

Abstract
Hepatocellular carcinoma (HCC) is a leading cause of cancer‐related death worldwide. Although HCC can respond to immune checkpoint inhibitors, such as monoclonal antibodies against programmed death 1 (PD‐1), many patients fail to respond or develop secondary resistance. Activation of Wnt/β‐catenin signaling can contribute to immune evasion. Mutations in β‐catenin are among the most frequent mutations associated with HCC. Thus, our aim was to directly target β‐catenin to enhance the therapeutic response to immune checkpoint inhibition. A synthetic transgenic mouse model of experimental HCC induced by tyrosine‐protein kinase Met/β‐catenin expression and extracellular vesicles (EVs) as a therapeutic delivery agent was used to evaluate the efficacy of directly targeting β‐catenin on the response to anti‐PD‐1. These studies showed that (1) oncogenic β‐catenin could be therapeutically targeted using a biological nanoparticle‐based delivery approach, (2) targeting β‐catenin using small interfering RNA (siRNA) delivered within EVs can reduce tumor growth, and (3) the therapeutic response to anti‐PD‐1 can be enhanced by concomitantly targeting β‐catenin using therapeutic EVs. These preclinical studies establish the efficacy of the use of biological nanoparticles as an endogenous delivery vehicle for therapeutic RNA delivery and support the use of therapeutic strategies targeting tumor‐intrinsic β‐catenin as an adjunct to anti‐PD‐1‐based therapy. Conclusion: Combination therapy with anti‐PD‐1 and β‐catenin siRNA delivered using biological nanoparticles provides an effective strategy for the treatment of HCC. This strategy could be further exploited into targeted approaches for immune potentiation by countering oncogene‐mediated resistance to immunotherapies.
Abbreviations
- AFP
alpha‐fetoprotein
- ANOVA
analysis of variance
- BCAT
β‐catenin
- CD
clusters of differentiation
- FDR
false discovery rate
- g‐luc
Gaussia luciferase
- HCC
hepatocellular carcinoma
- HDI
hydrodynamic injection
- H&E
hematoxylin and eosin
- LW/BW
liver weight/body weight
- MET
tyrosine‐protein kinase Met
- MNV
milk‐derived nanovesicle
- mRNA
messenger RNA
- NK
natural killer
- PBS
phosphate‐buffered saline
- PCR
polymerase chain reaction
- PD‐1
programmed death 1
- PD‐L1
programmed death ligand 1
- qRT‐PCR
real‐time quantitative polymerase chain reaction
- RLU
relative luminescence unit
- RNA‐seq
RNA sequencing
- RRID
research resource Identification
- RT
room temperature
- siRNA
small interfering RNA
- tMNV
therapeutic milk‐derived nanovesicle
Hepatocellular carcinoma (HCC) is the most common primary cancer of the liver. Patients with HCC have poor prognosis due in part to the lack of effective therapies for advanced cancers.1, 2 Treatment approaches for HCC and responses are further impacted by the heterogeneity of oncogenic drivers for these cancers. The benefits of targeting the immune system for cancer therapy are being increasingly recognized. Immunotherapy with checkpoint inhibitors targeting anti‐programmed death 1 (anti‐PD‐1), anti‐programmed death ligand 1 (anti‐PD‐L1), and cytotoxic T lymphocyte antigen 4 (CTLA4) has resulted in durable responses and is currently approved for use in several types of aggressive cancers.3, 4, 5 The PD‐1/PD‐L1 interaction enables tumor cells to escape from the attack of cytotoxic T cells.6 Recent studies have reported responses in some patients with HCC treated with nivolumab or tremelimumab.7, 8, 9, 10 Despite the demonstrated clinical activity of anti‐PD‐1/PD‐L1 antibodies in HCC and other cancer types, many patients with advanced cancer do not derive clinical benefit from these drugs. A subset of patients does not show any response, and in some patients who show an initial response, secondary resistance may occur, resulting in relapse.4, 11, 12
Sensitivity to anti‐PD‐1 requires the presence of tumor antigen‐specific T cells within tumor tissue. The absence of T‐cell infiltration contributes to an immune‐desert phenotype and poor response to immunotherapy. Consequently, immune‐suppressive mechanisms within the tumor microenvironment that facilitate T‐cell exclusion may reduce the benefit from immunotherapy.13, 14, 15 Emerging evidence shows that alterations in cancer cell autonomous signaling pathways can contribute to primary and/or secondary resistance to checkpoint inhibition. Oncogenic alterations are now being recognized as a contributor to tumor cell‐dependent stromal responses that can result in immune deserts characterized by the absence of T cells. The Wnt/β‐catenin pathway, in particular, has been identified as an important oncogenic contributor to immune evasion.16 Mutations in β‐catenin are among the most frequently observed alterations associated with HCC.17 Thus, we sought to evaluate the role of targeting Wnt/β‐catenin as a strategy to enhance the response to anti‐PD‐1 therapy in HCC.
Our approach involved the use of a biological nanoparticle‐mediated delivery system based on the use of extracellular vesicles (EVs) for intrahepatic delivery of small interfering RNA (siRNA) to directly target β‐catenin. Specifically, we demonstrated the efficacy of therapeutic EVs using a synthetic transgenic model in which HCC formation is driven by activated β‐catenin signaling. We identified that systemic administration of β‐catenin siRNA using EVs could enhance the effect of anti‐PD‐1 therapy. These effects were associated with an increase in T cells within the tumor microenvironment. These findings demonstrate the successful use of therapeutic EVs and further provide the rationale for the use of immunotherapy in combination with strategies to inhibit Wnt/β‐catenin signaling for additional benefits that can enhance treatment response and improve outcomes.
Materials and Methods
Animal Studies
All studies involving animals were performed in accordance with protocols approved by the Mayo Clinic Institutional Animal Care and Use Committee. All animals received humane care as outlined in the National Institutes of Health Guide for the Care and Use of Laboratory Animals. We obtained 5‐ to 6‐week‐old male and female Friend virus B (FVB) 21 mice from Jackson Laboratory (Bar Harbor, ME). A synthetic transgenic model of hepatocellular cancer was generated by co‐expression of c‐tyrosine‐protein kinase Met (cMET) and mutant ΔN90‐β‐catenin in mouse livers using a sleeping beauty (SB) transposon/transposase system.18, 19, 20 All mice underwent hydrodynamic injection (HDI) of DNA and plasmids to induce hepatocarcinogenesis as we have described.21 Plasmids were amplified using One Shot TOP10 chemically competent Escherichia coli cells (Thermo Fisher Scientific, Waltham, MA) and further purified using PureYield Plasmid Maxiprep System (Promega, Madison, WI). Before injection, endotoxin was removed using a MiraCLEAN endotoxin removal kit (Mirus Bio, Madison, WI) and plasmid DNAs were concentrated in 2 μg/μL. Briefly, 22.5 μg pT3‐EF1a–c‐MET (human), 22.5 μg pT3‐EF1a–ΔN90‐β‐catenin (human), 5 μg pT3–Gaussia luciferase (g‐luc), along with 5 μg SB transposase were diluted in 2 mL TransIT‐EE Hydrodynamic Delivery Solution (Mirus Bio) and injected hydrodynamically into the lateral tail veins.20 This mouse model recapitulates morphological, biochemical, and gene expression changes that occur in clinical HCC associated with concomitant c‐MET activation and β‐catenin mutations that are reported to occur in 9%‐12.5% of patients.
For treatment with anti‐PD‐1 alone, mice were randomized into two experimental groups that did not differ in sex, body weight, or serum luciferase expression before treatment. Three weeks after HDI, mice bearing HCC were treated with anti‐PD‐1 (clusters of differentiation [CD]279; Bio X Cell, West Lebanon, NH; Research Resource Identification [RRID], AB_0949053,) 250 μg/mouse (n = 12) or phosphate‐buffered saline (PBS) as control (n = 8) administered intraperitoneally 3 times per week for 2 weeks. For treatment with therapeutic EVs and anti‐PD‐1, mice were randomized into four experimental groups that were matched for sex, body weight, or serum luciferase expression before treatment. Treatment was administered for a 2‐week period starting at 3 weeks after HDI. Mice were maintained on standard chow and followed until their death. All mice were euthanized 12 weeks after HDI.
