

Abstract
The gut and the liver are anatomically and physiologically connected, and this “gut–liver axis” exerts various influences on liver pathology. The gut microbiota consists of various microorganisms that normally coexist in the human gut and have a role of maintaining the homeostasis of the host. However, once homeostasis is disturbed, metabolites and components derived from the gut microbiota translocate to the liver and induce pathologic effects in the liver. In this review, we introduce and discuss the mechanisms of liver inflammation, fibrosis, and cancer that are influenced by gut microbial components and metabolites; we include recent advances in molecular‐based therapeutics and novel mechanistic findings associated with the gut–liver axis and gut microbiota.
Abbreviations
- α‐SMA
α‐smooth muscle actin
- ACC
acetyl‐coenzyme A carboxylase
- ASK
apoptosis signal‐regulated kinase
- BA
bile acid
- CCL
chemokine (C‐C motif) ligand
- CCR
chemokine (C‐C motif) receptor
- CD
clusters of differentiation
- COL
collagen
- COX‐2
cyclooxygenase 2
- CX3CR1
chemokine (C‐X3‐C motif) receptor 1
- CXCL
chemokine (C‐X‐C motif) ligand
- CYGB
cytoglobin
- CYP7A1
cholesterol 7a‐hydroxylase
- DAMP
damage‐associated molecular pattern
- DCA
deoxycholic acid
- ECM
extracellular matrix
- FGF
fibroblast growth factor
- FXR
farnesoid X receptor
- HCC
hepatocellular carcinoma
- HE
hepatic encephalopathy
- HFD
high‐fat diet
- HSC
hepatic stellate cell
- IL
interleukin
- JNK
c‐jun N‐terminal kinase
- LPS
lipopolysaccharide
- LSEC
liver sinusoidal endothelial cell
- LTA
lipoteichoic acid
- Ly6C
lymphocyte antigen 6 complex, locus C
- MAMP
microbe‐associated molecular pattern
- MFB
myofibroblast
- MMP
matrix metalloproteinase
- NAFLD
nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- NKT
natural killer T
- NO
nitric oxide
- NTCP
Na+/taurocholate cotransporting polypeptide
- PAMP
pathogen‐associated molecular pattern
- PD‐1
programmed cell death protein 1
- PDGF
platelet‐derived growth factor
- PD‐L1
programmed cell death ligand 1
- PGE2
prostaglandin E2
- PPAR
peroxisome proliferator‐activated receptor
- ROS
reactive oxygen species
- SASP
senescence‐associated secretory phenotype
- TGF
transforming growth factor
- TGR5
Takeda G‐protein‐coupled receptor 5
- TLR
toll‐like receptor
- TMA
trimethylamine
- TMAO
trimethylamine oxide
- TNF
tumor necrosis factor
- TRAIL
tumor necrosis factor–related apoptosis‐inducing ligand
- VDR
vitamin D receptor
The intestinal tract and the liver are anatomically and physiologically connected. This relationship between the two has been called the “gut–liver axis,” and the effects of intestinal metabolites on the liver are considered very important for the onset and progression of liver diseases.1, 2, 3, 4 The gut microbiota, in particular, has recently emerged as an important gut–liver axis‐mediated factor. Attenuation of the gut barrier function by excessive intake of tissue damaging foods, such as alcohol and/or a high‐fat diet (HFD), renders large amounts of gut microbial components (so‐called microbe‐associated molecular patterns [MAMPs]) and bacterial metabolites or even the gut microbiota itself susceptible to transfer to the liver. This can promote serious liver diseases, such as hepatic inflammation, fibrosis, and cancer.3, 4 Therefore, these gut microbial components and metabolites affect not only the intestine where the gut microbes reside but also organs distant from the intestine through their systemic circulation5, 6 (Fig. 1).
Figure 1.
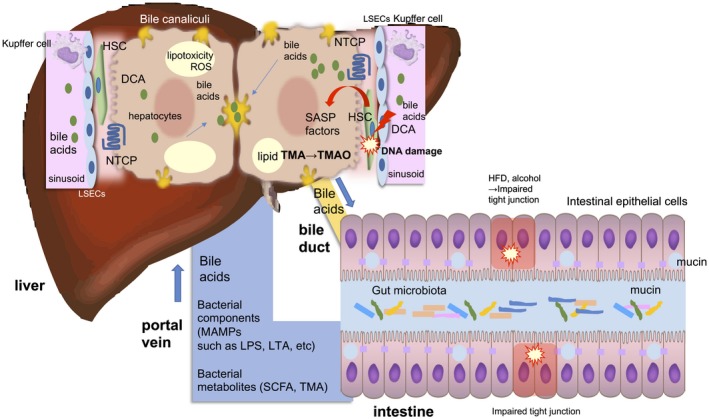
The gut–liver axis. The intestinal tract and the liver are anatomically and physiologically connected. This relationship between the intestine and liver has been called the “gut–liver axis.” Impaired tight junction results in the breakage of the gut barrier function and renders large amounts of MAMPs and bacterial metabolites or even the gut microbiota itself susceptible to transfer to the liver. BAs are actively absorbed by the BA transporter in terminal ileum and enter the colon epithelium through passive diffusion. Secondary BAs, such as DCA, are known to be toxic and elicit DNA damage and thereby producing SASP factors in the HSCs. The gut microbiota is also involved in choline metabolism by converting it into choline metabolites, such as TMA. It is transferred to liver and converted into TMAO, which causes liver inflammation and damage. Abbreviation: SCFA, short‐chain fatty acid.
There are between 500 and 1,000 gut microbes in the human intestine on average, with the total microbial composition being 100 trillion or more. Gut microbiota coexist with the host by metabolizing substances that cannot be metabolized by the host. With the development of analytic technologies, such as next‐generation sequencing and metabolome analysis, it has become possible to classify bacteria according to the DNA sequences of the 16S ribosomal RNA gene and other microbial genes.7, 8 Indeed, gut microbial metabolites have various effects on human physiology and pathology. For example, short‐chain fatty acids, such as butyric and acetic acids (the end products of dietary fiber fermentation by gut microbiota) can suppress inflammation through induction of regulatory T cells by an epigenetic mechanism.9 Moreover, short‐chain fatty acids bind to G protein‐coupled receptors and are involved in controlling obesity.10 On the other hand, lipopolysaccharide (LPS), an outer membrane component of gram‐negative bacteria, and lipoteichoic acid (LTA), a cell wall component of gram‐positive bacteria, interact with toll‐like receptor (TLR) 4 and TLR2, respectively, and induce inflammation by innate immune responses, facilitating liver fibrosis and cancer depending on the physiological context.11, 12, 13 Moreover, bile acids (BAs) modulate metabolic pathways in hepatocytes or intestinal epithelial cells through nuclear receptor transcription factors by acting as their ligands to maintain the homeostasis of the liver.14 However, the excess amount of secondary BAs, such as deoxycholic acid (DCA) and lithocholic acid (LCA) produced by gut microbiota, provokes liver damage and induces stress response signaling, thereby possibly promoting liver cancer.15
In this review, we introduce recent studies from the viewpoint of liver diseases through the gut–liver axis with a special focus on the effects of bacterial cell components and their metabolites not only in hepatocytes but also in stromal cells, such as hepatic stellate cells (HSCs) and Kupffer cells.
HSCs and Their Roles in Fibrosis and Liver Cancer
HSCs are one of the hepatic sinusoid‐constituent cells along with liver sinusoidal endothelial cells (LSECs), Kupffer cells, pit cells, dendritic cells, and natural killer T (NKT) cells and were originally discovered by Karl von Kupffer in 1876.16, 17 They are located in the space of Disse, a space between hepatocyte lineage cells and LSECs. Wake18 illustrated their striking three‐dimensional structure using Golgi staining, showing that HSCs wrap LSECs on one side and contact hepatocytes on the other side with their well‐developed cytoplasmic processes. According to this anatomic disposition, HSCs are liver‐specific “pericytes,” although an evident basement membrane is lacking in the case of hepatic sinusoids.
