

Abstract
The vasculature is a remarkably interesting, complex, and interconnected organ. It provides a conduit for oxygen and nutrients, filtration of waste products, and rapid communication between organs. Much remains to be learned about the specialized vascular beds that fulfill these diverse, yet vital functions. This review was prompted by the discovery that Notch signaling in mouse endothelial cells is crucial for the development of specialized vascular beds found in the heart, kidneys, liver, intestines, and bone. We will address the intriguing questions raised by the role of Notch signaling and that of its regulator, the metalloprotease ADAM10, in the development of specialized vascular beds. We will cover fundamentals of ADAM10/Notch signaling, the concept of Notch-dependent cell fate decisions, and how these might govern the development of organ-specific vascular beds through angiogenesis or vasculogenesis. We will also consider common features of the affected vessels, including the presence of fenestra or sinusoids and their occurrence in portal systems with two consecutive capillary beds. We hope to stimulate further discussion and study of the role of ADAM10/Notch signaling in the development of specialized vascular structures, which might help uncover new targets for the repair of vascular beds damaged in conditions like coronary artery disease and glomerulonephritis.
I. INTRODUCTION
Vasculogenesis and angiogenesis are essential for building the vascular tree with its many branches during development. The vasculature reaches into all parts of the body so that it can provide a steady supply of oxygen and nutrients, remove CO2 and metabolic waste products, and serve as a conduit for signaling molecules (30, 44). The vascular tree also feeds into a variety of highly specialized structures that add enhanced functionality to the circulatory system, such as the glomeruli in the kidney, the sinusoids in the liver, the vessels that absorb nutrients in the intestinal tract, or the coronary vessels of the heart. Each of these specialized vascular structures has unique morphological features, such as fenestrations or sinusoidal openings, that are crucial for their specific functions (5, 101, 200). There is a considerable amount of interest in understanding the basic principles of the development and maintenance of these specialized vascular beds, as this holds the promise of finding better approaches to preventing vessel damage or to helping rebuild and repair diseased vessels, for example, in coronary artery disease or glomerulonephritis.
Recent studies have uncovered a crucial role of the a disintegrin and metalloprotease 10 (ADAM10)/Notch signaling pathway in the development of specialized vascular structures, which is the main focus of this review. Notch receptors are key regulators of angiogenesis and have essential roles during the earliest stages of vasculogenesis and angiogenesis in the murine embryo and yolk sac (38, 44, 61, 154, 196, 200). The Notch receptors 1–4 are part of a family of membrane-anchored transcription factors that are activated by binding of membrane-anchored Notch ligands [e.g., Jagged 1 (Jag1) and Jagged 2, Delta-like 1 and Delta-like 4 (Dll4); see FIGURE 1, A and B, please note that Delta-like 3 is not considered a canonical Notch ligand (80)]. Binding of a Notch ligand to a Notch receptor triggers two highly coordinated and sequential proteolytic steps that release the Notch intracellular domain (NICD) from its membrane anchor, allowing it to enter the nucleus and activate Notch-dependent transcription. The first of these proteolytic processing events depends on the membrane-anchored metalloproteinase, ADAM10 (133, 134), and the next on the intramembrane proteases called presenilins/γ-secretases (20, 91, 134, 252) (FIGURE 1, B and C). The presenilins are unable to cleave Notch without prior processing by ADAM10 in the extracellular juxtamembrane domain, so ADAM10 is considered an essential regulator of physiological Notch signaling.
FIGURE 1.

Domain organization of Notch receptors and their membrane-anchored ligands and the role of proteolysis in Notch signaling. A and B: diagrammatic representation of the domain organization of the Drosophila Notch receptor and of the four human Notch receptors (A) and of the Drosophila and human Notch ligands (B). C: diagram depicting different steps involved in Notch signaling. A membrane-anchored Notch ligand, such as Delta, on the signal-sending cell, engages a membrane-anchored Notch receptor on the signal-receiving cell (1). This triggers endocytosis of the ligand, which provides the force to expose the Notch cleavage site to a disintegrin and metalloprotease 10 (ADAM10) for processing (2). This S2 processing step precedes the γ-secretase-dependent S3 processing event. This liberates the Notch intracellular domain (NICD) from its membrane anchor, allowing its translocation to the nucleus, where it enters into a transcriptional activation complex with CBF1/Su(H)/Lag-1 (CSL) and MAM, thereby activating transcription of Notch target genes. Please note that S1 processing by Furin in the secretory pathway is not shown in this diagram. ANK, ankyrin; HD, heterodimerization domain; LNR, Lin12/Notch repeat; NEC, Notch extracellular domain; NRR, negative regulatory region; PEST, proline-, glutamate-, serine-, and threonine-rich domain; RAM, RBP-Jκ-associated module; TM, transmembrane domain. [Adapted from Gordon et al. (83), with permission from J Cell Sci.]
A principal function of Notch signaling is the regulation of cell fate decisions in development and disease. A well-characterized, Notch-dependent cell fate decision occurs in angiogenesis in the developing retinal vasculature, where Notch is responsible for the “tip cell” vs. “stalk cell” decision (16, 65, 88, 99, 105, 113, 156, 232) (FIGURE 2A). In angiogenesis, tip cells are the first to bud from a previously quiescent vessel and migrate in response to a VEGF gradient. In contrast, stalk cells follow tip cells and generate a new vessel lumen to support blood flow. When endothelial Notch signaling was disrupted during retinal vascular development by inactivating endothelial ADAM10 with the endothelial-specific Tie2-Cre transgene (A10ΔEC) (82), an abundance of tip cells and increased vascular density was observed (FIGURE 2B), presumably caused by an abundance of tip cells (FIGURE 2C). In addition to the increased vascular density in the retinal vasculature, A10ΔEC mice also had defects in several other specialized vascular structures. These included enlarged vessels on the liver surface and under the epicardium of the heart, enlarged kidney glomeruli, intestinal polyps filled with endothelial cells, and defects in the developing bone vasculature and long bone growth (82). These additional vascular phenotypes were unexpected, since earlier studies had established that inactivation of Notch signaling in endothelial cells results in early embryonic lethality. The additional vascular defects observed in A10ΔEC mice thus raised interesting questions about their underlying cause. Specifically, why were A10ΔEC mice born and surviving into adulthood, despite these vascular defects, whereas previously described Notch1ΔEC mice died during early embryogenesis? Furthermore, were the defects in specialized vascular structures in A10ΔEC mice caused by a block in ADAM10-dependent Notch signaling, or by other functions of ADAM10 in endothelial cells not related to Notch signaling (82, 136, 152)?
FIGURE 2.

The tip cell vs. stalk cell fate decision in the developing retinal vascular tree. A: diagram of a tip cell (on the right) with filopodia that endow it with a migratory phenotype and that allow the newly developed vascular branch to follow a VEGF gradient. The stalk cells that follow the tip cells are able to generate a lumen for blood flow (adapted from Ref. 116). B: increased vascular density at the leading edge of the retinal vascular tree in mice lacking a disintegrin and metalloprotease 10 (ADAM10) in endothelial cells (A10ΔEC mice) compared with controls. C: this increased vascular density was associated with an increase in the number of tip cells with numerous filopodia, indicated by red dots, at the leading edge of the developing retina. DLL4, Delta-like 4. [B and C are from Glomski et al. (82), with permission from Blood.]
A few years later, a bone phenotype that morphologically resembled that observed in A10ΔEC animals was reported in mice with a temporal conditional inactivation of the Notch ligand Dll4 or of the Notch-dependent transcriptional regulator RBPJ in endothelial cells after birth. This strongly suggested that the development of the specialized bone vasculature depends on ADAM10/Notch1/RBPJ signaling (191). Moreover, when mice lacking Notch1 in endothelial cells that also lacked Notch4 systemically (N1ΔEC/N4−/−) were generated with the same Tie2-Cre transgene as A10ΔEC mice, these animals survived into postnatal life with essentially the same constellation of vascular defects as A10ΔEC mice. Importantly, all defects in A10ΔEC mice could be rescued by coexpression of the Notch1 intracellular domain (NICD) (6), supporting the interpretation that the vascular abnormalities in A10ΔEC mice were, in fact, caused by disruption of ADAM10/Notch signaling (6). In a separate study, postnatal inactivation of Notch1 or RBPJ in endothelial cells resulted in enlarged subcapsular liver vessels, which resembled those previously described in A10ΔEC mice, as well as the development of porto-systemic shunts (45). Taken together, these studies unveiled an essential role for endothelial ADAM10/Notch signaling pathway in the development of specialized vascular niches, raising a number of interesting questions about how ADAM10 and Notch control the process of vascular specialization.
Here, we will explore some of the intriguing questions that are raised by the studies showing a role for ADAM10/Notch signaling in the development of specialized vascular structures. We will first provide some background and context by briefly reviewing how Notch signaling controls cell fate decisions through a process called lateral inhibition, and how ADAM10 regulates this process. Although the related ADAM17 has also been implicated as a regulator of Notch signaling, we will present cell biological and functional studies that argue against a physiologically relevant role for ADAM17 in activating Notch receptors. We will then outline basic principles of vasculogenesis and angiogenesis in the early embryo, with an emphasis on the contribution of the Notch signaling pathway, and will summarize what is known about Notch signaling in the development of the specialized vascular structures that are affected in A10ΔEC mice. We will conclude by proposing several models and testable hypotheses for how inactivation of ADAM10/Notch signaling in endothelial cells might cause defects in the development of specialized vascular structures. Our hope is that this review will stimulate further discussion and studies to help to improve our understanding of the development of the endothelial cells that ensure the proper functioning of these fascinating specialized vascular beds.
II. BRIEF INTRODUCTION TO NOTCH SIGNALING
A. A Primer on Notch and Its Ligands
Notch was first discovered as a gene that is responsible for wing margin development in Drosophila melanogaster (54, 163, 165, 263). Flies with mutations in this receptor stood out because of a notch in their wing tip (FIGURE 3, A and B). Mutants with defects in other genes that are part of the Notch pathway were later identified because they caused similar phenotypes, and were named Delta and Serrate (49, 58, 73, 130, 242, 254) (FIGURE 3C). Over time, it became clear that the Notch signaling pathway is highly conserved through evolution, and that its main purpose is to control cell fate decisions during specialization of two different cell types from a common precursor. Notch signaling is repeatedly employed to make different cell fate decisions in diverse contexts and at distinct stages of development (22, 23, 134). Notch also has essential roles in tissue homeostasis, including in the lung (143) and the small intestine (209), where Notch signaling regulates the life-long self-renewal of the specialized cell types that populate intestinal crypts and villi from intestinal stem cells (188). Not surprisingly, mutations leading to dysregulated Notch signaling have also been implicated in cancer, including hematological malignancies and solid tumors (192, 208, 224, 253).
FIGURE 3.
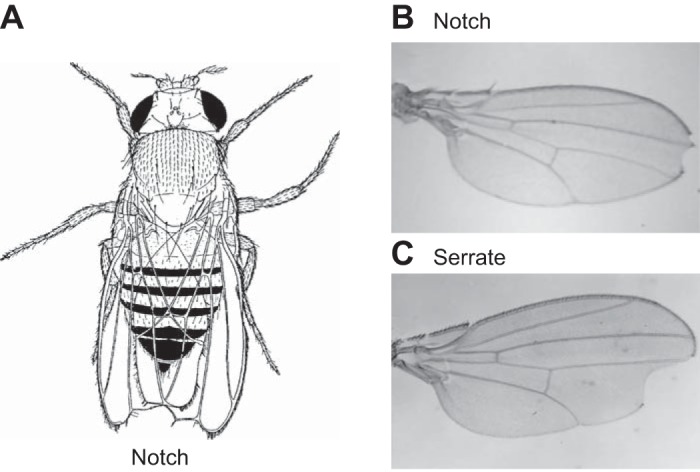
Phenotype of Drosophila with Notch pathway mutations. A: drawing of a fly with a notched wing tip from a seminal paper published by Dr. T. Morgan in 1917 (165), and a photo of a wing from a fly carrying a Notch mutation (B) (121) or a mutation in Serrate (C) (225).
B. Notch Processing by ADAMs and Presenilin
Important conceptual insights into the mechanism of Notch signaling came from genetic and cell biological studies that uncovered a role for the cytoplasmic domain of Notch as a transcriptional regulator in the nucleus (key papers to establish this concept were Refs. 52, 213, 229, 230; see also Refs. 133, 167, 168, 264). This was initially quite unexpected, since cell surface receptors usually transmit a signal by activating a cytoplasmic kinase domain to engage intracellular signaling pathways or by assembling a signaling complex at the plasma membrane. The studies on Notch showed that it is essentially a membrane-anchored transcription factor whose cytoplasmic domain is released and activated by ligand-induced proteolysis (23, 134) (FIGURE 1C). Binding of membrane-anchored Notch ligands (such as Delta or Jagged) that reside on one cell (the “signal sending” cell) to Notch on an adjacent cell (the “signal receiving” cell) triggers endocytosis, which leads to uncovering and exposure of a membrane-proximal cleavage site for ADAM10 (83, 135, 246) (FIGURE 4, A AND B). Processing of Notch by ADAM10 (FIGURE 4C) generates a membrane-anchored stalk domain that is recognized by the intramembrane γ-secretase protease complex, which proceeds to process Notch within its transmembrane domain (20, 52, 95, 133, 150, 203, 213, 230, 252, 264) (FIGURE 1C). This releases the NICD from the plasma membrane, allowing it to translocate into the nucleus. In the nucleus, the NICD binds to the cotranscriptional regulators CSL [CBF1/Su(H)/Lag-1] and mastermind, leading to the expression of Notch-dependent transcriptional targets (22, 23, 134) (FIGURE 1C). The exact transcriptional targets vary with cell type and epigenetic context (22, 66), but the ultimate downstream effect is typically to promote one out of two possible cell fates or cell types.
FIGURE 4.

Exposure of the Notch S2 cleavage site for a disintegrin and metalloprotease 10 (ADAM10) by endocytosis of the membrane-anchored Notch-ligand Delta. A: the negative regulatory region (NRR) of Notch, composed of three Lin12/Notch repeat domains (LNR; A, B, C) and a heterodimerization domain (HD-N, HD-C), which contains the buried S2 cleavage site for ADAM10 in HD-C. B: 1, endocytosis of a membrane-anchored Notch ligand such as Delta provides a force that is sufficient to unfold the NRR (85) (2–4), exposing the S2 ADAM cleavage site, indicated by an arrow in 4. C: once the cleavage site is uncovered, it can be processed by ADAM10 or ADAM17, generating a membrane-anchored stub that becomes a substrate for γ-secretase (see also FIGURE 1C). The structure of the extracellular domain of ADAM10, which is responsible for the processing of Notch under physiological conditions, is shown. The red box in the schematic of ADAM10 on the right indicates the domains shown in the structure of ADAM10. C, cysteine rich; Cyt, cytoplasmic tail; D, disintegrin; M, metalloproteinase; Pro, Pro domain; SS, signal sequence; TM, transmembrane. The colors in the schematic on the right correspond to those in the X-ray structure of ADAM10. The catalytic zinc ion is gray, and a bound calcium ion is shown in orange. Cysteine residues engaged in disulfide bonds are shown as sticks. [A is adapted from Gordon et al. (84), with permission from Blood; B and C from Gordon et al. (83), with permission from J Cell Sci; and C includes the structure of ADAM10, which was recently solved by Seegar et al. (215). We thank Stephen Blacklow for assistance in generating C.]
C. The Role of Notch in Lateral Inhibition and Cell Fate Decisions
A typical Notch-dependent cell fate decision involves a group of precursor cells that receive a signal as a prompt to change their cell fate (FIGURE 5). During this process, this group of cells responds to stimulation by an inductive signal, such as a growth factor, to begin a differentiation process that includes the production of Notch receptors and Notch ligands (FIGURE 5A, peach-colored cells). One cell in this group of differentiating cells will be the first to reach a threshold concentration of Notch ligands on the surface, allowing this “signal sending” cell (green cell in FIGURE 5B) to activate Notch signaling on the adjacent “signal receiving” cells (peach-colored cells in FIGURE 5B). This lateral inhibition (black arrows in FIGURE 5B) serves to prevent the neighboring cells (peach) from acquiring the new cell fate (green), which is referred to as the default cell fate (105, 200). In the absence of Notch signaling, many more cells that are responding to the growth factor would acquire this new default cell fate (FIGURE 5C). Notch thus determines the outcome of a binary cell fate decision, thereby regulating the ratio of cells that acquire one of the two cell fates. This type of a cell fate decision not only inhibits the default cell state (tip cell, green), but can also actively promote a Notch-dependent differentiation process in the signal receiving cells, indicated by a change of color from peach to red in FIGURE 5D.
FIGURE 5.

Diagram of Notch-dependent lateral inhibition of a cell fate decision. A: a small cluster of cells (peach) begins to differentiate in response to a local cue, such as a growth factor. B: expression of Notch and a Notch ligand [e.g., Delta-like 4 (Dll4) for an endothelial tip/stalk cell decision] increase until one cell, shown in green, reaches a threshold that can trigger Notch signaling in the adjacent, signal receiving cell (arrows). C: if Notch signaling is blocked, all peach-colored cells will acquire the default differentiation state (green). D: intact Notch signaling helps execute the proper cell fate decision in which a single green cell acquires one cell fate while the surrounding cells are prevented from acquiring the same fate through lateral inhibition. The change in color from peach to red indicates that Notch signaling can also promote further differentiation in the laterally inhibited cells.
A particularly good example to illustrate lateral inhibition in a Notch-dependent cell fate decision is the tip cell vs. stalk cell decision, which occurs in the context of angiogenesis during murine retinal vascular development (16, 65, 99, 226) (FIGURES 2 and 6). New branches in the developing vascular tree are thought to be elicited by retinal astrocytes that migrate ahead of the vascular tree (7, 228). As the retinal astrocytes move further away from the existing blood supply, they become hypoxic and release VEGF, which promotes development of, and recruitment of, new vessels (105, 200, 228) (FIGURE 6A). The increasing levels of VEGF prompt nearby endothelial cells to respond by ultimately yielding one tip cell among several stalk cells. As part of the tip cell differentiation process, the Notch ligand Dll4 and its receptor, Notch1, are upregulated in a patch of endothelial cells. Eventually, the levels of Dll4 in one cell reach a threshold that allows activation of Notch receptors on the adjacent endothelial cells (see FIGURES 2, 5, and 6B). Coupled with endocytosis of the Notch ligand and receptor, ADAM10-dependent processing ensues, followed by γ-secretase-dependent processing (FIGURES 1C and 4). This activates a Notch-dependent transcriptional program that prevents the endothelial cells adjacent to the tip cells from also acquiring this cell fate, resulting in tight control of the number of tip and stalk cells. Since tip cells are able to sense and migrate along a VEGF gradient, they guide the new vessel sprout by leading stalk cells in the right direction, toward the VEGF source. Stalk cells have very different properties from tip cells, including the ability to proliferate and form a lumen and thus build a conduit for blood to reach its target destination.
FIGURE 6.

The Tip/Stalk cell decision in the developing retinal vascular tree. A: during development, the retinal vasculature is recruited by retinal astrocytes to provide oxygen and nutrients. The immunofluorescence image shows astrocytes (green, red) that are not covered by blood vessels in the top part of the image, and the recruited vessels in blue in the middle to bottom left part of the image. B: diagrammatic representation of the outcome of a successful endothelial cell fate decision shows two tip cells with filopodia at the leading edge of the retinal vascular tree (see FIGURE 2C), which is followed by stalk cells that generate a vessel with a lumen (see also FIGURE 2A). The red, green, and orange dots in endothelial cells indicate differential expression of Delta-like 4 (Dll4) and Jag1 in endothelial cells (for a more detailed discussion, see Ref. 105). [A is from Fruttiger (77), with permission from Angiogensis; B is from Hofmann and Iruela-Arispe (105), with permission from Circ Res.]
If Notch or ADAM10 signaling is disrupted, then an excess of tip cells develops at the expense of stalk cells, resulting in an increased vascular density and disruption of vascular morphogenesis (FIGURE 2B). The quite detailed understanding of this particular example of a Notch-dependent endothelial cell fate decision is based on the results of several elegant studies that employed genetic and pharmacological approaches to modulate Notch signaling in the developing mouse retina (key initial papers were Refs. 99, 156, 195, 232; for additional key references and reviews, see Refs. 16, 65, 88, 105, 113). In addition, the Notch-ligand Jag1 has critical roles in vascular morphogenesis, by opposing the action of Dll4, as first shown by Ref. 15 (see also Refs. 18, 104, 105, 124, 179, 231, 271). Finally, glycosylation regulates Notch signaling by differentially modulating its interaction with Jag1 vs. Dll4 (28, 164). (Please see Ref. 157 for an elegant structural analysis of the mechanism underlying ligand discrimination.) This process has also been implicated in angiogenesis (211).
D. Distinct Roles of ADAM10 and ADAM17 in Physiological and Nonphysiological Notch Processing
When it first became clear that Notch is regulated through sequential proteolytic processing, the identity of the responsible S2 protease was somewhat controversial (FIGURE 7 shows Notch S1, S2, and S3 processing products). In genetic studies of Drosophila, C. elegans, and mice, ADAM10 (also referred to as Kuzbanian/KUZ in Drosophila, and SUP-17 in C. elegans) clearly emerged as the enzyme responsible for S2 processing of Notch (95, 203, 264). However, when biochemical approaches were used to purify the Notch processing activity from mammalian cells, ADAM17 was identified as the principal Notch processing enzyme (27). As a consequence, both ADAM10 and ADAM17 are frequently listed together as potential Notch S2 proteases (23, 148, 169, 241, 262), causing some confusion, particularly for those who are less familiar with the role of these proteases in Notch signaling. Thus, even though both ADAM17 and ADAM10 are, in principle, able to cleave Notch, it is important to emphasize that there is no conclusive evidence that inactivation of ADAM17 interferes with physiological Notch signaling in mice. [The first reported ADAM17 knockout mouse demonstrated a key role in epidermal growth factor receptor (EGFR) signaling (180). For additional studies on the role of ADAM17 in vivo, see Refs. 32, 74, 94, 107, 149, 176, 207, 227, 265). Regarding Ref. 171, which suggests a role of ADAM17 in processing Notch in vivo, please see Ref. 89 for a rebuttal and clarification.] Instead, numerous genetic studies using conditional inactivation of ADAM10 in different mouse tissues have recapitulated the effect of deleting one or more Notch family members (first shown by Ref. 95, see also Refs. 6, 81, 82, 117, 243, 262, 273). Moreover, in cell biological studies, two groups independently showed that ADAM10 is required for physiological, ligand-induced Notch1 signaling, whereas ADAM17 can only cleave Notch1 following nonphysiological activation, for example by chelating Ca2+ with EDTA (20, 252). (Please note that the Western blot in FIGURE 7 was generated using EDTA to activate Notch processing by ADAM17, although ADAM17 is not the physiological processing enzyme; FIGURE 8.) Similar results were reported for Notch2 and Notch3, which also require ADAM10 for canonical signaling (91).
FIGURE 7.

