

Abstract
Alcoholic liver disease (ALD) develops in a subset of heavy drinkers (HDs). The goals of our study were to (1) characterize the global serum metabolomic changes in well‐characterized cohorts of controls (Cs), HDs, and those with alcoholic cirrhosis (AC); (2) identify metabolomic signatures as potential diagnostic markers, and (3) determine the trajectory of serum metabolites in response to alcohol abstinence. Serum metabolic profiling was performed in 22 Cs, 147 HDs, and 33 patients with AC using ultraperformance liquid chromatography–tandem mass spectrometry. Hepatic gene expression was conducted in Cs (n = 16) and those with AC (n = 32). We found progressive changes in the quantities of metabolites from heavy drinking to AC. Taurine‐conjugated bile acids (taurocholic acid [TCA], 127‐fold; taurochenodeoxycholic acid [TCDCA], 131‐fold; and tauroursodeoxycholic acid, 56‐fold) showed more striking elevations than glycine‐conjugated forms (glycocholic acid [GCA], 22‐fold; glycochenodeoxycholic acid [GCDCA], 22‐fold; and glycoursodeoxycholic acid [GUDCA], 11‐fold). This was associated with increased liver cytochrome P450, family 7, subfamily B, member 1 and taurine content (more substrates); the latter was due to dysregulation of homocysteine metabolism. Increased levels of GCDCA, TCDCA, GCA, and TCA positively correlated with disease progression from Child‐Pugh A to C and Model for End‐Stage Liver Disease scores, whereas GCDCA, GCA, and GUDCA were better predictors of alcohol abstinence. The levels of glucagon‐like peptide 1 (GLP‐1) and fibroblast growth factor (FGF) 21 but not FGF19 were increased in HDs, and all three were further increased in those with AC. Conclusion: Serum taurine/glycine‐conjugated bile acids could serve as noninvasive markers to predict the severity of AC, whereas GLP‐1 and FGF21 may indicate a progression from heavy drinking to AC.
Abbreviations
- 1,5‐AG
1,5‐anhydroglucitol
- 2/3D
two/three dimensional
- ABCG
adenosine triphosphate binding cassette subfamily G
- AC
alcoholic cirrhosis
- AH
alcoholic hepatitis
- ALD
alcoholic liver disease
- ANOVA
analysis of variance
- BA
bile acid
- BAL
bile acid coenzyme A ligase
- BAT
bile acid coenzyme A:amino acid N‐acyltransferase
- BCAA
branched‐chain amino acid
- BCAT
branched‐chain amino acid transaminase
- BSEP
bile salt export pump
- BVRA
biliverdin reductase A
- C/CTRL
control
- CDCA
chenodeoxycholic acid
- CSAD
cysteine sulfinic acid decarboxylase
- CYP7A1
cytochrome P450 family 7, subfamily A, member 1
- CYP7B1
cytochrome P450 family 8, subfamily B, polypeptide 1
- ESI
electrospray ionization
- FGF
fibroblast growth factor
- FXR
farnesoid X receptor
- GCA
glycocholic acid
- GCDCA
glycochenodeoxycholic acid
- GLP‐1
glucagon‐like peptide 1
- GUDCA
glycoursodeoxycholic acid
- HD
heavy drinker
- HD1
heavy drinker with last alcohol consumption >10 days before enrollment
- HD2
heavy drinker with drinking up to the time of enrollment
- HNF4α
hepatocyte nuclear factor 4 alpha
- LRH1
liver receptor homolog 1
- LXR
liver X receptor
- MAT1A
methionine adenosyltransferase 1A
- MELD
Model for End‐Stage Liver Disease
- mRNA
messenger RNA
- MRP
multidrug resistance‐associated protein
- MS
mass spectroscopy
- MS/MS
tandem mass spectroscopy
- MTR
methionine synthase
- ns
not significant
- OATP
organic‐anion‐transporting polypeptide
- PCA
principal component analysis
- PXR
pregnane X receptor
- qRT‐PCR
quantitative real‐time polymerase chain reaction
- RF
random forest
- SULT
sulfotransferase
- TCA
taurocholic acid
- TCDCA
taurochenodeoxycholic acid
- TUDCA
tauroursodeoxycholic acid
- UPLC
ultrahigh performance liquid chromatography
Excessive alcohol use can lead to adverse health outcomes that result in significant social and economic burdens.1 Alcohol‐related health disorders are generally determined by the quantity of alcohol consumed and the pattern of drinking.2 Excessive alcohol use is associated with cancers, cardiovascular disease, pancreatitis, and alcoholic liver disease (ALD).3
ALD comprises a spectrum of disorders and histopathologic changes in individuals with acute and chronic alcohol consumption, ranging from alcoholic steatosis to alcoholic hepatitis (AH) and alcoholic cirrhosis (AC).4 Alcoholic steatosis occurs almost universally in those with excessive alcohol use.5 Once developed, it can progress to steatohepatitis, fibrosis, and cirrhosis.6, 7 Alcoholic steatosis is reversible with abstinence5; however, continued drinking can lead to AC in up to 20% to 30% of excessive drinkers.6, 7 Evidence of a genetic component for the susceptibility to ALD has been described,8, 9 but a large variability remains unexplained, suggesting that other factors in addition to a genetic component might be involved in the disease process.
Excessive alcohol use interferes with the absorption, metabolism, transport, and excretion of nutrients.10 It can also lead to electrolyte imbalance and metabolic changes.11 Advancement of the disease during progression of ALD is accompanied by alterations in carbohydrate and lipid metabolism, as demonstrated in different models of mice fed with ethanol.12, 13 Metabolomics is an emerging new area of basic and translational research. A comprehensive analysis of the changes in the levels of metabolites in patients with excessive alcohol use and those with ALD may provide mechanistic insights into why only a subset of patients develop ALD. Recently, Harada et al.14 reported global metabolomic profiling among patients with alcohol intake. Nineteen metabolites are associated with daily alcohol consumption. Among them, 13 metabolites are involved in amino acid metabolism, three in carbohydrate metabolism, two in lipid metabolism, and one in cofactor and vitamin metabolism.14 In addition, plasma levels of three metabolites, i.e., threonine, guanidinosuccinate, and glutamine, are strongly associated with markers of hepatic inflammation, such as aspartate aminotransferase, suggesting their potential link to alcohol‐induced liver injury14; however, the underlying mechanism and the trajectory of these metabolites with alcohol abstinence were unclear in that study. Furthermore, metabolomic alterations for ALD are based on the association of these metabolites with markers of hepatic enzymes in those with alcohol use and not from patients with a known diagnosis of ALD.14
The goals of our study are to (1) characterize the global metabolomic changes in a well‐characterized cohort of controls (Cs), heavy/excessive drinkers (HDs), and those with AC; (2) identify metabolomic signatures as potential diagnostic markers linking the changes in serum metabolomes with gene expression in the liver tissues of patients with AC; and (3) determine the trajectory of serum metabolites in response to alcohol abstinence.
Participants and Methods
Recruitment of the Study Cohort
The study design was approved by the institutional review board at Indiana University Purdue University Indianapolis, Fairbanks Alcohol Rehabilitation Center, and Roudebush Veterans Administration Medical Center. Recruitments were performed in accordance with the relevant guidelines and regulations.
