

Abstract
Many DNA methylome profiling methods cannot distinguish between 5-methylcytosine (5mC) and 5-hydroxymethylcytosine (5hmC). Because 5mC typically acts as a repressive mark whereas 5hmC is an intermediate form during active demethylation, the inability to separate their signals could lead to incorrect interpretation of the data. Is the extra information contained in 5hmC signals worth the additional experimental and computational costs? Here we combine whole-genome bisulfite sequencing (WGBS) and oxidative WGBS (oxWGBS) data in various human tissues to investigate the quantitative relationships between gene expression and the two forms of DNA methylation at promoters, transcript bodies, and immediate downstream regions. We find that 5mC and 5hmC signals correlate with gene expression in the same direction in most samples. Considering both types of signals increases the accuracy of expression levels inferred from methylation data by a median of 18.2% as compared to having only WGBS data, showing that the two forms of methylation provide complementary information about gene expression. Differential analysis between matched tumor and normal pairs is particularly affected by the superposition of 5mC and 5hmC signals in WGBS data, with at least 25%–40% of the differentially methylated regions (DMRs) identified from 5mC signals not detected from WGBS data. Our results also confirm a previous finding that methylation signals at transcript bodies are more indicative of gene expression levels than promoter methylation signals. Overall, our study provides data for evaluating the cost-effectiveness of some experimental and analysis options in the study of DNA methylation in normal and cancer samples.
DNA methylation, the methylation of the carbon 5 atom of cytosines, usually occurs within the CpG context in eukaryotes, and in some cell types, also within CpHpG and CpHpH contexts (Bird 2002; Cokus et al. 2008; Lister et al. 2009). It is involved in various biological processes, including embryonic development, genomic imprinting, X Chromosome inactivation, and genome stability maintenance (Lister et al. 2009). Aberrant DNA methylation is associated with a variety of human diseases, including cancer (Robertson 2005). Many types of cancer show global hypomethylation as compared to normal tissues, whereas specific loci could be hypermethylated (Ehrlich 2009; Klutstein et al. 2016).
DNA methylation is tightly related to gene expression. Methylation of CpG islands within promoter regions is associated with long-term gene silencing, whereas methylation in other regions is more dynamic and tissue-specific (Jones 2012). Sequences up to 2 kb away from CpG islands, termed CpG island shores, have been shown to display differential methylation in cancer that correlates with differential gene expression (Irizarry et al. 2009). DNA methylation at regulatory elements other than promoters is less studied, but some recent work has started to show correlations between enhancer methylation and gene silencing (Aran et al. 2013; Heyn et al. 2016; Cao et al. 2017). How gene body methylation is related to gene expression has been more controversial, with a positive correlation between them observed in some cell types but not in some others. Mechanistically, gene body methylation could be related to repression of antisense transcript, efficiency of transcription elongation, usage of alternative promoter, and RNA splicing (Rountree and Selker 1997; Lorincz et al. 2004; Choi et al. 2009; Maunakea et al. 2010).
The ambiguous relationship between gene body methylation and gene expression could be partly due to the presence of multiple forms of DNA methylation. 5-Methylcytosine (5mC) can be converted by the Ten-eleven translocation (TET) family of proteins into 5-hydroxymethylcytosine (5hmC), 5-formylcytosine (5fC), and 5-carboxylcytosine (5caC) during active demethylation (Song et al. 2012). Unlike 5mC's characteristic enrichment at promoters of repressed genes, 5hmC has been found enriched at active enhancers and around expressed genes, including gene body regions (Song et al. 2012; Yu et al. 2012). If an experimental method cannot distinguish between 5mC and 5hmC (or the other two intermediate forms), depending on their relative levels, methylation may appear to correlate with gene expression in different ways for different genes with the same total methylation level.
Unfortunately, that is exactly the situation with many commonly used experimental methods. In standard bisulfite conversion, which is used in whole-genome bisulfite sequencing (WGBS), reduced representation bisulfite sequencing (RRBS), and Infinium 27K/450K/EPIC arrays, both 5mC and 5hmC are unconverted. The resulting data can only tell whether a cytosine is methylated or not but cannot tell which form of methylation a cytosine takes (Jin et al. 2010). Some other methods such as MeDIP-seq and MBDCap-seq could specifically detect 5mC (Jin et al. 2010), but they do not offer single-base resolution and fail to provide information about the other forms of DNA methylation.
Recently, realizing the importance of 5hmC as a representative of the demethylation forms, a number of methods have been proposed to detect it at single-base resolution (Petterson et al. 2014; Li et al. 2016). Some of these methods are based on oxidative bisulfite sequencing (oxBS), which specifically detects 5mC. Conceptually, by subtracting the methylation level detected by oxBS from that detected by standard bisulfite sequencing (BS), the 5hmC level can also be deduced. In practice, this subtraction could lead to negative values due to errors and stochastic factors in the experimental and analysis procedures, which can be corrected computationally (Xu et al. 2016).
Currently, it is still not completely clear how 5mC and 5hmC at promoter, gene body, and downstream regions are related to gene expression, both separately and jointly. For instance, was the previously observed positive correlation between gene body methylation and gene expression purely due to 5hmC? Do 5mC and 5hmC correlate with gene expression in opposite directions? If 5mC level is already known, does 5hmC level provide extra information about gene expression in normal and disease samples?
Also, a lot of existing knowledge about DNA methylation is qualitative rather than quantitative. For example, strong promoter 5mC is known to be associated with gene silencing, but the expected amount of gene expression given a certain level of promoter methylation is usually not known. More generally, if the 5mC and 5hmC levels at the promoter, gene body, and downstream regions are measured, is it possible to tell the corresponding expression level of a gene? This question is particularly important in epigenomic studies of diseases, in which a common question is whether an observed expression level change of a gene can be attributed to the promoter methylation change alone or whether it is also affected by methylation at other regions or even by other regulatory mechanisms.
In addition to the methylation level (“β value”), defined as the proportion of methylated reads/signal intensity at a CpG site among the total number of reads/signal intensity, it has been proposed that the proportion of discordant reads (PDRs), defined as the ratio of reads having discordant methylation status at different CpGs, is a better indicator of gene expression level in chronic lymphocytic leukemia (Landau et al. 2014). Is PDR generally more informative than β values, especially when signals for 5mC and 5hmC are separately measured?
In this work, we use WGBS and oxidative WGBS (oxWGBS) data from normal liver and lung tissues and paired cancer samples to study the quantitative relationships between gene expression and the two DNA methylation forms, 5mC and 5hmC, quantified by both β values and PDRs, at different genic and regulatory elements associated with the transcripts. In addition to answering the above conceptual questions, another goal of this study is to provide practical guidelines as to whether both 5mC and 5hmC should be measured and whether both β values and PDRs should be computed, neither of which is a common practice currently.
