

SUMMARY
Circadian rhythms are a hallmark of physiology, but how such daily rhythms organize cellular catabolism is poorly understood. Here, we used proteomics to map daily oscillations in autophagic flux in mouse liver and related these rhythms to proteasome activity. We also explored how systemic inflammation affects the temporal structure of autophagy. Our data identified a globally harmonized rhythm for basal macroautophagy, chaperone-mediated autophagy, and proteasomal activity, which concentrates liver proteolysis during the daytime. Basal autophagy rhythms could be resolved into two antiphase clusters that were distinguished by the subcellular location of targeted proteins. Inflammation induced by lipopolysaccharide reprogrammed autophagic flux away from a temporal pattern that favors cytosolic targets and toward the turnover of mitochondrial targets. Our data detail how daily biological rhythms connect the temporal, spatial, and metabolic aspects of protein catabolism.
Graphical Abstract
In Brief
How circadian rhythms contribute to cellular quality control is poorly understood. Ryzhikov et al. mapped proteome-wide biological rhythms in liver autophagy and identified synchronous daily oscillations in autophagic and proteasomal activity. Diurnal rhythms play a role in autophagy substrate selection based on subcellular location and inflammation status.
INTRODUCTION
Autophagy represents a collection of catabolic pathways that deliver proteins and other cellular material to lysosomes for disposal (Kaur and Debnath, 2015). The most intensely studied form of autophagy, macroautophagy, is distinguished by a specialized vesicle called an autophagosome that forms around cytoplasmic material intended for removal (Kaur and Debnath, 2015). The degradative process is completed when autophagosomes fuse with endosomes and lysosomes. Another key form of autophagy, called chaperone-mediated autophagy (CMA), operates by directly translocating protein targets across the lysosomal membrane (Dice, 1992). Regardless of the specific mechanism, genetic interference with autophagy disrupts cellular quality control and leads to the accumulation of dysfunctional mitochondria, reactive oxygen species, metabolic defects, and faster cellular aging (Ezaki et al., 2011; Nakahira et al., 2011; Schneider et al., 2015).
Autophagy is usually conceptualized as a homeostatic process, operating at a constant rate unless modulated by external stimuli such as starvation. However, the activity of one autophagic mechanism, macroautophagy, is not constant but rather oscillates according to a circadian rhythm (Ma et al., 2011; Pfeifer, 1971). Circadian rhythms are daily variations in biological function that depend upon a group of conserved transcription factors called clock genes (Green et al., 2008). Clock gene deletion also leads to defects in cellular quality control, metabolism, and aging, similar to macroautophagy disruption (Cho et al., 2012; Gong et al., 2015; Kondratov et al., 2006). This suggests that circadian rhythms may optimize proteostasis by providing a temporal organization to cellular catabolism. However, the dynamics of protein turnover and its relation to cellular function remain poorly understood. This is partly because rhythms in macroautophagy have yet to be detailed at the protein level, and it is unknown whether these rhythms extend to other degradative pathways such as CMA or the ubiquitin-proteasome system.
Here, we used proteomics to examine daily oscillations in macroautophagic flux in mouse liver and related these rhythms to proteasome activity. We also explored how systemic inflammation induced by lipopolysaccharide (LPS) affects the temporal structure of autophagy. Our data identified a globally harmonized rhythm for basal macroautophagy, CMA, and proteasomal activity that coordinates the temporal, spatial, and metabolic aspects of protein catabolism.
RESULTS
Rhythms in Basal Macroautophagy
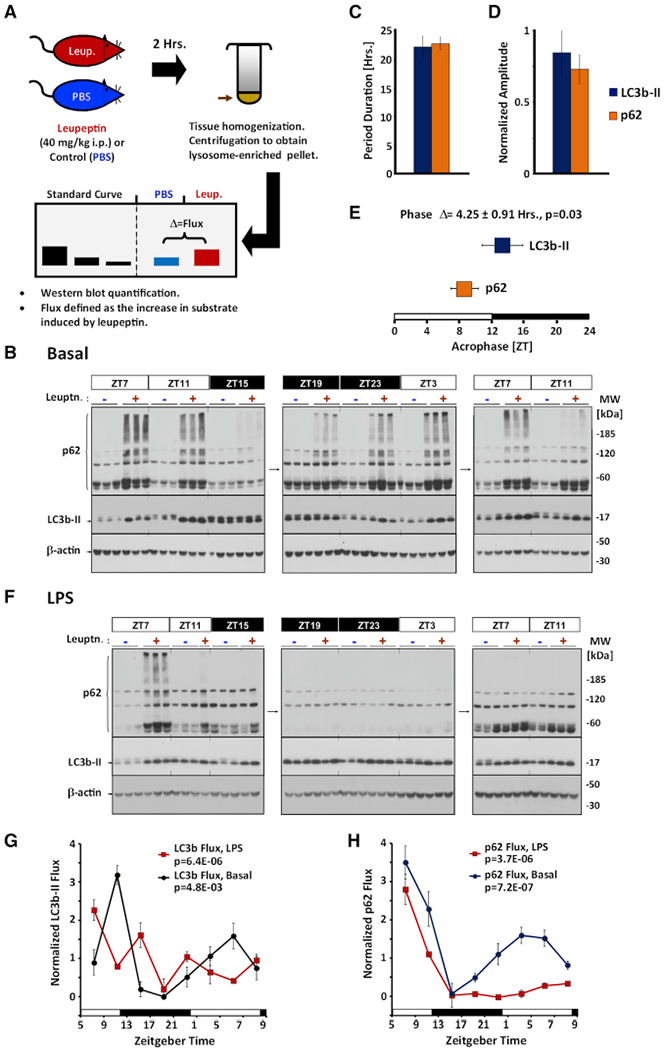
Macroautophagic activity (or flux) is typically measured in vivo using a turnover assay (Haspel et al., 2011; Klionsky et al., 2016) (Figure 1A). Mice are injected with leupeptin to suppress cathepsin activity within lysosomes, leading to an accumulation of protein substrates. This buildup can be quantified by fractionating lysosomes and using western blots to calculate the increase in the abundance of macroautophagy marker proteins like LC3b-II or p62 (Figure 1B). Collected as a time series, turnover assays can visualize circadian rhythms in macroautophagic flux (Ma et al., 2011).
Figure 1. Circadian Characteristics of Macroautophagic Flux in Basal Mouse Liver.
(A) Cartoon depicting protocol for measuring macroautophagic flux.
(B) Representative time series analysis of circadian rhythms in macroautophagy in basal mouse liver. Tiled images are representative western blots against p62 (top), LC3b-II (middle), and β-actin (bottom). Standards are shown to the left, and molecular weight markers (in kilodaltons) are depicted to the right of the images. Each lane represents lysosome-enriched protein fractions isolated from 126 μg of total liver homogenate (12 μL or 6% of the total fraction). Times of tissue harvest are depicted in units of zeitgeber time (ZT), where ZT0 represents lights on (6:00 a.m. local time) and ZT12 represents lights off (6:00 p.m. local time). Collection times that occurred during subjective night are highlighted in black. −, PBS control-injected animals; +, leupeptin-injected animals.
(C–E) Circadian rhythm parameter analysis of basal macroautophagic flux in mouse liver using LC3b-II (blue bars) and p62 (orange bars) as markers.
(C and D) Period duration (C) and amplitude (D) expressed as a fraction of the mean (mean ± SE, n = 6).
(E) Acrophase (mean ± SE, n = 6). The average phase difference ± SE between LC3b-II and p62 turnover is indicated above the graph (n = 6). The p value reflects the likelihood that the phase difference between LC3b-II and p62 acrophases is zero.
(F) Western blots depicting macroautophagic flux in livers of LPS-treated mice (12 mg/kg i.p. dose given at ZT5). Each lane represents lysosome-enriched fractions isolated from 126 μg of total liver homogenate (12 μL or 6%of the total fraction) (see STAR Methods). −, PBS control-injected animals; +, leupeptin-injected animals. Data are representative of 4 independent experiments.
(G and H) Quantification of macroautophagic flux using LC3b-II turnover (G) or p62 turnover (H) as a marker of macroautophagic activity (mean ± SE, n = 3 flux measurements). The p values derived by one-way ANOVA are provided in the graph.
Also see Figure S1.
Using this approach, we estimated that rhythms in basal LC3b-II and p62 turnover have a periodicity of 20.3 ± 1.7 and 22.6 ± 1.2 h, respectively (Figure 1C). The amplitude of these rhythms in flux was estimated at 0.80 ± 0.16 of the mean for LC3b-II and 0.76 ± 0.10 of the mean for p62, suggesting that, on average, macroautophagy undergoes daily swings in proteolytic activity of about 7- to 9-fold (Figure 1D). The time of maximum turnover (or acrophase) for LC3b-II and p62 turnover occurred around lights off, but there was a slight but consistent phase lag between the two marker proteins, suggesting the dynamics of these two proteins may be different (Figure 1E).
Rhythms in macroautophagic flux are generally regarded as cell intrinsic and directly coupled to the function of circadian clock genes (Ma et al., 2011). Consistent with this, we found that rhythms in the steady-state level of liver LC3b-II persist in the absence of light cues and food availability (Figures S1A and S1B). However, we found that the master clock gene bmal1 was genetically dispensable for maintaining rhythms in macroautophagic flux (Figure S1C), which contrasts with one prior report (Ma et al., 2011). Our bmal1−/− mice were maintained on a 12-h light-dark cycle that preserves normal feeding and activity rhythms in these animals (Storch et al., 2007). Thus, daily rhythms in liver macroautophagy might not directly arise from the molecular circadian oscillator but instead may be indirectly coupled to the molecular clock via feeding or other normally circadian-gated behaviors.
LPS Dissociates the Temporal Profile of LC3b-II and p62 Turnover
LPS is an inflammatory molecule that is known to disrupt clock gene expression (Haspel et al., 2014; Okada et al., 2008). We therefore examined how acute LPS challenge would affect diurnal rhythms in macroautophagic flux (Figure 1F). A single 12 mg/kg dose of LPS was enough to disrupt the normal circadian protein expression of Clock, RORα, and phosphorylated RS6 in mouse liver, as well as expression of the clock genes bmal1 and dbp (Figures S1D and S1E). However, the effect of LPS on the turnover of macroautophagy marker proteins was target specific. In the case of LC3b-II, LPS disrupted the temporal profile of its turnover, but the overall daily rate of degradation changed little (Figure 1G). In contrast, LPS produced delayed inhibition in p62 flux (Figure 1H). The differing patterns of LC3b-II and p62 turnover exemplified the need for proteome-wide data to define the global architecture of daily variations in macroautophagy, in health, and during inflammation.
A Proteomic Method for Analyzing Diurnal Rhythms in Autophagy
To examine daily rhythms in liver macroautophagy on a proteome-wide scale, we constructed a time series analysis of mice in the basal state and after treatment with LPS (Figure 2). Diurnal rhythms in macroautophagic flux were clearly demonstrated on western blots by oscillations in p62 turnover in the livers of basal mice, and this was suppressed in LPS-treated mice (Figure 2A). A flux composite score (FCS), defined as the difference in spectral counts between leupeptin and sham-treated livers adjusted for apparent protein abundance, was used to estimate turnover rate using proteomics data (see STAR Methods). FCS values for p62 strongly correlated with flux measurements obtained via western blot and successfully captured its diurnal rhythm (Figures 2A and 2B).
Figure 2. A Proteomics-Based Approach to Measuring Autophagic Flux.