We enrolled 147 HDs from Fairbanks Drug and Alcohol Treatment Center (Indianapolis, IN). All fulfilled the definition of heavy drinking as defined by the National Institutes of Health (NIH)/National Institute on Alcohol Abuse and Alcoholism: >4 standard drinks in a day (or >14 per week) in men and >3 drinks in a day (or >7 per week) in women.15 All participants were at least 21 years old and were excluded if they had active and serious medical diseases, past history of jaundice or complications from portal hypertension, concurrent infection with viral hepatitis B or C, history of any infection within 4 weeks, or recent surgeries within the past 3 months before enrollment.16, 17 We recruited 22 healthy Cs with no known history of heavy drinking from the Roudebush Veterans Administration Medical Center and enrolled 33 patients with known diagnosis of AC from the hepatology clinic at Indiana University. The detailed inclusion and exclusion criteria for AC have been described.18 Of importance, patients with AC with ongoing infection, sepsis, or active gastrointestinal bleeding were excluded. To determine the levels of serum metabolites in response to alcohol abstinence, we prospectively recruited 24 HDs participating in an alcohol treatment program. Blood was collected at baseline and at weeks 2 and 4 after alcohol abstinence. During this period, all participants complied with treatment, and none relapsed to alcohol use.
Human Liver Tissues
Coded human liver specimens from normal Cs (n = 16) and patients with AC (n = 32) were obtained through the Liver Tissue Cell Distribution System (Minneapolis, MN; NIH contract HSN276201200017C).19, 20, 21 The use of coded liver tissues was reviewed and approved by the University of Connecticut institutional review board.
Data and Biosample Collection
Demographic information, clinical characteristics, and laboratory tests were obtained at enrollment. Participants also completed self‐administered questionnaires, including Alcohol Use Disorders Identification Test‐Concise (AUDIT‐C) and Timeline Followback, to determine the quantity of alcohol consumed over the 30 days before enrollment. The date of last alcohol consumption was also recorded. For those with AC, baseline Child‐Pugh and Model for End Stage Liver Disease (MELD) scores were calculated. Blood was collected and centrifuged at 1,500g for 10 minutes at 4°C. Serum was stored at −80oC until analysis.
RNA Extraction and Quantitative Real‐time Polymerase Chain Reaction Analyses
Total RNA isolation of human liver tissues was performed using a commercially available RNA isolation kit. Complementary DNA synthesis was conducted as described.22 Specific primers for quantitative real‐time polymerase chain reaction (qRT‐PCR) are shown in Supporting Table S1.
Sample Preparation and Metabolic Profiling
Sample Preparation
Metabolic profiling of serum samples was performed, and analyses were carried out at Metabolon (Durham, NC). Samples from HDs were separated into two groups based on the duration of last alcohol consumption. Group one comprised HDs with the last alcohol consumption >10 days before enrollment (HD1); group two comprised HDs who continued to drink up to the time of enrollment (HD2). Samples were prepared using the automated MicroLab STAR system (Hamilton, Reno, NV). For quality control purposes, several recovery standards were added prior to the first step in the extraction process. To remove protein, small molecules bound to protein or trapped in the precipitated protein matrix were dissociated. To recover chemically diverse metabolites, proteins were precipitated with methanol under vigorous shaking for 2 minutes (Glen Mills GenoGrinder 2000), followed by centrifugation. The resulting extract was divided into five fractions: two for analysis by two separate reverse phase (RP)/ultrahigh performance liquid chromatography–tandem mass spectroscopy (UPLC‐MS/MS) methods with positive ion mode electrospray ionization (ESI), one for analysis by RP/UPLC‐MS/MS with negative ion mode ESI, one for analysis by hydrophilic interaction liquid chromatography (HILIC)/UPLC‐MS/MS with negative ion mode ESI, and one sample reserved for backup. Samples were placed briefly on a TurboVap (Zymark) to remove the organic solvent. Sample extracts were stored overnight in liquid nitrogen before preparation for analysis.
UPLC‐MS/MS
All methods used a Waters ACQUITY UPLC and a Thermo Scientific Q‐Exactive high resolution/accurate mass spectrometer interfaced with a heated ESI‐II source and Orbitrap mass analyzer operated at 35,000 mass resolution. The sample extract was dried and then reconstituted in solvents compatible with each of the four methods. Each reconstitution solvent contained a series of standards at fixed concentrations to ensure injection and chromatographic consistency. One aliquot was analyzed using acidic positive ion conditions and chromatographically optimized for more hydrophilic compounds. In this method, the extract was gradient eluted from a C18 column (Waters UPLC BEH C18‐2.1 × 100 mm, 1.7 µm) using water and methanol, containing 0.05% perfluoropentanoic acid (PFPA) and 0.1% formic acid (FA). Another aliquot was also analyzed using acidic positive ion conditions but chromatographically optimized for more hydrophobic compounds. In this method, the extract was gradient eluted from the same aforementioned C18 column using methanol, acetonitrile, water, 0.05% PFPA, and 0.01% FA and was operated at an overall higher organic content. Another aliquot was analyzed using basic negative ion‐optimized conditions using a separate dedicated C18 column. The basic extracts were gradient eluted from the column using methanol and water but with 6.5 mM ammonium bicarbonate at pH 8. The fourth aliquot was analyzed by negative ionization following elution from an HILIC column (Waters UPLC BEH Amide 2.1 × 150 mm, 1.7 µm) using a gradient consisting of water and acetonitrile with 10 mM ammonium formate, pH 10.8. The MS analysis alternated between MS and data‐dependent multistage scans using dynamic exclusion. The scan range varied slighted between methods but covered 70‐1,000 m/z. Raw data files were archived and extracted.
Data Extraction and Compound Identification
Metabolites were identified by automated comparison of ion features in the experimental samples to a reference library of chemical standard entries that included retention time, molecular weight (m/z), preferred adducts, and in‐source fragments as well as associated MS spectra. Metabolites were curated by visual inspection for quality control.
Measurement of Serum Fibroblast Growth Factor 19 and 21 and Glucagon‐like Peptide 1
Serum levels of fibroblast growth factor 19 (FGF19) (catalog #DF1900; R&D Systems, Minneapolis, MN), FGF21 (catalog #DF2100; R&D Systems), and glucagon‐like peptide 1 (GLP‐1) (catalog #EGLP‐35K; Millipore, Burlington, MS) were conducted per the manufacturers’ protocols.
Measurement of Hepatic Taurine and Glycine
Levels of hepatic taurine and glycine were measured using the taurine (catalog #MET‐5071) and glycine (catalog #MET‐5070) assay kits (Cell Biolabs, Inc., San Diego, CA).
Statistical Analysis
We presented basic characteristics as mean ± SEM for continuous variables and as numbers with percentages for categorical variables. Appropriate comparisons, including chi‐square test, Student t test, and analysis of variance (ANOVA), were used. For metabolomic data, we used significance tests and classification analysis. Standard statistical analyses were performed in ArrayStudio on log‐transformed data. For those analyses not standard in ArrayStudio, the programs R or JMP were used. Biochemicals that differed significantly between experimental groups were determined using one‐way and repeated measures ANOVA tests. Multiple comparisons were accounted for by estimating the false discovery rate using q values. Random forest (RF) analysis was performed to bin individual samples into groups based on the similarities and differences of metabolites. P ≤ 0.05 was considered statistically significant, and P < 0.10 was reported as trends.
Results
Baseline Demographics and Clinical Characteristics of the Study Cohort
The detailed demographic and clinical characteristics of the study cohort are shown in Supporting Table S2. Patients with AC were older (mean age, 53.7 years old) compared to other groups (P = 0.001). No differences in sex and races were observed among the four study cohorts. As expected, patients with AC had the lowest levels of hemoglobin (11.9 g/dL), platelet counts (135.1 × 103 cell/mm3), and serum albumin (3.2 g/dL) (P = 0.0001); this group also had the highest levels of serum bilirubin (3.3 mg/dL) and creatinine (1.14 mg/dL) (P = 0.0001).
