

Abstract
Numerous lethal stresses in bacteria including antibiotics, thymineless death, and MalE-LacZ expression trigger an increase in the production of reactive oxygen species. This results in the oxidation of the nucleotide pool by radicals produced by Fenton chemistry. Following the incorporation of these oxidized nucleotides into the genome, the cell’s unsuccessful attempt to repair these lesions through base excision repair (BER) contributes causally to the lethality of these stresses. We review the evidence for this phenomenon of incomplete BER-mediated cell death and discuss how better understanding this pathway could contribute to the development of new antibiotics.
Keywords: Base excision repair, cell death, antibiotics, reactive oxygen species, thymineless death, 8-oxo-dG
1. Introduction
Maintaining the integrity of the genome is vital to the survival of any organism. The DNA of cells is constantly subject to potentially mutagenic damage from many endogenous and exogenous sources such as reactive oxygen species (ROS), reactive nitrogen species (RNS), UV, and alkylating agents, so cells have evolved multiple mechanisms to repair or tolerate this damage. One of these pathways, base excision repair (BER), removes a single damaged or mispaired base from the genome, which can then be replaced with the appropriate nucleotide [1,2]. BER is a multistep process that requires several enzymes working in concert with the intermediate steps all being forms of DNA damage themselves that are potentially much more acutely dangerous to the cell than the original lesion [3]. While the potential toxicity of incomplete base excision repair has been extensively studied in eukaryotes [4], much less is known about this phenomenon in bacteria.
There is a large and growing body of evidence from multiple laboratories that numerous lethal stresses in bacteria produce ROS that causally contribute to cell death. These stresses are extremely varied and include multiple classes of bactericidal antibiotics [[5], [6], [7]], the P1vir phage, the type VI secretion system, antimicrobial peptides [8,9], the MalE-LacZ fusion protein [10], and thymineless death [11]. One consequence of this ROS production is the oxidation of guanine nucleotide pool to produce the mutagenic nucleotide 8-oxo-GTP, which is then incorporated into the genome. The resulting 8-oxo-dG can then be removed through BER and the DNA accurately repaired. However, the cell’s attempt at using BER to repair these lesions can also be a contributing factor to the lethality of these stresses.
This phenomenon of bacterial cell death caused by incomplete BER has been extensively documented [6,7,[10], [11], [12], [13], [14], [15], [16], [17]], but its relevance to antibiotic lethality has only recently been appreciated. In this review, we will summarize the evidence for this pathway of cell death in bacteria and draw parallels to similar results in eukaryotes.
2. 8-oxo-guanine: its processing and repair
Cells normally generate a low level of reactive oxygen species as a consequence of aerobic metabolism, with superoxide and hydrogen peroxide both being produced [18]. E. coli detoxifies superoxide by dismutating it to hydrogen peroxide using superoxide dismutases, while hydrogen peroxide is removed by catalases or peroxidases [19]. Despite these protective mechanisms, the basal level of endogenous ROS is still capable of damaging cellular components.
Due to its low redox potential, guanine is the most susceptible nucleotide to oxidative damage with the major product being 7,8-dihydro-8-oxoguanine (8-oxo-G) (Fig. 1B) [20]. Importantly, nucleotides are not directly damaged by the superoxide or hydrogen peroxide produced by the cell, instead they are damaged by •OH or other radicals produced through the Fenton reaction between Fe2+ and hydrogen peroxide [19,21]. Additionally, nucleotides are even more at risk as they naturally chelate Fe2+, which both strikingly increases the rate of the Fenton reaction as well as localizes the resultant highly reactive radical to the nucleotide [22], adjacent to the C8 carbon in purines [23]. The resulting 8-oxo-GTP can then be incorporated into the genome, where it is highly mutagenic [24].
Fig. 1. Nucleotides.
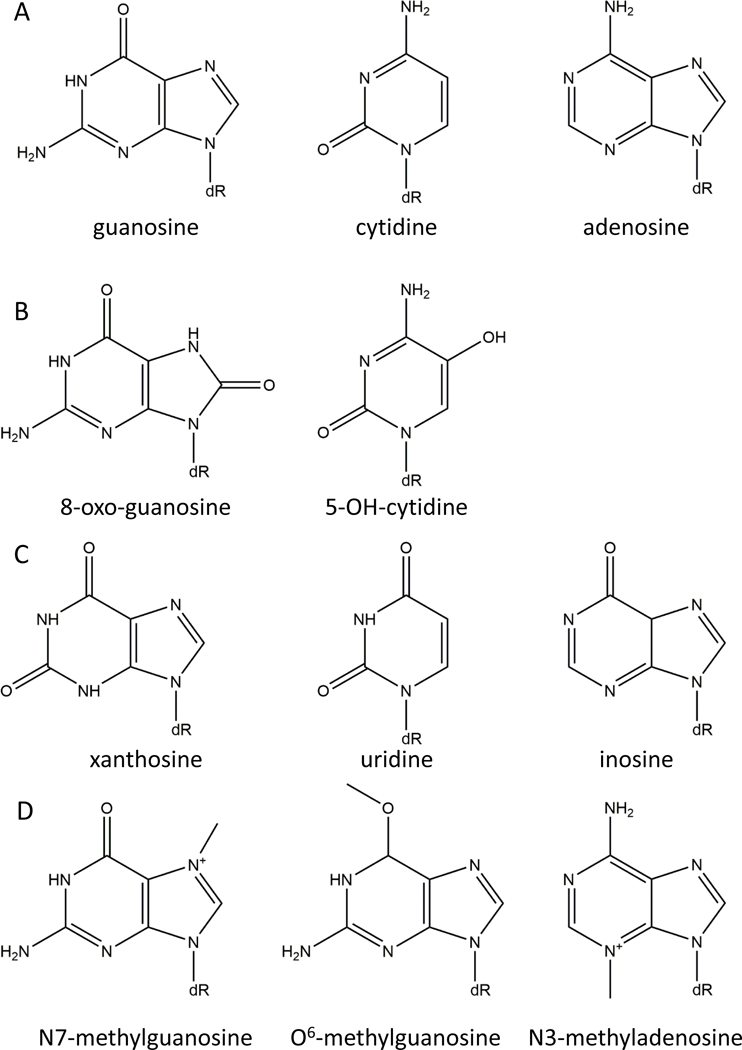
A. Undamaged. B. Oxidized. C. Deaminated. D. Alkylated.
8-oxo-dG is mutagenic because, like guanine, it can pair with dC in its anti conformation, but it is also capable of rotating at its glycosidic bond and pairing with dA in its syn conformation. Thus, 8-oxo-dG introduced into DNA through the use of 8-oxo-dGTP during DNA synthesis produces A:T to C:G transversions, whereas 8-oxo-dG resulting from oxidation of a dG in DNA produces G:C to T:A transversions. 8-oxo-dG is one of the most common kinds of DNA damage in the genome, but accurately measuring it has been challenging, with earlier estimates being too high due to oxidation of undamaged dG residues during sample preparation [25]. Older estimates in E. coli were around 2.5 8-oxo-dG/105 dG in E. coli [26] compared a more modern estimate of 0.5–4.24 8-oxo-dG/106 dG in human cells [27].
2.1. 8-oxo-dG repair
Bacterial cells possess multiple systems to mitigate the mutations caused by 8-oxo-dG [1,28]. In E. coli, the 8-oxo-dGTP diphosphatase MutT acts a nucleotide sanitizer, converting both 8-oxo-dGTP and 8-oxo-GTP to their monophosphates, thereby preventing the incorporation 8-oxo-dG and 8-oxo-G into DNA and RNA respectively [24,[29], [30], [31]]. Cells with a loss of function mutT allele are classic mutator strains, with a 1000 fold higher rate of A:T to C:G transversions, which further demonstrates how common this form of DNA damage is.
Once incorporated into the genome, E. coli uses BER DNA glycosylases to remove the damaged nucleotides. MutM, the formamidopyrimidine DNA glycosylase (Fpg), recognizes 8-oxo-dG and breaks the glycosidic bond connecting the base to the sugar, creating an apurinic site [26,30,[32], [33], [34]]. MutM is a bifunctional enzyme that can then act as a lyase to remove the sugar through β and δ elimination, resulting in both 3′ and 5′ phosphates [35,36]. Since DNA polymerase requires a 5′ phosphate and 3′ OH, the 3′ phosphate must then be removed by an AP endonuclease such as exodeoxyribonuclease III (XthA) or endonuclease IV (Nfo) [37]. A new base can then be inserted by DNA polymerase I, and finally DNA ligase reseals the nicked strand, completing the repair (Fig. 2).
Fig. 2. Diagram of the BER repair pathways. Bacterial proteins are in black and eukaryotic are in blue.