Processing of Notch at the S1, S2, and S3 cleavage sites. A: diagrammatic representation of the uncleaved Notch1 precursor, as it is synthesized in the endoplasmic reticulum (left) (89). The S1 processing is a constitutive processing event carried out by furin-type pro-protein convertases in the Golgi apparatus. In physiological, canonical Notch signaling, S2 processing occurs in a ligand-dependent manner and depends on a disintegrin and metalloprotease 10 (ADAM10) (see also FIGURES 1 and 4). The generation of the juxtamembrane stub triggers further processing by γ-secretase, which releases the Notch intracellular domain (NICD) from its membrane anchor (see also FIGURE 1). B: Western blot analysis of nonphysiological EDTA-dependent activation of Notch processing, which depends on ADAM17 (20, 252), shows the S1, S2, and S3 cleavage products on a Western blot probed with an antibody against the cytoplasmic domain of Notch1 (Notch1 antibody, EP1238Y, Millipore). ANK, ankyrin; EGF, epidermal growth factor repeat; FL, full length; LNR, Lin12/Notch repeat; NEXT, Notch extracellular domain; RAM, RBP-Jκ-associated module; PEST, proline-, glutamate-, serine-, and threonine-rich domain; TMIC, transmembrane intracellular fragment. [A adapted from Groot et al. (89), with permission from J Invest Dermatol.]
FIGURE 8.

A disintegrin and metalloprotease 10 (ADAM10) is the physiological Notch processing enzyme, whereas ADAM17 only processes Notch following nonphysiological activation of Notch. A: in principle, both ADAM10 and the related ADAM17 are able to process the Notch cleavage site. B: however, since physiological Notch signaling depends on ligand-dependent exposure of the Notch cleavage site, neither ADAM10 nor ADAM17 has access to the Notch cleavage site in the absence of ligand binding. C: addition of EDTA unfolds the negative regulatory region (NRR) to expose the cleavage site, which is preferably processed by ADAM17 under these conditions, for reasons that remain to be explained. D: genetic and cell biological studies have clearly established that ADAM10 is the physiologically relevant Notch processing enzyme. See FIGURE 4, B and C, for a model of the unfolding of the NRR domain by mechanical force. EGF, epidermal growth factor; HD, heterodimerization domain; LNR, Lin12/Notch repeat; NICD, Notch intracellular domain.
Interestingly, some mutant forms of Notch have been found to be oncogenic. Mutations in the Notch negative regulatory region (NRR), the structure that prevents processing of wild-type Notch in the absence of the pulling force of ligands like Dll4, are well-established drivers of T-cell acute lymphoblastic leukemia (crucial conceptual insights provided by Ref. 84; see also Refs. 83, 135, 233, 246) (FIGURE 9). These cancer-causing mutations apparently provide inappropriate access to the Notch cleavage site, which can then be cleaved by both ADAM10 and ADAM17 (233). However, in the absence of the appropriate ligand or stimuli, the cleavage site of wild-type Notch1 (and likely also other Notch receptors) appears to be well protected within the NRR, preventing processing by ADAM10 or ADAM17 (83, 84, 135). This serves to ensure that S2 processing and activation of Notch signaling strictly depends on ligand binding and the regulated exposure of the cleavage site (FIGURE 4A). It remains to be determined why ADAM10 is required for physiological S2 Notch processing, and why ADAM17, even though it is evidently able to process the Notch cleavage site upon nonphysiological activation by EDTA, cannot compensate in the absence of ADAM10 in genetic studies. Perhaps the ligand-induced conformational change in the Notch NRR differs from that induced by EDTA, such that only ligand-induced processing allows ADAM10 to access the Notch cleavage site. Further studies will be necessary to address this question.
FIGURE 9.

A major class of oncogenic Notch mutations in the Notch negative regulatory region in T-cell acute lymphoblastic leukemia. Diagram of the location of mutations in Notch that cause cancer, many of which map to the negative regulatory region (NRR) domain [particularly in the heterodimerization domain (HD)]. This presumably leads to destabilization of the interacting domain units and to constitutive and inappropriate exposure of the Notch cleavage site, which can then be processed by both a disintegrin and metalloprotease 10 (ADAM10) and ADAM17 (233). Other mutations, such as those found in the proline-, glutamate-, serine-, and threonine-rich (PEST) sequence, affect turnover and stability of the Notch intracellular domain (NICD), thereby leading to increased and dysregulated Notch signaling. ANK, ankyrin; EGF, epidermal growth factor; ICN, intracellular Notch1; LNR, Lin12/Notch repeat; NEC, Notch extracellular domain; NTM, Notch1 transmembrane subunit; RAM, RBP-Jκ-associated module; TAD, transactivation domain; TM, transmembrane domain.[Adapted from Malecki et al. (160), with permission from Mol Cell Biol.]
Notch ligands can also be processed, and this is thought to help fine-tune Notch signaling by controlling ligand availability. An excellent example is Kul, a Kuzbanian (ADAM10)-like protease in Drosophila that does not have an evident homolog in vertebrates. Genetic studies have implicated Kul in regulating Notch signaling by processing the Notch ligand Delta (210). In cell biological studies, ADAM17 has been reported to process the Notch ligand Jagged (145), and ADAM12 has been shown to process Delta-like 1 (63). Taken together, juxtamembrane and intramembrane proteolysis is essential for Notch receptor processing and signaling, and, in addition, can serve to fine-tune Notch signaling by controlling ligand availability.
III. ROLE OF NOTCH SIGNALING IN EARLY VASCULAR DEVELOPMENT
The first targeted deletion of Notch1 in mice revealed its essential role in development by demonstrating that knockout mice die during early embryogenesis before embryonic day 11.5 (E11.5) (235) with defects in somitogenesis (39). A similarly early embryonic lethality was observed in knockout mice for the Notch response gene RBPJ (122). Moreover, mice lacking ADAM10 (95) or both presenilins (59, 102) resembled Notch1-deficient mice, highlighting the conserved, essential function of ADAM10 and the presenilin proteases in Notch processing in vivo (presenilins are the proteolytic component of the γ-secretase complex). Insight into the role of Notch signaling in endothelial cells came from studies of mice with conditionally inactivated Notch1 (N1ΔEC) and conditionally inactivated RBPJ in endothelial cells. (Key studies highlighting the essential role of Notch in vascular morphogenesis and angiogenesis were Refs. 108, 136, 137, 152; see also Refs. 41, 86–88, 181, 200.) The endothelial Notch pathway mutant mice typically suffered from early embryonic lethality with severe defects in the embryonic and yolk sac vasculature.
The development of these two vascular beds differs in that the yolk sac vasculature is generated through blood islands that contain hematopoietic and EPCs (41, 61, 196, 197), whereas the intraembryonic vasculature depends on endothelial cells that are derived from mesodermal precursor cells (72, 197). Nevertheless, defects in Notch signaling in the yolk sac and in the early embryonic vasculature have conceptually similar consequences: a skewed artery/vein cell fate decision that yields more veinlike vessels at the expense of arteries (41, 128, 154). That inactivation of Notch signaling produces similar defects in the embryonic vasculature as in the yolk sac indicates common underlying principles in the development of these different vascular beds. To further explore this notion, we will briefly outline the role of Notch signaling during vasculogenesis in the embryo proper and then turn to extraembryonic vasculogenesis in the yolk sac.
A. Embryonic Vasculogenesis and Angiogenesis
The first uncommitted EPCs, also referred to as angioblasts, arise from the embryonic mesoderm at E7.5 and coalesce to form a vascular plexus (1, 5, 42, 72, 236) (FIGURE 10A). Expression of sonic hedgehog in mesodermal cells triggers the production of VEGF, which in turn binds to VEGFR2 and neuropilin 1 on arterial precursor cells (aPC), stimulating the expression of Dll4 and Notch (FIGURE 10B). Similar to the tip and stalk cell decision outlined above, once a given cell in this small patch of vascular precursor cells reaches a threshold level of Dll4 that can elicit robust Notch signaling in an adjacent cell, this sets up one successful cell fate decision (artery over vein) (reviewed in Refs. 5, 42, 72, 141, 185, 236). Inactivation of Dll4 in mice has shown that it is the principal Notch ligand responsible for promoting artery development (62, 248). Moreover, recent studies have implicated Sox17 upstream of Notch signaling in arterial differentiation (43). Notch signaling, in turn, upregulates the arterial marker ephrinB2 and suppresses the venous marker EphB4. Therefore, Notch signaling is crucial to initiate the sorting of ephrinB2-expressing aPCs from EphB4-expressing venous precursor cells (128) and for the establishment of arteries, veins, and a normal capillary network (2, 5, 128, 260). An elegant study from the Wang laboratory [Kim et al. (128)] showed that cell proliferation was not necessary for the Notch-dependent arterial vs. venous endothelial cell fate decision. Instead, Notch signaling appeared to ensure an approximately equal split between a preexisting number of precursor cells into these two cell fates (FIGURE 11A). Thus, if Notch signaling is blocked during early development, fewer aPCs differentiate into arterial endothelial cells, so fewer arterial cells are available to sort into the dorsal aortas, resulting in smaller dorsal aortas compared with cardinal veins.
FIGURE 10.

First stages of embryonic vascular development in mice and the role of Notch signaling in arterial cell fate determination. A: at embryonic day 7.5 (E7.5) (1−2 somite stage), the first angioblasts coalesce on both sides of the embryonic midline, posterior to endocardial precursor cells [cardiac crescent (CC), purple; anterior intestinal portal (aip)]. In the following stages of embryonic development, these arterial precursor cells (aPCs), formed through vasculogenesis, give rise to the paired dorsal aortas (DA). By the 2–3 somite stage (~E8.5), the first venous precursors appear adjacent to the aPCs and eventually coalesce to form the vitelline vein (VV), and ultimately give rise to the cardinal veins. B: sonic hedgehog (Shh) produced by the notochord and floorplate promotes production of high levels of VEGF in the somites, which activates angioblasts from the lateral plate mesoderm. Left: VEGF, in turn, activates the VEGFR2 and neuropilin 1 expressed on the surface of aPCs. This particular configuration of high VEGF coupled with VEGFR2/neuropilin 1 signaling in aPCs activates a signaling cascade, including phospholipase Cγ-1 (Plc-γ) and extracellular signal-regulated kinase (Erk) that ultimately stimulates production of Notch and Delta-like 4 (Dll4) in aPCs. Dll4-activated Notch signaling upregulates the arterial marker ephrinB2 while suppressing the venous marker EphB4. Right: in the presence of low levels of VEGF, venous fated angioblasts express the venous marker chicken ovalbumin upstream promoter transcription factor II (COUP-TFII), which suppresses the expression of neuropilin 1, a membrane-bound co-receptor for VEGF that is required to induce an arterial phenotype, and the downstream expression of Notch. This altered signaling pathway results in preferential expression of the vein marker EphB4. Please note that both Notch and Dll4 are membrane-anchored proteins present on the cell surface and are only indicated inside the cell to illustrate the control of their gene expression by key upstream regulators. Ephb4, B4 ephrin receptor; grl, gridlock; Hey, hairy-and-enhancer-of-split related; Mek, mitogen-activated protein kinase kinase; Pkc, protein kinase c. [We thank Kate Wythe for preparing the illustrations in A, which were adapted from Fish and Wythe (72), with permission from Dev Dyn; B was adapted from Lin et al. (153), with permission from EMBO Rep.]
FIGURE 11.

The role of Notch signaling in balancing the artery vs. vein decision during vasculogenesis in the embryo and in the yolk sac. A, top: the relative size of the dorsal aorta and cardinal vein in a wild-type embryo is schematized. In mice lacking Notch1, the endothelial arterial precursor cells (aPCs) fail to attain their arterial differentiation state, including the proper level of ephrinB2 (Efnb2) expression, which is necessary to allow sorting out of the aPCs and venous precursors (vPC). This results in a rudimentary dorsal aorta (da) with more cells attaining a vein-like fate [bottom left, cardinal vein (cv)]. In the absence of both ephrinB2 and EphB4, an enlarged dorsal aorta develops, most likely as a consequence of the improper sorting of both aPCs and vPCs into the dorsal aorta. Bottom: a seesaw model of the reciprocal relationships between appropriate arterial and venous cell sorting, which depends on Notch and ephrinB2/EphB4 signaling and controls the size of the dorsal aorta and cardinal vein. B: a similar situation is encountered in yolk sac vasculogenesis, where increased Notch signaling results in a predominance of large-caliber arterial vessels, whereas inactivation results in a predominance of small-caliber veinlike vessels. The normal wild-type situation results in development of both types of vessels that provide normal circulation in the yolk sac. [A is adapted from Kim et al. (128), with permission from Development; B is from Copeland et al. (41), with permission from BMC Dev Biol.]
Interestingly, the venous EPCs lack neuropilin 1 and respond to low levels of VEGF to induce expression of the vein marker COUP-TFII, which, in turn, is regulated by the chromatin remodeling enzyme BRG1 (47). COUP-TFII further represses VEGFR2, neuropilin 1, and Notch signaling to promote expression of the vein marker EphB4 and ensure maintenance of the venous state. In this context, it should be noted that Notch signaling controls the development of the full arterial identity in aPCs, which appear more veinlike if Notch signaling is blocked. However, the venous identity of vein-fated angioblasts develops separately and independently.
B. Yolk Sac Vasculogenesis and Angiogenesis
The development of the extraembryonic yolk sac vasculature via de novo vasculogenesis begins during early embryogenesis at E7–E7.5 (61, 196). In yolk sac vasculogenesis, hemangioblasts, which give rise to blood islands with hematopoietic precursors adjacent to endothelial cells, are the source of endothelial cell precursors. In this respect, yolk sac vasculogenesis differs from vasculogenesis in the embryo proper, where EPCs arise from mesenchymal cells (see above). Nevertheless, the basic Notch-dependent cell fate decision (i.e., artery vs. vein) is conceptually similar (for crucial early studies, see Refs. 62, 78, 136; see also Refs. 41, 128). Assembly of the initial vascular plexus and lumen formation is followed by intussusception and branching angiogenesis (56, 178, 196). Interestingly, while both loss- and gain-of-function of endothelial Notch signaling results in impaired remodeling of yolk sac vasculature, loss-of-function of endothelial Notch signaling (by inactivation of Rbpj) produces small-caliber, veinlike vessels, whereas gain-of-function of endothelial Notch signaling (by overexpression of NICD) promotes the development of large-caliber arterial vessels (41) (FIGURE 11B).
Overall, inactivation of Notch signaling during early development favors the development of veins over arteries in the embryo proper and in the yolk sac (62, 78, 128, 136) (FIGURE 11). This interpretation is further supported by gain-of-function experiments, in which overexpression of Dll4 or NICD has the opposite effect (preferential development of arteries over veins) from inactivation of Notch signaling (41, 248). The process of Notch-dependent lateral inhibition can contain dynamic components (113, 181, 182), but it ultimately gives rise to endothelial cells destined for an arterial vs. venous cell fate (128). This also provides a compelling explanation for the development of arteriovenous shunts upon inactivation of endothelial Notch signaling during development (45, 170, 172). Even more severe defects in early vasculogenesis are observed in mice lacking sonic hedgehog (256), VEGF (31, 70), the VEGFR2 (218), or Tie2 and its ligand Ang1 (14, 234), all of which are upstream regulators of Notch signaling.
Taken together, these and other studies have provided crucial insights into the role of Notch signaling and its upstream regulators and downstream effectors in the early stages of vasculogenesis and angiogenesis. However, the early embryonic lethality caused by the endothelial-specific deletions of key components of the Notch pathway presented an impediment to further analysis of the role of this signaling pathway at later embryonic and postnatal stages of vascular development.
IV. ROLE OF ADAM10/NOTCH SIGNALING IN THE DEVELOPMENT OF SPECIALIZED VASCULAR STRUCTURES
A. Mice Lacking ADAM10 in Endothelial Cells (A10ΔEC) Have Defects in Specialized Vascular Structures
In light of the very severe vascular defects that develop early during embryogenesis in mice lacking different components of the Notch pathway in endothelial cells, it was surprising to find that endothelial-specific inactivation of the major Notch processing enzyme, ADAM10, did not cause early embryonic lethality (82). Instead, A10ΔEC animals were born at the expected Mendelian ratio, and most survived into adulthood, although their life expectancy was shorter than that of wild-type controls. Moreover, A10ΔEC mice had striking defects in several organ-specific vascular niches, including in the retina, liver, heart, kidney, intestine, and bone. Since little was known about the role of Notch signaling in the development of organ-specific vascular beds at the time, this intriguing constellation of vascular defects raised questions about whether ADAM10 controls Notch-dependent endothelial cell fate decisions that are crucial for the development of organ-specific vascular niches. Alternatively, the defects in A10ΔEC mice could also have been caused by an essential contribution of ADAM10 to the processing of other molecules with essential roles in organ-specific vascular development.
B. Mice Lacking Notch1 in Endothelial Cells and Notch4 Systemically Recapitulate the Defects in Specialized Vascular Beds Observed in A10ΔEC Mice
Since then, it has become clear that all of the phenotypes observed in A10ΔEC mice are recapitulated in animals lacking Notch1 in endothelial cells and Notch4 systemically (N1ΔEC/N4−/−) mice (6). The majority of these defects were also at least partially recapitulated in animals lacking only Notch1 in endothelial cells (N1ΔEC mice). Specifically, N1ΔEC mice had similar defects in long-bone vasculature, less severe defects in the heart, kidney, and liver, and no evident abnormalities in the intestinal and retinal vasculature. In addition, all vascular defects observed in A10ΔEC could be rescued by overexpression of the NICD, further corroborating that they were caused by a lack of ADAM10-dependent Notch processing. Importantly, the N1ΔEC and N1ΔEC/N4−/− mice were generated with the same Tie2-Cre transgene that gave rise to viable A10ΔEC animals with abnormal organ-specific vascular structures (82, 129). The observation that the vascular defects in N1ΔEC/N4−/− animals resemble those found in A10ΔEC mice suggests that ADAM10 is also involved in processing Notch4, although there are conflicting reports on whether or not Notch4 participates in canonical Notch signaling (12, 115, 219). Since the N4−/− mouse strain used to generate the N1ΔEC/N4−/− double-mutant mice produces an N4 extracellular domain with potential dominant-negative activity on Notch1, we cannot rule out that the N1ΔEC/N4−/− double-mutant mice have a more severe vascular phenotype than the N1ΔEC mice because of potential dominant-negative effects of the N4−/− construct on residual Notch1 signaling in the N1ΔEC mice. The availability of a different N4−/− mouse line that does not express the dominant-negative extracellular domain of N4 could be used to address this possibility (115). On the other hand, when A10ΔEC mice were generated with a different Tie2-Cre driver, the one that had been used in previous studies to inactivate Notch1 or RBPJ in endothelial cells (132), the resulting mice resembled previously described endothelial-specific Notch-pathway knockout animals in that they died by E8.5 with defects in heart development and in the yolk sac vasculature (6). Analysis of the timing of excision of floxed ADAM10 by the two different Tie2-Cre transgenes showed earlier excision by the Tie2-Cre generated by Koni et al. (132) (referred to as Tie2-Cre Flv) than that generated by Kisanuki et al. (129) (referred to as Tie2-Cre Ywa), providing a plausible explanation for the aforementioned differences in phenotype and lethality (6).
In this context, it should be noted that conditional inactivation of both ADAM10 and ADAM17 by Tie2-Cre Ywa in endothelial cells (A10/17ΔEC) did not significantly exacerbate the phenotype observed in A10ΔEC mice. Moreover, previous studies had shown that conditional deletion of ADAM17 in endothelial cells with Tie2-Cre Ywa does not produce developmental phenotypes, although these animals are protected from pathological neovascularization, presumably because of the role of ADAM17 in EGFR signaling (265). Taken together, these findings argue against a major compensatory or redundant role of ADAM17 in the Notch signaling pathway during in vivo blood vessel development (6). Moreover, they underscore the conclusion that ADAM10 is required for physiological, ligand-induced Notch signaling, whereas ADAM17 is not (20, 252) (see above).
When RBPJ was knocked out in endothelial cells after birth, this produced very similar defects in the bone vasculature and in bone growth as those seen with inactivation of endothelial ADAM10 (190, 191). Moreover, postnatal conditional endothelial knockout of Notch1 produced defects in liver vessels, including enlarged, subcapsular, veinlike vessels and defective sinusoidal vessels, which were strikingly similar to A10ΔEC knockouts (45). The observation that the development of several specialized vascular beds depends on ADAM10/Notch signaling raises interesting questions about the underlying mechanisms. One question is whether cell fate decisions could be at play, perhaps similar to those that are well-characterized for the retina or for early embryonic or yolk sac vasculogenesis. Another question relates to what the affected vascular niches might have in common. In the next section, we will briefly summarize the current understanding of the role of Notch signaling and the origins of endothelial cells during the development and maturation of the affected vascular beds (113, 181). (For a review of the role of Notch signaling in the vasculature, see Ref. 87, and for more comprehensive reviews of organ-specific vascular development, see Refs. 3, 4, 13, 106, 189, 201.)
V. WHAT IS KNOWN ABOUT THE ROLE OF NOTCH SIGNALING AND THE ORIGIN OF ENDOTHELIAL CELLS IN THE DEVELOPMENT OF SPECIALIZED VASCULAR NICHES?
A. Coronary Vessels
The coronary vasculature (FIGURE 12A) supplies the heart muscle with oxygen and nutrients as it relaxes during diastole. Since coronary vessel disease is a major cause of morbidity and mortality, understanding the development of this vascular bed is both a fascinating topic and of utmost biomedical relevance. A10ΔEC and N1ΔEC/N4−/− mice have enlarged subepicardial vessels lined by endothelial cells and lacking mural cell ensheathment (sections from A10ΔEC hearts are shown in FIGURE 12B). Studies of avian and mammalian coronary vessel development support the concept that portions of the coronary vasculature develop separately from the main vascular tree (FIGURE 12C). Lineage tracing experiments in mice and chickens suggest that the coronaries are assembled from endothelial cells of three different origins: the sinus venosus, the endocardium, and the epicardium (33, 158, 244, 258, 267; for a recent review, see Ref. 245) (FIGURE 12D). Since these lineage tracing studies were done with distinct cell type-specific Cre driver lines, some disagreements remain regarding the relative contribution of these three sources of endothelial cells to the coronary vasculature [discussed by Tian et al. (245)]. Nevertheless, there seems to be a consensus that most, if not all, coronary vessels derive from de novo vasculogenesis. Ultimately, the coronary arteries connect to the aorta just above the aortic valve to establish a link to the main arterial circulation (FIGURE 12C) (158).
FIGURE 12.

