

SUMMARY
Innate immunity is central to the pathophysiology of neurodegenerative disorders, but it remains unclear why immunity is altered in the disease state and whether changes in immunity are a cause or a consequence of neuronal dysfunction. Here, we identify a molecular pathway that links innate immunity to age-dependent loss of dopaminergic neurons in Drosophila. We find, first, that altering the expression of the activating subunit of the Cdk5 protein kinase (Cdk5α) causes severe disruption of autophagy. Second, this disruption of autophagy is both necessary and sufficient to cause the hyperactivation of innate immunity, particularly expression of anti-microbial peptides. Finally, it is the upregulation of immunity that induces the age-dependent death of dopaminergic neurons. Given the dysregulation of Cdk5 and innate immunity in human neurodegeneration and the conserved role of the kinase in the regulation of autophagy, this sequence is likely to have direct application to the chain of events in human neurodegenerative disease.
Graphical Abstract
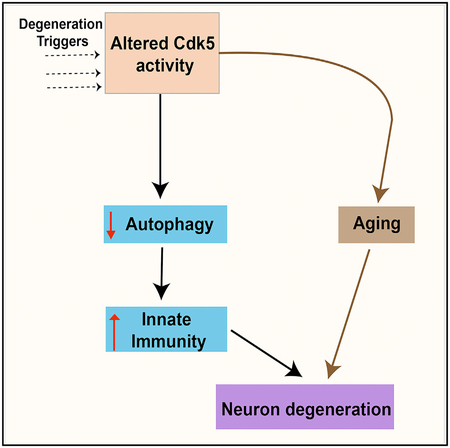
In Brief
How can one disentangle the many pathologies of neurodegeneration from one another and from normal aging? Shukla et al. show that a mutation in Drosophila kills neurons by impairing autophagy, which in turn stimulates neurotoxic levels of innate immunity, and this acts synergistically with a parallel pathway that accelerates aging.
INTRODUCTION
Neurodegenerative diseases (NDs) are a huge public health problem, and dissecting their pathophysiological mechanism is one of our greatest challenges. Neuroinflammation is an important component of many NDs, including Alzheimer disease (AD), frontotemporal dementia (FTD), Parkinson disease (PD), and others (Gjoneska et al., 2015; Heneka et al., 2014; Holmes et al., 2009; Richards et al., 2016; Zhang, 2015). However, the role of immune dysfunction in NDs remains paradoxical; there is evidence that the activation of microglia may induce neurotoxicity, but also evidence that it is protective, through the clearance of toxic protein aggregates (Clayton et al., 2017). Thus, it remains controversial whether neurodegeneration is the consequence of hyperactivation or inactivation of the immune response, and what the triggers are that induce its dysfunction.
The immune response in the nervous system is not only triggered by pathogens but also by its linkage to autophagy (Richards et al., 2016). Autophagy is essential for removing damaged proteins and organelles, safeguarding cellular energy balance, and maintaining cellular homeostasis (Wang and Qin, 2013). Autophagy is also an alternative route of cell death that is distinct from apoptosis, and it is implicated in a wide variety of NDs (Clarke, 1990; Nixon, 2013). Fundamental questions remain, however: is autophagy a pro-death program or a protective program that enhances survival, and does disruption of autophagy serve as an early, triggering event in ND, or is it a late-acting piece of the mechanism?
The greatest risk factor for most NDs is aging (Wyss-Coray, 2016). Aging broadly changes the physiology of the organism, in part by disrupting cellular homeostasis. The nervous system is particularly sensitive to the function and regulation of homeostatic mechanisms, including both autophagy and immunity, among many others (Nixon, 2013; Schwartz et al., 2013). One issue confounding our understanding of human ND is that the normal modulation of immunity and autophagy by aging has obscured whether changes in these processes reflect a direct role in pathogenesis or simply a correlation among the processes of normal aging.
The mechanisms of autophagy and innate immunity, as well as aging, are significantly conserved between mammals and Drosophila (Kimbrell and Beutler, 2001; Mulakkal et al., 2014). Drosophila has a well-regulated innate immune system that uses anti-microbial peptides (AMPs) as effector molecules, including several with clear mammalian orthologs. Two parallel pathways exist for the activation of AMP synthesis, under control of the receptors Toll and Imd (immune deficiency), and these are homologous to innate immune pathways in mammals (Lemaitre and Hoffmann, 2007). Toll and Imd, respectively, signal through the nuclear factor κB (NF-κB) transcription factors Dif and Relish, which promote the transcription of multiple classes of AMPs in Drosophila, including attacin, cecropin, diptericin, drosocin, drosomycin, defensin, and metchnikowin, as well as other immune effectors. Some recent studies in Drosophila have suggested a negative role for hyperactive innate immune response in neurodegeneration and aging (Cao et al., 2013; Kounatidis et al., 2017; Petersen et al., 2013), although other reports suggest a positive role for the overexpression of AMPs on aging (Loch et al., 2017). Therefore, in flies as in mammals, the relation among these processes in the progression to disease remains unclear.
We have shown previously that increased or decreased activity of cyclin-dependent kinase 5 (Cdk5), achieved by altered expression of its essential activating subunit, Cdk5α (also called D-p35), causes a neurodegenerative syndrome in Drosophila that has extensive similarities to human NDs, including adult-onset degeneration and the death of neurons that are associated with learning and memory (mushroom body [MB] neurons), impaired auto-phagy, sensitivity to oxidative stress, and progressive loss of motor function, along with an accelerated rate of aging (Spurrier et al., 2018; Trunova and Giniger, 2012). Cdk5 is a divergent member of the cyclin-dependent kinase family that does not associate with a classical cyclin for its activation and is not required for cell-cycle progression. Cdk5 is expressed ubiquitously; however, its function is limited to postmitotic neurons due to the restricted expression of its activating subunit (Connell-Crowley et al., 2000; Tsai et al., 1994). Cdk5 activation in mammals requires specific binding with either p35 or the related protein p39 (Ko et al., 2001), while in Drosophila there is only a single p35 ortholog (Connell-Crowley et al., 2007). Deregulated Cdk5 has been associated with different NDs in humans. For example, in the case of AD, Cdk5 causes hyperphosphorylation of tau and is involved in the formation of neurofibrillary tangles (Cruz et al., 2003). In the 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP) model of PD, Cdk5 causes phosphorylation of an antioxidant enzyme, Prx2, which is associated with cell death (Qu et al., 2007a). Notably, both gain and loss of function of Cdk5 induce neuronal death and neurodegeneration in cell culture and in mouse models, perhaps because of cross-regulatory interactions among a network of kinases with closely related target-site specificity (Cruz et al., 2003; Patrick et al., 1999; Takahashi et al., 2010).
Here, we unravel the relations among the core phenotypes of Cdk5-associated neurodegeneration in Drosophila. We show that increasing or decreasing the expression of Cdk5α impairs autophagy and that this in turn causes the degeneration of a sensitive neuronal population through hyperactivation of the innate immune response. We first show that the activation of innate immunity occurs independently of aging in Cdk5α-altered Drosophila. We then demonstrate that dopamine (DA) neurons, like MB neurons, undergo age-dependent degeneration when we increase or decrease the expression of Cdk5α. Furthermore, we show that the activation of innate immunity by altered Cdk5α is both necessary and sufficient to cause DA neuron death, and that the disruption of autophagy by altered Cdk5α is necessary and sufficient to account for the hyperactivation of immunity. Our data provide a clear picture of the relation among three of the key features of NDs, aging, autophagy, and immunity, revealing that autophagy and immunity make up a dependent genetic pathway that assaults neurons, in conjunction with the overall fragility induced by aging.
RESULTS
Increased or Decreased Expression of Cdk5α Causes Age-Dependent Degeneration of Dopaminergic Neurons in Drosophila
MB cell counting and global gene expression profiling in Drosophila that are null mutant for Cdk5α (Cdk5α null) or have mild (2.5- to 3-fold) overexpression of Cdk5α (Cdk5-OE; from the introduction of four extra copies of the wild-type Cdk5α genomic locus) revealed age-dependent degeneration of MB neurons (Spurrier et al., 2018). While comparing the gene expression profile obtained upon altering Cdk5α to that from other Drosophila models of degeneration, we noted a striking similarity with the profile of flies bearing a mutation in the fly ortholog of the human PD-associated gene Pink1 (Pearson r > 0.36; corrected p < 0.05 in three out of four conditions examined; Table S1). Pink1 mutant flies, like humans, lose DA neurons as they age (Park et al., 2006). Moreover, flies with altered Cdk5α show impaired motor function with age, reminiscent of Pink1 mutant flies. We therefore counted DA neurons using whole-mount immunostaining with anti-Drosophila tyrosine hydroxylase (DTH). We found that the number of neurons was not significantly changed in wild-type flies until 45 days of age, whereas both Cdk5α null and Cdk5α-OE show an earlier, age-dependent loss of DA neurons (Figures 1A, 1B, and S1; Table S2). Consistent results were observed using a different anti-TH antibody (Figure S2B) or by co-labeling with TH-GAL4; UAS-nls-mCherry and counting double-tagged neurons at 30 days old (Figure S2A). We verified the specificity of the mutant phenotype by restoring the DA number with a single copy of a Cdk5α genomic trans-gene in Cdk5α null flies (“rescue” hereafter: w+; Cdk5α/Cdk5α; P[w+,Tn Cdk5α]R157/+) (Figure 1C; Table S2). Note that we typically assay degeneration at 30 days, since by 45 days, only the last few percent of mutant flies survive and these are not representative of the starting population (Spurrier et al., 2018). These data show that the altered expression of Cdk5α causes age-associated degeneration of DA neurons.