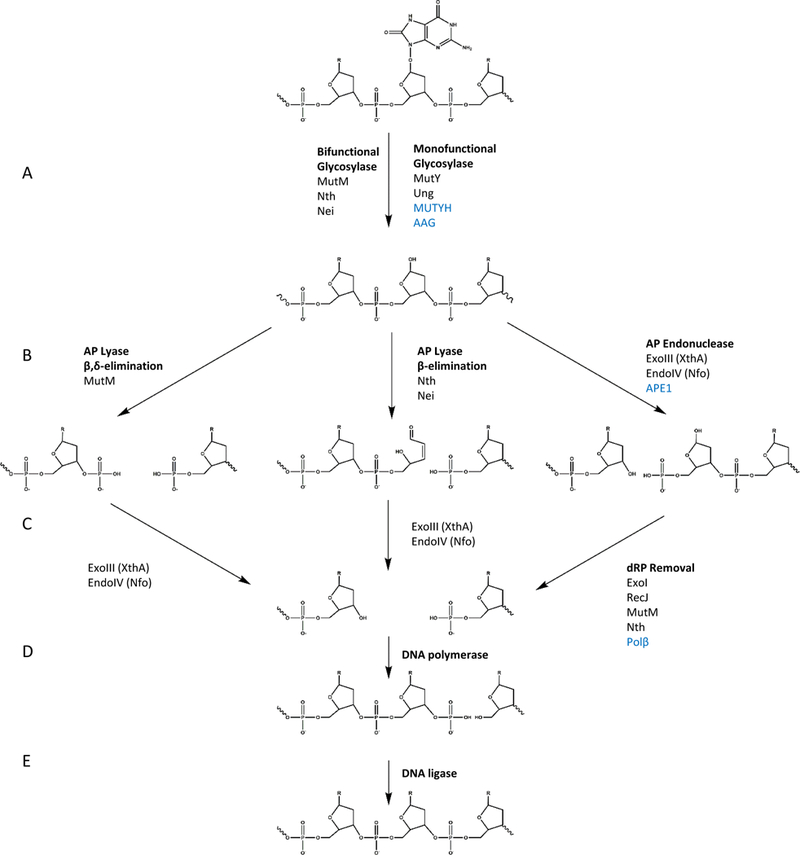
A. The damaged or mispaired nucleotide, represented here by 8-oxo-dG, is removed by a glycosylase to generate an AP site. B. Bifunctional glycosylases then act as a lyase and generate a single strand break. Some, such as Nth and Nei act through β-elimination, generating a 5’ phosphate and a 3’ 4-hydroxy-2-pentenal-5-phosphate. MutM, which acts through β,δ-elimination leaves both 3’ and 5’ phosphates. Monofunctional glycosylases including, MutY, Ung and AAG require an AP endonuclease, such as the E. coli ExoIII or eukaryotic APE1 and generate 5’ dRP and 3’ OH. C. The resulting ends then need to be cleaned up by different enzymes to generated the 5’ phosphate and 3’ OH required for a polymerase: in E. coli, either of the two AP endonucleases ExoII and EndoIV can process the 3’ end. 5’ dRP is processed by Polβ in eukaryotes and a variety of different proteins are proposed to have this activity in E. coli. D. The gap is then filled in by a DNA polymerase. E. And sealed by DNA ligase.
Complementing the actions of MutM is MutY, an adenine DNA glycosylase that removes undamaged but mispaired adenine residue across from 8-oxo-dG, thereby preventing G:C to T:A mutations that would otherwise occur from incorporation of an A opposite an 8-oxo-dG that has resulted from oxidation of a dG in cellular DNA [38]. In contrast to the bifunctional MutM, MutY is a monofunctional DNA glycosylase, which produces an AP site that must be further processed by an AP endonuclease or lyase [39]. AP endonucleases generate a 5′ deoxyribose phosphate (dRP) residue that must be processed by yet another enzyme. Several different proteins have been proposed to have this dRPase function in E. coli. These include Exonuclease I (SbcB) [40] and RecJ [41], as well as the bifunctional glycosylases MutM [42] and Nth [43] acting through their lyase activity, although more work is still needed to clarify which enzymes actually perform this activity. MutY also initially stays bound to the resulting AP lesion to exclude MutM, preventing the 8-oxo-dG from being removed until its corresponding base is properly repaired (Fig. 2) [44].
In addition to BER, E. coli can use the methyl-directed mismatch repair system (MMR) to repair oxidized lesions [45]. This system requires the MutS, MutL, and MutH proteins as well as the Dam methylase, which methylates the adenine at newly synthesized GATC sites after DNA replication. Methylation is much slower than replication, so recently replicated DNA is only hemimethylated, thereby creating a temporal window that allows the cell to differentiate the newly synthesized unmethylated daughter strand from the methylated parental template [46,47]. MutS binds to mismatched bases and recruits MutL and together they activate the nuclease MutH, which cuts the unmethylated strand at hemimethylated GATC sites [[48], [49], [50]]. The unmethylated strand is unwound and digested by an exonuclease and the gap is filled in by DNA polymerase III [[51], [52], [53]]. Additionally, MutS can interact with the DNA glycosylase MutY and increases its affinity for A:8-oxo-G mismatches, increasing the rate of repair and specifically targeting it to the newly synthesized strand [54].
Interestingly, these repair pathways are regulated independently of each other. The nucleotide sanitizer MutT is encoded within the secM secA mutT operon along with components of the Sec protein translocation system, so its expression is increased in response to secretion stress [55]. MutM is under the direct control of a σ32 heat-shock promoter, so that mutM is upregulated in response to high temperatures, misfolded proteins, and other related stresses [56]. MutY expression is downregulated under oxidative stress and upregulated under anaerobic conditions. It is positively regulated by Fur, Fnr and ArcA, although it is unknown whether this regulation is direct [57]. The MutHLS system is negatively regulated in stationary phase by the sRNA SdsR [58]. Importantly, none of these genes are part of the OxyR- and SoxRS-dependent responses to oxidative stress, despite the fact they are all involved in oxidized nucleotide repair. This suggests that increasing their expression under conditions of oxidative stress may not be advantageous to the cell, likely due to the risk of BER-mediated cell death.
2.2. Repair intermediates and strand breaks
While BER allows a cell to limit the number of mutations that become fixed in its genome under normal conditions, the act of repair itself is potentially dangerous. Although mutagenic, 8-oxo-dG is not immediately toxic to the cell since it is not a barrier to replication or transcription. However, the intermediate steps involved in BER are all potentially lethal to the cell because they introduce strand breaks.
MutY and MutM produce a transient AP site that is highly mutagenic. Without a base to serve as a template, RNA polymerase and replicative DNA polymerases are blocked and unable to continue synthesis, however error-prone translesion polymerases are able to bypass the lesion [1,59]. AP sites are also unstable and can form more complex DNA lesions such as interstrand crosslinks that require more extensive repair mechanisms [60].
To complete BER, the AP site then requires the action of an AP endonuclease or AP lysase which generates a single-strand break (SSB). Depending on the enzyme responsible, including MutM itself, the resulting break requires further processing by another protein, thereby slowing down the rate of repair. SSBs within 7 bp of each other on opposite strands will result in double strand break [61], which means that under conditions that cause a high amount of damage, these SSBs could become a DSB. This mechanism of double strand break formation might be especially important in stressed cells in which the 8-oxo-dG’s are being incorporated by a DNA polymerase such as DNA Pol IV (DinB), which is prone to using 8-oxo-dGTP and thus might incorporate them close together [14]. Alternatively, a replication fork encountering one of these SSB will result in a double-strand break [62], which can then be repaired using the RecA-dependent homologous recombination pathway [63]. Since a single unrepaired DSB in E. coli is lethal [64], the accumulation of repair intermediates poses a serious threat to the cell.
3. Lethal stresses and ROS
Under normal physiological conditions, the endogenous levels of ROS produced by E. coli are well tolerated despite contributing to spontaneous mutagenesis [65]. Increasing the amount of ROS by directly adding exogenous hydrogen peroxide to E. coli results in the cells dying. The exact mechanism of cell death is bimodal and dependent on the concentration of hydrogen peroxide added. At lower concentrations of exogenously added H2O2 (∼1–5 mM), the cell’s DNA is heavily damaged and recA mutants defective in DSB repair are more susceptible, indicating that DNA damage contributes to the mechanism of action. In contrast, high concentrations (>20 mM) kill though a non-DNA repair dependent manner [66,67]. It is important to remember that the “low” levels of H2O2 in these seminal exogenously added H2O2 experiments are orders of magnitude higher than the much lower intracellular levels of H2O2 that cells normally experience [68].
Base excision repair is a key part of E. coli’s response to H2O2 induced DNA damage. Null mutants lacking two of the AP endonucleases, ExoIII (XthA) and EndoIV (Nfo), are much more susceptible to killing by 5 mM of H2O2., and this phenotype can be partially suppressed by knocking out mutM [37]. In the absence of AP endonucleases, MutM would generate potentially lethal unrepairable SSB intermediates. In contrast, a single mutant ΔmutM strain that still possesses the AP endonucleases has wildtype susceptibility at the same concentrations [69].
While cells can be exposed to damagingly high levels of exogenous H2O2 in their environment, there are also a diverse range of conditions that result in an increase in their endogenous levels of H2O2. The increased production of ROS and the accumulation of its associated cellular damage is well described in eukaryotes and is associated with both cancer [70] and neurodegeneration [71], with the levels of both 8-oxo-G and 8-oxo-dG being commonly used as a biomarker for oxidative stress and cancer [25,72]. There is a growing body of evidence that numerous stresses can similarly induce ROS production in bacteria that contributes to their lethality.
3.1. Bactericidal antibiotics
The direct targets of most antibiotics have been extensively studied. However, aside from their classical mechanism of actions, antibiotics also trigger complex and diverse physiological changes as the cell attempts to respond to this potentially lethal stress [13]. Numerous reports have shown that this response includes the production of ROS, and that ROS contributes to the mechanism of killing in diverse classes of antibiotics [6,7,[10], [11], [12], [13], [14], [15]].
The exact molecular events that result in this antibiotic-induced ROS production are still incompletely understood. However, similar metabolic changes occur after treatment with three different classes of bactericidal antibiotics: β-lactams which inhibit cell wall synthesis, aminoglycosides which interfere with translation, and fluoroquinolones which block gyrase and topoisomerase, suggesting that the downstream effects are related [12,73]. In the case of β-lactams, they trigger a futile cycle of synthesis and degradation of the cell wall [74], while more generally such futile metabolic cycles have been shown to increase production of endogenous ROS [75]. Trimethoprim, an inhibitor of dihydrofolate reductase, also leads to an increase in ROS production [7] and will be discussed later.