Assessment of Tumor Burden
The cotransfection of g‐luc enables us to use serum g‐luc expression level to monitor tumor burden and evaluate the efficacy of therapeutic interventions.22 Blood was collected by tail vein venipuncture every 3 weeks. Serum was collected from blood kept at room temperature (RT) for 30 minutes after centrifugation at 1,200g for 10 minutes. Five microliters of serum was plated on a 96‐well, flat‐bottom, white, polystyrene assay plate, and serum luciferase levels were determined using the BioLux Gaussia Luciferase Assay Kit (New England BioLabs, Ipswich, MA) after the controlled injection of 50 μL of substrate mixture into plates containing the serum. Luminescence measurements were acquired using the FLUOstar Omega microplate reader (BMG Labtech, Cary, NC) and expressed as relative luminescence units (RLUs). Tumor growth rate was determined as change in luciferase over a defined time interval. Serum alpha‐fetoprotein (AFP) was quantitated with enzyme immunoassays using commercial kits (R&D Systems, Minneapolis, MN; Immunodiagnostic Systems, United Kingdom). Postmortem liver weights correlate with tumor burden in this model.
Histology and Immunohistochemistry
Tissue staining for hematoxylin and eosin (H&E) and immunohistochemistry were performed by the Mayo Clinic Florida Cancer Biology Histology Shared Resource. Briefly, slides were deparaffinized and hydrated with distilled water, and antigen retrieval was performed by soaking slides in ethylene diamine tetraacetic acid (EDTA) in a 100°C steamer for 25 minutes. A protein block was prepared using Protein Block Serum‐Free (Dako, Carpinteria, CA), and immunohistochemistry was performed using a primary antibody against anti‐β‐catenin E5 using a 1:200 dilution (Santa Cruz Biotechnology, Minneapolis, MN; RRID, AB_626807), polyclonal antibody CD8 rabbit using a 1:75 dilution (Cell Signaling, Danvers, MA; RRID, AB_2572861), or Ki67 at a 1:100 dilution (Thermo Fisher Scientific; RRID, AB_805388) and EnVision Plus System‐Horseradish Peroxidase‐Labeled Polymer Anti‐Rabbit (Dako; RRID, AB_2630375), or anti‐mouse secondary antibodies (Dako) along with 3,3‐diaminobenzidine (Dako).
Preparation of Therapeutic EVs
Milk‐derived nanovesicles (MNVs) were isolated following a procedure that we have described in detail.23 Fat‐free milk was diluted with an equal volume of PBS and centrifuged at 12,000g for 30 minutes at 4°C. The supernatant was collected and clarified using Ca2+‐free EDTA. The pH of the solution was adjusted to 4.2, and the solution was kept at 4°C for 10‐30 minutes followed by centrifugation at 3,000g for 30 minutes at 4°C to precipitate milk proteins. The supernatant was collected and filtered through a 0.45‐μM polyethersulfone vacuum filter to remove any residual cell debris. The pH of the solution was then adjusted to 7.0 with 3.0 M sodium hydroxide, filtered through a 0.22‐μM polyethersulfone vacuum filter, and then transferred into 25 × 89 mm polycarbonate tubes (Beckman Coulter Inc., Indianapolis, IN) and centrifuged at 100,000g using an Optima L‐80XP Ultracentrifuge (Beckman Coulter Inc.) and a Type 70 Ti fixed‐angle rotor for 70 minutes at 4°C. The pellet was resuspended in PBS and ultracentrifuged again at 100,000g for 70 minutes at 4°C. The resulting pellet consisting of MNVs was resuspended in sterile PBS. The number and size of MNVs were measured using Nanosight, LM10, NTA 3.2 software (Malvern, United Kingdom). Endotoxin was removed from isolated MNVs using an endotoxin removal kit (Pierce High‐Capacity Endotoxin Removal Resin; Thermo Fisher Scientific). An equal volume of β‐catenin or control siRNA (0.5 μM) (Qiagen, Germantown, MD) and 2.0 μL Lipofectamine 2000 (Invitrogen) was prepared in 25 μL Opti‐MEM (Thermo Fisher Scientific) and mixed and incubated for 10 minutes at RT. Lipofectamine‐siRNA complex solution 50 μL and MNVs (5 × 1012 particles/mL, 50 μL) were mixed in ultraclear centrifuge tubes (11 × 60 mm; Beckman Coulter, Inc.), gently pipetted, and incubated for 30 minutes at RT. The solution was diluted with 4 mL PBS and ultracentrifuged at 100,000g in a Type 60 Ti swing rotor for 70 minutes at 4°C. The supernatant was removed, and the pellet was resuspended in an appropriate volume of PBS for further studies.
Western Blotting
Protein concentrations were determined using a bicinchoninic acid (BCA) assay protein assay kit (Thermo Fisher Scientific). Samples were separated by NuPAGE Bis‐Tris Mini Gels (Novex; Thermo Fisher Scientific) and transferred to nitrocellulose membranes. Membranes were blocked with Odyssey blocking buffer (LI‐COR, Lincoln, NE) for 1 hour at RT and then incubated overnight with primary antibodies at 4°C and subsequently with corresponding secondary antibodies for 1 hour at RT. Antibodies and dilutions used were anti‐β‐catenin (1:500; Santa Cruz Biotechnology; RRID, AB_626807), anti‐actin (1:1,000; Santa Cruz Biotechnology; RRID, AB_630836), and anti‐mouse (1:15,000; LI‐COR; RRID, AB_2721181) or anti‐goat secondary antibody (1:15,000; Thermo Fisher Scientific; RRID, AB_2535744). Immunoreactive bands were detected and quantitated using the Odyssey Imaging system (LI‐COR).
Polymerase Chain Reaction
Total RNA was extracted using TRIzol (Life Technologies, Grand Island, NY) and treated with ribonuclease (RNase)‐free deoxyribonuclease (DNase) I (Qiagen, Valencia, CA). RNA concentration was measured using NanoDrop ND‐2000 (Nano‐Drop Technologies, Wilmington, DE). Complementary DNA (cDNA) was transcribed using the iScript cDNA synthesis kit (Bio‐Rad, Hercules, CA) in a 10‐μL reaction volume containing 10 U/μL RNase inhibitor. Real‐time quantitative polymerase chain reaction (qRT‐PCR) was performed using LightCycler 96 (Roche Diagnostics Corporation, Indianapolis, IN) and SYBR green I qPCR Premix (Clontech Laboratories, Inc., Mountain View, CA). The PCR primers used were β‐catenin, forward: 5′‐ATGGCCATGGAACCAGACAG‐3′, reverse: 5′‐TGGTAGTGGCACCAGAATGG‐3′; and cyclin D1, forward: 5′‐GCTGCTCCTGGTGAACAAGC‐3′, reverse: 5′‐CACAGAGGGCAACGAAGGTC‐3′. Droplet digital (dd) PCRs were performed using 10 µL ddPCR 2× Master Mix (Bio‐Rad), 1 μL 20× Primer Mix (5 µM), 5 μL nuclease free water, and 4 μL reverse‐transcribed product per reaction using a QX200 Droplet Digital PCR System (Bio‐Rad). The following PCR primers were used: retinoic acid early transcript 1 (Raet‐1), forward: 5′‐CAGGTGACCCAGGGAAGATG‐3′, reverse: 5′‐CTCAACTCCTGGCACAAATCG‐3′. Data were analyzed using QuantaSoft software with an automatic threshold setting defined by no template control.