Figure 1. Both Gain and Loss of Cdk5α Induce the Degeneration of Dopaminergic Neurons.

Brains of 3-, 10-, 30-, and 45-day-old flies of the indicated genotypes were dissected and immunostained with anti-DTH antibody.
(A) Representative projected confocal images of DA neurons labeled with anti-DTH antibody.
(B) The number of DTH+ DA neurons per hemisphere is presented as mean ± SEM, along with individual counts. The pooled DA neuron count includes neurons from PPL1, PPL2, PPM1/2, PPM3, and PAL clusters. For individual counts in these clusters, see Figure S1. For each genotype and age, the number of hemispheres examined is presented at the bottom of the bar. Error bars indicate SEM. The significance of differences is relative to the age-matched control (one-way ANOVA with Dunnett’s multiple correction). The DA neuron count in 45-day-old Cdk5α-OE shows an apparent rebound in numbers relative to 30 days; this reflects selective survival of only the fittest individuals at the oldest time point (Spurrier et al., 2018). For complete details of statistical analysis for this and all of the figures, see Table S2.
(C) Top: projected confocal images of 30-day-old brains of DTH-stained control, Cdk5α null, and Rescue {Cdk5α null; Tn[Cdk5α]/+}. Bottom: counts showing mean ± SEM along with individual values. For the rescue samples, the significance of differences was calculated between rescue and age-matched Cdk5α null using one-way ANOVA with Tukey’s multiple correction. In the rescue calculation, the same data for 30-day-old controls and Cdk5α null were used.
Altering the Expression of Cdk5α Hyperactivates the Expression of AMP Genes
Using global gene expression profiling, we previously developed a comprehensive, quantitative, and unbiased metric for the physiological age of wild-type flies, and applying it to young, 10-day-old Cdk5α null and Cdk5α-OE flies, we demonstrated that acceleration of the intrinsic rate of aging was a major contributor to Cdk5α-associated neurodegeneration (Spurrier et al., 2018). These data, however, suggested that there were also non-aging components of degeneration acting in concert. To identify those components, we applied principal-component analysis (PCA) to our expression profiling data. PCA cleanly apportions the effects of aging to PC1 and PC2, but reveals the presence of a third component, PC3, that contributes little variance to wild-type but separates the Cdk5α-altered genotypes roughly in proportion to the severity of the degeneration phenotypes that they will later display (Figure 2A). Identification of the genes that contribute to PC3 reveals that nearly half are components of the innate immune system, including members of several families of antimicrobial peptide (AMP) genes (Figure 2B).
Figure 2. Altered Expression of Cdk5α Causes Overexpression of Anti-microbial Peptides.

(A) Principal-component analysis (PCA) of micro-array expression profiling data. Colored ovals represent individual samples (N = 5 biological replicates), color coded as indicated. PC2 is found to separate samples essentially by effective age; PC3 separates by the severity of the neurodegenerative phenotype.
(B) Z scores of genes that make a statistically significant contribution to PC3. AMPs and other secreted innate immune proteins are represented by orange. Genes with predicted function as proteases and hydrolases are shown in blue.
(C) qRT-PCR for the expression level of the AMPs listed (color key at top). RNA was isolated from the heads of flies of the indicated genotypes and ages. Fold change of AMP expression was determined relative to 3-day-old controls, with rp49 as an endogenous control, and is displayed as mean ± SEM for three biological replicates. Student’s t test, as compared to 3-day-old control, was used to calculate significance (Table S2).
(D) qRT-PCR for AMPs was performed and quantified as above, using RNA from the heads of 30-day-old flies that were Cdk5α null, without (−) or with (+) Tn[Cdk5α+] rescuing genomic transgenes. Statistical significance was assessed by t test for each AMP. Note that this transgene expresses at a lower level than the endogenous locus, so that the rescue of the phenotype is only expected to be partial (Spurrier et al., 2018). Student’s t test, as compared to 30-day-old Cdk5α null was used to calculate significance. (Three biological replicates; error bars indicate SEM).
We validated and extended these expression profiling data by qRT-PCR of RNA isolated from fly heads in Cdk5α null, Cdk5α-OE, and control Drosophila at various ages. We found that Cdk5α-altered flies have a significant upregulation of AMPs at older ages (30 and 45 days; Figure 2C; Table S2) compared to controls. For example, the expression of individual AMPs in 45-day-old controls varies from 0.72 ± 0.08 to 7.37 ± 0.79 as compared to 3-day-old controls, while in Cdk5α null, expression ranges from 4.26 ± 0.20 to 173.76 ± 10.53 and 8.75 ±± 0.55 to 105.38 ± 6.49 for Cdk5α-OE. Expression of AMPs in 30-day-old Cdk5α null was partially rescued by restoring a single copy of the Cdk5α genomic transgene (Figure 2D; Table S2), demonstrating the specificity of the AMP-upregulation phenotype. Thus, the expression of AMP genes is enhanced in flies with an altered expression of Cdk5α as early as 10 days after eclosion, increasing to much greater levels as the flies age. Because the increase in AMP expression is even greater than the intrinsic activation of AMPs with aging, we refer to it as hyperactivation of AMP expression. A crucial question is which cells are making the AMPs in Cdk5α null. We assayed the tissue distribution of a Drosomycin-GFP gene trap and observed widespread expression in neurons, and some in glia, in addition to the expected expression in cells resembling circulating immune cells (Figure 3A).
Figure 3. Cdk5α-Altered Drosophila Have Neuronal Expression of AMPs.

(A) Projected confocal image showing drosomycin expression (green) in the brains of 30-day-old Cdk5α null Drosophila along with embryonic lethal, abnormal vision (ELAV) (red, neuronal marker) and reversed polarity (Repo) (magenta, glial marker). Yellow arrowheads highlight drosomycin in glial cells, while white arrows highlight expression in neurons. Some GFP+ cells were observed that were positive for neither neuronal nor glial markers (stars); some of these resemble hemocytes. Five biological replicates were examined.
(B) AMP expression in Cdk5α null flies with or without Elav-GAL4-mediated knock down of Relish expression. Fold change of AMPs was calculated versus 30-day-old controls and presented as mean ± SEM; significance in Elav-GAL4; Cdk5α null was assessed for each AMP by comparison to age-matched Elav-Gal4, while for Elav-Gal4;Cdk5α null; Relish RI, it was compared to Elav-Gal4;Cdk5α null, using Student’s t test for five biological replicates.
(C) DA neuron counts in 30-day-old Elav-Gal4, Elav-Gal4;Cdk5α null, and Elav-Gal4;Cdk5α null;Relish RI. Flies were aged at 25°C, and DA neurons were counted by staining with anti-TH antibody. Data are presented as mean ± SEM with individual counts shown. The number of brain hemispheres counted are at the bottom of each bar. Statistical significance was determined using one-way ANOVA with Tukey’s multiple correction.
Activation of Immunity Is Necessary and Sufficient to Promote DA Neuron Loss in the Brains of Flies with Altered Cdk5α Expression
Since overexpression of AMPs correlated with the severity of degeneration, and previous studies have reported generalized neurotoxicity upon the overexpression of AMPs in Drosophila, we hypothesized that the elevated AMPs in aged Cdk5α null and Ckd5α-OE flies are toxic to DA neurons. We first over-expressed various AMPs individually (attacin, drosocin, drosomycin, and metchnikowin; Figure S3) in DA neurons using TH-Gal4 and observed a significant loss of DA neurons even with individually overexpressed AMPs (Figure 4A; Table S2). We note that the magnitude of degeneration was not as high upon the overexpression of any single AMP as in Cdk5α null and Cdk5α-OE flies, which have a simultaneous overexpression of multiple AMPs. Note also that the UAS-Drosocin and UAS-Metchnikowin transgenes have significant GAL4-independent expression as assayed by qRT-PCR; consistent with this, these lines have reduced DA cell numbers even without GAL4. Nevertheless, these results demonstrate that an increase in AMP expression in wild-type Drosophila is sufficient to cause the loss of DA neurons.
Figure 4. Overexpression of AMPs Induces Loss of DA Neurons.

(A) TH-Gal4 was used to drive the indicated AMP genes in DA neurons, and the neuron number was counted at 3 and 30 days by immunostaining with anti-TH antibody. The graphs depict mean ± SEM for the four experimental conditions, and mean ± SD for the comparison of 3-day-old versus 30-day-old control; individual counts also are shown. The number of brain hemispheres counted is at the bottom of each bar. UAS-Drosocin and UAS-Metchnikowin flies show significant GAL4-independent expression and GAL4-independent loss of DA neurons at 30 days of age. For the expression level of these AMPs, see Figure S3. Significance analysis for TH-Gal4 was done using an unpaired t test, while significance was measured by one-way ANOVA with Tukey’s multiple correction for others.
(B) qRT-PCR for the AMPs shown was performed on the RNA from the heads of 30-day-old flies of the indicated genotypes. Fold change of AMPs was calculated relative to 3-day-old controls. Statistical significance for Cdk5α null was assessed as compared to 30-day-old controls, while significance for Cdk5α null;RelE20/TM6B was compared to the fold change in Cdk5α null. Statistical significance was assessed by Student’s t test (three biological replicates; error bars indicate SEM).
(C) Males of the indicated genotypes were aged at 25°C, and the brains were fixed, dissected, and examined for DA neuron number by immunostaining with anti-TH antibody at 30 days of age. Data are presented as mean ± SEMs, with individual values shown. The number of brain hemispheres examined is presented at the bottom of each bar. Statistical significance was determined using one-way ANOVA with Tukey’s multiple correction.