Results
5mC and 5hmC levels alone can partially infer transcript expression level
We obtained WGBS, oxWGBS, and RNA sequencing (RNA-seq) data for 12 samples, including three pairs of human normal liver tissues (Liver N1–N3) and matched tumors (Liver T1–T3) and three pairs of human normal lung tissues (Lung N1–N3) and matched tumors (Lung T1–T3) (Li et al. 2016). Based on these data, for each transcript, we computed the average raw WGBS and oxWGBS β values, as well as the inferred 5mC and 5hmC levels, at its 16 associated upstream, transcript body, and downstream regions in each sample (Fig. 1A; Materials and Methods). Heat maps of the resulting data set reveal some subtle correlations between these methylation features at the different associated regions and the corresponding expression levels of the transcripts, with lower methylation at promoters and some body features for transcripts with higher expression (Fig. 1B; Supplemental Fig. S1). A hierarchical clustering of the samples based on all their methylation features shows two main clusters corresponding to the two tissues of origin rather than cancer status (Fig. 1C). A related observation has recently been made based on gene expression data of 8000 patients of 17 cancer types, that liver tumors are more similar to normal liver tissues than to other types of tumors (Uhlen et al. 2017).
Figure 1.

Definition of the regions associated with each transcript and the resulting data set. (A) Genomic regions defined for each transcript at which the β values or PDR values were used to infer expression level of the transcript. Both the upstream (Up) and downstream (Down) regions were divided into five 400-bp bins (Up1–Up5 and Down1–Down5). The transcript body (Body) was divided into first exon (FirstEx), first intron (FirstIn), internal exons (IntEx), internal introns (IntIn), last exon (LastEx), and last intron (LastIn). (B) A heat map of the resulting large data set for sample Liver T1. Each row represents a transcript, and the transcripts are sorted in ascending order according to their expression levels. The four blocks of columns represent β values based on WGBS, oxWGBS, 5mC, and 5hmC, respectively. Within each block, the different columns are, respectively, Up5–Up1, FirstEx, FirstIn, IntEx, IntIn, LastEx, LastIn, and Down1–Down5. After the four methylation blocks, the last two columns show the log expression level and expression class, respectively. (C) Hierarchical clustering of the samples based on all their methylation features in the large data set using Ward's method.
Before proceeding to other analyses, we first verified the computed 5mC and 5hmC β values by comparing them with another data set with the two types of methylation signals profiled by combining standard RRBS and TET-assisted modification of RRBS (TAB-RRBS) (Hlady et al. 2019). After removing source-specific biases, in the first two principal components, the nontumor samples from the two data sets were found to form clusters together, which were largely separated from the tumor samples (Supplemental Fig. S2). These results show that the 5mC and 5hmC levels for the same group of samples are comparable across the two studies, despite the different biological samples and experimental protocols involved.
We also defined a similar data set with both β values and PDR values computed. Although β values can be computed from the raw WGBS and oxWGBS data, as well as the processed 5mC and 5hmC levels, PDR values can only be defined directly from sequencing reads and were thus computed from raw WGBS and oxWGBS data only. To ensure reliable calculations of PDR values, only regions with sufficient read coverage were considered (Methods), leading to a smaller number of transcripts included in this data set. Hereafter, we refer to this data set as the “small” set and the data set with only β-value features as the “large” set. In the following, we first focus on the analyses of the large data set.
To investigate whether β values in the regions associated with a transcript are indicative of its expression level, we performed statistical modeling of transcript expression classes (zero, low, and high expression) and evaluated the accuracy of the resulting models using a rigorous cross-validation procedure (Methods).
Considering methylation levels in all 16 regions associated with each transcript, the constructed models were fairly accurate in separating transcripts belonging to the different expression classes, with a median area under the receiver–operator characteristic (AUROC) of around 0.7 (Fig. 2A, “BS + oxBS + 5mC + 5hmC”). This value is close to the AUROC reported in a previous study that involved only WGBS features (Lou et al. 2014) despite a more rigorous evaluation procedure and a different way of quantifying methylation level used in the current study. We found that the modeling accuracy was not affected by the sequencing depth, in that transcripts with different read depths received similar AUROC values except when the depth was less than 10 (Supplemental Fig. S3), which involved only 0.25%–0.89% of transcripts based on the WGBS and oxWGBS data. We also derived methylation features further away from each transcript, covering 96% of CpG island shores, and found that the accuracy of the resulting models is similar to the ones covering only the 16 regions with the difference of average AUROC for each sample group not more than 0.006 (Supplemental Fig. S4), suggesting that these 16 regions already capture a substantial portion of information about gene expression.
Figure 2.

Accuracy of the models for inferring expression classes based on the large data set. (A–C) Each bar represents the distribution of AUROC values across the three expression classes of the three samples in each sample group. (A) Comparison of models involving different combinations of methylation features from all associated genomic regions of the transcripts. (B) Comparison of models involving both 5mC and 5hmC levels at different combinations of genomic regions associated with each transcript. (C) Comparison of several knowledge-driven models. (D) The most useful methylation feature blocks for inferring gene expression level based on the forward-search procedure of feature selection. For each sample, the top feature block was given a score of eight, the second given a score of seven, and so on, for the top eight feature blocks. The total score of each feature block across all 12 samples is shown as a percentage of the maximum possible score of 8 × 12 = 96.
Comparing the different expression classes, the DNA methylation features were more successful in identifying transcripts with zero or high expression than those with an intermediate expression level (Supplemental Fig. S5).
To evaluate whether these results are sensitive to the transcript annotation set, we repeated the above procedures considering only protein-coding transcripts and/or only the transcripts with experimental evidence or manual curation (from GENCODE levels 1 and 2). The resulting AUROC values were similar for all these settings (Supplemental Fig. S6), suggesting that the models constructed were general for both protein-coding and noncoding genes and for transcripts at different confidence levels.
5mC and 5hmC provide complementary information about gene expression
We then investigated the relative importance of the DNA methylation features by constructing models using only subsets of features. First, we compared methylation features derived from the four types of methylation data considering all 16 associated regions (Fig. 2A). Among the models involving only features derived from a single type of data, the models with WGBS, oxWGBS, or 5mC features had similar AUROC values, all higher than models with 5hmC features. On the other hand, models involving both 5mC and 5hmC features (“5mC + 5hmC”) performed better than models involving either 5mC or 5hmC features alone, most substantially for the normal liver samples, showing that these two forms of DNA methylation provide complementary information about gene expression. Similarly, combining both WGBS and oxWGBS features (“BS + oxBS”) slightly improved the modeling results as compared to having only WGBS or only oxWGBS features. Finally, models involving all four types of data (“BS + oxBS + 5mC + 5hmC”) had similar performance as models involving only the inferred 5mC and 5hmC levels (“5mC + 5hmC”), suggesting that these derived DNA methylation features successfully captured the essential information about gene expression contained in the raw WGBS and oxWGBS data. Overall, models involving all features had a median AUROC improvement of 0.7%–7.2% for the different samples compared with the models involving only WGBS features.