RNA Sequencing
Livers were frozen and stored in RNA‐later. Total RNA was extracted using TRIzol (Life Technologies). Samples were processed at Mayo Clinic Medical Genome Facility Sequencing Core. Illumina Tru‐Seq RNA‐sequencing (RNA‐seq) libraries were prepared, and samples were indexed using the NEBNext Small RNA Library prep kit for Illumina (San Diego, CA). Libraries were amplified 12 to 18 cycles. Amplified libraries were size selected using the Pippin prep (Sage Bioscience) on 2% agarose gels with a size range from 135 to 200 base pairs (bp). Quality control analysis for size and concentration of purified libraries was performed using an Agilent 2100 bioanalyzer high‐sensitivity DNA chip. Libraries were pooled in equal molar amounts for multiplex sequencing on an Illumina HiSeq 2000 sequencer for paired ends.
Bioinformatics Analysis
Raw sequencing reads (75 bp paired end) were quality checked for potential sequencing issues and contaminants using FastQC. Sequence reads were clipped from the invariant portions of both 5′ and 3′ adapters to remove the four randomized nucleotides in the adapters. Sequencing analysis was performed using MAP‐RSeq version 2.1.1. workflow developed at the Mayo Clinic Bioinformatics Core.24 RNA‐seq results were included if ≥100,000 reads mapped to the transcriptome and the number of reads mapping to annotated transcripts represented at least 50% of the number of reads mapping to the murine genome. Basically, reads were aligned and mapped against the mm10 reference genome using TopHat2, and the gene and exon counts were generated by FeatureCounts based on definitions from Ensembl GRCm38.79. All samples passed quality control (QC) according to RSeqQC and additional checks,25 and genes with lower that 2 counts per million were removed. An edgeR standard workflow (version 3.20.1)26 was used to determine differential expression between responders and nonresponders. The gene signature associated with response was identified from differentially expressed genes using a method adopted from Lee et al.,27 starting with a z‐scaled gene expression matrix and calculating F statistics for each gene across all samples. The genes were then sorted based on their F statistics, and the top ranked gene was included in the gene list, which was then iteratively expanded. At each iteration, the next highest ranked gene was added to the list and F statistics were recalculated based on the mean expression of the list genes. The iteration was continued until there was no further improvement in the gene set’s F statistics, at which point a final signature gene list was obtained. Gene ontological analysis was performed using Genevestigator.28 Gene enrichment analysis was performed using the Protein Analysis Through Evolutionary Relationships database (PANTHER) version 13.1.7, 29
Statistical Analysis
All analyses were performed using GraphPad Prism 7 (GraphPad Software, Inc., San Diego, CA). Data are presented as mean ± SEM, with the number of analyses for each mean indicated. The minimum sample size for animal studies (n = 6 per group) was determined to provide 90% power with alpha 0.05 based on an anticipated 20% reduction in mean tumor burden with treatment. The number of animals used was greater than the minimum in all groups to account for a variable success rate in HDI and tumor growth kinetics to ensure that tumor burden was similar across all groups at the start of treatment. The Student t test was used to compare the statistical difference between two groups, and one‐way analysis of variance (ANOVA) was used to compare three or more groups. P < 0.05 was considered significant. Correlation data were analyzed by Pearson's correlation coefficient. Survival data were analyzed by Kaplan‐Meier analysis and compared by log‐rank test with Bonferroni correlation.
Results
Anti‐PD‐1 Decreases Tumor Progression and Increases Overall Survival in Experimental Hepatocellular Cancer
We sought to evaluate the role of targeting β‐catenin as a strategy to enhance response to anti‐PD‐1 therapy in HCC. For these studies, we used a mouse model of experimental HCC that is driven by co‐expression of c‐MET and activated β‐catenin. These synthetic transgenic mice develop multifocal HCC, leading to death within 12 to 15 weeks.30 The lack of a therapeutic effect of c‐MET inhibitors in this model indicates that tumor growth is driven by β‐catenin.31 First, we evaluated the therapeutic effect of anti‐PD‐1. Beginning 3 weeks after HDI, tumor‐bearing mice were treated with anti‐PD‐1 (250 μg/mouse) or PBS for 2 weeks. Anti‐PD‐1 reduced the rate of tumor growth, with a significant reduction in the rate of change in g‐luc expression over a 6‐week period after the start of treatment compared with that observed in the control group (P = 0.017) (Fig. 1A). Furthermore, survival was extended in mice receiving anti‐PD‐1, with a median survival of 119 days compared with 96 days in controls (P = 0.0096) (Fig. 1B). Gross appearances of the liver (Fig. 1C) demonstrated a reduction in the number of visible nodules with anti‐PD‐1. A response was further confirmed by the reduced hepatic expression of Ki67 with anti‐PD‐1 treatment (Fig. 1D). These results demonstrating a reduction in tumor growth in experimental hepatocarcinogenesis provide a rationale for the use of anti‐PD‐1 in the treatment of HCC associated with aberrant β‐catenin activation in humans.
Figure 1.
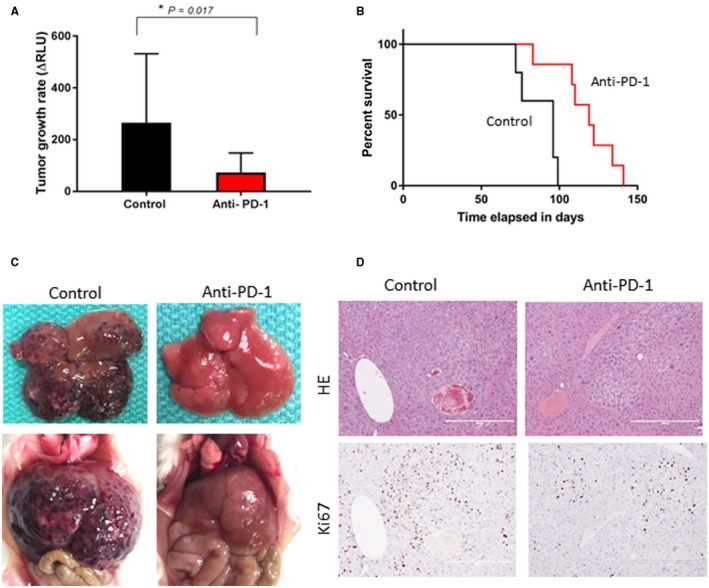
Efficacy of anti‐PD‐1 therapy. HCC was induced by HDI of MET/β‐catenin. Starting at 3 weeks after HDI, mice were treated with anti‐PD‐1 or control diluent for 2 weeks. (A) Tumor growth rate on treatment was determined as a change in g‐luc expression between 3 and 9 weeks after HDI (*P = 0.017). Data are presented as mean ± SEM from eight control and 12 anti‐PD‐1‐treated animals. (B) Survival curves of control or anti‐PD‐1‐treated groups estimated by Kaplan‐Meier analysis (P < 0.01 between groups by log‐rank test). (C) Gross appearance of livers ex vivo and in situ. (D) Representative liver sections showing H&E and immunohistochemistry for Ki67 (original magnification ×10). Abbreviation: HE, hematoxylin and eosin.
In Vitro Efficacy of β‐Catenin Sirna Delivery using EVs
Although activating mutations and accumulation of β‐catenin are frequently observed in liver tumors, controlling the production of β‐catenin has been challenging. For these studies, we developed a therapeutic approach involving hepatic delivery of siRNA using EVs as a biological nanoparticle delivery vehicle. We have established scalable procedures for production of EVs and have demonstrated the safety profile of these MNVs in vitro.3 Proof‐of‐principle studies have demonstrated the ability to generate therapeutic EVs (tMNVs) by loading these nanovesicles with siRNA and for these tMNVs to deliver their cargo into liver cells in vitro and in vivo.32 Thus, tMNVs provide an attractive option for the hepatic delivery of RNA‐based therapeutics to target β‐catenin. To evaluate their utility for cellular delivery of functionally effective β‐catenin‐targeting constructs, we generated tMNVs loaded with β‐catenin siRNA. HepG2 cells were incubated with tMNVs, and the functional end‐target effect was assessed after 48 hours by performing immunoblot analysis for β‐catenin. Delivery of β‐catenin siRNA by means of EVs decreased β‐catenin protein expression in both a tMNV concentration‐dependent manner as well as an siRNA concentration‐dependent manner (Fig. 2). β‐catenin expression was unchanged with siRNA alone, but a reduction was observed when Lipofectamine was included. To verify these results, we analyzed messenger RNA (mRNA) expression of β‐catenin and a downstream target gene (cyclin D1) using qRT‐PCR and observed a consistent and significant decrease in their mRNA expression after exposure to tMNVs (Fig. 2C,D). These results indicate that siRNA delivered within tMNVs retains its functional effectiveness following uptake by HCC cells.