Comparative Metabolomic Profiles in Cs, HDs, and Patients with ALD
The data set comprises a total of 773 compounds of known identity in serum. A summary of the numbers of biochemicals that achieved statistical significance (P ≤ 0.05) as well as those approaching significance (0.05 < P < 0.10) is shown in Supporting Table S3. Using one‐way ANOVA, we observed significant differences in metabolomic profiles among groups. We used hierarchical clustering to demonstrate the differentiation of metabolites in our study cohorts (Fig. 1A). A majority of AC samples clustered to the right of the plot. C samples tended to cluster more to the left but were interspersed with HD1 and HD2. Principal component analysis (PCA) was then performed using all serum samples with two views of the three‐dimensional (3D) plot (Fig. 1B). PCA differentiated the AC sera from Cs and HDs. There appeared to be some partial segregation of C sera from HD1 and HD2 samples. Differentiation of HD1 from HD2 sera was not evident in the 3D plot. To further explore potential differences in the HD1 and HD2 samples based on global metabolic profiles, a 2D PCA was conducted (Fig. 1C). Although not impressive, there appeared to be some skewing of the HD2 samples lower on the y axis. We then used RF analysis to bin individual samples into groups based on the similarities and differences of metabolites (Supporting Fig. S1). RF analysis of the serum groups resulted in an overall predictive accuracy of 63%. C and AC samples were binned with the greatest accuracy; HD1 and HD2 samples were frequently mis‐segregated to the alternate HD group or C group. The top 30 metabolites that most strongly contributed to binning of individual samples into groups are shown in Supporting Fig. S1. Metabolites with higher “mean decrease accuracy” values contributed more strongly to group differentiation. The top two metabolites in mean decrease accuracy were 7‐methylguanidine, a modified nucleobase that is possibly derived from nucleotide degradation, and cystine, an oxidized dimer of cysteine molecules. Metabolites in the bile acid (BA) pathway were also identified in the top 30 by using RF analysis (Supporting Fig. S1).
Figure 1.
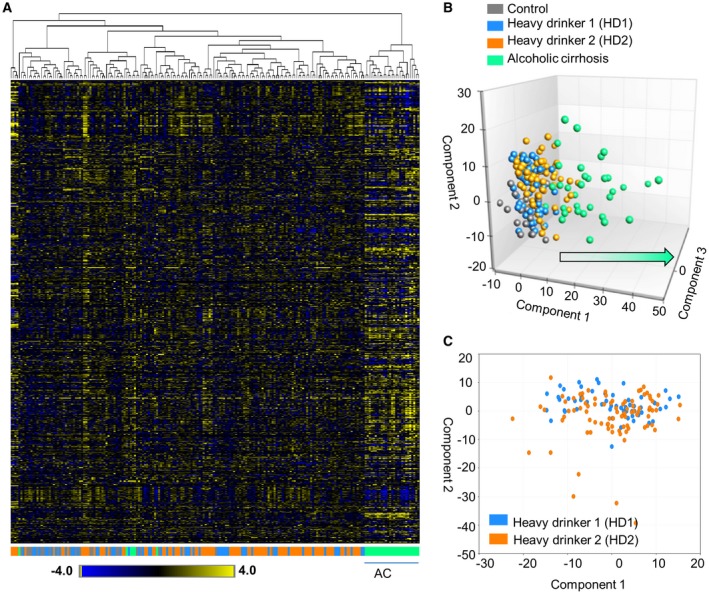
Metabolic profiling of sera. (A) Hierarchical clustering displays differentiation of serum metabolites among study groups. Color scale demonstrates relative expression of metabolites across all samples. A majority of AC samples cluster to the right; control samples tend to cluster more to the left but are interspersed with HD1 and HD2 samples. (B) PCA of all serum samples with two views of the 3D plot. PCA differentiated AC serum from control and HD sera. (C) 2D PCA based on global metabolite profiles in the HD1 and HD2 samples.
Serum Taurine‐conjugated BAs were Strikingly Elevated in HDs and Patients with AC
BAs are produced in the liver and secreted into the intestine where a significant portion are deconjugated and structurally modified into secondary BAs by intestinal bacteria. Both primary (Fig. 2A) and secondary (Fig. 2B) BAs were elevated in HD1 and HD2 sera relative to Cs, although not all increases achieved statistical significance (Supporting Table S4; Fig. 2). Although our HD cohort had no clinically apparent AC, we observed that a subset of these patients in fact had a slight elevation of transaminases (Supporting Table S2). Interestingly, we found that those with abnormal transaminases (using the laboratory cutoff of 40 U/L) had significantly higher levels of glycochenodeoxycholic acid (GCDCA) sulfate, taurocholenate sulfate, glycocholenate sulfate, and glycodeoxycholate (GDCA) sulfate compared to those with normal transaminases.
Figure 2.
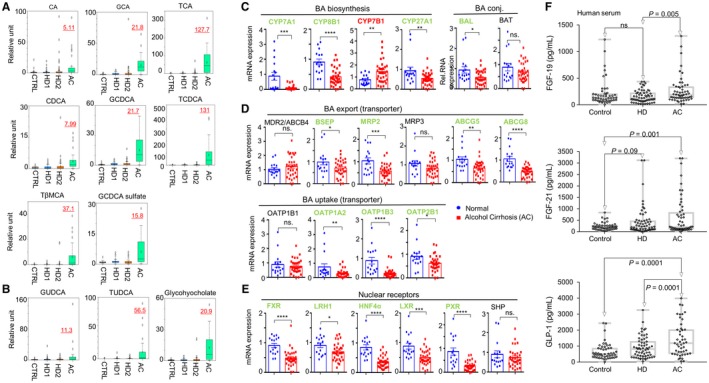
Primary and secondary BAs. Each Color represents a study group. Box plots demonstrating relative levels of (A) primary BAs and (B) secondary BAs among the study cohorts. (C‐E) qRT‐PCR of hepatic mRNA expression of genes related to (C) BA biosynthesis, (D) BA export and uptake, and (E) nuclear receptors regulating BA synthesis in controls and patients with AC. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. (F) Serum levels of FGF19, FGF21, and GLP‐1 in controls, HDs, and those with ALD. Abbreviations: CA, cholate; GCA, glycocholate; SHP, small heterodimer partner; TβMCA, tauro‐beta‐muricholate.
Serum levels of primary BAs (Fig. 2A), such as cholic acid (CA), glycocholic acid (GCA), taurocholic acid (TCA), chenodeoxycholic acid (CDCA), GCDCA, taurochenodeoxycholic acid (TCDCA), tauro‐beta‐muricholate, and GCDCA sulfate, were significantly higher in AC sera. Similar observations in the increase in levels of secondary BAs were also found in patients with AC (Fig. 2B). It was noted that taurine‐conjugated BAs (TCA, TCDCA, tauroursodeoxycholic acid [TUDCA]) showed a more striking elevation than glycine‐conjugated forms (GCA, GCDCA, glycoursodeoxycholic acid [GUDCA]). Increases in serum BAs may reflect increased synthesis because we found that the BA synthesis intermediate 7 alpha‐hydroxy‐3‐oxo‐4‐cholestenoic acid was sharply higher in AC sera relative to all other groups (Supporting Fig. S2).
Hepatic Cytochrome P450, Family 7, Subfamily B1 Expression in the Acidic Pathway of BA Synthesis was Significantly Elevated in Patients with ALD
To further determine the mechanism underlying the alterations in serum BA levels, we measured the levels of gene expression involved in BA synthesis in the liver of patients with AC. Expression of rate‐limiting enzymes in the neutral pathway of BA biosynthesis microsomal cholesterol 7α‐hydroxylase (cytochrome P450 [CYP], family 7, subfamily A, member 1 [CYP7A1] and CYP, family 8, subfamily B, polypeptide 1 [CYP8B1]) as well as the enzyme in the acidic pathway CYP27A1, was markedly down‐regulated in AC livers versus Cs (Fig. 2C, left). In contrast, hepatic messenger RNA (mRNA) expression of CYP7B1 in the acidic pathway was significantly higher than Cs. Unconjugated BAs are conjugated with taurine by the liver‐specific enzymes BA coenzyme A (CoA) ligase (BAL) and BA CoA:amino acid N‐acyltransferase (BAT). Interestingly, BAL but not BAT was down‐regulated in AC livers relative to Cs (Fig. 2C, right).