Regardless of its source, the ROS produced by ampicillin, kanamycin, and norfloxacin treatment oxidize many biomolecules, which leads to an increase in the amount of 8-oxo-dG present in the nucleotide pool and in the genome [12]. Several key observations indicate that incomplete BER of this 8-oxo-dG contributes causally to the lethality of these antibiotics. Many antibiotics are less lethal under anaerobic conditions, which would prevent the production of ROS [6]. Preventing the incorporation of 8-oxo-dG through overexpression of the 8-oxo-dGTP sanitizer MutT provides a protective effect [6,14], as does removing it immediately after replication through the MutHSL-dependent MMR system [76,77]. Interestingly, subinhibitory concentrations of β-lactam antibiotics induce the RpoS regulon, which causes a reduction in MMR mediated by SdsR, the RpoS-controlled small RNA that seems to directly repress the mutS mRNA [58]. Importantly, a ΔmutM ΔmutY double knockout of the two major DNA glycosylases is also more resistant to killing by bactericidal antibiotics [14]. Antibiotics, at least partially through the action of the MutM and MutY DNA glycosylases, induce breaks in the genome [6,14]. Additionally, ΔrecA mutants defective in homologous recombination, including DSB repair, are more susceptible to killing by bactericidal antibiotics [14,78]. Collectively these observations have led to a model in which incomplete base excision repair of 8-oxo-dG contributes to the lethality of antibiotics [6,12,14]. As discussed below, depending on the complement of nucleotide sanitizers, DNA glycosylases, and DNA polymerases possessed by a particular bacterium, other oxidized nucleotides such as 5-OH-dCTP can also cause cell death by being incorporated into DNA and then being processed to yield toxic BER intermediates [15]. Not considering the evidence for the role of incomplete BER in antibiotic lethality [14], appears to have contributed to the inference that ROS are not involved in cell death from antibiotics [79,80], a conclusion that has subsequently be shown to be inaccurate [6,7,[11], [12], [13],73,81,82]
3.2. MalE-LacZ
The historically important MalE-LacZ fusion protein also causes cell death through the incomplete BER death pathway [10]. This periplasmic-cytoplasmic fusion protein was used by John Beckwith and colleagues to identify and characterize the Sec-dependent secretion system [83].
The expression of MalE-LacZ is only lethal under aerobic conditions and induces the expression of the oxidative response gene soxS. The levels of endogenous hydrogen peroxide and superoxide are increased and the amount of 8-oxo-dG incorporated in the genome is elevated. Deletion mutants of genes that protect against oxidative stress are also more susceptible, showing that ROS is causative to cell death [10].
Importantly, increasing the level of expression of MutT, which degrades 8-oxo-dGTP, or the MMR protein MutS, which can restore MMR due to reduced MutS levels [6,58] both suppress the lethality of MalE-LacZ expression. Deletion of the genes encoding the DNA glycosylases mutM and mutY renders the fusion protein bacteriostatic, indicating that their action is essential for lethality. Additionally, a recA mutant or a double recB recF mutant have greatly increased susceptibility to MalE-LacZ [10], indicating that in wild type cells homologous recombination is able to repair DNA strand breaks that would otherwise prove lethal. Interestingly, the incorporation of 8-oxo-dG into the genome followed by incisions by BER DNA glycosylases that lead to lethal DNA problems is the primary mechanism of MalE-LacZ-induced cell death. This contrasts with bactericidal antibiotics, where this mechanism involving incomplete BER is instead only a contributing factor to cell death.
Time-resolved microarrays at multiple time points performed on cells expressing MalE-LacZ provided an interesting spotlight on the transcriptomic changes caused by this lethal stress. Despite soxS being induced, the rest of the OxyR and SoxRS oxidative stress systems were not activated, which would have limited the protective response against ROS. Instead, key genes responsible for 8-oxo-dG repair are all affected. Expression of mutT is increased, probably as a consequence of it sharing an operon with secM secA whose expression is increased in response to secretion stress. Both MutM and MutY are upregulated, with MutM induction in particular being strong due to it being a member of the heat shock regulon [10]. This increased production of MutM is thought to be a factor in why incomplete BER is the primary mechanism responsible for the death of cells upon MalE-LacZ induction.
3.3. Thymineless death
Thymineless death (TLD) is a phenomenon in which thymine auxotrophs, both eukaryotic and prokaryotic, that are provided every nutrient except thymine will lose viability rather than simply experiencing growth arrest [84,85]. The exact mechanism of cell death in TLD has remained elusive, but it is known to stall replication forks, induce the SOS response, and eventually lead to fragmentation of the genome [[86], [87], [88]]. Numerous drugs induce this effect primarily through targeting dihydrofolate reductase, including chemotherapeutics such as methotrexate [89], antiparasitics such as pyrimethamine [90], as well as antibacterials such as trimethoprim [91].
Treatment of bacteria with trimethoprim (TMP) is bactericidal in rich media and closely mimics, although is not identical to, thymine starvation of a thyA mutant [92,93]. These cells also experience oxidative stress due to production of ROS. Catalases and anaerobic growth provide some protective effect, indicating that ROS causally contribute to killing [7]. Additionally, a mutM mutY double mutant is less susceptible to TMP, indicating that 8-oxo-dG is involved [7]. Similarly, thymine starvation itself also leads to the production of ROS which contributes to the lethality of TLD and again the deletion of mutM mutY has a modest protective effect [11].
3.4. Other ROS inducing stresses
Other lethal stresses have also been linked to the production of ROS, but have not yet been investigated for a potential involvement of incomplete BER-mediated cell death. For example, the type VI secretion system (T6SS) of Vibrio cholera induces the production of ROS and increases the expression of the oxidative stress regulator SoxS in E. coli. This production of ROS is also causal to death, as a double knockout mutant of the sodAB superoxide dismutases is more susceptible to being killed. Similarly, the T6SS from Acinetobacter baylyi and Pseudomonas aeruginosa both increase soxS expression [8], suggesting this may be a common feature of killing via the T6SS. Additionally, the bacteriophage P1vir also causes the production of ROS and induces the expression of soxS [8].
Antimicrobial peptides are also capable of causing physiological and metabolic changes that result in ROS production. CM15, a hybrid antimicrobial peptide (AMP) derived from cecropin A (moth) and melittin (bee venom) [94], increases the autofluorescence of E. coli due to the oxidation of flavins and leads to the production of ROS. Its MIC is 20 fold higher under anaerobic conditions, suggesting that ROS production contributes to the mechanism of killing [9]. Additionally, the unrelated AMP polymyxin B (from Paenibacillus polymyxa) induces the expression of the oxidative response gene soxS [8].
4. Other lethal stresses
Increasing the amount of 8-oxo-dG through the production of ROS is not the only way for cells to die via incomplete BER. Any other stress or condition that would lead to an increase in repair intermediates present in the genome would potentially be susceptible.
4.1. Radiation
Ionizing radiation is capable of directly breaking the phosphodiester bonds of DNA and fragmenting the genome. However, the majority of DNA damage from irradiation actually results from oxidative damage caused by free radicals produced through the radiolysis of water [95]. The resultant damaged bases are the same as those produced through regular metabolism, primarily 8-oxo-dG, and are repaired through the standard BER pathway.
E. coli mutants lacking the MutM DNA glycosylase are significantly more resistant to x-ray radiation. Cells that are allowed to recover after irradiation normally generate more DSBs as they attempt to repair their DNA, while a ΔmutM strain produces far fewer DSBs. Conversely, expression of MutM from a plasmid results in an increase in susceptibility to radiation and an increase in the number of DSBs [96]. In this case, direct oxidation by ROS produced by ionizing radiation rather than endogenous ROS produces the same end result, DNA damage and cell death through incomplete BER.
4.2. DinB overexpression
Overexpression of the E. coli Y family DNA polymerase DinB (DNA pol IV) is lethal [14,25], and this can be suppressed by growing the cells under anaerobic conditions, expressing the 8-oxo-dGTP sanitizer MutT, or by knocking out the DNA glycosylases MutM and MutY. In this case, the increased incorporation of 8-oxo-dG is a result of DNA pol IV having a higher affinity for 8-oxo-dGTP than the replicative polymerases [97], rather than from an increased 8-oxo-dGTP level caused by an increase in ROS production. Overexpression of a mutant allele DinB F13V which has a lower affinity for 8-oxo-dGTP is not lethal [14]. Increasing the rate of 8-oxo-dG incorporation into the genome without increasing the levels of 8-oxo-dGTP is sufficient to cause bacterial cell death.
DinB also contributes to the lethality of antibiotics through this pathway. Subinhibitory concentrations of bactericidal antibiotics induce mutagenesis in an oxygen-dependent manner [78], and this pathway requires the induction of the SOS response [98] and specifically DinB [58,76].
4.3. Hyperinitiation
Hyperinitiation of DNA replication in E. coli leads to replication fork collapse and cell death [99,100]. This initiation is regulated by DnaA binding to origin of replication, oriC [101]. DnaA is an AAA + ATPase that binds both ADP and ATP, with the ATP bound form initiating replication [102]. The cell controls the rate of replication by altering the ratio of DnaAATP/DnaAADP [103]. The protein Hda lowers this ratio by promoting the hydrolysis of ATP, limiting the replication initiation [100,104]. hda null mutants hyperinitiate replication and have a severe growth defect that quickly leads to the generation of suppressor mutations [105,106].
However this phenotype is suppressed by growth under anaerobic conditions, by supplementing the media with the antioxidant glutathione, or by knocking out the MutM DNA glycosylase. [107]. The increase in the number of replication forks in this strain increases the probability that a fork will encounter a repair intermediate in the genome, as there is less time to complete BER. While 8-oxo-dG lesions would be one of the most common in the genome, some breaks are still detected in the Δhda ΔmutM ΔmutY strain, suggesting that there may be other potentially toxic repair intermediates present.
4.4. Other nucleotides
This pathway of bacterial cell death involving incomplete BER has been most extensively studied in E. coli with a focus on 8-oxo-dG, however, the same mechanism can apply to other oxidized nucleotides and to other bacteria.