Figure 2.
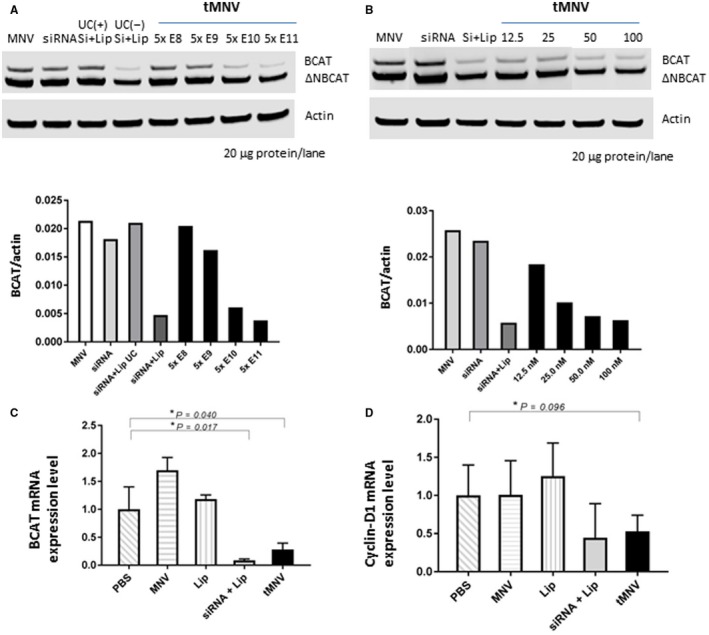
Therapeutic nanovesicles can deliver siRNA to β‐catenin into tumor cells in vitro. MNVs were isolated from fat‐free milk, and tMNVs were prepared by loading them with siRNA to β‐catenin using Lipofectamine. HepG2 cells were incubated with tMNVs. Cells were lysed and immunoblot performed after 48 hours for expression of wild‐type or ΔN90 β‐catenin and actin. (A,B) A representative blot (above) and average normalized expression (relative to actin; below) for wild‐type or ΔN90 β‐catenin from four separate studies are shown. (A) HepG2 cells were incubated with MNVs alone, β‐catenin‐siRNA at a final concentration of 50 nM, β‐catenin‐siRNA plus Lip alone (UC–) or followed by ultracentrifugation (UC+), or with the indicated number of tMNV particles. (B) HepG2 cells were incubated with MNVs alone (5 × 1011 particles), β‐catenin‐siRNA 100 nM alone or with Lip, or tMNVs loaded with the indicated concentration of β‐catenin‐siRNA. (C,D) HepG2 cells were incubated with PBS, MNVs, Lip, or β‐catenin‐siRNA plus Lip alone or with 0.50 μg/μL tMNVs. After 48 hours, cells were harvested, RNA isolated, and qRT‐PCR performed for (C) β‐catenin mRNA or (D) cyclin D1 mRNA expression. Data represent the mean ± SEM of 2‐ΔΔCt normalized expression from four separate studies for each. *P = <0.05. Abbreviations: Lip, Lipofectamine; ΔNBCAT, ΔN90 β‐catenin; UC+, ultracentrifugation; UC–, without ultracentrifugation.
Targeting β‐Catenin Enhances the Efficacy of Anti‐PD‐1
We next evaluated the therapeutic effect of targeting β‐catenin using siRNA in vivo on the response to anti‐PD‐1. As before, a 2‐week course of treatment was administered, starting 3 weeks after HDI (Fig. 3A). Mice were randomized to receive 250 μg anti‐PD‐1 injected intraperitoneally 3 times per week for 2 weeks (n = 11), tMNVs loaded with siRNA to β‐catenin (2 × 1012 particles/body) injected intravenously once every 3 days (n = 10) for five doses, or both anti‐PD‐1 and tMNVs (n = 10) for 2 weeks. Responses were compared with a control group (n = 8) that did not receive any treatment. All mice were euthanized after 12 weeks. Tumor burden as assessed by gross liver appearance was reduced in all treated animals compared with controls (Fig. 3B). In representative histologic sections, tumor growth was associated with an increase in Ki67 expression as well as membranous/cytoplasmic expression and strong nuclear accumulation of β‐catenin (Fig. 3C). Consistent with an increase in oncogene‐driven tumor growth, the expression of β‐catenin increased with age (Fig. 4). However, β‐catenin expression was reduced in responders to therapeutic intervention. Notably, in the combination treatment group (anti‐PD‐1/β‐catenin), both β‐catenin expression and the number of Ki67‐positive cells were reduced.
Figure 3.
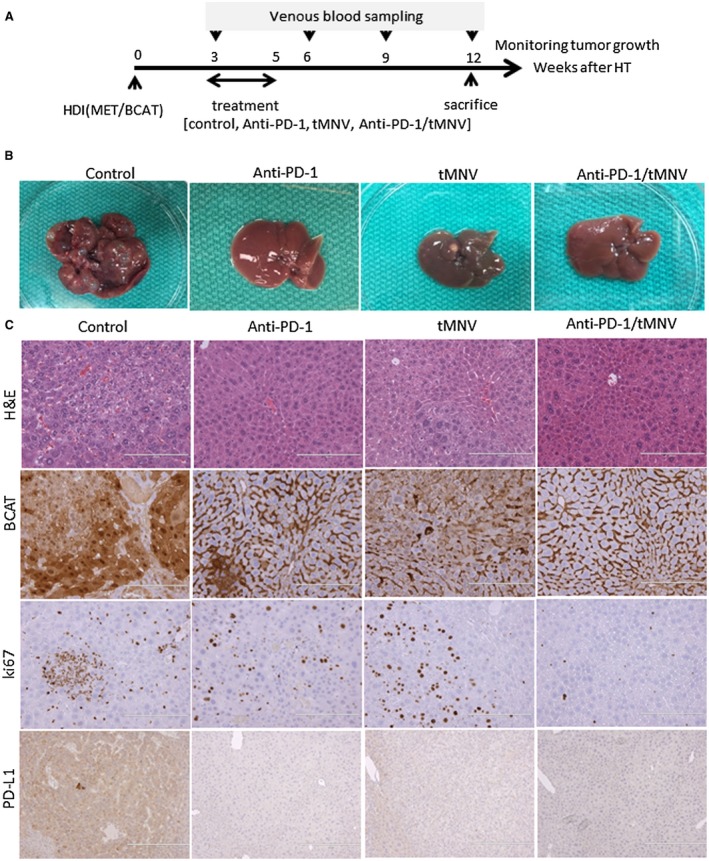
Single and combination use of anti‐PD‐1 and tMNV‐directed therapy targeting β‐catenin for experimental HCC. (A) Experimental schematic. HDI with MET/β‐catenin as performed to induce HCC. Three weeks after HDI, mice were randomized into one of four 2‐week treatment groups (control, anti‐PD‐1, tMNVs [siRNA to β‐catenin], or anti‐PD‐1 with tMNVs [siRNA to β‐catenin]). Blood samples were collected every 3 weeks to monitor tumor growth. All animals were euthanized at 12 weeks. (B,C) Representative livers from each group are shown to illustrate the (B) gross appearance and (C) histologic features of liver within each group with H&E and immunohistochemistry for β‐catenin and Ki67 and PD‐L1 (magnification ×20). Abbreviation: HDI, Hydrodynamic injection.
Figure 4.