Among BA transporters, both export (bile salt export pump [BSEP], multidrug resistance‐associated protein 2 [MRP], adenosine triphosphate binding cassette, subfamily G, member 5 and member 8 [ABCG5, ABCG8]) and uptake (organic‐anion‐transporting polypeptide [OATP] 1A2, OATP1B3, OATP2B1) genes were down‐regulated in AC versus Cs (Fig. 2D). In addition, the levels of several nuclear receptors, including farnesoid X receptor (FXR), hepatocyte nuclear factor 4α (HNF4α), liver X receptor (LXR), and pregnane X receptor (PXR), were markedly reduced in patients with AC (Fig. 2E).
Serum FGF21 and GLP‐1 Levels were Markedly Elevated in HDs and Patients with ALD
BAs in the intestine can also activate intestinal cells to release several hormones, such as FGF19 and GLP‐1. Levels of FGF19 in HDs and Cs were comparable; however, they were significantly increased in patients with AC versus HDs (Fig. 2F, top). Levels of GLP‐1 were significantly increased in HDs and patients with AC compared to Cs (Fig. 2F, bottom). We also measured FGF21 and found increased levels in HDs (442 ± 81.8 pg/mL) and in patients with AC (632.4 ± 111.8) compared to Cs (222.0 ± 24.0 pg/mL) (Fig. 2F, middle).
Serum Methionine/homocysteine Metabolites were Significantly Altered in Patients with AC
Serum of patients with AC and to a lesser extent HD2 and HD1 sera displayed increases in metabolites from the homocysteine pathway (Supporting Table S5), including methionine, S‐adenosylhomocysteine, cystathionine, and cysteine (Fig. 3A). The antioxidant glutathione is one of the primary molecules generated by cells to combat oxidative stress. We found levels of glutathione‐related metabolites were lower in the serum of patients with AC relative to Cs (Fig. 3B), whereas serum taurine and glycine levels remained unaltered (Fig. 3C).
Figure 3.
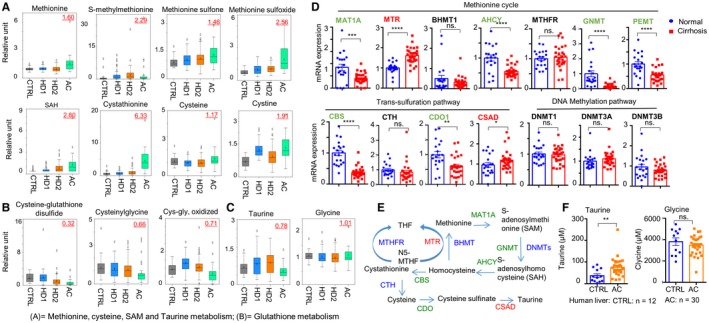
Box plots demonstrating relative levels of metabolites. (A) methionine metabolism; (B) glutathione metabolism. (C) Box plots of serum taurine and glycine. (D) qRT‐PCR of hepatic mRNA expression of genes related to methionine metabolism, trans‐sulfuration, and DNA methylation pathway in controls (n = 10, in duplicate) and those with ALD (n = 30). *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. (E) Schematic diagram illustrating the methionine metabolism pathway. (F) Hepatic taurine and glycine concentration from controls (n = 12) and from those with ALD (n = 30). Abbreviations: AHCY, adenosylhomocysteinase; BHMT1, betaine‐homocysteine S‐methyltransferase 1; CBS, cystathionine‐beta‐synthase; CDO1, cysteine dioxygenase type 1; CTH, cystathionine gamma‐lyase; DNMT, DNA methyltransferase; GNMT, glycine N‐methyltransferase; MTHFR, methylenetetrahydrofolate reductase; N5‐MTHF, N5‐methyltetrahydrofolate; PEMT, phosphatidylethanolamine methyltransferase; SAH, S‐adenosylhomocysteine; SAM, S‐adenosylmethionine; THF, tetrahydrofolate.
To establish potential correlations between the expression of genes involved in these pathways and levels of serum metabolites, we determined mRNA expression of major genes in the livers of Cs versus patients with AC. Interestingly, the majority of genes showed down‐regulation (methionine adenosyltransferase 1A [MAT1A], adenosylhomocysteinase [AHCY], glycine N‐methyltransferase, phosphatidylethanolamine methyltransferase [PEMT], cystathionine‐beta‐synthase, cysteine dioxygenase type 1), whereas methionine synthase (MTR) and cysteine sulfinic acid decarboxylase (CSAD) exhibited increased expression (Fig. 3D). Up‐regulation of MTR and CSAD may contribute to elevated methionine and taurine synthesis, respectively (Fig. 3E). Indeed, liver taurine content was markedly increased in the livers of patients with AC compared to C livers (Fig. 3F). It is plausible that dysregulation in homocysteine metabolism in the livers of patients with AC produced more taurine substrate for conjugation to BAs, resulting in substantial elevation of taurine‐conjugated BAs (Fig. 2A).
Significant Elevation in Heme Degradation Metabolites in Patients with AC
We also found that heme degradation metabolites were elevated in sera of patients with AC relative to Cs and the HD groups (Supporting Table S6). Unconjugated bilirubin, derived from heme degradation, circulates in the bloodstream and is removed from the circulation by the liver. Metabolomic analysis showed an elevation in serum bilirubin levels (Fig. 4A); this was consistent with patient characteristics (Supporting Table S2). In addition, we observed an increase in serum levels of biliverdin and urobilinogen in patients with ALD. Conversion of biliverdin to bilirubin is under the control of biliverdin reductase A (BVRA), and its expression was increased in patients with AC (Fig. 4B).
Figure 4.
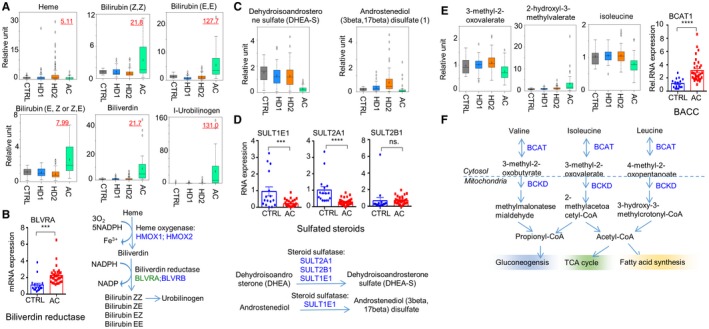
Metabolites in bilirubin, sulfated steroids, and branched‐chain amino acids. (A) Box plots demonstrating the relative levels of metabolites in hemoglobin and bilirubin metabolism. (B) qRT‐PCR of hepatic mRNA expression of the BVRA gene in controls and patients with AC and a schematic diagram illustrating heme metabolism. ***P < 0.001, ****P < 0.0001. (C) Box plots demonstrating relative levels of metabolites of the sulfated steroids dehydroisoandrosterone sulfate and androstenediol (3beta,17beta) disulfate.1 (D) Hepatic mRNA expression of genes related to sulfated steroid metabolism and diagram demonstrating the correlated pathway. (E) Box plots demonstrating relative levels of metabolites in leucine, isoleucine, and valine metabolism and hepatic mRNA expression of the BCAT1 gene in controls and those with AC. (F) Schematic diagram illustrating the pathway of leucine, isoleucine, and valine metabolism. Abbreviations: BCKD, branched‐chain alpha‐ketoacid dehydrogenase complex; BLVRA/BLVRB, biliverdin reductase; CoA, coenzyme A; NADP, nicotinamide adenine dinucleotide phosphate; NADPH, nicotinamide adenine dinucleotide phosphate reduced form; TCA cycle, tricarboxylic acid cycle.
