

Global quantitative mass spectrometry characterized tumor heterogeneity of lung metastatic site and eight different serially collected progressive metastatic lymph nodes over seven years from an exceptional responder lung adenocarcinoma patient. Specific signaling networks were enriched in lung compared with the lymph node metastatic sites. Fifty-five germline and 6 somatic variant peptides were identified and validated. MRM assays were developed for two novel somatic variant peptides. CDK12-G879V mutant specific to lung metastatic sites resulted in increased chemotherapy sensitivity of lung tumors.
Keywords: Lung cancer, Cancer Biology*, Database design, Multiple reaction monitoring, Mass Spectrometry, Tumor Heterogeneity
Graphical Abstract

Highlights
Serial biopsies and autopsy from a metastatic lung cancer patient over 7 years.
Tumor heterogeneity characterized by quantifying the proteome and phosphoproteome.
Patient-specific database built using whole genome sequencing data from tumors.
MRM assay and functional validation of a novel lung-specific CDK12-G879V mutant.
Abstract
Lung cancer is the leading cause of cancer death in both men and women. Tumor heterogeneity is an impediment to targeted treatment of all cancers, including lung cancer. Here, we sought to characterize tumor proteome and phosphoproteome changes by longitudinal, prospective collection of tumor tissue from an exceptional responder lung adenocarcinoma patient who survived with metastatic lung adenocarcinoma for over seven years while undergoing HER2-directed therapy in combination with chemotherapy. We employed “Super-SILAC” and TMT labeling strategies to quantify the proteome and phosphoproteome of a lung metastatic site and eight distinct metastatic progressive lymph nodes collected during these seven years, including five lymph nodes procured at autopsy. We identified specific signaling networks enriched in lung compared with the lymph node metastatic sites. We correlated the changes in protein abundance with changes in copy number alteration (CNA) and transcript expression. ERBB2/HER2 protein expression was higher in lung, consistent with a higher degree of ERBB2 amplification in lung compared with the lymph node metastatic sites. To further interrogate the mass spectrometry data, a patient-specific database was built by incorporating all the somatic and germline variants identified by whole genome sequencing (WGS) of genomic DNA from the lung, one lymph node metastatic site and blood. An extensive validation pipeline was built to confirm variant peptides. We validated 360 spectra corresponding to 55 germline and 6 somatic variant peptides. Targeted MRM assays revealed two novel variant somatic peptides, CDK12-G879V and FASN-R1439Q, expressed in lung and lymph node metastatic sites, respectively. The CDK12-G879V mutation likely results in a nonfunctional CDK12 kinase and chemotherapy susceptibility in lung metastatic sites. Knockdown of CDK12 in lung adenocarcinoma cells increased chemotherapy sensitivity which was rescued by wild type, but not CDK12-G879V expression, consistent with the complete resolution of the lung metastatic sites in this patient.
Lung cancer is the leading cause of cancer mortality in men and women. The identification of several actionable targets in lung adenocarcinoma, the most common non-small cell lung cancer (NSCLC)1 histology, has been exploited therapeutically. Although patients harboring those somatic mutational targets, such as the Epidermal growth factor receptor (EGFR) and EML4-ALK fusion respond well to the targeted agents, they eventually develop resistance. A key determinant of this acquired resistance is tumor evolution which generates intra-tumor and inter-metastatic heterogeneity (1–3). The degree of mutational heterogeneity within primary tumors and between primary and metastatic or recurrence sites is highly variable. In some tumor types, such as recurrent glioma, the emergence of heterogeneity may be therapy related. In certain cancer types driven by environmental mutagens such as ultraviolet light in melanoma and smoking in lung cancer, the extent of homogenous mutational burden is greater than in other cancer types (4). However, there is a subgroup of lung cancer patients that possess extreme intra-tumor and inter-metastatic genomic mutational heterogeneity (2, 3, 5).
We previously revealed the existence of unprecedented genomic heterogeneity in an exceptional responder lung adenocarcinoma patient who survived with metastatic lung adenocarcinoma for more than 7 years while receiving a combination of HER2-directed targeted treatment and chemotherapy. Although HER2-targeted therapy is well established in HER2 positive breast cancer, recently HER2 amplification or mutation was documented to be present in around 1–2% of lung adenocarcinoma patients (6). Less than 1% of the somatic alterations were common to both the lung and lymph node metastatic sites in this patient (5). ERBB2/HER2 was the predominant oncogene affected in both the lung and lymph node metastatic sites. However, although HER2 amplification was more pronounced in the lung sites, a novel ERBB2-L869R mutation was found only in the lymph node metastases. Hence, although both the lung and the lymph node metastases initially responded well to HER2-targeted treatment, the response was less prolonged in the lymph node sites and progression was primarily in the lymph nodes during these 7 years (5). Multidimensional genomic analyses of next-generation sequencing data including, single nucleotide variants (SNVs), insertion-deletions (in-dels), copy number alterations (CNA) and expressed variants from RNA-seq can give a comprehensive view of the genomic landscape and its contribution to tumor heterogeneity. However, how this genomic heterogeneity influences the proteome and phosphoproteome is an important question that can be addressed by global mass spectrometry-based proteomics analyses. Recently, the NCI Clinical Proteomic Tumor Analysis Consortium (CPTAC) has employed mass spectrometry to analyze the proteome and phosphoproteome of colon, serous ovarian and breast primary tumors which previously underwent extensive genomic characterization (7–9). However, understanding of temporal and spatial proteogenomic tumor evolution in metastatic lung adenocarcinoma is lacking.
Here, we used quantitative mass spectrometry strategies to identify and quantify the proteome and phosphoproteome of the sequentially acquired lung and lymph node metastatic sites of this patient. The metastatic sites were surgically excised at the time of site-specific disease progression and the final acquisition was at autopsy. Interrogation of patient-specific databases built using the whole genome sequencing data from the lung and lymph node metastatic sites identified key somatic variants at the peptide-level. We further validated the novel CDK12-G879V mutation by functional analyses in lung adenocarcinoma cells and provide evidence that this mutation unique to the lung, may have resulted in the chemotherapeutic sensitivity and a “cure” of the lung metastatic sites by HER2 directed targeted therapy combined with standard chemotherapy in this patient.
EXPERIMENTAL PROCEDURES
Tumor Tissue Collection by Serial Biopsies and At Autopsy
Index patient was treated with standard of care treatment regimens or specific IRB approved clinical protocols at the NIH Clinical Center or Walter Reed National Military Medical Center over a period of more than 7 years. At specific times when there was progression, patient underwent excisional biopsy of progressive lymph nodes (n = 8). Patient also underwent a surgery to remove a progressive lesion in the lung (see time line, Fig. 1A). A complete autopsy was performed on expiration and 5 lymph nodes were analyzed in this study. All tumors procured were surgical specimens and macro-dissection was performed to enrich tumor areas. Tumor tissue was frozen immediately in liquid nitrogen before transporting to the laboratory for storage in liquid nitrogen freezer at −196 °C. All tissues collected were analyzed by the Laboratory of Pathology at the NIH Clinical Center and tumor cellularity determined by a Pathologist. Tumor cellularity was at least 40%. About 10–15 mg of tumor tissue from 9 separate tumor samples was cut and lysed in 400 μl of urea lysis buffer using a TissueLyser (Qiagen, Germantown, MD). Lysates were centrifuged at 14,000 rpm at 4 °C for 10 mins and the clear supernatants were transferred to new tubes. Protein concentrations were determined by the Modified Lowry method (BioRad, Hercules, CA).
Fig. 1.

Treatment and tumor acquisition timeline, experimental workflow and summary of MS analyses. A, The timeline of treatment history is indicated by the vertical lines along the blue bar with names of the drug used at each approximate time point. Tumor biopsies were taken at each time point (red arrow) with the date, sample description, and sample names in parenthesis (orange box). B, Experimental workflow of super-SILAC based quantitative mass spectrometry. 12 heavy labeled (13C6 Arg, 13C6 Lys) lung adenocarcinoma cell lines were mixed in equal proportions and used as a standard. The three tumor biopsies (LN1, Lung, and LN2) were then combined with the standard in equal proportions, followed by tryptic digestion, high pH-RPLC fractionation, and then subjected to LC-MS/MS analysis on an Orbitrap Elite. C, Experimental workflow of TMT based quantitative mass spectrometry. Three patient derived tumor biopsies (LN1, Lung, and LN2), one PDX sample, and five tumor autopsies were used in this experiment. All samples were pooled in equal proportions to serve as the reference. The digested peptides or TiO2 enriched phosphopeptides were TMT labeled then combined in equal proportions, followed by high pH-RPLC fractionation and then LC-MS/MS analysis on an Orbitrap Elite. D, Bar graph showing the total number of proteins identified and quantified in both the super-SILAC experiment and TMT experiment. 24% of proteins identified by the super-SILAC experiment are not quantified. E, Venn diagram showing protein identification from both super-SILAC and TMT experiments. 3648 proteins were common between the two methods. F, Pie chart represents the distribution of proteins identified by both TMT and super-SILAC experiments together, characterized by their molecular functions, biological processes and cellular component. Categories were based on information provided by the online resource PANTHER classification system.
Cell Culture and SILAC Labeling
Human lung adenocarcinoma cells were obtained from ATCC. All cells were cultured in RPMI 1640 supplemented with 10% dialyzed FBS and Penicillin/Streptomycin at 37 °C and 5% CO2. The heavy amino acids-labeled super-SILAC (stable isotope labeling with amino acids in cell culture) mix constituted of 2 immortalized lung epithelial cell lines (HBEC3KT and HPL1D), 1 lung adenocarcinoma cell line with wild type EGFR and KRAS (H1648), 2 EGFRL858R mutant cell lines (H3255, 11–18), 1 EGFRL858R/T790M cell line (H1975), 3 EGFRDEL mutant cell lines (H1650, PC9, and HCC827), and 3 KRAS mutant cell lines (A549, H2030, and H358). These cell lines were labeled by culturing in RPMI with the natural lysine and arginine replaced by heavy isotope labeled amino acids, l-13C6-arginine (R6; 13C6 98%) and l-13C6-lysine (K6; 13C6 98%). Labeled amino acids were purchased from Cambridge Isotope Laboratories (Andover, MA). Cells were cultured for approximately seven passages in the SILAC medium for complete incorporation of the heavy isotopes. Labeling efficiency was measured by mass spectrometry analysis of tryptic peptides processed from lysates obtained from individual cell lines after 5–7 generations of growth.
Protein Extraction
Cells were lysed with urea lysis buffer (20 mM HEPES pH 8.0, 8 m urea, 1 mm sodium orthovanadate, 2.5 mM sodium pyrophosphate and 1 mM β-glycerophosphate). Protein concentrations were determined by the Modified Lowry method (BioRad). Equal amounts of protein from lysates of each heavy labeled cell line were mixed together to constitute pooled lysate and used as a reference for the super-SILAC experiments.