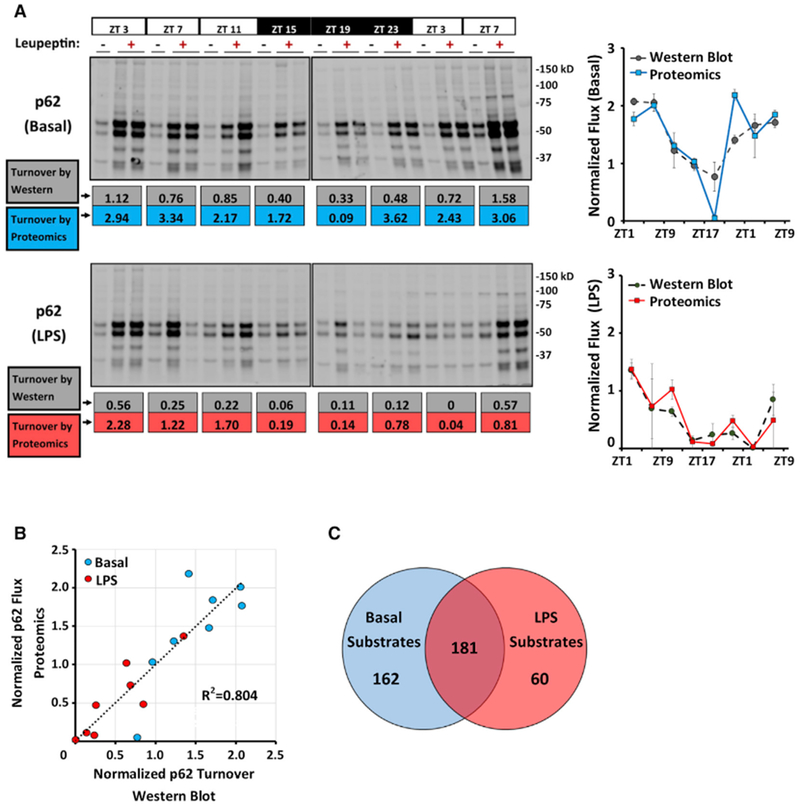
(A) Western blots depicting p62 turnover in samples used for label-free proteomics analysis (see STAR Methods). Each lane represents 12 μL or 6% of the total fraction (see STAR Methods). Top, basal livers. Bottom, LPS (12 mg/kg given at ZT1). Below each blot, p62 turnover is calculated using either densitometry from western blotting (in units of picomoles of p62 per milligram of total protein per hour, gray values), or using FCSs obtained from proteomics (blue values for basal and red values for LPS). These data are depicted graphically in normalized form to the right of the blots. See also Figure S2A.
(B) Correlation between proteomics-based and western blot-based estimates of macroautophagic flux using p62 turnover as a marker. Datapoints represent the mean of 2 turnover measurements obtained at various times of day. Blue circles, basal livers; red circles, LPS-treated mice.
(C) Macroautophagy substrates identified in our proteomics time series analysis using an FDR cutoff of <5% (see STAR Methods).
Using a false detection rate (FDR) < 5% (see STAR Methods), we identified as autophagy substrates 343 of 3,885 proteins (8.8%) in basal livers and 241 of 4,083 proteins (5.9%) in LPS-treated livers (Figure 2C; Data S1 and S2). Several known macroautophagy targets were detected in basal livers, including p62 (Ichimura et al., 2008), NBR1 (Lamark et al., 2009), FASN (Shpilka et al., 2015), and the ferritinophagy marker FTH1 (Mancias et al., 2014) (Data S1), thus validating our approach. In addition, hundreds of novel substrates were identified, two of which, FABP1 and CHI3L3, we validated by western blot (Figure S2A). However, LC3b-II turnover was not detected in this dataset, potentially because of low apparent protein abundance (5 spectral counts across all basal samples) (Data S1). In addition to macroautophagy-associated proteins, we detected the turnover of CMA substrates, including GAPDH, PK, and MDH1 (Agarraberes and Dice, 2001; Schneider et al., 2014) (Data S1). This was logical, because our approach used lysosome-enriched preparations for proteomic analysis, rather than purified autophagosomes.
To confirm our experimental approach, we used an orthogonal method to immunoaffinity isolate autophagosomes for proteomic analysis. GFP-LC3-expressing transgenic mice were injected with lysosome inhibitors, and vesicles were purified using an anti-GFP resin (Figure S2B). Electron micrographs of the eluted material showed a heterogeneous mixture of vesicular structures that included classical autophagosomes with double-unit membranes (Figure S2C). Analysis of these GFP-LC3+ structures confirmed the turnover of macroautophagy substrates LC3b-II, p62, and the GFP-LC3 fusion protein, as well as the suppression of their turnover by LPS. As before, proteomic estimates of macroautophagic flux correlated tightly with flux quantification by western blot (Figures S2D and S2E). The novel autophagic substrates FABP1 and CHI3L3 that we validated in our time series experiment could similarly be validated via western blot of affinity-purified GFP-LC3+ structures (Figure S2F). Applying an FDR threshold of less than 5% (see STAR Methods), we classified 282 of 3,407 proteins detected in GFP-LC3+ vesicles (8.3%) as basal autophagic substrates and 101 of 3,038 proteins (3.3%) as targets of LPS-associated autophagy (Figure S2G; Data S2 and S3). Comparing the basal substrate proteomes derived from lysosome-enriched fractions and immunoaffinity-purified autophagosomes, there was 45.3% overlap between the datasets at the previously established thresholds, which rose to more than 70% when higher-stringency thresholds were applied (Figure S2H; Data S2). This implied that observations of autophagic flux in lysosome-enriched material and in affinity-purified autophagosomes referred to the same biological phenomenon and should arrive at qualitatively similar conclusions.
We next asked what proportion of the autophagic substrates detected by our experimental approach could be ascribed specifically to macroautophagy. To this end, we compared our data with a macroautophagy dataset generated in cultured fibroblasts by Zhang et al. (2016), who used isotopic labeling to identify proteins whose half-life was increased when the macroautophagy genes atg5 and atg7 were deleted. Of the 343 proteins identified in our time series as basal autophagy substrates, 165 had decay rates measured by Zhang et al. (2016), of which 135 (81.2%) had half-lives sensitive to atg5 and atg7 deletion (Data S1). Based on this genetic standard, most of the autophagy substrates we identified are likely clients of macroautophagy, with a sizable minority dependent on CMA or other means to reach the lysosome. Thus, we defined a panel of several hundred macroautophagy and CMA substrates, which we then used to analyze the daily dynamics of autophagy in mouse liver.
Basal Autophagy Is Universally Rhythmic
To examine global temporal patterns in autophagy, we generated a heatmap of autophagic flux over time for all 403 substrates identified by proteomics (Figure 3A). In basal livers, all 343 substrates had observable temporal variations in protein turnover. Statistically, this variation was significant in 62.6% of substrates by one-way ANOVA (p < 0.05), 49.0% of substrates by COSOPT (FDR < 5%), 62.1% of substrates by Student’s t test of peak versus trough turnover (p < 0.05), and 88.9% of substrates by at least one method (Data S1). However, these statistics likely understate the prevalence of rhythmic autophagic flux given our sampling, and we found no clear evidence that any autophagic substrate was degraded at a constant rate in basal livers. Such a result could be explained through one of two scenarios: by direct oscillations in proteolytic activity or by oscillations in protein abundance that would lead to passive rhythms in protein removal. To differentiate between these two possibilities, we cross-referenced our basal autophagy substrate proteome with 4 datasets that report circadian rhythms in mouse liver protein abundance (Mauvoisin et al., 2014; Reddy et al., 2006; Robles et al., 2014; Wang et al., 2018). Of the 343 basal autophagy substrates identified in our time series, only 101 (29.4%) exhibited diurnal rhythms in steady-state abundance in the preceding datasets (Data S1). Therefore, rhythms in autophagic flux are unlikely to be a passive reflection of substrate abundance and more likely to be due to rhythmic modulation of proteolysis.
Figure 3. Circadian Analysis of the Liver Autophagic Flux.
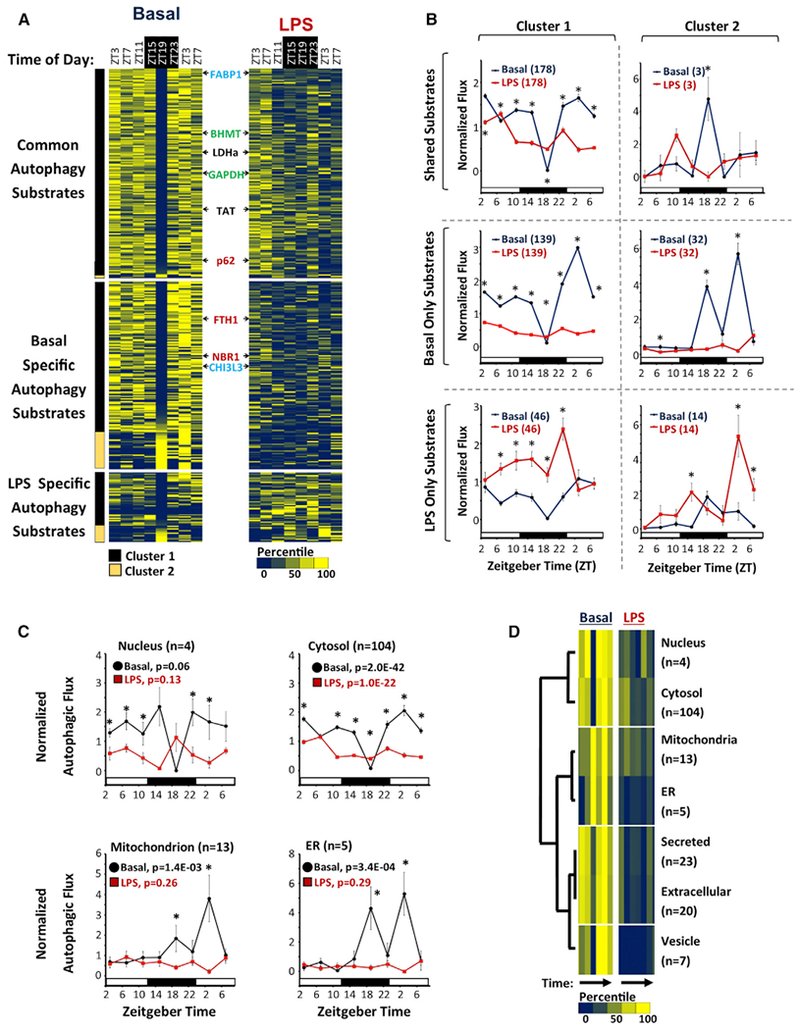
(A) Heatmap depicting autophagic flux in mouse liver as a function of time of day and treatment (basal versus LPS). FCS values were normalized and then expressed as a percentile from the mean, with dark blue representing low turnover and yellow representing high turnover. Substrates are grouped based on whether turnover was detected in both basal and LPS-treated livers (n = 181), basal alone (n = 162), or LPS alone (n = 60). The substrates are further subdivided into 2 clusters, based on whether they have a nadir in turnover at ZT19 (cluster 1, black bar, n = 308) or not (cluster 2, light brown bar, n = 35). The positions of known selective autophagy, bulk autophagy, and chaperone-mediated autophagy (CMA) substrates are depicted in red, black, and green text, respectively. The positions of FABP1 and CHI3L3 are noted in blue text.
(B) Circadian rhythms in mean normalized autophagic flux ± SE for different groups of substrates. Black circles, basal livers; red squares, LPS-treated livers. Sample sizes are specified in parentheses. **p < 0.05 basal versus LPS (Student’s two-tailed t test).
(C) Normalized autophagic flux (mean ± SE) for basal autophagy substrates with UNIPROT annotations exclusive for the nucleus (n = 4), cytosol (n = 104), mitochondria (n = 13), and ER (n = 5). Black circles, basal livers; red squares, LPS-treated livers. The p values calculated by one-way ANOVA are provided. *p < 0.05 basal versus LPS (Student’s two-tailed t test).
(D) Hierarchal clustering analysis of autophagic flux mapping to specific subcellular compartments. Colored bars represent a heatmap of autophagic flux as a function of time of day in basal and LPS-treated livers. Low flux, dark blue bars; high flux, bright yellow bars. Sample sizes are depicted to the right.
See also Figures S3 and S4 and Data S1.
Rhythms in basal autophagic flux were highly consolidated and could be divided into 2 patterns. The main group (cluster 1) contained 308 of 343, or roughly 90%, of the substrates and had a sharp nadir in autophagic flux at zeitgeber time (ZT) 19 (equivalent to 1:00 a.m. local time). Basal turnover of known macroautophagy and CMA substrates were congregated in cluster 1 (Figure 3A; Data S1). To our surprise, there was no obvious distinction in temporal profile between selective macroautophagy substrates like p62 (Ichimura et al., 2008) and proteins like TAT or LDHa that prior literature identified as clients of nonselective bulk sequestration (Figure 3A) (Kopitz et al., 1990). The second group of temporal profiles (cluster 2) was composed of the remaining 10% of substrates and was characterized by peak levels of autophagic flux at or near ZT19. Overall, LPS dampened the turnover of basal autophagy substrates while promoting the degradation of a smaller, novel set of proteins analogous to the reprogramming effect seen in lung circadian gene expression (Haspel et al., 2014) (Figures 3A and 3B).
