

Abstract
Regulation of protein activity is essential for revealing the molecular mechanisms of biological processes. DNA and RNA achieve many uniquely efficient functions, such as genetic expression and regulation. The chemical capability to synthesize artificial nucleotides can expand the chemical space of nucleic acid libraries and further increase the functional diversity of nucleic acids. Herein, a versatile method has been developed for modular expansion of the chemical space of nucleic acid libraries, thus enabling the generation of aptamers able to regulate protein activity. Specifically, we identify the aptamer targeting integrin alpha3 and show that it can inhibit cell adhesion and migration. Overall, this chemical design-assisted in vitro selection approach enables the generation of functional nucleic acids for elucidating the molecular basis of biological activities and uncovering a novel basis for the rational design of new protein-inhibitor pharmaceuticals.
Keywords: Aptamers, Cell adhesion, Inhibitors, Molecular evolution, Membrane proteins
Graphical Abstract
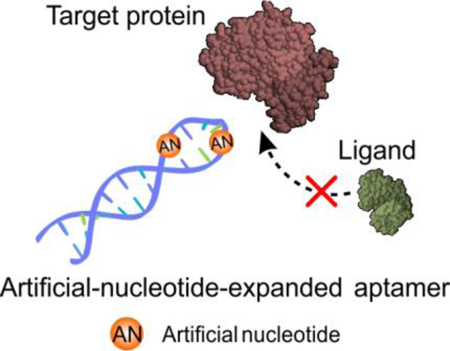
Artificial-nucleotide-expanded aptamer as a protein activity regulator: The chemical design-assisted in vitro selection approach reported in this paper enables the generation of a new class of functional nucleic acids for regulating the biologically relevant performance of its target molecular and uncovering a novel basis for the rational design of new protein-inhibitor pharmaceuticals.
Proteins are central to life activities, and most biological processes involve the accurate regulation of protein activity to perform specific tasks. The exploration of molecules for protein activity modulation is important for uncovering the molecular mechanisms of biological activities,[1] disease processes,[2] and disease treatment.[3] As a class of extensively used chemical molecules with specific recognition function, a few aptamers with strong binding affinity, high specificity and good reversibility have been selected with the ability to inhibit protein activity.[4] Aptamers are promising tools with the obvious advantages of strong binding affinity, high specificity and good reversibility.[4c, 5] Therefore, the exploitation of aptamers to regulate protein activity is of great significance for elucidating the molecular basis of biological processes and uncovering a novel basis for the rational design of new protein-regulator pharmaceuticals.
It has been reported that the biological functions of nucleic acids are strongly coupled to their chemical space and structures.[6] In addition to the canonical nucleobases, DNA/RNA molecules host various chemically diverse modifications. Over 100 naturally occurring modifications have been identified and most life activities involve the modified DNA/RNA to perform specific tasks.[7] Therefore, providing various modifications on nucleic acids is a promising strategy to achieve new properties and better functions. Molecularly designed artificial nucleotides could be an inexhaustible source of innovation and evolution to enrich the configurations and functions of nucleic acids for generating aptamers able to regulate protein activity.