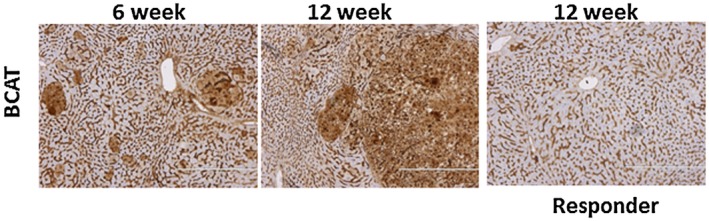
Tissue β‐catenin expression. Hepatic expression of β‐catenin was assessed by immunohistochemistry at 6 and 12 weeks after HDI with MET/β‐catenin. An increase in β‐catenin was observed at later time points but was reduced in the liver in responders to therapeutic intervention with anti‐PD‐1.
The response rate to tMNVs 1 week after end of treatment was 64%, identical to the response rate of anti‐PD‐1. Thus, directly targeting an oncogenic driver can be accomplished with the use of tMNVs. Treatment‐dependent effects were noted in both luciferase expression and AFP levels in some but not all subjects (Fig. 5A,B). The tumor growth rate between 3 and 6 weeks was reduced with anti‐PD‐1, tMNVs, or both (Fig. 5C). However, relapse as determined by an increase in g‐luc expression in prior responders was noted in 38% of the anti‐PD‐1 group and 100% of the tMNV group at 4 weeks after end of treatment. Thus, the effect of directly targeting β‐catenin using siRNA is transient. However, a sustained reduction of growth rate (between 3 and 12 weeks) was noted with the combination of anti‐PD‐1 and tMNVs and was greater than that with either treatment alone (Fig. 5D). Notably, mice that responded to the combination of anti‐PD‐1 and tMNVs had no relapse observed during the duration of follow‐up. Similarly, a reduction in liver weight/body weight (LW/BW) ratio was observed across groups. LW/BW was noted to have a strong correlation with g‐luc (r = 0.961), thereby supporting the use of g‐luc expression to serially monitor tumor burden and hence response to intervention (Fig. 5F). AFP was noted to show poor correlation with LW/BW, but there was a greater correlation noted between AFP and increased Ki67. These observations suggest that the AFP response could be modulated by enhanced hepatic regeneration, thus diminishing its utility as a measure of tumor burden (data not shown).
Figure 5.
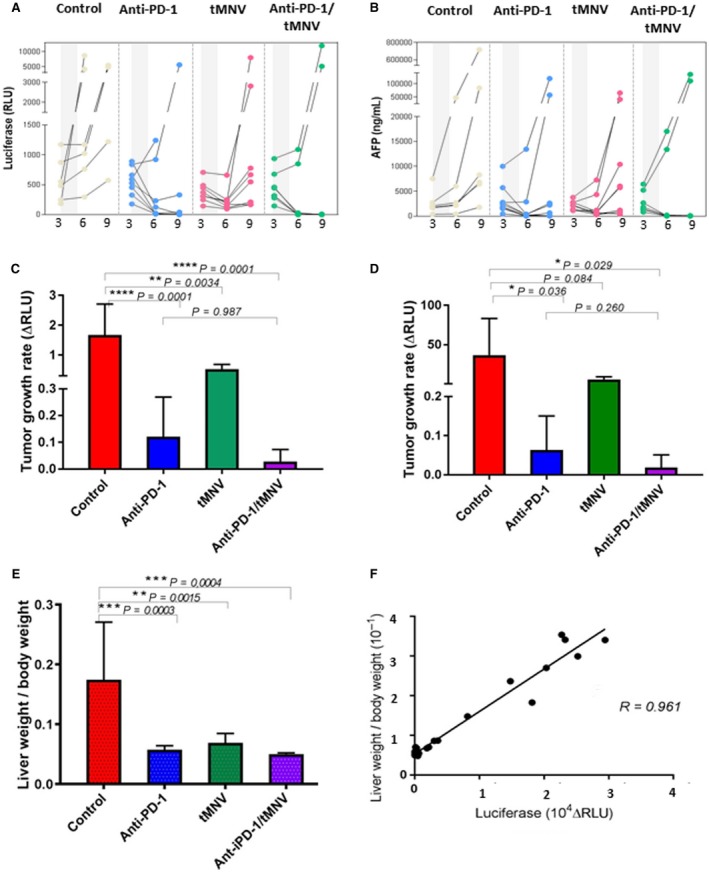
Therapeutic effect of anti‐PD‐1 and tMNV‐directed therapy targeting β‐catenin. HCC was induced by HDI of MET/β‐catenin. (A) Luciferase and (B) AFP expression at the indicated times in weeks for each subject in each of four experimental groups (control, anti‐PD‐1, tMNVs, and anti‐PD‐1/tMNVs). Gray areas depict time of treatment administration. (C) Tumor growth rate on treatment was assessed as the change in g‐luc expression level from 3 to 6 weeks after HDI. P values as indicated, and P < 0.01 for all groups compared with control. (D) Sustained effect on tumor growth was assessed by determining the change in g‐luc expression between start of treatment (3 weeks after HDI) and termination (12 weeks after HDI). P values for comparisons across groups are as indicated. (E) Tumor burden was assessed as LW/BW in each group. P values as indicated and were <0.01 for all groups compared with control. (F) LW/BW is strongly correlated with g‐luc expression (r = 0.961) and indicative of tumor burden. Data are presented as mean value ± SEM. One‐way ANOVA was used for statistical analysis between groups. The correlation data were analyzed by Pearson’s correlation coefficient.
CD8+ Infiltration Within the Tumor Environment
Clinical responses and improved survival to anti‐PD‐1 and other immune checkpoint inhibitors have been associated with a CD8+ T‐cell‐inflamed microenvironment.33, 34, 35 We examined the relationship between CD8+ cell infiltration in the tumor microenvironment and therapeutic response to anti‐PD‐1. The average number of CD8‐positive cells/mm2 was not significantly different among the groups (P = 0.12), indicating that the number of CD8+ cells itself in whole liver is not directly correlated with response or immune pathway suppression (Fig. 6A,B). In contrast, the average number of Ki67 staining cells depended on tumor growth (Fig. 6C). Notably, the ratio of the number of CD8+ cells to the number of Ki67/mm2 staining cells in the anti‐PD‐1/tMNV group was significantly higher than the control group, which is consistent with published literature indicating that distinct CD8+ T‐cell infiltration in the tumor microenvironment is associated with a clinical benefit to anti‐PD‐1. To evaluate for direct effects of MNVs on modulating CD8+ in vivo, CD8+ cell infiltration was assessed in heart, lungs, kidney, liver, and spleen (controls) 24 hours after a single intravenous administration or after every other day administration for 2 weeks of either MNVs or tMNVs (2 × 1012/mouse in 200 μL). Other than in the spleen, CD8+ cells were not detected in any other organ after acute or subacute administration of MNVs.
Figure 6.
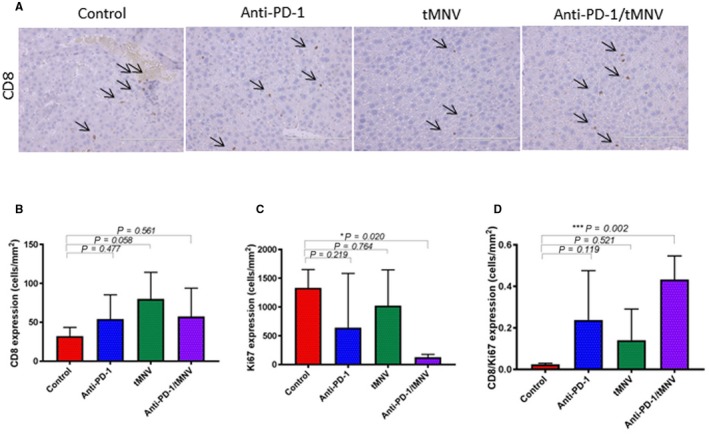
CD8 T‐cell infiltration and proliferation in the tumor microenvironment. (A) CD8 expression in representative liver sections by immunohistochemistry using CD8 rabbit polyclonal antibody (dilution 1:75). Arrows indicate CD8‐positive cells. Original magnification ×20; scale bars, 200 μm. (B) Quantitation of CD8‐positive cells. There is no significant difference in average density between groups (*P > 0.01). (C) Quantitation of Ki67‐positive cells. There is a reduction in Ki67 density in anti‐PD‐1‐ and anti‐PD‐1/MNV‐treated groups compared with controls. (D) Ratio of CD8:Ki67‐positive cells. There is an increase observed with the anti‐PD‐1/MNV group compared with controls. Data represent the mean value ± SEM; one‐way ANOVA was used for analysis.