Enzymatic Digestion
Protein lysates were reduced with 45 mM dithriothreitol (Sigma Aldrich, St. Louis, MO), alkylated with 100 mM iodoacetamide (Sigma Aldrich), and subsequently digested with modified sequencing grade Trypsin (Promega, Madison, WI) at 30 °C overnight. The digest was then acidified using 0.1% TFA and the peptides were desalted using solid phase extraction C18 columns (Supelco, Bellefonte, PA), and vacuum dried in a centrifugal evaporator.
TMT Labeling
TMT10plexTM amine reactive reagents (0.8 mg per vial) (Thermo Fisher Scientific) were resuspended in 41 μl of anhydrous acetonitrile (ACN), added to 200 ng peptides from each sample and mixed briefly on a vortexer. Reactions were proceeded at room temperature for 1 h, quenched by the addition of 8 μl of 5% hydroxylamine for 15 min and then tagged peptides were combined at equal amounts. Peptides from all tumors were pooled together to make the reference channel and labeled with TMT10–126.
Basic Reversed Phase Liquid Chromatography (RPLC) Fractionation
Basic RPLC separation was performed with a XBridge C18, 100 × 2.1 mm analytical column containing 5 μm particles and equipped with a 10 × 2.1 mm guard column (Waters, Milford, MA) with a flow rate of 0.25 ml/min. The solvent consisted of 10 mM triethylammonium bicarbonate (TEABC) as mobile phase A, and 10 mM TEABC in ACN as mobile phase B. Sample separation was accomplished using the following linear gradient: from 0 to 1% B in 5 min, from 1 to 10% B in 5 min, from 10 to 35% B in 30 min, and from 35 to 100% B in 5min, and held at 100% B for an additional 3 min. A total of 96 fractions were collected during the LC separation in a 96-well plate containing 12.5 μl of 1% formic acid for immediate acidification. The collected fractions were concatenated into 12 fractions and dried in a vacuum centrifuge. One tenth of the peptides was injected directly for LC-MS/MS analysis.
Titanium Dioxide (TiO2) Enrichment
Nine tenth of the dried peptides was dissolved in solution A containing 80% acetonitrile, 0.4% trifluoroacetic acid and 3% lactic acid and enriched with the TiO2 phosphopeptide enrichment spin column (Thermo Fisher Scientific, Rockford, IL). After binding, TiO2 spin columns were washed with solution A and thrice with 80% acetonitrile containing 0.4% trifluoroacetic acid. TiO2 bound peptides were eluted using 5% NH4OH and 5% pyrrolidine and immediately acidified using trifluoroacetic acid. The peptides were vacuum dried, cleaned on C18 stage-tips before LC-MS/MS analysis.
LC-MS/MS Analyses
Peptides separated/fractionated by basic reversed phase chromatography followed by TiO2 enrichment were analyzed on an LTQ-Orbitrap Elite interfaced with an UltimateTM 3000 RSLCnano System (Thermo Fisher Scientific, San Jose, CA). The dried peptides and the enriched phosphopeptides were loaded onto a nano-trap column (Acclaim PepMap100 Nano Trap Column, C18, 5 μm, 100 Å, 100 μm i.d. x 2 cm) and separated on an Easy-sprayTM C18 LC column (Acclaim PepMap100, C18, 2 μm, 100 Å, 75 μm i.d. × 25 cm). Mobile phases A and B consisted of 0.1% formic acid in water and 0.1% formic acid in 90% ACN, respectively. Peptides were eluted from the column at 300 nL/min using the following linear gradient: from 4 to 35% B in 60min, from 35 to 45% B in 5min, from 45 to 90% B in 5min, and held at 90% B for an additional 5min. The heated capillary temperature and spray voltage were 275 °C and 2kV, respectively. Full spectra were collected from m/z 350 to 1800 in the Orbitrap analyzer at a resolution of 120,000, followed by data-dependent HCD MS/MS scans of the fifteen most abundant ions at a resolution of 30,000, using 40% collision energy and dynamic exclusion time of 30s.
Patient-specific Database Construction
Customized patient-specific database was created by QUILTS (10) (quilts.fenyolab.org) using RefSeq as the reference protein sequence database. (1) a BED file containing RNA-Seq predicted junctions; VCF files containing (2) somatic variants and (3) germline variants; and (4) a fusion file containing all predicted fusion genes were used as inputs. QUILTS creates tumor-specific databases by enumerating all possible protein sequence variation that results from the genomic and transcriptomic sequence variation including single amino acid variants, introduced and removed stop codons, alternative splicing, fusion proteins, expression of unannotated parts of the genome. Our database for this study contained a total of 71,503 entries.
Data Analysis
Peptides and proteins were identified and quantified using the MaxQuant software package (version 1.5.3.30) with the Andromeda search engine (11, 12). MS/MS spectra were searched against the customized patient specific database and quantification was performed using default parameters for 3s-SILAC or TMT10plex in MaxQuant. The TMT data were also searched against both patient-specific database and the Uniprot mouse database (Version 20170207, 82253 entries) together to identify proteins contributed from the patient-derived xenograft (PDX) sample (channel 128C). For database search, trypsin was used as a protease with two missed cleavage sites allowed. Carbamidomethyl cysteine was specified as a fixed modification. Phosphorylation at serine, threonine and tyrosine; deamidation of asparagine and glutamine; oxidation of methionine; and protein N-terminal acetylation were specified as variable modifications. The precursor mass tolerance was set to 7 ppm and fragment mass tolerance to 20 ppm. False discovery rate was calculated using a decoy database and a 1% cut-off was applied to both the peptide and phosphosite tables. Proteins with single peptide identification were removed from the final tables.
Normalized ratios from super-SILAC experiment or corrected intensities of the reporter ions from TMT labels were obtained from the MaxQuant search. For the TMT experiment, relative ratios of each channel to the reference channel were calculated. Perseus (version 1.5.5.3) was used to view and further analyze the data. Hierarchical clustering of proteins and phosphosites were obtained in Perseus using log ratios or log intensities of protein and phosphorylation abundance. The protein-protein interaction (PPI) maps of the phosphosite clusters were imported from the “STRING: protein query” module of the Cytoscape software (San Diego, CA, version 3.4.0) (13) with the confidence cutoff of 0.80. These maps were analyzed for functional enrichment of the gene ontology biological process categories using the ClueGO 2.2.6 plugin (14) with the kappa statistic ≥ 0.4, a two-sided hypergeometric test for enrichment with Bonferroni step down method for correction of the multiple hypothesis testing. A p value of 0.001 was used as the cut-off criterion.
Experimental Design and Statistical Rationale
A patient diagnosed with metastatic lung adenocarcinoma with metastases to the lung and lymph nodes was treated with combination chemotherapy followed by HER2-directed therapy either alone or in combination with various chemotherapy regimens over 7 years. Two lymph node metastases (LN1 and LN2) obtained by sequential excisional lymph node biopsies, a progressive metastatic lung tumor obtained by wedge resection surgery (Lung), a PDX tumor generated from one of the lymph node biopsies from the left neck, and five distinct lymph node metastases obtained at autopsy (MET1–5) were analyzed in this study.
One super-SILAC experiment and one TMT10plex-based quantitative proteomics experiment were employed to explore the tumor-specific and temporal heterogeneity of the expressed proteomes. In the super-SILAC strategy, 12 “heavy” amino acids-labeled lung adenocarcinoma and immortalized lung epithelial cell lines were pooled at equal amounts and used as the standard for “spike-in.” Lysates from three distinct metastatic tumors (LN1, Lung and LN2) were combined with the “heavy” standard lysate at equal amounts for further sample processing and mass spectrometry analysis. The TMT10plex quantitative proteomics strategy was used for the quantification of the proteome and phosphoproteome of nine distinct tumor metastases. Lysates from the nine samples were pooled at equal amounts and labeled with TMT126 to be used as the reference channel.
Nine distinct metastatic sites, including one lung tumor obtained from wedge resection of lung metastasis, and 5 lymph nodes obtained at autopsy were used for the analysis. Each tumor from this patient was a distinct metastatic site. Histopathology was confirmed to identify samples for processing with at least 40% tumor content. All the tumors were obtained by longitudinal biopsies and autopsy and were used for clinical and protocol-specific assays, including histopathology. There was not enough tissue left to conduct biological replicates.
Histograms of the log2 SILAC ratios (normalized H/L of each experiment) were plotted for individual tumor tissue. Proteins with 3-fold changes of final normalized combined ratio in either up or down regulated direction were considered as significant changes, which corresponds to the ±1.5 SD of mean in the experiments. For the TMT experiment, relative ratios of each channel to the reference channel were calculated. Histograms of the log 2 ratios were plotted and mean ± 1.5 SD corresponded to the TMT ratios of 1.88 and 0.48, respectively.
Correlation Between mRNA Expression and Protein Abundance
We correlated mRNA expression and protein abundance within each of two metastatic tumor sites (Lung and LN1) and the PDX. We used RNA-seq FPKM values and protein intensity measurements from TMT and SILAC experiments to estimate mRNA and protein abundance, respectively. For each tumor site, we calculated the Spearman correlation coefficient between log2 values of FPKM and protein intensity values for genes that had measurements in both mRNA and protein (number of genes for TMT experiments were: L, 3853; LN1, 3880; PDX, 3791, and number of genes for SILAC measurements were: L, 5890; LN1, 5920).
13. Copy Number Variation Analysis
We used OncoScan CNV FFPE Assay Kit (Affymetrix) to perform copy number variation (CNV) analysis. Copy number data was processed and normalized using OSCHP-TuScan algorithm, which determines allele specific copy number and estimates ploidy. Copy number log2 ratios of each tumor site was estimated with Nexus Express for OncoScan. Regions of copy number gains or losses across all metastatic tumor sites were matched based on the location of genes in each region. A CNV heatmap was generated based on hierarchical clustering analysis using Euclidean distance.
Mutant Peptide Validation
Confirmatory analysis of MS2 spectra was performed separately and following MaxQuant analysis using mutant-specific database. First, Oribtrap Elite HCD spectra were converted to MGF format using the NIST program ReAdW4Mascot2.exe (v20130604a) (http://chemdata.nist.gov/dokuwiki/doku.php?id=peptidew:pepsoftware). Parameters for conversions were as follows: -c -ChargeMgfOrbi -FixPepmass -MaxPI -MonoisoMgfOrbi -NoPeaks1 -PIvsRT -sep1 -TolPPM 20 -NoMzXml. These settings include the -FixPepmass option, which reassess the monoisotopic peak, assigned by XCaliburTM, using the previous and next MS1 scans, changing the PEPMASS value if a better assignment can be made.
All fragmentation spectra from the Super SILAC experiments were searched using Mascot 2.5 (Matrix Science, Ltd., London, UK) and MS-GF+ (v20140716) against the patient-specific database created using the QUILTS pipeline. Mascot searches were run using a 20ppm precursor mass tolerance (monoisotopic) and a 0.05 Da fragment mass tolerance, allowing for up to 2 missed cleavages and considering only fully tryptic peptides. Quantitation mode was set to SILAC K+6; R+6. Carbamidomethylation of C was set as a fixed modification and oxidized methionine as variable. MS-GF+ settings were the following: 10 ppm precursor mass tolerance, -m 3, -inst 1, -ntt 2, -tda 1, -ti 0, 0 -max Length 60 -min Length 7. These settings were specific for Orbitrap HCD spectra, performed target-decoy-analysis, did not allow for isotopic matching, and excluded peptides longer than 60 and shorter than 7 amino acids long. Carbamodomethyl of C was used as a fixed modification and heavy versions of K and R (+6.020), oxidation of M, and protein N-terminal acetylation were used as variable modifications. MZID files were converted to TSV using the MzIDToTSV function of the program.