In recent years, a series of artificial nucleotides with different functional groups have been reported.[8] These artificial nucleotides can expand the chemical space of nucleic acid libraries. In addition, artificially expanded libraries have been successfully employed for the selection of aptamers with high affinity and specificity to target molecules.[9] Introducing chemical groups absent in natural nucleic acids can offer new kinds of binding interactions between aptamers and their targets, such as halogen bonding interactions and hydrophobic interactions.[10] Therefore, the use of chemical space expanded libraries can dramatically improve the binding affinity of aptamers, and accordingly increase the success rate of obtaining aptamers for protein activity regulation.[11]
Herein, an artificial-nucleotide-expanded (ANE) cell-SELEX was developed to generate aptamers for protein activity regulation (Scheme 1). As a proof-of-principle, nucleotides with a ferrocenyl group (Fe base), a trifluoromethyl group (F base), and a Z:P pair were employed to demonstrate the ANE libraries. Specifically, Fe base was selected since ferric ions can coordinate with metal binding-sites of proteins,[12] F base was chosen because fluorines can inhibit protein activity by forming halogen bonds or anion coordination,[13] and Z:P pair was introduced to offer a richer variety of conformations over complementary interaction.[14] Moreover, the nitro moiety in Z may act as a “universal weak binder” to enhance the interactions between aptamers and proteins. [9c, 9d, 14] Using these three types of ANE libraries, SELEX was performed to target triple-negative breast cancer cells MDA-MB-231. The aptamer obtained by ANE cell-SELEX using the ZP-containing library is shown to inhibit the biologically relevant performance of its target, integrin alpha 3 (denoted as ITGA3). In addition, this aptamer can reduce ligand binding, thereby inhibiting tumor cell adhesion and migration. Overall, this chemical design-assisted in vitro selection approach makes “biology-oriented synthesis” a guiding principle to harness the power of evolution in the quest for bioactive aptamers, which can, in turn, promote chemical biology research and drug discovery.
Scheme 1.
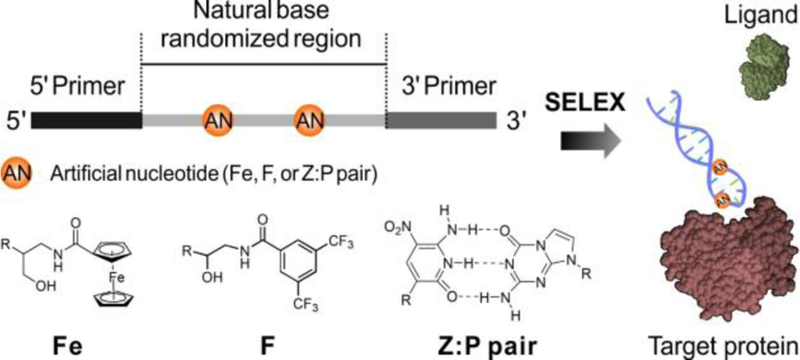
Molecular design of ANE aptamer as a protein activity regulator.
To evolve ANE aptamers targeting MDA-MB-231 cells, ANE cell-SELEX was established (Figure S1–S4, Table S1–S3). The aptamer of interest was chosen from the surviving library members (Table S4) on the basis of clonal dominance and demonstrable binding to MDA-MB-231 cells. This aptamer was named ZAP-1 with the sequence 5’-CCCGAGTGACGCAGC-CCCCGGZGGGATTPATCGGT-GGACACGGTGGCTGA-3’. It was reported that the Z:P pair is highly compatible with natural DNA, offering the six-nucleotide library a richer variety of conformations.[15] As seen from the molecular structures (Figure 1a), artificial bases Z and P are structurally complementary and are similar to the natural bases in DNA, except for the nitro group in Z. Therefore, the GCATZP library would be appropriate for aptamer evolution in this situation.[9d, 15]
Figure 1.
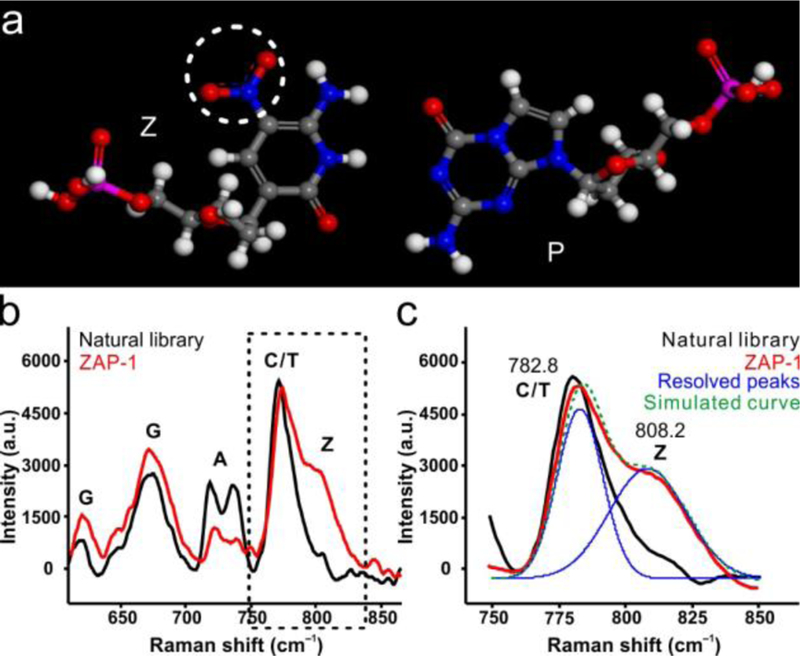
Characterization of ZAP-1. (a) Proposed base pairing model for Z:P pair. (b) Raman spectra of natural library (black) and ZAP-1 (red). (c) Peak-resolution of ZAP-1 Raman spectra (the dotted line in b).