Determinants of Response
In order to identify tissue or genetic markers predictive of response, we defined two groups (responders and nonresponders) based on g‐luc expression. Response was defined as a g‐luc expression level of <100 RLUs after 12 weeks. Among responders, there was an increase in CD8+ cells associated with Ki67 staining. Conversely, there were few CD8+ cells present among the nonresponder group (Fig. 7A). The ratio of the number of CD8‐positive cells to the number of Ki67 staining cells was significantly higher in responders than in nonresponders (P = 0.0003; Fig. 7B), indicating the utility of intratumoral CD8+ T cells for the prediction of tumor responses to immune checkpoint inhibition. To identify genetic predictors of immune responses, we performed RNA‐seq on total RNA obtained from the livers of two mice, each with or without a response to anti‐PD‐1. A total of 1,430 genes were identified that were >3‐fold differentially expressed between the two groups and with P < 0.05. Based on this, a gene signature of 31 genes was identified. This signature was able to accurately classify all responders and nonresponders. Gene ontology analysis of the genes in this signature identified 11 that were involved in the innate immune response, with P < 0.001 and a false discovery rate (FDR) <0.001 (Fig. 8). In a complementary analysis, we identified biological processes that were overrepresented in the gene set compared with a reference Mus musculus database using PANTHER (version 13.1). Of the 31 genes that mapped onto the database, 30 were compared with a reference group of 22,262 nonoverlapping genes in the database. Fisher’s exact test with FDR multiple test correction was used, and results with FDR <0.05 were selected. Genes involved in immune response were enriched 8.25‐fold, whereas those involved in response to interferon‐gamma were enriched 49.5‐fold (Table 1). Thus, genes involved in innate immune responses were highly enriched in this gene set (Fig. 8A). Among these, the gene with the highest expression was Raet‐1, a ligand for the natural killer group 2, member D (NK2GD) receptor, which is an activating receptor involved in innate and adaptive immunity. Expression of Raet‐1γ/Raet‐1δ showed a strong relationship to response (Fig. 8B). We postulate that enhanced expression of Raet‐1 leading to activation of NKGD2 could enhance antitumor response to anti‐PD‐1 through an activation of NK cells. Further studies to elucidate the contribution of NK cells in modulating the response to anti‐PD‐1 are warranted to evaluate this hypothesis. Genes associated with interferon signaling were also enriched and consistent with data that have associated interferon‐gamma‐related signatures with response to anti‐PD‐1 in clinical studies.
Figure 7.
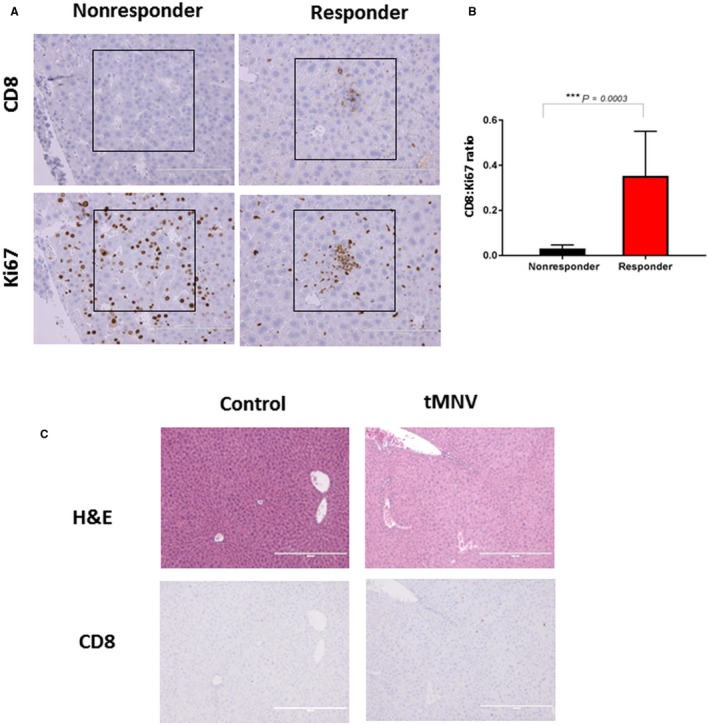
Tissue markers of response. (A) Representative data of CD8‐positive T cells and Ki67 staining cells in adjacent liver sections from responders and nonresponders to anti‐PD‐1. CD8‐positive cells were present in HCC regions with Ki67 positivity in responders but not in nonresponders. (B) Ratio of the number of CD8‐positive cells to the number of Ki67 staining cells in responders (n = 12) and nonresponders (n = 10, including control group); ***P = 0.0003. Data represent the mean value ± SEM. (C) Liver histology 24 hours after control or tMNV injection. Top panels show H&E; bottom panels show immunohistochemical staining with anti‐CD8.
Figure 8.
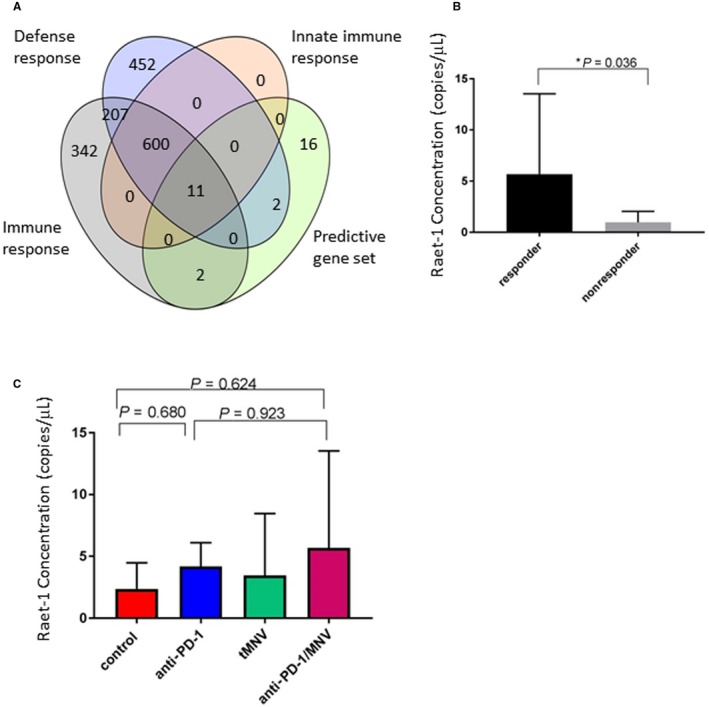
Gene expression signatures predictive of response to anti‐PD‐1. (A) Diagram depicts the relationship of genes within the predictive gene set (green) that are related to a predefined set of genes that are involved in processes involved with immune responses (gray), the innate immune response (peach), or defense responses (blue). Numbers represent the number of individual genes within each set. (B,C) Expression of hepatic Raet‐1 mRNA was assessed by digital PCR in (B) responders and nonresponders or (C) different experimental groups. Higher expression was noted in responders compared with nonresponders. There was no significant overall difference between groups receiving either anti‐PD‐1 or combined anti‐PD‐1/tMNVs.
Table 1.
Gene Expression Analysis on a 30‐Gene Set
| Genes in Reference Set | Genes in Gene Set | Expected | Fold Enrichment | Raw P Value | FDR | |
|---|---|---|---|---|---|---|
| Response to interferon‐gamma | 60 | 4 | 0.08 | 49.47 | 1.60E–06 | 3.91E–04 |
| Immune response | 630 | 7 | 0.85 | 8.25 | 1.73E–05 | 2.11E–03 |
Gene expression analysis on a 30‐gene set. Gene ontology biological processes that are overrepresented in the gene set compared with those in a nonoverlapping data set. Only biological processes with an FDR <0.05 are shown.