The changes that LPS imposed on autophagic flux occurred within the context of a metabolic realignment that accompanies severe inflammatory states (Pool et al., 2018). Such changes include mitochondrial dysfunction and a Warburg effect, in which glycolytic flux and lactate production increase despite oxygen availability (Pool et al., 2018). To understand how LPS affects the physiological contributions of autophagic flux, we performed unsupervised functional enrichment pathway analysis on the basal and LPS-associated autophagy substrate proteome (Figure S3). We noted that the top enriched Kyoto Encyclopedia of Genes and Genomes (KEGG) annotation terms were largely metabolic and included the glycolysis pathway (Figure S3A). In the basal state, the turnover of glycolytic enzymes by autophagy was highly organized as part of cluster 1, and turnover of these enzymes became disorganized after LPS challenge (Figure S3B). The net effect of LPS was to reduce the average daily turnover of most glycolytic enzymes, including LDHa and the terminal rate-limiting glycolytic enzyme PKLR (Figures S3B and S3C). Because reduced autophagic turnover should stabilize or augment levels of these enzymes, the suppression of the basal autophagy program by LPS would be predicted to support enhanced glycolytic activity and lactate production.
In summary, we found that daily rhythms are a near-universal feature of basal macroautophagy and basal CMA in mouse liver and can be separated into 2 distinct temporal profiles. In addition, LPS reprograming of autophagic flux likely supports the energetic shifts that occur during severe inflammation.
The Temporal Profile of Basal Autophagic Flux Correlates with Subcellular Location
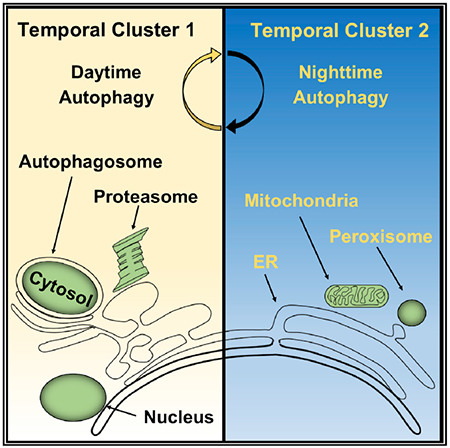
Autophagosome formation in mammalian cells occurs in multiple locations throughout the cytoplasm (Kaur and Debnath, 2015). However, cytoplasm is not homogeneous but is instead composed of numerous ultrastructural and functional subdomains. We therefore examined whether temporal profiles in autophagic flux might be connected to the subcellular location of substrates. To this end, we performed a frequency analysis of the basal autophagy proteome using UNIPROT annotation terms for subcellular localization (Data S4). For the dominant temporal profile (cluster 1), autophagy substrates localized primarily to the cytoplasm, with nucleus and mitochondria being the next most prevalent addresses (Figure S4A). Adjusting for the overall prevalence of each annotation term in our proteomics data, cluster 1 substrates were statistically over-enriched for proteins of likely cytosolic, nuclear, and extracellular location, while mitochondrially localized substrates were under-represented (Figure S4B; Data S4). In contrast, autophagy substrates belonging to the cluster 2 temporal profile were most frequently associated with mitochondria (Figure S4A). Moreover, mitochondrial, endoplasmic reticulum (ER), and peroxisome-associated proteins were over-represented in the cluster 2 autophagy substrate proteome, while nucleus and cytosol exhibited a trend toward under-enrichment (Figure S4B). These data raised the possibility that temporal patterns of basal autophagic flux may map to specific subcellular domains. To examine this theory, we analyzed the temporal profiles of proteins that had an exclusive UNIPROT subcellular localization. The temporal profiles of autophagic flux for nuclear and cytosolic proteins were highly correlated and were stereotypical of cluster 1 (Figure 3C). Meanwhile, mitochondrial and ER-specific substrates had similar rhythmic patterns in autophagic flux, which were characteristic of cluster 2. These conclusions were supported by hierarchal clustering analysis that organized nucleus-cytosol, ER-mitochondria, and secreted-extracellular substrates into distinct temporal groups (Figure 3D). Regardless of subcellular localization, LPS suppressed the turnover of basal autophagy targets (Figures 3C and 3D). At the same time, LPS induced the turnover of different proteins that as a group were biased toward a mitochondrial localization, both in the time series experiment and in immunoaffinity-purified autolysosomes (Figures S4C–S4F; Data S4). Altogether, our data suggest a relationship between the physical location of proteins within the cell and the temporal dynamics of autophagic turnover. Moreover, LPS shifts autophagy substrate preferences toward proteins associated with mitochondria, again consistent with the broader metabolic context of mitochondrial dysfunction that occurs during inflammation (Pool et al., 2018).
Selective Targeting of Proteasomes by Autophagy Is Explained by Subcellular Location
Proteasomal subunits were significantly enriched in the basal autophagy substrate proteome, constituting roughly 7% of all substrates identified (Figures 4A and 4C; Data S4). A total of 35 proteasome subunits were represented, derived from both the 20S catalytic particle and the 19S and 11S regulatory caps (Figure 4B). Autophagic flux of proteasome subunits had highly synchronous rhythms in the basal state as part of cluster 1, and the temporal profile of these subunits was similarly blunted by LPS (Figure 4B). This synchrony and the even distribution of hits among the 20S, 19S, and 11S complexes suggested that entire proteasomes were being targeted for autophagy. To confirm autophagic degradation of proteasomes, we separated total liver homogenates by density centrifugation and confirmed that the proteasomal subunits PSMB2 and PSMD11 accumulated in dense autophagosome-containing fractions after injecting mice with leupeptin (Figure 4D).
Figure 4. The Proteasome Is a Selective Target of Autophagy.
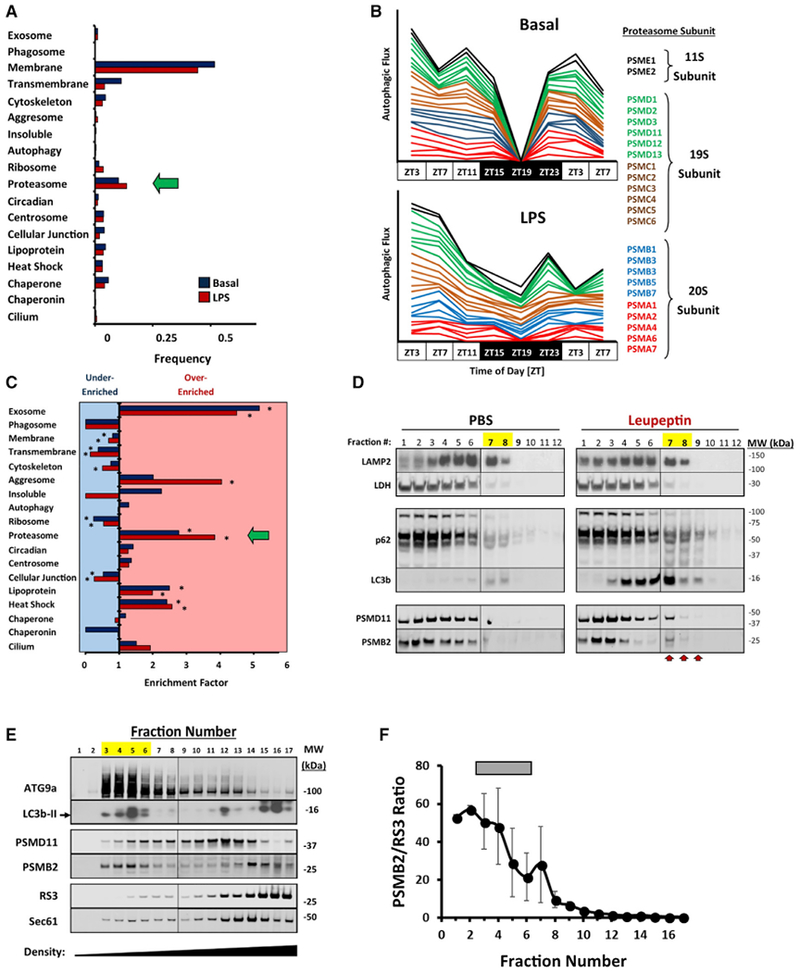
(A) Frequency of selected UNIPROT annotation terms in the basal (blue bars, n = 343) and LPS-associated (red bars, n = 241) autophagy substrate proteome. The annotation for proteasomes is marked by a green arrow.
(B) Stacked line graphs depicting the time structure of proteasome subunit turnover via autophagy (n = 24). Top graph, basal livers. Bottom graph, LPS-treated livers. As a visual aid, turnover profiles for 11S regulatory subunits are black, those for 19S subunits are green and brown, and those for 20S subunits are blue and red.
(C) Enrichment analysis for the UNIPROT annotation terms depicted in (A). Bars pointed to the right denote over-enrichment, and bars pointed to the left denote under-enrichment. See Data S4 for a tabular presentation of these data. *p < 0.05 (χ2 contingency table analysis).
(D) Western blot depicting subcellular fractionation of total liver protein using Nycodenz density gradient centrifugation (see STAR Methods). LAMP2 and LDH are provided as lysosomal and cytosolic markers, respectively. Higher fraction numbers denote increasing density. Left, sham (PBS)-treated mice. Right, leupeptin-treated mice. High-density fractions containing autophagic substrates are denoted by arrows and yellow highlights. Data are representative of 3 independent analyses.
(E) Western blots of mouse liver ER separated by OptiPrep density ultracentrifugation (see STAR Methods). Peak fractions for ATG9a and LC3b-II are highlighted yellow. Sec61 is provided as an ER marker. Data are representative of 3 independent analyses.
(F) Mean proteasome enrichment relative to ribosomes ± SE (n = 3), as reflected by PSMB2/RS3 ratios. Peak fractions for ATG9a and LC3b-II are denoted by a gray bar.
The enrichment of proteasomes among basal autophagy substrates stood in contrast to ribosomal proteins, which were significantly under-represented (Figure 4C). This occurred even though ribosomes are large, multi-subunit complexes that, like proteasomes, are highly abundant in the cytoplasm. A similar contrast between proteasome and ribosome turnover via autophagy was noted by Zhang and colleagues in immortalized human fibroblasts but was unexplained by their data (Zhang et al., 2016). Given our observation that autophagy substrate dynamics track with subcellular location, we asked whether the relative abundance of proteasomes among autophagy substrates could have a topological explanation. Because ER membranes are believed to be the primary site of autophagosome formation (Axe et al., 2008) and both proteasomes and ribosomes attach to ER membranes (Kalies et al., 2005), we separated rough and smooth ER by density centrifugation to segregate ribosome-rich from ribosome-poor ER subdomains (Figure 4E). As expected, ribosomes congregated in high-density fractions as indicated by RS3 protein abundance. In contrast, we found that the autophagosome initiation protein ATG9a, as well as the autophagosome marker LC3b-II, migrated in low-density ER fractions (Figure 4E). These same fractions were enriched in proteasome subunits PSMB2 and PSMD11 relative to the ribosomal subunit protein RS3 (Figures 4E and 4F). Thus, both the selective abundance of proteasomes and the scarcity of ribosomes among autophagy substrates can be explained by a preference for initiating autophagosome formation on smooth ER membranes.
Rhythms in Autophagic Flux and Proteasomal Flux Run in Tandem
Low-amplitude rhythms in proteasome activity were demonstrated in immortalized tissue culture cells using model peptide substrates (Desvergne et al., 2016). We asked whether similar rhythms in proteasomal activity could be detected in mouse liver and, if so, what temporal relationship exists between autophagic and proteasomal activity. To this end, we modified our turnover assay to measure proteasomal, rather than autophagic, degradation. We found that intraperitoneal (i.p.) injection of the proteasome inhibitor bortezomib could induce quantifiable cytosolic accumulation of proteasomal substrates like Lys48-linked polyubiquitinated proteins (K48-Ub) within the same 2-h time frame used to assay autophagic flux (Figures S5A and S5C). In addition, bortezomib induced a dose-dependent accumulation of p62 and LC3b-II in lysosome-enriched fractions (Figures S5B and S5D). Although induction of LC3b-II by proteasome inhibition was previously interpreted as induction of macroautophagy (Korolchuk et al., 2010), other research showed that LC3b is a direct proteasomal substrate, as well as a macroautophagy substrate (Gao et al., 2010). Using model peptides, we determined that a 1.6 μg/g i.p. dose of bortezomib was enough to reduce chymotrypsin-like and caspase-like proteasomal activities by 50%, while leupeptin administration had no effect (Figure S5E). To summarize, we adapted our turnover assay to enable parallel measurement of autophagic and proteasomal flux.