Coronary vasculature. A: diagram of the coronary vasculature, with coronary arteries emanating from the aorta, just above the aortic valves (arrow). B: A10ΔEC mice have enlarged subepicardial vascular structures (arrow in B, top) that are not observed in controls. In histopathological sections, these subepicardial structures are surrounded by thin-walled MECA32-positive endothelial cells (brown staining pointed to by arrow in B, bottom left). C: a diagram of the time course of mouse coronary artery development shows the pro-epicardial organ attaching to the heart at embryonic day 9.5 (E9.5), followed by the appearance of angioblasts that form a primitive vasculature by vasculogenesis by E11.5. Eventually, this primitive vasculature gives rise to a more organized network of large and small vessels through angiogenesis. Initially, these vessels are not connected to the aorta, although they connect to the ascending aorta above the aortic valves around E13. Subsequently, these endothelial cells (ECs) become ensheathed with smooth muscle cells (SMC) during the process of arteriogenesis. P0, postnatal day 0. D, top: diagram of the mouse embryo sinus venosus (SV) at E10.5 to illustrate the formation of the nascent coronary vessel plexus in the developing heart. At this stage of development, the SV is intimately associated with the pro-epicardium (PE), the endocardium (Endo), as well as the developing liver sinusoids. The letter “a” (green highlight) indicates the ventricular endocardium, “b” (blue highlight) indicates the subepicardium, and “c” (gray highlight) indicates the epicardium. Bottom: different sources of coronary vessels at distinct stages of development are indicated in the flow chart, which is based on lineage tracing experiments that are discussed in Ref. 245. The numbers at the bottom correspond to the migration events indicated at the top. [A is from Servier Medical Arts, with permission; B is from Glomski et al. (82), with permission from Blood; C is from Luttun and Carmeliet (158), with permission from Cardiovasc Res; and D is from Tian et al. (245), with permission from Circ Res.]
Previous studies have focused on the regulation of coronary vascular development by Notch signaling. During avian heart development, the NICD can be detected in endothelial cells in a nascent vascular plexus at the atrioventricular junction, suggesting a role for Notch in coronary endothelial progenitor cell differentiation (269). In mice, inactivation of Notch1 in the epicardium using Wt1-Cre leads to defects in the differentiation of coronary arteries, suggesting that Notch signaling controls coronary artery commitment, differentiation, and maturation (53). Moreover, the Notch regulator POFUT was recently reported to control coronary artery development from endocardially derived cells (261). Red-Horse et al. (193) provided evidence that venous endothelial cells from the sinus venosus must first de-differentiate so that they can give rise to different endothelial cell types in the myocardium, including arterial endothelial cells. Interestingly, if adult venous endothelial cells are used for coronary bypass surgery, they lose some of their vein-specific markers over time (139), but are not able to assume an arterial phenotype. In contrast, bypass grafts of arterial origin more closely resemble coronary arteries (206). This supports the notion that arterial cells are, in principle, able to acquire an arterial phenotype on transplantation, whereas vein-derived cells are not. Defects in Notch signaling were also found in mice that express only the 120-kDa isoform of VEGF-A; these mice exhibited decreased expression of arterial markers, such as EfnB2, and increased expression of venous markers in coronary endothelial cells (250). Finally, with respect to the role of Notch signaling in heart development, the endothelial Notch ligand Jagged is important for heart valve and coronary vascular development (104, 257) and homeostasis in adults (124), and mutations in Notch have been implicated in development of bicuspid aortic valves. (See Refs. 46, 50 for recent reviews on the role of Notch signaling in heart development.)
B. Liver Sinusoids
The liver contains intricate vascular structures that are fed from two sources, the hepatic artery, which branches from the aorta, and the portal vein, which delivers blood from the mesenteric veins after it has passed through the vessels of the digestive tract (FIGURE 13A). The sinusoids derive their name from their sinusoidal endothelial lining, which is characterized by large irregularly shaped openings that facilitate efficient transfer of nutrients and solutes between the vascular space and surrounding hepatocytes (FIGURE 13B, inset). Hepatocytes also produce bile, which drains through bile canaliculi into the bile duct, gall bladder, and, ultimately, duodenum. Cuervo and colleagues (45) have shown that postnatal inactivation of RBPJ or Notch1 in endothelial cells by VECad-ERT2-Cre results in formation of enlarged, abnormal vessels, resembling those described in A10ΔEC or N1ΔEC mice generated with the Tie2-Cre Ywa, under the liver capsule (6, 45, 82) (FIGURE 13C). In addition, Cuervo et al. (45) noted the presence of hepatic porto-systemic shunts in the mutant animals, suggesting that endothelial Notch/RBPJ signaling is required for proper assembly of the liver’s intricate capillary vascular bed. Overall, these studies support the conclusion that ADAM10/Notch/RBPJ signaling has an important role in the development of the sinusoidal vasculature. Moreover, the observation that the timing of conditional inactivation of Notch or RBPJ affects the severity of the phenotype, with later inactivation producing a less severe phenotype (45), suggests that the vascular defects in the liver are caused, at least in part, through a defect in maturation or maintenance of the hepatic sinusoidal endothelium.
FIGURE 13.

Liver vasculature. A: major blood conduits through the liver are the portal vein, which carries poorly oxygenated but nutrient-rich blood from the gastrointestinal tract to the liver, and the hepatic artery, which delivers well-oxygenated blood to the liver. Input from both vascular beds flows through the sinusoids and drains into the central vein, which feeds hepatic venules, then into larger hepatic veins that drain into the inferior vena cava. B: the building blocks of liver lobules include input from the portal venule and hepatic arteriole, which feed into the sinusoids separately, but merge toward the center, where blood from the sinusoidal vasculature collects into the central vein. Adjacent hepatocytes process the nutrients and lipids in portal vein blood to generate bile. The sinusoidal endothelium functions as a selective sieve and scavenger, together with luminal monocyte-derived Kupffer cells, and as a mediator of immune tolerance. Inset: the sinusoidal vasculature contains large fenestrations (100–200 nm), also called sinusoidal openings, to allow transport of solutes and macromolecules as well as lipids to the hepatocytes for further processing. C: A10ΔEC mice have enlarged vessels on their liver surface that often end blindly under the liver surface, without connection to similarly large-caliber vessels for drainage (82). Histochemical analysis showed enlarged subcapsular vessels surrounded by thin-walled endothelial cells lacking evident mural cell coverage. [A and B are from Aird (4), with permission from Circ Res; C is from Glomski et al. (82), with permission from Blood.]
C. Small Intestinal Vasculature
Small intestinal villi, which are lined by enterocytes responsible for absorption of nutrients into the bloodstream, contain individual capillary bed units. In mice, endothelial cells associated with mesodermal tissue sprout into the proximal (small) intestine by E9.5 to form a primary capillary plexus through vasculogenesis. A uniform submucosal capillary plexus is formed throughout the intestine by E11.5. By E15.5, it has undergone further angiogenic remodeling and reorganization to form the branching, hierarchical capillary network characteristic of intestinal vasculature (37, 96–98). In zebrafish, veinlike vascular precursors have been shown to contribute to the development of the intestinal vasculature; however, Notch signaling does not appear to be required for this process (100). Little is currently known about the role of Notch signaling in the development of the murine intestinal vasculature. However, Dll4/Notch signaling is required to maintain the correct proliferative and regenerative state of lymphatic capillaries (lacteals) in intestinal villi (17, 174) (FIGURE 14A). Since the intestinal polyps observed in A10ΔEC or N1ΔEC/N4−/− mice consist of abnormal clusters of CD31-positive endothelial cells, they appear to be distinct from the lacteals (FIGURE 14B). Instead, they appear more reminiscent of the large aggregation of PECAM+ (CD31+) cells found associated with endothelial sprouts in early intestinal vascular development, and could plausibly result from defective or arrested vasculogenesis or angiogenesis in individual villi (98). Additional studies will be necessary to provide further insight into this and other potential roles of endothelial Notch signaling in the development of the enteric vasculature.
FIGURE 14.

Intestinal villus vasculature. A: small intestinal villi are supplied by arterioles, which branch into a capillary system that drains into venules, then into larger veins, and ultimately into the portal vein. A separate system of lacteals functions as lymphatic vessels in the small intestine. B: A10ΔEC mice have large polyps in the small intestine that are characterized by abnormal clusters of MECA32-positive endothelial cells, which are not seen in wild-type intestines. [A is from Servier Medical Arts, with permission; B is from Glomski et al. (82), with permission from Blood.].
D. Glomerular Vasculature
The renal vasculature is composed of highly specialized vascular components critical to the filtration, secretory, and reabsorptive functions of the kidney. The first vessels of the mouse kidney start to form around E12.5, with the first glomeruli developing soon thereafter (103). Glomeruli contain specialized endothelial cells that have fenestrae, which allows filtration of molecules from the blood into the adjacent nephron (FIGURE 15, A AND B). The endothelial progenitors that form glomerular tufts are VEGFR2 positive and are thought to originate in the aortogonadal mesonephros, where early hematopoiesis occurs (255). However, questions about the exact origins of glomerular endothelial cells still remain. Once immature glomerular endothelial cells appear next to a developing glomerulus, podocytes release VEGF to recruit them into a structure known as the S-shaped body (255) (FIGURE 15C, left). The endothelial cells next form a single glomerular loop, which subsequently branches into additional loops (FIGURE 15C, middle and right), that then acquire fenestrations (68, 187). The enlarged glomeruli found in A10ΔEC (FIGURE 15D) and N1ΔEC/N4−/− mice indicate that endothelial Notch signaling plays a role in the development, maturation, or both of glomeruli. Notably, studies from the Kopan group have shown that normal Notch signaling in podocytes and mesangial cells is also required for proper glomerular development (19, 35, 36). Further analysis of glomeruli in A10ΔEC mice has revealed an increase in sites of intussusception and persistence of fenestral diaphragms (69), findings consistent with arrest at an immature stage of development (109, 159). In addition, the expression of several molecules that have been implicated in the control of vessel diameter (VEGFR3, apelin/AplnR, and CXCR4) was elevated, providing a possible explanation for the increased caliber of the glomerular loops in A10ΔEC kidneys (57, 69, 125, 126, 237, 239).
FIGURE 15.

Kidney vasculature. A: cross section of the cellular organization of a glomerulus depicts the afferent and efferent arteriole (AA, AE), the glomerular vascular loops lined by endothelial cells, as well as mesangial cells (MC) and podocytes (pod). Filtrate from the glomerular capillary tufts collects into Bowman’s space (BS). GEC, glomerular endothelial cell, PEC, parietal epithelial cell; PT, proximal tubule. B: a fenestrated GEC is shown surrounded by a podocyte with foot processes that touch the basal lamina/glomerular basement membrane. C: different stages of glomerular vascular development illustrated by sections of newborn kidneys stained with CD31 (brown staining) include the S-shaped body (left), the immature glomerulus (middle), and the mature glomerulus stage (right). D: the glomeruli in A10ΔEC mice are enlarged compared with controls and also contain enlarged glomerular capillaries (top, pointed to by arrows). Moreover, the sections of A10ΔEC glomeruli show stronger staining with MECA32, an antibody that recognizes the fenestral diaphragm protein PV-1. [A is from Scott and Quaggin (214), with permission from J Cell Biol; B is from Aird (4), with permission from Circ Res; C is from Farber et al. (69), with permission from Angiogenesis; D is from Glomski et al. (82), with permission from Blood.]
E. Bone Vasculature
Most bones contain highly vascularized cavities that harbor hematopoietic cells, as well as blood vessels (FIGURE 16A). Different types of vessels in the bone can be distinguished based on their expression profiles and markers and are defined as type H, type L, and type E vessels (142, 144, 190, 191) (FIGURE 16B). A crucial role for endothelial ADAM10 in the development of long bones was first described by Glomski and colleagues (82, 275), who found that A10ΔEC mice exhibited defective growth plates and significantly shortened long bones (FIGURE 16C). Bone defects resembling those in A10ΔEC mice were subsequently observed on inducible, postnatal inactivation of Rbpj or Dll4 in mouse endothelial cells (191) and in N1ΔEC/N4−/− mice (6).
FIGURE 16.

Bone vasculature. A: section through a femur shows a large artery entering and a large vein exiting the bone, whose cavity is filled with bone marrow, which consists of hematopoietic cells (including red blood cell, white blood cell, and platelets precursors), supporting stromal cells and angioblasts. B: the bone contains specialized vessels that are referred to as type H and type L vessels. Type H vessels typically reside in proximity to the growth plate, whereas type L vessels are found in the marrow cavity. The development of type H vessels is controlled by Notch signaling. Different types of perivascular cells (PVCs) are associated with blood vessels in the bone. PVCs surrounding type H capillaries express nestin, platelet-derived growth factor receptor (PDGFR)-β, and NG2, PVCs associated with type H-vessel columns express PDGFR-β and NG2. Type L vessel PVCs include LEPR and PDGFR-α positive mesenchymal cells and CXCL12-abundant reticular (CAR) cells. Red arrows indicate blood flow. dp, Diaphysis; mp, metaphysis. C: A10ΔEC mice have shorter long bones compared with controls, with aberrant growth plates and enlarged, abnormal vascular structures in the proximity of the growth plate. [A was provided with permission by the National Cancer Institute, 2014 Terese Winslow LLC, U.S. Govt. has certain rights; B is from Sivaraj and Adams (222), with permission from Development; C is from Glomski et al. (82), with permission from Blood.].
Studies from the Adams laboratory have been instrumental in defining the different types of vasculature in the bone. Specifically, the type H vessels are characterized by their high expression of endomucin and CD31 and their close proximity to the growth plate in long bones (142, 191). Type H vessels are in close contact with osteoblasts and participate in crosstalk with chondrocytes (142). The type L vessels are not associated with osteoblasts, are found in the bone marrow cavity, and are described as sinusoidal. Notch signaling is crucial for differentiation of type H endothelial cells and promotes the expression and secretion of the bone morphogenic protein family member Noggin, which is required for normal chondrocyte and osteoblast function, and, by extension, maintenance of the normal growth plate (191). Interestingly, blood flow has also been implicated in regulating Notch signaling, and thus in normal osteoblast and chondrocyte function in bone (190). The gradual impairment of blood flow that occurs naturally with aging impacts bone health by reducing angiogenesis and osteogenesis. These effects could be reversed in mice through activation of Notch signaling by overexpression of the NICD, thus uncovering a potential new approach to treatment of bone disease.
F. Retinal Developmental Angiogenesis
The specialized retinal vasculature differentiates to yield a blood-retinal barrier, which is similar to the blood-brain barrier. Many fundamental insights into the mechanisms of angiogenesis have come from studies of retinal vascular development. In mice, this process is initiated close to the optic nerve at birth, and the developing vessels follow in the path of astrocytes that migrate to the periphery of the retina (77) (FIGURE 17A, see also FIGURE 6). As discussed above, perturbation of Notch signaling causes defects in the tip/stalk cell fate decision during retinal vascular development, (key first studies were Refs. 99, 156, 232; see also Refs. 6, 16, 65, 82; reviewed in Refs. 88, 105, 113, 200). Both VEGFR2 and VEGFR3 have been implicated as upstream regulators of Notch signaling in the developing retinal vasculature (16), although a more recent study from the Alitalo group emphasizes that VEGFR2 has a crucial role in this process, whereas VEGFR3 does not (272). Interestingly, the defects in the retinal vasculature caused by blocking Notch signaling can later be remodeled and repaired. This explains why the retinal vessels in adult A10ΔEC mice appear normal, even though they are profoundly affected in 5-day-old mice (82) (FIGURE 17B). Leukocytes have been implicated in the remodeling of retinal vasculature, since their absence is sufficient to cause increased vascular density, resembling the phenotype seen in mice with defects in endothelial Notch signaling (111, 183).
FIGURE 17.

Retinal vascular development. A: the developing retinal vascular tree is a widely used system for the study of developmental angiogenesis and the tip/stalk cell decision (see also FIGURES 2 and 6). The retinal vasculature in mice develops postnatally and starts to grow out from the optic nerve head at postnatal day 1 (P1). The primary plexus of the developing retinal vascular tree then expands toward the periphery of the developing retina (left panel shows the growth after ∼1 wk), ultimately producing the extensively remodeled mature superficial plexus seen in adults (right). B: example of how defective a disintegrin and metalloprotease 10 (ADAM10)-dependent Notch signaling affects retinal vascular development can be seen in an isolectin B4-stained A10ΔEC retina at P5, which shows an increased vascular density compared with a wild-type control (an enlarged image of the vasculature within the white boxes is shown in FIGURE 2). [A is from Ruhrberg and Bautch (205), with permission from Cell Mol Life Sci; B is from Glomski et al. (82), with permission from Blood.]
In this context, it is important to note that many concepts regarding the properties and behavior of Notch signaling as a regulator of the cell fate decision between endothelial tip cells and stalk cells are largely based on in vivo studies. However, the basic cell biology of these phenomena remains incompletely understood and needs to be investigated in more detail. It is likely that many of these concepts may have to be revised over time based on new information. For example, studies in the Gerhardt laboratory reported that tip and stalk cells dynamically shuffle their position, resulting in a regular exchange of the leading tip cell (114). Moreover, recently reported cell lineage tracing experiments of endothelial cells using the Confetti mouse system (198, 212) suggest random integration or mixing of endothelial cells during mouse retinal angiogenesis (161), which is different from the current model of angiogenesis by branching from the existing vascular tree.
G. Other Vascular Beds
In light of the variety of defects in specialized vascular structures in A10ΔEC or N1ΔEC/N4−/− mice, it was remarkable that they had no evident major vascular defects on histopathological evaluation of hematoxylin-and-eosin- and CD31-stained sections in other tissues, including the lung, brain, and skeletal muscle. (For a recent review on the development of the brain vasculature and the blood-brain barrier, see Ref. 259.) This does not rule out the possibility that additional vascular defects in one or more of these other tissues might emerge with more detailed analysis. Moreover, it is possible that potential transient vascular defects are remodeled, like in the retina, so that these vascular beds appear normal in the adult A10ΔEC or N1ΔEC/N4−/− mice. Nevertheless, an intriguing alternative interpretation is that Notch signaling might not be uniformly required for tip/stalk cell decisions during vascular branching, and that the branch frequency and the cell fate decisions that determine vascular patterning in the muscle, for example, might depend on different pathways. In this context, it is worth noting other regulators of endothelial cell sprouting and branching. Endothelial cell metabolism, in particular glycolysis and fatty acid oxidation, has been implicated in endothelial cell sprouting from quiescent vessels (29, 48, 64, 184), and angiopoietin 1 can induce sprouting angiogenesis and acts synergistically with VEGF (127, 131). On the other hand, transforming growth factor (TGF)-β signaling has been shown to suppress vascular sprouting in the brain (8), and EphB4 can negatively regulate blood vessel branching (67). Clearly, the elucidation of the molecular mechanisms and pathways responsible for vascular branching in the development of different vascular structures and during pathological neovascularization represents a challenging, but also exciting, opportunity for future studies.
H. Pathological Neovascularization
In addition to its various roles in physiological vascular development, Notch signaling has also been implicated in pathological neovascularization. There are multiple mouse models to study this process, such as the oxygen-induced retinopathy (OIR) model for retinopathy of prematurity (ROP) or various tumor models that require angiogenesis and neovascularization. [A comprehensive review of different models to study angiogenesis and tissue vascularization can be found elsewhere (221).] The mouse model for OIR (also referred to as ROP model) has two key components: relative hyperoxia, which triggers vascular regression, and relative hypoxia, which triggers a strong VEGF-dependent neovascular response (34, 40, 223). In this model, which was developed by Smith et al. (223) and is now widely used, the Dll4/Notch pathway has been reported to promote vascular regression following hyperoxia (155). However, no difference in vascular regression was seen in A10ΔEC mice subjected to OIR, although these animals showed increased revascularization and developed unusual vascular sheets that were not seen in controls (82). Interestingly, there are reports that pathological retinal neovascularization could also be initiated, at least in part, by de novo vasculogenesis (76) by recruiting angioblasts, or endothelial cell precursors, from the bone marrow (60, 79, 151, 175). Recent lineage tracing experiments showed an increase in clonally expanded endothelial cells carrying gene signatures of endothelial-to-mesenchymal transition in the OIR model (161). The relative ease of visualizing the retinal vasculature makes this a particularly attractive model system to study pathological neovascularization.
Endothelial Notch signaling also has a crucial role in tumor angiogenesis. In a seminal paper, Noguera-Troise et al. (173) found that Dll4 controls vascular sprouting during tumor angiogenesis and reported that inactivation of Dll4 leads to an increased, nonproductive vascularity associated with poor perfusion. (For a review of the promise of Dll4/Notch signaling in tumor angiogenesis, see Refs. 92, 140.) On the other hand, transgenic Dll4 expression in endothelial cells reduces their response to VEGF, thereby affecting proliferation, vessel density, and tumor perfusion, and reducing metastatic potential (247). Therefore, concomitant blockade of the related Dll4/Notch and ephrinB2/EphB4 pathways has emerged as an effective approach to impede tumor angiogenesis (55).
Additional Notch pathway members have been implicated in tumor angiogenesis. Gain and loss-of-function studies with endothelial Jag1 revealed a role for this Notch ligand in regulating tumor angiogenesis and crosstalk with surrounding tumor cells (179). (For a review of Jagged as a tumor target, see Ref. 147.) Moreover, dysregulation of Jagged expression has also been suggested to disrupt physiological angiogenesis and promote pathological angiogenesis by interfering with the correct execution of the tip/stalk cell decision (18). Other studies have shown that the NICD is frequently found in tumor endothelial cells or in the pre-metastatic niche. This, in turn, promotes senescence in endothelial cells and secretion of proinflammatory cytokines that generate favorable conditions for cancer metastasis (202, 266).
Given the various contributions of endothelial Notch signaling to cancer, it is not surprising that this pathway is considered a potential target in oncology. Several different approaches have been explored to translate this concept into practice, such as the generation of an antibody that blocks ligand binding and affects tumor angiogenesis. A seminal paper by Wu et al. (268) provided proof of concept that antibodies could be used to target individual Notch receptors by stabilizing the NRR domain. Antibodies against Notch1 blocked cancer growth and deregulated angiogenesis in a mouse cancer model, without causing the toxicity seen with γ-secretase inhibitors that block several Notch receptors (268). This has prompted several attempts to translate these concepts into the clinic (e.g., Refs. 71, 186, 270). Binding of different Notch ligands can be differentially targeted, resulting in distinct outcomes [hypersprouting of nonfunctional vessels vs. inhibition of angiogenesis and pericyte coverage (119), reviewed in Refs. 25, 26]. Interestingly, a soluble Dll4 construct not only affected tumor angiogenesis and thus tumor growth, but also interfered with Notch-dependent microglia recruitment (120). Thus targeting Notch signaling may not only affect endothelial cells in a cell-autonomous manner, but may also influence the interaction of endothelial cells with other cell types, suggesting possible additive benefits for Notch signaling antagonists used in oncological treatment. In principle, deletion of ADAM10 in endothelial cells would be expected to effectively block all canonical Notch signaling in these cells, although it remains to be established whether Notch4 participates in canonical Notch signaling (91, 115). The resulting phenotype would, therefore, be predicted to equate to the combined blockade of inputs from all relevant Notch ligands, regardless of the ligand-expressing cell type (e.g., other endothelial cells or nonendothelial cell types such as pericytes).
VI. WHAT, IF ANYTHING, MIGHT THE SPECIALIZED VASCULAR NICHES THAT DEPEND ON ADAM10/NOTCH SIGNALING HAVE IN COMMON?
The unique constellation of vascular defects in A10ΔEC and N1ΔEC/N4−/− mice raises intriguing questions about potential common underlying endothelial ADAM10/Notch-dependent mechanisms in the development or maturation of these very different structures. As discussed above, the ADAM10/Notch pathway usually guides differentiating cells toward one of two possible cell fates. Therefore, the most likely common underlying mechanism is an ADAM10/Notch-dependent cell fate decision. This raises questions about the identity of the EPCs that participate in such cell fate decisions, and about the outcomes of these decisions, i.e., the specific molecular features and functional properties that define differentiated and specialized endothelial cell fates or their alternative, less differentiated states.
Typically, the Notch-dependent prevention of a default fate is referred to as lateral inhibition (FIGURE 5), but, in addition, ADAM10/Notch signaling most likely promotes the next differentiation state of endothelial cells in a context-specific manner. The abnormal organ-specific vasculature observed in adult A10ΔEC and N1ΔEC/N4−/− mice thus presumably represents the less differentiated default outcome of such a cell fate decision. Most likely these default-state endothelial cells no longer exist in a normal adult, because they acquired the appropriate next level of differentiation through ADAM10/Notch signaling during the course of normal development. Therefore, the abnormal specialized vascular beds in A10ΔEC and N1ΔEC/N4−/− mice might provide an attractive opportunity to learn more about transient, but crucial, intermediate stages of vascular differentiation. In this context, it should be emphasized that Notch signaling can promote differentiation or keep cells in a less differentiated state, such as in intestinal crypts (75, 251) or in airway epithelia (93, 143, 166, 249), so both scenarios should be considered as possible consequences of blocking Notch signaling.
The specific set of vascular defects in A10ΔEC and N1ΔEC/N4−/− mice also raises questions about the origin of the endothelial cells populating specialized vascular beds. In principle, there are at least three possible sources of these precursor cells: 1) they could branch from an existing vessel; 2) they could be recruited from the bone marrow or from a primitive hematopoietic organ; or 3) they could develop through de novo vasculogenesis in the affected organ. In either scenario, the Tie2-Cre transgene (Ywa) would have to be turned on before the ADAM10/Notch-dependent cell fate decision is made. In addition to a role for ADAM10/Notch signaling during vascular development, it is also possible that this pathway is required for the maturation and homeostatic maintenance of specialized vasculature beds. Finally, it is interesting to consider specific features that some of the affected vascular beds have in common, including occurrence within a portal system with two consecutive capillary beds and specialized endothelial linings with fenestral or sinusoidal openings.
A. Where Do the Precursor Cells for the Affected Vascular Niches Originate?
In one model, the vascular niches could develop through branching angiogenesis from the existing vasculature in response to an inductive event (FIGURE 18, model A). During or after the formation of the new branch, the cells in this developing structure would undergo their particular tissue-specific cell fate decision, perhaps following a de-differentiation step. The resulting specialized endothelial cell type would proceed to develop into the specialized vascular niche, e.g., a glomerular tuft or a sinusoidal vessel in the bone or liver. In A10ΔEC and N1ΔEC/N4−/− mice, the new vessel branch would lack ADAM10/Notch signaling because it would derive from the main vascular tree, where expression of Tie2-Cre would have already deleted ADAM10 or Notch.
FIGURE 18.