Discussion
Using a synthetic transgenic model of experimental HCC induced by enforced expression of MET and mutated β‐catenin, we showed that (1) oncogenic β‐catenin could be therapeutically targeted using a biological EV‐based delivery approach, (2) targeting β‐catenin using siRNA can reduce tumor growth, and (3) the effect of immune checkpoint inhibition using anti‐PD‐1 can be enhanced by concomitantly targeting β‐catenin. These studies provide preclinical evidence that support the use of therapeutic strategies targeting β‐catenin for HCC and as an adjunct to anti‐PD‐1‐based therapy.
In this study, we demonstrated the feasibility of using EVs for therapeutic delivery of siRNA to target β‐catenin. Although knockdown of β‐catenin in HCC can impact tumor cell survival and angiogenesis, β‐catenin is considered to be undruggable. Therapeutic strategies to modulate gene expression with the use of siRNA and microRNA are an attractive option for clinical applications. Their use, however, has been hampered by the instability of RNA molecules following systemic administration and the lack of effective methods for their delivery into their target tissues and cells. EVs can carry RNA and can transfer this RNA between cells to modulate gene expression.36, 37, 38 Compared with synthetic nanoparticles, they may have better biocompatibility and higher delivery efficiency.37, 39 Their use as a therapeutic delivery modality exploits these biological functions and provides an efficient and effective means of hepatic delivery of RNA‐based therapeutics. The need to be able to isolate sufficient amounts of EVs for clinical application has been a major challenge. The use of MNVs circumvents this limitation and represents a major advance in providing a scalable source of EVs, which is further complemented by a favorable short‐term safety profile after intravenous administration.
Oncogenic mutations within tumor cells may contribute to ineffective immune cell infiltration.6, 33, 34, 35 β‐catenin signaling is frequently activated in HCC and regulates diverse processes, such as cell growth and survival.40 β‐catenin signaling has been linked with immune evasion and absence of a T‐cell gene expression signature in metastatic melanoma. Activated β‐catenin can contribute to resistance to anti‐PD‐1 therapy by reducing dendritic cell migration and excluding tumor‐infiltrating lymphocytes.41 Based on these observations, our a priori hypothesis was that inhibition of activated β‐catenin signaling would improve CD8+ T‐cell infiltration and priming and thereby enhance the effect of immune checkpoint inhibition by decreasing immune evasion.13 Although these studies highlight the particular relevance of the interplay of oncogenic signaling and tumor immunity to HCC, the specific determinants of a lack of response or resistance to anti‐PD‐1 are not fully understood. In addition to genetic mutations in tumor cells, potential causes could include individual variabilities in germline genotype or microbiome, comorbidities, or immunosuppressive treatments. Targeting different checkpoints in combination with anti‐PD‐1 have demonstrated synergistic outcomes in a variety of preclinical models in other cancers.42, 43
Our results demonstrate a superior antitumor effect of combining anti‐PD‐1 with β‐catenin siRNA compared with targeting either anti‐PD‐1 or β‐catenin alone. Interestingly, combination treatment was associated with the greatest degree of CD8+ T‐cell infiltration within the tumor microenvironment and was consistent with a synergistic effect to anti‐PD‐1 monotherapy. Primary therapeutic resistance was unavoidable, even in combination therapy (anti‐PD‐1/β‐catenin), although the frequency of primary nonresponse was lower than that observed with anti‐PD‐1 monotherapy. Secondary resistance was not determined in the present study. Based on these observations, inhibition of β‐catenin signaling in HCC may enhance activation of antitumor‐specific T cells, inducing the generation of CD8+ effector T cells and their infiltration into the tumor microenvironment and preventing CD8+ T‐cell exhaustion following an initial response to anti‐PD‐1 therapy. Further research is needed to clarify the molecular mechanism by which β‐catenin can enhance tumoral CD8+ infiltration or those implicated in unresponsiveness to anti‐PD‐1 therapy.
The use of siRNA‐based therapeutics may be an important therapeutic strategy to target β‐catenin given the lack of β‐catenin inhibitors available for clinical translation. An initial response to β‐catenin siRNA as monotherapy was observed, but unlike that observed with the use of anti‐PD‐1 alone, this response was not sustained and was short lived. Future studies will be needed to examine whether response rates or durability of response could be improved using different dosing schedules. Other than in HCC, mutations of β‐catenin are commonly observed in many other types of cancers, such as breast, lung, colorectal, and glioblastoma. Indeed, up to 10% of all cancers may have mutations in the β‐catenin gene. These mutations result in increased β‐catenin protein expression by inhibiting degradation. Thus, the use of siRNA‐based therapeutics may have broader clinical utility beyond their use in HCC that warrants further assessment.
Characterization of cellular and molecular pathways involved in hepatocarcinogenesis and immunogenicity will enhance knowledge of the pathobiology of liver cancers. A search for genes or gene profiles associated with response to immunotherapy, singly or in combination with other strategies, may have potential utility in HCC. The use of gene expression profiling to characterize therapeutic responses to immunotherapy is garnering interest for other solid organ tumors. Computational analyses to characterize the immune cell contexture and identify different subsets of tumor‐associated immune cells will further generate new hypotheses regarding the interactions of immune cells, checkpoint inhibition, and therapy of HCC. Studies to examine whether an increased expression of β‐catenin in tumors could serve as a predictor of response to checkpoint inhibitors could be considered. Ultimately, such information may enable the development of clinically valuable predictors of therapeutic responses and more rational selection of therapies for HCC.
In conclusion, combining immunotherapy that targets the host immune system with an oncogene‐centric targeting approach provides a powerful strategy for the treatment of HCC associated with alterations in β‐catenin. Targeting is facilitated by the use of an EV‐based delivery of a therapeutic RNA. Further studies will be required to determine whether these effects will be efficacious in other settings, such as HCC, that are not driven by β‐catenin mutations; with other cancers; or with the use of other immunotherapies. This combinatorial approach using a biological nanotherapeutic to improve the efficacy of checkpoint inhibitors, such as anti‐PD‐1, provides optimism for countering resistance and improving responses of immune‐targeting therapies.
Acknowledgments
We thank Yuanhang Liu, PhD for assistance with bioinformatics analyses.
Supported by the National Institutes of Health Common Fund Extracellular RNA Communication Program (UH3‐TR000884 to T.P.) and the National Cancer Institute (CA217833 to T.P.).
Potential conflict of interest: Nothing to report.
References
Author names in bold designate shared co‐first authorship.