Next, spectra from identifications of variant peptides identified by MaxQuant were extracted following removal of peptides that did not contain variant sequences (e.g. those giving rise to a K or R adjacent to the start of the peptide sequence). In total, 833 spectra corresponding to 105 sequences (of the 198 identified by MaxQuant) were analyzed. All spectra were required to have corresponding identifications better than the Mascot Identity Threshold (95%) or a Q Value < 0.01 (MS-GF+) per file. We found no disagreements between Mascot and MS-GF+ for these spectra, and when both search engines agreed, the Mascot identification information was kept.
Because variant sequences, in particularly those that are unknown (i.e. not found in dbSNP) or somatic are rare, additional steps were taken to ensure the quality of the peptide-spectrum matches. These steps were performed according to (10) and in consideration of Nesvishskii et al. (15). First, all fragmentation spectra were labeled and the fraction of matched MS2 intensity was calculated for each. These values provide a good measure of the purity of a spectrum, and there is a correlation with a low unmatched intensity and correct assignments. Additionally, sequencing “gaps” were analyzed (10). A gap is defined as missing fragmentation evidence between adjacent residues. This analysis considers b- and y-type ions only (including neutral losses of water and ammonia) and is based on the observation that large gaps such as the complete absence of one or the other ion series can give rise to good matching identifications but often correspond to incorrect matches and cases where the correct peptide sequence is not in the target database. For our analysis, we removed all spectra with unmatched abundance of >50% and those with any sequence gap >2 (10).
In addition to Mascot and MS-GF+ searches, X! Tandem searches of the 833 putative variant peptide spectra were run. These searches were run to make use of X! Tandem's refinement mode options as well as search the common contaminants database cRAP. In particular, X!Tandem can search for single amino acid substitutions as well as Single Amino acid Polymorphism (SAPs) indexed from dbSNP. Settings for X! Tandem search were the following: version- Vengance (2015.12.15.2), 20 ppm precursor tolerance, 20 ppm fragment mass tolerance, fixed cabamidomethyl C, variable oxidized M and deamidated N. For refinement searching, carbamylation of K and/or the peptide N terminus was optionally allowed, as was dioxidation of M. Substitution of up to 1 amino acid and SAP searching were also turned on. Agreement between the X! Tandem analysis and the Mascot/MS-GF+ analysis was very good. However, X! Tandem did not confidently identify 51 of the 833 spectra, returning no match. Additionally, we found peptides where X! Tandem disagreed. One case, preferring a deamidated N rather than the N>D amino acid change. Because of this result all matches to N->D peptides were excluded from further analysis. Additionally, for another peptide, the top-ranking X! Tandem match was an amino acid change to an alternate reference peptide over the variant present in the database. Because the variant was found by genomic sequencing, the variant identification was kept over the substitution.
Targeted Analysis Using Multiple Reaction Monitoring (MRM)
Tier 2 level of the MRM assays were developed and applied. Heavy labeled peptides synthesized with a C-terminal 13C615N4-labeled arginine or 13C6 15N2 -labeled lysine were purchased from Thermo Fisher Scientific. The synthetic peptides were re-constituted in 0.1% formic acid and were analyzed on the nano-chip-LC using a 1260 Infinity Series HPLC-Chip cube interface (Agilent, Palo Alto, CA) coupled to a 6495-triple quadrupole mass spectrometer (Agilent). A large capacity chip system (G4240–62010) consisting of a 160 nl enrichment column and a 150 mm*75 μm analytical column (Zorbax 300SB-C18, 5 μm, 30 A pore size) was used. Mobile phase A consisted of 95% water and 0.1% FA, and mobile phase B consisted of 95% acetonitrile in 0.1% FA. A flow rate of 3 μl/min was applied for sample loading by the capillary pump and 600 nl/min for the analytical separation through the nano-pump. A 25-min gradient (0 min, 0% B; 1 min, 5% B; 17 min, 40% B; 20 min, 70% B; 21 min, 80% B; 25 min 0% B) was used for the chromatographic separation of the target peptides. A spray voltage of 1850 V was applied and quadrupoles 1 and 3 were run at 0.7 FWHM resolution. Individual injections were carried out to select optimum precursors and intense product ions for each synthetic peptide. The top transitions were selected based on the presence of intense y-ions at m/z greater than the precursor. In case high m/z y-ions were not seen, other more abundant ions were chosen. All the raw data were imported in Skyline 3.7 and manually reviewed to delete any poorly performing transitions. Five transitions were selected for each peptide. The optimization of collision energies was performed by using the values calculated by Skyline for the monoisotopic precursor and product masses for the Agilent 6495 system. The best performing transitions were combined in one scheduled MRM method with a 25-min gradient and a 2-min retention time window, using retention times extracted during the method refinement stage. For endogenous target peptide quantification, heavy labeled synthesized peptides were spiked-in as an internal standard to the tryptic peptides of different tissue samples. All the raw data files were imported in Skyline 3.7 and data annotations were manually inspected. Peak Area Ratios (PAR) of light endogenous signals to the heavy internal standards were exported to MS excel for further analysis of mean, standard deviation and % co-efficient of variation.
CRISPR-Cas9-mediated Knockdown of CDK12 and Cell Growth Analysis
A549 cells were transduced with lentivirus expressing Cas9. Clones were obtained after blastocidin selection and one of the clones with higher expression of Cas9 was selected for experiments based on Western blotting detection of Cas9. A549-Cas9 cells were then transduced with lentivuruses expressing CRISPR sgRNAs that target exon 1 or exon 2 of CDK12. After puromycin selection, clones were picked and expanded. Cells were lysed with 1x modified RIPA buffer, then immunoblotted for CDK12 expression. Two clones, A549-Cas9–261-7 and A549-Cas9–264-5, in which the sgRNAs target exon 1 and exon 2 of CDK12, respectively, were selected for further functional analysis based on the knockdown of CDK12 expression. Further, either wild type CDK12 or CDK12-G879V mutant cDNAs were overexpressed in the CRISP/Cas9 mediated CDK12 knockout cells to rescue the effects of knockdown of CDK12 and study the function of CDK12-G879V mutant.
For chemotherapy treatment and survival analysis, cells were trypsinized with 0.25% trypsin in EDTA, and centrifuged at 1000 rpm for 5 mins at room temperature. Cells were re-suspended in RPMI medium with 10% FBS, 1% pen/strep, counted with trypan blue exclusion and plated 4000 cells per well in a 96 well-plate. Cells were allowed to settle overnight before drug treatment. A 10× stock of camptothecin (Cell Signaling Technology) was prepared fresh for each experiment. 10μl of camptothecin was added to 90μl of RPMI media already present in each well. Cells were incubated at 37 °C, 5% CO2 for 48 or 72 h. After 48 or 72 h of treatment, all media was removed, and 50μl of 1x Promega CellTiter-Glo® Luminescent Cell Viability Assay reagent was added to each well. Luminescence was measured with a SpectraMax M5 microplate reader and recorded by SoftMax Pro 5.4.1. Raw luminescence was normalized and plotted with MS Excel.
γ-H2AX Foci Formation Assay
A549 cas9 (WT), A549 cas9 261-7 (exon 1 KO) and A549 cas9 264-5 (exon 2 KO) were plated onto 2-chambered borosilicate cover glass, and they were treated for 48 h with camptothecin. Cells were fixed in 4% formaldehyde at room temperature for 10 min, and membrane permeabilized with 0.25% Triton-X 100 in H2O for 10 min. The cells were blocked with 1% BSA in PBS for 1 h and then incubated with rabbit anti-γ-H2AX primary antibody (1:500 dilution with 1% BSA in PBS; Cell Signaling Technology) overnight at 4 °C. Incubation of goat anti-rabbit IgG-Alexa Fluor 488-conjugated (1:250 dilution with 1% BSA in PBS; Abcam) was performed for 1 h at room temperature in darkness for foci visualization. Nuclei were stained with ProLong™ Gold Antifade Mountant with DAPI (1:1000 dilution with PBS; ThermoFisher Scientific). Zeiss 800/Airyscan confocal microscope imaged 15–20 different fields for each condition. Images that overlap Alexa 488 channel and DAPI channel were processed through FoCo (https://www.ncbi.nlm.nih.gov/pmc/articles/PMC4654864/), which creates masks of nuclei from DAPI signal in order to identify true nuclear γ-H2AX foci (Alexa 488 signal). Calculation of number of γ-H2AX foci per nucleus and statistical analysis were performed automatically by FoCo, and values from biological replicates were plotted in MS Excel.
RESULTS
Brief Clinical History, Tumor Tissue Procurement and Summary of Protein Identification and Quantification
A 50-year old African American never smoker was diagnosed with metastatic lung adenocarcinoma with metastases to lung and lymph nodes. The patient was treated with combination chemotherapy followed by HER2-directed therapy either alone or in combination with various chemotherapy regimens over 7 years (Table I). Two case reports (16, 17) and a comprehensive genomics analysis of multiple tumor biopsies across his 7-year treatment course have been published. In the later report, we extensively characterized the extreme genomic heterogeneity between the lung and lymph node metastatic sites of this patient (5). HER2 was amplified in both the lung and lymph node metastatic sites, although the amplification was more extensive in the lung sites. The lymph node metastatic sites, in addition, harbored an ERBB2-L869R mutation that likely reduced the sensitivity to HER2-targeted therapy; hence the patient progressed primarily in the lymph nodes during the treatment (5). Here, we sought to determine the temporal and spatial heterogeneity of the proteome and conduct a comprehensive quantitative proteomics analysis on different metastatic lymph node tumors (LN1, LN2 and PDX) obtained by sequential excisional lymph node biopsies, 5 distinct lymph node metastases obtained at autopsy (MET1–5), and a progressive metastatic lung tumor obtained by wedge resection surgery (L) (Fig. 1A). The patient-derived xenograft (PDX) was generated from one of the lymph node biopsies from the left neck. The patient was treated with combination chemotherapy followed by HER2-targeted therapy alone or with chemotherapy throughout his course of over 7 years as depicted in the timeline (Fig. 1A). Both super-SILAC and TMT10plex-based quantitative proteomics strategies were employed to explore the tumor-specific and temporal heterogeneity of the expressed proteomes. For the super-SILAC strategy, we first made a novel mixture of “heavy” amino acid-labeled lysates from twelve cell lines with different mutational profiles: KRAS mutant, EGFR mutant, and wild type KRAS/EGFR lung adenocarcinoma, along with normal human bronchial epithelial cells (HBECs) and immortalized lung epithelial cells. The lung adenocarcinoma-specific super-SILAC mix that we generated can potentially be used in many laboratories for comparative quantitative analyses. Equal amounts of these twelve “heavy” labeled lung adenocarcinoma and immortalized lung epithelial cell lysates were pooled and used as the standard for “spike-in.” Equal amounts of lysates from three distinct metastatic tumors (LN1, Lung and LN2) were combined with the “heavy” standard lysate, for further sample processing and mass spectrometry analysis (Fig. 1B, supplemental Table S1). The TMT-based quantitative proteomics strategy was used for proteome and phosphoproteome quantification of nine distinct tumor metastases, including two lymph nodes (LN1 and LN2), one lung tumor, one PDX derived from a lymph node metastasis, and five lymph node metastases (MET1–5) procured at autopsy (Fig. 1C, supplemental Table S2). Equal amounts of lysates from the nine samples were pooled and labeled with TMT126 to be used as the reference. After trypsin digestion and basic reversed-phase HPLC fractionation, tandem mass spectrometry data with high accuracy and resolution was acquired on an Orbitrap Elite mass spectrometer. A total of 6214 and 4061 proteins were identified from super-SILAC and TMT experiments, respectively (Fig. 1D). 5295 and 3293 proteins were identified with 2 unique peptides or more, respectively. More than 4000 proteins were quantified from both experiments. 3648 proteins were identified and quantified in both experiments (Fig. 1E). PANTHER analysis, which distributes proteins based on their molecular functions, biological processes and cellular components determined that 2000 proteins had catalytic activity including kinases, phosphatases and metabolic enzymes (Fig. 1F).