Raman spectra of the synthesized nucleic acids were measured (Figure 1b). The baseline corrected Raman spectra of ZAP-1 (red) and natural library sample (black) are shown in the most informative spectral region, indexing to the general vibrational assignment of purine and pyrimidine residues.[16] Compared to the natural library, Raman spectra of ZAP-1 (Figure 1c) show a relative increase of peak intensity at about 810 cm−1, which is the nitro-related feature. The existence of Z nucleobase in the synthesized oligonucleotides can be proven using Raman spectroscopy, since Z and P bases are highly similar to natural bases except for the nitro group in Z. Confirmed by electrospray ionization mass spectrometry (Figure S5–S7) and Raman spectrometry, the above results demonstrate that Z and P bases have been successfully introduced into the synthesized ZAP-1 oligonucleotides.
The specific binding capacity of ZAP-1 aptamer to target cells was determined by flow cytometry. The results (Figure S8) show that ZAP-1 has obvious binding to the target cell line, but little binding to negative MCF-7 cells, suggesting that ZAP-1 aptamer is specific to MDA-MB-231 cells. The Kd value of aptamer ZAP-1 was 48.5 nM, demonstrating that ZAP-1 aptamer can bind to MDA-MB-231 cells with high affinity (Figure S9).[17] These results prove that ANE cell-SELEX holds great promise in developing aptamers for specific recognition.
It is known that any molecular differences between the target and control cells could lead to the selection of aptamers able to recognize such differences. Therefore, the target molecule of ZAP-1 was identified to find the unique molecular signatures expressed on the MDA-MB-231 cell surface. As shown in Figure 2a, for cells treated with proteinases, ZAP-1 exhibits a loss of its binding ability. From this, it can be deduced that the binding site of ZAP-1 had been removed by the proteinases, indicating that the target molecule is most likely an extracellular membrane protein. The affinity purified protein-aptamer complexes were resolved by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). Compared with protein profiles captured by the initial library, an aptamer-specific protein band is shown in lane 5. This characteristic protein band was then digested and submitted for liquid chromatography electrospray ionization tandem mass spectrometry identification. The MS results suggest that ITGA3 was the target of ZAP-1 (Table S5).
Figure 2.

Analysis and verification of ZAP-1-targeted protein. (a) Flow cytometry of FAM-labelled initial library (red) and ZAP-1 (blue) with MDA-MB-231 cells treated with proteinase K (orange) and trypsin (green). (b) SDS-PAGE was used to identify ZAP-1-specific band T1, marked as asterisk. 1, marker; 2, cell lysate; 3, beads only; 4, proteins captured by initial library; 5, proteins captured by ZAP-1. (c) Fluorescence confocal images of MDA-MB-231 cells incubated with ZAP-1 and anti-ITGA3. (Scale bar, 50 μm.)