- 1. Mittal S, El‐Serag HB. Epidemiology of hepatocellular carcinoma: consider the population. J Clin Gastroenterol 2013;47(Suppl):S2‐S6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Wallace MC, Preen D, Jeffrey GP, Adams LA. The evolving epidemiology of hepatocellular carcinoma: a global perspective. Expert Rev Gastroenterol Hepatol 2015;9:765‐779. [DOI] [PubMed] [Google Scholar]
- 3. Maji S, Yan IK, Parasramka M, Mohankumar S, Matsuda A, Patel T. In vitro toxicology studies of extracellular vesicles. J Appl Toxicol 2017;37:310‐318. [DOI] [PubMed] [Google Scholar]
- 4. Spranger S, Gajewski TF. Tumor‐intrinsic oncogene pathways mediating immune avoidance. Oncoimmunology 2015;5:e1086862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sharma P, Allison JP. The future of immune checkpoint therapy. Science 2015;348:56‐61. [DOI] [PubMed] [Google Scholar]
- 6. Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 2012;12:252‐264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mi H, Huang X, Muruganujan A, Tang H, Mills C, Kang D, et al. PANTHER version 11: expanded annotation data from Gene Ontology and Reactome pathways, and data analysis tool enhancements. Nucleic Acids Res 2017;45:D183‐D189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mahoney KM, Freeman GJ, McDermott DF. The next immune‐checkpoint inhibitors: PD‐1/PD‐L1 blockade in melanoma. Clin Ther 2015;37:764‐782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kudo M. Immune checkpoint blockade in hepatocellular carcinoma: 2017 update. Liver Cancer 2016;6:1‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Sangro B, Gomez‐Martin C, de la Mata M, Inarrairaegui M, Garralda E, Barrera P, et al. A clinical trial of CTLA‐4 blockade with tremelimumab in patients with hepatocellular carcinoma and chronic hepatitis C. J Hepatol 2013;59:81‐88. [DOI] [PubMed] [Google Scholar]
- 11. O'Donnell JS, Long GV, Scolyer RA, Teng MW, Smyth MJ. Resistance to PD1/PDL1 checkpoint inhibition. Cancer Treat Rev 2017;52:71‐81. [DOI] [PubMed] [Google Scholar]
- 12. Ramos RN, Piaggio E, Romano E. Mechanisms of resistance to immune checkpoint antibodies. Handb Exp Pharmacol 2018;249:109‐128. [DOI] [PubMed] [Google Scholar]
- 13. Chen DS, Mellman I. Oncology meets immunology: the cancer‐immunity cycle. Immunity 2013;39:1‐10. [DOI] [PubMed] [Google Scholar]
- 14. Ock CY, Keam B, Kim S, Lee JS, Kim M, Kim TM, et al. Pan‐cancer immunogenomic perspective on the tumor microenvironment based on PD‐L1 and CD8 T‐cell infiltration. Clin Cancer Res 2016;22:2261‐2270. [DOI] [PubMed] [Google Scholar]
- 15. Vilain RE, Menzies AM, Wilmott JS, Kakavand H, Madore J, Guminski A, et al. Dynamic changes in PD‐L1 expression and immune infiltrates early during treatment predict response to PD‐1 blockade in melanoma. Clin Cancer Res 2017;23:5024‐5033. [DOI] [PubMed] [Google Scholar]
- 16. Spranger S, Gajewski TF. A new paradigm for tumor immune escape: beta‐catenin‐driven immune exclusion. J Immunother Cancer 2015;3:43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Monga SP. beta‐Catenin signaling and roles in liver homeostasis, injury, and tumorigenesis. Gastroenterology 2015;148:1294‐1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Bell JB, Podetz‐Pedersen KM, Aronovich EL, Belur LR, McIvor RS, Hackett PB. Preferential delivery of the Sleeping Beauty transposon system to livers of mice by hydrodynamic injection. Nat Protoc 2007;2:3153‐3165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Tward AD, Jones KD, Yant S, Cheung ST, Fan ST, Chen X, et al. Distinct pathways of genomic progression to benign and malignant tumors of the liver. Proc Natl Acad Sci U S A 2007;104:14771‐14776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Shang N, Arteaga M, Zaidi A, Stauffer J, Cotler SJ, Zeleznik‐Le NJ, et al. FAK is required for c‐Met/beta‐catenin‐driven hepatocarcinogenesis. Hepatology 2015;61:214‐226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Matsuda A, Ishiguro K, Yan IK, Patel T. Therapeutic efficacy of vitamin D in experimental c‐MET‐beta‐catenin‐driven hepatocellular cancer. Gene Expr 2018; doi: 10.3727/105221618X15355518848281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Subleski JJ, Scarzello AJ, Alvord WG, Jiang Q, Stauffer JK, Kronfli A, et al. Serum‐based tracking of de novo initiated liver cancer progression reveals early immunoregulation and response to therapy. J Hepatol 2015;63:1181‐1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Matsuda A, Patel T. Milk‐derived extracellular vesicles for therapeutic delivery of small interfering RNAs. Methods Mol Biol 2018;1740:187‐197. [DOI] [PubMed] [Google Scholar]
- 24. Kalari KR, Nair AA, Bhavsar JD, O'Brien DR, Davila JI, Bockol MA, et al. MAP‐RSeq: Mayo analysis pipeline for RNA sequencing. BMC Bioinformatics 2014;15:224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Liu Y, Wu P, Zhou J, Johnson‐Pais TL, Lai Z, Chowdhury WH, et al. XBSeq2: a fast and accurate quantification of differential expression and differential polyadenylation. BMC Bioinformatics 2017;18(Suppl. 11):384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Robinson MD, McCarthy DJ, Smyth GK. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010;26:139‐140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lee E, Chuang HY, Kim JW, Ideker T, Lee D. Inferring pathway activity toward precise disease classification. PLoS Comput Biol. 2008;4:e1000217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Hruz T, Laule O, Szabo G, Wessendorp F, Bleuler S, Oertle L, et al. Genevestigator v3: a reference expression database for the meta‐analysis of transcriptomes. Adv Bioinformatics 2008;2008:420747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Mi H, Muruganujan A, Casagrande JT, Thomas PD. Large‐scale gene function analysis with the PANTHER classification system. Nat Protoc 2013;8:1551‐1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Chen X, Calvisi DF. Hydrodynamic transfection for generation of novel mouse models for liver cancer research. Am J Pathol 2014;184:912‐923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Zhan N, Michael AA, Wu K, Zeng G, Bell A, Tao J, et al. The effect of selective c‐MET inhibitor on hepatocellular carcinoma in the MET‐active, beta‐catenin‐mutated mouse model. Gene Expr 2018;18:135‐147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. George J, Yan IK, Patel T. Nanovesicle‐mediated delivery of anticancer agents effectively induced cell death and regressed intrahepatic tumors in athymic mice. Lab Invest 2018;98:895‐910. [DOI] [PubMed] [Google Scholar]
- 33. Spranger S, Spaapen RM, Zha Y, Williams J, Meng Y, Ha TT, et al. Up‐regulation of PD‐L1, IDO, and T(regs) in the melanoma tumor microenvironment is driven by CD8(+) T cells. Sci Transl Med 2013;5:200ra116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Tumeh PC, Harview CL, Yearley JH, Shintaku IP, Taylor EJ, Robert L, et al. PD‐1 blockade induces responses by inhibiting adaptive immune resistance. Nature 2014;515:568‐571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Herbst RS, Soria JC, Kowanetz M, Fine GD, Hamid O, Gordon MS, et al. Predictive correlates of response to the anti‐PD‐L1 antibody MPDL3280A in cancer patients. Nature 2014;515:563‐567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Ratajczak J, Miekus K, Kucia M, Zhang J, Reca R, Dvorak P, et al. Embryonic stem cell‐derived microvesicles reprogram hematopoietic progenitors: evidence for horizontal transfer of mRNA and protein delivery. Leukemia 2006;20:847‐856. [DOI] [PubMed] [Google Scholar]
- 37. Valadi H, Ekstrom K, Bossios A, Sjostrand M, Lee JJ, Lotvall JO. Exosome‐mediated transfer of mRNAs and microRNAs is a novel mechanism of genetic exchange between cells. Nat Cell Biol 2007;9:654‐659. [DOI] [PubMed] [Google Scholar]
- 38. Pegtel DM, Cosmopoulos K, Thorley‐Lawson DA, van Eijndhoven MA, Hopmans ES, Lindenberg JL, et al. Functional delivery of viral miRNAs via exosomes. Proc Natl Acad Sci U S A 2010;107:6328‐6333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Skog J, Wurdinger T, van Rijn S, Meijer DH, Gainche L, Sena‐Esteves M, et al. Glioblastoma microvesicles transport RNA and proteins that promote tumour growth and provide diagnostic biomarkers. Nat Cell Biol 2008;10:1470‐1476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Clevers H, Nusse R. Wnt/beta‐catenin signaling and disease. Cell 2012;149:1192‐1205. [DOI] [PubMed] [Google Scholar]
- 41. Spranger S, Bao R, Gajewski TF. Melanoma‐intrinsic beta‐catenin signalling prevents anti‐tumour immunity. Nature 2015;523:231‐235. [DOI] [PubMed] [Google Scholar]
- 42. Nguyen LT, Ohashi PS. Clinical blockade of PD1 and LAG3–potential mechanisms of action. Nat Rev Immunol 2015;15:45‐56. [DOI] [PubMed] [Google Scholar]
- 43. Chowdhury PS, Chamoto K, Honjo T. Combination therapy strategies for improving PD‐1 blockade efficacy: a new era in cancer immunotherapy. J Intern Med 2018;283:110‐120. [DOI] [PubMed] [Google Scholar]