Next, we sought to test whether blocking immune activation, including AMP induction, would protect DA neurons from degeneration in Cdk5α null. We generated Cdk5α mutant flies that were heterozygous for a null allele of Relish, an NF-κB transcription factor that is a central activator of humoral innate immunity (Hedengren et al., 1999). We found that Cdk5α null;RelE20/TM6Bs have significantly lower expressions of all of the AMPs examined (22%–90% rescue, depending on AMP) (Figure 4B; Table S2), along with significantly higher numbers of surviving DA neurons, as compared to 30-day-old Cdk5α null flies (Figure 4C; Table S2). The same result was obtained when the experiment was repeated without the balancer chromosome present (Figure S4). These results strongly suggest that hyperactivity of the innate immune response, including over-expression of AMPs, is necessary and sufficient to induce the degeneration of DA neurons in animals with reduced Cdk5α expression. We extended this experiment by performing RNAi knock down of Rel selectively in neurons of Cdk5α null, using an ELAV-GAL4 driver, and found significantly reduced AMP expression and rescue of DA neuron loss (Figures 3B and 3C). These data confirm the rescue of DA neurons by reduced levels of Rel and also show that immune activation is specifically required in neurons to produce lethality, which is consistent with the AMP expression pattern observed above.
Finally, we tested whether immune activation and consequent DA neuron loss may occur because of increased bacterial infection in mutant flies. First, we measured bacterial load by PCR of 16S rDNA among controls, Cdk5α null, and Cdk5α-OE and found no significant difference (data not shown). Second, we prepared axenic flies and found that the activation of AMP expression and DA neuron loss occur in 30-day-old Cdk5α-altered flies raised under sterile conditions just as they do under normal conditions (Figure S5). This argues for the idea that the activation of immunity is not due to exacerbated bacterial infection in Cdk5α-altered conditions.
Altered Expression of Cdk5α Disrupts Autophagy in Drosophila
If altering Cdk5α does not intensify microbial challenge, why does it activate the innate immune response? Defective autophagy can stimulate innate immunity, including AMP expression (Tusco et al., 2017; Wu et al., 2007), and data from us and others show that altering Cdk5α levels or inactivating Cdk5 kinase activity can disrupt autophagy (Nandi et al., 2017; Spurrier et al., 2018; Trunova and Giniger, 2012). Thus, we hypothesized that reduced autophagy in flies having an altered level of Cdk5α may be responsible for overactivated innate immunity. This conjecture was supported by our PCA of microarray data, as four out of the nine genes that were downregulated by altered Cdk5α expression were proteases, protease regulators, and a lipid hydrolase (CG12256, SPH93, CG3513, and Cyp4g1; Figure 2B).
Previous experiments demonstrated that Cdk5α-altered flies have distorted autophagy in the brain at an older age (Spurrier et al., 2018; Trunova and Giniger, 2012). To quantify autophagic flux specifically in DA neurons, we assayed the p62 ortholog, Ref(2)P (refractory to sigma P), and a tandem tagged Atg8 (GFP-mCherry-Atg8a). Ref(2)P accumulates if autophagy is disrupted (Nagy et al., 2015). The tandem tagged Atg8a assays autophagic flux per se; since its GFP fluorescence is quenched in acidified autolysosomes, functional lysosomes bearing the reporter contain red puncta, while autophagosomes that fail to acidify accumulate double-tagged (yellow) puncta (Kimura et al., 2007). Counting Ref(2)P puncta in the brains of 30-day old flies revealed that Cdk5α-altered flies have a significantly higher number of puncta than controls (Figures 5A and 5B). Three-dimensional (3D) reconstruction clearly revealed the accumulation of Ref(2)P inside TH+ DA neurons specifically, as well as in the brain overall (Figure 5A). To assay autophagy flux in Cdk5α null flies, we expressed tandem tagged Atg8a (UAS-GFP-mCherry-Atg8a) using TH-Gal4. At 30 days of age, the PPL1 cluster of DA neurons shows mostly double-positive autophagosomes (positive for both GFP-Atg8a and mCherry-Atg8a), while the age-matched control has mostly single-positive (mCherry only) autolysosomes (Figure 5C), demonstrating impaired autophagic flux in the mutant.
Figure 5. Altered Cdk5α Levels Reduce Autophagy Efficiency.

(A and B) Flies of the indicated genotypes were aged to 30 days, and brains were fixed, dissected, and labeled with DAPI (blue), anti-TH (green), and anti-Ref(2)P (red).
(A) Projected confocal images, including PPL1 cluster, showing separated channels and merged image, as well as a 3D surface rendering of a single TH+ cell (dotted yellow rectangle in merged image).
(B) Ref(2)P+ puncta were counted using the ImageJ (NIH) particle counting tool, and data are presented as mean ± SEM, with individual values shown. Brain region including PPL1 cluster was examined for six hemispheres per genotype, and puncta were counted in two size ranges, 0.02–0.40 μm2 and >0.40 μm2. Statistical significance was assessed by one-way ANOVA with Dunnett’s multiple correction. For a 3D view documenting the localization of Ref(2)P puncta inside the DA neuron, the 3D cropping tool of Imaris (Bitplane) was used, followed by surface rendering and manual pruning of puncta outside the DA neuron.
(C) TH-Gal4 was used to drive UAS-GFP-mCherry-Atg8a in DA neurons. Brains were dissected without fixation, and fluorescence was examined. DA neurons of control flies (w+;UAS-GFP-mCherry-Atg8a/CyO;TH-Gal4/TM6B) have mostly mCherry+ puncta, while Cdk5α null flies (w+; Cdk5α/DfC2,UAS-GFP-mCherry-Atg8a;TH-Gal4/TM6B) have mostly double-positive puncta (yellow). Six biological replicates were used for this experiment.
(D) Western immunoblot with anti-GFP antibody was used to quantify Vha13 protein in the extract of 30-day-old heads of flies of the indicated genotypes bearing a Vha-13-GFP gene trap. Left: typical immunoblot; numbers give the molecular weights of the markers. Right: quantification, displaying mean ± SEM (average of three biological replicates, normalized with anti-tubulin as loading control). Significance determined by paired t test.
(E) qRT-PCR was performed in biological triplicate, as above, to quantify Mitf expression in the RNA of the heads of the indicated genotypes. Error bars indicate SEM. UAS-Mitf was driven with a third chromosome insert of ELAV-GAL4 that has very low adult expression (BL8760).
(F) Western immunoblot (left) and quantification (right) of cysteine protease (Cp1), detected with anti-Cp1 antibody in the extract of the heads of the indicated genotypes isolated at 30 days of age. The Cp1 value presented is an average of three biological replicates, normalized with anti-tubulin as the loading control. Error bars indicate SEM. Raw intensity values from all of the trials are given in Table S4. Significance was assessed by one-way ANOVA with Dunnett’s multiple correction. The molecular weights of the markers are indicated next to the blots.
Since the experiments above revealed autophagosomes failing to convert to acidified autolysosomes in Cdk5α null, we hypothesized that there may be a defect in lysosome metabolism. We examined Vha-13-GFP, a reporter for the vacuolar (H+)-ATPase that has a prominent role in lysosome acidification (Zhang et al., 2015). Extract of the heads of 30-day-old Cdk5α null;Vha-13-GFP flies was analyzed by immunoblotting using anti-GFP antibody, and revealed that Cdk5α null have significantly lower amounts of Vha protein than controls (Figure 5D). This observation was extended by qRT-PCR assay showing reduced transcript of Mitf, a master regulator of the lysosomal-autophagy pathway, including v-ATPase expression (Bouché et al., 2016; Zhang et al., 2015), in 30-day-old mutants as compared to controls (Figure 5E). Along with the reduction in Vha, both Cdk5α null and Cdk5α-OE flies also had a significantly lower level of cysteine proteinase-1 (Cp1, also known as cathepsin L; Figure 5F), another marker for lysosomal metabolism. These data demonstrate that the Cdk5α mutation reduces autophagic flux generally, including in DA neurons, which is consistent with the reduced expression of proteins required for lysosomal metabolism in the Cdk5α mutant and of the transcription factor that promotes their expression.
Disruption of Autophagy Is Responsible for the Activation of Immunity and Consequent Loss of DA Neurons upon Altering the Level of Cdk5α
Given the disruption of autophagy by altered Cdk5α and the potential linkage of autophagy to immunity, we tested whether impairment of autophagy can cause overexpression of AMPs and progressive loss of DA neurons. We first quantified AMP expression and DA neuron number in an Atg8a mutant (Atg8a1/Y). Atg8a is one of the two ATG8 paralogs in Drosophila (Nagy et al., 2015). Atg8a acts in autophagosome formation, and while the Atg8a mutant is viable, it has poor autophagy efficiency (Figure 6A). We found that Atg8a1/Y flies have a significantly elevated expression of AMPs as compared to age-matched controls (5.46 ± 0.0.65- to 16.57 ± 1.18-fold depending upon the AMP; Figure 6B; Table S2). Moreover, while Atg8a mutant animals have the same number of DA neurons as controls at 3 days of age, they have significantly fewer DA neurons at 30 days of age (Figure 6C). These data demonstrate that the disruption of autophagy is sufficient to cause the overexpression of AMPs and the age-dependent loss of DA neurons.
Figure 6. Reduced Autophagy Induces AMP Expression and DA Neuron Loss in Aged Flies.