To evaluate whether these findings are specific to liver and lung tissues, we collected additional genome-wide 5mC, 5hmC, and gene expression data from four human kidney samples (Chen et al. 2016) and 16 human placenta samples (Green et al. 2016). Based on the same strategy of modeling transcript expression levels, we found that the models from different tissues had similar AUROC values (Supplemental Fig. S7A–E) except when the data were produced by RRBS. The different tissue types also showed the same trend that models involving 5mC features alone were more accurate than those involving 5hmC features alone, whereas combining the two types of methylation features led to even more accurate models (Supplemental Fig. S7A–E). The genome-wide average 5hmC level was higher in liver and lung samples than in kidney and placenta samples, but it did not correlate with the increment of modeling accuracy that was caused by including 5hmC features (Supplemental Fig. S7F). Instead, the increment was slightly larger in normal samples than in cancer samples (Supplemental Fig. S7F).
Next, we compared models involving methylation features at the different regions associated with each transcript (Fig. 2B). Among the upstream, transcript body, and downstream features, features at the transcript body were most indicative of the expression class, followed by those at the upstream regions. The higher accuracy of the transcript body models was partially, but not completely, due to the effect of the first exon, in that including the first exon always led to better modeling accuracy (comparing “Up + FirstEx” with “Up,” and comparing “Body” with “Body-FirstEx”), but models involving transcript body features were consistently more accurate than those involving upstream features regardless of whether the first exon was included or excluded in both sets (comparing “Body” with “Up + FirstEx,” and comparing “Body-FirstEx” with “Up”). Integrating features in both transcript body and upstream regions (“Up + Body”) or all three region types (“Up + Body + Down”) only improved the modeling accuracy slightly as compared to the models involving transcript body features alone.
It is well accepted that a high 5mC level at promoters is an indicator of gene repression (Miranda and Jones 2007; Suzuki and Bird 2008), whereas 5hmC has been shown to be associated with gene bodies (Stroud et al. 2011). We checked whether these knowledge-driven features are redundant and whether together they are sufficient for inferring gene expression level to the maximal accuracy. We found that combining 5mC features at upstream regions and 5hmC features at transcript bodies indeed improved modeling accuracy compared with having either set of features alone, but their combination was still not sufficient to reach the accuracy of models involving all types of methylation features at all associated regions of the transcripts (Fig. 2C), suggesting that methylation features other than promoter 5mC and transcript body 5hmC levels also contribute substantially to the understanding of transcript expression levels.
To make sure that the above observations are not specific to our definition of expression classes, we also constructed regression models to infer log expression levels of transcripts directly. The resulting correlation values (Supplemental Fig. S8) displayed trends highly similar to the AUROC values from the classification models, thereby confirming the generality of the results. For example, combining 5mC and 5hmC features led to better results than having either alone (Supplemental Fig. S8A,B), and transcript body features could infer expression levels more accurately than upstream features (Supplemental Fig. S8C,D).
Comparing models involving only WGBS features with those involving all methylation features (Supplemental Fig. S8A, “BS” vs. “BS + oxBS + 5mC + 5hmC”), the median Pearson's correlation (across the 12 samples) between the predicted and actual log expression values increased from 0.21 to 0.25, which is equivalent to a 18.2% improvement. Among the two tissue types, liver samples had a larger increment of 26.4%.
Feature importance and the smallest set of features with maximal information about gene expression
To systematically determine the most important methylation features for explaining expression variability, we defined a feature importance score based on the frequency of each feature being selected as one of the top features in a forward-searching procedure (Methods). When we grouped features into upstream, transcript body, and downstream feature blocks (Fig. 2D), WGBS and 5hmC signals at transcript bodies received the highest importance scores. This is particularly interesting because although 5hmC features alone could not infer expression levels accurately, they provided the best complementation to the WGBS features, whereas other individually strong features (such as 5mC-Body) appeared to provide less extra information not already contained in WGBS features at transcript bodies. Among the upstream features, as expected, 5mC was selected as most important.
We then further studied the 16 individual regions (Supplemental Fig. S9) and found that in addition to first exon (“FirstEx”) and the upstream region closest to the transcription start site (TSS) (“Up1”), which are important for transcription factor binding and transcription initiation, some other features also consistently showed up among the most important features, including the last exon (“LastEx”) and internal introns (“IntIn”). These regions may affect transcription through other independent mechanisms such as transcriptional elongation and splicing, and were therefore selected as the next most important features.
Using the models involving all features as the best case, we investigated how the model accuracy changed as we included each additional feature or feature block during the forward searching procedure. In terms of feature blocks (Supplemental Fig. S10), usually three to four blocks were sufficient to reach the best-case performance, and these top blocks were predominantly transcript body features. In terms of individual methylation features (Supplemental Fig. S11), usually 10 or more features were necessary to reach the best-case performance. Although the first three to four top features, mainly from transcript bodies, provided the most rapid improvement of modeling accuracy, the remaining six to eight features still provided nonnegligible improvements, and sometimes they also included upstream and downstream features.
The constructed models remain reasonably accurate when applied to other samples
All the results described above were obtained by training and testing on distinct subsets of transcripts from the same sample using a cross-validation procedure. This procedure was designed to avoid overfitting the training data, such that the models could capture the general relationships between DNA methylation and gene expression rather than trends specific to the training sample only. To confirm this generality, we applied models trained on a subset of transcripts from a sample to infer the expression class of a different subset of transcripts in another sample. The results (Fig. 3) reveal that except for normal liver samples that appear to be more distinct from the other samples, our constructed models could infer expression classes of transcripts in other samples as accurately as in the training sample, which can be seen by having off-diagonal AUROC values in the result matrix not substantially lower than the diagonal ones. A large portion of these models did not show tissue- or disease-state specificity, with similar AUROC values regardless of whether or not the testing sample had the same tissue type or disease state as the training sample.
Figure 3.
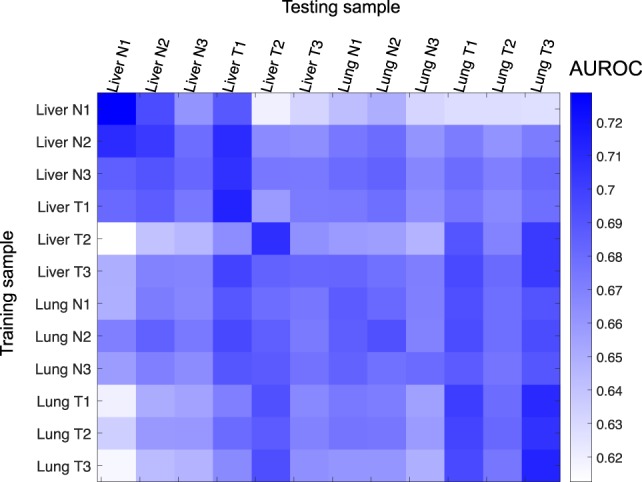
Generality of the models for predicting expression classes with all methylation features based on the large data set. Each row corresponds to a sample from which the model was trained, and each column represents a sample to which the model was applied, based on which evaluation measure was computed. The training and testing transcripts were disjointed regardless of whether the testing sample was the same as or different from the training sample.