To further confirm the target of ZAP-1, an aptamer pull-down experiment was performed. As shown in Figure S11, the band for ITGA3 is clearly displayed in the aptamer-enriched sample, indicating that ITGA3 can be substantially pulled down with the use of aptamer ZAP-1, but not library. In addition, the binding of aptamer to target cells and to ITGA3 knockdown cells was investigated with flow cytometry. For ITGA3 knockdown cells (Figure S12, S13), ZAP-1 exhibits a loss of its binding ability, suggesting that ITGA3 suppression results in the loss of ZAP-1 aptamer binding to the target cells. Furthermore, as shown in confocal microscopy images (Figure 2c), green signal from ZAP-1 and red signal from anti-ITGA3 were clearly present and merged in yellow, indicating the co-localization of ZAP-1 and anti-ITGA3.[18] However, for library-treated cells, such co-localization cannot be observed. The above results confirm that ITGA3 is the target protein of ZAP-1.
Integrins are heterodimeric adhesion receptors for extracellular matrix proteins, consisting of an α-subunit and a β-subunit.[19] Among these, integrin α3β1 can specifically bind to laminins, the major cell-adhesive proteins.[20] Much evidence indicates that integrin α3β1 displays a strong binding activity toward laminin 10 (LN10, Kd = 7.9 nM).[20–21] Therefore, the competitive binding ability of aptamer ZAP-1 and LN10 was investigated with confocal microscopy and flow cytometry. As shown in Figure 3a, for ZAP-1-pretreated cells incubated with LN10, the binding frequency decreased significantly, indicating a weaker affinity of LN10 to ZAP-1-pretreated cells than to untreated cells. In contrast, aptamer ZAP-1 can still effectively bind to LN10-pretreated cells. In addition, the binding ability of ZAP-1 and LN10 to MDA-MB-231 cells was characterized by confocal microscopy. As shown in Figure 3b, for ZAP-1-pretreated cells incubated with LN10, the green signal from ZAP-1 and the red signal from LN10 were both clearly present. Whereas, for LN10-pretreated cells incubated with ZAP-1, a strong red signal was detected, indicating a tight binding of LN10, which is not weakened by the post-treatment of ZAP-1. The quantified data of each sample measured by flow cytometry and confocal microscopy were well matched (Figure 3c,d). These results clearly demonstrate that the binding ability of LN10 can be reduced by the pre-treatment of ZAP-1, and for cells pre-treated with LN10, aptamer ZAP-1 can still bind to target cells.
Figure 3.
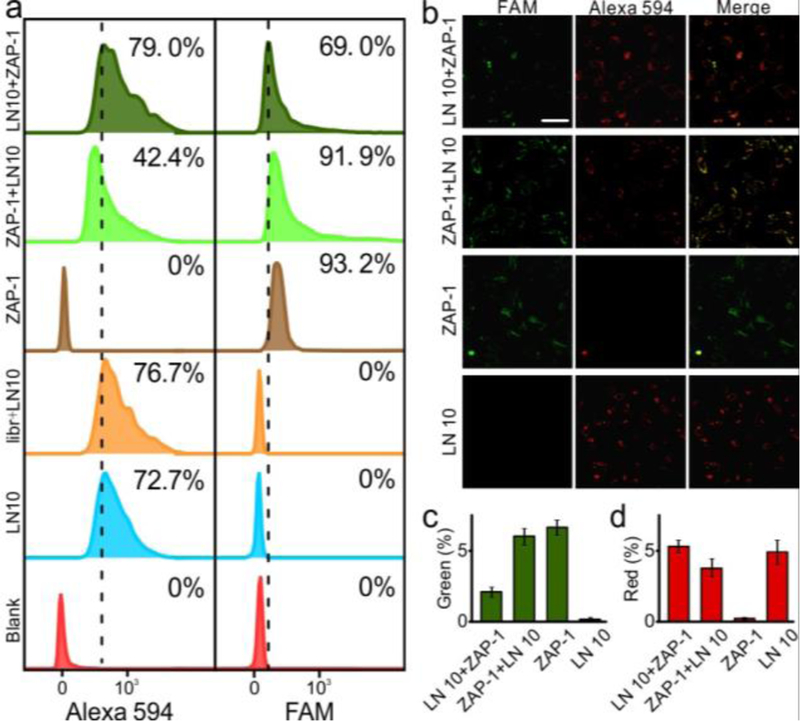
Flow cytometry analysis and fluorescence confocal images of MDA-MB-231 cells incubated with LN10 (Alexa 594) and aptamer ZAP-1 (FAM). (a) Flow cytometry analysis to monitor the binding ability of LN10 and ZAP-1. Percentage of cells above the autofluorescence of cells (dash line) was listed in each sample. (b) Fluorescence confocal images of cells incubated with ZAP-1 and LN 10. Quantified results were shown in c (green channel) and d (red channel). Each bar represents the mean ± SD. (Scale bar, 50 μm.)