(A) Ref(2)P accumulation in the brains of Atg8a1/Y flies. Flies were aged to 30 days; brains were fixed, dissected, and stained with DAPI (blue), anti-TH (green), and anti-Ref(2)P (red). Separated channels and merged image are shown as projected confocal images; the rightmost panel is a 3D rendering of a single TH+ cell showing Ref(2)P accumulation inside the cell (cell highlighted with yellow box in the merged image). Five biological replicates were examined for Ref(2)P accumulation.
(B) AMP expression in the heads of Atg8a1/Y flies. RNA was isolated from the heads of 30-day-old flies that were Atg8a1/Y or controls, and qRT-PCR was performed, as above. Bars show fold change of the expression level in mutants relative to age-matched controls (mean ± SEM; significance was calculated using Student’s t test; five biological replicates).
(C) DA neuron counts in the brains of Atg8a1/Y flies and controls. Flies were aged to 30 days, and the brains were fixed, dissected, and assayed for DA neuron number by fluorescence microscopy after immunostaining with anti-TH. Bars indicate mean ± SEM with individual values shown. The number of brain hemispheres examined is at the bottom of each bar, and significance was assessed relative to controls (t test).
(D) Ref(2)P accumulation in the brains of controls and 30-day-old Cdk5α null without and with low-level, GAL4-driven expression of Mitf. Top: projections of the brains of the indicated genotypes, which were fixed, dissected, and immunostained with anti-Ref(2)P. Bottom: quantification of Ref(2)P puncta from six to eight brain hemispheres of each condition. Data are presented as mean ± SEM, along with individual counts. Two-way ANOVA with Tukey’s multiple correction was used for statistical analysis. ELAV-Gal4 > UAS-Mitf expression generated general cytoplasmic background label with this antibody; background subtraction was performed by rolling circle (5.0 pixels) before counting the puncta. For an unprocessed image, see Figure S6.
(E) AMP expression levels in Cdk5α null flies without and with ELAV-driven restoration of Mitf. qRT-PCR for the AMP level was performed on the RNA from the heads of 30-day-old flies, as previously described. Fold change of AMPs was calculated versus 30-day-old controls and presented as mean ± SEM; significance was assessed for each AMP by comparison to age-matched Cdk5α null using three biological replicates and Student’s t test.
(F) DA neuron counts in 30-day-old Cdk5α null and Cdk5α null; ELAV-Gal4/UAS-Mitf. Flies were aged at 25°C, and DA neurons were counted, as before, by staining with anti-TH antibody. Data are presented as mean ± SD, with individual counts shown. The number of brain hemispheres counted are at the bottom of each bar, and significance is by comparison to Cdk5α null (Student’s t test).
Finally, we investigated whether restoring lysosomal gene expression and metabolism rescues autophagy, and if so, whether this would restore AMP gene expression and DA neuron survival. Since strong overexpression of Mitf is extremely toxic (Zhang et al., 2015), we took advantage of a third chromosome insert of ELAV-Gal4 (a pan-neuronal driver) with extremely low adult activity to restore the Mitf level in Cdk5α null flies (1.68 ± 0.10 fold versus Cdk5α null and 0.86 ± 0.05 fold versus controls; Figure 5E). We then counted Ref(2)P puncta in 30-day-old flies, which revealed a significant improvement in autophagy in flies with Mitf restoration (Cdk5α null; elav-GAL4 > UAS-Mitf) (Figure 6D). We next found significantly reduced expression of AMPs in Cdk5α null flies with restored Mitf expression as compared to age-matched Cdk5α null (Figure 6E; Table S2). Finally, we counted DA neurons and discovered significant restoration of DA neuron viability (Figure 6F). Thus, restoring autophagy by the mild expression of Mitf in Cdk5α null flies reduces AMP expression and rescues DA neuron loss. These data demonstrate that reduced auto-phagy efficacy in the Cdk5α mutant is necessary and sufficient to induce high-level AMP expression and is associated with DA neuron loss.
DISCUSSION
Here, we show that altering the level of the Cdk5 activating subunit, Cdk5α, impairs autophagy, which leads to the upregulation of the innate immune response, including antimicrobial peptides (AMPs), and this in turn causes age-dependent degeneration of dopamine neurons in Drosophila. PCA of transcriptome data revealed that the overexpression of AMPs drives much of the aging-independent component of the gene expression changes between flies with altered levels of Cdk5α and wild-type flies, even before the onset of overt degeneration. Directed overexpression of AMPs is sufficient to cause the death of DA neurons in otherwise wild-type flies, while blocking immune activation in Cdk5α mutants by reducing the expression of the NF-κB transcription factor, Rel, rescues DA neuron survival. The activation of immunity in turn is caused by Cdk5α-associated disruption of autophagy, as hindering autophagy with a mutation in Atg8a is sufficient to enhance AMP expression and kill DA neurons, while rescuing autophagy in a Cdk5α mutant by overexpression of the TFEB transcription factor, Mitf, restores the AMP level and rescues DA neurons. These data reveal a simple, linear, dependent genetic pathway, encompassing both autophagy and innate immunity, which, while rigorously separable from aging, interacts with the effects of aging to lead to the degeneration of DA neurons in vivo.
Two major variables have confounded our understanding of the relation of autophagy and immunity to degeneration. The first is age: neurodegeneration occurs with aging, and both auto-phagy and the immune response change with age, so how does one discriminate the effects of disease from the normal, variable course of aging? The second is that both autophagy and the immune response are homeostatic processes: how does one determine whether alterations in autophagy or immunity are part of the disease mechanism, or are part of the organism’s response to pathology? This second problem is particularly difficult because both autophagy and immunity are normally maintained within narrow limits. Therefore, robust experimental manipulation of these processes will certainly cause pathology, but not necessarily pathology that is relevant to the mechanism of NDs.
The data reported here allow us to answer these questions. First, statistical analysis of transcriptome profiling revealed that changes in innate immunity drive much of the non-aging component of gene expression differences between wild-type flies and flies destined to undergo degeneration. Thus, PCA reveals the existence of a large component of immune hyperactivation over and above that due to the natural progression of aging, and qRT-PCR quantification confirms that it correlates with the severity of degeneration. Second, multiple lines of evidence demonstrate that defective autophagy and hyperactive immunity are causal for DA neuron degeneration and furthermore, that they constitute a pathway, with defective autophagy being responsible for immune activation, and immune activation inducing neuron loss. Below, we consider each step of the degeneration pathway.
Altering the Level of Cdk5α Expression Causes Reduced Efficiency of Autophagy
Our data, together with published work, suggests that there are two parts to the association of autophagy with Cdk5/Cdk5α activity: the efficiency of autophagy changes with age, which is modulated by Cdk5, and there are mechanisms by which Cdk5 regulates autophagy more directly (Nagy et al., 2015; Spurrier et al., 2018). The autophagy-lysosomal system ensures continuous autophagic flux that promotes a healthy cellular environment (Bouché et al., 2016), and it requires the acidic lysosomal environment that is maintained by the vacuolar-type H+-ATPase (V-ATPase) complex (Mauvezin et al., 2015). We find that V-ATPase (Vha-13), as well as the Cp1 level, are reduced in the heads of Cdk5α null flies, and overexpression of a master regulator of lysosomal function, the TFEB Mitf, restores autophagy to these flies, supporting the hypothesis that the compromised autophagy in Cdk5α null flies is associated with abnormal lysosomal metabolism. It is not clear what molecular events are responsible for the reduction of Mitf transcript and reduced expression of V-ATPase upon altering the Cdk5α level, but expression profiling of wild-type Drosophila shows that most of the V-ATPase family genes are downregulated with age (Spurrier et al., 2018) and that Cdk5α-altered Drosophila have an accelerated aging rate (Spurrier et al., 2018). Thus, we hypothesize that the enhanced rate of aging from altered Cdk5α contributes to the reduced expression of the V-ATPase family, leading to lysosomal abnormality and compromised autophagy, probably in combination with more direct effects of Cdk5 on components of the autophagy machinery, such as the RNA-splicing factor acinus (Nandi et al., 2017).
Disruption of Autophagy Causes Hyperactivation of Innate Immunity
The reduced autophagy in animals with an altered level of Cdk5α was accompanied by an age-dependent increase in the expression of AMPs. While autophagy is regarded principally as a homeostatic process that removes harmful substances and maintains cellular metabolism by recycling substrates, it is not limited to these functions (Wang et al., 2013; Wang and Qin, 2013). Autophagy influences multiple aspects of immune system function and regulation in both vertebrates and invertebrates, although the mechanisms are not yet fully understood (Levine et al., 2011; Saitoh and Akira, 2010; Wu et al., 2007). One recent theory is that the disruption of various aspects of physiological homeostasis is monitored by the organism as prima facie evidence of microbial attack and used as a signal for the activation of immunity and detoxification pathways (Melo and Ruvkun, 2012). Mechanistically, there are at least two possible modes by which autophagy could influence inflammation–directly, by modifying the activity of the inflammasome complex (Lee et al., 2007), and indirectly, by autophagic clearance of molecules that activate an inflammatory response (Levine et al., 2011; Qu et al., 2007b).
We show here that the increase in AMP expression upon altering Cdk5α expression is a consequence of reduced autophagy. Reducing autophagy by the mutation of Atg8a was sufficient to induce the overexpression of AMPs. Moreover, rescuing autophagy using Mitf expression selectively in the neurons of Cdk5α null animals restored the expression of AMPs. These results strongly suggest that reduced autophagy efficiency is causal for the overactivation of innate immunity in Cdk5α null flies, reinforcing the idea that autophagy has a significant role in regulating immunity and inflammatory response. We exclude in two ways the model that increased immunity in Cdk5α-altered flies is a result of increased propensity for infection. First, bacterial load is not altered in flies with increased or decreased Cdk5α, and second, both activation of AMP expression and loss of DA neurons occurs in flies with altered Cdk5α when they are raised in germ-free conditions, just as it does under standard growth conditions.