With this assay in hand, we performed simultaneous time series analyses of autophagic and proteasomal flux in mouse liver using western blotting as a readout (Figure 5A). Diurnal variation in macroautophagic flux was evident in leupeptin-treated animals using lysosome-associated p62 and LC3b-II as markers (Figure 5B). In parallel, rhythms in proteasomal activity were observed in bortezomib-treated mice using cytosolic K48-Ub (Figure 5C). As alternative markers of proteasomal flux, we measured turnover using p62 and LC3b-II in the setting of bortezomib in the lysosome-enriched fraction (Figure 5B). Proteasomal flux exhibited strong diurnal rhythms in basal mouse liver, with an amplitude of 5.6-fold based on K48-Ub turnover (Figures 5C and 5D). In contrast, in vitro proteasome activity assays using small peptide substrates in molar excess did not capture the high-amplitude rhythms seen with endogenous substrates, perhaps because this assay is more a reflection of maximum degradative capacity in a sample than proteasome flux occurring in vivo (Figures S6A–S6C). However, the rate of K48-Ub protein turnover via the proteasome did correlate with the steady-state abundance of these proteins (Figure S6D and S6E). Altogether, these data suggested that rhythms in proteasomal substrate turnover may be driven by the availability of ubiquitinated substrates, ratherthan by autophagic pruning of proteasome capacity. In bmal1-null mice, rhythms in K48-Ub turnover were disturbed, suggesting clock gene sufficiency is important for proteasomal rhythms even under standard lighting and nutrition conditions (Figures S6F). Estimates of the temporal relationship between macroautophagic and proteasomal flux varied somewhat depending on what combination of substrates were used to construct best fit curves, with an absolute phase difference ranging from 2.2 to 4.6 h. The prevailing impression, regardless of marker protein, was that rhythms in macroautophagic and proteasomal flux roughly covaried. Because autophagy and the proteasome jointly account for the bulk of cellular proteolytic activity, our results suggest a global rhythm for basal protein catabolism in mice, in which overall protein turnover is elevated in the liver during the day and low at night.
Figure 5. Diurnal Rhythms in Autophagic and Proteasomal Activity.
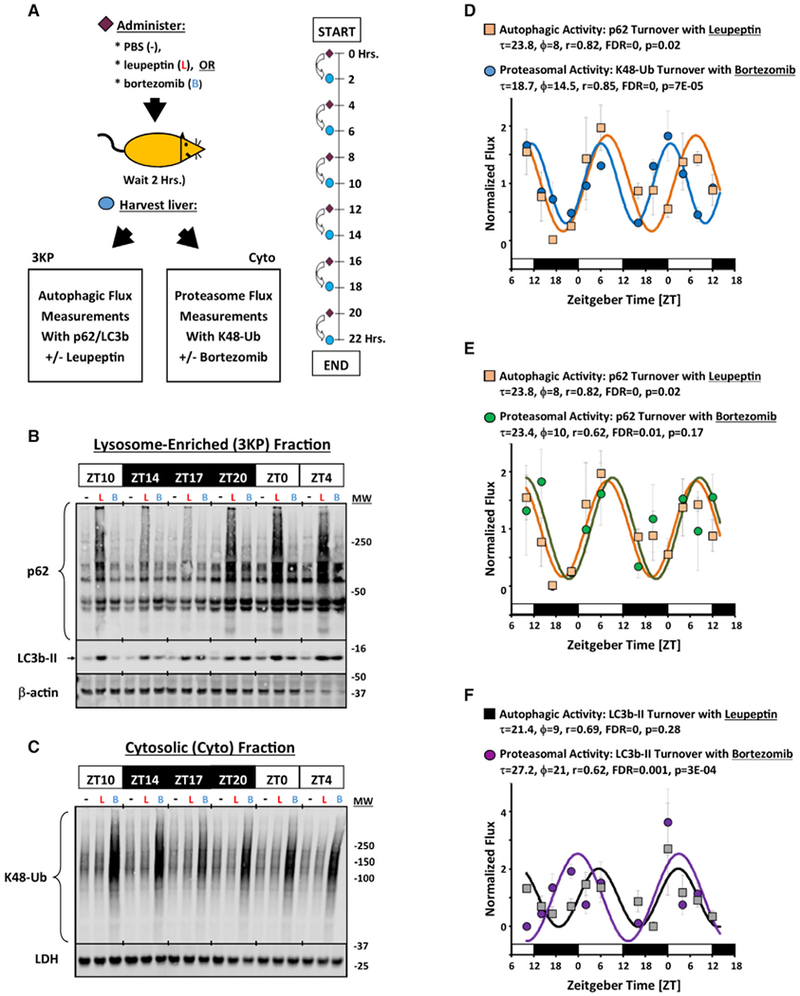
(A) Protocol for parallel time series analyses of autophagic and proteasomal flux. See STAR Methods and Figure S7A for descriptions of sample preparation.
(B and C) Representative western blots of p62 and LC3b-II in the lysosome-enriched (3KP) fraction (B) and Lys48-linked polyubiquitin (K48-Ub) chains in the Cyto fraction (C). β-actin and LDH are shown as loading controls (B and C, respectively). For (B), each lane represents 12 μL of the 3KP fraction pooled from n = 3–4 mice, representing the content obtained from 126 μg of total protein. For (C), each lane represents 19 μg of protein pooled from n = 3–4 mice. −, PBS-treated animals; L, leupeptin-treated animals; B, bortezomib-treated animals.
(D–F) Quantification of circadian rhythms in autophagic and proteasomal flux in normal mouse liver using various markers (mean ± SE, n = 3–4 measurements). Data presented were concatenated from 2 independent time series experiments. For each set of data, a best fit cosine curve and rhythm parameters were generated using COSOPT and are listed in the graphs. Statistical significance was determined using one-way ANOVA.
(D) Autophagic flux as measured by the increase in p62 with leupeptin in the 3KP fraction (orange squares), and proteasomal flux as measured by the increase in K48-Ub with bortezomib in the Cyto fraction (blue circles).
(E) Autophagic flux as measured by the increase in p62 with leupeptin in the 3KP fraction (orange squares), and proteasomal flux as measured by the uptick in p62 content with bortezomib in the 3KP fraction (green circles).
(F) Autophagic flux as measured by the increase in LC3b-II by leupeptin in the 3KP fraction (gray squares), and proteasomal flux as measured by the uptick in LC3b-II content with bortezomib in the 3KP fraction (purple circles).
Also see Figures S5 and S6.
If autophagic and proteasomal flux were coordinated, one might expect that an inhibitor of autophagic activity would also suppress proteasomal activity. We therefore measured autophagic and proteasomal flux in the setting of LPS challenge (Figure 6). As before, LPS suppressed macroautophagic flux, as indicated by lysosome-enriched p62 content ± leupeptin (Figures 6A and 6B). LPS treatment caused β-actin to more efficiently associate with the lysosome-enriched fraction, likely reflecting the known effect of LPS on actin polymerization and cytoskeletal structure (Kleveta et al., 2012). LPS also reduced proteasomal flux, as shown by cytosolic K48-Ub content ± bortezomib and p62 content ± bortezomib (Figures 6C and 6D). The results support our interpretation that proteasomal and autophagic flux are subject to coordinated, rather than reciprocal, regulation and that decreased flux through one catabolic pathway is not necessarily compensated for by increased flux through the other.
Figure 6. LPS Coordinately Represses Autophagic and Proteasomal Flux.
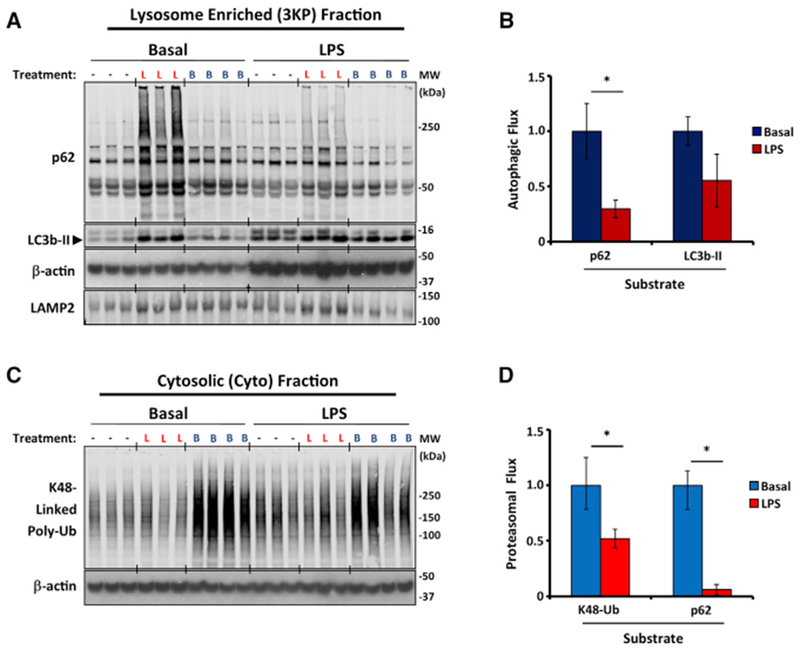
(A and C) Western blot analysis of macroautophagic (A) and proteasomal flux (C) in liver homogenates from PBS-treated (basal) and LPS-treated mice (12 mg/kg LPS given at ZT3, 19 h before harvest). −, PBS-treated animals; L, leupeptin-treated animals; B, bortezomib-treated animals. Data are representative of 2 independent experiments. Each lane in (A) represents 3KP fraction protein (12 μL or 6% of the total fraction), and each lane of (C) represents 19 mg of cytosolic protein.
(B and D) Quantification of macroautophagic (B) and proteasomal flux (D) using various markers depicted on the horizontal axis of each graph. Each bar represents the mean ± SE (n = 7 mice pooled from 2 independent experiments). To facilitate comparisons, data were normalized to results from basal mice. *p < 0.05 basal versus LPS (Student’s two-tailed t test).
DISCUSSION
Our data show how daily biological rhythms provide a framework for connecting the temporal, spatial, and meta-bolic aspects of protein catabolism. In the temporal domain, diurnal rhythms act to synchronize the proteolytic activity of autophagy and the ubiquitin-proteasome system in healthy liver, such that the bulk of protein catabolism occurs during the daytime. This is a period when food intake is low in mice and when hepatocytes contract their cytoplasmic volume (Echave Llanos et al., 1971; Sinturel et al., 2017). Basal protein turnover is conventionally assumed to be homeostatic: occurring at a constant rate that can be adjusted up or down in response to stimuli. However, our data suggest that this paradigm does not accurately describe protein catabolism in vivo, in which coherent biological rhythms pervade cellular physiology.
In the spatial domain, we found that diurnal rhythms influence where in the cell the substrates are obtained for autophagy. At the proteome-wide level, basal autophagy rhythms in mouse liver could be resolved into two clusters. In the dominant group (cluster 1), autophagy substrate preferences were directed toward cytosolic, nuclear, and secreted proteins. The dynamics of cluster 1 turnover were consistent in autophagosome number with previously published rhythms (Ma et al., 2011). However, proteomics revealed a second autophagy rhythm (cluster 2), in which substrate preferences were directed toward mitochondria, ER, and peroxisomes, all ROS-generating structures that are known to physically interact (Yoboue et al., 2018). Although these structures are thought to be targets of selective autophagy, the dynamics of cluster 2 protein degradation were not explained by known selective autophagy adaptor molecules (such as p62 and NBR1), whose turnover follows cluster 1. However, cluster 2 rhythms could be explained if different sites of autophagosome formation were subject to independent temporal control. In the basal state, autophagosomes are thought to arise from ER membranes at multiple sites throughout the cell (Axe et al., 2008; Polson et al., 2010). However, under starvation conditions, a second site of autophagosome assembly has been observed on mitochondria in proximity to ER-mitochondrial contact sites (Hailey et al., 2010). Our data argue for the existence of a separate site for basal autophagosome formation proximate to mitochondrial-ER-peroxisomal contacts that is temporally offset from other locations and is responsive to circadian regulation.