It has been proven that protein activity inhibition can be achieved by sterically blocking or altering the active domains.[1a] The ligand-binding activity of integrins has also been reported to be dependent on their conformation and available binding site.[22] As demonstrated above, ZAP-1 pretreatment can decrease the amount of cells bound to LN10 from about 80% to 40%, whereas excess LN10 pretreatment cannot block the binding of ZAP-1. Therefore, it can be concluded that ZAP-1 can regulate the ligand-binding activity of integrin α3β1 by sterically altering its active domains rather than blocking the binding sites. The conformation of integrin α3β1 is shown in Figure 4a, and the extracellular domains of the α3-(blue) and β1-(red) subunits are schematically depicted. Integrin α3β1 assumes both high-affinity (extended) and low-affinity (bent) conformers, which are in rapid equilibrium. Previous studies demonstrated that some molecules could regulate the binding performance of α3β1 by stabilizing one of the conformations.[21–22] In this case, it is speculated that association of ZAP-1 with α3β1 may stabilize the low-affinity conformers (Figure 4a, right), thereby inhibiting the ligand-binding activity of integrin α3β1.[21]
Figure 4.

Modulation of integrin α3β1 by ZAP-1 combination. (a) Schematic model for conformational modulation of integrin α3β1 by ZAP-1 combination. (b) MDA-MB-231 cell adhesion result. (Scale bar, 200 μm.) (c) Attached cell numbers in three separate fields expressed as percentages of the cell number after LN10-incubation. (d) Cell migration result. (e) Closure degree of the scratched area was expressed as percentages of the initial scratched area. Each bar represents the mean ± SD.
The effect of ZAP-1 on MDA-MB-231 cell adhesion and migration was examined. Figure 4b shows photographs of the adhesion of MDA-MB-231 cells. After incubation, cells pretreated with ZAP-1 showed conspicuously less adherence to LN10 than untreated cells. From statistical data of attached cell numbers (Figure 4c), significant inhibition of cell adhesion was observed with ZAP-1 pretreatment. These results demonstrate that ZAP-1 can inhibit MDA-MB-231 cell adhesion to LN10.
The effect of ZAP-1 on MDA-MB-231 cell migration ability was then investigated by a scratch-wound assay. After scratch, the wound area reduced vividly in the LN10-coated plates containing cells, whereas the cells still had a wide gap in the ZAP-1 treated plate (Figure 4d). The closure degree of the scratch area was expressed as percentage of the initial scratch area to quantify cell migration. Importantly, ZAP-1 pretreated cells significantly delayed wound closure compared to cells treated with initial library (Figure 4e). All the above results suggest that ZAP-1 can markedly reduce cell adhesion and migration ability by regulating integrin α3β1 activity, which stands in good agreement with the speculation mentioned before. Therefore, aptamer ZAP-1 could be a potential inhibitor of tumor metastasis.
In this work, we have established a versatile strategy for generating functional nucleic acids by introducing artificial nucleotides with specific chemical groups to the SELEX process. With this strategy, ZAP-1 aptamer, which can bind to and further regulate the activity of target protein, was developed. Once bound, it inhibits integrin α3β1-mediated adhesion and migration of MDA-MB-231 cells. Moreover, the aptamer obtained here shows potential for inhibiting metastatic activity in preclinical research. This study serves as a model for aptamer mediation of cellular and molecular interactions and demonstrates the capabilities of aptamers as protein activity regulators. In sum, this chemical design-assisted in vitro selection approach makes “biology-oriented synthesis” a guiding principle with which to harness the power of evolution in the quest for bioactive aptamers.