Hyperactivation of Innate Immunity Induces Age-Dependent Loss of DA Neurons
Three lines of evidence argue that the age-dependent loss of DA neurons in Drosophila with altered levels of Cdk5α is due to the hyperactivity of innate immunity, including the overexpression of AMPs. First, the age-dependent overexpression of AMPs in the heads of Cdk5α null and Cdk5α-OE flies is accompanied by the age-dependent loss of DA neurons, while blocking immune activation by reducing the Relish level in Cdk5α null flies restores DA neurons. Second, the overexpression of AMPs is sufficient to cause the age-associated loss of DA neurons in flies, even in the absence of an exogenous stressor. Neurotoxicity of AMPs has been shown previously in other neuronal subsets (Cao et al., 2013; Kounatidis et al., 2017; Petersen et al., 2013). Third, we also observed the overexpression of AMPs and age-dependent DA neuron loss in a different genetic paradigm, disruption of autophagy in a hemizygous Atg8a mutant. Therefore, we argue that overactive innate immune response is responsible for the degeneration of DA neurons in an age-dependent manner in Drosophila with altered Cdk5α. Our data regarding the neurotoxicity of AMPs, and the neuroprotective role of reduced Relish, are reminiscent of observations made in other experimental paradigms, and potentially explains the data obtained in previous studies of immunity and neurodegeneration (Cao et al., 2013; Kounatidis et al., 2017; Petersen et al., 2013). In future experiments, it will be interesting to examine the potential roles of the other NF-κB paralogs dif and dorsal.
In our PCA, eight of the nine circulating immune effectors that were hyperactivated by altered Cdk5α were AMPs. Taken together with the data above, this suggests that AMP overexpression likely plays a central role in the Cdk5α-associated loss of DA neurons. We note, however, that Rel induces many aspects of immunity aside from AMPs (Meyer et al., 2014). Moreover, we cannot rule out the possibility that the cell lethality of overexpressed AMPs could occur via a mechanism that is different from that of immune-correlated cell death in altered Cdk5α conditions (Sano and Reed, 2013). Therefore, it remains possible that other aspects of the immune response contribute to DA neuron loss in this model, in addition to or instead of AMP activation.
Two lines of experiments show that AMP expression is activated in neurons in the Cdk5α null: examination of an AMP reporter line and rescue by neuron-specific suppression of the immune response. The nature of AMP toxicity has been controversial, but our finding of enhanced AMP expression in neurons may be relevant. For example, it could be that the activation of endoplasmic reticulum (ER) stress–or the failure to activate protective mechanisms such as the ER stress response–may contribute to the toxicity of expressing secreted immune proteins such as AMPs in cells that do not normally produce them at high levels (Sano and Reed, 2013). These data also raise the question of whether activation of immune genes in non-immune cells, particularly neurons, may be part of the pathogenic mechanism in human NDs.
The overexpression of AMPs in DA neurons does not induce degeneration in 3-day-old flies; however, at 30 days old, it results in a significant loss of DA neurons. This is true of direct GAL4-driven overexpression of AMPs, overexpression due to an autophagy mutant, and overexpression due to the altered expression of Cdk5α. Conversely, aging to 30 days alone, in the absence of immune activation, is not sufficient to produce DA neuron loss. We therefore suggest that DA loss reflects a synergistic interaction between the direct, toxic effects of AMP overexpression and the general cellular fragility produced by aging (Herrup, 2010; Spurrier et al., 2018).
All of the genes we have manipulated and all of the interactions we have examined are conserved in mammals, and each individually has been implicated in human NDs. For example, the accumulation of autophagic vacuoles (AVs) has been observed in the brains of patients with PD (Anglade et al., 1997), and data from model organisms suggest the association of familial PD genes with autophagy (Narendra et al., 2010; Tong et al., 2010), although it remains unclear whether disease is associated with autophagy that is excessive, insufficient, or directed against inappropriate targets. Similarly, in recent years, inflammation has gained recognition as a factor promoting degeneration (Kannarkat et al., 2013; Schlachetzki and Winkler, 2015), although it has been controversial whether it is a cause or a consequence of cell loss and whether immune activation or insufficiency is to blame for ND. Moreover, epigenomic features in a mouse p25/Cdk5 model of degeneration and in human AD also highlight immunity as a predisposing process in degeneration (Gjoneska et al., 2015). Finally, just as our data indicate that degeneration arises from the synergistic interaction of inflammation and aging, Chakrabarty et al. (2011) have reported evidence for the age dependence of nigrostriatal degeneration upon the CNS-directed expression of the innate immune mediator interferon-γ in mice. However, while previous studies of degeneration have identified some of the same processes we describe here, those studies have not been able to establish whether disease arises from hyperactivity, inactivity, or the mistaken activity of common homeostatic mechanisms. Moreover, it has not been possible to connect those processes to mechanistic pathways, or to discriminate the progression of disease from the progression of normal aging. By contrast, using the precise genetic tools of the fly and our previous construction of a systems-level metric for physiological aging, we have shown here that a simple, dependent genetic pathway, comprising reduced autophagy and hyperactive immunity, interacts synergistically with disease-associated acceleration of the intrinsic rate of aging to produce the overall outcome of adult-onset neurodegeneration in the fly. Given the conservation of genes, pathways, and cellular phenotypes, it seems very likely that the processes we reveal here also play a central role in the development and progression of human ND.
STAR⋆METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
All requests for reagent and resources should be directed to the lead contact, Dr. Edward Giniger (ginigere@ninds.nih.gov).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Flies were maintained on standard cornmeal-molasses Drosophila media at 25°C and 50% humidity with 12h light/dark cycle. Oregon Red (w+) was used as the wild-type control unless otherwise stated. Male flies were used in all experiments. Cdk5α null, Cdk5α-OE and Cdk5α null; Tn[Cdk5+] (ie., rescue) stocks have been described previously (Connell-Crowley et al., 2000; Connell-Crowley et al., 2007; Spurrier et al., 2018; Trunova and Giniger, 2012). During aging, flies were transferred to new vials every 48h. Complete genotypes for all stocks are listed by Figure in Table S3, and sources of fly stocks are provided in Key Resources Table.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER | |
|---|---|---|---|
| Antibodies | |||
| Anti-DTH (rabbit) | Laboratory of W. Neckameyer (Neckameyer et al., 2000) | N/A | |
| Anti-Ref(2)P (rabbit) | Laboratory of I. Nezis (Nezis et al., 2008) | N/A | |
| Anti-TH (mouse) clone LNC1 | Milipore | Cat#:MAB318, RRID:AB_2201528 | |
| Anti-Cathepsin L(rabbit) Cp1 | abcam | Cat#:Ab58991, RRID:AB_940826 | |
| Anti-green fluorescent protein, rabbit IgG | Invitrogen | Cat#:A11122, RRID:AB_221569 | |
| Beta tubulin | Developmental studies hybridoma bank | Cat#:E7, RRID:AB_528499 | |
| Anti-Repo (Mouse) | Developmental studies hybridoma bank | Cat#:8D12, RRID:AB_528448 | |
| Anti-ELAV (Rat) | Developmental studies hybridoma bank | Cat#:7E8A10, RRID:AB_528218 | |
| Alexa Fluor®568 Goat anti-mouse IgG | Life technologies | Cat#:A11031, RRID:AB_144696 | |
| Alexa Fluor®488 Goat anti-mouse IgG | Life technologies | Cat#:A11029, RRID:AB_2534088 | |
| Alexa Fluor®568 Goat anti-rabbit IgG | Life technologies | Cat#:A11036, RRID:AB_10563566 | |
| Alexa Fluor™633 Goat anti-mouse IgG | Invitrogen | Cat#:A21126, RRID:AB_2535768 | |
| Alexa Fluor®568 Goat anti-rat IgG (H+L) | Molecular Probes | Cat#:A11077, RRID:AB_141874 | |
| IRDye700DX®Goat Anti-rabbit IgG | Rockland INC | Cat#:611-130-122, RRID:AB_220148 | |
| IRDye800DX®Goat Anti-mouse IgG | Rockland INC | Cat#:610-132-121, RRID:AB_220125 | |
| IRDye700DX®Goat Anti-mouse IgG | Rockland INC | Cat#:610-130-121, RRID:AB_220121 | |