Models for the potential sources of endothelial cells that contribute to specialized vascular structures. Model A: a branch forms from an existing vessel in response to a local cue, and differentiates into a specialized vascular niche, potentially following an initial de-differentiation step. Model B depicts a scenario in which bone marrow (BM)-derived precursor cells are recruited to integrate into an existing vessel so that they can participate in formation of a new vascular branch. Models C and D: de novo vasculogenesis gives rise to a small vascular plexus adjacent to an existing vessel. The source of these endothelial cells could be a hematopoietic organ that is adjacent to the vessel (model C) or, as also indicated in model B, endothelial cells recruited from the BM. Alternatively, angioblasts (green) could develop locally from mesenchymal precursors (blue) through a mechanism conceptually similar to the development of arterial precursor cells from mesenchymal precursors during embryonic vasculogenesis (model D). Model E: vascular defects caused by inactivation of endothelial Notch signaling could also be caused by disruption of blood vessel maturation (e.g., due to inadequate recruitment of mural and other nonendothelial cell types).
In a second model, endothelial precursors could be recruited from the bone marrow or from primitive hematopoietic organs during development (FIGURE 18, model B). This model is based in part on the seminal discovery of progenitor endothelial cells that can be incorporated into various sites of active neovascularization (9–11) and on studies reporting a role for bone marrow-derived endothelial precursors (EPCs) in pathological neovascularization of the retina, cornea, and tumors (9–11, 76, 118, 194, 220, 238). Endothelial cells recruited in this manner could contribute to branching angiogenesis by local integration into an existing vessel at the site of the new branch (FIGURE 18, model B). However, these vascular precursors could also engage in de novo vasculogenesis in situ during development. This could, for example, involve endothelial cell extravasation from an existing vessel to form a small cluster of endothelial cells or a small vascular plexus that would initially not be connected to the main vascular tree (FIGURE 18, model C) (variations of these models are also considered elsewhere, for example in Refs. 158, 177). The separate vascular plexus would further differentiate in response to local cues, such as VEGF-A, would expand by branching angiogenesis, and would ultimately invade the existing vasculature to establish a connection to the main vascular tree. In each of these scenarios, an ADAM10/Notch-dependent cell fate decision would be an essential part of the developmental program to ensure the proper differentiation of vasculogenic precursors into their specialized cell fate.
Pathological neovascularization in adults is supported by bone marrow EPCs (9–11, 76, 118, 194, 220, 238), raising questions about where such vasculogenic endothelial cells might originate during early development. As outlined above, the yolk sac vasculature contains blood islands, with hematopoietic cells and EPCs, which are considered the first and earliest source of angioblasts. During further development, these hematopoietic and vasculogenic precursor cells migrate through the aorta/gonadal ridge/mesonephros (AGM) in the early developing embryo (E7.5–E10) to the fetal liver (~E12.5), the site of fetal hematopoiesis (FIGURE 19, A AND B). At E10.5, the liver and heart are in close proximity to the sinus venosus (FIGURE 12D), an important source of endothelial cells for the coronary vasculature (33). Interestingly, some liver vessels have been shown to arise from the endocardium of the sinus venosus and thus apparently have a similar developmental origin to coronary arteries (274). Therefore, vasculogenic endothelial cells could, in principle, be at the right place at the right time to generate a vascular plexus next to several of the organs (i.e., heart and liver) affected in A10ΔEC and N1ΔEC/N4−/− mice. Finally, since hematopoietic and vasculogenic precursors ultimately take up residence in the bone marrow, it is possible that at least some of the previously noted bone vascular defects could depend on endothelial cells or angioblasts that undergo de novo vasculogenesis in the local environment of the bone marrow (FIGURE 19B). The spleen also harbors angioblasts, but, although the spleen of A10ΔEC mice is enlarged (82), it has not yet been analyzed for potential vascular defects.
FIGURE 19.

Migration of hematopoietic precursors during development. A: hematopoiesis begins with the development of blood islands surrounded by endothelial cells in the yolk sac at embryonic day 8.5 (E8.5). Around E10.5, the site of hematopoiesis has migrated to the aortogonadal ridge/mesonephros region (AGM), and by E11.5 it has moved to the fetal liver. B: the concept of multisite hematopoietic development is also outlined to indicate multiple potential sources in early development (the yolk sac, placenta, umbilical cord, and AGM), and eventual migration via the fetal liver to the spleen, thymus, and bone marrow. [A and B are from Medvinsky et al. (162), with permission from Development.]
In the third model, vasculogenesis could occur locally, driven by endothelial cells that derive de novo from mesenchymal precursors, in a process that would be a conceptually similar process to vasculogenesis in the early embryo proper (FIGURE 18D). There is evidence that at least some vessels are generated in this manner during development, such as in the intestinal and coronary vasculature, which have been reported to develop by local in situ differentiation from resident tissue vascular precursor cells (98, 138, 146, 158) (see above). In addition, during early development, the liver capsule, epicardium, and diaphragm originate in the septum transversum, which may also give rise to endothelial precursors or angioblasts in support of a vasculogenic process (204). It is important to note that this would be a distinct process, separate from the angioblasts coming from the sinus venosus, in that the endothelial cells coming from the septum transversum would be generated locally and de novo.
Finally, in a fourth model, some or all of the vascular abnormalities in A10ΔEC or N1ΔEC/N4−/− mice could be caused, at least in part, through a defect in the maintenance or maturation of the affected vessel beds (FIGURE 18, model E). Any aspect of vessel maturation, including pericyte ensheathment, could be disrupted in these mice (112). In principle, the relevant Notch ligand could be presented by other neighboring endothelial cells or by a different adjacent cell type like a mural cell (e.g., pericyte, mesangial cell) (65, 240). In both scenarios, a signal from the adjacent signal-sending cell would not be processed by the signal-receiving endothelial cells. A good example for the role of Notch signaling in vascular maturation is provided by the ability of Jagged1 to antagonize Dll4/Notch signaling during this process (179).
Taken together, it is quite possible that distinct vascular beds are assembled in a manner that utilizes different aspects of the proposed models. For example, defects in the vasculogenic or angiogenic development of one or more vascular structures could be further exacerbated by a lack of Notch-dependent maturation or adult homeostasis. However, it is also possible that some or all defects are only caused through misguided cell fate decisions during their early development, or only through a lack of maturation.
B. When Do the Relevant ADAM10/Notch-Dependent Cell Fate Decisions Occur?
1. Timing of Tie2-Cre expression
The phenotypes of A10ΔEC and N1ΔEC/N4−/− mice generated with different Tie2-Cre drivers differ dramatically in that the Tie2-Cre Flv mutants suffer from early embryonic lethality, whereas the Tie2-Cre Ywa mutants survive with organ-specific vascular defects (6, 82). The early lethality caused by Tie2-Cre Flv can most likely be explained by its earlier expression, which must precede the ADAM10/Notch-dependent endothelial cell fate decisions required during development of the yolk sac and the early embryonic vasculature [please note that the Tie2-Cre Flv can also lead to deletion of floxed genes in the female germline (51)]. On the other hand, Tie2-Cre Ywa apparently does not inactivate the endothelial ADAM10/Notch pathway early enough to significantly alter development of the yolk sac and embryonic vasculature. The recombination of floxed ADAM10 by Tie2-Cre Ywa is detected by qPCR later, starting at E11.5 (6). Presumably, this allows A10ΔEC and N1ΔEC/N4−/− (Tie2-Cre Ywa) mice to faithfully execute the first endothelial ADAM10/Notch-dependent artery/vein cell fate decisions in the yolk sac and embryo. Tie2-Cre Ywa expression must then turn on before the subsequent ADAM10/Notch-dependent cell fate decision in the affected vascular structures (unless the phenotypes are due to maturation defects). As a next step, it will be interesting to perform lineage-tracing studies to further explore the differences in spatial and/or temporal control of the expression of the Tie2-Cre Ywa compared with Flv recombinase. Moreover, additional studies would further our understanding of why the two Tie2-Cre transgenic mouse lines generate such different phenotypes, and whether the differences are due to subtle differences in their Tie2 promoter and enhancer sequences and targeting vector design (for details, see Refs. 129, 132), differences in accessibility of their chromatin integration sites during vascular development, or other causes.
2. Temporal-inducible deletion with Cdh5-CreERT2
As discussed above, vascular defects in the liver have been reported on postnatal inactivation of Rbpj or Notch1 using the temporal-inducible cadherin 5 (Cdh5)-CreERT2 recombinase. This implies that the expression of Cdh5-CreERT2 also precedes the activation of at least one critical postnatal Notch signaling event in the liver vasculature. Moreover, inactivation of Rbpj using the Cdh5-CreERT2 also resulted in changes in the bone vasculature (see above) (191). When RBPJ was deleted in endothelial cells at different postnatal time points, a series of vascular defects that included arterial/venous shunts and increased vascular density in the brain, as well as arterial/venous malformations in the intestine, heart, uterus, and skin were also observed (172). In the future, it will be interesting to compare the expression patterns of the Tie2-Cre transgenes and the inducible Cdh5-CreERT2 transgene, as well as the endothelial defects in Rbpj or Notch1 floxed mice generated with Cdh5-CreERT2 and those observed in A10ΔEC or N1ΔEC (Tie2-Cre Ywa) mice. Further analysis of mice in which ADAM10/Notch signaling is temporally inactivated at different points during development would help to better understand the importance of the timing of Notch signaling in the development of these specialized vascular beds. Moreover, the spectrum and severity of defects caused by postnatal or adult temporal conditional endothelial inactivation could help to further our understanding of the importance of ADAM10/Notch signaling in the maturation or maintenance of the specialized vascular structures (see FIGURE 18, model E).
C. Common Features of the Organ-Specific Vascular Defects in A10ΔEC or N1ΔEC/N4−/− Mice
1. Artery/vein decision
A remarkable aspect of the vascular phenotypes seen in A10ΔEC or N1ΔEC/N4−/− mice was the presence of large, ectopic, thin-walled vascular structures that are typically not seen in adult mice (FIGURE 20). Specifically, the enlarged vessels on the liver surface (FIGURE 20A), in the subepicardium (FIGURE 20B), in intestinal villi (FIGURE 20C), and in the bone marrow adjacent to the growth plate (FIGURE 20, D AND E) were lined by endothelial cells, but lacked any evident mural cell coverage. When A10ΔEC mice were subjected to OIR, they also developed large sheetlike vascular structures on top of the internal limiting membrane of the retina (FIGURE 20F). Such sheetlike vessels were not observed in wild-type controls, which developed the vascular tufts that are typical for this model (FIGURE 20G). The abnormal and enlarged vessels in different tissues were somewhat reminiscent of the enlarged vascular structures that form in early mouse embryos lacking one allele of VEGF, which is required to initiate Notch signaling (FIGURE 20H) (31). Moreover, the Wang laboratory [Kim et al. (128)] has shown that Notch-dependent control of EphB4 and EfnB2 expression in the early vascular plexus of the developing mouse embryo helps to sort out the vascular precursors into the embryonic aortas and the cardinal veins (see FIGURE 11A). Under normal circumstances, this cell fate decision is efficient and utilizes all precursor endothelial cells, so that ultimately no remnant structures are left behind. However, blocking Notch signaling results in more veinlike cells at the expense of arterial precursors, resulting in an enlarged embryonic vein, and a significantly smaller aorta compared with the cardinal vein (128). Perhaps a conceptually similar process occurs in the development of the affected specialized vascular structures in A10ΔEC or N1ΔEC/N4−/− mice. The enlarged veinlike vessels in several tissues in these animals could conceivably represent the remnants of aborted endothelial cell fate decisions, comprised of endothelial cell precursors that fail to acquire their next programmed cell fate and that cannot be sorted or recruited to participate in building normal vessels.
FIGURE 20.

Unusual large-caliber vessels in various tissues in mice lacking endothelial a disintegrin and metalloprotease 10 (ADAM10)/Notch signaling. Examples of enlarged vascular structures are observed in A10ΔEC or N1ΔEC/N4−/− mice, but not in controls. Subcapsular liver vessels (A), subepicardial vessels in the heart (B), enlarged fluid-filled vascular structures in intestinal villi (C) and in bone, adjacent to the growth plate (D and E, black arrows point to examples of enlarged vasculature structures), are shown. A10ΔEC mice also develop large sheets of endothelial cells in the retina after exposure to the oxygen-induced retinopathy model (F, see arrow), whereas wild-type controls usually contain tufts without lumens (G, see arrow). An embryonic day 8.5 embryo lacking VEGF, which induces endothelial Notch signaling, shows an enlarged cardinal vein (H, pointed at by arrows). (A–G are from Alabi et al. (6) with permission from Circ Res, and Glomski et al. (82), with permission from Blood; H is from Carmeliet et al. (31), with permission from Nature.]
2. Portal vessels
Another interesting shared feature of several of the defective vascular beds in A10ΔEC or N1ΔEC/N4−/− mice is their occurrence in portal systems (FIGURE 21A). The best known example of portal vessels is the hepatic system (FIGURE 21B), which forms a conduit for blood returning from the intestinal vasculature through the portal vein to the sinusoidal vessels of the liver. The development of portal vessels requires a departure from the normal process of branching angiogenesis, in which a large trunk, the aorta, branches into ever smaller arterial vessels or branches, which drain into capillary beds, with the returning venous blood flowing from capillaries into ever larger venous branches that merge into the vena cava (FIGURE 21A, top). Portal vessels differ in that the venous blood returning from the first capillary tree collects into a larger vein (portal vessel) that then branches out again into a second capillary network before returning to the main circulation (FIGURE 21A, bottom, see also FIGURE 13). The hypophysial and hypothalamic vasculature represent another portal system, which connects the hypothalamus with the anterior pituitary (FIGURE 21C; these have not yet been examined in A10ΔEC or N1ΔEC/N4−/− mice). Moreover, the renal vascular system can also be considered conceptually similar to a portal system in that it contains two consecutive capillary beds. Blood flowing into the glomeruli is filtered in the glomerular capillary tufts and collects into efferent arterioles that branch out again into a second capillary system surrounding the collecting ducts of the kidney (FIGURE 21D).
FIGURE 21.

Portal vessel systems. A: a typical circulatory system (top) consists of arteries that originate from the aorta and bifurcate into smaller arteriole branches until they reach the capillary bed. From there, blood begins its return to the heart via small venules, which feed into larger veins that ultimately drain into the vena cava and then the heart. In a portal system (bottom), blood from the first arterial system collects into a first capillary bed, which drains into portal vessels (yellow) that branch out again into a second capillary bed, before returning to the main circulatory system through the vena cava. B: the hepatic portal system is the largest portal system in the circulation. Blood flows from the aorta into mesenteric arteries, which feed into individual capillary beds in the stomach and intestine (the “first” capillary bed). Venules and veins that arise from these capillary beds drain into the portal vein, which delivers blood to the liver sinusoids (the “second” capillary bed). These ultimately collect to form the hepatic veins that drain into the vena cava. C: the hypophysial-hypothalamic circulation, which joins vasculature of the hypothalamus with vasculature of the anterior pituitary, represents another example of a portal system. D: the renal vascular system is conceptually similar to a portal system. Blood enters the glomerular tuft (the “first” capillary bed) via the afferent arteriole, and exits through the efferent arteriole, which feeds into the vasa recta (the “second” capillary bed) that supplies other portions of the nephron. [A is from the University of Cumberland Comparative Anatomy website, with permission; B is from Socratic Anatomy and Physiology (covered under creative commons license); C is from Servier Medical Arts, with permission; D is from Aird (4), with permission from Circ Res.]
It is tempting to speculate that portal vessels might arise through a de novo vasculogenic event, although they could plausibly also be formed by remodeling and selective pruning of a developing capillary network. Still, vasculogenesis remains an intriguing possibility. De novo vasculogenesis could produce a vascular plexus that later connects to the existing vasculature to divert the flow of blood through a second capillary bed, resulting in a portal system with two connected and consecutive capillary beds.
3. Specialized endothelium: fenestrated and sinusoidal vascular beds
Capillaries can be grouped into three general categories based on the characteristics of their endothelial lining: continuous, fenestrated, or sinusoidal (FIGURE 22). Fenestrated capillaries are thought to ensure high blood-tissue fluid transport and are found in vessel beds requiring this function, like kidney glomeruli, the gastrointestinal tract, exocrine and endocrine glands, and the choroid plexus (3, 199). Sinusoidal capillaries contain large openings to allow exchange of large molecules and are found in the liver (21), bone marrow, and spleen. Continuous, nonfenestrated capillaries are found in the central nervous system, muscle, skin, lungs, and heart (3). Since several of the vascular beds affected in A10ΔEC or N1ΔEC/N4−/− mice consist of fenestrated or sinusoidal capillaries, this raises questions about whether the ADAM10/Notch pathway has a conserved role in promoting capillary vessel specialization and/or maturation, resulting in vascular openings such as fenestra or sinusoids (112). A closer analysis of glomeruli in A10ΔEC mice revealed persistent fenestral diaphragms (69), which are typically found in immature glomeruli, implicating endothelial ADAM10/Notch signaling in the maturation of glomerular fenestra. In this context, it is noteworthy that VEGF-A, a principal driver of ADAM10/Notch signaling, was first discovered as a vascular permeability factor (123, 216, 217). VEGF can promote formation of fenestrae, which involves actin microfilament disassembly (110), and VEGF expression has been described adjacent to fenestrated vessel beds, such as glomeruli (24). These studies raise the possibility that VEGF could regulate the development or maturation of fenestrated endothelial cells by controlling ADAM10/Notch signaling. Further studies will be necessary to examine how ADAM10/Notch signaling affects the development of fenestrated and sinusoidal endothelia in other tissues in which either type of specialized endothelium can be found.
FIGURE 22.