Supplementary Material
Acknowledgements
This work was supported by NSFC grants (21521063, 21675120, 21804037, and 21327009) and by NIH GM R35 127130 and NSF 1645215. It is also supported by National Key R&D Program of China (2017YFA0208000), the start-up Research Fund for Prof. Q. Yuan (531107050973, 531109010053).
Footnotes
Supporting information for this article is given via a link at the end of the document.
Conflict of interest
The authors declare no conflict of interest.
Contributor Information
Jie Tan, Molecular Science and Biomedicine Laboratory (MBL), Institute of Chemical Biology and Nanomedicine, State Key Laboratory of Chemo/Biosensing and Chemometrics, College of Chemistry and Chemical Engineering, College of Biology, and Aptamer Engineering Center of Hunan Province, Hunan University, Changsha 410082 China, tan@chem.ufl.edu.
Mengmeng Zhao, Molecular Science and Biomedicine Laboratory (MBL), Institute of Chemical Biology and Nanomedicine, State Key Laboratory of Chemo/Biosensing and Chemometrics, College of Chemistry and Chemical Engineering, College of Biology, and Aptamer Engineering Center of Hunan Province, Hunan University, Changsha 410082 China.
Jie Wang, Key Laboratory of Analytical Chemistry for Biology and Medicine (Ministry of Education), College of Chemistry and Molecular Sciences, Wuhan University, Wuhan 430072, China.
Zhihao Li, Key Laboratory of Analytical Chemistry for Biology and Medicine (Ministry of Education), College of Chemistry and Molecular Sciences, Wuhan University, Wuhan 430072, China.
Ling Liang, Molecular Science and Biomedicine Laboratory (MBL), Institute of Chemical Biology and Nanomedicine, State Key Laboratory of Chemo/Biosensing and Chemometrics, College of Chemistry and Chemical Engineering, College of Biology, and Aptamer Engineering Center of Hunan Province, Hunan University, Changsha 410082 China.
Liqin Zhang, Department of Chemistry and Department of Physiology and Functional Genomics, Center for Research at the Bio/Nano Interface, Health Cancer Center, UF Genetics Institute and McKnight Brain Institute, University of Florida, Gainesville, Florida 32611-7200, United States.
Quan Yuan, Molecular Science and Biomedicine Laboratory (MBL), Institute of Chemical Biology and Nanomedicine, State Key Laboratory of Chemo/Biosensing and Chemometrics, College of Chemistry and Chemical Engineering, College of Biology, and Aptamer Engineering Center of Hunan Province, Hunan University, Changsha 410082 China, yuanquan@whu.edu.cn.
Weihong Tan, Molecular Science and Biomedicine Laboratory (MBL), Institute of Chemical Biology and Nanomedicine, State Key Laboratory of Chemo/Biosensing and Chemometrics, College of Chemistry and Chemical Engineering, College of Biology, and Aptamer Engineering Center of Hunan Province, Hunan University, Changsha 410082 China, Department of Chemistry and Department of Physiology and Functional Genomics, Center for Research at the Bio/Nano Interface, Health Cancer Center, UF Genetics Institute and McKnight Brain Institute, University of Florida, Gainesville, Florida 32611-7200, United States.