Deamination of nucleotides, either spontaneously through hydrolysis or caused by chemical agents such as nitric oxide [108,109], produces potentially mutagenic non-standard deoxynucleotides: hypoxanthine, xanthine, and uracil are produced by deamination of adenine, guanine, and cytosine, respectively (Fig. 1C). E. coli sanitizes inosine and xanthosine using the highly conserved ITP/XTP pyrophosphatase RdgB. While a ΔrdgB mutant is viable in a wildtype background, it is lethal in ΔrecA or ΔrecBC mutants, indicating a requirement for DSB repair. This can be suppressed with a knockout of Δnfi [110]. The nfi gene encodes endonuclease V, which removes incorporated inosine and xanthine nucleotides [111,112], indicating that it is the DNA breaks introduced by EndoV that cause the lethality, not the simple presence of the incorrect bases.
A similar system exists to prevent and repair any incorporation of uracil into DNA. The deoxyuridine triphosphatase Dut sanitizes dUTP [113] and the DNA glycosylase Ung removes uracil residues in the genome [114]. Again, mutations in the nucleotide sanitizer are lethal in a rec deficient background [115]. However, in this case a Δung mutant is not sufficient to suppress this lethality, possibly due to redundancy, as other enzymes including EndoV are also capable of removing uracil residues from DNA [116]. While BER-mediated cell death due to oxidized nucleotides can be caused through various stresses, BER-mediated cell death through deaminated nucleotides is currently only known to occur with gene knockouts of the nucleotide sanitizers. Whether reactive nitrogen species (RNS) or any deaminating agents can trigger this pathway is not yet known.
4.5. 5-OH-dCTP can result in lethality in stationary phase Mycobacteria
Most of what is known about bacterial death resulting from incomplete BER comes from E. coli, where 8-oxo-dGTP is the most important oxidized nucleotide. However, a mechanistically analogous example involving 5-OH-dCTP was recently reported for Mycobacteria. Mycobacterial MazG is a nucleotide pyrophosphatase specific for 5-OH-dCTP, an oxidized form of cytosine [117,118] (Fig. 1B). Deletions of this gene are more susceptible to antibiotic killing, but only during stationary phase, and they have an increase in the number of double-strand breaks. This increased susceptibility can be suppressed by knocking out Nth, the BER DNA glycosylase responsible for repairing 5-OH-dC lesions [15], indicating that the cell death does not result simply from the incorporation of 5-OH-dC into DNA, but rather from incisions introduced during BER.
The dependence of this pathway on stationary phase is due to the expression of the error-prone DNA polymerase DnaE3, which is only expressed under these conditions. Analogously to E. coli DinB, which has an increased propensity to use 8-oxo-GTP as a substrate, DnaE3 has a higher affinity for 5-OH-dCTP and deletion of this gene suppresses the increased susceptibility of a ΔmazG strain to antibiotic killing [15].
5. Eukaryotes
5.1. Incomplete BER in eukaryotic cells
Incomplete BER-mediated cell death is a phenomenon that is not limited to bacteria and has been extensively studied in eukaryotes, with the majority of work focusing on a alkylation-induced damage.
Alkylating agents are a diverse group of reactive compounds that include both carcinogens and chemotherapeutic agents [119,120]. These compounds act by transferring an alkyl group to one or more DNA bases, sugars, or phosphates and, depending on the alkylating agent, can produce a wide array of different lesions including strand crosslinks, large bulky adducts, or methylation of individual bases, all of which are repaired through a variety of different mechanisms [4].
The major methylation products (Fig. 1D) are: N7-methylguanine (7meG) which is comparatively innocuous although it can spontaneously convert to an AP site, N3-methyladenine (3meA) which is potentially cytotoxic due to its ability to block the replicative, but not translesion, DNA polymerases, and O6-methylguanine (O6meG), which can mispair with thymine and is highly mutagenic. Two less common methylation products 1meA and 3meC can be directly repaired through the direct oxidative removal of their methyl groups by a member of the AlkB homolog family, and O6-methylguanine can be directly repaired by O6-methylguanine-DNA methyltransferase. These types of direct removal of the damage do not introduce a strand break and are thus not potentially lethal. However, 3meA lesions must be repaired through BER by the alkyladenine-DNA glycosylase (AAG), thereby introducing a strand break [4].
As in bacteria, BER in eukaryotes is a multistep process that requires: i) a DNA glycosylase to remove the base generating an AP site, ii) AP endonuclease (APE) to cut the strand resulting in a 3′OH and 5′ deoxyribosephosphate (5′dRP), iii) DNA polymerase β (Polβ) to remove the 5′dRP and fill in the nucleotide, and iv) DNA ligase to ligate the ends (Fig. 2). It has been proposed that under normal conditions these repair proteins act in a coordinated manner as a relay to sequester the toxic repair intermediates [121].
Transgenic mice that are Aag−/− are viable and have been used extensively to study the role of BER following exposure to alkylating agents such as methyl methanesulfonate (MMS) [122,123]. Interestingly, wild type mice exposed to methylating agents experience severe degradation of their retinas due to apoptosis [[124], [125], [126]]. In contrast, retinas in Aag−/− mice are completely resistant to the cytotoxic effects of MMS treatment, and Aag+/− heterozygotes display an intermediate phenotype, while mice overexpressing Aag were even more sensitive [127]. Similarly, bone marrow cells isolated from Aag−/− mice were more resistant to alkylating agents [128], and Aag−/− mice are partially protected from developing diabetes after treatment with the β-cell specific alkylating agent streptozotocin [129,130] which normally results in the death of insulin producing cells through the incomplete BER-mediated death pathway.
However, the lack of Aag does not always provide protection. For example, Aag−/− mouse embryonic stem cells and RNAi knockdowns of Aag in human carcinoma cells both have an increased sensitivity to alkylating agents [131,132]. Additionally, while an Aag−/− mouse might be protected against the acute toxicity of alkylating agents in some tissues, it is at increased risk of colon cancer due to its inability to prevent alkylation-induced mutagenesis [133,134].
Alkylation is not the only kind of DNA damage that can contribute to incomplete BER-mediated cell death in eukaryotes. Retinitis pigmentosa (RP) is an inherited form of degenerative blindness caused by loss of photoreceptors that has been linked to oxidative stress. An RP mouse model using a mutation in PDE6β found in human patients recapitulates this retinal degradation. Transgenic overexpression of MTH1, the human homolog of the 8-oxo-dG sanitizer MutT, helps protect the photoreceptors from cell death [135]. Similarly, the knockout of Mutyh, the human homolog of the A:8-oxo-G DNA glycosylase MutY, results in mice that experience reduced retinal degradation. Interestingly, the Mutyh−/− mice also produce less ROS, suggesting that some sort of positive feedback loop is involved that exacerbates the damage [136].
The cells susceptible to this form of cell death from incomplete BER seem to be extremely specific. In the case of retinal photoreceptors, only a single layer of cells dies in both alkylation and oxidative stress, while neighboring cells are unaffected [127,136]. The exact reason for this specificity is currently unknown, but an imbalance between all the proteins involved in BER is a possibility. Blocking the function of APE, the next step of BER, increases the susceptibility to alkylating agents in many different cell types, and MEFs lacking the subsequent enzyme, Polβ, are also much more susceptible [137]. In contrast, Polβ and Aag double mutant cells are resistant to alkylating agents. More work will need to be done to determine if the cells susceptible to Aag or Mutyh mediated cell death display this sort of BER imbalance.
5.2. Cisplatin induction of ROS
There is an interesting example in eukaryotes that mirrors the results in bacteria treated with bactericidal antibiotics. The chemotherapeutic agent cisplatin causes interstrand crosslinks which results in cell death through blocking of replication and transcription [138]. Independent of the damage caused to the nuclear genome, cisplatin treatment also causes mitochondria to produce more ROS. This production of ROS contributes causally to the lethality of cisplatin in cancer cells as mitochondria that produce excess catalase and cells with dysfunctional mitochondria are both more resistant to cisplatin treatment. This increase in ROS production is likely a consequence of cisplatin damaging the mitochondrial genome and thus affecting the synthesis of the proteins in the electron transport chain [139]. It is not currently known which cellular components are damaged by this ROS that is responsible for the oxidative component of cisplatin lethality.
6. Conclusions
Incomplete base excision repair appears to be an important pathway of cell death in both eukaryotes and prokaryotes (Fig. 3). Importantly, it should be noted that it is not the only pathway, and that many of the lethal stresses discussed in this review would likely still be lethal even in the complete absence of BER, just with a different mechanism serving as the terminal cause of death.
Fig. 3.
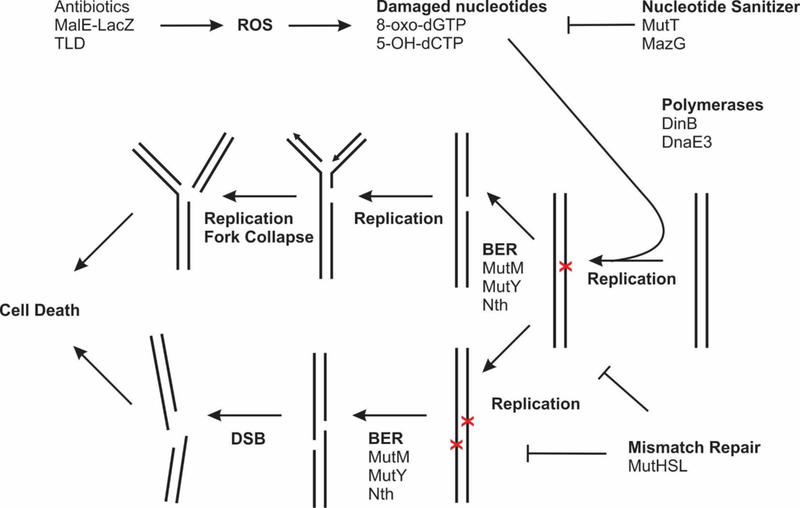
Model of BER-mediated cell death of oxidized nucleotides. Various lethal stresses induce ROS, which damages the nucleotide pool and nucleotide sanitizers such as E. coli MutT and mycobacterial MazG act to hydrolyze them. Damage nucleotides can be incorporated into the genome during replication, particularly through error-prone polymerases DinB and DnaE3 which have a higher affinity for oxidized nucleotides. The mismatch repair system repairs these lesions shortly after replication, protecting the cell. The BER pathway attempts to repair these lesions and generates SSBs as intermediates. If hit by a replication fork, a SSB would cause the fork to collapse and generate a DSB. Alternatively, clusters of SSBs are opposing strands can become a DSB. An unrepaired break is lethal to the cell.