Physiologically, the principal function of HSCs is their storage of lipid droplets consisting of retinyl esters, triglycerides, cholesterol esters, cholesterol, and phospholipids. Accordingly, HSCs were previously called “lipocytes,” “fat‐storing cells,” or “vitamin A‐storing cells.”19 HSCs also take part in regulating sinusoidal microcirculation through their contractility in response to vasoactive substances, including endothelin‐1, angiotensin II, and nitric oxide (NO),20, 21 and in storing gaseous mediators, such as O2, NO, and carbon monoxide, by expressing cytoglobin (CYGB).22
Following chronic liver injury or culture in vitro, HSCs undergo an “activation” process and transdifferentiate into myofibroblast‐like cells that express α‐smooth muscle actin (α‐SMA) and generate extracellular matrix (ECM) materials, including collagen (COL) I, collagen III, laminin, and fibronectin, contributing to the progression of fibrosis. Activation of HSCs is initiated by their exposure to extracellular stimuli derived from 1) damaged hepatocytes, which produce reactive oxygen species (ROS), lipid peroxides, and damage‐associated molecular patterns (DAMPs); 2) Kupffer cells and profibrotic macrophages, which produce ROS, NO, and cytokines (such as platelet‐derived growth factor [PDGF], transforming growth factor [TGF]‐β1, monocyte chemoattractant protein 1, and chemokines, such as chemokine (C‐C motif) ligand 2 [CCL2] and CCL18); and 3) platelets, which secrete PDGF subunit B, serotonin, 5‐hydroxytryptamine B2, and chemokine (C‐X‐C motif) ligand 4 (CXCL4). Activated HSCs are characterized by high expression of α‐SMA and the PDGF receptor and down‐regulation of peroxisome proliferator‐activated receptor gamma (PPARγ) and LIM homeobox 2 (Lhx2).16, 23, 24Activation of HSCs is perpetuated by proliferation, fibrogenic pathways, growth factors, such as 1) TGF‐β; 2) vascular endothelial growth factor; 3) PDGF; 4) connective tissue growth factor; 5) cytokines and/or adipocytokines (leptin, adiponectin, interleukin [IL]‐15 or IL‐17, IL‐20, IL‐22); 6) innate immune signaling (TLRs, LPS, DAMPs, and pathogen‐associated molecular patterns [PAMPs]); 7) nuclear receptors (liver X receptor, farnesoid X receptor [FXR], vitamin D receptor [VDR], PPARs, Rev‐erb, and nuclear receptor subfamily 4 group A member 1 [NR4A1]); 8) epigenetic factors (microRNAs, DNA methylation, histone modifications); and 9) oxidative/endoplasmic reticulum stress25, 26 (Fig. 2).
Figure 2.
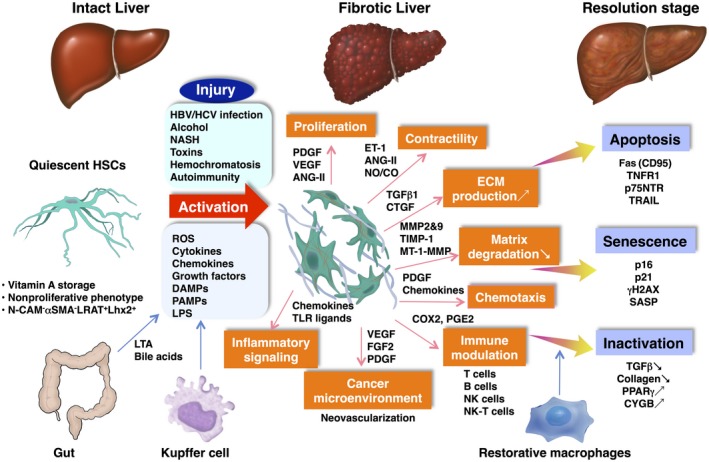
Pathways of HSC activation and their fate in the resolution stage. In intact liver, HSCs localize in the Disse space and contain lipid droplets consisting of mainly vitamin A. They show a nonproliferative phenotype and express quiescent markers, such as LRAT and Lhx2. HSCs take part in the regulation of sinusoidal microcirculation through their contractility. When liver injury occurs by, e.g., HBV/HCV infection, alcohol abuse, or obesity (NASH), HSCs are exposed to oxidative stress signals (reactive oxygen intermediates), DAMPs, PAMPs, LPS, and paracrine stimuli, including cytokines, chemokines, and growth factors (PDGF, VEGF, TGFβ1, FGF2, CTGF, ANG II) secreted from neighboring cells, such as hepatic Kupffer cells/bone marrow‐derived macrophages, sinusoidal endothelial cells, hepatocytes, and platelets, and undergo the process of “activation.” Activated HSCs exhibit a number of specific phenotypes, including proliferation, contractility (mediated by ET‐1, ANG‐II, NO/CO, and ECM production), altered matrix degradation, chemotaxis, immune modulation, inflammatory signaling, and contribution to the cancer microenvironment. In the resolution stage of the underling liver disease, activated HSCs undergo apoptosis through Fas (CD95), TNFR1, p75NTR, and TRAIL; senescence showing p16, p21, γH2AX, and SASP; and inactivation exhibiting low TGFβ and collagen production and high expression of PPARγ and CYGB. Restorative macrophages take part in fibrolysis basically by producing MMP‐9, MMP‐12, and MMP‐13. Abbreviations: ANG‐II, angiotensin II; CTGF, connective tissue growth factor; ET‐1, endothelin 1; γH2AX, gamma‐histone family member X; HBV, hepatitis B virus; HCV, hepatitis C virus; Lhx2, LIM homeobox 2; LRAT, lecithin:retinol acyltransferase; MT‐1‐MMP, membrane‐type matrix metalloproteinase 1; N‐CAM, neural cell adhesion molecule; p75NTR, p75neurotrophin receptor; TIMP‐1, tissue inhibitor of metalloproteinase 1; TNFR1, tumor necrosis factor receptor 1; VEGF, vascular endothelial growth factor.
The survival of activated HSCs depends on the paracrine and autocrine stimuli listed above. In the resolution stage of liver fibrosis, they are able to undergo (1) apoptosis by Fas (clusters of differentiation [CD]95), tumor necrosis factor (TNF) receptor 1, p75(neurotrophin receptor; p75NTR), and tumor necrosis factor–related apoptosis‐inducing ligand (TRAIL) receptors by combining with their ligands27; (2) senescence28; and (3) reversion to “inactivated” HSCs.29 We showed that senescent HSCs were present in the obesity‐associated liver tumor microenvironment.30 Senescent HSCs are α‐SMA positive, but they do not seem to proliferate or produce collagens (Fig. 2). Although reverted HSCs fail to resume the complete phenotype of quiescent HSCs, they exhibit lower expression of fibrosis‐related genes, such as actin alpha 2, smooth muscle (acta2) and transforming growth factor, beta receptor 1 (tgfbr1). Regarding “inactivated” HSCs, we recently observed that human recombinant fibroblast growth factor 2 (FGF2) was able to revert α‐SMA+ COL+ PPARγ– CYGB– ‐activated human HSCs to the α‐SMA– COL– PPARγ+ CYGB+ inactivated state.31 This suggests the occurrence of inactivation in humans as well as rodent HSCs.
HSCs also play important roles in the development of the tumor microenvironment. This microenvironment consists of inflammatory and immunologic reactions as well as neovascularization and fibrosis; this is considered to support the formation of an optimal growth milieu for cancer cells. For example, Nielsen et al.32 described a mechanism involving a tumor–stromal interaction in the metastatic progression of pancreatic ductal adenocarcinoma in the liver; metastasis‐associated macrophage‐derived granulin activated HSCs to produce periostin, which enhanced the deposition of ECM at the site of metastasis. Mogler et al.33 demonstrated that activated HSCs highly expressed the orphan receptor endosialin (CD248), which in turn attenuated the growth of hepatocellular carcinoma (HCC) and down‐regulated cell proliferation‐associated molecules, such as insulin‐like growth factor 2, retroviral binding protein 4, Dickkopf‐1, and CCL5 in HSCs. Duran et al.34 reported that p62, which is increased in hepatocytes but decreased in HSCs in HCC samples, played critical roles in the activation status of HSCs, resulting in the promotion of HCC development by reducing VDR–retinoid X receptor interactions, and in the impaired repression of fibrosis and inflammation by the VDR. We also demonstrated the susceptibility of cygb‐knockout mice to develop liver cancer when treated with diethylnitrosamine or a choline‐deficient L‐amino acid‐defined diet, implicating the regulatory role of CYGB in HSCs for the development of the tumor microenvironment.35, 36 In addition, senescent HSCs showing the senescence‐associated secretory phenotype (SASP) produce a variety of secreted proteins, such as cytokines, chemokines, and proteases,37, 38 that promote liver tumor progression and are present in an obesity‐associated liver tumor microenvironment13, 30 (Fig. 2). Thus, the interaction between HCC and stroma‐associated HSCs has attracted attention in the development of molecular‐targeted therapy for liver cancer.
Liver Macrophages and Their Roles in Inflammation and Fibrosis in the Liver
Liver macrophages consist of Kupffer cells (resident liver macrophages) and monocyte‐derived macrophages and function as scavengers and perform phagocytosis to remove DAMPs, PAMPs, bacteria, and fungi from portal blood flow.39, 40, 41 While LPS are derived from gut microbiota associated with the increased permeability of the intestine and in the liver, PAMPs are derived from damaged hepatocytes. These antimicrobial and phagocytotic functions of liver macrophages are largely impaired in patients with advanced liver disease. Human Kupffer cells are identified by their expression of CD14+, CD68+, TLR4+, and chemokine (C‐X3‐C motif) receptor 1 (CX3CR1)–, while mouse Kupffer cells are identified by their expression of CD11b+/low, F4/80+, CD68+, TLR4+, TLR9+, and CX3CR1–. Monocyte‐derived macrophages are positive for chemokine (C‐C motif) receptor (CCR)2+ (mice and human), CX3CR1+, and lymphocyte antigen 6 complex locus C (LY6C+/–) (mice).42
On inflammation, liver macrophages become activated and initiate the biological response. LPS binds to TLR4 with coreceptors CD14 and lymphocyte antigen 96 (MD‐2), leading to the activation of the myeloid differentiation primary response 88 (MyD88)‐dependent pathway to induce nuclear factor kappa B and p38/c‐jun N‐terminal kinase (JNK) activation. Activated liver macrophages produce PDGF, TGF‐β1, TNF‐α, IL‐1, IL‐6, IL‐10, IL‐18, CXCL1, CXCL2, CXCL8, macrophage chemotactic protein‐1 (also known as CCL2), regulated upon activation, normal T cell expressed, and secreted (RANTES, also known as CCL5), macrophage inflammatory protein (MIP)‐1α (CCL3), MIP‐1β (CCL4), ROS, and prostaglandins, among others, which influence HSC activation and lead to the infiltration of bone‐marrow‐derived monocytes and neutrophils into the damaged liver.43, 44
In vivo macrophage depletion or blockade experiments revealed a major role for macrophages in liver fibrogenesis. Ide et al.45 showed that depletion of Kupffer cells using GdCl3 suppresses α‐SMA‐positive myofibroblasts (MFBs) and ameliorates hepatic fibrosis by thioacetamide administration. A model of conditionally depleted macrophages (CD11b+F4/80+) in CD‐11b‐diphtheria toxin receptor transgenic mice showed marked reduction of ECM deposition and the number of α‐SMA‐positive MFBs under CCl4 intoxication for 12 weeks.46 Karlmark et al.47 demonstrated that Gr1+ (Ly6Chi) inflammatory monocytes are recruited into the liver in a CCR2‐dependent manner during CCl4‐induced chronic liver injury in mice and give rise to inducible NO synthase‐positive CD11b+F4/80+ intrahepatic macrophages that promote the activation of HSCs. Thus, infiltrating Ly6Chi macrophages play central roles in the profibrogenic response compared to mature resident Kupffer cells.