Endothelial cell types. Endothelial cells can be grouped into three major classes based on whether they assemble into a continuous endothelium (A), or whether they contain small pores to generate a fenestrated endothelium (B), or large irregularly shaped openings to form a sinusoidal endothelium (C). [Images are from https://commons.wikimedia.org/w/index.php?curid=30148246 (covered under creative commons).]
VII. CONCLUSIONS AND PERSPECTIVE
In conclusion, the discovery of a series of vascular defects in A10ΔEC mice had raised several questions about the underlying mechanism(s), some of which have now been addressed. Specifically, it is now clear that these organ-specific vascular defects result from a lack of Notch signaling. This is consistent with a large body of evidence that shows that the principal function of ADAM10 in vivo is as a key upstream regulator of canonical Notch signaling. On the other hand, it is important to emphasize that there is no convincing evidence, to our knowledge, that ADAM17 has any meaningful role in canonical Notch signaling in cell-based assays or in conditional knockout mice in vivo, even though it is frequently mentioned as a Notch processing enzyme.
Having established that endothelial ADAM10/Notch signaling is essential for the development of several very different specialized vascular structures, a number of new questions regarding the underlying mechanism and potential unifying features of the development of the affected organ-specific vessel beds arose, which were the main focus of this review. ADAM10/Notch signaling is used repeatedly during development to govern cell fate decisions. Thus different consequences of inactivating this pathway may depend on the timing and stage of development when ADAM10 or Notch is inactivated. It seems clear that at least two different stages of vascular development require ADAM10/Notch signaling. The first is the artery/vein decision in the yolk sac and embryonic vasculature. This endothelial cell fate decision apparently goes awry if Tie2-Cre Flv is used to inactivate the ADAM10/Notch pathway early in vascular development, resulting in early embryonic lethality. However, for reasons that remain to be established, Tie2-Cre Ywa does not seem to be activated until after this early endothelial cell fate decision has occurred. This allows A10ΔEC or N1ΔEC/N4−/− Tie2-Cre Ywa mice to circumvent the first cell fate decision, only to encounter a block in endothelial cell fate decision(s) at a later stage, at which specialized vascular beds are selectively affected.
Regarding the question of what the plethora of specific vascular structures affected in A10ΔEC or N1ΔEC/N4−/− mice might have in common, the presence of fenestrae or sinusoids, or of portal structures in several vascular beds, strikes us as a particularly compelling area for further research. We also look forward to learning more from a detailed molecular characterization of the vascular structures affected in A10ΔEC or N1ΔEC/N4−/− mice. At the very least, this should help define a default state of endothelial differentiation for each of the affected vascular structures that might usually be transient and only be seen if Notch signaling is blocked. This, in turn, promises to further our understanding of the individual steps of vascular differentiation, in general and in relation to Notch signaling.
From a basic research perspective, which is motivated by curiosity and the goal to better understand principles that may not have any obvious immediate practical applications, we look forward to learning more answers to the questions raised by the vascular phenotypes of A10ΔEC and N1ΔEC/N4−/− mice. How important is vasculogenesis vs. angiogenesis in their development? Do portal beds develop by vasculogenesis? Do angioblasts, which migrate from the yolk sac via the AGM and fetal liver to the bone, reside at the right place and right time to provide the basis for vasculogenic development of specialized vascular beds? Are other specialized vascular beds, such as those in the uterine endometrium, placenta, ovaries, the hypophysial system, pancreatic islets, and in other forms of pathological neovascularization (221), affected? If so, are there shared underlying principles in development of these vascular beds?
In addition to providing answers to these basic science questions, we hope that future studies focused on ADAM10/Notch-dependent development, differentiation, and maturation of specialized vessels will become translationally relevant. From a clinical point of view, we feel that any new insights into the development and differentiation of specialized vascular structures hold the promise of identifying new targets for therapy. Given the global burden of organ-specific vessel disease, including coronary artery disease and kidney glomerular disease, we feel that any new insights into the development and differentiation of specialized, organ-specific vessels hold great promise. While we realize that the development of actual treatments based on these findings might still be far in the future, we are hopeful that, with a better, more nuanced understanding of vascular development, it would be possible to devise novel and improved approaches to the repair and regeneration of damaged specialized vascular beds. We look forward to following further work on the role of ADAM10/Notch signaling in the development of specialized vascular structures and hope that it will ultimately translate into advancements in the care of patients suffering from vascular disease.
GRANTS
R. O. Alabi has received support from the Tri-Institutional MD-PhD program (T32GM007739), the Tri-Institutional Vision Research Training Program (T32EY00713820), and the Weill Cornell Medicine Developmental Biology Program (T32HD606006). G. Farber is currently supported by a predoctoral Fellowship from the American Heart Association and was previously supported by the Molecular and Cellular Biology Training Grant (5T32GM008539). C. P. Blobel is supported, in part, by the National Institute of General Medical Sciences Grant R01 GM-64750.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
ACKNOWLEDGMENTS
Address for reprint requests and other correspondence: C. P. Blobel, Arthritis and Tissue Degeneration Program, HSS Research Institute, Room 702, Hospital for Special Surgery, 535 East 70th St., New York, NY 10021 (e-mail: blobelc@hss.edu).
REFERENCES
- 1.Adams RH, Alitalo K. Molecular regulation of angiogenesis and lymphangiogenesis. Nat Rev Mol Cell Biol 8: 464–478, 2007. doi: 10.1038/nrm2183. [DOI] [PubMed] [Google Scholar]
- 2.Adams RH, Wilkinson GA, Weiss C, Diella F, Gale NW, Deutsch U, Risau W, Klein R. Roles of ephrinB ligands and EphB receptors in cardiovascular development: demarcation of arterial/venous domains, vascular morphogenesis, and sprouting angiogenesis. Genes Dev 13: 295–306, 1999. doi: 10.1101/gad.13.3.295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Aird WC. Phenotypic heterogeneity of the endothelium. I. Structure, function, and mechanisms. Circ Res 100: 158–173, 2007. doi: 10.1161/01.RES.0000255691.76142.4a. [DOI] [PubMed] [Google Scholar]
- 4.Aird WC. Phenotypic heterogeneity of the endothelium. II. Representative vascular beds. Circ Res 100: 174–190, 2007. doi: 10.1161/01.RES.0000255690.03436.ae. [DOI] [PubMed] [Google Scholar]
- 5.Aitsebaomo J, Portbury AL, Schisler JC, Patterson C. Brothers and sisters: molecular insights into arterial-venous heterogeneity. Circ Res 103: 929–939, 2008. doi: 10.1161/CIRCRESAHA.108.184937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Alabi RO, Glomski K, Haxaire C, Weskamp G, Monette S, Blobel CP. ADAM10-dependent signaling through Notch1 and Notch4 controls development of organ-specific vascular beds. Circ Res 119: 519–531, 2016. doi: 10.1161/CIRCRESAHA.115.307738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alon T, Hemo I, Itin A, Pe’er J, Stone J, Keshet E. Vascular endothelial growth factor acts as a survival factor for newly formed retinal vessels and has implications for retinopathy of prematurity. Nat Med 1: 1024–1028, 1995. doi: 10.1038/nm1095-1024. [DOI] [PubMed] [Google Scholar]
- 8.Arnold TD, Niaudet C, Pang MF, Siegenthaler J, Gaengel K, Jung B, Ferrero GM, Mukouyama YS, Fuxe J, Akhurst R, Betsholtz C, Sheppard D, Reichardt LF. Excessive vascular sprouting underlies cerebral hemorrhage in mice lacking αVβ8-TGFβ signaling in the brain. Development 141: 4489–4499, 2014. doi: 10.1242/dev.107193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Asahara T, Masuda H, Takahashi T, Kalka C, Pastore C, Silver M, Kearne M, Magner M, Isner JM. Bone marrow origin of endothelial progenitor cells responsible for postnatal vasculogenesis in physiological and pathological neovascularization. Circ Res 85: 221–228, 1999. doi: 10.1161/01.RES.85.3.221. [DOI] [PubMed] [Google Scholar]
- 10.Asahara T, Murohara T, Sullivan A, Silver M, van der Zee R, Li T, Witzenbichler B, Schatteman G, Isner JM. Isolation of putative progenitor endothelial cells for angiogenesis. Science 275: 964–966, 1997. doi: 10.1126/science.275.5302.964. [DOI] [PubMed] [Google Scholar]
- 11.Asahara T, Takahashi T, Masuda H, Kalka C, Chen D, Iwaguro H, Inai Y, Silver M, Isner JM. VEGF contributes to postnatal neovascularization by mobilizing bone marrow-derived endothelial progenitor cells. EMBO J 18: 3964–3972, 1999. doi: 10.1093/emboj/18.14.3964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Aste-Amézaga M, Zhang N, Lineberger JE, Arnold BA, Toner TJ, Gu M, Huang L, Vitelli S, Vo KT, Haytko P, Zhao JZ, Baleydier F, L’Heureux S, Wang H, Gordon WR, Thoryk E, Andrawes MB, Tiyanont K, Stegmaier K, Roti G, Ross KN, Franlin LL, Wang H, Wang F, Chastain M, Bett AJ, Audoly LP, Aster JC, Blacklow SC, Huber HE. Characterization of Notch1 antibodies that inhibit signaling of both normal and mutated Notch1 receptors. PLoS One 5: e9094, 2010. doi: 10.1371/journal.pone.0009094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Augustin HG, Koh GY. Organotypic vasculature: from descriptive heterogeneity to functional pathophysiology. Science 357: eaal2379, 2017. doi: 10.1126/science.aal2379. [DOI] [PubMed] [Google Scholar]
- 14.Augustin HG, Koh GY, Thurston G, Alitalo K. Control of vascular morphogenesis and homeostasis through the angiopoietin-Tie system. Nat Rev Mol Cell Biol 10: 165–177, 2009. doi: 10.1038/nrm2639. [DOI] [PubMed] [Google Scholar]
- 15.Benedito R, Roca C, Sörensen I, Adams S, Gossler A, Fruttiger M, Adams RH. The notch ligands Dll4 and Jagged1 have opposing effects on angiogenesis. Cell 137: 1124–1135, 2009. doi: 10.1016/j.cell.2009.03.025. [DOI] [PubMed] [Google Scholar]
- 16.Benedito R, Rocha SF, Woeste M, Zamykal M, Radtke F, Casanovas O, Duarte A, Pytowski B, Adams RH. Notch-dependent VEGFR3 upregulation allows angiogenesis without VEGF-VEGFR2 signalling. Nature 484: 110–114, 2012. doi: 10.1038/nature10908. [DOI] [PubMed] [Google Scholar]
- 17.Bernier-Latmani J, Cisarovsky C, Demir CS, Bruand M, Jaquet M, Davanture S, Ragusa S, Siegert S, Dormond O, Benedito R, Radtke F, Luther SA, Petrova TV. DLL4 promotes continuous adult intestinal lacteal regeneration and dietary fat transport. J Clin Invest 125: 4572–4586, 2015. doi: 10.1172/JCI82045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Boareto M, Jolly MK, Ben-Jacob E, Onuchic JN. Jagged mediates differences in normal and tumor angiogenesis by affecting tip-stalk fate decision. Proc Natl Acad Sci USA 112: E3836–E3844, 2015. doi: 10.1073/pnas.1511814112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Boyle SC, Liu Z, Kopan R. Notch signaling is required for the formation of mesangial cells from a stromal mesenchyme precursor during kidney development. Development 141: 346–354, 2014. doi: 10.1242/dev.100271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Bozkulak EC, Weinmaster G. Selective use of ADAM10 and ADAM17 in activation of Notch1 signaling. Mol Cell Biol 29: 5679–5695, 2009. doi: 10.1128/MCB.00406-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Braet F, Wisse E. Structural and functional aspects of liver sinusoidal endothelial cell fenestrae: a review. Comp Hepatol 1: 1, 2002. doi: 10.1186/1476-5926-1-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bray SJ. Notch signalling in context. Nat Rev Mol Cell Biol 17: 722–735, 2016. doi: 10.1038/nrm.2016.94. [DOI] [PubMed] [Google Scholar]
- 23.Bray SJ. Notch signalling: a simple pathway becomes complex. Nat Rev Mol Cell Biol 7: 678–689, 2006. doi: 10.1038/nrm2009. [DOI] [PubMed] [Google Scholar]
- 24.Breier G, Albrecht U, Sterrer S, Risau W. Expression of vascular endothelial growth factor during embryonic angiogenesis and endothelial cell differentiation. Development 114: 521–532, 1992. [DOI] [PubMed] [Google Scholar]
- 25.Bridges E, Oon CE, Harris A. Notch regulation of tumor angiogenesis. Future Oncol 7: 569–588, 2011. doi: 10.2217/fon.11.20. [DOI] [PubMed] [Google Scholar]
- 26.Briot A, Iruela-Arispe ML. Blockade of specific NOTCH ligands: a new promising approach in cancer therapy. Cancer Discov 5: 112–114, 2015. doi: 10.1158/2159-8290.CD-14-1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Brou C, Logeat F, Gupta N, Bessia C, LeBail O, Doedens JR, Cumano A, Roux P, Black RA, Israël A. A novel proteolytic cleavage involved in Notch signaling: the role of the disintegrin-metalloprotease TACE. Mol Cell 5: 207–216, 2000. doi: 10.1016/S1097-2765(00)80417-7. [DOI] [PubMed] [Google Scholar]
- 28.Brückner K, Perez L, Clausen H, Cohen S. Glycosyltransferase activity of Fringe modulates Notch-Delta interactions. Nature 406: 411–415, 2000. doi: 10.1038/35019075. [DOI] [PubMed] [Google Scholar]
- 29.Cantelmo AR, Brajic A, Carmeliet P. Endothelial metabolism driving angiogenesis: emerging concepts and principles. Cancer J 21: 244–249, 2015. doi: 10.1097/PPO.0000000000000133. [DOI] [PubMed] [Google Scholar]
- 30.Carmeliet P. Angiogenesis in life, disease and medicine. Nature 438: 932–936, 2005. doi: 10.1038/nature04478. [DOI] [PubMed] [Google Scholar]
- 31.Carmeliet P, Ferreira V, Breier G, Pollefeyt S, Kieckens L, Gertsenstein M, Fahrig M, Vandenhoeck A, Harpal K, Eberhardt C, Declercq C, Pawling J, Moons L, Collen D, Risau W, Nagy A. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature 380: 435–439, 1996. doi: 10.1038/380435a0. [DOI] [PubMed] [Google Scholar]
- 32.Chalaris A, Adam N, Sina C, Rosenstiel P, Lehmann-Koch J, Schirmacher P, Hartmann D, Cichy J, Gavrilova O, Schreiber S, Jostock T, Matthews V, Häsler R, Becker C, Neurath MF, Reiss K, Saftig P, Scheller J, Rose-John S. Critical role of the disintegrin metalloprotease ADAM17 for intestinal inflammation and regeneration in mice. J Exp Med 207: 1617–1624, 2010. doi: 10.1084/jem.20092366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen HI, Sharma B, Akerberg BN, Numi HJ, Kivelä R, Saharinen P, Aghajanian H, McKay AS, Bogard PE, Chang AH, Jacobs AH, Epstein JA, Stankunas K, Alitalo K, Red-Horse K. The sinus venosus contributes to coronary vasculature through VEGFC-stimulated angiogenesis. Development 141: 4500–4512, 2014. doi: 10.1242/dev.113639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen J, Smith LE. Retinopathy of prematurity. Angiogenesis 10: 133–140, 2007. doi: 10.1007/s10456-007-9066-0. [DOI] [PubMed] [Google Scholar]
- 35.Cheng HT, Kim M, Valerius MT, Surendran K, Schuster-Gossler K, Gossler A, McMahon AP, Kopan R. Notch2, but not Notch1, is required for proximal fate acquisition in the mammalian nephron. Development 134: 801–811, 2007. doi: 10.1242/dev.02773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cheng HT, Kopan R. The role of Notch signaling in specification of podocyte and proximal tubules within the developing mouse kidney. Kidney Int 68: 1951–1952, 2005. doi: 10.1111/j.1523-1755.2005.00627.x. [DOI] [PubMed] [Google Scholar]
- 37.Chin AM, Hill DR, Aurora M, Spence JR. Morphogenesis and maturation of the embryonic and postnatal intestine. Semin Cell Dev Biol 66: 81–93, 2017. doi: 10.1016/j.semcdb.2017.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Chong DC, Koo Y, Xu K, Fu S, Cleaver O. Stepwise arteriovenous fate acquisition during mammalian vasculogenesis. Dev Dyn 240: 2153–2165, 2011. doi: 10.1002/dvdy.22706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Conlon RA, Reaume AG, Rossant J. Notch1 is required for the coordinate segmentation of somites. Development 121: 1533–1545, 1995. [DOI] [PubMed] [Google Scholar]
- 40.Connor KM, Krah NM, Dennison RJ, Aderman CM, Chen J, Guerin KI, Sapieha P, Stahl A, Willett KL, Smith LE. Quantification of oxygen-induced retinopathy in the mouse: a model of vessel loss, vessel regrowth and pathological angiogenesis. Nat Protoc 4: 1565–1573, 2009. doi: 10.1038/nprot.2009.187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Copeland JN, Feng Y, Neradugomma NK, Fields PE, Vivian JL. Notch signaling regulates remodeling and vessel diameter in the extraembryonic yolk sac. BMC Dev Biol 11: 12, 2011. doi: 10.1186/1471-213X-11-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Corada M, Morini MF, Dejana E. Signaling pathways in the specification of arteries and veins. Arterioscler Thromb Vasc Biol 34: 2372–2377, 2014. doi: 10.1161/ATVBAHA.114.303218. [DOI] [PubMed] [Google Scholar]
- 43.Corada M, Orsenigo F, Morini MF, Pitulescu ME, Bhat G, Nyqvist D, Breviario F, Conti V, Briot A, Iruela-Arispe ML, Adams RH, Dejana E. Sox17 is indispensable for acquisition and maintenance of arterial identity. Nat Commun 4: 2609, 2013. doi: 10.1038/ncomms3609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Coultas L, Chawengsaksophak K, Rossant J. Endothelial cells and VEGF in vascular development. Nature 438: 937–945, 2005. doi: 10.1038/nature04479. [DOI] [PubMed] [Google Scholar]
- 45.Cuervo H, Nielsen CM, Simonetto DA, Ferrell L, Shah VH, Wang RA. Endothelial notch signaling is essential to prevent hepatic vascular malformations in mice. Hepatology 64: 1302–1316, 2016. doi: 10.1002/hep.28713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.D’Amato G, Luxán G, de la Pompa JL. Notch signalling in ventricular chamber development and cardiomyopathy. FEBS J 283: 4223–4237, 2016. doi: 10.1111/febs.13773. [DOI] [PubMed] [Google Scholar]
- 47.Davis RB, Curtis CD, Griffin CT. BRG1 promotes COUP-TFII expression and venous specification during embryonic vascular development. Development 140: 1272–1281, 2013. doi: 10.1242/dev.087379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.De Bock K, Georgiadou M, Carmeliet P. Role of endothelial cell metabolism in vessel sprouting. Cell Metab 18: 634–647, 2013. doi: 10.1016/j.cmet.2013.08.001. [DOI] [PubMed] [Google Scholar]
- 49.de Celis JF, Garcia-Bellido A, Bray SJ. Activation and function of Notch at the dorsal-ventral boundary of the wing imaginal disc. Development 122: 359–369, 1996. [DOI] [PubMed] [Google Scholar]
- 50.de la Pompa JL, Epstein JA. Coordinating tissue interactions: Notch signaling in cardiac development and disease. Dev Cell 22: 244–254, 2012. doi: 10.1016/j.devcel.2012.01.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.de Lange WJ, Halabi CM, Beyer AM, Sigmund CD. Germ line activation of the Tie2 and SMMHC promoters causes noncell-specific deletion of floxed alleles. Physiol Genomics 35: 1–4, 2008. doi: 10.1152/physiolgenomics.90284.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.De Strooper B, Annaert W, Cupers P, Saftig P, Craessaerts K, Mumm JS, Schroeter EH, Schrijvers V, Wolfe MS, Ray WJ, Goate A, Kopan R. A presenilin-1-dependent gamma-secretase-like protease mediates release of Notch intracellular domain Nature 398: 518–522, 1999. doi: 10.1038/19083. [DOI] [PubMed] [Google Scholar]