Table I. Sequence of treatment regimens used, the rationale for each regimen, duration of treatment and progressive sites in our patient.
| Sequence | Therapy regimen | Dates; duration (months) | Progression |
|---|---|---|---|
| 1 | Carboplatin/Paclitaxel/Bevacizumab (combination chemotherapy/anti-angiogenic therapy; front line NSCLC treatment-standard of care) | 1/2008–5/2008; 4 | Lung |
| 2 | Erlotinib (1st generation EGFR inhibitor was approved in 2008 for 2nd line treatment of NSCLC-that indication changed in 2016 to limit only to EGFR mutant patients) | 8/2008–8/2008; <1 | Lung |
| 3 | Dacomitinib (Clinical study-NIH) (dual HER2/EGFR inhibitor-initially enrolled for EGFR and HER2 expression in original diagnostic biopsy; later biopsy proven HER2 amplification) | 8/2008–4/2009; 8 | Lung, Lymph node |
| 4 | Trastuzumab (Herceptin)/Vinorelbine (anti-HER2 mAb, FDA approved for HER2-positive breast cancer in 1998, here used for HER2 amplified NSCLC along with chemotherapy) | 5/2009–1/2011; 20 | Neck lymph nodes |
| 5 | Lapatinib (small molecule HER2 inhibitor, approved for HER2 amplified breast cancer in 2007) | 2/2011–5/2011; 3 | Neck lymph nodes |
| Right cervical lymph node excision (5/2011)-LN1 | |||
| 6 | Crizotinib (ALK inhibitor, patient's tumor from 1/27/11 biopsy showed polysomy of ALK (>4 copies) in 40% cells) | 6/2011–7/2011; <1 | Lymph nodes |
| 7 | Lapatinib and Capecitabine (combination HER2-targeted and chemotherapy) | 7/2011–7/2012; 12 | Lung |
| Left lower lobe wedge resection of progressive lung metastasis (11/2011)-L | |||
| 8 | Lapatinib/nab-paclitaxel (Abraxane) (combination HER2-targeted and chemotherapy) | 4/2012–8/2012; 4 | Lymph nodes |
| 9 | Lapatinib/Pemetrexed (combination HER2-targeted and chemotherapy) | 8/2012–10/2012; 2 | Lymph nodes |
| 10 | Pertuzumab/Trastuzumab/Vinorelbine (pertuzumab-a HER2-dimerization inhibitor mAb, approved in 2012 for HER2-positive breast cancer here the HER2-targeted combination along with chemotherapy used) | 10/2012–11/2013; 13 | Lymph nodes |
| Right cervical lymph node excision (11/2013)-LN2 | |||
| 11 | T-DM1 (Trastuzumab-entansine) (FDA approved for HER2-positive breast cancer in 2013) | 11/2013–1/2014; 2 | Neck Lymph nodes |
| 12 | T-DM1/pertuzumab (Combination HER2-targeted therapy) | 1/2014–3/2014 | Neck Lymph nodes |
| 13 | Radiation treatment (local ablative treatment) | 4/2014 | Neck |
| 14 | Trastuzumab/vinorelbine (HER2-targeted and chemotherapy combination) | 4/2014–7/2014 | Neck and axillary lymph nodes |
| Left jugular and right axillary lymph node excision (9/2014)-PDX | |||
| Autopsy (2/2015)-5 metastatic lymph nodes (MET1–5) procured/no lung metastasis found | |||
For the TMT experiment, a total of 4191 proteins were identified when searching against patient-specific database and mouse database together, and 3405 proteins with more than 2 unique peptides were identified (supplemental Table S3). 2729 protein groups contained both human and mouse proteins and 138 proteins were identified from the mouse database only. To determine whether the mouse proteome contributed significantly to the quantitation results from the PDX sample (channel 128C), we plotted the correlation of the ratios of the PDX channel to the pool from the two searches (patient-specific database only and patient-specific along with the mouse database combined). We observed high correlation (R2 = 0.992) between the two analyses (supplemental Fig. S1), suggesting the mouse proteins from the tumor microenvironment of the PDX did not significantly alter the overall quantitative analysis from the PDX tumor. We also tabulated the ratios of PDX/Pool for common proteins identified from both the databases (supplemental Table S4). This further confirmed that the difference of ratios obtained on searching the mouse along with the patient-specific database was insignificant compared with searching the patient-specific database alone. This was likely a result of the fact that only one of nine tumor samples was a PDX containing mouse proteins. In summary, we identified and quantified the human proteome to further characterize the heterogeneity of protein expression and the associated signaling pathways between the lung and lymph node metastatic sites as described below.
Correlation and Distribution of Protein Abundance Ratios and Clustering Analysis Reveals Greater Similarity of Protein Abundances Between the Lung and LN2 Metastases
We employed super-SILAC and TMT-labeling strategies to quantify tumor tissue proteins. It was also our goal to compare both strategies. Correlations between the two approaches and three samples were plotted. The protein ratios across three samples (LN1, Lung and LN2) were well correlated between super-SILAC and TMT approaches (Fig. 2A). Analysis of the protein SILAC ratios demonstrated that the protein abundance of LN2 correlated better with lung and LN1 (R2 = 0.68 and 0.67, respectively) compared with the correlation of protein abundance ratios of LN1 to that of the lung (R2 = 0.53). This indicates that the lung tumor was more similar to the LN2 lymph node procured 2 years after the lung surgery than LN1 procured 6 months before the surgery (Fig. 2B). The SILAC protein ratio distribution, indicated that a large majority of proteins were unchanged (Fig. 2C). Approximately 12% of proteins from each tumor biopsy sample were differentially expressed > 2-fold relative to the heavy labeled standard lysate mixture. 8–9% of the proteins were differentially expressed < 2-fold in LN2 and lung; however, in LN1, 19% of the proteins were differentially expressed < 2-fold. The number of proteins differentially expressed <2-fold in LN1 is twice that of LN2 and lung, indicating that more proteins were less abundant in LN1 compared with LN2 and lung. Hierarchical clustering of proteins based on their “Super-SILAC” abundance ratios clustered LN2 and lung together, providing further evidence of greater proteomic heterogeneity between LN1 and either Lung or LN2 (supplemental Fig. S2A). We identified many extracellular proteins and proteins involved in immune effector processes with elevated expression compared with the standard lysate. Some of the membrane proteins involved in protein glycosylation and GTPase activity were expressed at lower levels in LN1 compared with LN2 and lung.
Fig. 2.
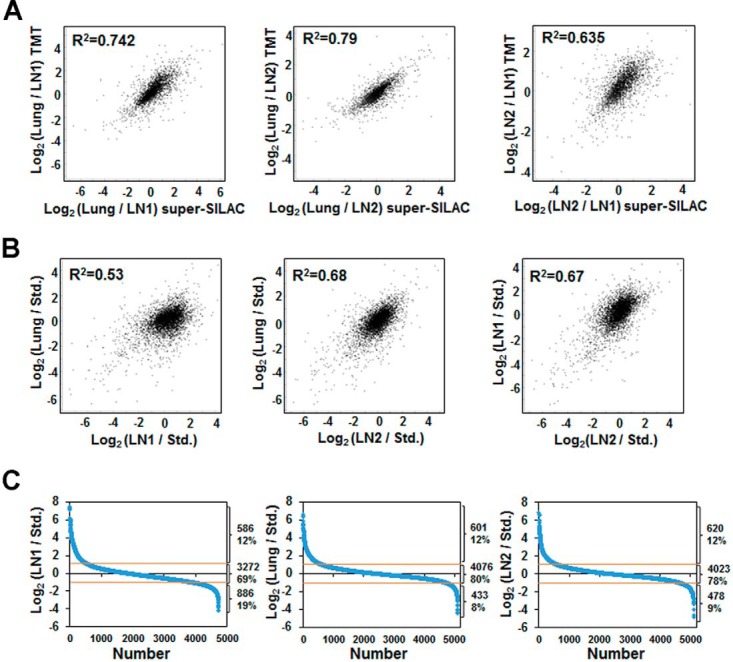
Correlation of the protein quantification. (A) Correlation between TMT and super-SILAC approaches. X-axis is the Log 2 ratio between two samples from super-SILAC experiments, and y axis is the Log 2 ratio between the same two samples from TMT experiments. (B) Correlation between the three tumor biopsies from the results obtained by super-SILAC experiments. Values are based on the Log 2 ratio of the tumor samples and the heavy labeled super-SILAC standard. (C) Distribution of proteins quantified for the three biopsies (LN1, Lung, and LN2) based on the ratio to the heavy labeled standard in the super-SILAC experiment. 19% of proteins downregulated and ∼70% of proteins remain unchanged in LN1, whereas 8–9% of proteins downregulated and ∼80% proteins remain unchanged in Lung and LN2.
PCA analysis of TMT protein ratios across all metastatic sites revealed that lung and LN2 metastatic sites were more like each other and LN1, the first progressive metastatic lesion excised. Further, LN1 was different than all other metastatic sites (supplemental Fig. S3A). Pair-wise correlation analysis of the TMT protein ratios showed a strong association between specific autopsy lymph nodes, MET4 and MET5 (R2 value 0.845). Overall, there was poor correlation of TMT protein ratios across all metastatic sites except between lung and LN2 and between autopsy lymph nodes (supplemental Fig. S3B). Hierarchical clustering of protein abundance ratios of each tumor lysate against the pooled lysate from the TMT experiment clustered LN2 and lung together. LN1 protein abundance ratios clustered with three lymph nodes obtained at autopsy but was quite distinct from the other two lymph node metastases (supplemental Fig. S2B). In summary, the global protein quantification was similar using the two strategies. Further, pairwise correlations across the entire data set and hierarchical clustering revealed that the lung and the LN2, procured approximately two years after the lung surgery, were more like each other than to LN1, obtained prior to the surgery. Instead, LN1 was more like lymph nodes MET2, 4, 5 which were procured at autopsy, demonstrating proteomic heterogeneity at specific metastatic sites.