There are still many unanswered questions about this pathway, with the most obvious being what the identity of the lethal DNA problem caused by these repair intermediates actually is. In the most extreme cases such as exposure to ionizing radiation, it seems perfectly plausible that the accumulation of adjacent SSBs could easily result in a DSB, however, in other instances it seems unlikely enough 8-oxo-dG would be incorporated for this to be the primary lethal mechanism. The contribution of MutM and MutY to hyperinitiation lethality [107] suggests a replication fork encountering a single strand break caused by incomplete BER is the potential “cause of death.” This interpretation is supported by the protective effect of the MutHSL system [13,76]. BER and MMR both require generating breaks to repair their various lesions, however, because it is specifically targeted to recently synthesized hemimethylated DNA, the intermediate steps in MMR are much less likely to be encountered by a replication fork.
Another direction for future research is to investigate what conditions make bacteria susceptible to this pathway. The data from both bacteria and eukaryotes suggest it is the imbalance of BER components that results in toxicity and that multiple factors may be involved. For example, simply increasing the amount of endogenous ROS alone may not produce enough 8-oxo-dG to be lethal under normal conditions [140], but antibiotics also increase its incorporation into the genome through induction of DinB, while MalE-LacZ expression strongly induces the DNA glycosylase MutM. The relative imbalance of the various genes involved in BER should be investigated in more of these lethal stresses. There is also the possibility of a positive feedback loop involved. DNA damage has been reported to cause ROS in both mice and yeast [136,141], and in E. coli TLD alone can trigger ROS production [11]. These complex interactions suggest that global changes in the cell’s physiology will have to be considered to further investigate this mechanism.
The commonality of this BER-mediated death pathway makes it a potential target for drug discovery. With the rise of antibiotic resistance and the relative lack of new drugs being discovered, there is an interest in finding ways to extend the usefulness of our current repertoire of antibiotics and increase their effectiveness, and a BER targeting adjuvant drug is an intriguing possibility. Currently, other bacterial proteins involved in DNA repair are also being investigated as drug targets, such as the single-strand binding protein (Ssb) which is a major coordinator of repair pathways including double strand break repair and uracil BER [68,142]. DNA repair pathways are also currently being investigated as potential targets for adjuvants in chemotherapy [143] with APE itself being a promising target [144]. Much work will need to be done to better characterize this common pathway of bacterial cell death and hopefully use it to better treat bacterial infections.
Acknowledgments
This work was supported by NIH Grant R01CA021615 and R35ES028303. G.C.W. is an American Cancer Society Professor.
Abbreviations:
- ROS
reactive oxygen species
- RNS
reactive nitrogen species
- BER
base excision repair
- 8-oxo-G
7,8-dihydro-8-oxoguanine
- MMR
mismatch repair
- DSB
double strand break
- SSB
single strand break
- AP
apurinic/apyrimidinic
- TLD
thymineless death
- MMS
methyl methanesulfonate
Footnotes
Conflict of interest
The authors declare no conflict of interest.
References
- [1].Friedberg EC, Walker GC, Siede W, Wood RD, Schultz RA, Ellenberger T, DNA Repair and Mutagenesis, Second Edition, American Society of Microbiology, 2006. [Google Scholar]
- [2].Krokan HE, Bjoras M, Base excision repair, Cold Spring Harbor perspectives in biology, 5 (2013) a012583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Sobol RW, Kartalou M, Almeida KH, Joyce DF, Engelward BP, Horton JK, Prasad R, Samson LD, Wilson SH, Base excision repair intermediates induce p53-independent cytotoxic and genotoxic responses, The Journal of biological chemistry, 278 (2003) 39951–39959. [DOI] [PubMed] [Google Scholar]
- [4].Fu D, Calvo JA, Samson LD, Balancing repair and tolerance of DNA damage caused by alkylating agents, Nature reviews. Cancer, 12 (2012) 104–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Kohanski MA, Dwyer DJ, Hayete B, Lawrence CA, Collins JJ, A common mechanism of cellular death induced by bactericidal antibiotics, Cell, 130 (2007) 797–810. [DOI] [PubMed] [Google Scholar]
- [6].Dwyer DJ, Belenky PA, Yang JH, MacDonald IC, Martell JD, Takahashi N, Chan CT, Lobritz MA, Braff D, Schwarz EG, Ye JD, Pati M, Vercruysse M, Ralifo PS, Allison KR, Khalil AS, Ting AY, Walker GC, Collins JJ, Antibiotics induce redox-related physiological alterations as part of their lethality, Proceedings of the National Academy of Sciences of the United States of America, 111 (2014) E2100–2109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Giroux X, Su WL, Bredeche MF, Matic I, Maladaptive DNA repair is the ultimate contributor to the death of trimethoprim-treated cells under aerobic and anaerobic conditions, Proceedings of the National Academy of Sciences of the United States of America, 114 (2017) 11512–11517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Dong TG, Dong S, Catalano C, Moore R, Liang X, Mekalanos JJ, Generation of reactive oxygen species by lethal attacks from competing microbes, Proceedings of the National Academy of Sciences of the United States of America, 112 (2015) 2181–2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Choi H, Yang Z, Weisshaar JC, Single-cell, real-time detection of oxidative stress induced in Escherichia coli by the antimicrobial peptide CM15, Proceedings of the National Academy of Sciences of the United States of America, 112 (2015) E303–310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Takahashi N, Gruber CC, Yang JH, Liu X, Braff D, Yashaswini CN, Bhubhanil S, Furuta Y, Andreescu S, Collins JJ, Walker GC, Lethality of MalE-LacZ hybrid protein shares mechanistic attributes with oxidative component of antibiotic lethality, Proceedings of the National Academy of Sciences of the United States of America, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Hong Y, Li L, Luan G, Drlica K, Zhao X, Contribution of reactive oxygen species to thymineless death in Escherichia coli, Nature microbiology, 2 (2017) 1667–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Belenky P, Ye JD, Porter CB, Cohen NR, Lobritz MA, Ferrante T, Jain S, Korry BJ, Schwarz EG, Walker GC, Collins JJ, Bactericidal Antibiotics Induce Toxic Metabolic Perturbations that Lead to Cellular Damage, Cell reports, 13 (2015) 968–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Dwyer DJ, Collins JJ, Walker GC, Unraveling the physiological complexities of antibiotic lethality, Annual review of pharmacology and toxicology, 55 (2015) 313–332. [DOI] [PubMed] [Google Scholar]
- [14].Foti JJ, Devadoss B, Winkler JA, Collins JJ, Walker GC, Oxidation of the guanine nucleotide pool underlies cell death by bactericidal antibiotics, Science, 336 (2012) 315–319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Fan XY, Tang BK, Xu YY, Han AX, Shi KX, Wu YK, Ye Y, Wei ML, Niu C, Wong KW, Zhao GP, Lyu LD, Oxidation of dCTP contributes to antibiotic lethality in stationary-phase mycobacteria, Proceedings of the National Academy of Sciences of the United States of America, 115 (2018) 2210–2215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Matic I, The major contribution of the DNA damage-triggered reactive oxygen species production to cell death: implications for antimicrobial and cancer therapy, Current genetics, (2017). [DOI] [PubMed] [Google Scholar]
- [17].Ter Kuile BH, Hoeksema M, Antibiotic Killing through Incomplete DNA Repair, Trends in microbiology, (2017). [DOI] [PubMed] [Google Scholar]
- [18].Li X, Imlay JA, Improved measurements of scant hydrogen peroxide enable experiments that define its threshold of toxicity for Escherichia coli, Free radical biology & medicine, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Imlay JA, The molecular mechanisms and physiological consequences of oxidative stress: lessons from a model bacterium, Nature reviews. Microbiology, 11 (2013) 443–454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Neeley WL, Essigmann JM, Mechanisms of formation, genotoxicity, and mutation of guanine oxidation products, Chemical research in toxicology, 19 (2006) 491–505. [DOI] [PubMed] [Google Scholar]
- [21].Winterbourn CC, The biological chemistry of hydrogen peroxide, Methods in enzymology, 528 (2013) 3–25. [DOI] [PubMed] [Google Scholar]
- [22].Rush JD, Maskos Z, Koppenol WH, Reactions of iron(II) nucleotide complexes with hydrogen peroxide, FEBS Letters, 261 (1990) 121–123. [Google Scholar]
- [23].Richter Y, Fischer B, Characterization and elucidation of coordination requirements of adenine nucleotides complexes with Fe(II) ions, Nucleosides, nucleotides & nucleic acids, 22 (2003) 1757–1780. [DOI] [PubMed] [Google Scholar]
- [24].Fowler RG, Schaaper RM, The role of the mutT gene of Escherichia coli in maintaining replication fidelity, FEMS microbiology reviews, 21 (1997) 43–54. [DOI] [PubMed] [Google Scholar]
- [25].Valavanidis A, Vlachogianni T, Fiotakis C, 8-hydroxy-2’ -deoxyguanosine (8-OHdG): A critical biomarker of oxidative stress and carcinogenesis, Journal of environmental science and health. Part C, Environmental carcinogenesis & ecotoxicology reviews, 27 (2009) 120–139. [DOI] [PubMed] [Google Scholar]
- [26].Alhama J, Ruiz-Laguna J, Rodriguez-Ariza A, Toribio F, Lopez-Barea J, Pueyo C, Formation of 8-oxoguanine in cellular DNA of Escherichia coli strains defective in different antioxidant defences, Mutagenesis, 13 (1998) 589–594. [DOI] [PubMed] [Google Scholar]
- [27].Gedik CM, Collins A, Escodd, Establishing the background level of base oxidation in human lymphocyte DNA: results of an interlaboratory validation study, FASEB journal : official publication of the Federation of American Societies for Experimental Biology, 19 (2005) 82–84. [DOI] [PubMed] [Google Scholar]
- [28].Lu AL, Li X, Gu Y, Wright PM, Chang DY, Repair of oxidative DNA damage: mechanisms and functions, Cell biochemistry and biophysics, 35 (2001) 141–170. [DOI] [PubMed] [Google Scholar]