Macrophages are also crucial in the resolution process of liver fibrosis. They are a source of fibrolytic matrix metalloproteinase (MMP)‐9, MMP‐12, and MMP‐13 and also express TRAIL, which promotes apoptosis of activated HSCs/MFBs.48 Ramachandran et al.49 illustrated that the Ly6ClowCD11bhiF4/80int subset of macrophages is the most abundant in the liver in the resolution stage and represents the principle MMP‐expressing subset. Ly6Clow macrophages are derived from a phenotypic transition of the profibrogenic Ly6Chi macrophages.
There have been several reports to translate these functions of macrophages into antifibrotic therapy. Bone marrow‐derived cell therapies, basically with monocytes, together with granulocyte colony‐stimulating factor (G‐CSF) accelerate the resolution stage of liver fibrosis by CCl4 in mice, while administration of G‐CSF with or without CD133‐positive hematopoietic stem cells failed to improve the hepatic function in patients with compensated cirrhosis.50, 51 Ceniciviroc, a dual antagonist for CCR2 and CCR5, is currently being evaluated in a clinical trial phase IIb (CENTAUR study) for its efficacy against nonalcoholic steatohepatitis (NASH) with fibrosis.52, 53, 54 In a proof‐of‐concept study, selonsertib, an inhibitor for apoptosis signal‐regulated kinase 1 (ASK1) (discussed later),55, 56 improved fibrosis in patients with NASH after only 24 weeks of therapy.57 Thus, research on macrophage function will give rise to the development of novel antifibrotic therapy available in human chronic liver disease and cirrhosis.
Gut Microbiota and Liver Cirrhosis (Including Cirrhosis‐Associated HCC)
Chronic hepatitis, caused by continuous damage to the liver, leads to liver cirrhosis and often liver cancer. The translocation of intestinal bacteria and their components is known to be increased in patients with chronic liver diseases because of the associated dysfunction of the intestinal barrier.58 Indeed, LPS levels in the portal vein were reported to increase according to the Child‐Turcotte‐Pugh cirrhosis stage and also in alcoholic liver disease.27, 58 In mouse models, hepatic translocation of LPS has been shown to be important for the development of liver cirrhosis and liver cancer.11, 12 Seki et al.11 reported that continuous signals from TLR4 induced by LPS promoted liver cirrhosis. TLR4‐mediated innate immunity activates the TGF‐β signal in HSCs by inhibiting the expression of the TGF‐β pseudoreceptor Bmp and activin membrane‐bound inhibitor (BAMBI), thereby promoting liver fibrosis (Fig. 3). Moreover, a gut microbiota profiling analysis in patients with liver cirrhosis was recently reported.59 In this report, gram‐negative bacteria, such as Prevotella and Veillonella, were increased in patients with cirrhosis.59, 60 Therefore, it is possible that the mechanism associated with the LPS–TLR4 axis11 is involved in liver cirrhosis. Furthermore, the same group showed that continuous exposure to a low concentration of LPS promoted hepatocarcinogenesis in a CCl4‐induced liver cirrhosis mouse model.12 They found that the LPS–TLR4 pathway promotes HCC by increasing proliferative and anti‐apoptotic signals by hepatocyte growth factor and epiregulin in resident hepatocytes.12 Based on these findings, inflammation by the LPS–TLR4 pathway could be involved in the induction of hepatocarcinogenesis associated with liver cirrhosis (Fig. 1). It was also shown that CD14, a cofactor of the innate immune receptor TLR4, which recognizes LPS as a ligand, is up‐regulated in fatty liver along with activated LPS–TLR4 signaling, also suggesting that long‐term exposure to low concentrations of LPS may cause fatty liver‐associated chronic hepatitis (Fig. 1).61 According to these key roles of the TLR4 pathway in liver fibrogenesis and NASH pathogenesis, TLR4 antagonists, such as TAK‐242 and E5564, have been discussed as therapeutic drugs.2, 62
Figure 3.
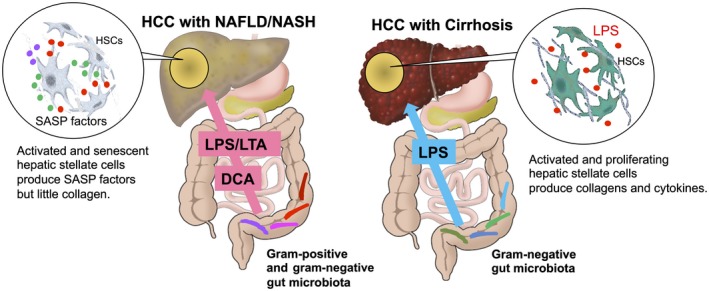
The role of the gut microbiota in liver cancer. In the leaky gut situation, a large amount of gut microbial components and metabolites are transferred to the liver and are likely to promote liver cancer progression. HCCs in the cirrhotic liver were associated with continuous exposure of LPS from gram‐negative bacteria of the gut microbiota. The development of NASH/NAFLD is also associated with LPS exposure. In addition, LTA from gram‐positive bacteria of the gut microbiota is likely to be involved in noncirrhotic NASH‐associated HCC. The HSCs in this type of HCC tumor region undergo cellular senescence and SASP. These HSCs do not appear to be proliferating or producing collagens.
Alcohol is known to be injurious to the intestinal epithelium, and secondary BAs, such as DCA, is known to have membrane destabilizing effects on the intestinal epithelium.63, 64 DCA may influence early stage liver cirrhosis when 7α‐dehydroxylating (DCA producing) bacteria, such as Lachonospiraceae and Ruminococcaceae, are fairly abundant60; these bacteria have been shown to be greatly decreased in the decompensated stage of cirrhosis.60, 65 The excessive intake of an HFD increases blood DCA level,13, 30 and an HFD promotes the destruction of tight junctions of gut epithelial cells.66 These effects promote a leaky gut, thereby rendering gut microbial components and metabolites more likely to be absorbed.67 Clinically, the significant increase in blood DCA level in patients with NASH has also been shown.68
Gut Microbial Metabolites and Hepatic Encephalopathy
Hepatic encephalopathy (HE) is a serious complication of liver cirrhosis associated with neuropsychiatric impairment.69 HE is characterized by a high level of blood ammonia, which is known to be produced abundantly by gut microbiota expressing deaminating enzymes and/or urease operons and also by the host's organs, such as the liver and kidney.70 HE is caused by liver dysfunction, such as liver fibrosis, leading to impaired detoxication of ammonia and creation of portosystemic shunts, thereby facilitating the direct efflux of ammonia into the brain. In the aspect of gut microbiota, gut microbial dysbiosis and small intestinal bacterial overgrowth are often observed in patients with cirrhosis, where the dominant species have been shown to be in the families Streptococcaceae and Veillonellaceae.59, 60, 71, 72 These species correlated significantly with blood ammonia levels and decreased cognitive function. Several reports have shown that patients with cirrhosis using proton‐pump inhibitors (PPIs) are more likely to develop HE,73, 74 presumably because PPIs cause destruction of the stomach acid barrier, leading to gut dysbiosis, which promotes HE. Moreover, by their comparative metagenomic analysis, Qin et al.59, 63 reported that ammonia production and gamma‐aminobutyric acid biosynthesis by gut microbiota were enriched in patients with liver cirrhosis. Other HE‐associated neurotransmitters, such as indoles, glutamine, and serotonin, known to be produced by the gut microbiota, are thought to be associated with HE onset. Recently, to target these HE‐associated gut bacteria, unabsorbed antibiotics, such as rifaximin and norfloxacin, have been used for patients with HE. Rifaximin reportedly changes the gut microbial profile to a gut barrier‐protective profile wherein Lactobacillus is the most abundant genus.75 Moreover, probiotic therapies, such as VSL#3 (a mixture of eight different strains of bacteria including four strains of Lactobacilli, three strains of Bifidobacteria, and one strain of Streptococcus thermophilus), are reportedly effective for patients with HE.76
Gut Microbiota and NASH
Recently, the development of therapies and methods for controlling hepatitis B virus and hepatitis C virus infections has led to a reduction in the mortality rate of viral HCC worldwide.77, 78 On the other hand, the incidence of NASH, the aggressive inflammatory form of nonalcoholic fatty liver disease (NAFLD) without viral etiology, and NASH‐associated cirrhosis and HCC is rapidly increasing.78, 79, 80, 81 Moreover, molecular‐based therapies for NASH have been developed, and these therapies could also help us understand the progression mechanisms of NASH and NASH‐associated HCC. FXR agonists, apoptosis acetyl‐coenzyme A carboxylase (ACC) inhibitors, ASK‐1 inhibitors, and other therapeutic drugs are being developed to treat NASH,82, 83, 84, 85, 86 and the gut–liver axis and gut microbiota are, at least in part, involved in these pathways (Fig. 4).