Differentially Expressed Classes of Proteins and Enrichment of Functional Pathways Between the Lung and the Two Lymph Node Metastases, LN1 and LN2
The natural history of this patient's disease was complicated by the fact that the lymph node metastatic sites were more aggressive and repeatedly progressed on multiple therapeutic regimens whereas the lung responded quite well to the therapies administered. At autopsy, we did not detect any lung metastasis, whereas the lymph nodes were laden with tumor cells. Hence, we further analyzed the results of our “super-SILAC” quantitative mass spectrometry experiments to compare the proteins and pathways that were differentially activated between the lung and the lymph node metastatic sites. We identified many oncoproteins, kinases, phosphatases, as well as transcriptional and translational regulators which were differentially expressed in the lung, LN1 and LN2 metastatic sites (Table II). The oncoproteins ERBB2, CTNNB1, IDH2, MLH1, NDRG1, and NF1 were up-regulated, whereas B2M, CDKN2A, FNBP1, MAX and SYK were significantly downregulated in the lung compared with the lymph nodes. Among the kinases with increased abundance in the lung compared with the lymph nodes were CCNK, MAPK3, MAP2K3, PAK1, PDK3, and YES1. Kinases with decreased abundance in the lung were AURKA, CKB, STK26 and STK4. DUSP23 and PPFIA1. Phosphatase levels were elevated in the lung metastatic sites. JUNB and SMARCD2 were two of the transcriptional regulators significantly up-regulated in the lung metastatic site compared with LN1. Interestingly, the lung and LN2 that was excised more than two years after the removal of the lung metastasis had similar levels of these two proteins.
Table II. Selected proteins with 3-fold change between any of the two biopsy tumor samples as determined by a super-SILAC experiment.
| Gene | Ratio lung/LN1 | Ratio lung/LN2 | Ratio LN2/LN1 |
|---|---|---|---|
| Cancer genes | |||
| B2M | 0.2 | 0.5 | 0.4 |
| CDH1 | 3.5 | 1.6 | 2.2 |
| CDK12 | 13.8 | 6.8 | 2.0 |
| CDKN2A | 0.2 | 0.07 | 3.1 |
| CTNNB1 | 3.1 | 2.2 | 1.4 |
| ERBB2 | 70.3 | 21.8 | 3.2 |
| FNBP1 | 0.2 | 0.6 | 0.3 |
| IDH2 | 7.1 | 2.6 | 2.8 |
| LCP1 | 0.2 | 0.4 | 0.5 |
| MAX | 0.3 | 0.5 | 0.5 |
| MLH1 | 3.6 | 1.8 | 2.0 |
| MYH11 | 0.1 | 0.6 | 0.2 |
| NDRG1 | 2.8 | 3.3 | 0.8 |
| NF1 | 10.9 | 3.7 | 3.0 |
| NFIB | 0.9 | 0.2 | 5.5 |
| SYK | 0.3 | 0.5 | 0.5 |
| TP53 | 0.5 | 2.4 | 0.2 |
| Kinases | |||
| AURKA | 0.3 | 0.3 | 0.8 |
| CCNK | 4.9 | 2.6 | 1.9 |
| CDK2 | 3.7 | 0.8 | 4.9 |
| CHKB | 3.4 | 2.6 | 1.3 |
| CKB | 0.2 | 0.2 | 1.1 |
| CKMT1A | 0.3 | 0.04 | 7.0 |
| CSNK1D | 5.1 | 2.6 | 1.9 |
| DGKA | 0.3 | 0.7 | 0.4 |
| MAP2K3 | 3.8 | 1.9 | 2.0 |
| MAP4K5 | 4.9 | 1.0 | 5.1 |
| MAPK3 | 4.5 | 3.2 | 1.4 |
| MPP1 | 0.3 | 0.4 | 0.8 |
| NME3 | 5.3 | 3.4 | 1.6 |
| PAK1 | 4.9 | 3.9 | 1.3 |
| PDK3 | 4.3 | 1.8 | 2.5 |
| PI4K2A | 3.0 | 0.1 | 25.5 |
| PRKAR2B | 0.2 | 0.4 | 0.4 |
| PRKCA | 0.3 | 0.6 | 0.5 |
| SRPK2 | 0.9 | 0.2 | 4.6 |
| STK26 | 0.3 | 0.5 | 0.7 |
| TK1 | 0.05 | 1.0 | 0.05 |
| YES1 | 4.4 | 2.8 | 1.6 |
| STK4 | 0.1 | 0.5 | 0.3 |
| Translation regulators | |||
| EEF1A2 | 2.6 | 3.7 | 0.7 |
| IGF2BP2 | 0.4 | 2.2 | 0.2 |
| PATL1 | 0.05 | 0.1 | 0.4 |
| Phosphatases | |||
| BPGM | 0.3 | 0.4 | 0.7 |
| DUSP23 | 4.5 | 1.5 | 3.1 |
| NT5C | 1.2 | 4.1 | 0.3 |
| NT5C3A | 0.3 | 0.5 | 0.6 |
| NUDT2 | 0.6 | 0.3 | 2.1 |
| NUDT3 | 0.6 | 0.2 | 3.2 |
| NUDT9 | 2.2 | 3.3 | 0.7 |
| OCRL | 0.2 | 0.6 | 0.3 |
| PPFIA1 | 3.7 | 6.4 | 0.6 |
| PPP1R12C | 0.2 | 0.6 | 0.4 |
| PPP1R1B | 3.4 | 2.3 | 1.5 |
| PPP1R7 | 0.3 | 0.4 | 0.6 |
| PTPN6 | 0.1 | 0.3 | 0.3 |
| TIMM50 | 0.4 | 1.3 | 0.3 |
| Transcription regulator | |||
| ANKLE2 | 6.7 | 6.5 | 1.0 |
| ARHGAP35 | 3.5 | 1.2 | 2.9 |
| ASCC1 | 2.9 | 0.9 | 3.4 |
| BASP1 | 0.4 | 1.6 | 0.2 |
| BTAF1 | 0.6 | 2.9 | 0.2 |
| CDKN2A | 0.2 | 0.1 | 3.1 |
| CGGBP1 | 0.2 | 0.6 | 0.4 |
| CTNNB1 | 3.1 | 2.2 | 1.4 |
| GTF2A1 | 0.4 | 1.3 | 0.3 |
| IFI16 | 0.3 | 0.8 | 0.3 |
| IRF9 | 0.8 | 0.1 | 5.9 |
| KANK2 | 0.3 | 1.2 | 0.2 |
| LBH | 0.2 | ||
| LPXN | 0.1 | 0.3 | 0.3 |
| MAX | 0.3 | 0.5 | 0.5 |
| MECP2 | 0.1 | 0.6 | 0.2 |
| MED27 | 3.7 | 1.0 | 3.8 |
| NFIB | 0.9 | 0.2 | 5.5 |
| PTGES2 | 2.2 | 0.7 | 3.4 |
| PTRF | 0.2 | 0.9 | 0.2 |
| RELB | 0.2 | 0.3 | 0.6 |
| SMARCD2 | 4.8 | 1.1 | 4.6 |
| TEFM | 3.0 | 0.9 | 3.4 |
| TFB1M | 4.3 | ||
| THOC1 | 0.5 | 1.8 | 0.3 |
| TLE1 | 4.1 | 2.0 | 2.1 |
| UXT | 1.6 | 0.5 | 3.5 |
| YBX3 | 0.04 | 0.1 | 0.3 |
Next, we examined the function of the differentially expressed proteins between either of the two lymph node metastases (LN1 versus LN2) or between either lymph node and lung metastases in the super-SILAC experiment. Ingenuity Pathway Analysis (IPA) was performed on proteins that had a “Super-SILAC” ratio of 3-fold. The top activated canonical pathways, whose proteins had at least a 3-fold change in lung compared with either of the lymph nodes, include activation of IRF by cytosolic pattern recognition receptors, Rac signaling, HGF signaling, ERBB and Ephrin B signaling. Among the top inhibited pathways were Interferon, Sirtuin, PKA and TP53 signaling pathways (Fig. 3A). The heatmaps of protein ratios of lung versus either of the lymph nodes in three selected pathways namely, ERBB, Sirtuin and PKA signaling showed differences in protein abundances for these specific pathways (Fig. 3B–3D). The expression of ERBB signaling pathway proteins, ERBB2, MAPK3, MAP2K2 and PAK1 was higher in lung than in the lymph nodes. Global proteome quantification by the super-SILAC strategy enabled us to compare proteins that were differentially expressed in the lung and lymph node metastatic sites. Increased expression of ERBB signaling pathway proteins, including ERBB2 may have promoted increased responsiveness to combination HER2-targeted therapy in the lung metastatic sites compared with the lymph nodes.
Fig. 3.

IPA analysis of significantly changed proteins. A, Selected top canonical pathways represented by proteins with 3-fold changes between lung and either lymph nodes. X-axis is the Z-score of the enriched pathways. Positive Z-score means the pathway is activated and negative Z-score means the pathway is inhibited. B, Heatmap of the significantly changed ERBB signaling. C, Proteins Heatmap of the significantly changed Sirtuin signaling proteins. D, Heatmap of the significantly changed PKA signaling proteins.
Clustering of Metastases Based on Copy Number Alteration and Correlation of Gene Expression and Protein Abundance Data
Genomic copy number changes may alter transcript and protein expression. However, post-transcriptional and post-translational events determine the observed abundance of proteins. We sought to correlate copy number alteration (CNA) and gene expression data, when available, with the protein estimation data described above. Hierarchical clustering of the CNA data showed that gene copy number across many chromosomal locations was, for the lung tumor, quite distinct from that of the lymph nodes, confirming significant metastatic site-specific copy number heterogeneity (Fig. 4A). Interestingly, certain lymph node metastases were more similar in their CNA than others, and hence clustered together. The lymph node PDX was more like LN2 than to the other lymph nodes. Transcriptome sequencing (RNA-seq) was performed to quantify expression in LN1, lung and PDX. We performed a Spearman's correlation of the RNA expression values with either the super-SILAC or TMT protein abundance ratios for these three metasteses. The correlation coefficients (R2) between the RNA expression values and protein abundance ratios ranged between 0.5162 for lung using the super-SILAC ratio (Fig. 4B) and 0.281 for LN1 using the TMT protein ratio (Fig. 4C). The CNA and gene expression data were complementary to the protein abundance data described here to characterize the tumor heterogeneity among the lung and lymph node metastatic sites. Further, this analysis highlighted the fact that there is modest correlation of gene expression and protein abundance data, reiterating the impact of post-transcriptional and post-translational regulation on tumor heterogeneity.
Fig. 4.

