

Abstract
Circular bivalent aptamers (cb-apt) comprise an emerging class of chemically engineered aptamers with substantially improved stability and molecular recognition ability. Its therapeutic application, however, is challenged by the lack of functional modules to control the interactions of cb-apt with therapeutics. We present the design of a β-cyclodextrin-modified cb-apt (cb-apt-βCD) and its supramolecular interaction with molecular therapeutics via host−guest chemistry for targeted intra-cellular delivery. The supramolecular ensemble exhibits high serum stability and enhanced intracellular delivery efficiency compared to a monomeric aptamer. The cb-apt-βCD ensemble delivers green fluorescent protein into targeted cells with efficiency as high as 80%, or cytotoxic saporin to efficiently inhibit tumor cell growth. The strategy of conjugating βCD to cb-apt, and subsequently modulating the supramolecular chemistry of cb-apt-βCD, provides a general platform to expand and diversify the function of aptamers, enabling new biological and therapeutic applications.
Aptamers, also known as the “chemical antibodies”, are oligonucleotides that bind with targeted biomolecules or cells with high affinity and selectivity.1–11 The excellent molecular recognition property of aptamers has led to their expanding use in biomedical applications,1,2 for example, as conjugates with cancer chemotherapy drugs.12–15 The practical application of aptamer-drug conjugates (ApDCs), however, requires tailor-made covalent chemistry to prepare aptamer conjugates.16,17 These conjugations not only pose a substantial synthetic barrier to aptamer usage, but also may compromise the aptamer’s targeting properties, due to the loss of control over its stability and conformational flexibility. Therefore, new chemistries that modulate aptameric structure with improved stability and recognition capabilities are highly desired.
To meet this need, we recently reported a cyclization strategy to stabilize aptamers by ligating two aptamer-based oligonucleotide sequences.11 The ligated circular bivalent aptamer (cb-apt) exhibited substantially improved thermal and physical stability in biological media, and showed enhanced target binding affinity, both in vitro and in vivo. However, the therapeutic potential of cb-apt has not yet been fully studied, mostly due to the lack of functional modules on cb-apt to control its interactions with biomedicines.18 To improve this, a facile and efficient chemical approach to functionalize cb-apt and control its interactions with molecular therapeutics is required. Recently, use of the supramolecular chemistry of oligonucleotides has proved to be an efficient approach to prepare aptamer conjugates for biomedical applications. For instance, the host−guest interaction of cucurbituril- or cyclodextrin-conjugated DNA has been used to expand aptamer functionality for molecular sensing,19,20,22 catalysis,21 and biomedicine.23,24 Therefore, we hypothesized that installing a supramolecular module onto cb-apt and designing its interaction with therapeutics would lead to a new generation of potentially paradigm-shifting aptamer-based molecular medicine.
In this paper, we report the design of β-cyclodextrin (βCD)-conjugated cb-apt, and the study of its supramolecular interaction with molecular therapeutics for targeted intracellular delivery. As shown in Scheme 1, we first conjugated a molecular container, βCD, onto a single-stranded sgc8 aptamer that targets protein tyrosine kinase-7 (PTK-7) on the cell surface.25,26 We then cyclized it with a complementary sequence (monoaptc) to form cb-apt-βCD. Incorporating βCD with cb-apt not only retained the high serum stability of the circular bivalent aptamer but also enabled encapsulation of a hydrophobic small-molecule, or adamantane-modified protein, into cb-apt, forming a supramolecular ensemble for enhanced intracellular delivery. We demonstrate that cb-apt-βCD can efficiently deliver not only small-molecule chemotherapeutics but also a cytotoxic macromolecule (protein) to inhibit tumor cell growth, highlighting the potency of using the supramolecularly engineered cb-apt as a general platform for the delivery of various therapeutics and for new biomedical applications.
Scheme 1.
Equipping Circular Bivalent Aptamers with Supramolecular βCD for Enhanced Intracellular Protein Deliverya

a(A) Synthesis route of cb-apt-βCD and its supramolecular interaction with adamantane-modified protein. (B) Schematic illustration of cb-apt-βCD-mediated enhanced protein delivery.