- [29].Akiyama M, Horiuchi T, Sekiguchi M, Molecular cloning and nucleotide sequence of the mutT mutator of Escherichia coli that causes A:T to C:G transversion, Molecular & general genetics : MGG, 206 (1987) 9–16. [DOI] [PubMed] [Google Scholar]
- [30].Maki H, Sekiguchi M, MutT protein specifically hydrolyses a potent mutagenic substrate for DNA synthesis, Nature, 355 (1992) 273–275. [DOI] [PubMed] [Google Scholar]
- [31].Taddei F, Hayakawa H, Bouton M, Cirinesi A, Matic I, Sekiguchi M, Radman M, Counteraction by MutT protein of transcriptional errors caused by oxidative damage, Science, 278 (1997) 128–130. [DOI] [PubMed] [Google Scholar]
- [32].Boiteux S, Gajewski E, Laval J, Dizdaroglu M, Substrate specificity of the Escherichia coli Fpg protein (formamidopyrimidine-DNA glycosylase): excision of purine lesions in DNA produced by ionizing radiation or photosensitization, Biochemistry, 31 (1992) 106–110. [DOI] [PubMed] [Google Scholar]
- [33].O’Connor TR, Laval J, Physical association of the 2,6-diamino-4-hydroxy-5N-formamidopyrimidine-DNA glycosylase of Escherichia coli and an activity nicking DNA at apurinic/apyrimidinic sites, Proceedings of the National Academy of Sciences of the United States of America, 86 (1989) 5222–5226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Tchou J, Kasai H, Shibutani S, Chung MH, Laval J, Grollman AP, Nishimura S, 8-oxoguanine (8-hydroxyguanine) DNA glycosylase and its substrate specificity, Proceedings of the National Academy of Sciences of the United States of America, 88 (1991) 4690–4694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Sugahara M, Mikawa T, Kumasaka T, Yamamoto M, Kato R, Fukuyama K, Inoue Y, Kuramitsu S, Crystal structure of a repair enzyme of oxidatively damaged DNA, MutM (Fpg), from an extreme thermophile, Thermus thermophilus HB8, The EMBO journal, 19 (2000) 3857–3869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Fromme JC, Verdine GL, Structural insights into lesion recognition and repair by the bacterial 8-oxoguanine DNA glycosylase MutM, Nature structural biology, 9 (2002) 544–552. [DOI] [PubMed] [Google Scholar]
- [37].Galhardo RS, Almeida CE, Leitao AC, Cabral-Neto JB, Repair of DNA lesions induced by hydrogen peroxide in the presence of iron chelators in Escherichia coli: participation of endonuclease IV and Fpg, Journal of bacteriology, 182 (2000) 1964–1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Au KG, Clark S, Miller JH, Modrich P, Escherichia coli mutY gene encodes an adenine glycosylase active on G-A mispairs, Proceedings of the National Academy of Sciences of the United States of America, 86 (1989) 8877–8881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Williams SD, David SS, Evidence that MutY is a monofunctional glycosylase capable of forming a covalent Schiff base intermediate with substrate DNA, Nucleic acids research, 26 (1998) 5123–5133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Sandigursky M, Franklin WA, DNA deoxyribophosphodiesterase of Escherichia coli is associated with exonuclease I, Nucleic acids research, 20 (1992) 4699–4703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Dianov G, Sedgwick B, Daly G, Olsson M, Lovett S, Lindahl T, Release of 5’-terminal deoxyribose-phosphate residues from incised abasic sites in DNA by the Escherichia coli RecJ protein, Nucleic acids research, 22 (1994) 993–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Graves RJ, Felzenszwalb I, Laval J, O’Connor TR, Excision of 5’-terminal deoxyribose phosphate from damaged DNA is catalyzed by the Fpg protein of Escherichia coli, The Journal of biological chemistry, 267 (1992) 14429–14435. [PubMed] [Google Scholar]
- [43].Piersen CE, McCullough AK, Lloyd RS, AP lyases and dRPases: commonality of mechanism, Mutation research, 459 (2000) 43–53. [DOI] [PubMed] [Google Scholar]
- [44].Michaels ML, Tchou J, Grollman AP, Miller JH, A repair system for 8-oxo-7,8-dihydrodeoxyguanine, Biochemistry, 31 (1992) 10964–10968. [DOI] [PubMed] [Google Scholar]
- [45].Wyrzykowski J, Volkert MR, The Escherichia coli methyl-directed mismatch repair system repairs base pairs containing oxidative lesions, Journal of bacteriology, 185 (2003) 1701–1704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Boye E, Marinus MG, Lobner-Olesen A, Quantitation of Dam methyltransferase in Escherichia coli, Journal of bacteriology, 174 (1992) 1682–1685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Szyf M, Avraham-Haetzni K, Reifman A, Shlomai J, Kaplan F, Oppenheim A, Razin A, DNA methylation pattern is determined by the intracellular level of the methylase, Proceedings of the National Academy of Sciences of the United States of America, 81 (1984) 3278–3282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Schofield MJ, Nayak S, Scott TH, Du C, Hsieh P, Interaction of Escherichia coli MutS and MutL at a DNA mismatch, The Journal of biological chemistry, 276 (2001) 28291–28299. [DOI] [PubMed] [Google Scholar]
- [49].Su SS, Modrich P, Escherichia coli mutS-encoded protein binds to mismatched DNA base pairs, Proceedings of the National Academy of Sciences of the United States of America, 83 (1986) 5057–5061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Au KG, Welsh K, Modrich P, Initiation of methyl-directed mismatch repair, The Journal of biological chemistry, 267 (1992) 12142–12148. [PubMed] [Google Scholar]
- [51].Marinus MG, DNA Mismatch Repair, EcoSal Plus, 5 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Cooper DL, Lahue RS, Modrich P, Methyl-directed mismatch repair is bidirectional, The Journal of biological chemistry, 268 (1993) 11823–11829. [PubMed] [Google Scholar]
- [53].Grilley M, Griffith J, Modrich P, Bidirectional excision in methyl-directed mismatch repair, The Journal of biological chemistry, 268 (1993) 11830–11837. [PubMed] [Google Scholar]
- [54].Heinze RJ, Giron-Monzon L, Solovyova A, Elliot SL, Geisler S, Cupples CG, Connolly BA, Friedhoff P, Physical and functional interactions between Escherichia coli MutL and the Vsr repair endonuclease, Nucleic acids research, 37 (2009) 4453–4463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Schmidt MG, Dolan KM, Oliver DB, Regulation of Escherichia coli secA mRNA translation by a secretion-responsive element, Journal of bacteriology, 173 (1991) 6605–6611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Nonaka G, Blankschien M, Herman C, Gross CA, Rhodius VA, Regulon and promoter analysis of the E. coli heat-shock factor, sigma32, reveals a multifaceted cellular response to heat stress, Genes & development, 20 (2006) 1776–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Yoon SH, Lee HS, Choi JY, Kang HK, Lee JJ, Hyun JW, Choi J, Ye SK, Chung MH, MutY is down-regulated by oxidative stress in E. coli, Free radical research, 37 (2003) 873–879. [DOI] [PubMed] [Google Scholar]
- [58].Gutierrez A, Laureti L, Crussard S, Abida H, Rodriguez-Rojas A, Blazquez J, Baharoglu Z, Mazel D, Darfeuille F, Vogel J, Matic I, beta-Lactam antibiotics promote bacterial mutagenesis via an RpoS-mediated reduction in replication fidelity, Nature communications, 4 (2013) 1610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Boiteux S, Guillet M, Abasic sites in DNA: repair and biological consequences in Saccharomyces cerevisiae, DNA repair, 3 (2004) 1–12. [DOI] [PubMed] [Google Scholar]
- [60].Greenberg MM, Abasic and oxidized abasic site reactivity in DNA: enzyme inhibition, cross-linking, and nucleosome catalyzed reactions, Accounts of chemical research, 47 (2014) 646–655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].D’Souza DI, Harrison L, Repair of clustered uracil DNA damages in Escherichia coli, Nucleic acids research, 31 (2003) 4573–4581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Kuzminov A, Single-strand interruptions in replicating chromosomes cause double-strand breaks, Proceedings of the National Academy of Sciences of the United States of America, 98 (2001) 8241–8246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Kuzminov A, Recombinational repair of DNA damage in Escherichia coli and bacteriophage lambda, Microbiology and molecular biology reviews : MMBR, 63 (1999) 751–813, table of contents. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Bonura T, Town CD, Smith KC, Kaplan HS, The influence of oxygen on the yield of DNA double-strand breaks in x-irradiated Escherichia coli K-12, Radiation research, 63 (1975) 567–577. [PubMed] [Google Scholar]
- [65].Sakai A, Nakanishi M, Yoshiyama K, Maki H, Impact of reactive oxygen species on spontaneous mutagenesis in Escherichia coli, Genes to cells : devoted to molecular & cellular mechanisms, 11 (2006) 767–778. [DOI] [PubMed] [Google Scholar]
- [66].Imlay JA, Chin SM, Linn S, Toxic DNA damage by hydrogen peroxide through the Fenton reaction in vivo and in vitro, Science, 240 (1988) 640–642. [DOI] [PubMed] [Google Scholar]
- [67].Imlay JA, Linn S, Bimodal pattern of killing of DNA-repair-defective or anoxically grown Escherichia coli by hydrogen peroxide, Journal of bacteriology, 166 (1986) 519–527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Voter AF, Killoran MP, Ananiev GE, Wildman SA, Hoffmann FM, Keck JL, A High-Throughput Screening Strategy to Identify Inhibitors of SSB Protein-Protein Interactions in an Academic Screening Facility, SLAS discovery : advancing life sciences R & D, 23 (2018) 94–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Asad NR, de Almeida CE, Asad LM, Felzenszwalb I, Leitao AC, Fpg and UvrA proteins participate in the repair of DNA lesions induced by hydrogen peroxide in low iron level in Escherichia coli, Biochimie, 77 (1995) 262–264. [DOI] [PubMed] [Google Scholar]