Reduced Levels of Sulfated Steroids Observed in Patients with AC
Interestingly, we found that sulfated steroids, such as dehydroisoandrosterone sulfate, were markedly decreased in AC sera relative to Cs (Supporting Table S7; Fig. 4C). Levels of androstenediol (3beta,7beta) disulfate,1 on the other hand, were elevated in HD2 but not in ALD sera. Expression of genes related to sulfuration (sulfotransferase [SULT] 1E1 and SULT2A1) was significantly decreased in liver tissues from patients with AC (Fig. 4D).
Alterations in Levels of Serum Branched‐Chain Amino Acid Metabolites in Patients with AC
We found that serum levels of branched‐chain amino acids (BCAAs) were reduced in patients with AC (Fig. 4E; Supporting Table S8) along with the α‐ketoacids produced by BCAA transaminase (BCAT) in the first step of BCAA catabolism. Each BCAA was significantly reduced (e.g., isoleucine) along with its respective α‐ketoacids, such as 3‐methyl‐2‐oxovalerate (generated from isoleucine) (Fig. 4E,F). Hepatic expression of BCAT1 was significantly higher in patients with AC compared to Cs (Fig. 4E, right). Each BCAA was reduced to a statistically significant level (e.g., isoleucine) along with its α‐ketoacid derivative 3‐methyl‐2‐oxovalerate. The 2‐hydroxycarboxylic acids, such as 2‐hydroxy‐3‐methylvalerate, were statistically increased in AC sera (Supporting Table S8; Supporting Fig. S3).
Alterations in the Levels of Serum Glucose Metabolites in Patients with AC
Glycolysis intermediates were altered in the serum of HDs and patients with AC compared to Cs (Supporting Fig. S4A,B). We found that glucose levels were statistically higher in AC sera. Coincident with the higher glucose levels, levels of 1,5‐anhydroglucitol (1,5‐AG) were also lower relative to other groups. 1,5‐AG, a noninvasive marker indicative of the levels of serum glucose, circulates in the blood and is filtered by renal glomeruli. Renal reabsorption of 1,5‐AG is inhibited by excess glucose, as shown by the reduced levels of 1,5‐AG in AC. Pyruvate levels were slightly increased in AC sera (although not statistically significant), whereas serum lactate was significantly elevated in AC (Supporting Fig. S4B). We next determined the changes in metabolites related to the mitochondrial tricarboxylic acid cycle and found a significant increase in the metabolites (citrate, alpha‐ketoglutarate, succinate, fumarate, and malate) in this pathway in AC and HD sera relative to Cs (Supporting Fig. S4C).
Alterations in the Levels of Serum Fatty Acid Metabolites in Patients with ALD
Serum levels of several long‐chain fatty acids were significantly elevated in the serum of patients with AC; these included serum palmitate, palmitoleate, and eicosenoate (Supporting Table S9). Acylcarnitines are generated to permit the transfer of free fatty acids across the mitochondrial membrane for β‐oxidation. Interestingly, we found a significant elevation of serum acylcarnitines in patients with AC.
Trajectory of Serum Metabolites in Response to Alcohol Abstinence
We demonstrated alterations of several serum metabolites in several metabolic pathways in HDs and patients with AC (Figs. 1, 2, 3, 4). To further test that changes in the levels of these metabolites were in response to alcohol, we prospectively followed the trajectory of their levels in HDs participating in an alcohol treatment program. Serum metabolomic analyses were performed at baseline (immediately before starting the program, week 0) and at the indicated times shown in Fig. 5. Among the metabolites being measured, we found that levels of GCDCA, GCA, GDCA, glycolithocholic acid, and GUDCA decreased significantly with alcohol abstinence compared to baseline (Fig. 5A,B). On the other hand, homocysteine metabolites and heme degradation metabolites were not markedly altered by alcohol abstinence (Fig. 5C,D).
Figure 5.
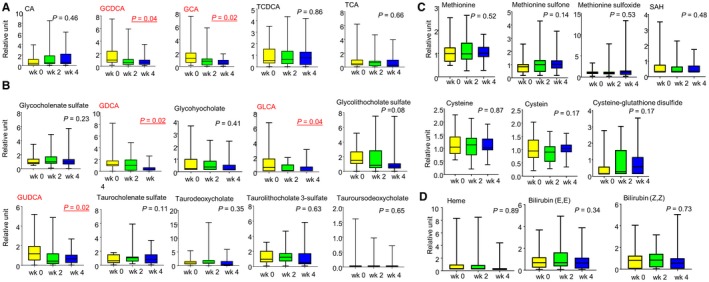
Trajectory of metabolites after alcohol abstinence. Trajectory of metabolites in the metabolism of (A) primary BAs, (B) secondary BAs, (C) methionine and glutathione, and (D) hemoglobin and bilirubin metabolism at baseline and at week 2 and week 4 after alcohol abstinence. Abbreviations: CA, cholate; GDCA, glycodeoxycholate; GLCA, glycolithocholic acid; SAH, S‐adenosylhomocysteine; wk, week.
Serum BAs Among Patients with AC Stratified by Disease Severity
To determine the levels of BAs in patients with AC by severity, we compared their levels stratified by Child‐Pugh classification (Supporting Table S10). Among primary BA metabolites, we found that only the levels of conjugated GCA (P = 0.02), TCA (P = 0.01), GCDCA (P = 0.0001), and TCDCA (P = 0.007) were significantly increased during progression of the disease from Child‐Pugh A to C. Interestingly, we did not observe any changes in the levels of secondary BAs by Child‐Pugh classification except for glycohyocholate (P = 0.04). The same trend was observed when we examined the levels of these metabolites in correlation with MELD scores (Fig. 6). Serum GCDCA (P = 0.003) and TCDCA (P = 0.04) levels significantly correlated with MELD scores, whereas GCA (P = 0.08) and TCA (P = 0.07) were less correlative.
Figure 6.
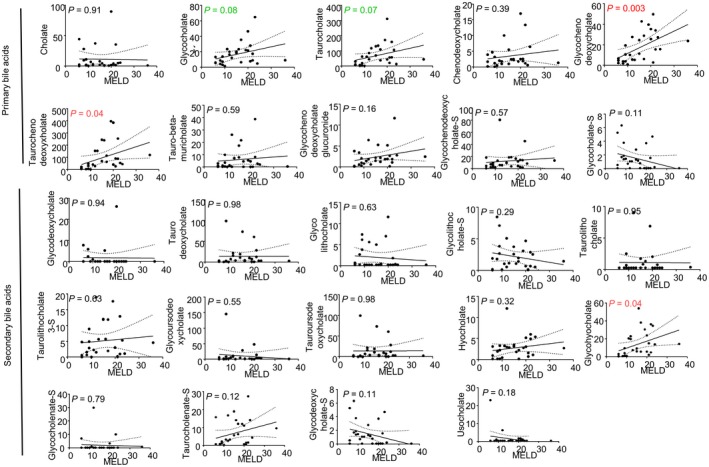
Linear regression analysis between the levels of BA and MELD scores.
Discussion
This study represents the first complete and comprehensive metabolic profiling of the largest cohort of human sera collected from well‐characterized HDs and HDs with AC. It is also the first study to determine trajectory changes of metabolites in this cohort following abstinence. Overall, metabolites produced from a wide range of pathways, including BAs, heme degradation products, sulfated steroids, fatty acids, tricarboxylic acid cycle, and BCAA metabolites, were significantly altered in HDs and in HDs with AC. We observed progressive changes in the quantities of metabolites from different metabolic pathways from HD1 to HD2 and AC. The total number of significantly increased metabolites rose from 89 to 195 when the analyses were performed in HDs whose last alcohol consumption was >10 days before enrollment compared to those who continued to drink up to the time of enrollment. This observation is of interest as we have previously shown that the timing of last alcohol consumption, especially within 10 days before enrollment, affects levels of circulating neutrophils and serum transaminase23. Once AC developed, alterations in the number of metabolites were more profound, with a total of 282 and 138 metabolites being increased and decreased, respectively, when compared to Cs.