- 53.del Monte G, Casanova JC, Guadix JA, MacGrogan D, Burch JB, Pérez-Pomares JM, de la Pompa JL. Differential Notch signaling in the epicardium is required for cardiac inflow development and coronary vessel morphogenesis. Circ Res 108: 824–836, 2011. doi: 10.1161/CIRCRESAHA.110.229062. [DOI] [PubMed] [Google Scholar]
- 54.Dexter JS. The analysis of a case of continuous variation in Drosophila by a study of its linkage relationships. Am Nat 48: 712–758, 1914. doi: 10.1086/279446. [DOI] [Google Scholar]
- 55.Djokovic D, Trindade A, Gigante J, Badenes M, Silva L, Liu R, Li X, Gong M, Krasnoperov V, Gill PS, Duarte A. Combination of Dll4/Notch and Ephrin-B2/EphB4 targeted therapy is highly effective in disrupting tumor angiogenesis. BMC Cancer 10: 641, 2010. doi: 10.1186/1471-2407-10-641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Djonov V, Schmid M, Tschanz SA, Burri PH. Intussusceptive angiogenesis: its role in embryonic vascular network formation. Circ Res 86: 286–292, 2000. doi: 10.1161/01.RES.86.3.286. [DOI] [PubMed] [Google Scholar]
- 57.Djonov VG, Kurz H, Burri PH. Optimality in the developing vascular system: branching remodeling by means of intussusception as an efficient adaptation mechanism. Dev Dyn 224: 391–402, 2002. doi: 10.1002/dvdy.10119. [DOI] [PubMed] [Google Scholar]
- 58.Doherty D, Feger G, Younger-Shepherd S, Jan LY, Jan YN. Delta is a ventral to dorsal signal complementary to Serrate, another Notch ligand, in Drosophila wing formation. Genes Dev 10: 421–434, 1996. doi: 10.1101/gad.10.4.421. [DOI] [PubMed] [Google Scholar]
- 59.Donoviel DB, Hadjantonakis AK, Ikeda M, Zheng H, Hyslop PS, Bernstein A. Mice lacking both presenilin genes exhibit early embryonic patterning defects. Genes Dev 13: 2801–2810, 1999. doi: 10.1101/gad.13.21.2801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Dorrell MI, Otani A, Aguilar E, Moreno SK, Friedlander M. Adult bone marrow-derived stem cells use R-cadherin to target sites of neovascularization in the developing retina. Blood 103: 3420–3427, 2004. doi: 10.1182/blood-2003-09-3012. [DOI] [PubMed] [Google Scholar]
- 61.Drake CJ, Fleming PA. Vasculogenesis in the day 6.5 to 9.5 mouse embryo. Blood 95: 1671–1679, 2000. [PubMed] [Google Scholar]
- 62.Duarte A, Hirashima M, Benedito R, Trindade A, Diniz P, Bekman E, Costa L, Henrique D, Rossant J. Dosage-sensitive requirement for mouse Dll4 in artery development. Genes Dev 18: 2474–2478, 2004. doi: 10.1101/gad.1239004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Dyczynska E, Sun D, Yi H, Sehara-Fujisawa A, Blobel CP, Zolkiewska A. Proteolytic processing of delta-like 1 by ADAM proteases. J Biol Chem 282: 436–444, 2007. doi: 10.1074/jbc.M605451200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Eelen G, Cruys B, Welti J, De Bock K, Carmeliet P. Control of vessel sprouting by genetic and metabolic determinants. Trends Endocrinol Metab 24: 589–596, 2013. doi: 10.1016/j.tem.2013.08.006. [DOI] [PubMed] [Google Scholar]
- 65.Ehling M, Adams S, Benedito R, Adams RH. Notch controls retinal blood vessel maturation and quiescence. Development 140: 3051–3061, 2013. doi: 10.1242/dev.093351. [DOI] [PubMed] [Google Scholar]
- 66.Endo K, Karim MR, Taniguchi H, Krejci A, Kinameri E, Siebert M, Ito K, Bray SJ, Moore AW. Chromatin modification of Notch targets in olfactory receptor neuron diversification. Nat Neurosci 15: 224–233, 2012. doi: 10.1038/nn.2998. [DOI] [PubMed] [Google Scholar]
- 67.Erber R, Eichelsbacher U, Powajbo V, Korn T, Djonov V, Lin J, Hammes HP, Grobholz R, Ullrich A, Vajkoczy P. EphB4 controls blood vascular morphogenesis during postnatal angiogenesis. EMBO J 25: 628–641, 2006. doi: 10.1038/sj.emboj.7600949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Eremina V, Baelde HJ, Quaggin SE. Role of the VEGF–a signaling pathway in the glomerulus: evidence for crosstalk between components of the glomerular filtration barrier. Nephron Physiol 106: p32–p37, 2007. doi: 10.1159/000101798. [DOI] [PubMed] [Google Scholar]
- 69.Farber G, Hurtado R, Loh S, Monette S, Mtui J, Kopan R, Quaggin S, Meyer-Schwesinger C, Herzlinger D, Scott RP, Blobel CP. Glomerular endothelial cell maturation depends on ADAM10, a key regulator of Notch signaling. Angiogenesis 21: 335–347, 2018. doi: 10.1007/s10456-018-9599-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ferrara N, Carver-Moore K, Chen H, Dowd M, Lu L, O’Shea KS, Powell-Braxton L, Hillan KJ, Moore MW. Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature 380: 439–442, 1996. doi: 10.1038/380439a0. [DOI] [PubMed] [Google Scholar]
- 71.Ferrarotto R, Mitani Y, Diao L, Guijarro I, Wang J, Zweidler-McKay P, Bell D, William WN Jr, Glisson BS, Wick MJ, Kapoun AM, Patnaik A, Eckhardt G, Munster P, Faoro L, Dupont J, Lee JJ, Futreal A, El-Naggar AK, Heymach JV. Activating NOTCH1 mutations define a distinct subgroup of patients with adenoid cystic carcinoma who have poor prognosis, propensity to bone and liver metastasis, and potential responsiveness to Notch1 inhibitors. J Clin Oncol 35: 352–360, 2017. doi: 10.1200/JCO.2016.67.5264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fish JE, Wythe JD. The molecular regulation of arteriovenous specification and maintenance. Dev Dyn 244: 391–409, 2015. doi: 10.1002/dvdy.24252. [DOI] [PubMed] [Google Scholar]
- 73.Fleming RJ, Scottgale TN, Diederich RJ, Artavanis-Tsakonas S. The gene Serrate encodes a putative EGF-like transmembrane protein essential for proper ectodermal development in Drosophila melanogaster. Genes Dev 4, 12A: 2188–2201, 1990. doi: 10.1101/gad.4.12a.2188. [DOI] [PubMed] [Google Scholar]
- 74.Franzke CW, Cobzaru C, Triantafyllopoulou A, Löffek S, Horiuchi K, Threadgill DW, Kurz T, van Rooijen N, Bruckner-Tuderman L, Blobel CP. Epidermal ADAM17 maintains the skin barrier by regulating EGFR ligand-dependent terminal keratinocyte differentiation. J Exp Med 209: 1105–1119, 2012. [Erratum in J Exp Med 209: 2517, 2012.] doi: 10.1084/jem.20112258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fre S, Huyghe M, Mourikis P, Robine S, Louvard D, Artavanis-Tsakonas S. Notch signals control the fate of immature progenitor cells in the intestine. Nature 435: 964–968, 2005. doi: 10.1038/nature03589. [DOI] [PubMed] [Google Scholar]
- 76.Friedlander M, Dorrell MI, Ritter MR, Marchetti V, Moreno SK, El-Kalay M, Bird AC, Banin E, Aguilar E. Progenitor cells and retinal angiogenesis. Angiogenesis 10: 89–101, 2007. doi: 10.1007/s10456-007-9070-4. [DOI] [PubMed] [Google Scholar]
- 77.Fruttiger M. Development of the retinal vasculature. Angiogenesis 10: 77–88, 2007. doi: 10.1007/s10456-007-9065-1. [DOI] [PubMed] [Google Scholar]
- 78.Gale NW, Dominguez MG, Noguera I, Pan L, Hughes V, Valenzuela DM, Murphy AJ, Adams NC, Lin HC, Holash J, Thurston G, Yancopoulos GD. Haploinsufficiency of delta-like 4 ligand results in embryonic lethality due to major defects in arterial and vascular development. Proc Natl Acad Sci USA 101: 15949–15954, 2004. doi: 10.1073/pnas.0407290101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Gao F, Hou H, Liang H, Weinreb RN, Wang H, Wang Y. Bone marrow-derived cells in ocular neovascularization: contribution and mechanisms. Angiogenesis 19: 107–118, 2016. doi: 10.1007/s10456-016-9497-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Geffers I, Serth K, Chapman G, Jaekel R, Schuster-Gossler K, Cordes R, Sparrow DB, Kremmer E, Dunwoodie SL, Klein T, Gossler A. Divergent functions and distinct localization of the Notch ligands DLL1 and DLL3 in vivo. J Cell Biol 178: 465–476, 2007. doi: 10.1083/jcb.200702009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gibb DR, El Shikh M, Kang DJ, Rowe WJ, El Sayed R, Cichy J, Yagita H, Tew JG, Dempsey PJ, Crawford HC, Conrad DH. ADAM10 is essential for Notch2-dependent marginal zone B cell development and CD23 cleavage in vivo. J Exp Med 207: 623–635, 2010. doi: 10.1084/jem.20091990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Glomski K, Monette S, Manova K, De Strooper B, Saftig P, Blobel CP. Deletion of Adam10 in endothelial cells leads to defects in organ-specific vascular structures. Blood 118: 1163–1174, 2011. doi: 10.1182/blood-2011-04-348557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gordon WR, Arnett KL, Blacklow SC. The molecular logic of Notch signaling—a structural and biochemical perspective. J Cell Sci 121: 3109–3119, 2008. doi: 10.1242/jcs.035683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gordon WR, Roy M, Vardar-Ulu D, Garfinkel M, Mansour MR, Aster JC, Blacklow SC. Structure of the Notch1-negative regulatory region: implications for normal activation and pathogenic signaling in T-ALL. Blood 113: 4381–4390, 2009. doi: 10.1182/blood-2008-08-174748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gordon WR, Zimmerman B, He L, Miles LJ, Huang J, Tiyanont K, McArthur DG, Aster JC, Perrimon N, Loparo JJ, Blacklow SC. Mechanical allostery: evidence for a force requirement in the proteolytic activation of notch. Dev Cell 33: 729–736, 2015. doi: 10.1016/j.devcel.2015.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gridley T. Notch signaling during vascular development. Proc Natl Acad Sci USA 98: 5377–5378, 2001. doi: 10.1073/pnas.101138098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Gridley T. Notch signaling in the vasculature. Curr Top Dev Biol 92: 277–309, 2010. doi: 10.1016/S0070-2153(10)92009-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Gridley T. Notch signaling in vascular development and physiology. Development 134: 2709–2718, 2007. doi: 10.1242/dev.004184. [DOI] [PubMed] [Google Scholar]
- 89. Groot AJ, Cobzaru C, Weber S, Saftig P, Blobel CP, Kopan R, Vooijs M, Franzke CW. Epidermal ADAM17 is dispensable for notch activation. J Invest Dermatol 133: 2286–2288, 2013. doi: 10.1038/jid.2013.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Groot AJ, Habets R, Yahyanejad S, Hodin CM, Reiss K, Saftig P, Theys J, Vooijs M. Regulated proteolysis of NOTCH2 and NOTCH3 receptors by ADAM10 and presenilins. Mol Cell Biol 34: 2822–2832, 2014. doi: 10.1128/MCB.00206-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Gurney A, Hoey T. Anti-DLL4, a cancer therapeutic with multiple mechanisms of action. Vasc Cell 3: 18, 2011. doi: 10.1186/2045-824X-3-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Guseh JS, Bores SA, Stanger BZ, Zhou Q, Anderson WJ, Melton DA, Rajagopal J. Notch signaling promotes airway mucous metaplasia and inhibits alveolar development. Development 136: 1751–1759, 2009. doi: 10.1242/dev.029249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hall KC, Hill D, Otero M, Plumb DA, Froemel D, Dragomir CL, Maretzky T, Boskey A, Crawford HC, Selleri L, Goldring MB, Blobel CP. ADAM17 controls endochondral ossification by regulating terminal differentiation of chondrocytes. Mol Cell Biol 33: 3077–3090, 2013. doi: 10.1128/MCB.00291-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Hartmann D, de Strooper B, Serneels L, Craessaerts K, Herreman A, Annaert W, Umans L, Lübke T, Lena Illert A, von Figura K, Saftig P. The disintegrin/metalloprotease ADAM 10 is essential for Notch signalling but not for alpha-secretase activity in fibroblasts. Hum Mol Genet 11: 2615–2624, 2002. doi: 10.1093/hmg/11.21.2615. [DOI] [PubMed] [Google Scholar]
- 96.Hashimoto H, Ishikawa H, Kusakabe M. Development of vascular networks during the morphogenesis of intestinal villi in the fetal mouse. Kaibogaku Zasshi 74: 567–576, 1999. [PubMed] [Google Scholar]
- 97.Hashimoto T, Izumi Y, Oota M, Kawada K, Miura A, Kato T, Momma K [Superficial small cell carcinoma of esophagus treated by endoscopic mucosal resection combined with chemoradiotherapy—a case report]. Gan To Kagaku Ryoho 34: 81–84, 2007. [PubMed] [Google Scholar]
- 98.Hatch J, Mukouyama YS. Spatiotemporal mapping of vascularization and innervation in the fetal murine intestine. Dev Dyn 244: 56–68, 2015. doi: 10.1002/dvdy.24178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hellström M, Phng LK, Hofmann JJ, Wallgard E, Coultas L, Lindblom P, Alva J, Nilsson AK, Karlsson L, Gaiano N, Yoon K, Rossant J, Iruela-Arispe ML, Kalén M, Gerhardt H, Betsholtz C. Dll4 signalling through Notch1 regulates formation of tip cells during angiogenesis. Nature 445: 776–780, 2007. doi: 10.1038/nature05571. [DOI] [PubMed] [Google Scholar]
- 100.Hen G, Nicenboim J, Mayseless O, Asaf L, Shin M, Busolin G, Hofi R, Almog G, Tiso N, Lawson ND, Yaniv K. Venous-derived angioblasts generate organ-specific vessels during zebrafish embryonic development. Development 142: 4266–4278, 2015. doi: 10.1242/dev.129247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Herbert SP, Stainier DY. Molecular control of endothelial cell behaviour during blood vessel morphogenesis. Nat Rev Mol Cell Biol 12: 551–564, 2011. doi: 10.1038/nrm3176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Herreman A, Hartmann D, Annaert W, Saftig P, Craessaerts K, Serneels L, Umans L, Schrijvers V, Checler F, Vanderstichele H, Baekelandt V, Dressel R, Cupers P, Huylebroeck D, Zwijsen A, Van Leuven F, De Strooper B. Presenilin 2 deficiency causes a mild pulmonary phenotype and no changes in amyloid precursor protein processing but enhances the embryonic lethal phenotype of presenilin 1 deficiency. Proc Natl Acad Sci USA 96: 11872–11877, 1999. doi: 10.1073/pnas.96.21.11872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Herzlinger D, Hurtado R. Patterning the renal vascular bed. Semin Cell Dev Biol 36: 50–56, 2014. doi: 10.1016/j.semcdb.2014.08.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hofmann JJ, Briot A, Enciso J, Zovein AC, Ren S, Zhang ZW, Radtke F, Simons M, Wang Y, Iruela-Arispe ML. Endothelial deletion of murine Jag1 leads to valve calcification and congenital heart defects associated with Alagille syndrome. Development 139: 4449–4460, 2012. doi: 10.1242/dev.084871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hofmann JJ, Iruela-Arispe ML. Notch signaling in blood vessels: who is talking to whom about what? Circ Res 100: 1556–1568, 2007. doi: 10.1161/01.RES.0000266408.42939.e4. [DOI] [PubMed] [Google Scholar]
- 106.Holm A, Heumann T, Augustin HG. Microvascular mural cell organotypic heterogeneity and functional plasticity. Trends Cell Biol 28: 302–316, 2018. doi: 10.1016/j.tcb.2017.12.002. [DOI] [PubMed] [Google Scholar]
- 107.Horiuchi K, Kimura T, Miyamoto T, Takaishi H, Okada Y, Toyama Y, Blobel CP. Cutting edge: TNF-alpha-converting enzyme (TACE/ADAM17) inactivation in mouse myeloid cells prevents lethality from endotoxin shock. J Immunol 179: 2686–2689, 2007. doi: 10.4049/jimmunol.179.5.2686. [DOI] [PubMed] [Google Scholar]
- 108.Huppert SS, Le A, Schroeter EH, Mumm JS, Saxena MT, Milner LA, Kopan R. Embryonic lethality in mice homozygous for a processing-deficient allele of Notch1. Nature 405: 966–970, 2000. [Erratum in Nature 408: 616, 2000.] doi: 10.1038/35016111. [DOI] [PubMed] [Google Scholar]
- 109.Ichimura K, Stan RV, Kurihara H, Sakai T. Glomerular endothelial cells form diaphragms during development and pathologic conditions. J Am Soc Nephrol 19: 1463–1471, 2008. doi: 10.1681/ASN.2007101138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Ioannidou S, Deinhardt K, Miotla J, Bradley J, Cheung E, Samuelsson S, Ng YS, Shima DT. An in vitro assay reveals a role for the diaphragm protein PV-1 in endothelial fenestra morphogenesis. Proc Natl Acad Sci USA 103: 16770–16775, 2006. doi: 10.1073/pnas.0603501103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ishida S, Yamashiro K, Usui T, Kaji Y, Ogura Y, Hida T, Honda Y, Oguchi Y, Adamis AP. Leukocytes mediate retinal vascular remodeling during development and vaso-obliteration in disease. Nat Med 9: 781–788, 2003. doi: 10.1038/nm877. [DOI] [PubMed] [Google Scholar]
- 112.Jain RK. Molecular regulation of vessel maturation. Nat Med 9: 685–693, 2003. doi: 10.1038/nm0603-685. [DOI] [PubMed] [Google Scholar]
- 113.Jakobsson L, Bentley K, Gerhardt H. VEGFRs and Notch: a dynamic collaboration in vascular patterning. Biochem Soc Trans 37: 1233–1236, 2009. doi: 10.1042/BST0371233. [DOI] [PubMed] [Google Scholar]
- 114.Jakobsson L, Franco CA, Bentley K, Collins RT, Ponsioen B, Aspalter IM, Rosewell I, Busse M, Thurston G, Medvinsky A, Schulte-Merker S, Gerhardt H. Endothelial cells dynamically compete for the tip cell position during angiogenic sprouting. Nat Cell Biol 12: 943–953, 2010. doi: 10.1038/ncb2103. [DOI] [PubMed] [Google Scholar]
- 115.James AC, Szot JO, Iyer K, Major JA, Pursglove SE, Chapman G, Dunwoodie SL. Notch4 reveals a novel mechanism regulating Notch signal transduction. Biochim Biophys Acta 1843: 1272–1284, 2014. doi: 10.1016/j.bbamcr.2014.03.015. [DOI] [PubMed] [Google Scholar]
- 116.Jones CA, Li DY. Common cues regulate neural and vascular patterning. Curr Opin Genet Dev 17: 332–336, 2007. doi: 10.1016/j.gde.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Jorissen E, Prox J, Bernreuther C, Weber S, Schwanbeck R, Serneels L, Snellinx A, Craessaerts K, Thathiah A, Tesseur I, Bartsch U, Weskamp G, Blobel CP, Glatzel M, De Strooper B, Saftig P. The disintegrin/metalloproteinase ADAM10 is essential for the establishment of the brain cortex. J Neurosci 30: 4833–4844, 2010. doi: 10.1523/JNEUROSCI.5221-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Kalka C, Masuda H, Takahashi T, Kalka-Moll WM, Silver M, Kearney M, Li T, Isner JM, Asahara T. Transplantation of ex vivo expanded endothelial progenitor cells for therapeutic neovascularization. Proc Natl Acad Sci USA 97: 3422–3427, 2000. doi: 10.1073/pnas.97.7.3422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Kangsamaksin T, Murtomaki A, Kofler NM, Cuervo H, Chaudhri RA, Tattersall IW, Rosenstiel PE, Shawber CJ, Kitajewski J. NOTCH decoys that selectively block DLL/NOTCH or JAG/NOTCH disrupt angiogenesis by unique mechanisms to inhibit tumor growth. Cancer Discov 5: 182–197, 2015. doi: 10.1158/2159-8290.CD-14-0650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Kangsamaksin T, Tattersall IW, Kitajewski J. Notch functions in developmental and tumour angiogenesis by diverse mechanisms. Biochem Soc Trans 42: 1563–1568, 2014. doi: 10.1042/BST20140233. [DOI] [PubMed] [Google Scholar]
- 121.Kankel MW, Hurlbut GD, Upadhyay G, Yajnik V, Yedvobnick B, Artavanis-Tsakonas S. Investigating the genetic circuitry of mastermind in Drosophila, a notch signal effector. Genetics 177: 2493–2505, 2007. doi: 10.1534/genetics.107.080994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kato H, Taniguchi Y, Kurooka H, Minoguchi S, Sakai T, Nomura-Okazaki S, Tamura K, Honjo T. Involvement of RBP-J in biological functions of mouse Notch1 and its derivatives. Development 124: 4133–4141, 1997. [DOI] [PubMed] [Google Scholar]
- 123.Keck PJ, Hauser SD, Krivi G, Sanzo K, Warren T, Feder J, Connolly DT. Vascular permeability factor, an endothelial cell mitogen related to PDGF. Science 246: 1309–1312, 1989. doi: 10.1126/science.2479987. [DOI] [PubMed] [Google Scholar]
- 124.Kerr BA, West XZ, Kim YW, Zhao Y, Tischenko M, Cull RM, Phares TW, Peng XD, Bernier-Latmani J, Petrova TV, Adams RH, Hay N, Naga Prasad SV, Byzova TV. Stability and function of adult vasculature is sustained by Akt/Jagged1 signalling axis in endothelium. Nat Commun 7: 10960, 2016. doi: 10.1038/ncomms10960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kidoya H, Takakura N. Biology of the apelin-APJ axis in vascular formation. J Biochem 152: 125–131, 2012. doi: 10.1093/jb/mvs071. [DOI] [PubMed] [Google Scholar]