- [70].Liou GY, Storz P, Reactive oxygen species in cancer, Free radical research, 44 (2010) 479–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Kim GH, Kim JE, Rhie SJ, Yoon S, The Role of Oxidative Stress in Neurodegenerative Diseases, Experimental neurobiology, 24 (2015) 325–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Roszkowski K, Jozwicki W, Blaszczyk P, Mucha-Malecka A, Siomek A, Oxidative damage DNA: 8-oxoGua and 8-oxodG as molecular markers of cancer, Medical science monitor : international medical journal of experimental and clinical research, 17 (2011) CR329–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Lobritz MA, Belenky P, Porter CB, Gutierrez A, Yang JH, Schwarz EG, Dwyer DJ, Khalil AS, Collins JJ, Antibiotic efficacy is linked to bacterial cellular respiration, Proceedings of the National Academy of Sciences of the United States of America, 112 (2015) 8173–8180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Cho H, Uehara T, Bernhardt TG, Beta-lactam antibiotics induce a lethal malfunctioning of the bacterial cell wall synthesis machinery, Cell, 159 (2014) 1300–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Adolfsen KJ, Brynildsen MP, Futile cycling increases sensitivity toward oxidative stress in Escherichia coli, Metabolic engineering, 29 (2015) 26–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Cohen NR, Ross CA, Jain S, Shapiro RS, Gutierrez A, Belenky P, Li H, Collins JJ, A role for the bacterial GATC methylome in antibiotic stress survival, Nature genetics, 48 (2016) 581–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Dwyer DJ, Camacho DM, Kohanski MA, Callura JM, Collins JJ, Antibiotic-induced bacterial cell death exhibits physiological and biochemical hallmarks of apoptosis, Molecular cell, 46 (2012) 561–572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Kohanski MA, DePristo MA, Collins JJ, Sublethal antibiotic treatment leads to multidrug resistance via radical-induced mutagenesis, Molecular cell, 37 (2010) 311–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Liu Y, Imlay JA, Cell death from antibiotics without the involvement of reactive oxygen species, Science, 339 (2013) 1210–1213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Keren I, Wu Y, Inocencio J, Mulcahy LR, Lewis K, Killing by bactericidal antibiotics does not depend on reactive oxygen species, Science, 339 (2013) 1213–1216. [DOI] [PubMed] [Google Scholar]
- [81].Mosel M, Li L, Drlica K, Zhao X, Superoxide-mediated protection of Escherichia coli from antimicrobials, Antimicrobial agents and chemotherapy, 57 (2013) 5755–5759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Mi H, Wang D, Xue Y, Zhang Z, Niu J, Hong Y, Drlica K, Zhao X, Dimethyl Sulfoxide Protects Escherichia coli from Rapid Antimicrobial-Mediated Killing, Antimicrobial agents and chemotherapy, 60 (2016) 5054–5058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Brickman E, Silhavy TJ, Bassford PJ Jr., Shuman HA, Beckwith JR, Sites within gene lacZ of Escherichia coli for formation of active hybrid beta-galactosidase molecules, Journal of bacteriology, 139 (1979) 13–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Khodursky A, Guzman EC, Hanawalt PC, Thymineless Death Lives On: New Insights into a Classic Phenomenon, Annual review of microbiology, 69 (2015) 247–263. [DOI] [PubMed] [Google Scholar]
- [85].Cohen SS, Barner HD, Studies on Unbalanced Growth in Escherichia Coli, Proceedings of the National Academy of Sciences of the United States of America, 40 (1954) 885–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Fonville NC, Bates D, Hastings PJ, Hanawalt PC, Rosenberg SM, Role of RecA and the SOS response in thymineless death in Escherichia coli, PLoS genetics, 6 (2010) e1000865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Kuong KJ, Kuzminov A, Stalled replication fork repair and misrepair during thymineless death in Escherichia coli, Genes to cells : devoted to molecular & cellular mechanisms, 15 (2010) 619–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Kuong KJ, Kuzminov A, Disintegration of nascent replication bubbles during thymine starvation triggers RecA- and RecBCD-dependent replication origin destruction, The Journal of biological chemistry, 287 (2012) 23958–23970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].McGuire JJ, Anticancer antifolates: current status and future directions, Current pharmaceutical design, 9 (2003) 2593–2613. [DOI] [PubMed] [Google Scholar]
- [90].Stone SR, Morrison JF, Mechanism of inhibition of dihydrofolate reductases from bacterial and vertebrate sources by various classes of folate analogues, Biochimica et biophysica acta, 869 (1986) 275–285. [DOI] [PubMed] [Google Scholar]
- [91].Bushby SR, Hitchings GH, Trimethoprim, a sulphonamide potentiator, British journal of pharmacology and chemotherapy, 33 (1968) 72–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Hitchings GH, Mechanism of action of trimethoprim-sulfamethoxazole. I, The Journal of infectious diseases, 128 (1973) Suppl:433–436 p. [DOI] [PubMed] [Google Scholar]
- [93].Sangurdekar DP, Zhang Z, Khodursky AB, The association of DNA damage response and nucleotide level modulation with the antibacterial mechanism of the anti-folate drug trimethoprim, BMC genomics, 12 (2011) 583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Schlamadinger DE, Wang Y, McCammon JA, Kim JE, Spectroscopic and computational study of melittin, cecropin A, and the hybrid peptide CM15, The journal of physical chemistry. B, 116 (2012) 10600–10608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Ward JF, Biochemistry of DNA lesions, Radiation research. Supplement, 8 (1985) S103–111. [PubMed] [Google Scholar]
- [96].Blaisdell JO, Wallace SS, Abortive base-excision repair of radiation-induced clustered DNA lesions in Escherichia coli, Proceedings of the National Academy of Sciences of the United States of America, 98 (2001) 7426–7430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Yamada M, Nunoshiba T, Shimizu M, Gruz P, Kamiya H, Harashima H, Nohmi T, Involvement of Y-family DNA polymerases in mutagenesis caused by oxidized nucleotides in Escherichia coli, Journal of bacteriology, 188 (2006) 4992–4995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Thi TD, Lopez E, Rodriguez-Rojas A, Rodriguez-Beltran J, Couce A, Guelfo JR, Castaneda-Garcia A, Blazquez J, Effect of recA inactivation on mutagenesis of Escherichia coli exposed to sublethal concentrations of antimicrobials, The Journal of antimicrobial chemotherapy, 66 (2011) 531–538. [DOI] [PubMed] [Google Scholar]
- [99].Simmons LA, Breier AM, Cozzarelli NR, Kaguni JM, Hyperinitiation of DNA replication in Escherichia coli leads to replication fork collapse and inviability, Molecular microbiology, 51 (2004) 349–358. [DOI] [PubMed] [Google Scholar]
- [100].Kato J, Katayama T, Hda, a novel DnaA-related protein, regulates the replication cycle in Escherichia coli, The EMBO journal, 20 (2001) 4253–4262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Leonard AC, Mechali M, DNA replication origins, Cold Spring Harbor perspectives in biology, 5 (2013) a010116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Skarstad K, Katayama T, Regulating DNA replication in bacteria, Cold Spring Harbor perspectives in biology, 5 (2013) a012922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Kurokawa K, Nishida S, Emoto A, Sekimizu K, Katayama T, Replication cycle-coordinated change of the adenine nucleotide-bound forms of DnaA protein in Escherichia coli, The EMBO journal, 18 (1999) 6642–6652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Katayama T, Kubota T, Kurokawa K, Crooke E, Sekimizu K, The initiator function of DnaA protein is negatively regulated by the sliding clamp of the E. coli chromosomal replicase, Cell, 94 (1998) 61–71. [DOI] [PubMed] [Google Scholar]
- [105].Charbon G, Riber L, Cohen M, Skovgaard O, Fujimitsu K, Katayama T, Lobner-Olesen A, Suppressors of DnaA(ATP) imposed overinitiation in Escherichia coli, Molecular microbiology, 79 (2011) 914–928. [DOI] [PubMed] [Google Scholar]
- [106].Riber L, Olsson JA, Jensen RB, Skovgaard O, Dasgupta S, Marinus MG, Lobner-Olesen A, Hda-mediated inactivation of the DnaA protein and dnaA gene autoregulation act in concert to ensure homeostatic maintenance of the Escherichia coli chromosome, Genes & development, 20 (2006) 2121–2134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Charbon G, Bjorn L, Mendoza-Chamizo B, Frimodt-Moller J, Lobner-Olesen A, Oxidative DNA damage is instrumental in hyperreplication stress-induced inviability of Escherichia coli, Nucleic acids research, 42 (2014) 13228–13241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Caulfield JL, Wishnok JS, Tannenbaum SR, Nitric oxide-induced deamination of cytosine and guanine in deoxynucleosides and oligonucleotides, The Journal of biological chemistry, 273 (1998) 12689–12695. [DOI] [PubMed] [Google Scholar]
- [109].Shapiro R, DiFate V, Welcher M, Deamination of cytosine derivatives by bisulfite. Mechanism of the reaction, Journal of the American Chemical Society, 96 (1974) 906–912. [DOI] [PubMed] [Google Scholar]
- [110].Bradshaw JS, Kuzminov A, RdgB acts to avoid chromosome fragmentation in Escherichia coli, Molecular microbiology, 48 (2003) 1711–1725. [DOI] [PubMed] [Google Scholar]
- [111].He B, Qing H, Kow YW, Deoxyxanthosine in DNA is repaired by Escherichia coli endonuclease V, Mutation research, 459 (2000) 109–114. [DOI] [PubMed] [Google Scholar]
- [112].Yao M, Hatahet Z, Melamede RJ, Kow YW, Purification and characterization of a novel deoxyinosine-specific enzyme, deoxyinosine 3’ endonuclease, from Escherichia coli, The Journal of biological chemistry, 269 (1994) 16260–16268. [PubMed] [Google Scholar]
- [113].Shlomai J, Kornberg A, Deoxyuridine triphosphatase of Escherichia coli. Purification, properties, and use as a reagent to reduce uracil incorporation into DNA, The Journal of biological chemistry, 253 (1978) 3305–3312. [PubMed] [Google Scholar]
- [114].Lindahl T, Ljungquist S, Siegert W, Nyberg B, Sperens B, DNA N-glycosidases: properties of uracil-DNA glycosidase from Escherichia coli, The Journal of biological chemistry, 252 (1977) 3286–3294. [PubMed] [Google Scholar]
- [115].Kouzminova EA, Kuzminov A, Chromosomal fragmentation in dUTPase-deficient mutants of Escherichia coli and its recombinational repair, Molecular microbiology, 51 (2004) 1279–1295. [DOI] [PubMed] [Google Scholar]
- [116].Gates FT 3rd, Linn S, Endonuclease V of Escherichia coli, The Journal of biological chemistry, 252 (1977) 1647–1653. [PubMed] [Google Scholar]
- [117].Lu LD, Sun Q, Fan XY, Zhong Y, Yao YF, Zhao GP, Mycobacterial MazG is a novel NTP pyrophosphohydrolase involved in oxidative stress response, The Journal of biological chemistry, 285 (2010) 28076–28085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Lyu LD, Tang BK, Fan XY, Ma H, Zhao GP, Mycobacterial MazG safeguards genetic stability via housecleaning of 5-OH-dCTP, PLoS pathogens, 9 (2013) e1003814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].D.W. Kufe, Holland, James F., Frei, Emil (eds), Cancer Medicine, 6th ed., BC Decker, Hamilton, Canada, 2003.