| IRDye®800Goat Anti-rabbit IgG | Rockland INC | Cat#:611-132-002, RRID:AB_1660971 | |
| VECTASHIELD Antifade Mounting Medium | Vector Laboratories, Burlingame, CA | Cat#: H-1000, RRID:AB_2336789 | |
| VECTASHIELD Antifade Mounting Medium with DAPI | Vector Laboratories, Burlingame, CA | Cat#: H-1200, RRID:AB_2336790 | |
| Chemicals, Reagents and Kit | |||
| PowerUp SYBR Green Master Mix | Applied biosystems | Cat#:A25742, N/A | |
| High capacity cDNA Reverse Transcription Kit | Applied biosystems | Cat#:4368814, N/A | |
| TRIzol® reagent | Life technologies | Cat#:15596026, N/A | |
| Paraformaldehyde 16% Solution, EM Grade | Electron Microscopy Sciences | Cat#: 15710, N/A | |
| Schneider’s Drosophila Medium | Life technologies | Cat#:21720-024, N/A | |
| Halt Protease Inhibitor Cocktail | ThermoFisher Scientific | Cat#:87785, N/A | |
| iBlot®2NC Regular Stacks | Invitrogen/ThermoFisher Scientific | Cat#:IB23001, N/A | |
| Bolt 4–12% Bis-Tris Plus gel | Invtrogen/ThermoFisher Scientific | Cat#:NW04120BOX, N/A | |
| Nutri-Fly® BF, 10 × 1L Packets | Genesee Scientific | Cat#: 66-112, N/A | |
| GIBCO Penicillin-Streptomycin (10,000 U/mL) | Life technologies | Cat#: 15140-122, N/A | |
| Fly media, CT modified + green dye | K.D Medicals | Cat#: IMT-0756, N/A | |
| Oligonucleotides | |||
| Table S5 | N/A | ||
| Experimental Models: Organism/Strains | |||
| D. melanogaster: w[*]; P{w[+mC] = ple-GAL4.F}3 | Bloomington Drosophila Stock Center | BDSC:8848, FlyBase: FBst0008848 | |
| D. melanogaster: w[1118]; Rel[E20] e[s] | Bloomington Drosophila Stock Center | BDSC:9457, FlyBase: FBst0009457 | |
| D. melanogaster: y[1] w[1118]; P{w[+mC] = UASp-GFP-mCherry-Atg8a}2 | Bloomington Drosophila Stock Center | BDSC:37749, FlyBase: FBst0037749 | |
| D. melanogaster: w[1118] P{w[+mC] = EP}Atg8a[EP362] | Laboratory of K. D. Finley (Simonsen et al., 2008) | BDSC:10107, FlyBase: FBst0010107 | |
| D. melanogaster: w*; P{UAS-mCherry.NLS}3 | Bloomington Drosophila Stock Center | BDSC:38424, FlyBase: FBst0038424 | |
| D. melanogaster: y[1] v[1]; P{y[+t7.7]v [+t1.8] = TRiP.HM05154}attP2 | Bloomington Drosophila Stock Center | BDSC:28943, FlyBase: FBst0028943 | |
| D. melanogaster: pStinger-Vha13–3 | Laboratory of F. Pignoni (Zhang et al., 2015) | N/A | |
| D. melanogaster: pUASTattB-Mitf (96E) / SM6::TM6B | Laboratory of F. Pignoni (Zhang et al., 2015) | N/A | |
| D. melanogaster:UAS-Attacin C | Laboratory of B. Ganetzky (Cao et al., 2013) | N/A | |
| D. melanogaster:UAS-Droscin/CyO | Laboratory of B. Ganetzky (Cao et al., 2013) | N/A | |
| D. melanogaster:UAS-Drosomycin | Laboratory of B. Ganetzky (Cao et al., 2013) | N/A | |
| D. melanogaster:UAS-Mtchnikowin/TM3Sb | Laboratory of B. Ganetzky (Cao et al., 2013) | N/A | |
| D. melanogaster:Oregon R+ (w+) | Laboratory of E. Giniger (Connell-Crowley et al., 2000; Spurrier et al., 2018) | N/A | |
| D. melanogaster:w+; Cdk5α-null | Laboratory of E. Giniger (Connell-Crowley et al., 2000; Spurrier et al., 2018) | N/A | |
| D. melanogaster: w+; P[w+,TnCdk5α]R244/P[w+,TnCdk5α]R244; P[w+,Tn Cdk5α] R157/P[w+,TnCdk5α]R157 | Laboratory of E. Giniger (Connell-Crowley et al., 2000; Spurrier et al., 2018) | N/A | |
| D. melanogaster: Df(Cdk5α)C2 | Laboratory of E. Giniger (Connell-Crowley et al., 2000; Spurrier et al., 2018) | N/A | |
| D. melanogaster: w+; Cdk5α / Cdk5α; P[w+,Tn Cdk5α] R157/+ | Laboratory of E. Giniger (Connell-Crowley et al., 2000; Spurrier et al., 2018) | N/A | |
| D. melanogaster: ELAV Gal4 (on X chromosome) | Laboratory of E. Giniger (Connell-Crowley et al., 2000; Spurrier et al., 2018) | N/A | |
| D. melanogaster: w[*]; P{w[+mC] = GAL4-elav.L}3 | Bloomington Drosophila Stock Center | BDSC:8760, FlyBase: FBst0008760 | |
| D. melanogaster: Drs-GFP | Laboratory of Jean-Marc Reichhart (Ferrandon et al., 1998) | N/A | |
| Software | |||
| Fiji | https://fiji.sc/; RRID:SCR_002285 | ||
| Zeiss LSM | Carl Zeiss Microscopy |
https://www.zeiss.com/microscopy/int/products/confocal-microscopes/lsm-800-with-airyscan.html; RID:SCR_015963 |
|
| Imaris version 8.2.1 | Bitplane |
http://www.bitplane.com/imaris/imaris; RID:SCR_007370 |
|
| Prism 7 | GraphPad |
https://www.graphpad.com; RRID:SCR_002798 |
|
| AutoQuant X | Media Cybernetics |
http://www.mediacy.com/autoquantx3; RRID:SCR_002465 |
|
METHOD DETAILS
Immunohistochemistry and Imaging
Whole brains were dissected after fixation with 8% paraformaldehyde (PFA) in PBS for 10min. Brains were blocked with 5% heat inactivated Fetal Bovine Serum (FBS) in PBS+0.2% Triton X-100 (1XPBS-T) for 2hr at room temperature (RT) followed by two-night incubation at 4°C with primary antibodies at appropriate dilution. After antibody staining, brains were mounted on slides with VectaShield mounting medium. Microscopy was performed on a Zeiss NLO510 confocal microscope or with the Zeiss LSM880.
For counting dopaminergic neurons, control, Cdk5α null, and Cdk5α-OE male flies were collected at 3d-age and aged to 10-, 30-, or 45-days. Brains were incubated with Anti-DTH or Anti-TH antibody (1:200) at 4°C for two nights followed by washing and staining with appropriate secondary antibody (1:400). Images were acquired using a 20-X objective. Individual brain hemispheres were analyzed using ImageJ (National Institute of Health) cell counter plugin. Note that there is not perfect agreement in the number of presumptive DA neurons identified by anti-TH versus anti-DTH. However, the mean difference in cell number produced by each genetic manipulation reported here was similar using either antibody.
For Ref(2)P accumulation and counting, staining of 30d-old brain of male flies was performed using Anti-Ref(2)P (1:100) and Anti-TH (1:200) as primary antibodies and respective secondary antibodies (1:400) were used. Images were acquired using a 40-X objective. Post-acquisition, images were processed for deconvolution using AutoQuant X2. Maximum intensity projection (MIP) of Deconvoluted images were used for counting of Ref(2)P puncta using ImageJ. In brief, MIP of Ref(2)P stained brains were processed for thresholding followed by binarization and watershed. Analyze Particle tool was then used to count puncta in two different size ranges (0.02–0.40μm2 and > 0.40μm2). Number of puncta was presented as mean ± SEM. The visualization of Ref(2)P puncta was done by Imaris software using 3D cropping and surface modeling using Ref(2)P- and TH- stained brain.
For examination of autophagy flux using TH-GAL4, UAS-GFP-mCherry-atg8a flies, unfixed brains of desired genotypes at 30d-age were dissected and mounted in Schneider′s Drosophila Medium. Images were acquired using Zeiss LSM880 confocal microscope using a 40-X objective, focusing on PPL1 clusters of DA neurons.
For examination of Drosomycin-GFP expression in Cdk5α null flies, brains of desired genotypes were fixed as above, dissected, and stained with Anti-GFP (1:200), Anti-ELAV (1:50) and Anti-Repo (1:50) antibodies and appropriate secondary antibodies (1:400). Stained brains were imaged using 40X objective. Images were processed for deconvolution using AutoQuant X3 and relevant planes of the Z stack were projected to provide representative images.
Western immunoblotting
Preparation of extract from flash frozen 30d-old flies and western blotting were performed as described previously (Spurrier et al., 2018). In brief, the heads of 20 male flies were separated from the rest of the body and homogenized in lysis buffer (2% SDS, 150mMNaCl, 50mM Tris, pH 7.5) containing protease inhibitors. Protein homogenate was centrifuged at 5000 rpm for 5min at 4°C. Protein supernatant was collected and transferred to fresh 1.5ml Eppendorf tube followed by mixing with BoldLDS sample buffer and Boldreducing agent (for 40μl total volume 10μl BoldLDS sample buffer and 4μl Boldreducing agent was used). Mixture was then incubated for 10min at 70°C followed by 2min incubation at 85°C. Proteins were resolved on a 4%–12% Bis-Tris plus gel and transferred onto iBlot2 NC membranes using the iBlot 2 system (Invitrogen/Thermo Fisher Scientific). After transfer, membranes were incubated in blocking solution (5% milk in 1XTBS-0.2% Triton X-100) for 2 hours at room temperature. Membranes were probed with primary antibodies overnight at 4°C. Primary antibodies included anti-GFP (1:1000), anti-Cp1 (1:500) and anti-β-Tubulin (1:500). Infrared fluorescent IRDye secondary antibodies, were applied for 20 minutes at room temperature (1:5000) followed by washing with 1XTBS-0.2% Triton X-100. Visualization and quantification was carried out using the LI-COR Odyssey scanner and software (LI-COR), with tubulin as a loading control.