To make the cb-aptamer-βCD construct, we modified the fifth thymine nucleotide (relative to 3′) of an sgc8 aptamer precursor with a primary amine (detailed sequence is provided in Table S1, Supporting Information), which could react with dibenzocyclooctyne-sulfo-N-hydroxysuccinimidyl ester (DBCO-NHS) to afford DBCO-conjugated sgc8 aptamer (monoapt-DBCO). As shown in Figure 1A, mixing monoapt-DBCO and 6-monoazido-6-monodeoxy-βCD at room temperature yields βCD-conjugated monoapt-βCD via click conjugation. After purification using RP-HPLC (Figure 1B), the monoapt-βCD was then hybridized with the complementary aptamer component and ligated using T4 DNA ligase to afford circular cb-apt-βCD (Scheme 1A). The successful ligation and formation of cb-apt-βCD was confirmed by denatured polyacrylamide gel electrophoresis (PAGE). As shown in Figure 1C, the click conjugation of βCD onto monoapt-DBCO slightly retarded its migration, while the cyclization of two aptamers resulted in a significantly higher degree of retardation.
Figure 1.

(A) Synthesis of βCD-modified monomeric aptamer via DBCO conjugation and copper-free click reaction. (B) RP-HPLC trace of amine, DBCO, and βCD-modified sgc8 aptamer. (C) PAGE analysis of cb-apt-βCD.
To evaluate whether cb-apt-βCD retains the targeting capability of sgc8 aptamer after chemical conjugation and cyclization, we first set out to study the capability of cb-apt-βCD for targeted anticancer drug delivery by making use of the host−guest chemistry of βCD to encapsulate a hydrophobic small-molecule drug. To this end, an anticancer agent that we have previously identified,13,27 N-heterocyclic carbene (NHC)−gold(I) (AuNHC), was selected and loaded into cb-apt-βCD, mainly through the hydrophobic interaction between AuNHC and βCD. AuNHC-encapsulated in cb-apt-βCD (cb-apt-AuNHC) was then exposed to CEM cells, a PTK-7-positive cell line, at different concentrations of AuNHC. Cell viability after treatment was measured and compared to that of cells treated with AuNHC encapsulated in monoapt-βCD. As shown in Figure S1, monoapt-βCD can deliver AuNHC to induce cytotoxicity against CEM cells in Opti-MEM, while showing a significantly decreased efficiency in the presence of serum, suggesting a profound effect from serum degradation of the monomeric aptamer conjugate.11 Notably, however, cb-apt-AuNHC exhibited a 5-fold increased cytotoxicity against CEM cells compared to that of mon-apt-AuNHC in the presence of serum, indicating the enhanced stability and retention of binding ability of cb-apt-βCD in serum for intracellular delivery. Moreover, we observed only minimal cytotoxicity of cb-apt-AuNHC against Ramos cells (Figure S2), a PTK-7 low-expressing cell line, again confirming that the targeting capability of sgc8 is retained in cb-apt-βCD, a critical prerequisite for using cb-apt-βCD for targeted intracellular delivery.
Having demonstrated the capability of cb-apt-βCD to encapsulate hydrophobic AuNHC for targeted delivery based on the host−guest chemistry of cb-apt-βCD, we next studied whether cb-apt-βCD could be engineered to deliver protein-based therapeutics, which are generally cell-impermeable biomacromolecules. Many proteins are attractive “biologics” for disease treatment, but the lack of approaches for efficient and effective intracellular delivery restricts their therapeutic potential.28–31 Addressing this in the present study, we chemically modified protein with adamantane (AdA), a hydrophobic moiety that forms a tight host−guest inclusion complex with β-CD.21,23 As a proof of concept, we first chose green fluorescent protein (GFP) as a model protein. The chemical conjugation of GFP with AdA ligand (Figure 2A; see experimental details in SI) resulted in GFP-AdA with, on average, one AdA group conjugated onto GFP. GFP-AdA was characterized using matrix-assisted laser desorption/ionization-time-of-flight (MALDI-TOF) mass spectroscopy, as shown in Figure S3. To study and confirm the efficient host−guest inclusion between apt-βCD and GFP-AdA, we fluorescently labeled apt-βCD with TMR (TMR-apt-βCD) to titrate the fluorescence of GFP-AdA. The efficient Förster resonance energy transfer (FRET) between TMR and GFP provides direct evidence for the inclusion complex of apt-βCD and GFP-AdA. As shown in Figure 2B, mixing GFP-AdA with TMR-apt-βCD at a 2:1 molar ratio resulted in ample fluorescence quenching of GFP-AdA and enhanced TMR emission, indicating the occurrence of efficient FRET between GFP-AdA and TMR-apt-βCD and, hence, the formation of the inclusion complex. Importantly, when TMR-apt, an aptamer without β-CD conjugation, was mixed with GFP-AdA, no occurrence of FRET was observed (Figure 2B), again highlighting the necessity of the AdA-βCD interaction to form the supramolecular aptamer and protein inclusion ensemble. A systematic titration of TMR-apt-βCD with GFP-AdA showed a concentration-dependent FRET ratio (Figure 2C). Moreover, the supramolecular GFP-AdA and TMR-apt-βCD complex were highly stable in the presence of serum, as evidenced by the minimal FRET change of the GFP-AdA and TMR-cb-apt-βCD in the presence of serum (Figure 2C). This stability is essential for intracellular protein delivery.