Relationship between transcript body methylation and expression
Whether DNA methylation at transcript body correlates positively or negatively with gene expression has been controversial (Ball et al. 2009; Lister et al. 2009; Rauch et al. 2009; Lou et al. 2014). Besides differences in measuring and quantifying methylation levels in the previous studies that could have led to discrepancies in their results, it has not been clear whether the relative 5mC and 5hmC levels could also be a key factor because many of these studies did not consider the two forms of DNA methylation separately. From our data, we found that at regions closest to the TSS (Up1 and FirstEx), both 5mC and 5hmC features correlated negatively with gene expression, although the correlation was stronger for 5mC (Supplemental Fig. S12). This is consistent with a recent report that both types of DNA methylation could repress gene expression by affecting transcription factor binding at the promoter (Kitsera et al. 2017). Inside the transcript body, 5hmC tended to correlate more positively with gene expression than did 5mC in normal liver samples, but the reverse is observed for some liver and lung tumor samples.
Although models involving both 5mC and 5hmC features were more accurate than those involving either only 5mC or only 5hmC features, exactly how the two forms of DNA methylation complement each other in indicating expression level is still unclear. For instance, expression level could be related to either a linear or nonlinear function of the two forms of DNA methylation. When we plotted these three variables at the same time, considering the whole transcript body as a single region (Supplemental Fig. S13), we could not observe any obvious functional form that explains how 5mC and 5hmC jointly indicate expression level, except that highly expressed transcripts usually did not have very high 5mC or 5hmC levels at their bodies. These results reiterate that transcript body methylation has more subtle relationships with gene expression than promoter methylation.
Comparisons between β-value and PDR features
After exploring properties of 5mC and 5hmC levels using the large data set, we then switched to the small data set to study PDR values (Supplemental Fig. S14). Overall, the AUROC values were generally lower than those obtained from the large data set (Fig. 2), likely due to the much smaller number of transcripts in the small data set that forbade reliable modeling. Nevertheless, this data set still allowed us to explore the relative importance of β-value and PDR features. When comparing the models involving these two types of features (Supplemental Fig. S14A), models involving β-value features had higher AUROC values regardless of whether WGBS, oxWGBS, or both types of data were used. Combining the two types of features resulted in more accurate models in all cases. Compared with having β-value features alone, incorporating PDR features led to 1.0%–5.2%, 0.6%–3.7%, and 0.0%–3.5% AUROC improvements across the samples when WGBS, oxWGBS, or both types of data were used, respectively. The same trends were also obtained from the regression results (Supplemental Fig. S15), with the median Pearson's correlation improved up to 9.5%, 9.8%, and 4.8% by incorporating PDR features when WGBS, oxWGBS, or both types of data were used, respectively.
Because PDR values could only be computed from the raw WGBS and oxWGBS data, we checked whether it would be beneficial to also incorporate β-value features of the derived 5mC and 5hmC levels. The results (Supplemental Fig. S14B) show that adding these features [“(BS + oxBS)Beta+PDR (5mC + 5hmC)Beta”] only led to a small increase of AUROC in normal liver samples compared with not adding them [“(BS + oxBS)Beta+PDR”], and did not lead to any clear improvements in other samples. These results again show that it is sufficient to define β-value features using either the raw WGBS and oxWGBS data alone or the processed 5mC and 5hmC data alone.
Necessity of integrating 5mC and 5hmC in differential analyses
The presence of both tumor and matched normal samples in our data enabled us to investigate the necessity of measuring both 5mC and 5hmC in studying differential methylation in cancer. We first checked whether differential expression class could be inferred by β-value features (Methods). The results (Fig. 4A) show that, as expected, transcripts with strong differential expression were more easily identified than those with only weak differential expression. In general, the β-value features were more successful in detecting differentially expressed transcripts in the liver sample pairs than in the lung sample pairs (Fig. 4A), which could be due to a more substantial reduction of 5hmC levels around transcripts from normal liver to liver cancer than in the case of lung (Li et al. 2016). Modeling accuracy was also higher when the four classes contained transcripts with more distinct differential expression profiles (Supplemental Fig. S16A). Again, combining both 5mC and 5hmC data led to the best modeling accuracy (Supplemental Fig. S16B), and methylation levels at transcript bodies were more useful than those at promoters or downstream regions in inferring the differential expression classes (Supplemental Fig. S16C).
Figure 4.

Relationship between methylation and differential expression in cancer. (A) Accuracy of the models for inferring differential expression class, involving all β-value features, based on the large data set with an interclass gap percentage of 80%. Each bar represents the AUROC values of the three pairs of samples in the group. (B) Overlap of DMRs identified using only WGBS data, only oxWGBS data, only 5mC levels, or only 5hmC levels, for liver (left) and lung (right) samples using metilene. The lower plots are zoomed-in views of the bottom parts of the upper plots.
In the above models, the methylation features in the individual samples were used to infer the differential expression class. Another common way to analyze cancer methylome and transcriptome data is to determine differentially methylated regions (DMRs) among the tumor and normal samples and look for differentially expressed transcripts potentially caused by them. To evaluate how this standard analysis procedure might be affected by the mixture of 5mC and 5hmC levels in the data, we determined DMRs genome-wide using only BS data, only oxBS data, only 5mC levels, or only 5hmC levels (Methods). When comparing the overlap of these four sets of DMRs at different stringency thresholds, we found them to differ substantially (Fig. 4B). First, we noticed that almost no DMRs were identified based on 5hmC levels, indicating that the 5hmC levels were not sufficiently different between the tumor and normal groups to be considered statistically significant DMRs by the standard DMR calling method. When considering the other three types of data, 5mC consistently gave the highest number of DMRs in both liver and lung samples, suggesting that compared with the mixture of 5mC and 5hmC signals in BS data, the inferred “clean” 5mC levels were more capable of capturing differential methylation events. When comparing the DMRs identified from BS, oxBS, and 5mC, if two DMRs were considered the same as long as their genomic locations had a small overlap (minimum overlap ratio close to zero), >90% of the oxBS DMRs were also identified from the 5mC data, whereas DMRs identified from standard BS data could only cover 60%–75% of the DMRs identified from oxBS data or 5mC levels. On the other hand, when two DMRs were considered the same only if they had substantial overlaps (with a large overlap ratio), few DMRs identified from these different types of data remained in common. These results show that standard DMR analysis is heavily affected by the type of methylation data involved.
We have also used an additional method for calling DMRs (Korthauer et al. 2018) from BS and oxBS data. The results (Supplemental Fig. S16D–F) contain trends that are consistent with the first method after removing an outlier sample.
Discussion
In this study, we have found that 5mC and 5hmC signals provide nonredundant information about gene expression. The median Pearson's correlation between the actual log expression levels and the expression levels inferred from methylation data was increased by 18.2% by having separate 5mC and 5hmC signals as compared to having only standard BS data. Whether this amount of extra information about expression variability is worth the extra cost of producing the additional experimental data (such as oxBS) is a practical decision to be made when designing methylome studies. On the other hand, if the study goal is to identify the most significant DMRs between tumor and normal samples and associate them with differential expression events, our results suggest that it is necessary to have separate measurements of 5mC and 5hmC signals because the DMRs identified from WGBS data could only cover 60%–75% of the DMRs identified from “pure” 5mC signals.