Heterogeneity in copy number alterations and correlation of RNA-seq and protein estimation data. A, Hierarchical clustering by copy number across the genome across all tumors (at cytoband resolution). Losses (purple) and gains (red) in log 2 scale are depicted relative to mean ploidy. B, Correlation between super-SILAC-derived proteome and RNA-seq results for LN1 and Lung metastases. X axis is the Log 2 intensity; Y axis is the Log 2 RPKM value. C, Correlation between TMT-derived proteome and RNA-seq results for LN1, Lung, and PDX samples. X axis is the Log 2 intensity; Y axis is the Log 2 RPKM value.
Heterogeneity Across Metastases Based on Global Protein Phosphorylation Levels
The functional consequences of tumor heterogeneity are largely dependent on signaling pathway alterations that are modulated, in part, by protein phosphorylation. Hence, we examined global protein phosphorylation of TMT-labeled lysates of different metastases using a TiO2-based enrichment strategy. Pair-wise correlation analysis of the phosphorylation ratios showed a strong association among autopsy lymph nodes (R2 values ranging from 0.486 to 0.835) (supplemental Fig. S4A). The PCA plot of phosphorylation ratios shows that the autopsy lymph nodes cluster together. Like what we saw for the protein quantitation ratios, LN2 was again closer to lung than to LN1 (supplemental Fig. S4B), although at the phosphorylation level, still quite apart. Hierarchical clustering using the quantitative phosphorylation data across the tissue samples confirmed the similarity of the 5 autopsy lymph nodes. LN1 clustered with LN2, and the lung and the lymph node PDX were distinct (Fig. 5A). There was a distinct cluster of proteins whose phosphorylation was inhibited in the autopsy lymph nodes (Cluster 662, Fig. 5A). Network analysis of these proteins showed that regulation of Rho protein signal transduction, cell-cell junction organization, regulation of mRNA splicing via spliceosome, spliceosome complex activity, and nuclear-transcribed mRNA catabolic process were the top networks represented by the hypo-phosphorylated autopsy lymph node proteins (Fig. 5B). There were several proteins that had increased phosphorylation in the autopsy lymph nodes (Clusters 633, Fig. 5A). Enriched networks among these proteins included cellular component disassembly involved in execution phase of apoptosis, regulation of mRNA processing and mRNA export from nucleus (Fig. 5C). There were proteins that were hyper-phosphorylated across all the tissue samples (Cluster 616, Fig. 5A) and the networks that were enriched among these proteins included spliceosome complex assembly, cellular component disassembly involved in execution phase of apoptosis, establishment of spindle orientation and mRNA export from nucleus (Fig. 5D).
Fig. 5.

Comparison of lung and lymph node metastases phosphoproteomes. A, Hierarchical clustering of protein phosphorylation by tissue shows that autopsy samples are distinct from the other tissues. Columns represent different tissue samples. Rows represent quantified phosphosites. 3 clusters are highlighted and labeled. B, Network of the proteins from cluster 662. C, Network of the proteins from cluster 633. D, Network grouping of the proteins from cluster 616. E, Top canonical pathways represented by phosphosites changed between lung and either lymph node as determined by the TMT experiment. X-axis is the Z-score of the enriched pathways.
Ingenuity Pathway Analysis (IPA) was performed on all identified phosphorylated proteins. Among the top canonical pathways activated in the lung compared with lymph nodes based on the TMT phosphorylation ratio (Lung/LN2 or Lung/LN1) were CDK5, PKA, ERK/MAPK, chemokine, androgen and RhoA signaling pathways (Fig. 5E). Among the pathways inhibited in lung compared with the lymph nodes were apoptosis, AMPK, VEGF, PPAR, SAPK/JNK and p38/MAPK signaling pathways. Global phosphorylation levels of proteins identified from each metastatic site characterized the tumor heterogeneity at greater depth. Differences in phosphorylation across the metastatic sites likely result in altered signaling pathways governing tumor aggressiveness and therapeutic response.
Identification of Variant Peptides and Mutant Proteins by Integrating the Genomics Sequencing and Mass Spectrometry Data
Next-generation sequencing studies (NGS) identify hundreds of somatic mutations in a tumor; however, the determination of driver versus passenger mutations remains a challenge. The first step to such analyses, which is often overlooked in genomics studies, is the confirmation of mutant protein expression. Because we performed an integrated proteogenomics analysis of the metastatic tumors from this patient, we sought to interrogate mutant protein expression from the mass spectrometry data. We constructed a patient-specific protein database based on the whole genome sequencing (5) of LN1, lung (L) and blood of this patient. We identified 78 and 23 mutant peptides from super-SILAC and TMT experiments, respectively. We further validated all the mutant peptides identified by the patient-specific database search. The mutant peptides were validated by searching through different search engines and applying a series of criteria (Fig. 6A). In total, we validated 360 spectra corresponding to 55 germline and 6 somatic variants (supplemental Table S5). In addition, we searched all variant peptides for matching reference sequences, allowing for I/L isobaric substitution, but none were found.
Fig. 6.

Confirmation of the mutant peptides identified from DDA data searched by the patient-specific database. A, Flow chart of the analysis of the MS2 spectra of all the mutant peptides. Two steps of filtering criteria were applied. B, MS/MS spectra of two of the validated variant peptides harboring the somatic mutations CDK12-G879V and FASN-R1439Q. On the left side are spectra of the endogenous “light” peptides identified in the lung (CDK12-G879V) and lymph node (FASN-R1439Q) lysates; on the right side are spectra of the synthetic “heavy” peptides labeled with 13C6 Arg and 13C6 Lys. C, Relative abundance of the two mutant peptides in lung and lymph node lysates by MRM. CDK12-G879V peptide (LADFVLAR) was highly abundant in lung and not detected in lymph node (LN2) whereas the FASN-R1439Q peptide (GILADEDSSQPVWLK) was highly abundant in LN2 and not in the lung. D, TICs of MRM transitions of the CDK12-G879V (top) and FASN-R1439Q (bottom) mutant peptides identified in LN2 (left) and lung (right) lysates. The MRM transitions of CDK12-G879V mutant peptide are identified only in the lung and those of the FASN-R1439Q mutant peptide are identified only in the lymph node.
We confirmed and then annotated spectra for the CDK12-G879V, FASN-R1439Q and HNRNPF-A105T somatic variants in the tumor lysates (Fig. 6B, supplemental Fig. S5, left panel). For further validation, heavy labeled mutant peptides were also synthesized then analyzed by mass spectrometry to match the annotated MS2 spectra (Fig. 6B, supplemental Fig. S5, right panel). The mutant peptide identification and validation pipeline developed to interrogate the data dependent acquisition (DDA) data, including MS2 spectra matching from selected heavy labeled peptides identified key somatic variant and many germline variant peptides confirming mutant protein expression in this patient's tumor and germline.
Targeted Quantitative Mass Spectrometry to Identify CDK12-G879V and FASN-R1439Q in Corresponding Metastatic Lysates
Identification of genomic alterations by NGS has been incorporated into routine clinical practice. However, identification and accurate quantification of variant peptides and proteins are not routine inpatient care. We sought to develop targeted mass spectrometry-based assays to identify and quantify two novel mutant peptides discovered in our study, with an overall goal of clinical application. We employed multiple reaction monitoring (MRM) in a triple quadrupole (QQQ) mass spectrometer to identify and relatively quantify the CDK12-G879V and FASN-R1439Q mutant variant tryptic peptides, in the lung and lymph node (LN2) metastatic sites, respectively. Two mutant peptides CDK12-G879V (LADFVLAR) and FASN-R1439Q (GILADEDSSQPVWLK) labeled with “heavy” amino acids were spiked into the tryptic peptide mix from the Lung and LN2 samples as internal standards for relative quantitation of their endogenous counterparts. Each sample spiked with the corresponding heavy peptide was analyzed in QQQ mass spectrometer in triplicate using the scheduled MRM method using five transitions for each variant peptide (supplemental Fig. S6). The normalized peak area ratio of the CDK12-G879V peptide shows that this mutant CDK12 peptide was significantly more abundant in Lung than in LN2 (Fig. 6C). The TICs of the transitions from the endogenous mutated peptide can clearly be seen in L, but only at background levels in LN2 (Fig. 6D–6E). In contrast, the FASN-R1439Q peptide was more abundant in LN2 than in L (Fig. 6F) and the TICs of the transitions were only detected in LN2, but not in L (Fig. 6G–6H). The MRM assays for these novel variant peptides confirmed the identity and differential expression of the mutant proteins in the lung and lymph node metastatic sites.
Functional Characterization of CDK12-G879V Mutant
The G879V mutation in CDK12 occurs in the DFG motif of the kinase domain and is expected to destabilize the active structure resulting in a non-functional kinase. CDK12 kinase activity is required for the expression of long transcripts, many of which are DNA damage response (DDR) genes. Hence, we hypothesized that the non-functional G879V mutant CDK12 will increased chemotherapy sensitivity in lung adenocarcinoma cells because of a diminished DNA damage response. Carboplatin, the chemotherapy drug used in this patient as a component of the first line of treatment causes DNA damage by multiple mechanisms. In addition, other agents, including pemetrexed and capecitabine affect DNA synthesis. To test our hypothesis, we ablated CDK12 gene expression in A549 cells, a human lung adenocarcinoma cell line that lacks both the ERBB2 and CDK12 mutations, stably expressing Cas9 using two CRISPR sgRNAs targeting exon 1 and exon 2. CDK12 protein expression was significantly lower in both clones compared with the parental cells (Fig. 7A). Further, the cell lines with diminished CDK12 expression were more sensitive, than the parental cells, to camptothecin, a DNA topoisomerase I inhibitor that inhibits DNA replication (Fig. 7B), consistent with CDK12 being essential for DNA damage repair. To further assay the extent of DNA damage because of camptothecin treatment, we performed the γH2AX assay (Fig. 7C). Quantitation of γH2AX foci showed that, compared with parental cells, on camptothecin treatment, CDK12 knockdown increased DNA damage significantly (Fig. 7D). Further, we could rescue the cell viability on camptothecin treatment of CDK12 knockout cells by overexpressing wild type CDK12, but not with the CDK12-G879V mutant (Fig. 7E–7F), demonstrating that this mutation ablates CDK12 DNA damage repair function.
Fig. 7.

CRISPR-Cas9-mediated knockdown of CDK12 increases chemotherapy sensitivity in A549 human lung adenocarcinoma cells. A, Western blotting of CDK12 in parental A549-Cas9 control cells and stable clones expressing CRISPR sgRNA targeting exon 1 (261-7) and exon 2 (264–5). CDK12 expression is ablated in both clones. B, Camptothecin sensitivity of the parental and CDK12 knockdown cell lines. The data is normalized to mock (PBS) treatment (left) or the with parental control cells (right) (*** p < 0.001; ** p < 0.01; * p < 0.05). C–D, γH2AX foci formation on treatment with camptothecin (C) and their quantitation (D) in parental and CDK12 exon 1 and exon 2 knockout cells. E, Wild type CDK12 and CDK12-G879V mutant were overexpressed in CDK12 exon 2 knockout cells for rescue experiments. Western blotting for CDK12 expression (upper panel) shows overexpression of corresponding CDK12 protein in the CDK12 exon 2 knockout cells. Rho-GDI was used as a loading control and relative quantitation of CDK12 expression is shown in lower panel. F, Camptothecin sensitivity of CDK12 exon 2 knockout cells compared with parental cells and its rescue by wild type CDK12, but not CDK12-G879V mutant (*** p < 0.001; ** p < 0.01; * p < 0.05).