Structurally distinct locations for autophagosome assembly can explain a mysterious finding that basal autophagy selectively degrades proteasomes but seems to exclude ribosomes (Zhang et al., 2016). Both proteasomes and ribosomes are highly abundant, and both interact with ER membranes, which are believed to be the primary site of autophagosome formation. If autophagosomes targeting cluster 1 substrates were to form on smooth ER membranes, it would automatically give advantage to the sequestration of ER-bound proteasomes while simultaneously placing the sequestration of ribosomes at a disadvantage. Thus, a bias toward proteasome degradation would be achieved without the need for selective autophagy adaptor molecules. In support of this idea, we found that the early autophagosome assembly protein ATG9a associates with proteasome-containing smooth ER membranes, but not ribosome-rich rough ER. If the goal of macroautophagy were merely to sequester cytosolic contents in bulk, one would expect autophagosome formation to equally advantageous at any point on the ER surface. However, localizing a major site of autophagosome formation to smooth ER, as our data suggest, could confer benefits to cellular quality control, because it would orient macroautophagy toward the digestion of misfolded proteins that are interacting with proteasomes through the Sec61 translocation pore but cannot fully transit to the cytoplasm as part of normal ER-associated degradation.
Proteasomes were previously shown to be degraded in lysosomes (Cuervo et al., 1995), and this was theorized to represent a form of cross-talk that would enable downturns in autophagic activity to be compensated for by increased proteasome activity (Rubinsztein, 2007; Wang et al., 2013). However, we found that rhythms in proteasomal and autophagic activity were roughly synchronous rather than reciprocal, at least in the basal state. Paradigms of proteasome regulation do not place proteasome number as the key factor in controlling degradation via this process (Collins and Goldberg, 2017). Moreover, proteasomes are highly abundant (Tanaka and Ichihara, 1989), they have a half-life of 12-15 days (Tanaka and Ichihara, 1989), and only a minority of proteasomes engage in degradation at any one time (Asano et al., 2015), all of which make it difficult for 24-h rhythms in proteasome breakdown via autophagy to be a major determinant of hour-to-hour activity. Although our data do not support bulk cross-talk between basal autophagic and proteasomal activity as previously theorized, it may be possible for rhythms in autophagic flux to impart a temporal structure to proteasome function at specific subcellular compartments, such as the ER. More research will be needed to investigate this possibility.
Finally, our observations of LPS-associated autophagic flux suggest that diurnal rhythms align autophagic activity with the energetic and metabolic needs of the cell. After LPS challenge, liver autophagic activity shifted to target mitochondria-associated proteins while simultaneously downregulating the turnover of glycolytic enzymes. Both actions are predicted to support cardinal metabolic changes that accompany inflammation and sepsis: namely, the need to manage mitochondrial dysfunction and increased aerobic glycolysis. While our data by themselves do not prove that autophagy is critical to these metabolic changes, our conclusions are supported by studies in quiescent cells, in which macroautophagy gene depletion resulted in the accumulation of dysfunctional mitochondria and increased glycolysis (Nakahira et al., 2011; Watson et al., 2015). Our data also suggest that basal rhythms in liver CMA are suppressed after LPS treatment. Genetic disruption of CMA was found to induce hepatic steatosis in mice (Schneider et al., 2014). Fatty liver disease is also a cardinal feature of systemic inflammation and sepsis (Koskinas et al., 2008). Collectively, these observations explain how autophagy reprogramming can mediate the metabolic phenotype of sepsis.
In summary, we report a proteome-wide view of autophagy dynamics that captures its diurnal variation and its relationship to proteasomal function during health and inflammation. Future advances in proteomics technologies will shed further light on the relationships among circadian regulation, protein catabolism, and cellular housekeeping.
STAR*METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jeffrey A. Haspel (jhaspel@wustl.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
Male C57BL/6J mice (4-8 weeks old, 20-25 g) were obtained from Jackson Laboratories and acclimatized for two weeks before use. GFP-LC3 transgenic mice (Mizushima, 2009) were obtained from RIKEN labs (Strain number RBRC00806). Bmal1 null mice were generated by crossing Jackson Laboratory strains B6.129S4(Cg)-Arntltm1Weit/J and B6.C-Tg(CMV-cre)1Cgn/J. Heterozygote progeny were mated to each other to obtain wt and bmal1 null littermates as described (Ehlers et al., 2018). To maximize sample size for circadian time series analysis, equal numbers of male and female bmal1−/− and bmal1+/+ littermates were used, but for all other experiments only male mice were employed. Mice were housed in a standard 12-hour light-dark cycle (lights-on at 6:00 AM and lights-off at 6:00 PM local time), with food and water available ad libitum. We followed the convention of expressing time of day in “Zeitgeber Time” (ZT), where ZT0 indicated lights-on and ZT12 lights-off. In some experiments mice were injected with 12 mg/kg LPS (E. coli. lipopolysaccharide O127:B8, Sigma Cat# L3129 Lot# 029K4055). All experiments were approved by the Washington University School of Medicine and the Harvard Medical School Animal Care and Use Committees.
METHOD DETAILS
Autophagic flux measurement
Our method for measuring LC3b-II and p62 autophagic turnover was reported previously (Haspel et al., 2011). Briefly, mice were i.p. injected with 40 mg/kg leupeptin (Sigma) in sterile PBS and were euthanized for analysis 2 hours later by isoflurane anesthesia followed by cervical dislocation. For time series analysis of autophagic flux, this process was repeated every 4 hours for 24-32 hours, with each set of samples time stamped based on when the mice were euthanized. The left lobe of the liver was manually dissected and Dounce homogenized in 7 mL homogenization buffer (HB; 10 mM Tris, 250 mM sucrose, 5 mM EDTA, 1 SigmaFast protease inhibitor tablet/100mL (Sigma-Alrdrich)). The fractionation strategy used in experiments is summarized in Figure S7. Briefly, samples were centrifuged at 700 × g for 10 minutes at 4°C to remove debris, and the protein concentration of the supernatant (termed the total homogenate fraction) was normalized to 2.1 mg/mL with HB. For data in Figure 1, we adhered to our original published protocol by centrifuging 1.5 mL of total homogenate fraction at 20,000 × g for 20 minutes (Haspel et al., 2011). The pellet was washed twice with cold HB, resuspended in 200 μL NuPAGE LDS sample buffer (Life Technologies), and boiled at 95°C for 5 minutes.
For data presented in all other figures, we modified our protocol by centrifuging 1.5 mL total homogenate at 3,000 × g for 15 minutes to produce a “3KP“ fraction (Figure S7A). The 3KP fraction was then washed and used for autophagic flux analysis by western blot or proteomics (see below). The change in this sedimentation step enabled us to reduce the protein complexity of our samples, which affects the sensitivity of label-free proteomics. The 3KP fraction was chosen because: (1) most of the leupeptin-inducible LC3b-II and p62 protein content in a leupeptin-treated protein sample was recoverable in this fraction, particularly the high MW forms of p62 (Figure S7B); (2) Enrichment of LC3b-II, p62, and the lysosomal marker LAMP2 was similar in the 3KP fraction and after pelleting lysates at 20,000 × g (20KP fraction) (Figure S7C); (3) most of the of the rhythmic turnover of LC3b-II and p62 segregated with the 3KP fraction (Figure S7D) and produced similar temporal profiles compared to time series generated via our original protocol (Figure 1), and; (4) pilot proteomic analysis demonstrated that the turnover of 5 known autophagy substrates was more easily detected in the 3KP fraction than in the 20KP fraction (Figure S7E). Thus, we confirmed that the 3KP fraction allowed us to reduce the sample complexity of liver homogenates in advance of proteomics without altering the detection of autophagic flux rhythms.
The post-3KP supernatant was further clarified by centrifugation at 20,000 × g for 20 minutes. The pellet (20KP fraction) was resuspended in 100 μL LDS sample buffer as above, while the supernatant (“Cyto” fraction), was brought up in sample buffer to a 1× final concentration. To analyze macroautophagic flux by western blot, 12 μL of the lysosome enriched pellet was separated via SDS-PAGE alongside 1:3 serial dilutions of purified p62 (Novus) and GST-LC3 (Abnova) protein as described (Haspel et al., 2011). To allow data to be combined across multiple gels (to accommodate the large number of samples in a given time series) each individual gel contained its own standard curve generated from the same stock of purified p62 and LC3b. Protein was transferred to PVDF membrane and probed with LC3b, p62, or β-actin antibodies. Over the duration of this project, western blots were imaged on a LiCOR Odyssey LCx, a BioRad Gel Doc XR, or on Fuji Medical X-Ray film using a Kodak X-OMAT 2000 processor. Densitometric analysis of western blots was done using Image Studio 4.0 Software (LiCOR). For p62 densitometry we included all bands ranging from the monomeric form (which migrates at approximately 50-60 kDa) to the top of the gel as described (Haspel et al., 2011). For each protein, densitometric units were converted to ng of purified protein by extrapolating from the purified protein standard curve run in each individual gel (Haspel et al., 2011). For conversion to molar equivalents, we assumed a MW of 16 kDa for LC3b-II, 62kDa for p62, and 14 kDa for Lys48-linked poly-ubiquitin (K48-Ub). As quantified by western blots, autophagic flux was defined as the quantity of LC3b-II or p62 (expressed as ng, pmol, or normalized values) in the leupeptin treated sample minus the average quantity in the sham treated samples (Haspel et al., 2011). Although other groups adopt the practice of normalizing densitometric data to β-actin, we found that β-actin normalization had only minimal effects on our autophagic flux readouts (data not shown).
Proteomic time series
Male C57BL/6J mice were treated with either 0.5mL PBS or 12 mg/kg LPS i.p (E. Coli O157:B8) at ZT1. At four-hour intervals, mice were injected with PBS or leupeptin as above and harvested 2 hours later (n = 4 mice per treatment group received leupeptin, and 2 mice were treated with 0.5 mL PBS). Samples were homogenized to generate a post-700 × g total protein fraction as above, and then equal protein from n = 2 samples were pooled to yield 2 leupeptin treated pooled samples and 1 sham control sample for each time point. 3KP pellets were prepared as above and analyzed through label-free, bottom-up shotgun proteomics. Briefly, 3KP pellets were dried and then solubilized in SDT buffer (4% SDS, 100mM Tris-HCl pH 7.6,100 mM DTT) using sonication (Covaris S220X). Subsequent sample processing and proteomics was conducted at the NIH / NIGMS Biomedical Mass Spectrometry Resource. As negative controls for autophagic flux, each sample was first spiked with 100 ng Horseradish peroxidase (Sigma), and yeast Enolase 1 (Sigma). Samples were trypsin digested as previously described (Wiśniewski et al., 2009). Briefly, lysates in SDT buffer were reduced by heating to 95°C for 10 minutes followed by buffer exchange into 8M urea/100mM Tris buffer pH 8.5 on top of a 30K filter. Reduced proteins were alkylated by addition of 50mM Iodoacetamide and incubation at room temperature in the dark for 20 minutes. Buffer was exchanged to 100 mM ammonium bicarbonate buffer pH 8 prior to addition of endoprotease trypsin at a ratio of 1:100 trypsin to protein. Digestion with trypsin was carried out overnight at 37°C. Peptides were spun through the filter and collected in a fresh collection tube. The filter was rinsed once with 0.5 M NaCl and rinse collected on top of the peptides. Samples were acidified with 5% formic acid and desalted on three each Glygen C4 and graphite Nutips. Eluates from the tips were combined for each sample, dried and resuspended in water for peptide concentration determination by fluorescent peptide assay (Pierce). Peptide concentration was normalized to 0.35 μg/mL prior to injection on LC-MS. LC-electrospray ionization tandem MS data acquisition was performed on a TripleTOF 5600+ mass spectrometer (AB SCIEX) coupled with a nano LC 2D+ (Eksigent) and nanoflex (Eksigent) dual column setup. Digested peptides were chromatographically separated on a 15 cm × 200 μm ChromXP LC column (Eksigent) at flow rate 800 nL/min using a linear gradient from 2%–30% B (1% formic acid in acetonitrile) in 715 minutes. Column eluates were introduced into the mass spectrometer via emitter tip (New Objective) using a Nanospray® III ion source (ABSciex). The mass spectrometer was operated in positive ion mode and acquired data in data dependent mode. MS1 scan range 350-1250 for 250 msec with 100MS2 spectra for 100 msec each collected per cycle.