The advantage of having separate measurements of 5mC and 5hmC signals compared with having 5mC or WGBS signals alone was clearest for the normal liver samples. This is consistent with the higher correlation between 5hmC and gene expression and the lower correlation between 5mC and gene expression of these samples compared with liver cancer samples reported in Li et al. (2016). This difference could be a combination of biological phenomena and technical biases in 5hmC quantification. More investigations are needed to determine the major factor.
In the investigation of PDR features, we found that they were not more informative than β values in indicating transcript expression levels based on the tumor and normal samples we studied. One limitation of this comparison is that PDR values could only be computed reliably when there were a reasonable number of CpG sites appearing on the same sequencing read and the whole region was covered by a reasonable number of reads. These requirements made the number of transcripts qualified for inclusion very small, because for many transcripts PDR values could not be computed in at least some of the 16 associated genomic regions. An additional difficulty of studying PDR values is that they can only be computed from the raw reads but not from the derived 5mC and 5hmC levels, because the correlation between different CpG sites on the same reads would be lost during the process. It would be useful to further check the usefulness of PDR features in inferring expression levels using additional data sets.
Results in the current study also confirm our previous finding (Lou et al. 2014) that transcript body methylation features are more indicative of gene expression level than promoter methylation features. The highly consistent results from the two studies is remarkable because they involved very different analysis details, including the way of quantifying methylation levels, the cross-validation procedures, the use of gene or transcript as the basic unit, and whether regression of log expression levels is performed. Based on these results, we strongly recommend that when DNA methylation data are used to study transcriptional regulation, methylation signals in the gene body should be included in the analysis, especially the signals at the first exon, last exon, and internal introns.
In this study, we investigated methylation of each transcript based on its promoter, body, and immediate downstream regions. It would be interesting to extend the study to include enhancer and other distal regulatory elements. Recently, a number of methods have been proposed for identifying target genes of enhancers in a cell-type–specific manner (Corradin et al. 2014; He et al. 2014; Roy et al. 2015; Whalen et al. 2016; Cao et al. 2017). However, the accuracy of these methods for cell types without genome contact data such as Hi-C or ChIA-PET is still not high enough for constructing models that can infer expression reliably. The exploration of the general quantitative relationships between enhancer methylation and gene expression levels will need to wait for the availability of more genome contact data or more accurate enhancer-target identification methods.
It has been shown that 5hmC levels are highly variable among tissue types (Nestor et al. 2012). Limited by data availability, we have only included a few tissue types each with only a small number of samples in our study. Whether 5hmC levels are more indicative of transcript expression levels in other tissue types and whether more DMRs can be identified based on 5hmC levels in other cancer types are questions to be answered when more genome-wide 5mC and 5hmC measurements become available.
Methods
Construction of the data sets
For the main data set used in this study, we downloaded raw sequencing read files (.sra) and alignment files (.bam) of the WGBS, oxWGBS, and RNA-seq data from the Sequence Read Archive (SRA; GSE70091, subseries GSE70089 for RNA-seq alignment files and GSE70090 for WGBS and oxWGBS raw read files) (Leinonen et al. 2010) using the SRA toolkit (https://www.ncbi.nlm.nih.gov/sra/docs/sradownload/). The original data set contained four normal–tumor pairs of liver, but since only three of them had the corresponding RNA-seq data, we excluded this fourth pair from all our analyses. Following Li et al. (2016), we aligned the WGBS and oxWGBS raw reads to the human reference genome hg19 using BSmooth (Hansen et al. 2012). Read pairs having identical alignments of both mates were considered potential duplicates due to PCR artifacts, and only one read pair was retained for each set of duplicate read pairs.
For each CpG site, we computed the β value as the number of reads supporting an unconverted cytosine divided by the total number of reads covering the site, for both WGBS and oxWGBS data sets. To ensure the reliability of the input data, following the data processing in Li et al. (2016), we excluded reads with a mapping quality less than 20, bases on a read with a base quality less than 10, and bases within the 10 5′-most positions of both mates of each read pair. To reduce effects of sampling errors, CpG sites with fewer than five aligned reads were also excluded. We further computed 5mC and 5hmC levels of each site using a maximum likelihood method (Xu et al. 2016).
For WGBS and oxWGBS data, we further computed the average methylation level of each associated region defined for a transcript as
where i loops through all CpG sites in the region, mi is the number of reads that support site i to be methylated, and ni is the total number of reads covering site i. 5mC and 5hmC levels of these regions were then determined by the corresponding WGBS and oxWGBS levels. These associated regions included 16 regions overlapping or immediately next to the transcript (Lou et al. 2014), namely, five consecutive 400-bp bins upstream of the TSS (Up1–Up5, with Up1 closest to the TSS), first exon (FirstEx), first intron (FirstIn), internal exons (IntEx), internal introns (IntIn), last exon (LastEx), last intron (LastIn), and five consecutive 400-bp bins downstream from the transcription termination site (TTS) (Down1-Down5, with Down1 closest to the TTS). By default, we included all annotated protein-coding and noncoding transcripts of levels 1, 2, and 3 in GENCODE (Harrow et al. 2012) version 19, whereas in some analyses, we only considered a subset of these transcripts to see how the results differed.
In order to have all 16 regions defined, we always considered only transcripts with at least four exons, because this is the minimum number of exons for which the first intron, internal introns, and last intron regions are all distinct. To avoid unreliable methylation levels caused by low read coverage, if the whole transcript body region had fewer than three CpG sites each with at least five aligned reads in a sample, the whole transcript was discarded from that sample.
In addition, for each region associated to a gene, we calculated its PDR value in each sample as the ratio of reads having discordant methylation status. Specifically, we considered reads with an alignment that overlapped the region, with reads having a mapping quality less than 20 or covering less than three CpG sites excluded. For each of the remaining reads, it was considered a concordant read if <10% or >90% of the CpG sites it covered had the same methylation status. The other reads were considered discordant, and the number of them was used to compute the PDR value. To ensure the robustness of the computed PDR values, transcripts with any one of the 16 associated regions having fewer than three aligned reads were discarded. This filtering was the main reason that the resulting small data set had a much smaller number of transcripts than the big data set, which only had β values as features. Because the calculations of PDR values required information of sequencing reads rather than individual CpG sites, they could not be computed for the processed 5mC and 5hmC data sets, which did not have read-level information anymore.
We also computed transcript expression levels, defined as fragments per kilobase per million mapped reads (FPKM), using Cufflinks (Trapnell et al. 2010) version 2.2.1 using the -G option.