References
- [1].a) Wells JA, McClendon CL, Nature 2007, 450, 1001–1009 [DOI] [PubMed] [Google Scholar]; b) Tzeng SR, Kalodimos CG, Nature 2012, 488, 236. [DOI] [PubMed] [Google Scholar]
- [2].a) Kuenzi BM, Rix LLR, Stewart PA, Fang B, Kinose F, Bryant AT, Boyle TA, Koomen JM, Haura EB, Rix U, Nat. Chem. Biol 2017, 13, 1222. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Fan YF, Zhang W, Zeng L, Lei ZN, Cai CY, Gupta P, Yang DH, Cui Q, Qin ZD, Chen ZS, Trombetta LD, Cancer Lett 2018, 421, 186. [DOI] [PubMed] [Google Scholar]
- [3].a) Zhang J, Jiang XD, Jiang YN, Guo MR, Zhang SY, Li JJ, He J, Liu J, Wang JH, Ouyang L, Eur. J. Med. Chem 2016, 108, 495. [DOI] [PubMed] [Google Scholar]; b) Cerchia C, Lavecchia A, Curr. Med. Chem 2017, 24, 2312. [DOI] [PubMed] [Google Scholar]; c) Vucic D, Fairbrother WJ, Clin. Cancer Res 2007, 13, 5995. [DOI] [PubMed] [Google Scholar]
- [4].a) Ellington AD, Szostak JW, Nature 1992, 355, 850. [DOI] [PubMed] [Google Scholar]; b) Bock LC, Griffin LC, Latham JA, Vermaas EH, Toole JJ, Nature 1992, 355, 564. [DOI] [PubMed] [Google Scholar]; c) Kim YM, Phillips JA, Liu HP, Kang HZ, Tan WH, Proc. Natl. Acad. Sci. USA 2009, 106, 6489. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Wang J, Wei YR, Hu XX, Fang YY, Li XY, Liu J, Wang SF, Yuan Q, J. Am. Chem. Soc 2015, 137, 10576. [DOI] [PubMed] [Google Scholar]
- [5].a) Lao YH, Phua KKL, Leong KW, Acs Nano 2015, 9, 2235. [DOI] [PubMed] [Google Scholar]; b) Simona C, Laura C, Cent. Nerv. Syst. Agents Med. Chem 2015, 15, 126. [DOI] [PubMed] [Google Scholar]; c) Zhou JH, Rossi J, Nat. Rev. Drug Discov 2017, 16, 181. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Zhuo ZJ, Yu YY, Wang ML, Li J, Zhang ZK, Liu J, Wu XH, Lu AP, Zhang G, Zhang BT, Int. J. Mol. Sci 2017, 18, 2430. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Röthlisberger P, Gasse C, Hollenstein M, Int. J. Mol. Sci 2017, 18, 2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Carell T, Brandmayr C, Hienzsch A, Muller M, Pearson D, Reiter V, Thoma I, Thumbs P, Wagner M, Angew. Chem. Int. Ed 2012, 51, 7110; Angew. Chem. 2012, 124, 7220. [DOI] [PubMed] [Google Scholar]
- [7].a) Frye M, Jaffrey SR, Pan T, Rechavi G, Suzuki T, Nat. Rev. Genet 2016, 17, 365. [DOI] [PubMed] [Google Scholar]; b) Harcourt EM, Kietrys AM, Kool ET, Nature 2017, 541, 339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].a) Megger DA, Megger N, Muller J, Met. Ions Life Sci 2012, 10, 295. [DOI] [PubMed] [Google Scholar]; b) Benner SA, Karalkar NB, Hoshika S, Laos R, Shaw RW, Matsuura M, Fajardo D, Moussatche P, Cold Spring Harb. Perspect. Biol 2016, 8, 23770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].a) Vaught JD, Bock C, Carter J, Fitzwater T, Otis M, Schneider D, Rolando J, Waugh S, Wilcox SK, Eaton BE, J. Am. Chem. Soc 2010, 132, 4141. [DOI] [PubMed] [Google Scholar]; b) Hili R, Niu J, Liu DR, J. Am. Chem. Soc 2013, 135, 98. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Sefah K, Yang ZY, Bradley KM, Hoshika S, Jimenez E, Zhang LQ, Zhu GZ, Shanker S, Yu FH, Turek D, Tan WH, Benner SA, Proc. Natl. Acad. Sci. USA 2014, 111, 1449. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Zhang LQ, Yang ZY, Sefah K, Bradley KM, Hoshika S, Kim MJ, Kim HJ, Zhu GZ, Jimenez E, Cansiz S, Teng IT, Champanhac C, McLendon C, Liu C, Zhang W, Gerloff DL, Huang Z, Tan WH, Benner SA, J. Am. Chem. Soc 2015, 137, 6734. [DOI] [PMC free article] [PubMed] [Google Scholar]; e) Dunn MR, Jimenez RM, Chaput JC, Nat. Rev. Chem 2017, 1, 76. [Google Scholar]