Principal Component analysis using microarray data
PCA was performed on our previously-published microarray-based gene expression profiling dataset from the heads of flies with altered Cdk5α-expression, specifically on the set of age-classifier genes we identified (Spurrier et al., 2018). In that study, selection as an “age classifier gene” was restricted to those genes having a significant difference of expression between ages (ANOVA) under multiple comparison correction condition (BH FDR p < 0.05) that also survived a leave-one-out (LOO) selection challenge in k-nearest neighbor (knn) analysis 100% of the time. For PCA, modeling of expression for these genes was accomplished using principal component regression followed by AIC-step optimization. Principal component loadings for the genes modeled were subset by component and a standard z-score calculated per gene. Genes having a standard z-score magnitude > 2 were subset as those most contributing to the percent variance explained by each component.
qRT-PCR of head RNA
RNA was isolated from 25 Drosophila heads using TRI reagent and synthesis of cDNA performed with High Capacity cDNA Reverse Transcription Kit using 1000ng RNA, following the manufacturer’s instructions. The expression of genes was quantified on a QuantStudio 6 Flex Real-Time PCR System using PowerUp SYBR Green Master Mix. Primer sequences are provided in Table S5. PCR was performed by the method of (Cao et al., 2013) with PCR conditions as follows: 35 cycles: step 1: 95°C for 10 s, step 2: 60°C for 30 s, step 3: 72°C for 40 s each cycle. The quantification of each gene, relative to rp49, was calculated using the DDCt method relative to 3d- or 30d-old control, as indicated.
Comparison of Cdk5α expression profile to published datasets
Raw expression files representing mutant conditions other than Cdk5α-OE and Cdk5α null in fly were downloaded from NCBI GEO: GSE23802, GSE9571, GSE20202, GSE10940, GSE26246, GSE25009 and EMBL-EBI ArrayExpress: E-MEXP-3645. Pre-processing of these files was performed by set, with noise modeling, noise filtering, and significance testing performed as described previously (Spurrier et al., 2018). Thus, noise-biased expression values were removed using lowess modeling to look for a relationship between mean gene expression and the corresponding coefficient of variation (CV). Lowess fits were then over-plotted to identify the common low-end expression value where the relationship between mean expression (signal) and CV (noise) deviated from linearity (mean expression value = 7.5). Expression values less than this value were set to equal 7.5, while gene probes not having at least one sample greater than 7.5 were discarded as non-informative. Fold changes observed for genes deemed to have differential expression between a mutation condition and its respective control were coded +1 or −1, depending on direction of change. These coded fold-changes were then intersected by gene symbol with similarly coded fold- changes observed for 10D Cdk5α-OE versus 10D Control and 10D Cdk5α null versus 10D Control. Spearman correlation was next applied to these intersections; providing for both a Rho estimate and an uncorrected p value per gene, and p values were then corrected via Benjamini-Hochberg procedure.
Generation of Axenic culture of Drosophila
Axenic culture of control, Cdk5α null and Cdk5α-OE flies were generated using eggs collected from sterile grape plates. Eggs from the grape plate were harvested using PBS followed by rinse with 70% ethanol. Washed eggs were dechorionated by bleaching using 3% Sodium hypochlorite for 7 minutes, followed by rinse with sterile water. Dechorionated embryo were transferred to axenic fly food supplemented with propionic acid (0.48ml/100ml) and Penicillin Streptomycin (Pen Strep) at 1ml/100ml concentration. The embryos were cultured at 25°C and validation of germ free state was done by homogenizing flies under aseptic condition and culturing the homogenate on Luria-Bertani medium plates. For axenic culture, all genotypes were grown together on axenic food and male progeny were collected at 3d-age to grow them to 30d-days for experiments.
QUANTIFICATION AND STATISTICAL ANALYSIS
All data were analyzed with GraphPad Prism 7.0, except PC analysis and comparison of Cdk5α null and Cdk5α-OE expression profiles with other mutant conditions and qRT-PCR. These two analyses were done using R: The R Project for Statistical Computing, and qRT-PCR analyses using Microsoft Excel. Error bars (SEM or SD) were calculated using Prism 7.0 as specified in figure legends. The number of replicates and brain hemispheres used per experiments are provided in figure legends. t test was performed on 3 or 5 replicates, hence normality of data distribution was not calculated due to limited sample size, while ANOVA was used for most analyses, which is considered as robust against normality assumption. Statistical test used for each figure is provided in figure legend and detailed statistical data for all figures are compiled in Table S2.
DATA AND SOFTWARE AVAILABILITY
Any data not included in Table S2 are available on request from the lead contact (EG). All R code used for analysis of microarray data is available through https://data.ninds.nih.gov.
Supplementary Material
Highlights.
Altering Cdk5 activity causes degeneration of dopamine neurons in Drosophila
Increasing or decreasing Cdk5 activity impairs autophagic flux
Impairing autophagic flux hyperactivates innate immunity
The hyperactivity of immunity, in turn, causes the age-dependent neuron loss
ACKNOWLEDGMENTS
We wish to thank all of the members of our lab for their advice and assistance in the design, execution, and interpretation of these experiments. We would also like to thank Chi-Hon Lee and Ela Serpe for helpful discussions and Mark Cookson and Richard Youle for comments on the manuscript. We particularly thank Kory Johnson for indispensable assistance in performing all of the statistical analysis of the expression profiling data. We also thank Karen Plevock Haase and Mary Dasso for their advice and assistance preparing axenic flies and Xiaoyi Li for helping us in generating axenic flies. We thank Kim D. Finley, Barry Ganetzky, Jean-Marc Reichhart, and Francesca Pignoni for sharing fly stocks and Ioannis Nezis and Wendi S. Neckameyer for antibodies. In addition, numerous essential Drosophila stocks were provided by the Bloomington Drosophila Stock Center and antibodies by the Developmental Studies Hybrid-oma Bank. We thank Stephen Wincovitch of the National Human Genome Research Institute (NHGRI) Cytogenetics and Microscopy Core Facility for assistance with confocal microscopy and Carsten Bonnemann and Eleonora Guadagnin for the use of their qRT-PCR machine. We are grateful to Joy Gu for outstanding technical assistance. This work was supported by the Basic Neuroscience Program of the Intramural Research Program of the National Institute of Neurological Disorders and Stroke (NINDS)/NIH (Z01 NS003106).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes five tables and six figures and can be found with this article online at https://doi.org/10.1016/j.celrep.2018.12.025.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Anglade P, Vyas S, Javoy-Agid F, Herrero MT, Michel PP, Marquez J, Mouatt-Prigent A, Ruberg M, Hirsch EC, and Agid Y (1997). Apoptosis and autophagy in nigral neurons of patients with Parkinson’s disease. Histol. Histopathol 12, 25–31. [PubMed] [Google Scholar]
- Bouché V, Espinosa AP, Leone L, Sardiello M, Ballabio A, and Botas J (2016). Drosophila Mitf regulates the V-ATPase and the lysosomal-autophagic pathway. Autophagy 12, 484–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao Y, Chtarbanova S, Petersen AJ, and Ganetzky B (2013). Dnr1 mutations cause neurodegeneration in Drosophila by activating the innate immune response in the brain. Proc. Natl. Acad. Sci. USA 110, E1752–E1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chakrabarty P, Ceballos-Diaz C, Lin WL, Beccard A, Jansen-West K, McFarland NR, Janus C, Dickson D, Das P, and Golde TE (2011). Interferon-γ induces progressive nigrostriatal degeneration and basal ganglia calcification. Nat. Neurosci 14, 694–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clarke PG (1990). Developmental cell death: morphological diversity and multiple mechanisms. Anat. Embryol. (Berl.) 181, 195–213. [DOI] [PubMed] [Google Scholar]
- Clayton KA, Van Enoo AA, and Ikezu T (2017). Alzheimer’s Disease: The Role of Microglia in Brain Homeostasis and Proteopathy. Front. Neurosci 11, 680. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connell-Crowley L, Le Gall M, Vo DJ, and Giniger E (2000). The cyclin-dependent kinase Cdk5 controls multiple aspects of axon patterning in vivo. Curr. Biol 10, 599–602. [DOI] [PubMed] [Google Scholar]
- Connell-Crowley L, Vo D, Luke L, and Giniger E (2007). Drosophila lacking the Cdk5 activator, p35, display defective axon guidance, age-dependent behavioral deficits and reduced lifespan. Mech. Dev 124, 341–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz JC, Tseng HC, Goldman JA, Shih H, and Tsai LH (2003). Aberrant Cdk5 activation by p25 triggers pathological events leading to neurode-generation and neurofibrillary tangles. Neuron 40, 471–483. [DOI] [PubMed] [Google Scholar]
- Ferrandon D, Jung AC, Criqui M, Lemaitre B, Uttenweiler-Joseph S, Michaut L, Reichhart J, and Hoffmann JA (1998). A drosomycin-GFP reporter transgene reveals a local immune response in Drosophila that is not dependent on the Toll pathway. EMBO J. 17, 1217–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gjoneska E, Pfenning AR, Mathys H, Quon G, Kundaje A, Tsai LH, and Kellis M (2015). Conserved epigenomic signals in mice and humans reveal immune basis of Alzheimer’s disease. Nature 518, 365–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedengren M, Asling B, Dushay MS, Ando I, Ekengren S, Wihlborg M, and Hultmark D (1999). Relish, a central factor in the control of humoral but not cellular immunity in Drosophila. Mol. Cell 4, 827–837. [DOI] [PubMed] [Google Scholar]