The additional data sets used were downloaded from Gene Expression Omnibus (GEO) (Barrett et al. 2013) or SRA. Specifically, for the four kidney samples (Chen et al. 2016), the processed 5mC and 5hmC β values and raw RNA-seq data were downloaded from GEO (GSE63183). For the 10 liver samples (Hlady et al. 2019), the processed 5mC and 5hmC β values and raw RNA-seq data were downloaded from GEO (GSE112221). For the 16 placenta samples (Green et al. 2016), the processed 5mC and 5hmC β values were downloaded from GEO (GSE71719), and the raw RNA-seq data were downloaded from SRA (SRP068290). All RNA-seq data were aligned to the human reference genome GRCh37/hg19 using TopHat2 v2.0.13 (Kim et al. 2013) for computing transcript expression levels. We used GRCh37/hg19 rather than the latest GRCh38/hg38 reference because the processed 5mC and 5hmC β values from these studies were based on the GRCh37/hg19 reference. Because our study focused on the well-annotated regions around genes and all analyses were performed based on aggregating data across genomic regions, changing the reference to GRCh38/hg38 would not significantly affect our results and conclusions, since the two references mainly differ by single-nucleotide variants, alternate locus scaffolds, centromeres, and mitochondrial DNA (Bhattacharyya et al. 2017; Schneider et al. 2017).
Statistical modeling of expression classes
In each sample, we defined a high-expression class of transcripts as those having an FPKM value of one or more, a low-expression class of transcripts as those having an FPKM value larger than 10−10 but smaller than 10−2, and a zero-expression class of transcripts as those having an FPKM value <10−100.
We modeled the expression class using either all or a subset of the methylation features. We chose random forest models (bagging with 50 random trees as the base classifiers) because they were previously shown to perform well for modeling the quantitative roles of DNA methylation (Lou et al. 2014). All expression classes were included in the same model.
We designed a cross-validation procedure for evaluating the performance of the models as follows. We paired up short and long autosomes, namely, Chromosome 1 with Chromosome 22, Chromosome 2 with Chromosome 21, and so on, leading to 11 chromosome pairs. Each time one of the chromosome pairs was left out for testing, whereas the other 10 pairs were used for training a model. The model was then applied to the transcripts in the left-out chromosome pair, either from the same sample (within-sample test) or from another sample (across-sample test). Finally, the predictions from the 11 left-out sets were combined to compute the performance metric AUROC using a one-class-against-all strategy. This design of the cross-validation procedure avoids two types of trivial memorization. First, in the within-sample test, if all transcripts were randomly distributed to the training and testing sets instead, two transcripts from the same gene could be respectively assigned to the training and testing sets, leading to a simple memorization of the training transcript's expression level when predicting the testing transcript's expression level, because they would share very similar methylation features. Second, in the between-sample test, if all transcripts from a sample were used to construct the model instead, when applying the model to another sample, again the predictions could be simply memorization of the expression levels of the same transcripts in the training sample when the training and testing samples were highly similar, such as those from the same tissue type and disease state. In addition to avoiding such memorization, the pairing of long and short chromosomes in our procedure also led to a similar number of transcripts in each chromosome pair. To make our results more reliable, we also repeated each classification task 10 times with different random seeds used to construct the random forest models, and reported the average performance.
Feature importance evaluation
To evaluate the importance of different features in explaining transcript expression levels, we used a forward-searching procedure to construct models with only subsets of the most useful features. Specifically, we started with constructing models having only one feature and compared their performance. The feature used in the most accurate model was then added to the set of selected features, and new models were constructed by having this feature plus one of the remaining features. This procedure was repeated iteratively, with the feature leading to the best performance in each iteration added to the set of selected features. The whole procedure ended when all features had been selected. Finally, we gave the first x features selected a score of x, x−1,…, 1, respectively, where x was chosen to be 1/2 of the total number of features when all 64 features were considered separately, and 2/3 of the total number of features/feature blocks in all other settings.
We performed this feature importance evaluation with each sample and also summed the scores across all samples to define a single importance score for each feature.
In addition to individual features, we also used this procedure to evaluate the importance of different feature blocks.
Statistical modeling of expression levels
Because the grouping of transcripts into expression classes relied on a specific way of defining the classes and a specific number of classes, to ensure that our findings were not affected by our specific choices, we also constructed regression models to predict the log expression level of the transcripts based on their methylation features. Specifically, for a transcript with a FPKM value of y, we used log10(y + 1) as the prediction target. We used support vector regression with a radial basis function (RBF) kernel to construct the models. Model performance was evaluated using the same cross-validation procedure as in the case of predicting expression classes, quantified by both Pearson's correlation and Spearman's correlation coefficients.
Definition of differential expression classes
To define differential expression classes, we first calculated a differential expression level of each transcript by subtracting its FPKM value in a normal sample from its FPKM value in the corresponding tumor sample, FPKMdiff = FPKMtumor − FPKMnormal. We kept only transcripts with no missing data in all six sample pairs. Then for each tissue type, we used the median differential expression level of a transcript among the three sample pairs to determine its differential expression class. Transcripts with a median differential expression level between −10−5 and 10−5 were considered to have no significant change of expression and were not included in any of the classes. For the remaining transcripts with a positive differential expression level (i.e., higher expression in tumor), we took the top and bottom x% of transcripts with the largest and smallest absolute differential expression values to define the strongly up-regulated and weakly up-regulated classes, respectively, where x is a variable, and we called 1 – 2x% the gap percentage between the two classes. We tried different values of the gap percentage from 10 to 90, with 80 used as the default as a tradeoff between the clear separation of the two classes and the number of transcripts that can be included in them. In the same way, we also defined the strongly down-regulated and weakly down-regulated classes.
Statistical modeling of differential expression classes
For each tumor–normal pair of samples, we compared the sizes of the four classes and randomly down-sampled the larger ones until all four classes had the same number of transcripts. This random down-sampling was repeated 10 times to generate 10 different data sets. We then trained and tested random forest models for the four differential expression classes together using the union of methylation features from individual samples and the same cross-validation procedure as in the case of modeling expression classes.
Analysis of DMRs
DMRs were first identified by metilene v0.2-6 (Jühling et al. 2016) based on the β values of CpG sites from the four types of methylation data. For each tissue type, the three tumor samples were compared with the three normal samples to identify the DMRs. We further filtered the DMRs by retaining only those at least 100 bp long with at least three CpG sites having 3× read coverage, as well as an average difference of β value between the samples in the tumor and nontumor groups of at least 0.1. We also tried five other sets of values for these filtering parameters, but the resulting trends were all highly similar. We considered a DMR called from one data set to overlap a DMR called from another data set if the intersection of them constitutes at least x% of both DMRs, where x is the minimum overlap ratio, and we tried a range of values for it. To count the number of DMRs commonly called from two data sets, we first obtained two numbers: namely, the number of DMRs in the first set that overlaps one or more DMRs in the second set, and the number of DMRs in the second set that overlaps one or more DMRs in the first set. It turns out that these two sets of numbers were usually identical and differed at most by a small number. We therefore used their average in our report.