Figure 2.

(A) Chemical structure of GFP-AdA. (B) Fluorescence spectra of GFP-AdA alone (1 μM), GFP-AdA (1 μM) in the presence of TMR-apt (2 μM) or TMR-cb-apt-βCD (0.5 μM) in PBS. All spectra were measured with excitation at 470 nm. The inset photographs show the fluorescence of images of GFP-AdA and TMR-apt-βCD alone, or their mixture under ultraviolet light. (C) Fluorescence titration of GFP-AdA with TMR-apt-βCD from 0 to 1.5 μM with and without serum.
The efficient and stable supramolecular interaction between cb-apt-βCD and GFP-AdA prompted us to study whether the complex could transport proteins into cells via aptamer-mediated uptake of cb-apt-βCD complex. Our preliminary study of the cytotoxicity of the cb-apt-βCD/GFP-AdA complex (Figure S4) ensured the high biocompability of cb-apt-β-CD for intracellular delivery. The cellular delivery of GFP to human cervical carcinoma HeLa cells, which also overexpress tyrosine kinase-7 on their cell surfaces, was studied by treating cells with cb-apt-βCD/GFP-AdA, or monoapt-βCD/GFP-AdA, as a negative control. Confocal laser scanning microscopy (CLSM) and flow cytometry were used to quantify and compare GFP delivery efficiency. As shown in Figure 3, and Figure S5, GFP-AdA alone could not enter cells, and monoapt-β-CD only facilitated the delivery of GFP-AdA with very low efficiency in the presence of serum. In contrast, the cb-apt-βCD/GFP-AdA complex delivered GFP into cells with efficiency as high as 80% in cell culture medium (Figure S5), showing dependence on protein concentration (Figure 3B). The inefficient cellular uptake and delivery of GFP-AdA by monoapt-βCD probably resulted from the lower stability of the monomeric aptamer in the presence of serum, leading to decreased binding to the cell surface receptor for intracellular delivery.11,15
Figure 3.
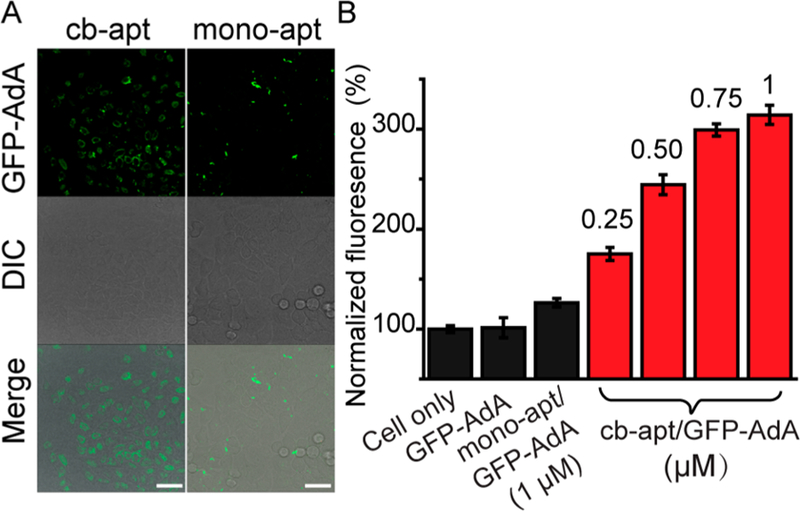
Cb-apt-βCD-mediated enhanced GFP delivery into HeLa cells. (A) CLSM images of HeLa cells treated with 600 nM GFP-AdA complexed with cb-apt-βCD (left panel) or monoapt-βCD (right panel). Scale bar: 40 μm. (B) Flow cytometry analysis of HeLa cells treated with PBS (cell only), GFP-AdA (1 μM), monoapt/GFP-AdA (1 μM), and the indicated concentration of cb-apt/GFP-AdA. The fluorescence intensity was normalized to cells with PBS treatment. Error bars represent the standard deviations of three parallel measurements.