- 126.Kidoya H, Ueno M, Yamada Y, Mochizuki N, Nakata M, Yano T, Fujii R, Takakura N. Spatial and temporal role of the apelin/APJ system in the caliber size regulation of blood vessels during angiogenesis. EMBO J 27: 522–534, 2008. doi: 10.1038/sj.emboj.7601982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kim I, Kim HG, Moon SO, Chae SW, So JN, Koh KN, Ahn BC, Koh GY. Angiopoietin-1 induces endothelial cell sprouting through the activation of focal adhesion kinase and plasmin secretion. Circ Res 86: 952–959, 2000. doi: 10.1161/01.RES.86.9.952. [DOI] [PubMed] [Google Scholar]
- 128.Kim YH, Hu H, Guevara-Gallardo S, Lam MT, Fong SY, Wang RA. Artery and vein size is balanced by Notch and ephrin B2/EphB4 during angiogenesis. Development 135: 3755–3764, 2008. doi: 10.1242/dev.022475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kisanuki YY, Hammer RE, Miyazaki J, Williams SC, Richardson JA, Yanagisawa M. Tie2-Cre transgenic mice: a new model for endothelial cell-lineage analysis in vivo. Dev Biol 230: 230–242, 2001. doi: 10.1006/dbio.2000.0106. [DOI] [PubMed] [Google Scholar]
- 130.Knust E, Dietrich U, Tepass U, Bremer KA, Weigel D, Vässin H, Campos-Ortega JA. EGF homologous sequences encoded in the genome of Drosophila melanogaster, and their relation to neurogenic genes. EMBO J 6: 761–766, 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Koblizek TI, Weiss C, Yancopoulos GD, Deutsch U, Risau W. Angiopoietin-1 induces sprouting angiogenesis in vitro. Curr Biol 8: 529–532, 1998. doi: 10.1016/S0960-9822(98)70205-2. [DOI] [PubMed] [Google Scholar]
- 132.Koni PA, Joshi SK, Temann UA, Olson D, Burkly L, Flavell RA. Conditional vascular cell adhesion molecule 1 deletion in mice: impaired lymphocyte migration to bone marrow. J Exp Med 193: 741–754, 2001. doi: 10.1084/jem.193.6.741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kopan R, Cagan R. Notch on the cutting edge. Trends Genet 13: 465–467, 1997. doi: 10.1016/S0168-9525(97)01318-8. [DOI] [PubMed] [Google Scholar]
- 134.Kopan R, Ilagan MX. The canonical Notch signaling pathway: unfolding the activation mechanism. Cell 137: 216–233, 2009. doi: 10.1016/j.cell.2009.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kovall RA, Blacklow SC. Mechanistic insights into Notch receptor signaling from structural and biochemical studies. Curr Top Dev Biol 92: 31–71, 2010. doi: 10.1016/S0070-2153(10)92002-4. [DOI] [PubMed] [Google Scholar]
- 136.Krebs LT, Shutter JR, Tanigaki K, Honjo T, Stark KL, Gridley T. Haploinsufficient lethality and formation of arteriovenous malformations in Notch pathway mutants. Genes Dev 18: 2469–2473, 2004. doi: 10.1101/gad.1239204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Krebs LT, Xue Y, Norton CR, Shutter JR, Maguire M, Sundberg JP, Gallahan D, Closson V, Kitajewski J, Callahan R, Smith GH, Stark KL, Gridley T. Notch signaling is essential for vascular morphogenesis in mice. Genes Dev 14: 1343–1352, 2000. [PMC free article] [PubMed] [Google Scholar]
- 138.Kubota Y, Takubo K, Hirashima M, Nagoshi N, Kishi K, Okuno Y, Nakamura-Ishizu A, Sano K, Murakami M, Ema M, Omatsu Y, Takahashi S, Nagasawa T, Shibuya M, Okano H, Suda T. Isolation and function of mouse tissue resident vascular precursors marked by myelin protein zero. J Exp Med 208: 949–960, 2011. doi: 10.1084/jem.20102187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kudo FA, Muto A, Maloney SP, Pimiento JM, Bergaya S, Fitzgerald TN, Westvik TS, Frattini JC, Breuer CK, Cha CH, Nishibe T, Tellides G, Sessa WC, Dardik A. Venous identity is lost but arterial identity is not gained during vein graft adaptation. Arterioscler Thromb Vasc Biol 27: 1562–1571, 2007. doi: 10.1161/ATVBAHA.107.143032. [DOI] [PubMed] [Google Scholar]
- 140.Kuhnert F, Kirshner JR, Thurston G. Dll4-Notch signaling as a therapeutic target in tumor angiogenesis. Vasc Cell 3: 20, 2011. doi: 10.1186/2045-824X-3-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Kume T. Novel insights into the differential functions of Notch ligands in vascular formation. J Angiogenes Res 1: 8, 2009. doi: 10.1186/2040-2384-1-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Kusumbe AP, Ramasamy SK, Adams RH. Coupling of angiogenesis and osteogenesis by a specific vessel subtype in bone. Nature 507: 323–328, 2014. [Erratum in Nature 513: 574, 2014.] doi: 10.1038/nature13145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Lafkas D, Shelton A, Chiu C, de Leon Boenig G, Chen Y, Stawicki SS, Siltanen C, Reichelt M, Zhou M, Wu X, Eastham-Anderson J, Moore H, Roose-Girma M, Chinn Y, Hang JQ, Warming S, Egen J, Lee WP, Austin C, Wu Y, Payandeh J, Lowe JB, Siebel CW. Therapeutic antibodies reveal Notch control of transdifferentiation in the adult lung. Nature 528: 127–131, 2015. doi: 10.1038/nature15715. [DOI] [PubMed] [Google Scholar]
- 144.Langen UH, Pitulescu ME, Kim JM, Enriquez-Gasca R, Sivaraj KK, Kusumbe AP, Singh A, Di Russo J, Bixel MG, Zhou B, Sorokin L, Vaquerizas JM, Adams RH. Cell-matrix signals specify bone endothelial cells during developmental osteogenesis. Nat Cell Biol 19: 189–201, 2017. doi: 10.1038/ncb3476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.LaVoie MJ, Selkoe DJ. The Notch ligands, Jagged and Delta, are sequentially processed by alpha-secretase and presenilin/gamma-secretase and release signaling fragments. J Biol Chem 278: 34427–34437, 2003. doi: 10.1074/jbc.M302659200. [DOI] [PubMed] [Google Scholar]
- 146.Leri A, Hosoda T, Kajstura J, Anversa P, Rota M. Identification of a coronary stem cell in the human heart. J Mol Med (Berl) 89: 947–959, 2011. doi: 10.1007/s00109-011-0769-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Li D, Masiero M, Banham AH, Harris AL. The notch ligand JAGGED1 as a target for anti-tumor therapy. Front Oncol 4: 254, 2014. doi: 10.3389/fonc.2014.00254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Li JL, Harris AL. Crosstalk of VEGF and Notch pathways in tumour angiogenesis: therapeutic implications. Front Biosci (Landmark Ed) 14: 3094–3110, 2009. doi: 10.2741/3438. [DOI] [PubMed] [Google Scholar]
- 149.Li Y, Brazzell J, Herrera A, Walcheck B. ADAM17 deficiency by mature neutrophils has differential effects on L-selectin shedding. Blood 108: 2275–2279, 2006. doi: 10.1182/blood-2006-02-005827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Lieber T, Kidd S, Young MW. kuzbanian-mediated cleavage of Drosophila Notch. Genes Dev 16: 209–221, 2002. doi: 10.1101/gad.942302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Lima e Silva R, Shen J, Hackett SF, Kachi S, Akiyama H, Kiuchi K, Yokoi K, Hatara MC, Lauer T, Aslam S, Gong YY, Xiao WH, Khu NH, Thut C, Campochiaro PA. The SDF-1/CXCR4 ligand/receptor pair is an important contributor to several types of ocular neovascularization. FASEB J 21: 3219–3230, 2007. doi: 10.1096/fj.06-7359com. [DOI] [PubMed] [Google Scholar]
- 152.Limbourg FP, Takeshita K, Radtke F, Bronson RT, Chin MT, Liao JK. Essential role of endothelial Notch1 in angiogenesis. Circulation 111: 1826–1832, 2005. doi: 10.1161/01.CIR.0000160870.93058.DD. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Lin FJ, Tsai MJ, Tsai SY. Artery and vein formation: a tug of war between different forces. EMBO Rep 8: 920–924, 2007. doi: 10.1038/sj.embor.7401076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Lindskog H, Kim YH, Jelin EB, Kong Y, Guevara-Gallardo S, Kim TN, Wang RA. Molecular identification of venous progenitors in the dorsal aorta reveals an aortic origin for the cardinal vein in mammals. Development 141: 1120–1128, 2014. doi: 10.1242/dev.101808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Lobov IB, Cheung E, Wudali R, Cao J, Halasz G, Wei Y, Economides A, Lin HC, Papadopoulos N, Yancopoulos GD, Wiegand SJ. The Dll4/Notch pathway controls postangiogenic blood vessel remodeling and regression by modulating vasoconstriction and blood flow. Blood 117: 6728–6737, 2011. doi: 10.1182/blood-2010-08-302067. [DOI] [PubMed] [Google Scholar]
- 156.Lobov IB, Renard RA, Papadopoulos N, Gale NW, Thurston G, Yancopoulos GD, Wiegand SJ. Delta-like ligand 4 (Dll4) is induced by VEGF as a negative regulator of angiogenic sprouting. Proc Natl Acad Sci USA 104: 3219–3224, 2007. doi: 10.1073/pnas.0611206104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Luca VC, Kim BC, Ge C, Kakuda S, Wu D, Roein-Peikar M, Haltiwanger RS, Zhu C, Ha T, Garcia KC. Notch-Jagged complex structure implicates a catch bond in tuning ligand sensitivity. Science 355: 1320–1324, 2017. doi: 10.1126/science.aaf9739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Luttun A, Carmeliet P. De novo vasculogenesis in the heart. Cardiovasc Res 58: 378–389, 2003. doi: 10.1016/S0008-6363(03)00258-X. [DOI] [PubMed] [Google Scholar]
- 159.Makanya AN, Stauffer D, Ribatti D, Burri PH, Djonov V. Microvascular growth, development, and remodeling in the embryonic avian kidney: the interplay between sprouting and intussusceptive angiogenic mechanisms. Microsc Res Tech 66: 275–288, 2005. doi: 10.1002/jemt.20169. [DOI] [PubMed] [Google Scholar]
- 160.Malecki MJ, Sanchez-Irizarry C, Mitchell JL, Histen G, Xu ML, Aster JC, Blacklow SC. Leukemia-associated mutations within the NOTCH1 heterodimerization domain fall into at least two distinct mechanistic classes. Mol Cell Biol 26: 4642–4651, 2006. doi: 10.1128/MCB.01655-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Manavski Y, Lucas T, Glaser SF, Dorsheimer L, Günther S, Braun T, Rieger MA, Zeiher AM, Boon RA, Dimmeler S. Clonal expansion of endothelial cells contributes to ischemia-induced neovascularization. Circ Res 122: 670–677, 2018. doi: 10.1161/CIRCRESAHA.117.312310. [DOI] [PubMed] [Google Scholar]
- 162.Medvinsky A, Rybtsov S, Taoudi S. Embryonic origin of the adult hematopoietic system: advances and questions. Development 138: 1017–1031, 2011. doi: 10.1242/dev.040998. [DOI] [PubMed] [Google Scholar]
- 163.Mohr OL. Character changes caused by mutation of an entire region of a chromosome in Drosophila. Genetics 4: 275–282, 1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Moloney DJ, Panin VM, Johnston SH, Chen J, Shao L, Wilson R, Wang Y, Stanley P, Irvine KD, Haltiwanger RS, Vogt TF. Fringe is a glycosyltransferase that modifies Notch. Nature 406: 369–375, 2000. doi: 10.1038/35019000. [DOI] [PubMed] [Google Scholar]
- 165.Morgan TH. The theory of the gene. Am Nat 51: 513–544, 1917. doi: 10.1086/279629. [DOI] [Google Scholar]
- 166.Mori M, Mahoney JE, Stupnikov MR, Paez-Cortez JR, Szymaniak AD, Varelas X, Herrick DB, Schwob J, Zhang H, Cardoso WV. Notch3-Jagged signaling controls the pool of undifferentiated airway progenitors. Development 142: 258–267, 2015. doi: 10.1242/dev.116855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Mumm JS, Kopan R. Notch signaling: from the outside in. Dev Biol 228: 151–165, 2000. doi: 10.1006/dbio.2000.9960. [DOI] [PubMed] [Google Scholar]
- 168.Mumm JS, Schroeter EH, Saxena MT, Griesemer A, Tian X, Pan DJ, Ray WJ, Kopan R. A ligand-induced extracellular cleavage regulates gamma-secretase-like proteolytic activation of Notch1. Mol Cell 5: 197–206, 2000. doi: 10.1016/S1097-2765(00)80416-5. [DOI] [PubMed] [Google Scholar]
- 169.Murphy G, Murthy A, Khokha R. Clipping, shedding and RIPping keep immunity on cue. Trends Immunol 29: 75–82, 2008. doi: 10.1016/j.it.2007.10.009. [DOI] [PubMed] [Google Scholar]
- 170.Murphy PA, Kim TN, Lu G, Bollen AW, Schaffer CB, Wang RA. Notch4 normalization reduces blood vessel size in arteriovenous malformations. Sci Transl Med 4: 117ra8, 2012. doi: 10.1126/scitranslmed.3002670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Murthy A, Shao YW, Narala SR, Molyneux SD, Zúñiga-Pflücker JC, Khokha R. Notch activation by the metalloproteinase ADAM17 regulates myeloproliferation and atopic barrier immunity by suppressing epithelial cytokine synthesis. Immunity 36: 105–119, 2012. doi: 10.1016/j.immuni.2012.01.005. [DOI] [PubMed] [Google Scholar]
- 172.Nielsen CM, Cuervo H, Ding VW, Kong Y, Huang EJ, Wang RA. Deletion of Rbpj from postnatal endothelium leads to abnormal arteriovenous shunting in mice. Development 141: 3782–3792, 2014. doi: 10.1242/dev.108951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Noguera-Troise I, Daly C, Papadopoulos NJ, Coetzee S, Boland P, Gale NW, Lin HC, Yancopoulos GD, Thurston G. Blockade of Dll4 inhibits tumour growth by promoting non-productive angiogenesis. Nature 444: 1032–1037, 2006. doi: 10.1038/nature05355. [DOI] [PubMed] [Google Scholar]
- 174.Nurmi H, Saharinen P, Zarkada G, Zheng W, Robciuc MR, Alitalo K. VEGF-C is required for intestinal lymphatic vessel maintenance and lipid absorption. EMBO Mol Med 7: 1418–1425, 2015. doi: 10.15252/emmm.201505731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Otani A, Dorrell MI, Kinder K, Moreno SK, Nusinowitz S, Banin E, Heckenlively J, Friedlander M. Rescue of retinal degeneration by intravitreally injected adult bone marrow-derived lineage-negative hematopoietic stem cells. J Clin Invest 114: 765–774, 2004. doi: 10.1172/JCI200421686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Palazuelos J, Crawford HC, Klingener M, Sun B, Karelis J, Raines EW, Aguirre A. TACE/ADAM17 is essential for oligodendrocyte development and CNS myelination. J Neurosci 34: 11884–11896, 2014. doi: 10.1523/JNEUROSCI.1220-14.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Parera MC, van Dooren M, van Kempen M, de Krijger R, Grosveld F, Tibboel D, Rottier R. Distal angiogenesis: a new concept for lung vascular morphogenesis. Am J Physiol Lung Cell Mol Physiol 288: L141–L149, 2005. doi: 10.1152/ajplung.00148.2004. [DOI] [PubMed] [Google Scholar]
- 178.Patan S. Vasculogenesis and angiogenesis as mechanisms of vascular network formation, growth and remodeling. J Neurooncol 50: 1–15, 2000. doi: 10.1023/A:1006493130855. [DOI] [PubMed] [Google Scholar]
- 179.Pedrosa AR, Trindade A, Fernandes AC, Carvalho C, Gigante J, Tavares AT, Diéguez-Hurtado R, Yagita H, Adams RH, Duarte A. Endothelial Jagged1 antagonizes Dll4 regulation of endothelial branching and promotes vascular maturation downstream of Dll4/Notch1. Arterioscler Thromb Vasc Biol 35: 1134–1146, 2015. doi: 10.1161/ATVBAHA.114.304741. [DOI] [PubMed] [Google Scholar]
- 180.Peschon JJ, Slack JL, Reddy P, Stocking KL, Sunnarborg SW, Lee DC, Russell WE, Castner BJ, Johnson RS, Fitzner JN, Boyce RW, Nelson N, Kozlosky CJ, Wolfson MF, Rauch CT, Cerretti DP, Paxton RJ, March CJ, Black RA. An essential role for ectodomain shedding in mammalian development. Science 282: 1281–1284, 1998. doi: 10.1126/science.282.5392.1281. [DOI] [PubMed] [Google Scholar]
- 181.Phng LK, Gerhardt H. Angiogenesis: a team effort coordinated by notch. Dev Cell 16: 196–208, 2009. doi: 10.1016/j.devcel.2009.01.015. [DOI] [PubMed] [Google Scholar]
- 182.Phng LK, Potente M, Leslie JD, Babbage J, Nyqvist D, Lobov I, Ondr JK, Rao S, Lang RA, Thurston G, Gerhardt H. Nrarp coordinates endothelial Notch and Wnt signaling to control vessel density in angiogenesis. Dev Cell 16: 70–82, 2009. doi: 10.1016/j.devcel.2008.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Pollard JW. Trophic macrophages in development and disease. Nat Rev Immunol 9: 259–270, 2009. doi: 10.1038/nri2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Potente M, Carmeliet P. The link between angiogenesis and endothelial metabolism. Annu Rev Physiol 79: 43–66, 2017. doi: 10.1146/annurev-physiol-021115-105134. [DOI] [PubMed] [Google Scholar]
- 185.Potente M, Gerhardt H, Carmeliet P. Basic and therapeutic aspects of angiogenesis. Cell 146: 873–887, 2011. doi: 10.1016/j.cell.2011.08.039. [DOI] [PubMed] [Google Scholar]
- 186.Proia T, Jiang F, Bell A, Nicoletti R, Kong L, Kreuter K, Poling L, Winston WM, Flaherty M, Weiler S, Perino S, O’Hagan R, Lin J, Gyuris J, Okamura H. 23814, an inhibitory antibody of ligand-mediated Notch1 activation, modulates angiogenesis and inhibits tumor growth without gastrointestinal toxicity. Mol Cancer Ther 14: 1858–1867, 2015. doi: 10.1158/1535-7163.MCT-14-1104. [DOI] [PubMed] [Google Scholar]
- 187.Quaggin SE, Kreidberg JA. Development of the renal glomerulus: good neighbors and good fences. Development 135: 609–620, 2008. doi: 10.1242/dev.001081. [DOI] [PubMed] [Google Scholar]
- 188.Radtke F, Clevers H. Self-renewal and cancer of the gut: two sides of a coin. Science 307: 1904–1909, 2005. doi: 10.1126/science.1104815. [DOI] [PubMed] [Google Scholar]
- 189.Ramasamy SK, Kusumbe AP, Adams RH. Regulation of tissue morphogenesis by endothelial cell-derived signals. Trends Cell Biol 25: 148–157, 2015. doi: 10.1016/j.tcb.2014.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Ramasamy SK, Kusumbe AP, Schiller M, Zeuschner D, Bixel MG, Milia C, Gamrekelashvili J, Limbourg A, Medvinsky A, Santoro MM, Limbourg FP, Adams RH. Blood flow controls bone vascular function and osteogenesis. Nat Commun 7: 13601, 2016. doi: 10.1038/ncomms13601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Ramasamy SK, Kusumbe AP, Wang L, Adams RH. Endothelial Notch activity promotes angiogenesis and osteogenesis in bone. Nature 507: 376–380, 2014. doi: 10.1038/nature13146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Ranganathan P, Weaver KL, Capobianco AJ. Notch signalling in solid tumours: a little bit of everything but not all the time. Nat Rev Cancer 11: 338–351, 2011. doi: 10.1038/nrc3035. [DOI] [PubMed] [Google Scholar]
- 193.Red-Horse K, Ueno H, Weissman IL, Krasnow MA. Coronary arteries form by developmental reprogramming of venous cells. Nature 464: 549–553, 2010. doi: 10.1038/nature08873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Ribatti D. The discovery of endothelial progenitor cells. An historical review. Leuk Res 31: 439–444, 2007. doi: 10.1016/j.leukres.2006.10.014. [DOI] [PubMed] [Google Scholar]
- 195.Ridgway J, Zhang G, Wu Y, Stawicki S, Liang WC, Chanthery Y, Kowalski J, Watts RJ, Callahan C, Kasman I, Singh M, Chien M, Tan C, Hongo JA, de Sauvage F, Plowman G, Yan M. Inhibition of Dll4 signalling inhibits tumour growth by deregulating angiogenesis. Nature 444: 1083–1087, 2006. doi: 10.1038/nature05313. [DOI] [PubMed] [Google Scholar]
- 196.Risau W. Mechanisms of angiogenesis. Nature 386: 671–674, 1997. doi: 10.1038/386671a0. [DOI] [PubMed] [Google Scholar]
- 197.Risau W, Flamme I. Vasculogenesis. Annu Rev Cell Dev Biol 11: 73–91, 1995. doi: 10.1146/annurev.cb.11.110195.000445. [DOI] [PubMed] [Google Scholar]
- 198.Ritsma L, Ellenbroek SIJ, Zomer A, Snippert HJ, de Sauvage FJ, Simons BD, Clevers H, van Rheenen J. Intestinal crypt homeostasis revealed at single-stem-cell level by in vivo live imaging. Nature 507: 362–365, 2014. doi: 10.1038/nature12972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Roberts W, Palade G. Morphogenesis of Endothelium, edited by Risau W, Rubanyi GM. Amsterdam: Hardwood, 2000, p. 23–41. [Google Scholar]
- 200.Roca C, Adams RH. Regulation of vascular morphogenesis by Notch signaling. Genes Dev 21: 2511–2524, 2007. doi: 10.1101/gad.1589207. [DOI] [PubMed] [Google Scholar]
- 201.Rocha SF, Adams RH. Molecular differentiation and specialization of vascular beds. Angiogenesis 12: 139–147, 2009. doi: 10.1007/s10456-009-9132-x. [DOI] [PubMed] [Google Scholar]
- 202.Rodriguez-Vita J, Fischer A. Notch1 induces endothelial senescence and promotes tumor progression. Cell Cycle 16: 911–912, 2017. doi: 10.1080/15384101.2017.1316575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Rooke J, Pan D, Xu T, Rubin GM. KUZ, a conserved metalloprotease-disintegrin protein with two roles in Drosophila neurogenesis. Science 273: 1227–1231, 1996. doi: 10.1126/science.273.5279.1227. [DOI] [PubMed] [Google Scholar]
