phosphoproteomics. JProteome Res 16, 852–861 [DOI] [PubMed] [Google Scholar]
- 31. Tallant C., Garcia-Castellanos R., Seco J., Baumann U., and Gomis-Ruth F. X. (2006) Molecular analysis of ulilysin, the structural prototype of a new family of metzincin metalloproteases. J. Biol. Chem. 281, 17920–17928 [DOI] [PubMed] [Google Scholar]
- 32. Huesgen P. F., et al. (2015) LysargiNase mirrors trypsin for protein C-terminal and methylation-site identification. Nat Methods 12, 55–58 [DOI] [PubMed] [Google Scholar]
- 33. Xu P., et al. (2009) Quantitative proteomics reveals the function of unconventional ubiquitin chains in proteasomal degradation. Cell 137, 133–145 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Zhao M., Wu F., and Xu P. (2015) Development of a rapid high-efficiency scalable process for acetylated Sus scrofa cationic trypsin production from Escherichia coli inclusion bodies. Protein expression and purification 116, 120–126 [DOI] [PubMed] [Google Scholar]
- 35. Zhao M. Z., et al. (2016) Recombinant expression, refolding, purification and characterization of Pseudomonas aeruginosa protease IV in Escherichia coli. Protein Expression and Purification 126, 69–76 [DOI] [PubMed] [Google Scholar]
- 36. Gao Y., et al. (2016) Enhanced Purification of Ubiquitinated Proteins by Engineered Tandem Hybrid Ubiquitin-binding Domains (ThUBDs). Molecular & cellular proteomics : MCP 15, 1381–1396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Ding Chen, et al. (2013) AFast Workflow for Identification and Quantification of Proteomes. Molecular & cellular proteomics : MCP 12, 2370–2380 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li Y., et al. (2017) A rapid and easy protein N-terminal profiling strategy using (N-Succinimidyloxycarbonylmethyl)tris(2,4,6-trimethoxyphenyl)phosphonium bromide (TMPP) labeling and StageTip. Proteomics 17 [DOI] [PubMed] [Google Scholar]
- 39. Wang L. H., et al. (2007) pFind 2.0: a software package for peptide and protein identification via tandem mass spectrometry. Rapid communications in mass spectrometry : RCM 21, 2985–2991 [DOI] [PubMed] [Google Scholar]
- 40. Chi H., et al. (2015) pFind-Alioth: A novel unrestricted database search algorithm to improve the interpretation of high-resolution MS/MS data (Reprinted from vol 125, pg 89–97, 2015). Journal of Proteomics 129, 33–41 [DOI] [PubMed] [Google Scholar]
- 41. Elias J. E., and Gygi S. P. (2007) Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nature methods 4, 207–214 [DOI] [PubMed] [Google Scholar]
- 42. Chi H., et al. (2018) Comprehensive identification of peptides in tandem mass spectra using an efficient open search engine. Nat Biotechnol [DOI] [PubMed] [Google Scholar]
- 43. Olson M. T., Epstein J. A., and Yergey A. L. (2006) De novo peptide sequencing using exhaustive enumeration of peptide composition. Journal of the American Society for Mass Spectrometry 17, 1041–1049 [DOI] [PubMed] [Google Scholar]
- 44. Alfaro J. A., et al. (2017) Detecting protein variants by mass spectrometry: a comprehensive study in cancer cell-lines. Genome Med 9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Blank-Landeshammer B., et al. (2017) Combining De Novo Peptide Sequencing Algorithms ASynergistic Approach to Boost Both Identifications and Confidence in Bottom-up Proteomics. JProteome Res 16, 3209–3218 [DOI] [PubMed] [Google Scholar]
- 46. Fu Q., and Li L. J. (2005) De novo sequencing of neuropeptides using reductive isotopic methylation and investigation of ESI QTOF MS/MS fragmentation pattern of neuropeptides with N-terminal dimethylation. Analytical chemistry 77, 7783–7795 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw data from the three samples mentioned in this study have been deposited to the ProteomeXChange Consortium via the PRIDE partner repository with the accession numbers PXD008688, PXD008690 and PXD011562. The database search results of all datasets investigated in this study are shown in supplemental Tables S6–S13. pNovoM was developed in the C/C++ language, and a standalone executable file can be accessed at http://pfind.ict.ac.cn/software/pNovoM/index.html.