The samples were run in 8 sessions corresponding to the eight time points in the series. Within each MS session, 7 samples were run separated by 1-2 MS runs of blank samples to mitigate the possibility of data carryover from one sample to the next. The identity of each biological sample was blinded and the order randomized with exception of a “QC” sample consisting of mouse total protein, which was run first in each block. The QC sample was used to confirm instrument consistency from session to session and to generate a negative control distribution (“Control Distribution 1,” see below) that we used to estimate experimental error. Tandem mass spectra were analyzed using Mascot (Matrix Science, version 2.4.1). Mascot was set up to search a custom database (HASPEL_UNI_MOUSE_SOME_HUM_20150805,17065 entries), consisting of the whole mouse proteome, plus horse radish peroxidase, yeast enolase, and 156 selected human contaminant proteins. Scaffold software (Proteome Software Inc., version 4.4.1) was used to validate MS/MS-based peptide and protein identifications. Peptide identifications were made if they could be identified with > 90% probability by the Peptide Prophet algorithm (Keller et al., 2002) with Scaffold delta-mass correction. Protein identifications were established by > 95% probability in at least 1 identified peptide. Protein probabilities were assigned by the Protein Prophet algorithm (Nesvizhskii et al., 2003). Proteins that contained similar peptides and could not be differentiated based on MS/MS analysis alone were grouped. Spectral count data that met an FDR threshold of 1% was used for autophagic flux measurement (see below). Spectral count data and sample preparation details are presented in Data S1.
Proteomics of GFP-LC3+ vesicles
Our method for isolating GFP-LC3+ vesicles was based on Dengjel et al. (Dengjel et al., 2012) but utilized livers from GFP-LC3 transgenic mice rather than tissue culture cells as the source material. Briefly, GFP-LC3 mice were injected with 0.5 mL PBS or 12 mg/kg LPS at ZT11. The following day autophagic flux was measured by injecting mice at ZT4 with PBS or a protease inhibitor cocktail (40 mg/kg leupeptin, 100 mg/kg Chloroquine (Sigma)). Three hours later, mice were euthanized, and the left lobe of the liver was Dounce homogenized in 7 mL HB as above. Homogenates were filtered through 205 μm mesh (Fischer Scientific), and 20KP pellets (see Figure S7A) were prepared from the entire volume of homogenate. The 20KP pellet was washed twice and resuspended in 2 mL HB to a final protein concentration of 0.6 mg/mL as determined by Bradford assay (BioRad). The 20KP slurry was then incubated with 75 μL of anti-GFP magnetic beads (Miltenyi Biotec) at 4°C overnight on a rotator. The beads were next magnetically immobilized on a MACS LS column, washed twice with 5mL of HB, and then eluted into 2mL of HB by removing the magnet. 50 μL of autophagosome-bound beads were then processed for transmission EM imaging as described (Haspel et al., 2011) and the remainder were pelleted by centrifugation and resuspended in 200 μL NuPAGE LDS sample buffer (Life Technologies). The slurry was boiled, spun at 13,000 × g for 5 minutes to sediment the magnetic beads and 30 μL was then separated by SDS-PAGE. The gel was stained with Coomassie Brilliant blue (BioRad), and each lane was cut into 8 equal slices, de-stained, and alkylated with iodoacetamide. Gel extraction, trypsin-digestion, and proteomics analysis of samples was performed at the Harvard Mass Spectrometry and Proteomics Resource Laboratory, FAS Center for Systems Biology by reverse-phase HPLC nano-electrospray tandem mass spectrometry (mLC/MS/MS) on an LTQ-Orbitrap mass spectrometer (ThermoFisher). MS/MS spectra were analyzed using SEQUEST against the database uniprotmus_frc as described (Chittum et al., 1998; Eng et al., 1994; Taniguchi et al., 2002). Spectral counts that met an FDR threshold of 1%, based on a reverse-sequence match strategy, were then used for estimation of autophagic flux (see below). We analyzed 2 independent experiments in which Basal and LPS conditions were compared and one experiment where the basal condition alone was examined. Spectral count data are presented in Data S3.
Proteomic estimation of autophagic flux
To develop a proteomics-based estimate of autophagic flux we utilized protein spectral counts, since this means of peptide quantification is conceptually straightforward and correlates linearly with changes in relative protein abundance (Collier et al., 2010). Moreover, spectral counting has been used successfully in other proteomic analyses of autophagy where leupeptin was used to enhance substrate abundance (Cudjoe et al., 2017; Mancias et al., 2014; Schneider et al., 2014). We defined autophagic flux as the increase in spectral counts in the leupeptin-treated animals compared to sham injected animals normalized to apparent protein abundance. To this end, we defined a flux composite score (FCS) for any given protein ID j as:
Where:
Subscript j is any given protein ID, Sp is spectral counts, I is protease inhibitor-treated mice, P is pbs-treated mice, and tot is the total spectral counts for a given sample. Note negative values were treated as an FCS score of 0.
Prior proteomic analyses adopted various standards for judging proteins as targets of autophagy as opposed to stable constituents of the autolysosome (Cudjoe et al., 2017; Mancias et al., 2014; Schneider et al., 2014; Wong et al., 2017). We derived our standard for autophagy substrate detection based on the technical variability of our discovery platform, as estimated by two negative control datasets. The first dataset (Control Distribution 1, Data S5), consisted of FCS scores that were synthetically generated from 8 independent proteomics runs of the same biological sample of total liver homogenate. The second dataset (Control Distribution 2, Data S5) consisted of human contaminating proteins identified in our samples, as well as 2 exogenous proteins (HRPand yeast Enolase, see above) that were spiked in equal quantity into each of our biological samples prior to processing. Since FCS scores generated from either dataset should ideally equal 0, these distributions provided a means of estimating the contribution of technical error to our autophagic flux measurements. Applying sensitivity analysis to both control distributions, we determined that a global mean FCS score >0.3 plus at least 2 time points with average FCS scores above the 95th percentile relative to the control distribution (i.e., Data Consistency Threshold > 1) would produce a false detection rate (FDR) of < 5% for identifying autophagic substrates (Data S2). For proteomics data derived from GFP-LC3+ vesicles we established a negative control distribution consisting of human contaminant proteins and false-positive hits identified from a reverse-sequence database (Data S5). In this distribution too, a global average FCS score > 0.3 plus at least 2 time points with FCS scores above the 95th percentile relative to the control distribution (i.e., Data Consistency Threshold > 1) produced a false detection rate (FDR) of < 5% for identifying autophagic substrates (Data S2). These thresholds were therefore used throughout this paper to identify autophagy substrates. To facilitate comparisons of autophagic flux rhythms between different groupings of substrates (Figure 3), FCS values were normalized by dividing by the global mean FCS value for each identified substrate (Basal and LPS groups combined).
Endoplasmic Reticulum isolation
Purified smooth and rough endoplasmic reticulum (ER) were isolated from liver microsomes using the Endoplasmic Reticulum Isolation Kit (Sigma-Aldrich) according to the manufacturer’s instructions. Briefly, microsomes were prepared from Dounce homogenized mouse liver, floated in a discontinuous Optiprep gradient and sedimented at 150,000 × g for 3 hours in a Beckman L8-80M ultracentrifuge. Fractions were collected from the top of the gradient and analyzed via SDS-PAGE and western blot.
In vitro proteasome activity assay
For measuring proteasome activity in vitro from liver extracts we utilized fluorogenic model substrates as described (Cui et al., 2014). Briefly, liver was Dounce homogenized in proteasome reaction buffer (50 mM Tris, 1mM EDTA, 150 mM NaCl, 5 mM MgCl2, 0.5 mM DTT, pH 7.5) and clarified by centrifugation. 20 μg of liver protein was then incubated with 100 μM of Suc-LLVY-AMC (Fisher Scientific), Z-LLE-AMC (Fisher Scientific), or Boc-LSTR-AMC (Sigma) to measure chymotrypsin, caspase, and trypsin-like proteasomal activities, respectively, plus freshly added 0.1 mM ATP and 0.5 mM DTT. As a negative control duplicate samples were incubated with mg132 (Sigma) as described (Cui et al., 2014). The fluorescence accumulation was monitored for 2 hours at 37°C and an excitation/emission of 380/460 using a BioTek Synergy 4 instrument. Fluorescence was converted to nmol of AMC cleaved per minute per mL and normalized by dividing individual activities by the average assay activity.
In vivo measurement of proteasomal activity
For measuring proteasome activity in vivo, mice were injected with either 0.5 mL PBS or 1.6 μg/g bortezomib (EMD Millipore) in PBS, and the left lobe of the liver was harvested 2 hours later. Livers were then processed to obtain 3KP and Cyto fractions as described above for measuring autophagic flux (Figure S7A). Proteasomal content was quantified by analyzing Lys48-linked poly-Ubiquitin (K48-Ub) in Cyto fractions as extrapolated from a standard curve of purified protein (Boston Biochem). Analogous to autophagy, proteasomal flux was defined as the difference in K48-Ub content between Bortezomib-treated and control-treated samples. As alternative markers for proteasomal flux, p62 content ± bortezomib and LC3b-II content ± bortezomib was analyzed in the 3KP fraction.
Quantitative PCR (qPCR)
Measurement of clock gene expression in mouse liver tissue via qPCR was conducted as described (Ehlers et al., 2018). Briefly, liver RNA was extracted in Trizol, and reverse transcription was carried out using the Applied Biosystems High Capacity cDNA Reverse Transcription Kit. Real Time PCR was performed on the ABI 7500 Fast Real-Time PCR system. For all analyses tbp was used as the housekeeping gene. For determining statistical significance, gene expression data were log normalized prior to applying the Students 2-tailed t-Test.
Methodological Limitations
Our approach to measuring autophagic flux has several limitations. We utilized liver homogenates which means our method does not detect variations in autophagy or proteasome activity between cell types (hepatocytes or Kupfer cells), or between different anatomical regions within the liver.
Because time must elapse after leupeptin injection to allow lysosomal substrates to accumulate, our readout of flux represents mean activity over a 2-hour window rather than a point estimate. As a result, there is a limit to the resolution our assay can provide for visualizing diurnal rhythms, and the precision of our period and phase estimates should be treated as approximations.
In order to ensure the consistent execution of autophagic flux time series experiments, data were derived from time series shorter than 48 hours and exclusively under standard lighting conditions (LD 12:12, food ad lb.). This choice limited our sensitivity for detecting low amplitude rhythms compared to longer periods of observation (> = 48 hours). It also meant our proteomics data cannot distinguish between cell intrinsic circadian rhythms in autophagy that are independent of environmental cues, and those that are merely “diurnal” (i.e., depend on a natural light-dark cycle for expression). For circadian rhythm parameter estimation, we utilized COSOPT because of its simplicity and long history of use. However nonparametric algorithms such as JTK CYCLE have become more popular for estimating rhythm parameters (Hughes et al., 2017). In some of our early time series (Figure 1), datapoints were not all evenly spaced across the day, which is incompatible with JTK CYCLE analysis (Hughes et al., 2010). Because COSOPT is tolerant of unevenly spaced datapoints and produces similar period and phase estimates to JTK CYCLE we deemed it a reasonable choice given our data.
Our analysis does not distinguish between the turnover of proteins bearing covalent modifications (such as acetylation) and those that do not. This represents a fertile avenue for future research.
Our proteomic approach to measuring autophagic flux detects lysosomal proteolysis in the aggregate, and our interpretation of whether a given protein was a client of macroautophagy, CMA, or other pathways is dependent on prior published literature. Moreover, many proteins arrive at the lysosome via more than one mechanism, and our approach was not designed to quantitate what proportion of turnover is dependent on CMA versus macroautophagy. However, given our data that rhythms in putative macroautophagy and CMA substrates were synchronous, our conclusions are not likely to be affected by this limitation.