- 204.Rossi JM, Dunn NR, Hogan BL, Zaret KS. Distinct mesodermal signals, including BMPs from the septum transversum mesenchyme, are required in combination for hepatogenesis from the endoderm. Genes Dev 15: 1998–2009, 2001. doi: 10.1101/gad.904601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Ruhrberg C, Bautch VL. Neurovascular development and links to disease. Cell Mol Life Sci 70: 1675–1684, 2013. doi: 10.1007/s00018-013-1277-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 206.Sabik JF III, Lytle BW, Blackstone EH, Houghtaling PL, Cosgrove DM. Comparison of saphenous vein and internal thoracic artery graft patency by coronary system. Ann Thorac Surg 79: 544–551, 2005. doi: 10.1016/j.athoracsur.2004.07.047. [DOI] [PubMed] [Google Scholar]
- 207.Sahin U, Weskamp G, Kelly K, Zhou HM, Higashiyama S, Peschon J, Hartmann D, Saftig P, Blobel CP. Distinct roles for ADAM10 and ADAM17 in ectodomain shedding of six EGFR ligands. J Cell Biol 164: 769–779, 2004. doi: 10.1083/jcb.200307137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Sanchez-Martin M, Ferrando A. The NOTCH1-MYC highway toward T-cell acute lymphoblastic leukemia. Blood 129: 1124–1133, 2017. doi: 10.1182/blood-2016-09-692582. [DOI] [PubMed] [Google Scholar]
- 209.Sancho R, Cremona CA, Behrens A. Stem cell and progenitor fate in the mammalian intestine: Notch and lateral inhibition in homeostasis and disease. EMBO Rep 16: 571–581, 2015. doi: 10.15252/embr.201540188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Sapir A, Assa-Kunik E, Tsruya R, Schejter E, Shilo BZ. Unidirectional Notch signaling depends on continuous cleavage of Delta. Development 132: 123–132, 2005. doi: 10.1242/dev.01546. [DOI] [PubMed] [Google Scholar]
- 211.Sawaguchi S, Varshney S, Ogawa M, Sakaidani Y, Yagi H, Takeshita K, Murohara T, Kato K, Sundaram S, Stanley P, Okajima T. O-GlcNAc on NOTCH1 EGF repeats regulates ligand-induced Notch signaling and vascular development in mammals. eLife 6: 6, 2017. doi: 10.7554/eLife.24419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Schepers AG, Snippert HJ, Stange DE, van den Born M, van Es JH, van de Wetering M, Clevers H. Lineage tracing reveals Lgr5+ stem cell activity in mouse intestinal adenomas. Science 337: 730–735, 2012. doi: 10.1126/science.1224676. [DOI] [PubMed] [Google Scholar]
- 213.Schroeter EH, Kisslinger JA, Kopan R. Notch-1 signalling requires ligand-induced proteolytic release of intracellular domain Nature 393: 382–386, 1998. doi: 10.1038/30756. [DOI] [PubMed] [Google Scholar]
- 214.Scott RP, Quaggin SE. The cell biology of renal filtration. J Cell Biol 209: 199–210, 2015. doi: 10.1083/jcb.201410017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Seegar TCM, Killingsworth LB, Saha N, Meyer PA, Patra D, Zimmerman B, Janes PW, Rubinstein E, Nikolov DB, Skiniotis G, Kruse AC, Blacklow SC. Structural basis for regulated proteolysis by the α-secretase ADAM10. Cell 171: 1638–1648.e7, 2017. doi: 10.1016/j.cell.2017.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Senger DR, Galli SJ, Dvorak AM, Perruzzi CA, Harvey VS, Dvorak HF. Tumor cells secrete a vascular permeability factor that promotes accumulation of ascites fluid. Science 219: 983–985, 1983. doi: 10.1126/science.6823562. [DOI] [PubMed] [Google Scholar]
- 217.Senger DR, Perruzzi CA, Feder J, Dvorak HF. A highly conserved vascular permeability factor secreted by a variety of human and rodent tumor cell lines. Cancer Res 46: 5629–5632, 1986. [PubMed] [Google Scholar]
- 218.Shalaby F, Rossant J, Yamaguchi TP, Gertsenstein M, Wu XF, Breitman ML, Schuh AC. Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature 376: 62–66, 1995. doi: 10.1038/376062a0. [DOI] [PubMed] [Google Scholar]
- 219.Shawber CJ, Das I, Francisco E, Kitajewski J. Notch signaling in primary endothelial cells. Ann N Y Acad Sci 995: 162–170, 2003. doi: 10.1111/j.1749-6632.2003.tb03219.x. [DOI] [PubMed] [Google Scholar]
- 220.Shi Q, Rafii S, Wu MH, Wijelath ES, Yu C, Ishida A, Fujita Y, Kothari S, Mohle R, Sauvage LR, Moore MA, Storb RF, Hammond WP. Evidence for circulating bone marrow-derived endothelial cells. Blood 92: 362–367, 1998. [PubMed] [Google Scholar]
- 221.Simons M, Alitalo K, Annex BH, Augustin HG, Beam C, Berk BC, Byzova T, Carmeliet P, Chilian W, Cooke JP, Davis GE, Eichmann A, Iruela-Arispe ML, Keshet E, Sinusas AJ, Ruhrberg C, Woo YJ, Dimmeler S; American Heart Association Council on Basic Cardiovascular Sciences and Council on Cardiovascular Surgery and Anesthesia . State-of-the-art methods for evaluation of angiogenesis and tissue vascularization: a scientific statement from the American Heart Association. Circ Res 116: e99–e132, 2015. doi: 10.1161/RES.0000000000000054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Sivaraj KK, Adams RH. Blood vessel formation and function in bone. Development 143: 2706–2715, 2016. doi: 10.1242/dev.136861. [DOI] [PubMed] [Google Scholar]
- 223.Smith LE, Wesolowski E, McLellan A, Kostyk SK, D’Amato R, Sullivan R, D’Amore PA. Oxygen-induced retinopathy in the mouse. Invest Ophthalmol Vis Sci 35: 101–111, 1994. [PubMed] [Google Scholar]
- 224.South AP, Cho RJ, Aster JC. The double-edged sword of Notch signaling in cancer. Semin Cell Dev Biol 23: 458–464, 2012. doi: 10.1016/j.semcdb.2012.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Speicher SA, Thomas U, Hinz U, Knust E. The Serrate locus of Drosophila and its role in morphogenesis of the wing imaginal discs: control of cell proliferation. Development 120: 535–544, 1994. [DOI] [PubMed] [Google Scholar]
- 226.Stahl A, Connor KM, Sapieha P, Willett KL, Krah NM, Dennison RJ, Chen J, Guerin KI, Smith LE. Computer-aided quantification of retinal neovascularization. Angiogenesis 12: 297–301, 2009. doi: 10.1007/s10456-009-9155-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Sternlicht MD, Sunnarborg SW, Kouros-Mehr H, Yu Y, Lee DC, Werb Z. Mammary ductal morphogenesis requires paracrine activation of stromal EGFR via ADAM17-dependent shedding of epithelial amphiregulin. Development 132: 3923–3933, 2005. doi: 10.1242/dev.01966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Stone J, Itin A, Alon T, Pe’er J, Gnessin H, Chan-Ling T, Keshet E. Development of retinal vasculature is mediated by hypoxia-induced vascular endothelial growth factor (VEGF) expression by neuroglia. J Neurosci 15: 4738–4747, 1995. doi: 10.1523/JNEUROSCI.15-07-04738.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Struhl G, Adachi A. Nuclear access and action of notch in vivo. Cell 93: 649–660, 1998. doi: 10.1016/S0092-8674(00)81193-9. [DOI] [PubMed] [Google Scholar]
- 230.Struhl G, Greenwald I. Presenilin is required for activity and nuclear access of Notch in Drosophila. Nature 398: 522–525, 1999. doi: 10.1038/19091. [DOI] [PubMed] [Google Scholar]
- 231.Suchting S, Eichmann A. Jagged gives endothelial tip cells an edge. Cell 137: 988–990, 2009. doi: 10.1016/j.cell.2009.05.024. [DOI] [PubMed] [Google Scholar]
- 232.Suchting S, Freitas C, le Noble F, Benedito R, Bréant C, Duarte A, Eichmann A. The Notch ligand Delta-like 4 negatively regulates endothelial tip cell formation and vessel branching. Proc Natl Acad Sci USA 104: 3225–3230, 2007. doi: 10.1073/pnas.0611177104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Sulis ML, Saftig P, Ferrando AA. Redundancy and specificity of the metalloprotease system mediating oncogenic NOTCH1 activation in T-ALL. Leukemia 25: 1564–1569, 2011. doi: 10.1038/leu.2011.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Suri C, Jones PF, Patan S, Bartunkova S, Maisonpierre PC, Davis S, Sato TN, Yancopoulos GD. Requisite role of angiopoietin-1, a ligand for the TIE2 receptor, during embryonic angiogenesis. Cell 87: 1171–1180, 1996. doi: 10.1016/S0092-8674(00)81813-9. [DOI] [PubMed] [Google Scholar]
- 235.Swiatek PJ, Lindsell CE, del Amo FF, Weinmaster G, Gridley T. Notch1 is essential for postimplantation development in mice. Genes Dev 8: 707–719, 1994. doi: 10.1101/gad.8.6.707. [DOI] [PubMed] [Google Scholar]
- 236.Swift MR, Weinstein BM. Arterial-venous specification during development. Circ Res 104: 576–588, 2009. doi: 10.1161/CIRCRESAHA.108.188805. [DOI] [PubMed] [Google Scholar]
- 237.Takabatake Y, Sugiyama T, Kohara H, Matsusaka T, Kurihara H, Koni PA, Nagasawa Y, Hamano T, Matsui I, Kawada N, Imai E, Nagasawa T, Rakugi H, Isaka Y. The CXCL12 (SDF-1)/CXCR4 axis is essential for the development of renal vasculature. J Am Soc Nephrol 20: 1714–1723, 2009. doi: 10.1681/ASN.2008060640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Takahashi T, Kalka C, Masuda H, Chen D, Silver M, Kearney M, Magner M, Isner JM, Asahara T. Ischemia- and cytokine-induced mobilization of bone marrow-derived endothelial progenitor cells for neovascularization. Nat Med 5: 434–438, 1999. doi: 10.1038/7434. [DOI] [PubMed] [Google Scholar]
- 239.Takakura N, Kidoya H. Maturation of blood vessels by haematopoietic stem cells and progenitor cells: involvement of apelin/APJ and angiopoietin/Tie2 interactions in vessel caliber size regulation. Thromb Haemost 101: 999–1005, 2009. [PubMed] [Google Scholar]
- 240.Tattersall IW, Du J, Cong Z, Cho BS, Klein AM, Dieck CL, Chaudhri RA, Cuervo H, Herts JH, Kitajewski J. In vitro modeling of endothelial interaction with macrophages and pericytes demonstrates Notch signaling function in the vascular microenvironment. Angiogenesis 19: 201–215, 2016. doi: 10.1007/s10456-016-9501-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Teodorczyk M, Schmidt MH. Notching on cancer’s door: Notch signaling in brain tumors. Front Oncol 4: 341, 2015. doi: 10.3389/fonc.2014.00341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 242.Thomas U, Speicher SA, Knust E. The Drosophila gene Serrate encodes an EGF-like transmembrane protein with a complex expression pattern in embryos and wing discs. Development 111: 749–761, 1991. [DOI] [PubMed] [Google Scholar]
- 243.Tian L, Wu X, Chi C, Han M, Xu T, Zhuang Y. ADAM10 is essential for proteolytic activation of Notch during thymocyte development. Int Immunol 20: 1181–1187, 2008. doi: 10.1093/intimm/dxn076. [DOI] [PubMed] [Google Scholar]
- 244.Tian X, Hu T, Zhang H, He L, Huang X, Liu Q, Yu W, He L, Yang Z, Zhang Z, Zhong TP, Yang X, Yang Z, Yan Y, Baldini A, Sun Y, Lu J, Schwartz RJ, Evans SM, Gittenberger-de Groot AC, Red-Horse K, Zhou B. Subepicardial endothelial cells invade the embryonic ventricle wall to form coronary arteries. Cell Res 23: 1075–1090, 2013. doi: 10.1038/cr.2013.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Tian X, Pu WT, Zhou B. Cellular origin and developmental program of coronary angiogenesis. Circ Res 116: 515–530, 2015. doi: 10.1161/CIRCRESAHA.116.305097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 246.Tiyanont K, Wales TE, Aste-Amezaga M, Aster JC, Engen JR, Blacklow SC. Evidence for increased exposure of the Notch1 metalloprotease cleavage site upon conversion to an activated conformation. Structure 19: 546–554, 2011. doi: 10.1016/j.str.2011.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Trindade A, Djokovic D, Gigante J, Mendonça L, Duarte A. Endothelial Dll4 overexpression reduces vascular response and inhibits tumor growth and metastasization in vivo. BMC Cancer 17: 189, 2017. doi: 10.1186/s12885-017-3171-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 248.Trindade A, Kumar SR, Scehnet JS, Lopes-da-Costa L, Becker J, Jiang W, Liu R, Gill PS, Duarte A. Overexpression of delta-like 4 induces arterialization and attenuates vessel formation in developing mouse embryos. Blood 112: 1720–1729, 2008. doi: 10.1182/blood-2007-09-112748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 249.Tsao PN, Vasconcelos M, Izvolsky KI, Qian J, Lu J, Cardoso WV. Notch signaling controls the balance of ciliated and secretory cell fates in developing airways. Development 136: 2297–2307, 2009. doi: 10.1242/dev.034884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 250.van den Akker NM, Caolo V, Wisse LJ, Peters PP, Poelmann RE, Carmeliet P, Molin DG, Gittenberger-de Groot AC. Developmental coronary maturation is disturbed by aberrant cardiac vascular endothelial growth factor expression and Notch signalling. Cardiovasc Res 78: 366–375, 2008. doi: 10.1093/cvr/cvm108. [DOI] [PubMed] [Google Scholar]
- 251.van Es JH, van Gijn ME, Riccio O, van den Born M, Vooijs M, Begthel H, Cozijnsen M, Robine S, Winton DJ, Radtke F, Clevers H. Notch/gamma-secretase inhibition turns proliferative cells in intestinal crypts and adenomas into goblet cells. Nature 435: 959–963, 2005. doi: 10.1038/nature03659. [DOI] [PubMed] [Google Scholar]
- 252.van Tetering G, van Diest P, Verlaan I, van der Wall E, Kopan R, Vooijs M. Metalloprotease ADAM10 is required for Notch1 site 2 cleavage. J Biol Chem 284: 31018–31027, 2009. doi: 10.1074/jbc.M109.006775. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 253.Van Vlierberghe P, Ferrando A. The molecular basis of T cell acute lymphoblastic leukemia. J Clin Invest 122: 3398–3406, 2012. doi: 10.1172/JCI61269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 254.Vässin H, Bremer KA, Knust E, Campos-Ortega JA. The neurogenic gene Delta of Drosophila melanogaster is expressed in neurogenic territories and encodes a putative transmembrane protein with EGF-like repeats. EMBO J 6: 3431–3440, 1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Vaughan MR, Quaggin SE. How do mesangial and endothelial cells form the glomerular tuft? J Am Soc Nephrol 19: 24–33, 2008. doi: 10.1681/ASN.2007040471. [DOI] [PubMed] [Google Scholar]
- 256.Vokes SA, Yatskievych TA, Heimark RL, McMahon J, McMahon AP, Antin PB, Krieg PA. Hedgehog signaling is essential for endothelial tube formation during vasculogenesis. Development 131: 4371–4380, 2004. doi: 10.1242/dev.01304. [DOI] [PubMed] [Google Scholar]
- 257.Volz KS, Jacobs AH, Chen HI, Poduri A, McKay AS, Riordan DP, Kofler N, Kitajewski J, Weissman I, Red-Horse K. Pericytes are progenitors for coronary artery smooth muscle. eLife 4: 4, 2015. doi: 10.7554/eLife.10036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Wada AM, Willet SG, Bader D. Coronary vessel development: a unique form of vasculogenesis. Arterioscler Thromb Vasc Biol 23: 2138–2145, 2003. doi: 10.1161/01.ATV.0000098645.38676.CC. [DOI] [PubMed] [Google Scholar]
- 259.Wälchli T, Wacker A, Frei K, Regli L, Schwab ME, Hoerstrup SP, Gerhardt H, Engelhardt B. Wiring the Vascular Network with Neural Cues: A CNS Perspective. Neuron 87: 271–296, 2015. doi: 10.1016/j.neuron.2015.06.038. [DOI] [PubMed] [Google Scholar]
- 260.Wang HU, Chen ZF, Anderson DJ. Molecular distinction and angiogenic interaction between embryonic arteries and veins revealed by ephrin-B2 and its receptor Eph-B4. Cell 93: 741–753, 1998. doi: 10.1016/S0092-8674(00)81436-1. [DOI] [PubMed] [Google Scholar]
- 261.Wang Y, Wu B, Lu P, Zhang D, Wu B, Varshney S, Del Monte-Nieto G, Zhuang Z, Charafeddine R, Kramer AH, Sibinga NE, Frangogiannis NG, Kitsis RN, Adams RH, Alitalo K, Sharp DJ, Harvey RP, Stanley P, Zhou B. Uncontrolled angiogenic precursor expansion causes coronary artery anomalies in mice lacking Pofut1. Nat Commun 8: 578, 2017. doi: 10.1038/s41467-017-00654-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 262.Weber S, Niessen MT, Prox J, Lüllmann-Rauch R, Schmitz A, Schwanbeck R, Blobel CP, Jorissen E, de Strooper B, Niessen CM, Saftig P. The disintegrin/metalloproteinase Adam10 is essential for epidermal integrity and Notch-mediated signaling. Development 138: 495–505, 2011. doi: 10.1242/dev.055210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Welshons WJ. A preliminary investigation of pseudoallelism at the Notch locus of Drosophila melanogaster. Proc Natl Acad Sci USA 44: 254–258, 1958. doi: 10.1073/pnas.44.3.254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Wen C, Metzstein MM, Greenwald I. SUP-17, a Caenorhabditis elegans ADAM protein related to Drosophila KUZBANIAN, and its role in LIN-12/NOTCH signalling. Development 124: 4759–4767, 1997. [DOI] [PubMed] [Google Scholar]
- 265.Weskamp G, Mendelson K, Swendeman S, Le Gall S, Ma Y, Lyman S, Hinoki A, Eguchi S, Guaiquil V, Horiuchi K, Blobel CP. Pathological neovascularization is reduced by inactivation of ADAM17 in endothelial cells but not in pericytes. Circ Res 106: 932–940, 2010. doi: 10.1161/CIRCRESAHA.109.207415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 266.Wieland E, Rodriguez-Vita J, Liebler SS, Mogler C, Moll I, Herberich SE, Espinet E, Herpel E, Menuchin A, Chang-Claude J, Hoffmeister M, Gebhardt C, Brenner H, Trumpp A, Siebel CW, Hecker M, Utikal J, Sprinzak D, Fischer A. Endothelial Notch1 activity facilitates metastasis. Cancer Cell 31: 355–367, 2017. doi: 10.1016/j.ccell.2017.01.007. [DOI] [PubMed] [Google Scholar]
- 267.Wu B, Zhang Z, Lui W, Chen X, Wang Y, Chamberlain AA, Moreno-Rodriguez RA, Markwald RR, O’Rourke BP, Sharp DJ, Zheng D, Lenz J, Baldwin HS, Chang CP, Zhou B. Endocardial cells form the coronary arteries by angiogenesis through myocardial-endocardial VEGF signaling. Cell 151: 1083–1096, 2012. doi: 10.1016/j.cell.2012.10.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Wu Y, Cain-Hom C, Choy L, Hagenbeek TJ, de Leon GP, Chen Y, Finkle D, Venook R, Wu X, Ridgway J, Schahin-Reed D, Dow GJ, Shelton A, Stawicki S, Watts RJ, Zhang J, Choy R, Howard P, Kadyk L, Yan M, Zha J, Callahan CA, Hymowitz SG, Siebel CW. Therapeutic antibody targeting of individual Notch receptors. Nature 464: 1052–1057, 2010. doi: 10.1038/nature08878. [DOI] [PubMed] [Google Scholar]
- 269.Yang K, Doughman YQ, Karunamuni G, Gu S, Yang YC, Bader DM, Watanabe M. Expression of active Notch1 in avian coronary development. Dev Dyn 238: 162–170, 2009. doi: 10.1002/dvdy.21811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Yen WC, Fischer MM, Axelrod F, Bond C, Cain J, Cancilla B, Henner WR, Meisner R, Sato A, Shah J, Tang T, Wallace B, Wang M, Zhang C, Kapoun AM, Lewicki J, Gurney A, Hoey T. Targeting Notch signaling with a Notch2/Notch3 antagonist (tarextumab) inhibits tumor growth and decreases tumor-initiating cell frequency. Clin Cancer Res 21: 2084–2095, 2015. doi: 10.1158/1078-0432.CCR-14-2808. [DOI] [PubMed] [Google Scholar]
- 271.Yoon C-H, Choi Y-E, Cha YR, Koh S-J, Choi JI, Kim T-W, Woo SJ, Park Y-B, Chae I-H, Kim H-S. Diabetes-Induced Jagged1 overexpression in endothelial cells causes retinal capillary regression in a murine model of diabetes mellitus: insights into diabetic retinopathy. Circulation 134: 233–247, 2016. doi: 10.1161/CIRCULATIONAHA.116.014411. [DOI] [PubMed] [Google Scholar]
- 272.Zarkada G, Heinolainen K, Makinen T, Kubota Y, Alitalo K. VEGFR3 does not sustain retinal angiogenesis without VEGFR2. Proc Natl Acad Sci USA 112: 761–766, 2015. doi: 10.1073/pnas.1423278112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 273.Zhang C, Tian L, Chi C, Wu X, Yang X, Han M, Xu T, Zhuang Y, Deng K. Adam10 is essential for early embryonic cardiovascular development. Dev Dyn 239: 2594–2602, 2010. doi: 10.1002/dvdy.22391. [DOI] [PubMed] [Google Scholar]
- 274.Zhang H, Pu W, Tian X, Huang X, He L, Liu Q, Li Y, Zhang L, He L, Liu K, Gillich A, Zhou B. Genetic lineage tracing identifies endocardial origin of liver vasculature. Nat Genet 48: 537–543, 2016. doi: 10.1038/ng.3536. [DOI] [PubMed] [Google Scholar]
- 275.Zhao R, Wang A, Hall KC, Otero M, Weskamp G, Zhao B, Hill D, Goldring MB, Glomski K, Blobel CP. Lack of ADAM10 in endothelial cells affects osteoclasts at the chondro-osseus junction. J Orthop Res 32: 224–230, 2014. doi: 10.1002/jor.22492. [DOI] [PMC free article] [PubMed] [Google Scholar]