DISCUSSION
The overall goal of our study was to examine tumor heterogeneity at the level of the proteome and phosphoproteome in our patient who survived with metastatic lung adenocarcinoma for over 7 years while undergoing combination HER2-directed therapy along with chemotherapy. We analyzed nine different tumor metastases sequentially acquired, on progression, by multiple biopsies and surgeries and at autopsy. We have previously demonstrated unprecedented mutational heterogeneity, in this patient, using whole genome and targeted NGS. Less than 1% of the somatic variants were common between the lymph node and lung metastatic sites (5). However, we demonstrated that similarities in key hallmarks of carcinogenesis, such as proliferation were manifest by different mechanisms in the lung and lymph node metastatic sites. We hypothesized that analysis of the proteome and the phosphorpoteome will provide more granular details of tumor evolution and give additional insight into the natural history of tumor progression in this patient. To our knowledge, this is one of the initial studies to perform comprehensive genomics and mass spectrometry-based quantitative proteomics across multiple sequential biopsies of progressive lesions and at autopsy in an “exceptional responder” lung adenocarcinoma patient. Our patient responded to combination chemotherapy along with HER2-directed targeted therapy over the span of 7 years, during which time his lymph node metastases repeatedly progressed. At autopsy we did not see any lung metastatic sites, suggesting the lung metastases were “cured” in this “exceptional responder” patient. We had demonstrated before that our patient harbored an L869R ERBB2 mutation in the lymph node metastatic sites, but not the lung metastatic sites. Although ERBB2 was amplified in both sites, the degree of amplification was far greater in the lung compared with the lymph node (5). Here, we use quantitative mass spectrometry to demonstrate that ERBB2 protein levels were 20–70-fold greater in the lung compared with the two lymph nodes. Interestingly, there was also tumor evolution and selection for higher ERBB2 protein levels in LN2 (LN2/LN1 fold change 3.2) that had been procured over 2 years after the progressive lung metastasis and 2.5 years after the progressive lymph node LN1. The control of multiple metastases using HER2-directed combination therapy suggests HER2 was the major driver of carcinogenesis in this patient; through amplification and increased protein levels in the lung and a somatic kinase domain mutation in the lymph node sites.
One major reason the patient may have survived such a long time with metastatic disease was that his lung metastases were essentially “cured” after the surgical removal of the progressive lung tumor, which was likely the primary lesion. At autopsy, we did not detect any lung tumor metastases. One reason could be the significantly increased ERBB2 protein levels in the lung metastases as shown in this study, and the absence of the ERBB2-L869R mutation. We have previously shown that this novel ERBB2 mutant is more resistant to lapatinib, a HER2-targeting small molecule inhibitor, compared with wild type ERBB2. Further, cells expressing ERBB2-L869R developed greater resistance to lapatinib on prolonged exposure compared with wild type ERBB2 (5). Hence the lung tumor metastases were more dependent on HER2 inhibition by combination targeted therapy, whereas the lymph node metastases were relatively resistant and hence, continuously progressed. We also demonstrate the effect of the novel CDK12-G879V mutation in promoting chemotherapy sensitivity of the lung metastases. Here, we show convincingly that the CDK12-G879V mutant is expressed at the protein-level in the lung metastasis, but not the lymph nodes (Fig. 6). This mutation falls within the conserved DFG motif in the kinase domain of CDK12. A CDK12-D877N mutation within the DFG motif has been documented, by TCGA, in a cholangiocarcinoma patient. Further, this CDK12-D877N mutation has been used as a kinase dead control in various functional studies (18). CDK12, in association with cyclin K, has been shown to regulate transcriptional elongation of long transcript genes, many of which are involved in DNA damage response (DDR) (19, 20). CDK12 was shown to be necessary for the expression of BRCA1, ATR, FANCI and FANCD2 on DNA damage. This was attributed to phosphorylation of Ser-2 in the heptapeptide repeat located in the C-terminal domain (CTD) of RNA polymerase II (21). CDK12 kinase domain mutations are seen in about 3% of serous ovarian cancer patients in the TCGA. In another study depletion of CDK12 resulted in deceased BRCA1 levels, reduced RAD51 foci formation and HR repair in ovarian cancer cells (20). CDK12 inhibition by pharmacological inhibition has been shown to circumvent intrinsic and acquired resistance to PARP inhibitors in BRCA wild type and mutated models of triple negative breast cancer (22). We hypothesized that the G879V mutation within the conserved DFG motif in the kinase domain of CDK12 will result in a non-functional kinase and increase chemotherapy sensitivity of tumors. We engineered CRSPR-Cas9 mediated knockdown of CDK12 in the A549 lung adenocarcinoma cell line. Complete ablation of CDK12 protein expression indeed increased chemotherapy sensitivity of these cells (Fig. 7). This correlated with increased DNA damage foci in the knock-down cells treated with chemotherapeutic agents, suggesting more DNA damage and less repair. Further, this effect could be rescued on expression of wild type CDK12, but not the CDK12-G879V mutant.
We compared two methodologies for performing quantitative mass spectrometry using DDA data, namely the “super-SILAC” and TMT labeling method. The “super-SILAC” method has been previously used to quantify the proteome in human breast cancer by spiking “super-SILAC” mix of heavy labeled lysates from breast cancer cell lines (23). We used a similar strategy, and to our knowledge, for the first time made a lung adenocarcinoma-specific “super-SILAC” mix of heavy labeled lysates. We developed a pooled sample of heavy labeled lysates from 12 cell lines, including 2 immortalized normal lung epithelial cell lines, 1 lung adenocarcinoma (LUAD) cell line lacking EGFR or KRAS mutations, 6 EGFR mutant LUAD cell lines harboring the most common EGFR mutations, and 3 KRAS mutant LUAD cell lines. Because this pool contains some of the major subtypes of lung adenocarcinoma, it can be used to compare mass spectrometry-based proteomics experiments performed in different laboratories. The problem with “super-SILAC” data is that of missing data. Our super-SILAC strategy identified slightly more proteins, however, quantified proteins were similar to the TMT labeling strategy (Fig. 1D, 7E). More importantly, the correlation of the quantitation data overall was quite good between the two strategies with R2 values between 0.64 and 0.79 (Fig. 2A).
One purpose of this study was to correlate genomic and proteomic data using copy number analysis (CNA), transcriptome analysis (RNA-seq) and mass spectrometry-based quantitative proteomics. CNA clearly showed that the lung metastatic site was significantly different than the 8 lymph node metastases (Fig. 4A). This reinforces the unprecedented genomic tumor heterogeneity already seen in this patient with respect to somatic variants. We had demonstrated less than 1% similarity of somatic variants between the lung and lymph node metastases based on whole WGS data (5). RNA of adequate quality was available from, LN1, L and PDX, three distinct metastases. Spearman's correlation (range 0.28–0.52) of the RNA-seq-based FPKM values for transcript expression compared with either the super-SILAC or the TMT protein ratio across datasets demonstrated low correlation of transcript and protein data across a single sample, consistent with results obtained in various other systems.
We constructed a patient-specific database using the WGS data we previously published (5) using all germline and somatic variants identified in the lung and lymph node metastases and normal blood combined with the normal human RefSeq database. We used an extensive validation pipeline to confirm the identification of the variant MS/MS spectra. We also matched the spectra obtained from the patient's tumor tissue with heavy labeled variant synthetic peptides. Although, we identified a total ∼2000 germline and somatic variant peptides, these represented only 55 germline and 6 somatic variants. Interestingly, we identified the CDK12-G879V mutant tryptic peptide in metastatic lung tumor lysates without enrichment. This could be because of overexpression of the mutant protein from the amplified ERBB2/CDK12 locus. The overall low percentage of variant peptide detection by mass spectrometry has been previously documented (7–10). This is likely a result of limited peptide coverage of the individual proteins by current mass spectrometry technologies in global proteomics experiments. Regardless of this limitation, any somatic variant peptide detected by mass spectrometry is likely to be important because the variant is expressed at the protein level specifically in the tumor. Here, we also developed an MRM assay for the novel CDK12-G879V variant to perform quantitative estimation of variant peptide/protein expression. Such assays using variant peptides would allow the quantitation of mutant proteins from a limited amount of biopsy sample.
Proteomic and phosphoproteomic heterogeneity has been demonstrated before by quantitative mass spectrometry of metastatic pancreatic cancer sites procured from a single patient at autopsy. Interestingly, an AXL inhibitor was more effective in lung and liver metastatic sites than in peritoneal sites, suggesting that there was heterogeneity in signaling pathway activation in different metastatic sites (24). Our study involves multiple metastatic sites procured during the treatment course over 7 years, including at autopsy. We also reveal the importance of integrated proteogenomic analyses in such studies to identify variant peptides in mass spectrometry data. We demonstrate how the CDK12-G879V mutant specific to the lung metastatic site contributed to the enhanced sensitivity of the lung metastatic sites to a combination of chemotherapy along with HER2-targeted therapy. The knockdown and rescue experiments (Fig. 7) further validate our hypothesis that CDK12-G879V mutation results in a non-functional CDK12 kinase, particularly in relation to the role of CDK12 in DNA damage repair. We also developed an MRM assay to quantify the CDK12-G879V mutant peptide. We could identify this variant peptide in the lung lysates without any enrichment, likely because of high mutant protein expression, thus confirming the importance of CDK12 amplification detected by whole genome sequencing and CNV-seq.
In summary, we have shown the feasibility of examining multiple biopsies obtained from an “exceptional responder” lung adenocarcinoma patient throughout treatment and culminating in an autopsy. The global proteomics and phosphoproteomics, CNA and transcript expression described in this study together with the NGS analyses published previously illuminate the unique tumor evolution of this patient. Most importantly, our study highlights the functional significance of one of the most relevant somatic variants, the CDK12-G879V mutation, identified directly at the peptide-level by both DDA and MRM mass spectrometry. Most importantly, this variant may have influenced the overall prognosis and treatment response of this patient.
Data Availability
The MS proteomics data in this study has been deposited in ProteomeXchange Consortium (http://proteomecentral.proteomeexchange.org) via the PRIDE partner repository with the dataset identifier PXD010779.
Supplementary Material
Acknowledgments
Clinical study statement: This study was performed with written consent from our patient enrolled in a Molecular Profiling study at the National Institutes of Health (NIH) Clinical Center (NCT01306045). The NIH-institutional review board approved the study under study number 11-C-0096.
Footnotes
* This work was supported by the U.S. National Cancer Institute, Center for Cancer Research (NCI-CCR) Intramural Research Program support (U.G.) and partly from the CPTAC grant U24 CA210972 (D.F.).