Figure 4.
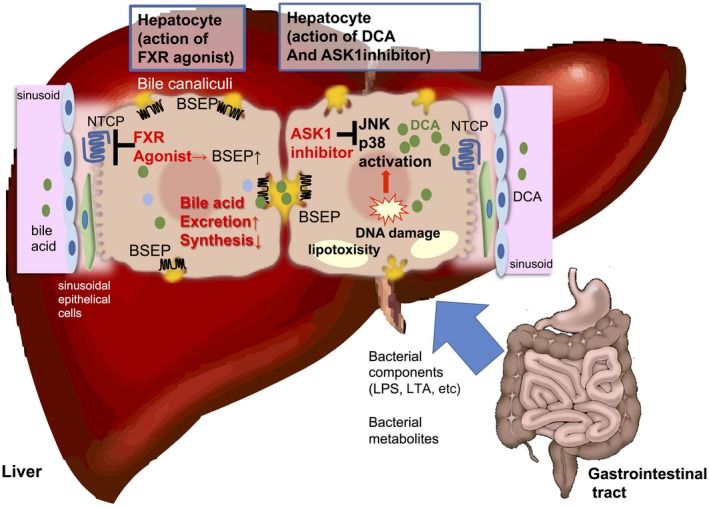
BA‐mediated signaling pathways and related therapeutic drugs for NASH. In the leaky gut situation, a large amount of BAs as well as MAMPs, such as LPS and LTA, transfer to the liver through the portal vein. FXR agonists reduce the expression of CYP7A1, a rate limiting enzyme for BA synthesis, and NTCP, one of the key transporters in hepatocytes that uptake BAs from the sinusoids but up‐regulate the expression of BSEP, the major transporter for the excretion of BAs from hepatocytes into bile canaliculi. FXR activation reduces the excess BA pool in the hepatocyte, thereby preventing cholestasis and/or accumulation of toxic secondary BAs, such as DCA (drawn in the hepatocyte on the left). The secondary BA, DCA, the level of which is known to be increased in individuals with NASH, provokes DNA damage as well as stress response signals, such as JNK and p38‐mediated signaling pathways. Lipotoxicity derived from lipid storage in hepatocytes in NASH also activate these signals. ASK‐1 inhibitors suppress these stress response signals and improve NASH‐associated inflammation (drawn in the hepatocyte on the right). Abbreviation: BSEP, bile salt export pump.
BA and its Mediated Signaling
BAs are mainly synthesized in hepatocytes from cholesterol by cholesterol 7a‐hydroxylase (CYP7A1), a rate‐limiting enzyme of BA biosynthesis. De novo synthesized BAs are called primary BAs. In humans, the primary BAs are mainly cholic acid (CA) and chenodeoxycholic acid (CDCA).87, 88 These BAs are subsequently conjugated with glycine or taurine to render them less toxic in the liver. BAs are transported from the hepatocytes into the bile canaliculi and released into the duodenum. In the intestine, deconjugation and 7‐α‐dehydroxylation of the primary BAs are facilitated by gut microbiota to produce secondary BAs. DCA and LCA, formed from CA and CDCA, respectively, are major secondary BAs in humans. DCA has been reported to activate ß‐catenin signaling to promote cell proliferation89 and to enhance the production of ROS,15 both of which could provoke oncogenic signaling. In the ileum, active uptake of conjugated BAs occurs by the apical sodium‐dependent BA transporter. In the colon, however, unconjugated BAs enter the colon epithelium through passive diffusion. In total, about 95% of the BAs are reabsorbed into intestinal epithelial cells. The remaining 5% is excreted through feces, and this loss is compensated by de novo BA synthesis in the liver. BAs can be recycled 4‐12 times per day between hepatocytes in the liver and enterocytes in the intestine; this is called the enterohepatic circulation.90, 91, 92, 93, 94 BAs can act as ligands for nuclear receptors, such as FXR, and Takeda G‐protein‐coupled receptor 5 (TGR5).87
FXR agonists reduce the expression of CYP7A1 and Na+/taurocholate cotransporting polypeptide (NTCP), one of the key transporters in hepatocytes that uptake BAs from the sinusoids, but up‐regulate the expression of bile salt export pump, the major transporter for the excretion of BAs from hepatocytes into bile canaliculi95, 96 (Fig. 4). FXR activation reduces the excess BA pool in the hepatocyte, thereby preventing cholestasis and/or accumulation of toxic secondary BAs, such as DCA and LTA.97 FXR also stimulates the production of FGF15 (mice) or 19 (human) which, after binding to FGF receptor 4 in liver cells, represses BA synthesis and promotes hepatic glycogen storage and fatty acid oxidation.14 BAs also activate TGR5 in muscle and adipose tissues, increasing thermogenesis and energy expenditure. Moreover, activation of TGR5 in the intestine promotes glucagon‐like peptide 1 release from L cells, promoting insulin release from pancreatic β cells.14 Signaling from another nuclear receptor, constitutive androstane receptor (CAR), was shown to activate ß‐catenin signaling, thereby promoting NASH‐associated liver cancer.98 Furthermore, these nuclear receptors are strongly linked to the circadian rhythms in the liver. Therefore, disorders of circadian rhythm facilitate the progression of NAFLD to fibrotic NASH, presumably through CAR.98
Trimethylamine Oxide‐Mediated Hepatic Steatosis and ACC
There are several reports mentioning that gut microbial‐dependent metabolites from choline are strongly associated with NAFLD/NASH. Choline facilitates lipid transport in hepatocytes and prevents the abnormal accumulation of lipids in the liver, while choline deficiency usually leads to hepatic steatosis.99, 100 The gut microbiota is also involved in choline metabolism by converting it into choline metabolites (dimethylamine, trimethylamine [TMA], dimethylglycine, betaine), which is transferred to liver and converted into trimethylamine oxide (TMAO), which causes liver inflammation and damage101, 102 (Fig. 1). TMA production results in choline deficiency, thereby facilitating hepatic steatosis.101, 102 ACC inhibitors reduce lipogenesis in the liver by inhibiting the production of maronyl coenzyme A, thereby reducing hepatic steatosis,103 and are thus expected to be promising therapeutic drugs for NASH/NAFLD. In mouse models of hepatic steatosis induced by a choline‐deficient diet, ACC up‐regulation is usually observed, suggesting the efficacy of ACC inhibitors in TMAO‐mediated hepatic steatosis.104, 105 Moreover, gut microbial fatty acids, such as 10‐hydroxy‐12(Z)‐octadecenoic acid (18:1) and its derivative Keto A, mainly produced by Lactobacillus species,106, 107 have been reported to reduce ACC expression108 and could therefore be potential probiotic candidates.
Stress Responses and the ASK‐1 Pathway
DCA is known to increase intracellular ROS and is involved in liver damage and induces stress responses.15 Inhibitors of ASK‐1, a serine/threonine kinase, improve NASH pathology by inhibiting stress response pathways, such as phosphorylation of p38 mitogen‐activated kinase and JNK, which leads to hepatic inflammation, apoptosis, and fibrosis57, 109 (Fig. 4).
DCA and NASH‐Associated Liver Cancer
Similar to viral hepatitis‐associated liver cancer, NASH‐associated liver cancer in many cases also results from a state of chronic inflammation, hepatic fibrosis, and cirrhosis. However, there are some cases of liver cancer (approximately 10%‐20%) that develop with little fibrotic background.110, 111, 112, 113 These NASH‐associated liver cancers with less fibrosis could have their own carcinogenic mechanism. Recently, we and others reported that DCA could be associated with noncirrhotic NASH‐associated HCC as described below.