Finally, we used label-free proteomics and spectral counting to measure autophagy due to its comparatively low cost and flexibility. However, this approach likely biased our data toward the detection of highly abundant proteins, and low abundance autophagy substrates may have been missed. In future studies, newer on-label and multiplexed proteomics systems, as well as quantification schemes such as iBAQ, may afford greater sensitivity in detecting low frequency autophagy substrates.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistics
Circadian rhythm analysis was conducted using COSOPT as described (Haspel et al., 2014). COSOPT was set up to return best fit curves within a period range of 18-28 hours. Statistical tests including ANOVA, Students t-Test, and 2×2 χ2-square contingency tables for functional enrichment analyses were done using Microsoft Excel. We used the Excel function NORMDIST to determine the probability that the difference in LC3b-II and p62 fluxacrophases was 0 (Figure 1E). As a cutoff for assigning temporal profiles to basal autophagy substrates, proteins whose FCS scores was above the 60th percentile at ZT19 were classified assigned to Cluster 2. The rest were classified as Cluster 1. To obtain lists of mouse proteins associated with particular annotations, we queried UNIPROT in March, 2016 with the following terms: Nucleus, Cytosol, Mitochondrion, ER, Golgi, Lysosome, Peroxisome, Vesicle, Endosome, Secreted, Extracellular, Exosome, Phagosome, Membrane, Transmembrane, Multi-pass Membrane Protein, Cytoskeleton, Aggresome, Insoluble, Autophagy, Ribosome, Proteasome, Circadian, Centrosome, Cellular Junction, Lipoprotein, Heat Shock, Glycogen Granule, Chaperone, Chaperonin, and Cilium. Hierarchal clustering analysis and visualization were done using open source Cluster 3.0 and Java TreeView software (de Hoon et al., 2004), available at http://bonsai.hgc.jp/~mdehoon/software/cluster/software.htm. For KEGG pathway functional annotation enrichment analysis we used DAVID version 6.8 (Huangda et al., 2009a, 2009b), available at https://david.ncifcrf.gov/content.jsp?file=citation.htm. Using a database of 6 independent time series experiments in which LC3b-II and p62 turnover was measured, we found statistically significant temporal variations in macroautophagic flux could be observed via 1-way ANOVA in 10/12 datasets using at least one marker protein, yielding a statistical power of about 83% (data not shown). We employed blinding in the processing of samples for western blot and proteomic readouts of autophagic flux.
DATA AND SOFTWARE AVAILABILITY
Proteomics data have been deposited in the MassIVE repository under the accession number MSV000083314.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| LC3b antibody | Novus | Cat#NB100-2220; RRID:AB_10003146 |
| LC3b antibody | Cell Signaling Technology | Cat#2775; RRID:AB_915950 |
| Rabbit Anti-p62/SQSTM1 | Sigma-Aldrich | Cat#P0067; RRID:AB_1841064 |
| K48-linkage Specific Polyubiquitin (D9D5) Rabbit mAb | Cell Signaling Technology | Cat#8081S; RRID:AB_10859893 |
| beta Actin Loading Control Monoclonal Antibody (BA3R) | Thermo Fisher Scientific | Cat#MA5-15739; RRID:AB_10979409 |
| beta-Actin Antibody (AC-15) [HRP] | Novus | Cat# NB600-501H; RRID:AB_1216153 |
| LDH Antibody (H-160) | Santa Cruz Biotechnology | Cat# sc-33781; RRID:AB_2134947 |
| LAMP-2 Antibody (M3/84) | Santa Cruz Biotechnology | Cat# sc-19991; RRID:AB_626855 |
| PSMD11 (D1T1R) Rabbit mAb | Cell Signaling Technology | Cat#14303 |
| 20S Proteasome β2 Antibody (MCP165) | Santa Cruz Biotechnology | Cat# sc-58410; RRID:AB_785340 |
| FABP1 (D2A3X) XP® Rabbit mAb | Cell Signaling Technology | Cat#13368 |
| Mouse YM1/Chitinase 3-like 3 Antibody | R and D Systems | Cat# AF2446; RRID:AB_2079008 |
| IRDye® 800CW Donkey anti-Rabbit IgG (H + L) | LI-COR | Cat#926-32213; RRID:AB_621848 |
| IRDye 680RD Donkey anti-Mouse IgG (H + L) | LI-COR | Cat# 926-68072; RRID:AB_10953628 |
| CLOCK (D45B10) Rabbit mAb | Cell Signaling Technology | Cat# 5157s; RRID:AB_10695411 |
| Phospho-S6 Ribosomal Protein (Ser235/236) (D57.2.2E) XP® Rabbit mAb | Cell Signaling Technology | Cat# 4858; RRID:AB_916156 |
| Ribosomal Protein S6 Antibody (E-13) | Santa Cruz Technology | Cat# sc-13007; RRID:AB_655867 |
| RORα (H-65) rabbit polyclonal antibody | Santa Cruz Technology | Cat# sc-28612; RRID:AB_2180141 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Bortezomib | EMD Millipore | Cat# 5.04314.0001; CAS: 179324-69-7 |
| Leupeptin | Sigma | Cat# L2884; CAS: 103476-89-7 |
| Lipopolysaccharides from Escherichia coli O127:B8 | Sigma | Cat# L3129 |
| Chloroquine | Sigma | Cat# C6628 |
| Histodenz | Sigma | Cat# D2158; CAS: 66108-95-0 |
| SIGMAFAST Protease Inhibitor Tablets | Sigma | Cat# S8820 |
| NuPAGE 4-12% Bis-Tris Midi Protein Gels | Thermo Fisher Scientific | Cat# WG1403BOX |
| Horseradish peroxidase | Sigma | Cat# P-6782; CAS: 9003-99-0 |
| Enolase from baker’s yeast (S. cerevisiae) | Sigma | Cat# E6126; CAS: 9014-08-8 |
| QC Colloidal Coomassie Stain | Bio-Rad | Cat# 1610803 |
| Suc-LLVY-AMC, fluorogenic substrate | Anaspec | Cat# AS-63892 |
| Z-Leu-Leu-Glu-AMC (Z-LLE-AMC) | R&D Systems | Cat# S-230-05M |
| Boc-Leu-Ser-Thr-Arg-7-amido-4-methylcoumarin protein C substrate | Sigma | Cat# B4636 |
| 7-Amino-4-methylcoumarin, 98% | Alfa Aesar | Cat# A15017-MD |
| MG132 | Sigma | Cat# C2211; CAS 133407-82-6 |
| GST-LC3b | Novus | Cat# H00081631-Q01 |
| P62-his | Novus | Cat# NBP1-44490 |
| LC3a | Boston Biochem | Cat# UL-430 |
| rhPoly-Ub WT (2-7) (K48) | Boston Biochem | Cat# UC-230 |
| Critical Commercial Assays | ||
| uMACS GFP Isolation Kit | Miltenyi Biotec | Cat# 130-091-125 |
| Deposited Data | ||
| Proteomics Data | This paper | MassIVE: MSV000083314 |
| Experimental Models: Organisms/Strains | ||
| Mouse: C57BL/6J | Jackson Laboratories | JAX: 000664 |
| Mouse: GFP-LC3 transgenic mice | RIKEN Labs | RBRC00806 |
| Mouse: B6.129S4(Cg)-Arntltm1Weit/J | Jackson Laboratories | JAX: 007668 |
| Mouse: B6.C-Tg(CMV-cre)1Cgn/J | Jackson Laboratories | JAX: 006054 |
| Oligonucleotides | ||
| PrimeTime Std® qPCR Assay for dbp, mouse, Assay ID Mm.PT.58.16911772 | IDT | Cat# Mm.PT.58.16911772 |
| TaqMan® Gene Expression Assay for arntl, mouse, Assay ID Mm00500223_m1 | ThermoFisher | Cat# Mm00500223_m1 |
| PrimeTime Std® qPCR Assay for tbp, mouse, Assay ID Mm.PT.39a.22214839 | IDT | Cat#Mm.PT.39a.22214839 |
| Software and Algorithms | ||
| COSOPT | Haspel et al., 2014 | N/A |
| Cluster 3.0 | de Hoon et al., 2004 | http://bonsai.hgc.jp/~mdehoon/software/cluster/software.htm |
| Java TreeView software | de Hoon et al., 2004 | http://bonsai.hgc.jp/~mdehoon/software/cluster/software.htm |
| Other | ||
| Immobilon-FL PVDF Membrane | EMD Millipore | Cat# IPFL00010 |
Highlights.
Autophagy exhibits proteome-wide rhythms in activity in mouse liver
Rhythms globally harmonize autophagic and proteasomal activity
Biological rhythms inform the subcellular location of autophagy substrates
Inflammation polarizes autophagy toward the breakdown of mitochondria
ACKNOWLEDGMENTS
We thank Erik Herzog, Michael Hughes, Steven Brody, Amjad Horani, Robyn Haspel, Renee Robinson, John Neveu, and the Clocks & Sleep Club at the Hope Center for Neurological Disorders, Washington University School of Medicine, for their scientific input. This work was funded by K08GM102694, RO1HL135846, a Children’s Development Institute grant (PD-II-2016-529), ATS Recognition Award for Outstanding Early Career Investigators, VA (VISN1) Career Development Award I, and a Parker B. Francis Scientific Opportunity Award (to J.A.H.). It was also funded by P01HL114501 and R01HL133801 (to A.M.K.C.) and T32HL007317 (to M.R.).
Footnotes
DECLARATION OF INTERESTS
A.M.K.C. is a cofounder, stock holder, and serves on the Scientific Advisory Board for Proterris, which develops therapeutic uses for carbon monoxide. A.M.K.C. also has a use patent on CO. A.M.K.C. served as a consultant for Teva Pharmaceuticals in July 2018.