- Heneka MT, Kummer MP, and Latz E (2014). Innate immune activation in neurodegenerative disease. Nat. Rev. Immunol 14, 463–477. [DOI] [PubMed] [Google Scholar]
- Herrup K (2010). Reimagining Alzheimer’s disease–an age-based hypothesis. J. Neurosci 30, 16755–16762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmes C, Cunningham C, Zotova E, Woolford J, Dean C, Kerr S, Cull-iford D, and Perry VH (2009). Systemic inflammation and disease progression in Alzheimer disease. Neurology 73, 768–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannarkat GT, Boss JM, and Tansey MG (2013). The role of innate and adaptive immunity in Parkinson’s disease. J. Parkinsons Dis 3, 493–514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimbrell DA, and Beutler B (2001). The evolution and genetics of innate immunity. Nat. Rev. Genet 2, 256–267. [DOI] [PubMed] [Google Scholar]
- Kimura S, Noda T, and Yoshimori T (2007). Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy 3, 452–460. [DOI] [PubMed] [Google Scholar]
- Ko J, Humbert S, Bronson RT, Takahashi S, Kulkarni AB, Li E, and Tsai LH (2001). p35 and p39 are essential for cyclin-dependent kinase 5 function during neurodevelopment. J. Neurosci 21, 6758–6771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kounatidis I, Chtarbanova S, Cao Y, Hayne M, Jayanth D, Ganetzky B, and Ligoxygakis P (2017). NF-κB Immunity in the Brain Determines Fly Lifespan in Healthy Aging and Age-Related Neurodegeneration. Cell Rep. 19, 836–848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee HK, Lund JM, Ramanathan B, Mizushima N, and Iwasaki A (2007). Autophagy-dependent viral recognition by plasmacytoid dendritic cells. Science 315, 1398–1401. [DOI] [PubMed] [Google Scholar]
- Lemaitre B, and Hoffmann J (2007). The host defense of Drosophila melanogaster. Annu. Rev. Immunol 25, 697–743. [DOI] [PubMed] [Google Scholar]
- Levine B, Mizushima N, and Virgin HW (2011). Autophagy in immunity and inflammation. Nature 469, 323–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loch G, Zinke I, Mori T, Carrera P, Schroer J, Takeyama H, and Hoch M (2017). Antimicrobial peptides extend lifespan in Drosophila. PLoS One 12, e0176689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauvezin C, Nagy P, Juhász G, and Neufeld TP (2015). Autophagosomelysosome fusion is independent of V-ATPase-mediated acidification. Nat. Commun 6, 7007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melo JA, and Ruvkun G (2012). Inactivation of conserved C. elegans genes engages pathogen- and xenobiotic-associated defenses. Cell 149, 452–466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meyer SN, Amoyel M, Bergantiños C, de la Cova C, Schertel C, Basler K, and Johnston LA (2014). An ancient defense system eliminates unfit cells from developing tissues during cell competition. Science 346, 1258236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mulakkal NC, Nagy P, Takats S, Tusco R, Juhász G, and Nezis IP (2014). Autophagy in Drosophila: from historical studies to current knowledge. BioMed Res. Int 2014, 273473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy P, Varga Á, Kovács AL, Takáts S, and Juhász G (2015). How and why to study autophagy in Drosophila: it’s more than just a garbage chute. Methods 75, 151–161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nandi N, Tyra LK, Stenesen D, and Krämer H (2017). Stress-induced Cdk5 activity enhances cytoprotective basal autophagy in Drosophila melanogaster by phosphorylating acinus at serine437. eLife 6, e30760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, and Youle RJ (2010). PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 8, e1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neckameyer WS, Woodrome S, Holt B, and Mayer A (2000). Dopamine and senescence in Drosophila melanogaster. Neurobiol. Aging 21, 145–152. [DOI] [PubMed] [Google Scholar]
- Nezis IP, Simonsen A, Sagona AP, Finley K, Gaumer S, Contamine D, Rusten TE, Stenmark H, and Brech A (2008). Ref(2)P, the Drosophila melanogaster homologue of mammalian p62, is required for the formation of protein aggregates in adult brain. J. Cell Biol 180, 1065–1071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixon RA (2013). The role of autophagy in neurodegenerative disease. Nat. Med 19, 983–997. [DOI] [PubMed] [Google Scholar]
- Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, Bae E, Kim J, Shong M, Kim JM, and Chung J (2006). Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 441, 1157–1161. [DOI] [PubMed] [Google Scholar]
- Patrick GN, Zukerberg L, Nikolic M, de la Monte S, Dikkes P, and Tsai LH (1999). Conversion of p35 to p25 deregulates Cdk5 activity and promotes neurodegeneration. Nature 402, 615–622. [DOI] [PubMed] [Google Scholar]
- Petersen AJ, Katzenberger RJ, and Wassarman DA (2013). The innate immune response transcription factor relish is necessary for neurodegeneration in a Drosophila model of ataxia-telangiectasia. Genetics 194, 133–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qu D, Rashidian J, Mount MP, Aleyasin H, Parsanejad M, Lira A, Haque E, Zhang Y, Callaghan S, Daigle M, et al. (2007a). Role of Cdk5-mediated phosphorylation of Prx2 in MPTP toxicity and Parkinson’s disease. Neuron 55, 37–52. [DOI] [PubMed] [Google Scholar]
- Qu X, Zou Z, Sun Q, Luby-Phelps K, Cheng P, Hogan RN, Gilpin C, and Levine B (2007b). Autophagy gene-dependent clearance of apoptotic cells during embryonic development. Cell 128, 931–946. [DOI] [PubMed] [Google Scholar]
- Richards RI, Robertson SA, O’Keefe LV, Fornarino D, Scott A, Lardelli M, and Baune BT (2016). The Enemy Within: Innate Surveillance-Mediated Cell Death, the Common Mechanism of Neurodegenerative Disease. Front. Neurosci 10, 193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saitoh T, and Akira S (2010). Regulation of innate immune responses by autophagy-related proteins. J. Cell Biol. 189, 925–935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sano R, and Reed JC (2013). ER stress-induced cell death mechanisms. Biochim. Biophys. Acta 1833, 3460–3470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlachetzki JC, and Winkler J (2015). The innate immune system in Parkinson’s disease: a novel target promoting endogenous neuroregeneration. Neural Regen. Res 10, 704–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schwartz M, Kipnis J, Rivest S, and Prat A (2013). How do immune cells support and shape the brain in health, disease, and aging? J. Neurosci 33, 17587–17596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simonsen A, Cumming RC, Brech A, Isakson P, Schubert DR, and Finley KD (2008). Promoting basal levels of autophagy in the nervous system enhances longevity and oxidant resistance in adult Drosophila. Autophagy 4, 176–184. [DOI] [PubMed] [Google Scholar]
- Spurrier J, Shukla AK, McLinden K, Johnson K, and Giniger E (2018). Altered expression of the Cdk5 activator-like protein, Cdk5α, causes neurode-generation, in part by accelerating the rate of aging. Dis. Model. Mech 11, dmm031161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takahashi S, Ohshima T, Hirasawa M, Pareek TK, Bugge TH, Morozov A, Fujieda K, Brady RO, and Kulkarni AB (2010). Conditional deletion of neuronal cyclin-dependent kinase 5 in developing forebrain results in micro-glial activation and neurodegeneration. Am. J. Pathol 176, 320–329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong Y, Yamaguchi H, Giaime E, Boyle S, Kopan R, Kelleher RJ 3rd, and Shen J (2010). Loss of leucine-rich repeat kinase 2 causes impairment of protein degradation pathways, accumulation of alpha-synuclein, and apoptotic cell death in aged mice. Proc. Natl. Acad. Sci. USA 107, 9879–9884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trunova S, and Giniger E (2012). Absence of the Cdk5 activator p35 causes adult-onset neurodegeneration in the central brain of Drosophila. Dis. Model. Mech 5, 210–219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai LH, Delalle I, Caviness VS Jr., Chae T, and Harlow E (1994). p35 is a neural-specific regulatory subunit of cyclin-dependent kinase 5. Nature 371, 419–423. [DOI] [PubMed] [Google Scholar]
- Tusco R, Jacomin AC, Jain A, Penman BS, Larsen KB, Johansen T, and Nezis IP (2017). Kenny mediates selective autophagic degradation of the IKK complex to control innate immune responses. Nat. Commun 8, 1264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, and Qin ZH (2013). Coordination of autophagy with other cellular activities. Acta Pharmacol. Sin 34, 585–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Y, Li YB, Yin JJ, Wang Y, Zhu LB, Xie GY, and Pan SH (2013). Autophagy regulates inflammation following oxidative injury in diabetes. Autophagy 9, 272–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Randle KE, and Wu LP (2007). ird1 is a Vps15 homologue important for antibacterial immune responses in Drosophila. Cell. Microbiol 9, 1073–1085. [DOI] [PubMed] [Google Scholar]
- Wyss-Coray T (2016). Ageing, neurodegeneration and brain rejuvenation. Nature 539, 180–186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J (2015). Mapping neuroinflammation in frontotemporal dementia with molecular PET imaging. J. Neuroinflammation 12, 108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang T, Zhou Q, Ogmundsdottir MH, Möller K, Siddaway R, Larue L, Hsing M, Kong SW, Goding CR, Palsson A, et al. (2015). Mitf is a master regulator of the v-ATPase, forming a control module for cellular homeostasis with v-ATPase and TORC1. J. Cell Sci 128, 2938–2950. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Any data not included in Table S2 are available on request from the lead contact (EG). All R code used for analysis of microarray data is available through https://data.ninds.nih.gov.