In brief, obesity‐associated liver cancer was promoted by increased DCA in serum.30 We confirmed that almost all mice fed an HFD and treated with 7,12‐dimethylbenzathracene in the neonatal stage developed HCC. The enterohepatic circulation of DCA provoked cellular senescence and an SASP that could create a tumor‐promoting microenvironment,13, 30 which in turn facilitated HCC progression. In this system, the promoted excretion of BAs by ursodeoxycholic acid reduced HCC development.30
Moreover, we found that long‐term HFD feeding altered the gut microbial profile to increase gram‐positive bacteria, such as Clostridium species. The resulting hepatic translocation of LTA, a gram‐positive microbial component, and the enhanced LTA–TLR2 axis facilitated HCC development. LTA enhanced the SASP of HSCs in collaboration with DCA to up‐regulate the expression of not only SASP factors but also cyclooxygenase‐2 (COX‐2) and TLR2 (Fig. 3). Interestingly, COX‐2‐mediated prostaglandin E2 (PGE2) production suppresses antitumor immunity through PGE2 receptor 4 (EP4), thereby contributing to HCC progression. Pretreatment of the EP4 antagonist was shown to be effective to reduce HCC development. COX‐2 overexpression and excessive PGE2 production were detected in HSCs of human noncirrhotic NASH, indicating that a similar mechanism could function in humans.13 A recent report showed that DCA also promotes HCC progression by activating the mammalian target of rapamycin pathway.114 It was found that a new class of a steatohepatitis‐inducing HFD, STHD‐01, could promote HCC without chemical carcinogen administration.114
Moreover, it was reported that short‐chain fatty acids, such as butyrate fermented by gut bacteria from dietary soluble fibers, which are considered broadly health promoting, strongly promoted HCC while improving metabolic dysfunction. An inulin‐enriched HFD intake induced both dysbiosis and HCC in wild‐type mice and was accompanied by cholestasis and increased BA levels, including DCA. Interestingly, administrating soluble fibers, such as inulin, but not insoluble fibers, such as cellulose, induced HCC. This report shows a dark side of fermentation by gut microbiota and a deleterious role of cholestasis for HCC development.115
Gut Microbiota and Antitumor Immunity
In addition to our findings that an enhanced LTA–TLR2 axis up‐regulates PGE2 production and suppresses antitumor immunity,13 Ma et al.116 found that altering the commensal gut bacteria by antibiotics in mice induced a liver‐specific antitumor effect, with an increase of hepatic chemokine (C‐X‐C motif) receptor 6 (CXCR6)‐positive NKT cells and heightened interferon‐γ production following antigen stimulation. The accumulation of NKT cells was regulated by CXCL16 expression of LSECs, which was controlled by gut microbiome‐mediated BAs by the gut–liver axis. Antibiotic administration decreased the amount of secondary BAs; however, many primary BAs were increased. NKT cells were strongly activated, particularly by one of the primary BAs, tauro‐β‐muricholic acid.116
Furthermore, gut microbiota species facilitating the effect of immune checkpoint therapy have recently been identified. Antitumor immunity becomes ineffective as cancer progresses because immune checkpoints are activated by cancer cells. Although the immune checkpoint mechanism is a necessary system for suppressing excessive inflammation, it is disadvantageous in cancer tissues as it allows the progression of cancer. Therefore, in recent years, immune checkpoint inhibitors have been developed as promising molecular targeting agents for the treatment of many types of cancer. For example, antibodies against immunosuppressive molecules, such as programmed cell death protein 1 (PD‐1) and its ligand PD‐L1, and cytotoxic T‐lymphocyte associated protein 4 (CTLA‐4), are used clinically as immune checkpoint inhibitors.
Unfortunately, immune checkpoint inhibitors are not effective in all cancer patients. It has recently been reported that certain species of the gut microbiota enhance antitumor immunity and assist in the activity of immune checkpoint inhibitors.117, 118 In a mouse model, Bifidobacteria activated dendritic cells to enhance the antitumor effect of an anti‐PD‐L1 antibody.117 Another report showed that Bacteroides fragilis increased the effectiveness of an anti‐CTLA‐4 antibody in a colon cancer model.118 More recently, the blockage of the PD‐1/PD‐L1 axis was facilitated by gut microbiota in humans. In patients with malignant melanoma who showed an effect of immune checkpoint inhibitors, members of the Ruminococcaceae family119 and Bifidobacterium longum 120 increased significantly in the gut. Moreover, in epithelial cancers, such as colorectal and non‐small cell lung cancer, Akkermansia species were increased.121 Gut microbiota derived from patients in whom immune checkpoint inhibitors were effective was transplanted into germ‐free mice.121 These mice also showed a significant antitumor effect of immune checkpoint inhibitors. These findings suggest that alterations of the gut microbiota profile can be expected to synergistically increase the therapeutic effect of immune checkpoint inhibitors, which may be applicable to liver cancer, judging from the preclinical and clinical study of immune checkpoint therapy for liver cancer.122 However, the precise molecular mechanism (in which gut‐microbial metabolites are involved) that facilitates antitumor immunity is still unclear.
Concluding Remarks
The important role of gut microbiota in modulating liver diseases, including cancer, has been emerging. Thus, the mechanisms by which liver cancer development are promoted or suppressed through the gut–liver axis have become attractive research topics. Gut microbial components, such as LPS and LTA, are associated with hepatic fibrosis and HCC progression. Furthermore, gut microbial metabolites, including secondary BAs and fatty acids, as well as unknown microbial metabolites and toxins could influence liver pathology. We have recently elucidated the mechanism of HCC progression by DCA and LTA, at least in part.13 DCA causes DNA damage in HSCs, thereby inducing cellular senescence. This is accompanied by the expression of a series of cytokines and COX‐2, creating liver tumor‐promoting microenvironments. Recently, NKT cells were shown to be strongly activated, particularly by the primary BA tauro‐β‐muricholic acid.116 Moreover, FXR agonists have now been approved as therapeutic agents for NASH, and it is expected that signals from this nuclear transcription factor suppress lipogenic and fibrotic signals in the liver.14, 97 Elucidation of the detailed molecular mechanisms linked to gut microbial metabolites by the gut–liver axis may lead to the development of promising preventive and therapeutic drugs using advanced techniques for liver diseases. Moreover, liver cancer‐specific gut microbiota and the associated therapeutics using gut microbial species might be uncovered in the near future.
Supported in part by the Japan Society for the Promotion of Science Grants‐in‐Aid for Scientific Research (KAKENHI) (16H04699 to N.O.; 16H05290 to N.K.) and the Japan Agency for Medical Research and Development (AMED) (17929503 to N.O.; 18fk0210004h0003 to N.K.).
Potential conflict of interest: Nothing to report.
Contributor Information
Naoko Ohtani, Email: ohtani.naoko@med.osaka-cu.ac.jp.
Norifumi Kawada, Email: kawadanori@med.osaka-cu.ac.jp.
References
Author names in bold designate shared co‐first authorship.
- 1. Tripathi A, Debelius J, Brenner DA, Karin M, Loomba R, Schnabl B, et al. The gut‐liver axis and the intersection with the microbiome. Nat Rev Gastroenterol Hepatol 2018;15:397‐411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Yu LX, Schwabe RF. The gut microbiome and liver cancer: mechanisms and clinical translation. Nat Rev Gastroenterol Hepatol 2017;14:527‐539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Seo YS, Shah VH. The role of gut‐liver axis in the pathogenesis of liver cirrhosis and portal hypertension. Clin Mol Hepatol 2012;18:337‐346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Wiest R, Albillos A, Trauner M, Bajaj JS, Jalan R. Targeting the gut‐liver axis in liver disease. J Hepatol 2017;67:1084‐1103. Erratum. J Hepatol 2018;68:1336. [DOI] [PubMed] [Google Scholar]
- 5. Kasubuchi M, Hasegawa S, Hiramatsu T, Ichimura A, Kimura I. Dietary gut microbial metabolites, short‐chain fatty acids, and host metabolic regulation. Nutrients 2015;7:2839‐2849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Burcelin R, Serino M, Chabo C, Blasco‐Baque V, Amar J. Gut microbiota and diabetes: from pathogenesis to therapeutic perspective. Acta Diabetol 2011;48:257‐273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Garrett WS. Cancer and the microbiota. Science 2015;348:80‐86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Dzutsev A, Badger JH, Perez‐Chanona E, Roy S, Salcedo R, Smith CK, et al. Microbes and cancer. Annu Rev Immunol 2017;35:199‐228. [DOI] [PubMed] [Google Scholar]