We also used a second method, dmrseq (Korthauer et al. 2018) v1.0.14, to call DMRs. Because dmrseq requires read coverage as input, which is not well defined for the derived 5mC and 5hmC levels, we only used it to call DMRs from the WGBS and oxWGBS data. For each tissue type, we discarded CpG sites with fewer than three reads in any sample and then ran dmrseq to call DMRs using default settings. For the liver samples, no DMRs were called from the WGBS data. We found that this was due to differences in the methylome profiles between liver tumor sample T2 and the other two liver tumor samples. We therefore removed this sample and reran DMR calling using dmrseq.
Supplementary Material
Acknowledgments
Q.W., A.S.L.C., and K.Y.Y. were partially supported by the Hong Kong Research Grants Council Collaborative Research Fund (C4017-14G). K.Y.Y. was partially supported by the Hong Kong Research Grants Council General Research Fund (14170217).
Footnotes
[Supplemental material is available for this article.]
Article published online before print. Article, supplemental material, and publication date are at http://www.genome.org/cgi/doi/10.1101/gr.240036.118.
References
- Aran D, Sabato S, Hellman A. 2013. DNA methylation of distal regulatory sites characterizes dysregulation of cancer genes. Genome Biol 14: R21 10.1186/gb-2013-14-3-r21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ball MP, Li JB, Gao Y, Lee JH, LeProust EM, Park IH, Xie B, Daley GQ, Church GM. 2009. Targeted and genome-scale strategies reveal gene-body methylation signatures in human cells. Nat Biotechnol 27: 361–368. 10.1038/nbt.1533 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett T, Wilhite SE, Ledoux P, Evangelista C, Kim IF, Tomashevsky M, Marshall KA, Phillippy KH, Sherman PM, Holko M, et al. 2013. NCBI GEO: archive for functional genomics data sets—update. Nucleic Acids Res 41: D991–D995. 10.1093/nar/gks1193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya S, Pradhan K, Campbell N, Mazdo J, Vasantkumar A, Maqbool S, Bhagat TD, Gupta S, Suzuki M, Yu Y, et al. 2017. Altered hydroxymethylation is seen at regulatory regions in pancreatic cancer and regulates oncogenic pathways. Genome Res 27: 1830–1842. 10.1101/gr.222794.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird A. 2002. DNA methylation patterns and epigenetic memory. Genes Dev 16: 6–21. 10.1101/gad.947102 [DOI] [PubMed] [Google Scholar]
- Cao Q, Anyansi C, Hu X, Xu L, Xiong L, Tang W, Mok MTS, Cheng C, Fan X, Gerstein M, et al. 2017. Reconstruction of enhancer–target networks in 935 samples of human primary cells, tissues and cell lines. Nat Genet 49: 1428–1436. 10.1038/ng.3950 [DOI] [PubMed] [Google Scholar]
- Chen K, Zhang J, Guo Z, Ma Q, Xu Z, Zhou Y, Xu Z, Li Z, Liu Y, Ye X, et al. 2016. Loss of 5-hydroxymethylcytosine is linked to gene body hypermethylation in kidney cancer. Cell Res 26: 103–118. 10.1038/cr.2015.150 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi JK, Bae JB, Lyu J, Kim TY, Kim YJ. 2009. Nucleosome deposition and DNA methylation at coding region boundaries. Genome Biol 10: R89 10.1186/gb-2009-10-9-r89 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cokus SJ, Feng S, Zhang X, Chen Z, Merriman B, Haudenschild CD, Pradhan S, Nelson SF, Pellegrini M, Jacobsen SE. 2008. Shotgun bisulphite sequencing of the Arabidopsis genome reveals DNA methylation patterning. Nature 452: 215–219. 10.1038/nature06745 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corradin O, Saiakhova A, Akhtar-Zaidi B, Myeroff L, Willis J, Cowper-Sal·lari R, Lupien M, Markowitz S, Scacheri PC. 2014. Combinatorial effects of multiple enhancer variants in linkage disequilibrium dictate levels of gene expression to confer susceptibility to common traits. Genome Res 24: 1–13. 10.1101/gr.164079.113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ehrlich M. 2009. DNA hypomethylation in cancer cells. Epigenomics 1: 239–259. 10.2217/epi.09.33 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Green BB, Houseman EA, Johnson KC, Guerin DJ, Armstrong DA, Christensen BC, Marsit CJ. 2016. Hydroxymethylation is uniquely distributed within term placenta, and is associated with gene expression. FASEB J 30: 2874–2884. 10.1096/fj.201600310R [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansen KD, Langmead B, Irizarry RA. 2012. BSmooth: from whole genome bisulfite sequencing reads to differentially methylated regions. Genome Biol 13: R83 10.1186/gb-2012-13-10-r83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrow J, Frankish A, Gonzalez JM, Tapanari E, Diekhans M, Kokocinski F, Aken BL, Barrell D, Zadissa A, Searle S, et al. 2012. GENCODE: the reference human genome annotation for the ENCODE project. Genome Res 22: 1760–1774. 10.1101/gr.135350.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- He B, Chen C, Teng L, Tan K. 2014. Global view of enhancer–promoter interactome in human cells. Proc Natl Acad Sci 111: E2191–E2199. 10.1073/pnas.1320308111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyn H, Vidal E, Ferreira HJ, Vizoso M, Sayols S, Gomez A, Moran S, Boque-Sastre R, Guil S, Martinez-Cardus A, et al. 2016. Epigenomic analysis detects aberrant super-enhancer DNA methylation in human cancer. Genome Biol 17: 11 10.1186/s13059-016-0879-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hlady R, Sathyanarayan A, Thompson JJ, Zhou D, Wu Q, Pham K, Lee J, Liu C, Robertson KD. 2019. Integrating the epigenome to identify novel drivers of hepatocellular carcinoma. Hepatology 69: 639–652. 10.1002/hep.30211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry RA, Ladd-Acosta C, Wen B, Wu Z, Montano C, Onyango P, Cui H, Gabo K, Rongione M, Webster M, et al. 2009. The human colon cancer methylome shows similar hypo- and hypermethylation at conserved tissue-specific CpG island shores. Nat Genet 41: 178–186. 10.1038/ng.298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin SG, Kadam S, Pfeifer GP. 2010. Examination of the specificity of DNA methylation profiling techniques towards 5-methylcytosine and 5-hydroxymethylcytosine. Nucleic Acids Res 38 e125 10.1093/nar/gkq223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones PA. 2012. Functions of DNA methylation: islands, start sites, gene bodies and beyond. Nat Rev Genet 13: 484–492. 10.1038/nrg3230 [DOI] [PubMed] [Google Scholar]