Encouraged by the efficient delivery of GFP using cb-apt-βCD, we finally investigated its potency for functional protein delivery for therapeutic interventions. To this end, saporin, a cytotoxic protein that inhibits protein synthesis by disrupting ribosomes, was selected, because its potent toxicity has been harnessed for protein-based cancer therapy.32,33 We similarly conjugated saporin with an AdA handle (Figure S6) and loaded it into β-CD-conjugated aptamer before exposure to HeLa cells. As shown in Figure 4, treatment of HeLa cells with 100 nM cb-apt-βCD and saporin-AdA complex (cb-apt-saporin) reduced cell viability to 20%, whereas no obvious cytotoxicity was observed for the cells treated with the same concentration of free saporin or saporin-AdA alone, indicating that cb-apt-βCD effectively enhanced the intracellular delivery of saporin (Figure 4 and Figure S7). Significantly, cb-apt-βCD delivered saporin to inhibit cell proliferation with much higher efficiency than that of monoapt-saporin at any protein concentration level, again confirming a superior protein delivery efficiency using cb-apt-βCD in biological settings.
Figure 4.
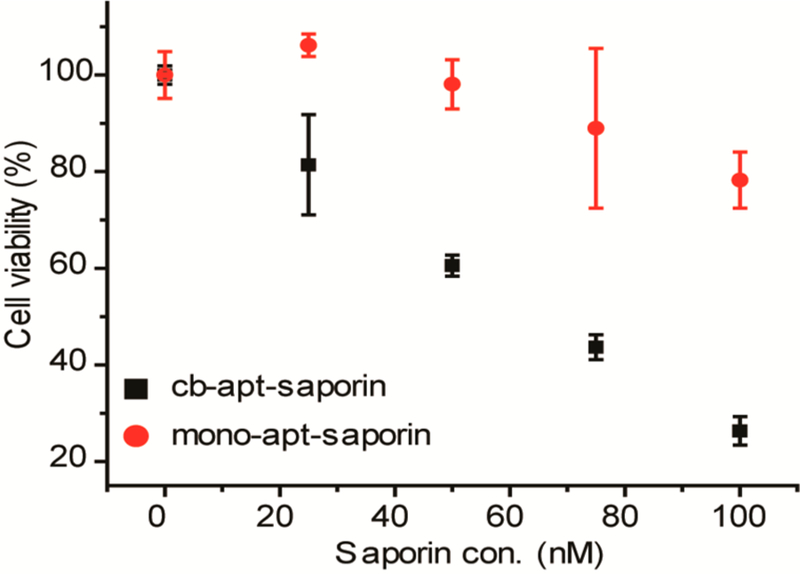
Cell viability of HeLa cells treated with different apt-βCD-saporin complexes. HeLa cells treated with cb-apt-saporin or monoapt-saporin in 10% FBS containing medium at indicated saporin concentrations for 36 h, followed by determining cell viability by MTS assay. The error bars represent the standard deviations of three parallel measurements.
In conclusion, we report herein a supramolecular approach to modulate the interaction of circular bivalent aptamer with molecular therapeutics for enhanced intracellular protein delivery. By controlling the host−guest chemistry of cb-apt-βCD, we are able to efficiently deliver a small-molecule or cytotoxic protein for targeted cancer therapy. The supramolecularly engineered circular bivalent aptamer ensemble provides a general platform for creating a functional and stable synthetic aptamer construct to solve biological and physiological challenges associated with the clinical translation of aptamer-based molecular medicine.
Supplementary Material
ACKNOWLEDGMENTS
This work is financially supported by NSFC grants 21521063, 21778056, and 21405041, and NIH GM R35 127130, and NSF 1645215.
Footnotes
Notes
The authors declare no competing financial interest.
ASSOCIATED CONTENT
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/jacs.8b03442.