- 9. Furusawa Y, Obata Y, Fukuda S, Endo TA, Nakato G, Takahashi D, et al. Commensal microbe‐derived butyrate induces the differentiation of colonic regulatory T cells. Nature 2013;504:446‐450. [DOI] [PubMed] [Google Scholar]
- 10. Kimura I, Inoue D, Maeda T, Hara T, Ichimura A, Miyauchi S, et al. Short‐chain fatty acids and ketones directly regulate sympathetic nervous system via G protein‐coupled receptor 41 (GPR41). Proc Natl Acad Sci U S A 2011;108:8030‐8035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Seki E, De Minicis S, Osterreicher CH, Kluwe J, Osawa Y, Brenner DA, et al. TLR4 enhances TGF‐beta signaling and hepatic fibrosis. Nat Med 2007;13:1324‐1332. [DOI] [PubMed] [Google Scholar]
- 12. Dapito DH, Mencin A, Gwak GY, Pradere JP, Jang MK, Mederacke I, et al. Promotion of hepatocellular carcinoma by the intestinal microbiota and TLR4. Cancer Cell 2012;21:504‐516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Loo TM, Kamachi F, Watanabe Y, Yoshimoto S, Kanda H, Arai Y, et al. Gut microbiota promotes obesity‐associated liver cancer through PGE2‐mediated suppression of antitumor immunity. Cancer Discov 2017;7:522‐538. [DOI] [PubMed] [Google Scholar]
- 14. Shapiro H, Kolodziejczyk AA, Halstuch D, Elinav E. Bile acids in glucose metabolism in health and disease. J Exp Med 2018;215:383‐396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Payne CM, Weber C, Crowley‐Skillicorn C, Dvorak K, Bernstein H, Bernstein C, et al. Deoxycholate induces mitochondrial oxidative stress and activates NF‐kappaB through multiple mechanisms in HCT‐116 colon epithelial cells. Carcinogenesis 2007;28:215‐222. [DOI] [PubMed] [Google Scholar]
- 16. Tsuchida T, Friedman SL. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol 2017;14:397‐411. [DOI] [PubMed] [Google Scholar]
- 17. Blomhoff R, Wake K. Perisinusoidal stellate cells of the liver: important roles in retinol metabolism and fibrosis. FASEB J 1991;5:271‐277. [DOI] [PubMed] [Google Scholar]
- 18. Wake K. Perisinusoidal stellate cells (fat‐storing cells, interstitial cells, lipocytes), their related structure in and around the liver sinusoids, and vitamin A‐storing cells in extrahepatic organs. Int Rev Cytol 1980;66:303‐353. [DOI] [PubMed] [Google Scholar]
- 19. Moriwaki H, Blaner WS, Piantedosi R, Goodman DS. Effects of dietary retinoid and triglyceride on the lipid composition of rat liver stellate cells and stellate cell lipid droplets. J Lipid Res 1988;29:1523‐1534. [PubMed] [Google Scholar]
- 20. Kawada N, Tran‐Thi TA, Klein H, Decker K. The contraction of hepatic stellate (Ito) cells stimulated with vasoactive substances. Possible involvement of endothelin 1 and nitric oxide in the regulation of the sinusoidal tonus. Eur J Biochem 1993;213:815‐823. [DOI] [PubMed] [Google Scholar]
- 21. Pinzani M, Failli P, Ruocco C, Casini A, Milani S, Baldi E, et al. Fat‐storing cells as liver‐specific pericytes. Spatial dynamics of agonist‐stimulated intracellular calcium transients. J Clin Invest 1992;90:642‐646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sugimoto H, Makino M, Sawai H, Kawada N, Yoshizato K, Shiro Y. Structural basis of human cytoglobin for ligand binding. J Mol Biol 2004;339:873‐885. [DOI] [PubMed] [Google Scholar]
- 23. She H, Xiong S, Hazra S, Tsukamoto H. Adipogenic transcriptional regulation of hepatic stellate cells. J Biol Chem 2005;280:4959‐4967. [DOI] [PubMed] [Google Scholar]
- 24. Wandzioch E, Kolterud A, Jacobsson M, Friedman SL, Carlsson L. Lhx2‐/‐ mice develop liver fibrosis. Proc Natl Acad Sci U S A 2004;101:16549‐16554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Koo JH, Lee HJ, Kim W, Kim SG. Endoplasmic reticulum stress in hepatic stellate cells promotes liver fibrosis via PERK‐mediated degradation of HNRNPA1 and up‐regulation of SMAD2. Gastroenterology 2016;150:181‐193.e188. [DOI] [PubMed] [Google Scholar]
- 26. Kawasaki K, Ushioda R, Ito S, Ikeda K, Masago Y, Nagata K. Deletion of the collagen‐specific molecular chaperone Hsp47 causes endoplasmic reticulum stress‐mediated apoptosis of hepatic stellate cells. J Biol Chem 2015;290:3639‐3646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Schwabe RF, Luedde T. Apoptosis and necroptosis in the liver: a matter of life and death. Nat Rev Gastroenterol Hepatol 2018;15:738‐752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Kim KH, Chen CC, Monzon RI, Lau LF. Matricellular protein CCN1 promotes regression of liver fibrosis through induction of cellular senescence in hepatic myofibroblasts. Mol Cell Biol 2013;33:2078‐2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Kisseleva T, Cong M, Paik Y, Scholten D, Jiang C, Benner C, et al. Myofibroblasts revert to an inactive phenotype during regression of liver fibrosis. Proc Natl Acad Sci U S A 2012;109:9448‐9453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yoshimoto S, Loo TM, Atarashi K, Kanda H, Sato S, Oyadomari S, et al. Obesity‐induced gut microbial metabolite promotes liver cancer through senescence secretome. Nature 2013;499:97‐101. [DOI] [PubMed] [Google Scholar]
- 31. Sato‐Matsubara M, Matsubara T, Daikoku A, Okina Y, Longato L, Rombouts K, et al. Fibroblast growth factor 2 (FGF2) regulates cytoglobin expression and activation of human hepatic stellate cells via JNK signaling. J Biol Chem 2017;292:18961‐18972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nielsen SR, Quaranta V, Linford A, Emeagi P, Rainer C, Santos A, et al. Macrophage‐secreted granulin supports pancreatic cancer metastasis by inducing liver fibrosis. Nat Cell Biol 2016;18:549‐560. Erratum. In: Nat Cell Biol 2016;18:822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mogler C, Konig C, Wieland M, Runge A, Besemfelder E, Komljenovic D, et al. Hepatic stellate cells limit hepatocellular carcinoma progression through the orphan receptor endosialin. EMBO Mol Med 2017;9:741‐749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Duran A, Hernandez ED, Reina‐Campos M, Castilla EA, Subramaniam S, Raghunandan S, et al. p62/SQSTM1 by binding to vitamin D receptor inhibits hepatic stellate cell activity, fibrosis, and liver cancer. Cancer Cell 2016;30:595‐609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. le Thuy TT, Morita T, Yoshida K, Wakasa K, Iizuka M, Ogawa T, et al. Promotion of liver and lung tumorigenesis in DEN‐treated cytoglobin‐deficient mice. Am J Pathol 2011;179:1050‐1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. le Thuy TT, Matsumoto Y, Thuy TT, Hai H, Suoh M, Urahara Y, et al. Cytoglobin deficiency promotes liver cancer development from hepatosteatosis through activation of the oxidative stress pathway. Am J Pathol 2015;185:1045‐1060. [DOI] [PubMed] [Google Scholar]
- 37. Rao SG, Jackson JG. SASP: Tumor suppressor or promoter? Yes! Trends Cancer 2016;2:676‐687. [DOI] [PubMed] [Google Scholar]
- 38. Rodier F, Campisi J. Four faces of cellular senescence. J Cell Biol 2011;192:547‐556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Smith K. Liver disease: Kupffer cells regulate the progression of ALD and NAFLD. Nat Rev Gastroenterol Hepatol 2013;10:503. [DOI] [PubMed] [Google Scholar]
- 40. Petrasek J, Bala S, Csak T, Lippai D, Kodys K, Menashy V, et al. IL‐1 receptor antagonist ameliorates inflammasome‐dependent alcoholic steatohepatitis in mice. J Clin Invest 2012;122:3476‐3489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Olefsky JM, Glass CK. Macrophages, inflammation, and insulin resistance. Annu Rev Physiol 2010;72:219‐246. [DOI] [PubMed] [Google Scholar]
- 42. Krenkel O, Tacke F. Liver macrophages in tissue homeostasis and disease. Nat Rev Immunol 2017;17:306‐321. [DOI] [PubMed] [Google Scholar]
- 43. Tsutsui H, Matsui K, Okamura H, Nakanishi K. Pathophysiological roles of interleukin‐18 in inflammatory liver diseases. Immunol Rev 2000;174:192‐209. [DOI] [PubMed] [Google Scholar]
- 44. Kazankov K, Jorgensen SMD, Thomsen KL, Moller HJ, Vilstrup H, George J, et al. The role of macrophages in nonalcoholic fatty liver disease and nonalcoholic steatohepatitis. Nat Rev Gastroenterol Hepatol 2018; 10.1038/s41575-018-0082-x. [DOI] [PubMed] [Google Scholar]
- 45. Ide M, Kuwamura M, Kotani T, Sawamoto O, Yamate J. Effects of gadolinium chloride (GdCl(3)) on the appearance of macrophage populations and fibrogenesis in thioacetamide‐induced rat hepatic lesions. J Comp Pathol 2005;133:92‐102. [DOI] [PubMed] [Google Scholar]