REFERENCES
- Agarraberes FA, and Dice JF (2001). A molecular chaperone complex at the lysosomal membrane is required for protein translocation. J. Cell Sci 774, 2491–2499. [DOI] [PubMed] [Google Scholar]
- Asano S, Fukuda Y, Beck F, Aufderheide A, Förster F, Danev R, and Baumeister W (2015). Proteasomes. A molecular census of 26S proteasomes in intact neurons. Science 347, 439–442. [DOI] [PubMed] [Google Scholar]
- Axe EL, Walker SA, Manifava M, Chandra P, Roderick HL, Habermann A, Griffiths G, and Ktistakis NT (2008). Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J. Cell Biol 182, 685–701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chittum HS, Lane WS, Carlson BA, Roller PP, Lung FD, Lee BJ, and Hatfield DL (1998). Rabbit beta-globin is extended beyond its UGA stop codon by multiple suppressions and translational reading gaps. Biochemistry 37, 10866–10870. [DOI] [PubMed] [Google Scholar]
- Cho H, Zhao X, Hatori M, Yu RT, Barish GD, Lam MT, Chong LW, DiTacchio L, Atkins AR, Glass CK, et al. (2012). Regulation of circadian behaviour and metabolism by REV-ERB-α and REV-ERB-β. Nature 485, 123–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collier TS, Sarkar P, Franck WL, Rao BM, Dean RA, and Muddiman DC (2010). Direct comparison of stable isotope labeling by aminoacids in cell culture and spectral counting for quantitative proteomics. Anal. Chem 82, 8696–8702. [DOI] [PubMed] [Google Scholar]
- Collins GA, and Goldberg AL (2017). The logic of the 26S proteasome. Cell 169, 792–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cudjoe EK Jr., Saleh T, Hawkridge AM, and Gewirtz DA (2017). Proteomics insights into autophagy. Proteomics 17, 1700022. [DOI] [PubMed] [Google Scholar]
- Cuervo AM, Palmer A, Rivett AJ, and Knecht E (1995). Degradation of proteasomes by lysosomes in rat liver. Eur. J. Biochem 227, 792–800. [DOI] [PubMed] [Google Scholar]
- Cui Z, Gilda JE, and Gomes AV (2014). Crude and purified proteasome activity assays are affected by type of microplate. Anal. Biochem 446, 44–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Hoon MJ, Imoto S, Nolan J, and Miyano S (2004). Open source clustering software. Bioinformatics 20, 1453–1454. [DOI] [PubMed] [Google Scholar]
- Dengjel J, Høyer-Hansen M, Nielsen MO, Eisenberg T, Harder LM, Schandorff S, Farkas T, Kirkegaard T, Becker AC, Schroeder S, et al. (2012). Identification of autophagosome-associated proteins and regulators by quantitative proteomic analysis and genetic screens. Mol. Cell, Proteomics 11, M111.014035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Desvergne A, Ugarte N, Radjei S, Gareil M, Petropoulos I, and Friguet B (2016). Circadian modulation of proteasome activity and accumulation of oxidized protein in human embryonic kidney HEK 293 cells and primary dermal fibroblasts. Free Radic. Biol. Med 94, 195–207. [DOI] [PubMed] [Google Scholar]
- Dice JF (1992). Selective degradation of cytosolic proteins by lysosomes. Ann. N Y Acad. Sci 674, 58–64. [DOI] [PubMed] [Google Scholar]
- Echave Llanos JM, Aloisso MD, Souto M, Balduzzi R, and Surur JM (1971). Circadian variations of DNA synthesis, mitotic activity, and cell size of hepatocyte population in young immature male mouse growing liver. Virchows Arch. B Cell Pathol. Incl. Mol. Pathol 8, 309–317. [DOI] [PubMed] [Google Scholar]
- Ehlers A, Xie W, Agapov E, Brown S, Steinberg D, Tidwell R, Sajol G, Schutz R, Weaver R, Yu H, et al. (2018). BMAL1 linksthe circadian clock to viral airway pathology and asthma phenotypes. Mucosal Immunol. 11, 97–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eng JK, McCormack AL, and Yates JR (1994). An approach to correlate tandem massspectral data of peptides with amino acid sequences in a protein database. J. Am. Soc. Mass Spectrom 5, 976–989. [DOI] [PubMed] [Google Scholar]
- Ezaki J, Matsumoto N, Takeda-Ezaki M, Komatsu M, Takahashi K, Hiraoka Y, Taka H, Fujimura T, Takehana K, Yoshida M, et al. (2011). Liver autophagy contributes to the maintenance of blood glucose and amino acid levels. Autophagy 7, 727–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao Z, Gammoh N, Wong PM, Erdjument-Bromage H, Tempst P, and Jiang X (2010). Processing of autophagic protein LC3 by the 20S proteasome. Autophagy 6, 126–137. [DOI] [PubMed] [Google Scholar]
- Gong C, Li C, Qi X, Song Z, Wu J, Hughes ME, and Li X (2015). The daily rhythms of mitochondrial gene expression and oxidative stress regulation are altered by aging in the mouse liver. Chronobiol. Int 32, 1254–1263. [DOI] [PubMed] [Google Scholar]
- Green CB, Takahashi JS, and Bass J (2008). The meter of metabolism. Cell 134, 728–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hailey DW, Rambold AS, Satpute-Krishnan P, Mitra K, Sougrat R, Kim PK, and Lippincott-Schwartz J (2010). Mitochondria supply membranes for autophagosome biogenesis during starvation. Cell 141, 656–667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haspel J, Shaik RS, Ifedigbo E, Nakahira K, Dolinay T, Englert JA, and Choi AM (2011). Characterization of macroautophagic flux in vivo using a leupeptin-based assay. Autophagy 7, 629–642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haspel JA, Chettimada S, Shaik RS, Chu JH, Raby BA, Cernadas M, Carey V, Process V, Hunninghake GM, Ifedigbo E, et al. (2014). Circadian rhythm reprogramming during lung inflammation. Nat. Commun 5, 4753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W, Sherman BT, and Lempicki RA (2009a). Bioinformatics enrichment tools: paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 37, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang da W., Sherman BT, and Lempicki RA (2009b). Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc 4, 44–57. [DOI] [PubMed] [Google Scholar]
- Hughes ME, Hogenesch JB, and Kornacker K (2010). JTK_CYCLE: an efficient nonparametric algorithm for detecting rhythmic components in genome-scale data sets. J. Biol. Rhythms 25, 372–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hughes ME, Abruzzi KC, Allada R, Anafi R, Arpat AB, Asher G, Baldi P, de Bekker C, Bell-Pedersen D, Blau J, et al. (2017). Guidelines for genome-scale analysis of biological rhythms. J. Biol. Rhythms 32, 380–393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ichimura Y, Kominami E, Tanaka K, and Komatsu M (2008). Selective turnover of p62/A170/SQSTM1 by autophagy. Autophagy 4, 1063–1066. [DOI] [PubMed] [Google Scholar]
- Kalies KU, Allan S, Sergeyenko T, Kröger H, and Römisch K (2005). The protein translocation channel binds proteasomes to the endoplasmic reticulum membrane. EMBO J. 24, 2284–2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaur J, and Debnath J (2015). Autophagy at the crossroads of catabolism and anabolism. Nat. Rev. Mol. Cell Biol 16, 461–472. [DOI] [PubMed] [Google Scholar]
- Keller A, Nesvizhskii AI, Kolker E, and Aebersold R (2002). Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal. Chem 74, 5383–5392. [DOI] [PubMed] [Google Scholar]
- Kleveta G, Borzęcka K, Zdioruk M, Czerkies M, Kuberczyk H, Sybirna N, Sobota A, and Kwiatkowska K (2012). LPS induces phosphorylation of actin-regulatory proteins leading to actin reassembly and macrophage motility. J. Cell. Biochem 113, 80–92. [DOI] [PubMed] [Google Scholar]
- Klionsky DJ, Abdelmohsen K, Abe A, Abedin MJ, Abeliovich H, Acevedo Arozena A, Adachi H, Adams CM, Adams PD, Adeli K, et al. (2016). Guidelines for the use and interpretation of assays for monitoring autophagy (3rd edition). Autophagy 12, 1–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondratov RV, Kondratova AA, Gorbacheva VY, Vykhovanets OV, and Antoch MP (2006). Early aging and age-related pathologies in mice deficient in BMAL1, the core component of the circadian clock. Genes Dev. 20, 1868–1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kopitz J, Kisen GO, Gordon PB, Bohley P, and Seglen PO (1990). Nonselective autophagy of cytosolic enzymes by isolated rat hepatocytes. J. Cell Biol 111, 941–953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korolchuk VI, Menzies FM, and Rubinsztein DC (2010). Mechanisms of cross-talk between the ubiquitin-proteasome and autophagy-lysosome systems. FEBS Lett. 584, 1393–1398. [DOI] [PubMed] [Google Scholar]
- Koskinas J, Gomatos IP, Tiniakos DG, Memos N, Boutsikou M, Garatzioti A, Archimandritis A, and Betrosian A (2008). Liver histology in ICU patients dying from sepsis: a clinico-pathological study. World J. Gastroenterol 14, 1389–1393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lamark T, Kirkin V, Dikic I, and Johansen T (2009). NBR1 and p62 as cargo receptors for selective autophagy of ubiquitinated targets. Cell Cycle 8, 1986–1990. [DOI] [PubMed] [Google Scholar]
- Ma D, Panda S, and Lin JD (2011). Temporal orchestration of circadian autophagy rhythm by C/EBPβ. EMBO J. 30, 4642–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancias JD, Wang X, Gygi SP, Harper JW, and Kimmelman AC (2014). Quantitative proteomics identifies NCOA4 as the cargo receptor mediating ferritinophagy. Nature 509, 105–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauvoisin D, Wang J, Jouffe C, Martin E, Atger F, Waridel P, Quadroni M, Gachon F., and Naef F (2014). Circadian clock-dependent and -independent rhythmic proteomes implement distinct diurnal functions in mouse liver. Proc. Natl. Acad. Sci. USA 111, 167–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N (2009). Methods for monitoring autophagy using GFP-LC3 transgenic mice. Methods Enzymol. 452, 13–23. [DOI] [PubMed] [Google Scholar]
- Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim HP, et al. (2011). Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat. Immunol 12, 222–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nesvizhskii AI, Keller A, Kolker E, and Aebersold R (2003). A statistical model for identifying proteins by tandem mass spectrometry. Anal. Chem 75, 4646–658. [DOI] [PubMed] [Google Scholar]
- Okada K, Yano M, Doki Y, Azama T, Iwanaga H, Miki H, Nakayama M, Miyata H, Takiguchi S, Fujiwara Y, et al. (2008). Injection of LPS causes transient suppression of biological clock genes in rats. J. Surg. Res 145, 5–12. [DOI] [PubMed] [Google Scholar]
- Pfeifer U (1971). [Circadian rhythm of cellularautophagy]. Naturwissenschaften 58, 152. [PubMed] [Google Scholar]
- Polson HE, de Lartigue J, Rigden DJ, Reedijk M, Urbé S, Clague MJ, and Tooze SA (2010). Mammalian Atg18 (WIPI2) localizes to omegasome-anchored phagophores and positively regulates LC3 lipidation. Autophagy 6, 506–522. [DOI] [PubMed] [Google Scholar]
- Pool R, Gomez H, and Kellum JA (2018). Mechanisms of organ dysfunction in sepsis. Crit. Care Clin 34, 63–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy AB, Karp NA, Maywood ES, Sage EA, Deery M, O’Neill JS, Wong GK, Chesham J, Odell M, Lilley KS, et al. (2006). Circadian orchestration of the hepatic proteome. Curr. Biol 16, 1107–1115. [DOI] [PubMed] [Google Scholar]
- Robles MS, Cox J, and Mann M (2014). In-vivo quantitative proteomics reveals a key contribution of post-transcriptional mechanisms to the circadian regulation of liver metabolism. PLoS Genet. 10, e1004047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubinsztein DC (2007). Autophagy induction rescues toxicity mediated by proteasome inhibition. Neuron 54, 854–856. [DOI] [PubMed] [Google Scholar]
- Schneider JL, Suh Y, and Cuervo AM (2014). Deficient chaperone-mediated autophagy in liver leads to metabolic dysregulation. Cell Metab 20, 417–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider JL, Villarroya J, Diaz-Carretero A, Patel B, Urbanska AM, Thi MM, Villarroya F, Santambrogio L, and Cuervo AM (2015). Loss of hepatic chaperone-mediated autophagy accelerates proteostasis failure in aging. Aging Cell 14, 249–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shpilka T, Welter E, Borovsky N, Amar N, Shimron F, Peleg Y, and Elazar Z (2015). Fatty acid synthase is preferentially degraded by autophagy upon nitrogen starvation in yeast. Proc. Natl. Acad. Sci. USA 112, 1434–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinturel F, Gerber A, Mauvoisin D, Wang J, Gatfield D, Stubblefield JJ, Green CB, Gachon F, and Schibler U (2017). Diurnal oscillations in liver mass and cell size accompany ribosome assembly cycles. Cell 169, 651–663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storch KF, Paz C, Signorovitch J, Raviola E, Pawlyk B, Li T, and Weitz CJ (2007). Intrinsic circadian clock of the mammalian retina: importance for retinal processing of visual information. Cell 130, 730–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tanaka K, and Ichihara A (1989). Half-life of proteasomes (multiprotease complexes) in rat liver. Biochem. Biophys. Res. Commun 159, 1309–1315. [DOI] [PubMed] [Google Scholar]
- Taniguchi T, Garcia-Higuera I, Xu B, Andreassen PR, Gregory RC, Kim ST, Lane WS, Kastan MB, and D’Andrea AD (2002). Convergence of the fanconi anemia and ataxia telangiectasia signaling pathways. Cell 109, 459–472. [DOI] [PubMed] [Google Scholar]
- Wang XJ, Yu J, Wong SH, Cheng AS, Chan FK, Ng SS, Cho CH, Sung JJ, and Wu WK (2013). A novel crosstalk between two major protein degradation systems: regulation of proteasomal activity by autophagy. Autophagy 9, 1500–1508. [DOI] [PubMed] [Google Scholar]
- Wang Y, Song L, Liu M, Ge R, Zhou Q, Liu W, Li R, Qie J, Zhen B, Wang Y, et al. (2018). A proteomics landscape of circadian clock in mouse liver. Nat. Commun 9, 1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson AS, Riffelmacher T, Stranks A, Williams O, De Boer J, Cain K, MacFarlane M, McGouran J, Kessler B, Khandwala S, et al. (2015). Autophagy limits proliferation and glycolytic metabolism in acute myeloid leukemia. Cell Death Discov. 1, 15008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wisniewski JR, Zougman A, Nagaraj N, and Mann M (2009). Universal sample preparation method for proteome analysis. Nat. Methods 6, 359–362. [DOI] [PubMed] [Google Scholar]
- Wong YK, Zhang J, Hua ZC, Lin Q, Shen HM, and Wang J (2017). Recent advances in quantitative and chemical proteomics for autophagy studies. Autophagy 13, 1472–1486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoboue ED, Sitia R, and Simmen T (2018). Redox crosstalk at endoplasmic reticulum (ER) membrane contact sites (MCS) uses toxic waste to deliver messages. Cell Death Dis. 9, 331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Shen S, Qu J, and Ghaemmaghami S (2016). Global analysis of cellular protein flux quantifies the selectivity of basal autophagy. Cell Rep. 14, 2426–2439. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Proteomics data have been deposited in the MassIVE repository under the accession number MSV000083314.