 This article contains supplemental material. Authors declare that they have no conflicts of interest.
This article contains supplemental material. Authors declare that they have no conflicts of interest.
1 The abbreviations used are:
- NSCLC
- nonsmall cell lung cancer
- EGFR
- epidermal growth factor receptor
- SILAC
- stable isotope labelling with amino acids in cell culture
- PDX
- patient derived xenograft
- PPI
- protein-protein interaction
- MRM
- multiple reaction monitoring.
REFERENCES
- 1. Gerlinger M., Horswell S., Larkin J., Rowan A. J., Salm M. P., Varela I., Fisher R., McGranahan N., Matthews N., Santos C. R., Martinez P., Phillimore B., Begum S., Rabinowitz A., Spencer-Dene B., Gulati S., Bates P. A., Stamp G., Pickering L., Gore M., Nicol D. L., Hazell S., Futreal P. A., Stewart A., and Swanton C. (2014) Genomic architecture and evolution of clear cell renal cell carcinomas defined by multiregion sequencing. Nat. Genet. 46, 225–233 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. de Bruin E. C., McGranahan N., Mitter R., Salm M., Wedge D. C., Yates L., Jamal-Hanjani M., Shafi S., Murugaesu N., Rowan A. J., Gronroos E., Muhammad M. A., Horswell S., Gerlinger M., Varela I., Jones D., Marshall J., Voet T., Van Loo P., Rassl D. M., Rintoul R. C., Janes S. M., Lee S. M., Forster M., Ahmad T., Lawrence D., Falzon M., Capitanio A., Harkins T. T., Lee C. C., Tom W., Teefe E., Chen S. C., Begum S., Rabinowitz A., Phillimore B., Spencer-Dene B., Stamp G., Szallasi Z., Matthews N., Stewart A., Campbell P., and Swanton C. (2014) Spatial and temporal diversity in genomic instability processes defines lung cancer evolution. Science 346, 251–256 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Zhang J., Fujimoto J., Zhang J., Wedge D. C., Song X., Zhang J., Seth S., Chow C. W., Cao Y., Gumbs C., Gold K. A., Kalhor N., Little L., Mahadeshwar H., Moran C., Protopopov A., Sun H., Tang J., Wu X., Ye Y., William W. N., Lee J. J., Heymach J. V., Hong W. K., Swisher S., Wistuba I. I., and Futreal P. A. (2014) Intratumor heterogeneity in localized lung adenocarcinomas delineated by multiregion sequencing. Science 346, 256–259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. McGranahan N., and Swanton C. (2015) Biological and therapeutic impact of intratumor heterogeneity in cancer evolution. Cancer Cell 27, 15–26 [DOI] [PubMed] [Google Scholar]
- 5. Biswas R., Gao S., Cultraro C. M., Maity T. K., Venugopalan A., Abdullaev Z., Shaytan A. K., Carter C. A., Thomas A., Rajan A., Song Y., Pitts S., Chen K., Bass S., Boland J., Hanada K. I., Chen J., Meltzer P. S., Panchenko A. R., Yang J. C., Pack S., Giaccone G., Schrump D. S., Khan J., and Guha U. (2016) Genomic profiling of multiple sequentially acquired tumor metastatic sites from an “exceptional responder” lung adenocarcinoma patient reveals extensive genomic heterogeneity and novel somatic variants driving treatment response. Cold Spring Harb. Mol. Case Stud. 2, a001263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. TCGA. (2014) Comprehensive molecular profiling of lung adenocarcinoma. Nature 511, 543–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhang B., Wang J., Wang X., Zhu J., Liu Q., Shi Z., Chambers M. C., Zimmerman L. J., Shaddox K. F., Kim S., Davies S. R., Wang S., Wang P., Kinsinger C. R., Rivers R. C., Rodriguez H., Townsend R. R., Ellis M. J., Carr S. A., Tabb D. L., Coffey R. J., Slebos R. J., Liebler D. C., and Nci C. (2014) Proteogenomic characterization of human colon and rectal cancer. Nature 513, 382–387 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Zhang H., Liu T., Zhang Z., Payne S. H., Zhang B., McDermott J. E., Zhou J. Y., Petyuk V. A., Chen L., Ray D., Sun S., Yang F., Chen L., Wang J., Shah P., Cha S. W., Aiyetan P., Woo S., Tian Y., Gritsenko M. A., Clauss T. R., Choi C., Monroe M. E., Thomas S., Nie S., Wu C., Moore R. J., Yu K. H., Tabb D. L., Fenyo D., Bafna V., Wang Y., Rodriguez H., Boja E. S., Hiltke T., Rivers R. C., Sokoll L., Zhu H., Shih I. M., Cope L., Pandey A., Zhang B., Snyder M. P., Levine D. A., Smith R. D., Chan D. W., Rodland K. D., and Investigators C. (2016) Integrated proteogenomic characterization of human high-grade serous ovarian cancer. Cell 166, 755–765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mertins P., Mani D. R., Ruggles K. V., Gillette M. A., Clauser K. R., Wang P., Wang X., Qiao J. W., Cao S., Petralia F., Kawaler E., Mundt F., Krug K., Tu Z., Lei J. T., Gatza M. L., Wilkerson M., Perou C. M., Yellapantula V., Huang K. L., Lin C., McLellan M. D., Yan P., Davies S. R., Townsend R. R., Skates S. J., Wang J., Zhang B., Kinsinger C. R., Mesri M., Rodriguez H., Ding L., Paulovich A. G., Fenyo D., Ellis M. J., Carr S. A., and Nci C. (2016) Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 534, 55–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Ruggles K. V., Tang Z., Wang X., Grover H., Askenazi M., Teubl J., Cao S., McLellan M. D., Clauser K. R., Tabb D. L., Mertins P., Slebos R., Erdmann-Gilmore P., Li S., Gunawardena H. P., Xie L., Liu T., Zhou J. Y., Sun S., Hoadley K. A., Perou C. M., Chen X., Davies S. R., Maher C. A., Kinsinger C. R., Rodland K. D., Zhang H., Zhang Z., Ding L., Townsend R. R., Rodriguez H., Chan D., Smith R. D., Liebler D. C., Carr S. A., Payne S., Ellis M. J., and Fenyo D. (2016) An analysis of the sensitivity of proteogenomic mapping of somatic mutations and novel splicing events in cancer. Mol. Cell Proteomics 15, 1060–1071 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cox J., and Mann M. (2008) MaxQuant enables high peptide identification rates, individualized p.p.b.-range mass accuracies and proteome-wide protein quantification. Nature Biotechnol. 26, 1367–1372 [DOI] [PubMed] [Google Scholar]
- 12. Cox J., Neuhauser N., Michalski A., Scheltema R. A., Olsen J. V., and Mann M. (2011) Andromeda: a peptide search engine integrated into the MaxQuant environment. J Proteome Res. 10, 1794–1805 [DOI] [PubMed] [Google Scholar]
- 13. Cline M. S., Smoot M., Cerami E., Kuchinsky A., Landys N., Workman C., Christmas R., Avila-Campilo I., Creech M., Gross B., Hanspers K., Isserlin R., Kelley R., Killcoyne S., Lotia S., Maere S., Morris J., Ono K., Pavlovic V., Pico A. R., Vailaya A., Wang P. L., Adler A., Conklin B. R., Hood L., Kuiper M., Sander C., Schmulevich I., Schwikowski B., Warner G. J., Ideker T., and Bader G. D. (2007) Integration of biological networks and gene expression data using Cytoscape. Nat. Protoc. 2, 2366–2382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bindea G., Mlecnik B., Hackl H., Charoentong P., Tosolini M., Kirilovsky A., Fridman W. H., Pages F., Trajanoski Z., and Galon J. (2009) ClueGO: a Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 25, 1091–1093 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Nesvizhskii A. I. (2014) Proteogenomics: concepts, applications and computational strategies. Nat. Methods 11, 1114–1125 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kelly R. J., Carter C., and Giaccone G. (2010) Personalizing therapy in an epidermal growth factor receptor-tyrosine kinase inhibitor-resistant non-small-cell lung cancer using PF-00299804 and trastuzumab. J. Clin. Oncol. 28, e507–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kelly R. J., Carter C. A., and Giaccone G. (2012) HER2 mutations in non-small-cell lung cancer can be continually targeted. J. Clin. Oncol. 30, 3318–3319 [DOI] [PubMed] [Google Scholar]
- 18. Bosken C. A., Farnung L., Hintermair C., Merzel Schachter M., Vogel-Bachmayr K., Blazek D., Anand K., Fisher R. P., Eick D., and Geyer M. (2014) The structure and substrate specificity of human Cdk12/Cyclin K. Nat. Commun. 5, 3505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Blazek D., Kohoutek J., Bartholomeeusen K., Johansen E., Hulinkova P., Luo Z., Cimermancic P., Ule J., and Peterlin B. M. (2011) The Cyclin K/Cdk12 complex maintains genomic stability via regulation of expression of DNA damage response genes. Genes Dev. 25, 2158–2172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Joshi P. M., Sutor S. L., Huntoon C. J., and Karnitz L. M. (2014) Ovarian cancer-associated mutations disable catalytic activity of CDK12, a kinase that promotes homologous recombination repair and resistance to cisplatin and poly(ADP-ribose) polymerase inhibitors. J. Biol. Chem. 289, 9247–9253 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Blazek D. (2012) The cyclin K/Cdk12 complex: an emerging new player in the maintenance of genome stability. Cell Cycle 11, 1049–1050 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Johnson S. F., Cruz C., Greifenberg A. K., Dust S., Stover D. G., Chi D., Primack B., Cao S., Bernhardy A. J., Coulson R., Lazaro J. B., Kochupurakkal B., Sun H., Unitt C., Moreau L. A., Sarosiek K. A., Scaltriti M., Juric D., Baselga J., Richardson A. L., Rodig S. J., D'Andrea A. D., Balmana J., Johnson N., Geyer M., Serra V., Lim E., and Shapiro G. I. (2016) CDK12 inhibition reverses de novo and acquired PARP inhibitor resistance in BRCA wild-type and mutated models of triple-negative breast cancer. Cell Rep. 17, 2367–2381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Geiger T., Cox J., Ostasiewicz P., Wisniewski J. R., and Mann M. (2010) Super-SILAC mix for quantitative proteomics of human tumor tissue. Nat. Methods 7, 383–385 [DOI] [PubMed] [Google Scholar]
- 24. Kim M. S., Zhong Y., Yachida S., Rajeshkumar N. V., Abel M. L., Marimuthu A., Mudgal K., Hruban R. H., Poling J. S., Tyner J. W., Maitra A., Iacobuzio-Donahue C. A., and Pandey A. (2014) Heterogeneity of pancreatic cancer metastases in a single patient revealed by quantitative proteomics. Mol. Cell Proteomics 13, 2803–2811 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The MS proteomics data in this study has been deposited in ProteomeXchange Consortium (http://proteomecentral.proteomeexchange.org) via the PRIDE partner repository with the dataset identifier PXD010779.