- 46. Duffield JS, Forbes SJ, Constandinou CM, Clay S, Partolina M, Vuthoori S, et al. Selective depletion of macrophages reveals distinct, opposing roles during liver injury and repair. J Clin Invest 2005;115:56‐65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Karlmark KR, Weiskirchen R, Zimmermann HW, Gassler N, Ginhoux F, Weber C, et al. Hepatic recruitment of the inflammatory Gr1+ monocyte subset upon liver injury promotes hepatic fibrosis. Hepatology 2009;50:261‐274. [DOI] [PubMed] [Google Scholar]
- 48. Fallowfield JA, Mizuno M, Kendall TJ, Constandinou CM, Benyon RC, Duffield JS, et al. Scar‐associated macrophages are a major source of hepatic matrix metalloproteinase‐13 and facilitate the resolution of murine hepatic fibrosis. J Immunol 2007;178:5288‐5295. [DOI] [PubMed] [Google Scholar]
- 49. Ramachandran P, Pellicoro A, Vernon MA, Boulter L, Aucott RL, Ali A, et al. Differential Ly‐6C expression identifies the recruited macrophage phenotype, which orchestrates the regression of murine liver fibrosis. Proc Natl Acad Sci U S A 2012;109:E3186‐E3195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Thomas JA, Pope C, Wojtacha D, Robson AJ, Gordon‐Walker TT, Hartland S, et al. Macrophage therapy for murine liver fibrosis recruits host effector cells improving fibrosis, regeneration, and function. Hepatology 2011;53:2003‐2015. [DOI] [PubMed] [Google Scholar]
- 51. Newsome PN, Fox R, King AL, Barton D, Than NN, Moore J, et al. Granulocyte colony‐stimulating factor and autologous CD133‐positive stem‐cell therapy in liver cirrhosis (REALISTIC): an open‐label, randomised, controlled phase 2 trial. Lancet Gastroenterol Hepatol 2018;3:25‐36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Ambade A, Lowe P, Kodys K, Catalano D, Gyongyosi B, Cho Y, et al. Pharmacological inhibition of CCR52/5 signaling prevents and reverses alcohol‐induced liver damage, steatosis and inflammation in mice. Hepatology 2018; 10.1002/hep.30249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Krenkel O, Puengel T, Govaere O, Abdallah AT, Mossanen JC, Kohlhepp M, et al. Therapeutic inhibition of inflammatory monocyte recruitment reduces steatohepatitis and liver fibrosis. Hepatology 2018;67:1270‐1283. [DOI] [PubMed] [Google Scholar]
- 54. Friedman SL, Ratziu V, Harrison SA, Abdelmalek MF, Aithal GP, Caballeria J, et al. A randomized, placebo‐controlled trial of cenicriviroc for treatment of nonalcoholic steatohepatitis with fibrosis. Hepatology 2018;67:1754‐1767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Nakagawa H, Maeda S, Hikiba Y, Ohmae T, Shibata W, Yanai A, et al. Deletion of apoptosis signal‐regulating kinase 1 attenuates acetaminophen‐induced liver injury by inhibiting c‐Jun N‐terminal kinase activation. Gastroenterology 2008;135:1311‐1321. [DOI] [PubMed] [Google Scholar]
- 56. Zhang P, Wang PX, Zhao LP, Zhang X, Ji YX, Zhang XJ, et al. The deubiquitinating enzyme TNFAIP3 mediates inactivation of hepatic ASK1 and ameliorates nonalcoholic steatohepatitis. Nat Med 2018;24:84‐94. [DOI] [PubMed] [Google Scholar]
- 57. Loomba R, Lawitz E, Mantry PS, Jayakumar S, Caldwell SH, Arnold H, et al.; GS‐US‐384‐1497 Investigators . The ASK1 inhibitor selonsertib in patients with nonalcoholic steatohepatitis: a randomized, phase 2 trial. Hepatology 2017; 10.1002/hep.29514. Erratum. In: Hepatology 2018;67:2063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Lin RS, Lee FY, Lee SD, Tsai YT, Lin HC, Lu RH, et al. Endotoxemia in patients with chronic liver diseases: relationship to severity of liver diseases, presence of esophageal varices, and hyperdynamic circulation. J Hepatol 1995;22:165‐172. [DOI] [PubMed] [Google Scholar]
- 59. Qin N, Yang F, Li A, Prifti E, Chen Y, Shao L, et al. Alterations of the human gut microbiome in liver cirrhosis. Nature 2014;513:59‐64. [DOI] [PubMed] [Google Scholar]
- 60. Bajaj JS, Betrapally NS, Gillevet PM. Decompensated cirrhosis and microbiome interpretation. Nature 2015;525:E1‐E2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Imajo K, Fujita K, Yoneda M, Nozaki Y, Ogawa Y, Shinohara Y, et al. Hyperresponsivity to low‐dose endotoxin during progression to nonalcoholic steatohepatitis is regulated by leptin‐mediated signaling. Cell Metab 2012;16:44‐54. [DOI] [PubMed] [Google Scholar]
- 62. Peri F, Piazza M. Therapeutic targeting of innate immunity with Toll‐like receptor 4 (TLR4) antagonists. Biotechnol Adv 2012;30:251‐260. [DOI] [PubMed] [Google Scholar]
- 63. Fukui H. Gut Microbiome‐based Therapeutics in liver cirrhosis: basic consideration for the next step. J Clin Transl Hepatol 2017;5:249‐260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Stenman LK, Holma R, Eggert A, Korpela R. A novel mechanism for gut barrier dysfunction by dietary fat: epithelial disruption by hydrophobic bile acids. Am J Physiol Gastrointest Liver Physiol 2013;304:G227‐G234. [DOI] [PubMed] [Google Scholar]
- 65. Kakiyama G, Pandak WM, Gillevet PM, Hylemon PB, Heuman DM, Daita K, et al. Modulation of the fecal bile acid profile by gut microbiota in cirrhosis. J Hepatol 2013;58:949‐955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Rahman K, Desai C, Iyer SS, Thorn NE, Kumar P, Liu Y, et al. Loss of junctional adhesion molecule a promotes severe steatohepatitis in mice on a diet high in saturated fat, fructose, and cholesterol. Gastroenterology 2016;151:733‐746.e712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Ilan Y. Leaky gut and the liver: a role for bacterial translocation in nonalcoholic steatohepatitis. World J Gastroenterol 2012;18:2609‐2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Puri P, Daita K, Joyce A, Mirshahi F, Santhekadur PK, Cazanave S, et al. The presence and severity of nonalcoholic steatohepatitis is associated with specific changes in circulating bile acids. Hepatology 2017; 10.1002/hep.29359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Acharya C, Bajaj JS. Current management of hepatic encephalopathy. Am J Gastroenterol 2018;113:1600‐1612. [DOI] [PubMed] [Google Scholar]
- 70. Swaminathan M, Ellul MA, Cross TJ. Hepatic encephalopathy: current challenges and future prospects. Hepat Med 2018;10:1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Mancini A, Campagna F, Amodio P, Tuohy KM. Gut: liver: brain axis: the microbial challenge in the hepatic encephalopathy. Food Funct 2018;9:1373‐1388. [DOI] [PubMed] [Google Scholar]
- 72. Inoue T, Nakayama J, Moriya K, Kawaratani H, Momoda R, Ito K, et al. Gut dysbiosis associated with hepatitis C virus infection. Clin Infect Dis 2018;67:869‐877. [DOI] [PubMed] [Google Scholar]
- 73. Tsai CF, Chen MH, Wang YP, Chu CJ, Huang YH, Lin HC, et al. Proton pump inhibitors increase risk for hepatic encephalopathy in patients with cirrhosis in a population study. Gastroenterology 2017;152:134‐141. [DOI] [PubMed] [Google Scholar]
- 74. Dam G, Vilstrup H, Watson H, Jepsen P. Proton pump inhibitors as a risk factor for hepatic encephalopathy and spontaneous bacterial peritonitis in patients with cirrhosis with ascites. Hepatology 2016;64:1265‐1272. [DOI] [PubMed] [Google Scholar]
- 75. Xu D, Gao J, Gillilland M 3rd, Wu X, Song I, Kao JY, et al. Rifaximin alters intestinal bacteria and prevents stress‐induced gut inflammation and visceral hyperalgesia in rats. Gastroenterology 2014;146:484‐496.e484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76. Alisi A, Bedogni G, Baviera G, Giorgio V, Porro E, Paris C, et al. Randomised clinical trial: the beneficial effects of VSL#3 in obese children with non‐alcoholic steatohepatitis. Aliment Pharmacol Ther 2014;39:1276‐1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Bertuccio P, Turati F, Carioli G, Rodriguez T, La Vecchia C, Malvezzi M, et al. Global trends and predictions in hepatocellular carcinoma mortality. J Hepatol 2017;67:302‐309. [DOI] [PubMed] [Google Scholar]
- 78. Tateishi R, Uchino K, Fujiwara N, Takehara T, Okanoue T, Seike M, et al. A nationwide survey on non‐B, non‐C hepatocellular carcinoma in Japan: 2011‐2015 update. J Gastroenterol 2018; 10.1007/s00535-018-1532-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Mak LY, Cruz‐Ramon V, Chinchilla‐Lopez P, Torres HA, LoConte NK, Rice JP, et al. Global epidemiology, prevention, and management of hepatocellular carcinoma. Am Soc Clin Oncol Educ Book 2018;262‐279. [DOI] [PubMed] [Google Scholar]
- 80. Arrese M, Feldstein AE. NASH‐related cirrhosis: an occult liver disease burden. Hepatol Commun 2017;1:84‐86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Friedman SL, Neuschwander‐Tetri BA, Rinella M, Sanyal AJ. Mechanisms of NAFLD development and therapeutic strategies. Nat Med 2018;24:908‐922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Rotman Y, Sanyal AJ. Current and upcoming pharmacotherapy for non‐alcoholic fatty liver disease. Gut 2017;66:180‐190. [DOI] [PubMed] [Google Scholar]