- [10].a) Yin H, Hamilton AD, Angew. Chem. Int. Ed 2005, 44, 4130; Angew. Chem. 2005, 117, 4200 [DOI] [PubMed] [Google Scholar]; b) Gasse C, Zaarour M, Noppen S, Abramov M, MarliHre P, Liekens S, De Strooper B, Herdewijn P, ChemBioChem 2018, 19, 754. [DOI] [PubMed] [Google Scholar]
- [11].Voth AR, Ho PS, Curr. Top. Med. Chem 2007, 7, 1336. [DOI] [PubMed] [Google Scholar]
- [12].a) Mangani S, Orioli PL, Scozzafava A, Messori L, Carloni P, BioMetals 1994, 7, 104. [DOI] [PubMed] [Google Scholar]; b) Terron A, Fiol JJ, Garcia-Raso A, Barcelo-Oliver M, Moreno V, Coord. Chem. Rev 2007, 251, 1973 [Google Scholar]; c) Stadtman ER, Free Radical Biol. Med 1990, 9, 315. [DOI] [PubMed] [Google Scholar]
- [13].a) Lau KH, Farley JR, Freeman TK, Baylink DJ, Metabolism 1989, 38, 858. [DOI] [PubMed] [Google Scholar]; b) Bollen M, Stalmans W, Biochem. J 1988, 255, 327. [PMC free article] [PubMed] [Google Scholar]; c) Pollock J, Borkin D, Lund G, Purohit T, Dyguda-Kazimierowicz EY, Grembecka J, Cierpicki T, J. Med. Chem 2015, 58, 7465. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Guo FM, Li Q, Zhou CZ, Org. Biomol. Chem 2017, 15, 9552. [DOI] [PubMed] [Google Scholar]
- [14].Biondi E, Lane JD, Das D, Dasgupta S, Piccirilli JA, Hoshika S, Bradley KM, Krantz BA, Benner SA, Nucleic Acids Res 2016, 44, 9565. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Georgiadis MM, Singh I, Kellett WF, Hoshika S, Benner SA, Richards NG, J. Am. Chem. Soc 2015, 137, 6947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Morla-Folch J, Xie HN, Gisbert-Quilis P, Gomez-de Pedro S, Pazos-Perez N, Alvarez-Puebla RA, Guerrini L, Angew. Chem. Int. Ed 2015, 54, 13650; Angew. Chem. 2015, 127, 13854. [DOI] [PubMed] [Google Scholar]
- [17].Jellinek D, Green LS, Bell C, Lynott CK, Gill N, Vargeese C, Kirschenheuter G, McGee DP, Abesinghe P, Pieken WA, et al. , Biochemistry 1995, 34, 11363. [DOI] [PubMed] [Google Scholar]
- [18].Shangguan D, Cao Z, Meng L, Mallikaratchy P, Sefah K, Wang H, Li Y, Tan W, J. Proteome Res 2008, 7, 2133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Hynes RO, Cell 2002, 110, 673. [DOI] [PubMed] [Google Scholar]
- [20].a) Nishiuchi R, Takagi J, Hayashi M, Ido H, Yagi Y, Sanzen N, Tsuji T, Yamada M, Sekiguchi K, Matrix Biol 2006, 25, 189. [DOI] [PubMed] [Google Scholar]; b) Parsons SF, Lee G, Spring FA, Willig TN, Peters LL, Gimm JA, Tanner MJA, Mohandas N, Anstee DJ, Chasis JA, Blood 2001, 97, 312. [DOI] [PubMed] [Google Scholar]
- [21].Nishiuchi R, Sanzen N, Nada S, Sumida Y, Wada Y, Okada M, Takagi J, Hasegawa H, Sekiguchi K, Proc. Natl. Acad. Sci. USA 2005, 102, 1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].a) Takagi J, Petre BM, Walz T, Springer TA, Cell 2002, 110, 599. [DOI] [PubMed] [Google Scholar]; b) Takagi J, Strokovich K, Springer TA, Walz T, EMBO J 2003, 22, 4607. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.