Can Serum‐conjugated BAs Serve as Noninvasive Diagnostic Markers for AC?
Three taurine‐conjugated BAs were strikingly elevated more than 50‐fold in AC serum relative to Cs; these were TCA (127‐fold), TCDCA (131‐fold), and TUDCA (56‐fold) (Fig. 2A,B). Glycine‐conjugated BAs (GCA, GCDCA, GUDCA) were elevated about 10‐fold to 20‐fold in AC versus C sera. Nonconjugated BAs (CA, CDCA) were elevated less than 10‐fold in AC serum. The results suggest increased liver BA synthesis, conjugation, or export.
Synthesis of BAs is initiated by oxidation of cholesterol by two different pathways, the neutral pathway and the acidic pathway.24 Major BA synthesis enzymes CYP7A1 and CYP8B1 (neutral pathway) and CYP27A1 (acidic pathway)25 were significantly down‐regulated in AC livers (Fig. 2C, left), which could have been due to feedback inhibition of their expression by BAs. On the contrary, CYP7B1 (acidic pathway) showed a marked induction in the livers of patients with AC. Thus, CYP7B1 may be the key contributor to increased BA synthesis in patients, an assumption that should be mechanistically tested in a future study. In line with this observation, we found a progressive increase mainly in the subsets of conjugated primary (GCDCA, TCDCA, GCA, TCA) but not secondary (except for glycohyocholate) BA metabolites in parallel with disease severity in patients with AC (Fig. 6; Supporting Table S10).
BAL and BAT mediate the conjugation of CA and CDCA with taurine and glycine, respectively. Surprisingly, BAL expression was down‐regulated whereas BAT expression showed no changes in the livers of patients with AC (Fig. 2C, right). Therefore, increased taurine‐ and glycine‐conjugated BAs are unlikely to result from changes of BAL and BAT. Instead, the significantly increased liver taurine content (Fig. 3F) due to increased CSAD and taurine synthesis (Fig. 3E) would provide more substrates to generate taurine‐conjugated BAs.
Another interesting observation was that most BA transporters, including export (BSEP, MRP2, ABCG5, ABCG8) and uptake (OATP1A2, OATP1B3, OATP2B1), were all down‐regulated in the livers of patients with AC (Fig. 2D), suggesting an overall dysregulation. In addition, BAs serve as signaling molecules by activating nuclear receptor FXR in the liver.26, 27 Activation of FXR leads to inhibition of genes related to BA synthesis (CYP7A1, CYP8B1, CYP27A1, and CYP7B1)28 through activation of the small heterodimer partner.29, 30 Expression of liver receptor homolog 1 (LRH1), HNF4a, LXR, and PXR was down‐regulated in the livers of patients with AC (Fig. 2E), which may contribute to the decreased expression of CYP enzymes.
Can Serum FGF21 and GLP‐1 Serve as Predictive Markers from Heavy Drinking to AC Transition?
BAs can activate FXR in the intestine, leading to the release of FGF19 from enterocytes31; this, in turn, inhibits BA synthesis through CYP7A1. Although FXR expression did not occur in the intestine, levels of FGF19 were highly elevated in patients with AC versus Cs or HDs (Fig. 2F).
Another closely related nuclear receptor‐regulated FGF negatively regulating BA synthesis is FGF21.26, 32 BAs also act as the ligands for the G protein‐coupled receptor (GPCR) TGR5.33 Once activated, TGR5 stimulates the release of GLP‐1 from enterochromaffin L cells in the intestine.34 LXR negatively regulates FGF21 expression. The increase in serum levels of FGF21 may be secondary to the inhibition of LXR in ALD35 (Fig. 2E). Both FGF21 and GLP‐1 were markedly elevated in HDs and further elevated in ALD relative to Cs. Thus, the increased levels of FGF21 and GLP‐1 but not FGF19 are associated with the severity of alcoholic liver injury from heavy drinking to ALD.
At the time of this manuscript preparation, dysregulation of serum BAs and FGF19 was reported in patients with AH.36 Interestingly, one common observation is the significant reduction in de novo BA synthesis gene CYP7A1 among patients with AH.36 Our results (focusing on AC) provide complementary support that alterations of BA homeostasis may be a key factor driving the progression of ALD. Modulation of BA signaling and metabolism could represent a promising therapeutic strategy for the treatment of ALD.
Homocysteine Metabolism is the Second Most Severely Disrupted Pathway in Patients with AC
Heavy alcohol consumption is known to disturb the methionine metabolic pathway.37 High levels of serum methionine (Fig. 3A) are likely due to inhibition of hepatic MAT1A (Fig. 3D), similar to the results of a previous report.38 A number of studies have shown that alcohol consumption reduces hepatic glutathione.38, 39, 40 Interestingly, we observed significantly lower levels of glutathione‐related metabolites in patients with ALD (Fig. 3B). Production of reactive oxygen species can lead to oxidation of metabolites and cellular countermeasures to reduce oxidative stress. We found that levels of the oxidized methionine species methionine sulfoxide and methionine sulfone were significantly increased in patients with AC (Fig. 3A). Cystine, representing an oxidized dimer of cysteine molecules, was also increased in HDs and patients with AC relative to Cs (Fig. 3A). Glutathione can be generated from methionine by cystathionine and cysteine synthesis through the trans‐sulfuration process (Fig. 3E). Although cystathionine was significantly increased in AC serum (Fig. 3A), this did not translate into elevated cysteine. If cystathionine and cysteine levels in sera reflect production of cysteine in the liver, there may be evidence for some restriction in cysteine production from cystathionine, although we did not observe an increase in the expression of hepatic cystathionine gamma‐lyase in patients with AC (Fig. 3D,E). Dysregulation of methionine/homocysteine metabolism may lead to increased production of taurine, resulting in substantial elevation of taurine‐conjugated instead of glycine‐conjugated BAs (Fig. 2A). Indeed, a significant increase in the hepatic expression of CSAD (Fig. 3D,E) and hepatic taurine levels (Fig. 3F) was observed in patients with AC.
Disruption of other Metabolic Pathways in Patients with AC
Levels of total bilirubin in our study cohort (Supporting Table S2) were significantly higher than those of Cs and HDs; thus, alterations of several metabolites related to heme metabolism in patients with ALD (Fig. 4A) were expected. We also found that hepatic BVRA, a multifunctioning protein primarily responsible for the reduction of biliverdin to bilirubin, was significantly up‐regulated in patients with ALD (Fig. 4B). BVRA has been shown to attenuate hepatic steatosis41; whether our observation is indicative of the protective mechanism against alcohol‐induced hepatic steatosis/liver injury in patients with AC should be further determined. Sulfation and desulfation are vital biological processes that regulate steroidogenesis and thus steroid hormone action in a variety of tissue.42 We found that levels of several sulfated steroids were significantly lower in patients with AC (Fig. 4C). Because steroid sulfation can occur in the liver, a reduction may reflect hepatic dysfunction as indicated by decreased hepatic expression of SULT (Fig. 4D). However, a recent study reported the important role of SULT enzymes, notably SULT1E1, in regulating glucose and lipid metabolism.43 Loss of SULT1E1 function results in improving insulin sensitivity, which decreases hepatic gluconeogenesis and lipogenesis. It is also plausible that inhibition of SULT1E1 is a compensatory mechanism to counter the alteration of insulin signaling and derangement in glucose metabolism in patients with ALD (Supporting Fig. S4).