- 83. Banini BA, Sanyal AJ. Current and future pharmacologic treatment of nonalcoholic steatohepatitis. Curr Opin Gastroenterol 2017;33:134‐141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Neuschwander‐Tetri BA, Loomba R, Sanyal AJ, Lavine JE, Van Natta ML, Abdelmalek MF, et al; NASH Clinical Research Network . Farnesoid X nuclear receptor ligand obeticholic acid for non‐cirrhotic, non‐alcoholic steatohepatitis (FLINT): a multicentre, randomised, placebo‐controlled trial. Lancet 2015;385:956‐965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Carr RM, Reid AE. FXR agonists as therapeutic agents for non‐alcoholic fatty liver disease. Curr Atheroscler Rep 2015;17:500. [DOI] [PubMed] [Google Scholar]
- 86. Sumida Y, Yoneda M. Current and future pharmacological therapies for NAFLD/NASH. J Gastroenterol 2018;53:362‐376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87. Zhou H, Hylemon PB. Bile acids are nutrient signaling hormones. Steroids 2014;86:62‐68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88. Hylemon PB, Zhou H, Pandak WM, Ren S, Gil G, Dent P. Bile acids as regulatory molecules. J Lipid Res 2009;50:1509‐1520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Ridlon JM, Alves JM, Hylemon PB, Bajaj JS. Cirrhosis, bile acids and gut microbiota: unraveling a complex relationship. Gut Microbes 2013;4:382‐387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Mertens KL, Kalsbeek A, Soeters MR, Eggink HM. Bile acid signaling pathways from the enterohepatic circulation to the central nervous system. Front Neurosci 2017;11:617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. Russell DW. The enzymes, regulation, and genetics of bile acid synthesis. Annu Rev Biochem 2003;72:137‐174. [DOI] [PubMed] [Google Scholar]
- 92. Thomas C, Pellicciari R, Pruzanski M, Auwerx J, Schoonjans K. Targeting bile‐acid signalling for metabolic diseases. Nat Rev Drug Discov 2008;7:678‐693. [DOI] [PubMed] [Google Scholar]
- 93. Hofmann AF. The enterohepatic circulation of bile acids in man. Adv Intern Med 1976;21:501‐534. [PubMed] [Google Scholar]
- 94. Mok HY, Von Bergmann K, Grundy SM. Regulation of pool size of bile acids in man. Gastroenterology 1977;73:684‐690. [PubMed] [Google Scholar]
- 95. Al‐Khaifi A, Rudling M, Angelin B. An FXR agonist reduces bile acid synthesis independently of increases in FGF19 in healthy volunteers. Gastroenterology 2018;155:1012‐1016. [DOI] [PubMed] [Google Scholar]
- 96. Modica S, Gadaleta RM, Moschetta A. Deciphering the nuclear bile acid receptor FXR paradigm. Nucl Recept Signal 2010;8:e005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Tanaka N, Aoyama T, Kimura S, Gonzalez FJ. Targeting nuclear receptors for the treatment of fatty liver disease. Pharmacol Ther 2017;179:142‐157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Kettner NM, Voicu H, Finegold MJ, Coarfa C, Sreekumar A, Putluri N, et al. Circadian homeostasis of liver metabolism suppresses hepatocarcinogenesis. Cancer Cell 2016;30:909‐924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Corbin KD, Zeisel SH. Choline metabolism provides novel insights into nonalcoholic fatty liver disease and its progression. Curr Opin Gastroenterol 2012;28:159‐165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Wang Z, Klipfell E, Bennett BJ, Koeth R, Levison BS, Dugar B, et al. Gut flora metabolism of phosphatidylcholine promotes cardiovascular disease. Nature 2011;472:57‐63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Chu H, Duan Y, Yang L, Schnabl B. Small metabolites, possible big changes: a microbiota‐centered view of non‐alcoholic fatty liver disease. Gut 2019;68:359‐370. [DOI] [PubMed] [Google Scholar]
- 102. He X, Ji G, Jia W, Li H. Gut microbiota and nonalcoholic fatty liver disease: insights on mechanism and application of metabolomics. Int J Mol Sci 2016;17:300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Kim CW, Addy C, Kusunoki J, Anderson NN, Deja S, Fu X, et al. Acetyl CoA carboxylase inhibition reduces hepatic steatosis but elevates plasma triglycerides in mice and humans: a bedside to bench investigation. Cell Metab 2017;26:394‐406.e396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104. Minami S, Miura K, Ishioka M, Morimoto N, Isoda N, Yamamoto H, et al. Homocysteine supplementation ameliorates steatohepatitis induced by a choline‐deficient diet in mice. Hepatol Res 2019;49:189‐200. [DOI] [PubMed] [Google Scholar]
- 105. Honda T, Ishigami M, Luo F, Lingyun M, Ishizu Y, Kuzuya T, et al. Branched‐chain amino acids alleviate hepatic steatosis and liver injury in choline‐deficient high‐fat diet induced NASH mice. Metabolism 2017;69:177‐187. [DOI] [PubMed] [Google Scholar]
- 106. Kishino S, Takeuchi M, Park SB, Hirata A, Kitamura N, Kunisawa J, et al. Polyunsaturated fatty acid saturation by gut lactic acid bacteria affecting host lipid composition. Proc Natl Acad Sci U S A 2013;110:17808‐17813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Miyamoto J, Mizukure T, Park SB, Kishino S, Kimura I, Hirano K, et al. A gut microbial metabolite of linoleic acid, 10‐hydroxy‐cis‐12‐octadecenoic acid, ameliorates intestinal epithelial barrier impairment partially via GPR40‐MEK‐ERK pathway. J Biol Chem 2015;290:2902‐2918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Nanthirudjanar T, Furumoto H, Zheng J, Kim YI, Goto T, Takahashi N, et al. Gut microbial fatty acid metabolites reduce triacylglycerol levels in hepatocytes. Lipids 2015;50:1093‐1102. [DOI] [PubMed] [Google Scholar]
- 109. Ferreira DM, Afonso MB, Rodrigues PM, Simao AL, Pereira DM, Borralho PM, et al. c‐Jun N‐terminal kinase 1/c‐Jun activation of the p53/microRNA 34a/sirtuin 1 pathway contributes to apoptosis induced by deoxycholic acid in rat liver. Mol Cell Biol 2014;34:1100‐1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110. Labenz C, Huber Y, Kalliga E, Nagel M, Ruckes C, Straub BK, et al. Predictors of advanced fibrosis in non‐cirrhotic non‐alcoholic fatty liver disease in Germany. Aliment Pharmacol Ther 2018;48:1109‐1116. [DOI] [PubMed] [Google Scholar]
- 111. Kawada N, Imanaka K, Kawaguchi T, Tamai C, Ishihara R, Matsunaga T, et al. Hepatocellular carcinoma arising from non‐cirrhotic nonalcoholic steatohepatitis. J Gastroenterol 2009;44:1190‐1194. [DOI] [PubMed] [Google Scholar]
- 112. Perumpail RB, Wong RJ, Ahmed A, Harrison SA. Hepatocellular carcinoma in the setting of non‐cirrhotic nonalcoholic fatty liver disease and the metabolic syndrome: US experience. Dig Dis Sci 2015;60:3142‐3148. [DOI] [PubMed] [Google Scholar]
- 113. Takuma Y, Nouso K. Nonalcoholic steatohepatitis‐associated hepatocellular carcinoma: our case series and literature review. World J Gastroenterol 2010;16:1436‐1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114. Yamada S, Takashina Y, Watanabe M, Nagamine R, Saito Y, Kamada N, et al. Bile acid metabolism regulated by the gut microbiota promotes non‐alcoholic steatohepatitis‐associated hepatocellular carcinoma in mice. Oncotarget 2018;9:9925‐9939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115. Singh V, Yeoh BS, Chassaing B, Xiao X, Saha P, Aguilera Olvera R, et al. Dysregulated microbial fermentation of soluble fiber induces cholestatic liver cancer. Cell 2018;175:679‐694.e622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116. Ma C, Han M, Heinrich B, Fu Q, Zhang Q, Sandhu M, et al. Gut microbiome‐mediated bile acid metabolism regulates liver cancer via NKT cells. Science 2018;360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117. Sivan A, Corrales L, Hubert N, Williams JB, Aquino‐Michaels K, Earley ZM, et al. Commensal Bifidobacterium promotes antitumor immunity and facilitates anti‐PD‐L1 efficacy. Science 2015;350:1084‐1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118. Vetizou M, Pitt JM, Daillere R, Lepage P, Waldschmitt N, Flament C, et al. Anticancer immunotherapy by CTLA‐4 blockade relies on the gut microbiota. Science 2015;350:1079‐1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119. Gopalakrishnan V, Spencer CN, Nezi L, Reuben A, Andrews MC, Karpinets TV, et al. Gut microbiome modulates response to anti‐PD‐1 immunotherapy in melanoma patients. Science 2018;359:97‐103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120. Matson V, Fessler J, Bao R, Chongsuwat T, Zha Y, Alegre ML, et al. The commensal microbiome is associated with anti‐PD‐1 efficacy in metastatic melanoma patients. Science 2018;359:104‐108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121. Routy B, Le Chatelier E, Derosa L, Duong CPM, Alou MT, Daillere R, et al. Gut microbiome influences efficacy of PD‐1‐based immunotherapy against epithelial tumors. Science 2018;359:91‐97. [DOI] [PubMed] [Google Scholar]
- 122. Xu F, Jin T, Zhu Y, Dai C. Immune checkpoint therapy in liver cancer. J Exp Clin Cancer Res 2018;37:110. [DOI] [PMC free article] [PubMed] [Google Scholar]