- Jühling F, Kretzmer H, Bernhart SH, Otto C, Stadler PF, Hoffmann S. 2016. metilene: fast and sensitive calling of differentially methylated regions from bisulfite sequencing data. Genome Res 26: 256–262. 10.1101/gr.196394.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Pertea G, Trapnell C, Pimentel H, Kelley R, Salzberg SL. 2013. TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol 14: R36 10.1186/gb-2013-14-4-r36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitsera N, Allgayer J, Parsa E, Geier N, Rossa M, Carell T, Khobta A. 2017. Functional impacts of 5-hydroxymethylcytosine, 5-formylcytosine, and 5-carboxycytosine at a single hemi-modified CpG dinucleotide in a gene promoter. Nucleic Acids Res 45: 11033–11042. 10.1093/nar/gkx718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klutstein M, Nejman D, Greenfield R, Cedar H. 2016. DNA methylation in cancer and aging. Cancer Res 76: 3446–3450. 10.1158/0008-5472.CAN-15-3278 [DOI] [PubMed] [Google Scholar]
- Korthauer K, Chakraborty S, Benjamini Y, Irizarry RA. 2018. Detection and accurate false discovery rate control of differentially methylated regions from whole genome bisulfite sequencing. Biostatistics 10.1093/biostatistics/kxy007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landau DA, Clement K, Ziller MJ, Boyle P, Fan J, Gu H, Stevenson K, Sougnez C, Wang L, Li S, et al. 2014. Locally disordered methylation forms the basis of intra-tumor methylome variation in chronic lymphocytic leukemia. Cancer Cell 26: 813–825. 10.1016/j.ccell.2014.10.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leinonen R, Sugawara H, Shumway M. 2010. The Sequence Read Archive. Nucleic Acids Res 39: D19–D21. 10.1093/nar/gkq1019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li X, Liu Y, Salz T, Hansen KD, Feinberg A. 2016. Whole-genome analysis of the methylome and hydroxymethylome in normal and malignant lung and liver. Genome Res 26: 1730–1741. 10.1101/gr.211854.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lister R, Pelizzola M, Dowen RH, Hawkins RD, Hon G, Tonti-Filippini J, Nery JR, Lee L, Ye Z, Ngo QMM, et al. 2009. Human DNA methylomes at base resolution show widespread epigenomic differences. Nature 462: 315–322. 10.1038/nature08514 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorincz MC, Dickerson DR, Schmitt M, Groudine M. 2004. Intragenic DNA methylation alters chromatin structure and elongation efficiency in mammalian cells. Nat Struct Mol Biol 11: 1068–1075. 10.1038/nsmb840 [DOI] [PubMed] [Google Scholar]
- Lou S, Lee HM, Qin H, Li JW, Gao Z, Liu X, Chan LL, Lam VK, So WY, Wang Y, et al. 2014. Whole-genome bisulfite sequencing of multiple individuals reveals complementary roles of promoter and gene body methylation in transcriptional regulation. Genome Biol 15: 408 10.1186/s13059-014-0408-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maunakea AK, Nagarajan RP, Bilenky M, Ballinger TJ, D'Souza C, Fouse SD, Johnson BE, Hong C, Nielsen C, Zhao Y, et al. 2010. Conserved role of intragenic DNA methylation in regulating alternative promoters. Nature 466: 253–257. 10.1038/nature09165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miranda TB, Jones PA. 2007. DNA methylation: the nuts and bolts of repression. J Cell Physiol 213: 384–390. 10.1002/jcp.21224 [DOI] [PubMed] [Google Scholar]
- Nestor CE, Ottaviano R, Reddington J, Sproul D, Reinhardt D, Dunican D, Katz E, Dixon JM, Harrison DJ, Meehan RR. 2012. Tissue type is a major modifier of the 5-hydroxymethylcytosine content of human genes. Genome Res 22: 467–477. 10.1101/gr.126417.111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petterson A, Chung TH, Tan D, Sun X, Jia XY. 2014. RRHP: a tag-based approach for 5-hydroxymethylcytosine mapping at single-site resolution. Genome Biol 15: 456 10.1186/s13059-014-0456-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rauch TA, Wu X, Zhong X, Riggs AD, Pfeifer GP. 2009. A human B cell methylome at 100–base pair resolution. Proc Natl Acad Sci 106: 671–678. 10.1073/pnas.0812399106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robertson KD. 2005. DNA methylation and human disease. Nat Rev Genet 6: 597–610. 10.1038/nrg1655 [DOI] [PubMed] [Google Scholar]
- Rountree MR, Selker EU. 1997. DNA methylation inhibits elongation but not initiation of transcription in Neurospora crassa. Genes Dev 11: 2383–2395. 10.1101/gad.11.18.2383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy S, Siahpirani AF, Chasman D, Knaack S, Ay F, Stewart R, Wilson M, Sridharan R. 2015. A predictive modeling approach for cell line-specific long-range regulatory interactions. Nucleic Acids Res 43: 8694–8712. 10.1093/nar/gkv865 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider VA, Graves-Lindsay T, Howe K, Bouk N, Chen H-C, Kitts PA, Murphy TD, Pruitt KD, Thibaud-Nissen F, Albracht D, et al. 2017. Evaluation of GRCh38 and de novo haploid genome assemblies demonstrates the enduring quality of the reference assembly. Genome Res 27: 849–864. 10.1101/gr.213611.116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song CX, Yi C, He C. 2012. Mapping recently identified nucleotide variants in the genome and transcriptome. Nat Biotechnol 30: 1107–1116. 10.1038/nbt.2398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stroud H, Feng S, Kinney SM, Pradhan S, Jacobsen SE. 2011. 5-Hydroxymethylcytosine is associated with enhancers and gene bodies in human embryonic stem cells. Genome Biol 12: R54 10.1186/gb-2011-12-6-r54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki MM, Bird A. 2008. DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet 9: 465–476. 10.1038/nrg2341 [DOI] [PubMed] [Google Scholar]
- Trapnell C, Williams BA, Pertea G, Mortazavi A, Kwan G, van Baren MJ, Salzberg SL, Wold BJ, Pachter L. 2010. Transcript assembly and quantification by RNA-seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 28: 511–515. 10.1038/nbt.1621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uhlen M, Zhang C, Lee S, Sjostedt E, Fagerberg L, Bidkhori G, Benfeitas R, Arif M, Liu Z, Edfors F, et al. 2017. A pathology atlas of the human cancer transcriptome. Science 357: eaan2507 10.1126/science.aan2507 [DOI] [PubMed] [Google Scholar]
- Whalen S, Truty RM, Pollard KS. 2016. Enhancer–promoter interactions are encoded by complex genomic signatures on looping chromatin. Nat Genet 48: 488–496. 10.1038/ng.3539 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Taylor JA, Leung YK, Ho SM, Niu L. 2016. oxBS-MLE: an efficient method to estimate 5-methylcytosine and 5-hydroxymethylcytosine in paired bisulfite and oxidative bisulfite treated DNA. Bioinformatics 32: 3667–3669. 10.1093/bioinformatics/btw527 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu M, Hon GC, Szulwach KE, Song CX, Zhang L, Kim A, Li X, Dai Q, Shen Y, Park B, et al. 2012. Base-resolution analysis of 5-hydroxymethylcytosine in the mammalian genome. Cell 149: 1368–1380. 10.1016/j.cell.2012.04.027 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.