Our data also suggest that changes in the serum metabolic profiles depend on the timing of alcohol consumption, as indicated by the number of changes in metabolites in HD1 and HD2 samples (Supporting Table S3). To further test this hypothesis, we found that levels of several metabolites, notably in the BA pathway, demonstrated a downward trajectory following alcohol abstinence (Fig. 5A,B). Whether we can use these metabolites as biomarkers to confirm alcohol abstinence should be further studied. The mechanistic illustration of the effect of alcohol on the dysregulation of genes involving several metabolic pathways leading to alterations in serum metabolomic profiles was summarized in Fig. 7.
Figure 7.
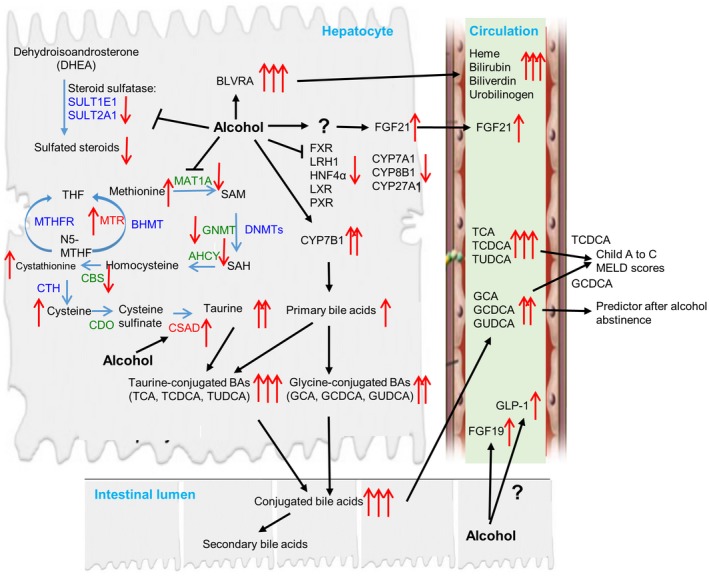
Mechanistic illustration of the effect of alcohol on the dysregulation of genes involving several metabolic pathways leading to alterations in serum metabolomic profiles. The levels of taurine‐conjugated BAs (TCA, TCDCA, and TUDCA) are markedly increased compared to glycine‐conjugated forms (GCA, GCDCA, and GUDCA) in patients with AC. This is associated with an increase in hepatic CYP7B1 expression in the BA synthesis pathway and taurine content secondary to the dysregulation of homocysteine metabolism. Increased levels of GCDCA, TCDCA, GCA, and TCA are positively correlated with disease progression from Child‐Pugh A to C and MELD scores, whereas GCDCA, GCA, and GUDCA are better predictors following alcohol abstinence. Alcohol also inhibits the expression of several nuclear receptors, such as FXR, HNF4α, LXR, and PXR, and affects the serum levels of FGF19, FGF21, and GLP‐1. An increase in the by‐product of heme metabolism in the serum is associated with the induction of BVRA, whereas the inhibition of genes related to sulfuration, such as SULT1E1 and SULT2A1, leads to a reduction in serum‐sulfated steroids. Abbreviations: AHCY, adenosylhomocysteinase; BHMT, betaine‐homocysteine S‐methyltransferase; CBS, cystathionine‐beta‐synthase; CDO, cysteine dioxygenase; CTH, cystathionine gamma‐lyase; DNMT, DNA methyltransferase; GNMT, glycine N‐methyltransferase; MTHFR, methylenetetrahydrofolate reductase; N5‐MTHF, N5‐methyltetrahydrofolate; PEMT, phosphatidylethanolamine methyltransferase; SAH, S‐adenosylhomocysteine; SAM, S‐adenosylmethionine; THF, tetrahydrofolate.
The strengths of our study are its prospective design and the inclusion of well‐characterized Cs, HDs, and patients with AC. We acknowledge several limitations. First, we did not perform paired analyses of samples from the same patients to determine the association between gene expression and serum metabolome. In clinical practice, it may be ethically impossible to conduct liver biopsy among healthy Cs, and it was impractical to perform a liver biopsy for all patients in our cohort. Second, although our data clearly showed metabolic signatures in patients with AC stratified by severity, the majority of our patients were those with decompensation (Child‐Pugh B and C). Future studies with a larger sample size for patients with compensated AC (Child‐Pugh A) may be needed to validate our results. Nonetheless, results from our studies may provide a new noninvasive diagnostic approach for ALD using serum metabolome profiling, especially conjugated BAs.
Supporting information
Acknowledgment
We thank the team at Metabolon for conducting the metabolomics analysis.
Supported by the National Institutes of Health (grants K01 AA026385 to Z.Y. and U01AA021840, R01 DK107682, R01 AA025208, and UH2AA026903 to S.L.), VA Merit Awards (1I01CX000361 to S.L.), and the U.S. Department of Defense (grant W81XWH‐12‐1‐0497 to S.L.).
Potential conflict of interest: Dr. Liangpunsakul consults for Durect Corporation. The other authors have nothing to report.
Contributor Information
Zhihong Yang, Email: yangjoe@iu.edu.
Suthat Liangpunsakul, Email: sliangpu@iu.edu.
References
- 1. Rehm J, Samokhvalov AV, Shield KD. Global burden of alcoholic liver diseases. J Hepatol 2013;59:160‐168. [DOI] [PubMed] [Google Scholar]
- 2. Bellentani S, Saccoccio G, Costa G, Tiribelli C, Manenti F, Sodde M, et al. Drinking habits as cofactors of risk for alcohol induced liver damage. The Dionysos Study Group. Gut 1997;41:845‐850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Rehm J. The risks associated with alcohol use and alcoholism. Alcohol Res Health 2011;34:135‐143. [PMC free article] [PubMed] [Google Scholar]
- 4. Liangpunsakul S, Haber P, McCaughan GW. Alcoholic liver disease in Asia, Europe, and North America. Gastroenterology 2016;150:1786‐1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rubin E, Lieber CS. Alcohol‐induced hepatic injury in nonalcoholic volunteers. N Engl J Med 1968;278:869‐876. [DOI] [PubMed] [Google Scholar]
- 6. Gao B, Bataller R. Alcoholic liver disease: pathogenesis and new therapeutic targets. Gastroenterology 2011;141:1572‐1585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Mandrekar P, Bataller R, Tsukamoto H, Gao B. Alcoholic hepatitis: translational approaches to develop targeted therapies. Hepatology 2016;64:1343‐1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Tian C, Stokowski RP, Kershenobich D, Ballinger DG, Hinds DA. Variant in PNPLA3 is associated with alcoholic liver disease. Nat Genet 2010;42:21‐23. [DOI] [PubMed] [Google Scholar]
- 9. Stickel F, Buch S, Lau K, Meyer zu Schwabedissen H, Berg T, Ridinger M, et al. Genetic variation in the PNPLA3 gene is associated with alcoholic liver injury in caucasians. Hepatology 2011;53:86‐95. [DOI] [PubMed] [Google Scholar]
- 10. Lieber CS. Relationships between nutrition, alcohol use, and liver disease. Alcohol Res Health 2003;27:220‐231. [PMC free article] [PubMed] [Google Scholar]
- 11. Palmer BF, Clegg DJ. Electrolyte disturbances in patients with chronic alcohol‐use disorder. N Engl J Med 2017;377:1368‐1377. [DOI] [PubMed] [Google Scholar]
- 12. Tran M, Yang Z, Liangpunsakul S, Wang L. Metabolomics analysis revealed distinct cyclic changes of metabolites altered by chronic ethanol‐plus‐binge and Shp deficiency. Alcohol Clin Exp Res 2016;40:2548‐2556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Yang Z, Tsuchiya H, Zhang Y, Lee S, Liu C, Huang Y, et al. REV‐ERBalpha activates C/EBP homologous protein to control small heterodimer partner‐mediated oscillation of alcoholic fatty liver. Am J Pathol 2016;186:2909‐2920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Harada S, Takebayashi T, Kurihara A, Akiyama M, Suzuki A, Hatakeyama Y, et al. Metabolomic profiling reveals novel biomarkers of alcohol intake and alcohol‐induced liver injury in community‐dwelling men. Environ Health Prev Med 2016;21:18‐26. Erratum. In: Environ Health Prev Med 2016;21:283‐284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. US Department of Health and Human Services; National Institute of Health; National Institute on Alcohol Abuse and Alcoholism . Helping patients who drink too much. A clinician’s guide. 2005 ed http://pubs.niaaa.nih.gov/publications/Practitioner/CliniciansGuide2005/guide.pdf. Accessed May 2018. [Google Scholar]