Experimental details (PDF)
REFERENCES
- (1).Sefah K; Shangguan D; Xiong XL; O’Donoghue MB; Tan WH Nat. Protoc 2010, 5, 1169. [DOI] [PubMed] [Google Scholar]
- (2).Ariga K; Ito H; Hill JP; Tsukube H Chem. Soc. Rev 2012, 41, 5800. [DOI] [PubMed] [Google Scholar]
- (3).Veneziano R; Ratanalert S; Zhang K; Zhang F; Yan H; Chiu W; Bathe M Science 2016, 352, 1534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Komiyama M; Yoshimoto K; Sisido M; Ariga K Bull. Chem. Soc. Jpn 2017, 90, 967. [Google Scholar]
- (5).Tuerk C; Gold L Science 1990, 249, 505. [DOI] [PubMed] [Google Scholar]
- (6).Hansen TB; Jensen TI; Clausen BH; Bramsen JB; Finsen B; Damgaard CK; Kjems J Nature 2013, 495, 384. [DOI] [PubMed] [Google Scholar]
- (7).Tian C; Kim H; Sun W; Kim Y; Yin P; Liu H ACS Nano 2017, 11, 227. [DOI] [PubMed] [Google Scholar]
- (8).Zhao Z; Fan H; Zhou G; Bai H; Liang H; Wang R; Zhang X; Tan WH J. Am. Chem. Soc 2014, 136, 11220. [DOI] [PubMed] [Google Scholar]
- (9).Li J; Zheng C; Cansiz S; Wu C; Xu J; Cui C; Liu Y; Hou W; Wang Y; Zhang L; Teng IT; Yang HH; Tan WH J. Am. Chem. Soc 2015, 137, 1412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Wan S; Zhang L; Wang S; Liu Y; Wu C; Cui C; Sun H; Shi M; Jiang Y; Li L; Qiu L; Tan WJ Am. Chem. Soc 2017, 139, 5289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Kuai HL; Zhao ZL; Mo LT; Liu H; Hu XX; Fu T; Zhang XB; Tan WH J. Am. Chem. Soc 2017, 139, 9128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Zhu GZ; Niu G; Chen XY Bioconjugate Chem 2015, 26, 2186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Niu WJ; Chen XG; Tan WH; Veige AS Angew. Chem., Int. Ed 2016, 55, 8889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Chen K; Liu B; Yu B; Zhong W; Lu Y; Zhang JN; Liao J; Liu J; Pu Y; Qiu LP; Zhang LQ; Liu HX; Tan WH Wires Nanomed. Nanobi 2017, 9, e1438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Tan WH; Donovan MJ; Jiang J Chem. Rev 2013, 113, 2842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Zhou J; Rossi J Nat. Rev. Drug Discovery 2017, 16, 181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Tan ZS; Feagin TA; Heemstra JM J. Am. Chem. Soc 2016, 138, 6328. [DOI] [PubMed] [Google Scholar]
- (18).Skakuj K; Wang SY; Qin L; Lee A; Zhang B; Mirkin CA J. Am. Chem. Soc 2018, 140, 1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Zhou X; Su XY; Pathak P; Vik R; Vinciguerra B; Isaacs L; Jayawickramarajah JJ Am. Chem. Soc 2017, 139, 13916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Yu G; Yang Z; Fu X; Yung BC; Yang J; Mao Z; Shao L; Hua B; Liu Y; Zhang F; Fan Q; Wang S; Jacobson O; Jin A; Gao C; Tang X; Huang F; Chen X Nat. Commun 2018, 9, 766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Garcia MAA; Hu YW; Willner I Chem. Commun 2016, 52, 2153. [DOI] [PubMed] [Google Scholar]
- (22).Ihara T; Uemura A; Futamura A; Shimizu M; Baba N; Nishizawa S; Teramae N; Jyo AJ Am. Chem. Soc 2009, 131, 1386. [DOI] [PubMed] [Google Scholar]
- (23).Crini G Chem. Rev 2014, 114, 10940. [DOI] [PubMed] [Google Scholar]
- (24).Webber MJ; Langer R Chem. Soc. Rev 2017, 46, 6600. [DOI] [PubMed] [Google Scholar]
- (25).Shangguan D; Li Y; Tang ZW; Cao ZHC; Chen HW; Mallikaratchy P; Sefah K; Yang CYJ; Tan WH Proc. Natl. Acad. Sci. U. S. A 2006, 103, 11838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Jiang Y; Shi ML; Liu Y; Wan S; Cui C; Zhang LQ; Tan WH Angew. Chem., Int. Ed 2017, 56, 11916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Hickey JL; Ruhayel RA; Barnard PJ; Baker MV; Berners-Price SJ; Filipovska AJ Am. Chem. Soc 2008, 130, 12570. [DOI] [PubMed] [Google Scholar]
- (28).Ray M; Lee YW; Scaletti F; Yu RJ; Rotello VM Nanomedicine 2017, 12, 941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Tang R; Wang M; Ray M; Jiang Y; Jiang ZW; Xu QB; Rotello VM J. Am. Chem. Soc 2017, 139, 8547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Gu Z; Biswas A; Zhao M; Tang Y Chem. Soc. Rev 2011, 40, 3638. [DOI] [PubMed] [Google Scholar]
- (31).Erazo-Oliveras A; Najjar K; Dayani L; Wang TY; Johnson GA; Pellois JP Nat. Methods 2014, 11, 861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Wang M; Alberti K; Sun S; Arellano CL; Xu QB Angew. Chem., Int. Ed 2014, 53, 2893. [DOI] [PubMed] [Google Scholar]
- (33).Stirpe F; Barbieri L; Battelli MG; Soria M; Lappi DA Bio/Technology 1992, 10, 405. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.