- [120].Weisburger EK, Carcinogenicity of alkylating agents, Public health reports, 81 (1966) 772–776. [PMC free article] [PubMed] [Google Scholar]
- [121].Prasad R, Shock DD, Beard WA, Wilson SH, Substrate channeling in mammalian base excision repair pathways: passing the baton, The Journal of biological chemistry, 285 (2010) 40479–40488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Engelward BP, Weeda G, Wyatt MD, Broekhof JL, de Wit J, Donker I, Allan JM, Gold B, Hoeijmakers JH, Samson LD, Base excision repair deficient mice lacking the Aag alkyladenine DNA glycosylase, Proceedings of the National Academy of Sciences of the United States of America, 94 (1997) 13087–13092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Elder RH, Jansen JG, Weeks RJ, Willington MA, Deans B, Watson AJ, Mynett KJ, Bailey JA, Cooper DP, Rafferty JA, Heeran MC, Wijnhoven SW, van Zeeland AA, Margison GP, Alkylpurine-DNA-N-glycosylase knockout mice show increased susceptibility to induction of mutations by methyl methanesulfonate, Molecular and cellular biology, 18 (1998) 5828–5837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Yuge K, Nambu H, Senzaki H, Nakao I, Miki H, Uyama M, Tsubura A, N-methyl-N-nitrosourea-induced photoreceptor apoptosis in the mouse retina, In vivo, 10 (1996) 483–488. [PubMed] [Google Scholar]
- [125].Nakajima M, Yuge K, Senzaki H, Shikata N, Miki H, Uyama M, Tsubura A, Photoreceptor apoptosis induced by a single systemic administration of N-methyl-N-nitrosourea in the rat retina, The American journal of pathology, 148 (1996) 631–641. [PMC free article] [PubMed] [Google Scholar]
- [126].Ogino H, Ito M, Matsumoto K, Yagyu S, Tsuda H, Hirono I, Wild CP, Montesano R, Retinal degeneration induced by N-methyl-N-nitrosourea and detection of 7-methyldeoxyguanosine in the rat retina, Toxicologic pathology, 21 (1993) 21–25. [DOI] [PubMed] [Google Scholar]
- [127].Meira LB, Moroski-Erkul CA, Green SL, Calvo JA, Bronson RT, Shah D, Samson LD, Aag-initiated base excision repair drives alkylation-induced retinal degeneration in mice, Proceedings of the National Academy of Sciences of the United States of America, 106 (2009) 888–893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Roth RB, Samson LD, 3-Methyladenine DNA glycosylase-deficient Aag null mice display unexpected bone marrow alkylation resistance, Cancer research, 62 (2002) 656–660. [PubMed] [Google Scholar]
- [129].Cardinal JW, Margison GP, Mynett KJ, Yates AP, Cameron DP, Elder RH, Increased susceptibility to streptozotocin-induced beta-cell apoptosis and delayed autoimmune diabetes in alkylpurine-DNA-N-glycosylase-deficient mice, Molecular and cellular biology, 21 (2001) 5605–5613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Burns N, Gold B, The effect of 3-methyladenine DNA glycosylase-mediated DNA repair on the induction of toxicity and diabetes by the beta-cell toxicant streptozotocin, Toxicological sciences : an official journal of the Society of Toxicology, 95 (2007) 391–400. [DOI] [PubMed] [Google Scholar]
- [131].Engelward BP, Dreslin A, Christensen J, Huszar D, Kurahara C, Samson L, Repair-deficient 3-methyladenine DNA glycosylase homozygous mutant mouse cells have increased sensitivity to alkylation-induced chromosome damage and cell killing, The EMBO journal, 15 (1996) 945–952. [PMC free article] [PubMed] [Google Scholar]
- [132].Paik J, Duncan T, Lindahl T, Sedgwick B, Sensitization of Human Carcinoma Cells to Alkylating Agents by Small Interfering RNA Suppression of 3-Alkyladenine-DNA Glycosylase, Cancer research, 65 (2005) 10472–10477. [DOI] [PubMed] [Google Scholar]
- [133].Meira LB, Bugni JM, Green SL, Lee CW, Pang B, Borenshtein D, Rickman BH, Rogers AB, Moroski-Erkul CA, McFaline JL, Schauer DB, Dedon PC, Fox JG, Samson LD, DNA damage induced by chronic inflammation contributes to colon carcinogenesis in mice, The Journal of clinical investigation, 118 (2008) 2516–2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Wirtz S, Nagel G, Eshkind L, Neurath MF, Samson LD, Kaina B, Both base excision repair and O6-methylguanine-DNA methyltransferase protect against methylation-induced colon carcinogenesis, Carcinogenesis, 31 (2010) 2111–2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Murakami Y, Ikeda Y, Yoshida N, Notomi S, Hisatomi T, Oka S, De Luca G, Yonemitsu Y, Bignami M, Nakabeppu Y, Ishibashi T, MutT homolog-1 attenuates oxidative DNA damage and delays photoreceptor cell death in inherited retinal degeneration, The American journal of pathology, 181 (2012) 1378–1386. [DOI] [PubMed] [Google Scholar]
- [136].Nakatake S, Murakami Y, Ikeda Y, Morioka N, Tachibana T, Fujiwara K, Yoshida N, Notomi S, Hisatomi T, Yoshida S, Ishibashi T, Nakabeppu Y, Sonoda KH, MUTYH promotes oxidative microglial activation and inherited retinal degeneration, JCI insight, 1 (2016) e87781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [137].Sobol RW, Horton JK, Kuhn R, Gu H, Singhal RK, Prasad R, Rajewsky K, Wilson SH, Requirement of mammalian DNA polymerase-beta in base-excision repair, Nature, 379 (1996) 183–186. [DOI] [PubMed] [Google Scholar]
- [138].Deans AJ, West SC, DNA interstrand crosslink repair and cancer, Nature reviews. Cancer, 11 (2011) 467–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Marullo R, Werner E, Degtyareva N, Moore B, Altavilla G, Ramalingam SS, Doetsch PW, Cisplatin induces a mitochondrial-ROS response that contributes to cytotoxicity depending on mitochondrial redox status and bioenergetic functions, PloS one, 8 (2013) e81162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Imlay JA, Diagnosing oxidative stress in bacteria: not as easy as you might think, Current opinion in microbiology, 24 (2015) 124–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Rowe LA, Degtyareva N, Doetsch PW, DNA damage-induced reactive oxygen species (ROS) stress response in Saccharomyces cerevisiae, Free radical biology & medicine, 45 (2008) 1167–1177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [142].Handa P, Acharya N, Varshney U, Chimeras between single-stranded DNA-binding proteins from Escherichia coli and Mycobacterium tuberculosis reveal that their C-terminal domains interact with uracil DNA glycosylases, The Journal of biological chemistry, 276 (2001) 16992–16997. [DOI] [PubMed] [Google Scholar]
- [143].Yamanaka K, Chatterjee N, Hemann MT, Walker GC, Inhibition of mutagenic translesion synthesis: A possible strategy for improving chemotherapy?, PLoS genetics, 13 (2017) e1006842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Shah F, Logsdon D, Messmann RA, Fehrenbacher JC, Fishel ML, Kelley MR, Exploiting the Ref-1-APE1 node in cancer signaling and other diseases: from bench to clinic, NPJ precision oncology, 1 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]