- 16. Gough G, Heathers L, Puckett D, Westerhold C, Ren X, Yu Z, et al. The utility of commonly used laboratory tests to screen for excessive alcohol use in clinical practice. Alcohol Clin Exp Res 2015;39:1493‐1500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Liangpunsakul S, Toh E, Ross RA, Heathers LE, Chandler K, Oshodi A, et al. Quantity of alcohol drinking positively correlates with serum levels of endotoxin and markers of monocyte activation. Sci Rep 2017;7:4462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Yang Z, Ross RA, Zhao S, Tu W, Liangpunsakul S, Wang L. LncRNA AK054921 and AK128652 are potential serum biomarkers and predictors of patient survival with alcoholic cirrhosis. Hepatol Commun 2017;1:513‐523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang L, Yang Z, Trottier J, Barbier O, Wang L. Long noncoding RNA MEG3 induces cholestatic liver injury by interaction with PTBP1 to facilitate shp mRNA decay. Hepatology 2017;65:604‐615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Zhang Y, Xu N, Xu J, Kong B, Copple B, Guo GL, et al. E2F1 is a novel fibrogenic gene that regulates cholestatic liver fibrosis through the Egr‐1/SHP/EID1 network. Hepatology 2014;60:919‐930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zhao Y, Yang Z, Wu J, Wu R, Keshipeddy SK, Wright D, et al. High‐mobility‐group protein 2 regulated by microRNA‐127 and small heterodimer partner modulates pluripotency of mouse embryonic stem cells and liver tumor initiating cells. Hepatol Commun 2017;1:816‐830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Choiniere J, Wu J, Wang L. Pyruvate dehydrogenase kinase 4 deficiency results in expedited cellular proliferation through E2F1‐mediated increase of cyclins. Mol Pharmacol 2017;91:189‐196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Li M, He Y, Zhou Z, Ramirez T, Gao Y, Gao Y, et al. MicroRNA‐223 ameliorates alcoholic liver injury by inhibiting the IL‐6‐p47phox‐oxidative stress pathway in neutrophils. Gut 2017;66:705‐715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Rudraiah S, Zhang X, Wang L. Nuclear receptors as therapeutic targets in liver disease: are we there yet? Annu Rev Pharmacol Toxicol 2016;56:605‐626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Song Y, Liu C, Liu X, Trottier J, Beaudoin M, Zhang L, et al. H19 promotes cholestatic liver fibrosis by preventing ZEB1‐mediated inhibition of epithelial cell adhesion molecule. Hepatology 2017;66:1183‐1196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Fuchs C, Claudel T, Trauner M. Bile acid‐mediated control of liver triglycerides. Semin Liver Dis 2013;33:330‐342. [DOI] [PubMed] [Google Scholar]
- 27. Zhang Y, Liu C, Barbier O, Smalling R, Tsuchiya H, Lee S, et al. Bcl2 is a critical regulator of bile acid homeostasis by dictating Shp and lncRNA H19 function. Sci Rep 2016;6:20559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chiang JY. Negative feedback regulation of bile acid metabolism: impact on liver metabolism and diseases. Hepatology 2015;62:1315‐1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Zhang Y, Hagedorn CH, Wang L. Role of nuclear receptor SHP in metabolism and cancer. Biochim Biophys Acta 2011;1812:893‐908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wang L, Lee YK, Bundman D, Han Y, Thevananther S, Kim CS, et al. Redundant pathways for negative feedback regulation of bile acid production. Dev Cell 2002;2:721‐731. [DOI] [PubMed] [Google Scholar]
- 31. Jung D, York JP, Wang L, Yang C, Zhang A, Francis HL, et al. FXR‐induced secretion of FGF15/19 inhibits CYP27 expression in cholangiocytes through p38 kinase pathway. Pflugers Arch 2014;466:1011‐1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Nishimura T, Nakatake Y, Konishi M, Itoh N. Identification of a novel FGF, FGF‐21, preferentially expressed in the liver. Biochim Biophys Acta 2000;1492:203‐206. [DOI] [PubMed] [Google Scholar]
- 33. Kawamata Y, Fujii R, Hosoya M, Harada M, Yoshida H, Miwa M, et al. A G protein‐coupled receptor responsive to bile acids. J Biol Chem 2003;278:9435‐9440. [DOI] [PubMed] [Google Scholar]
- 34. Ripken D, van der Wielen N, Wortelboer HM, Meijerink J, Witkamp RF, Hendriks HF. Nutrient‐induced glucagon like peptide‐1 release is modulated by serotonin. J Nutr Biochem 2016;32:142‐150. [DOI] [PubMed] [Google Scholar]
- 35. Uebanso T, Taketani Y, Yamamoto H, Amo K, Tanaka S, Arai H, et al. Liver X receptor negatively regulates fibroblast growth factor 21 in the fatty liver induced by cholesterol‐enriched diet. J Nutr Biochem 2012;23:785‐790. [DOI] [PubMed] [Google Scholar]
- 36. Brandl K, Hartmann P, Jih LJ, Pizzo DP, Argemi J, Ventura‐Cots M, et al. Dysregulation of serum bile acids and FGF19 in alcoholic hepatitis. J Hepatol 2018;69:396‐405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Kharbanda KK. Methionine metabolic pathway in alcoholic liver injury. Curr Opin Clin Nutr Metab Care 2013;16:89‐95. [DOI] [PubMed] [Google Scholar]
- 38. Lee TD, Sadda MR, Mendler MH, Bottiglieri T, Kanel G, Mato JM, et al. Abnormal hepatic methionine and glutathione metabolism in patients with alcoholic hepatitis. Alcohol Clin Exp Res 2004;28:173‐181. [DOI] [PubMed] [Google Scholar]
- 39. Jewell SA, Di Monte D, Gentile A, Guglielmi A, Altomare E, Albano O. Decreased hepatic glutathione in chronic alcoholic patients. J Hepatol 1986;3:1‐6. [DOI] [PubMed] [Google Scholar]
- 40. Vogt BL, Richie JP Jr. Glutathione depletion and recovery after acute ethanol administration in the aging mouse. Biochem Pharmacol 2007;73:1613‐1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Hinds TD Jr, Burns KA, Hosick PA, McBeth L, Nestor‐Kalinoski A, Drummond HA, et al. Biliverdin reductase A attenuates hepatic steatosis by inhibition of glycogen synthase kinase (GSK) 3beta phosphorylation of serine 73 of peroxisome proliferator‐activated receptor (PPAR) alpha. J Biol Chem 2016;291:25179‐25191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Mueller JW, Gilligan LC, Idkowiak J, Arlt W, Foster PA. The regulation of steroid action by sulfation and desulfation. Endocr Rev 2015;36:526‐563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Gao J, He J, Shi X, Stefanovic‐Racic M, Xu M, O'Doherty RM, et al. Sex‐specific effect of estrogen sulfotransferase on mouse models of type 2 diabetes. Diabetes 2012;61:1543‐1551. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials