

Abstract
The central functions fulfilled by mitochondria as both energy generators essential for tissue homeostasis and gateways to programmed apoptotic and necrotic cell death mandate tight control over the quality and quantity of these ubiquitous endosymbiotic organelles. Mitophagy, the targeted engulfment and destruction of mitochondria by the cellular autophagy apparatus, has conventionally been considered as the mechanism primarily responsible for mitochondrial quality control. However, our understanding of how, why, and under what specific conditions mitophagy is activated has grown tremendously over the past decade. Evidence is accumulating that nonmitophagic mitochondrial quality control mechanisms are more important to maintaining normal tissue homeostasis whereas mitophagy is an acute tissue stress response. Moreover, previously unrecognized mitophagic regulation of mitochondrial quantity control, metabolic reprogramming, and cell differentiation suggests that the mechanisms linking genetic or acquired defects in mitophagy to neurodegenerative and cardiovascular diseases or cancer are more complex than simple failure of normal mitochondrial quality control. Here, we provide a comprehensive overview of mitophagy in cellular homeostasis and disease and examine the most revolutionary concepts in these areas. In this context, we discuss evidence that atypical mitophagy and nonmitophagic pathways play central roles in mitochondrial quality control, functioning that was previously considered to be the primary domain of mitophagy.
I. INTRODUCTION AND BACKGROUND
Autophagy is an evolutionary conserved degradation pathway responsible for delivering cytoplasmic components to the lysosome in vesicles called autophagosomes. This process was described over 50 years ago, when researchers observed cytoplasmic material being engulfed inside double-membrane vesicles and subsequently degraded (332). The subsequent refinement of our understanding of autophagy has led to its implication in compensatory management of metabolic stress, as a mechanism for programmed cell death by self-cannibalization, and as the primary mechanism responsible for mitochondrial quality control, i.e., mitophagy. Our conceptual understanding of the pathophysiological relevance of mitophagy itself has expanded, evolving from simply the canonical mechanism by which mitochondrial fitness is maintained, to being one part of a complex orchestrated group of interactive processes that determine mitochondrial number, turnover rate, metabolic transitioning, and mitochondrial fitness in the broadest sense. This overview examines the most recent concepts for all of these areas and considers accumulating evidence that atypical mitophagy or nonmitophagic pathways play major roles in mitochondrial quality control, previously considered the canonical function of mitophagy.
It is useful to review major historical landmarks of a biological process before examining recent advances: In the early 1960s, Christian de Duve named autophagosomal delivery of cellular components to lysosomes “autophagy,” derived from the Greek words for eat (“phagy”) and oneself (“auto”) (49). Originally regarded to be a nonselective bulk degradation process, autophagy of organelles, protein aggregates, and even pathogens was observed under defined conditions, indicating that degradation could be a selective rather than always stochastic. It was subsequently observed that mammalian cells specifically identified and removed damaged mitochondria via autophagy in a highly context-dependent manner that spared other, healthy organelles, so-called “mitochondrial quality control” (67, 327). Thus Elmore et al. (67) reported that exposing mitochondria to photo-damage to induce opening of the mitochondrial permeability transition pore and depolarization led to their selective engulfment by autophagosomes in hepatocytes, and Xue et al. (327) observed that stimulation of apoptosis in the presence of caspase inhibitors led to the selective removal of mitochondria in neurons and HeLa cells. It has since been appreciated that mitophagy has many other functions in cells and that other complementary and/or redundant mechanisms exist for mitochondrial quality control (selectively culling damaged, potentially cytotoxic organelles) and quantity control (generally removing excessive numbers of mitochondria). Because it is essential that mitochondrial content and metabolic programming match the needs of cells in response to changing developmental, bioenergetic, and environmental conditions, mitophagy is highly regulated and interacts with the related process of mitochondrial biogenesis. The functional complexity of mitophagy contrasts with the comparatively straightforward purpose of nonselective (macro) autophagy, which is nutrient recycling to replenish macromolecules and maintain ATP levels during nutrient deprivation.
Induction of mitophagy relies on exquisite spatiotemporal coordination of two distinct events: 1) damaged or redundant mitochondria must be identified and labeled for degradation, and 2) vesicular structures required to sequester the mitochondrion and transport it to a lysosome for degradation need to be formed. Macroautophagy (herein referred to as autophagy) is the dominant pathway involved in nonspecific breakdown and recycling of intracellular proteins and organelles, which can include mitochondria. In either autophagy or mitophagy, double membrane structures called autophagosomes are formed that sequester mitochondria marked for elimination and deliver their cargo to lysosomes where contents are degraded by lysosomal enzymes (160) (FIGURE 1).
FIGURE 1.

Overview of nonselective autophagy and selective autophagy. The phagophore engulfs cargo in the cytosol nonselectively or selectively to form a double membrane autophagosome. The autophagsome fuses with a lysosome leading to the formation of the autolysosome where the cargo is degraded and then recycled.
II. MITOPHAGY: THE CONVENTIONAL VIEW
The early studies reviewed above described removal of dysfunctional mitochondria by autophagosomes, but the mechanisms by which damaged mitochondrial were identified and then selectively targeted by autophagosomes were unknown. Surprisingly, ubiquitin was identified as a critical signal for selective autophagy: the specific type of ubiquitin chain that was added to misfolded proteins determined the degradative pathway to which they would be delivered (136). Ubiquitin chains adopt different conformations that can be recognized by different ubiquitin-binding proteins. In general, proteins destined for proteasomal degradation are polyubiquitinated predominantly through K48 linkage, whereas K63-linked ubiquitin chains direct the cargo to autophagosomes (79, 206). Identification of autophagy adaptors that simultaneously bind both ubiquitin and autophagy-specific proteins Atg8/LC3/GABARAP on the autophagosome provided a molecular link between ubiquitination and autophagy. The prototypical selective autophagy adaptor in mammalian cells is p62/SQSTM1 (sequestosome 1) (222), but additional adaptor proteins have been identified conferring mechanistic richness that is a central characteristic of mitochondrial quality control (102, 156). A common feature of autophagy adaptor proteins is the presence of an ubiquitin binding domain (UBD) and a LC3-interacting region (LIR) motif, which allows them to link ubiquitinated cargo to the autophagosome (FIGURE 2A).
FIGURE 2.
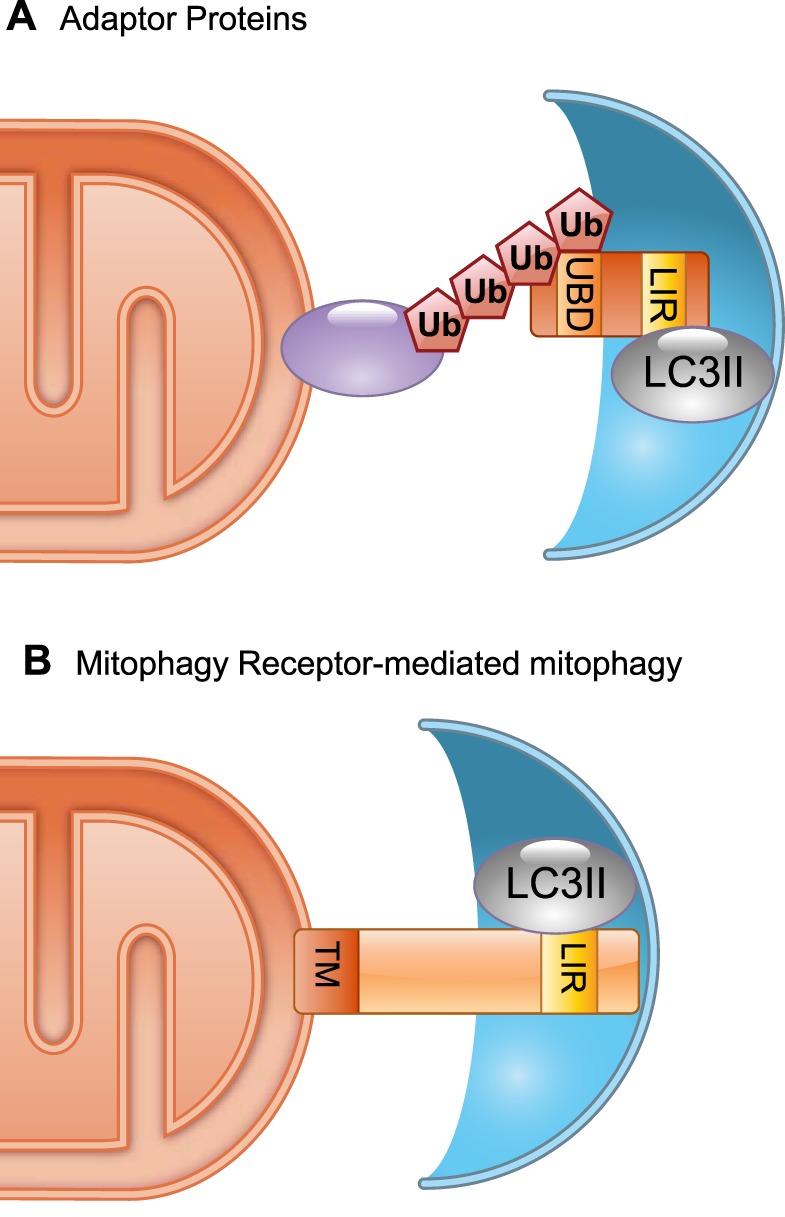
Selective mitophagy. A: an adaptor protein containing a ubiquitin binding domain (UBD) recognizes a polyubiquitinated protein in the outer mitochondrial membrane (OMM). It physically connects the mitochondrion to the autophagosome membrane via its LC3 interacting region (LIR) that binds to lipidated LC3 (GABARAP/Atg8). B: mitophagy receptors, such as Atg32, BNIP3, and Nix, are anchored in the outer mitochondrial membrane via a COOH-terminal transmembrane domain. A LIR motif in the NH2-terminal domain interacts directly with lipidated LC3/GABARAP/Atg8.
Mitophagy became a process of great interest after the unexpected convergence of two seemingly independent lines of investigation. On the clinical side, mutations in PINK1 [PTEN-induced putative kinase; encoded by the PINK1 (formerly PARK6) gene] and Parkin (encoded by the PARK2 gene) were the first genetic events linked to autosomal recessive early-onset Parkinson’s disease (134, 300). It was unclear how damaging mutations in the mitochondrial kinase PINK1 and the cytosolic E3 ubiquitin ligase Parkin caused similar clinical syndromes. This quandary was resolved by gene disruption studies in Drosophila, which revealed that PINK1 is upstream of Parkin in mitophagy signaling (42, 223). A similar mitophagy signaling pathway was subsequently found in mammalian systems (70). The PINK1-Parkin mitophagy signaling paradigm originally established in Drosophila has had a major influence over how we conceive the underlying pathology of both normal homeostatic mitochondrial quality control and hereditary Parkinson’s disease (93). However, it is important to recognize that although PINK1- and Parkin-deficient flies had very similar phenotypes consisting of abnormal mitochondrial morphology and function, locomotor deficits, muscle degeneration, and loss of neurons (42, 223), mitophagic activity was not assessed in these studies. Thus whether the accumulation of dysfunctional mitochondria is due to a defect in mitophagy or disruption of another key mitochondrial process such as fission or fusion is unknown. Thus the studies in flies raise several important questions, and the idea that defective PINK-Parkin-mediated mitophagy underlies the clinical pathology in human Parkinson’s disease still needs to be thoroughly validated.
Parkin was recognized as an E3 ubiquitin ligase localized in the cytosol (261), and the mitochondrial role for Parkin remained unknown for another decade, until 2008 when Youle’s group discovered that Parkin rapidly translocated from the cytosol to damaged mitochondria after cells were treated with a mitochondrial uncoupler that completely dissipates the normal electrochemical gradient maintained across the inner mitochondrial membrane to fuel oxidative phosphorylation, ΔΨm (202). Shortly thereafter, multiple groups demonstrated that recruitment of Parkin to mitochondria was regulated by the serine-threonine kinase PINK1 (85, 181, 203, 306). Subsequent studies linked Parkin-mediated ubiquitination to recruitment of the adaptor protein p62/SQSTM1 (85, 201, 214). PINK1/Parkin-mediated mitophagy, the original mechanism of mitochondrial quality control, has been more widely studied than other mitophagy pathways. It is now accepted that PINK1 coordinates the selective clearance of defective mitochondria and that ubiquitination of mitochondrial proteins in the outer mitochondrial membrane by Parkin and other potential E3 ubiquitin ligases generates a cargo recognition signal for autophagy adaptors and autophagosomes.
Other mechanisms of mitophagy, independent of Parkin, ubiquitination, and adaptor proteins and transduced by so-called “mitophagy receptors” (328), coexist with the PINK1-Parkin pathway mechanism. Studies in Saccharomyces cerevisiae provided the first evidence that mitophagy could be selective, but no Parkin or autophagy adaptor homologues were identified in yeast cells. Instead, genetic studies in yeast led to the identification of mitophagy receptors, defined as proteins anchored in outer membrane of mitochondria and containing a WXXL-like motif that mediates the interaction with autophagosomal Atg8/LC3/GABARAP to promote selective autophagy (FIGURE 2B). Atg32 was identified as a mitophagy receptor in yeast through two independent genome-wide screens for nonessential gene deletion mutants defective in mitochondrial degradation (123, 213). Although atg32 mutant cells had functional autophagy, they were defective in mitophagy, pointing to a specific Atg32 function in mitochondrial removal. Subsequent studies revealed Atg32 to be a single transmembrane domain spanning outer mitochondrial membrane protein acting as a mitochondrial receptor for Atg8 on the autophagosome. The pro-apoptotic Bcl-2 family member Nix was the first mammalian mitophagy receptor to be identified when it was discovered that Nix-deficient erythrocytes were unable to clear mitochondria during their maturation (55, 244, 257). Subsequent studies identified Nix functioning as a mitochondrial receptor that directly interacted with the Atg8 homologues LC3 and GABARAP on the autophagosome (53, 211, 256), and uncovered roles for Nix-mediated mitophagy in retinal development and macrophage activation (69). Several additional mitophagy receptors, including BNIP3 (99), Fundc1 (169), BCL2L13 (195), and FKBP8 (17), have since then been recognized in mammals, and more are likely to be detected in the future.
In summary, our understanding of mitophagy and its functions has grown to include at least two distinct pathways, the PINK1/Parkin and mitophagy receptor pathways. It seems clear that mitophagy plays a role in mitochondrial quality and quantity control, metabolic reprogramming, and differentiation. Because timely mitophagic elimination of dysfunctional mitochondria and maintenance of a contextually appropriate population of mitochondria are both essential for normal cellular health, it is not surprising that mitophagic defects have been implicated in a variety of diseases.
III. PHYSIOLOGICAL FUNCTIONS OF MITOPHAGY
A. Mitochondrial Quality Control
Aerobic eukaryotic life depends on mitochondrial oxidative phosphorylation to produce chemical fuel in the form of ATP. However, life is placed at risk when damaged mitochondria produce proteo- and nucleotoxic reactive oxygen species (ROS). Mitochondrial ATP synthesis is driven by the electrochemical gradient maintained across the inner mitochondrial membrane, the electron transport system. Transfer of electrons between different complexes of the electron transport chain promotes extrusion of protons (hydrogen ions) across the inner mitochondrial membrane and into the mitochondrial intermembrane space. Passive reversal of proton flow through complex V (mitochondrial ATP synthase) powers ATP synthesis from ADP (199). Oxygen serves as the terminal electron acceptor, producing (in sequence due to enzymatic reactions) superoxide anion (O2−), hydrogen peroxide (H2O2), and water (H2O). Electrons that escape these terminal reactions, frequently by leaking from complexes I or III of the electron transport chain (36, 196), permit damaging O2− or H2O2 to attack the mitochondrion and its host cell.
Although the range of mitochondrial fitness is a continuum (from the healthy ATP producer to the toxic ROS generator), the fateful decision regarding a given mitochondrion is necessarily dichotomous; it is either retained and repaired or it is removed. This apparent mismatch is resolved through the process of asymmetric mitochondrial fission (299). Mitochondria targeted for mitophagy are hypopolarized (299) and tend not to undergo fusion (290). The time between mitochondrial depolarization and autophagosomal engulfment is variable, suggesting that some mitochondria exist in a gray-zone state of recognizably impaired, but not sufficient to trigger mitophagy, a pre-autophagic pool (299, 316, 331). This is where the decision regarding mitochondrial fate is exercised; asymmetric mitochondrial fission produces two functionally dissimilar daughter mitochondria. In contrast to replicative fission where both daughter organelles are healthy, after asymmetric fission the two daughters of fission have different membrane potentials, one fully polarized and one depolarized. The depolarized daughter is fusion defective and joins a static population of pre-autophagic mitochondria destined to be removed from the cell, thereby protecting the rest of the mitochondrial pool and the host cell from ROS damage (1, 299).
The requisite for mitochondrial fission in mitophagy points to coordination between mitochondrial dynamics (fission and fusion) and mitochondrial quality control. Indeed, not only are the processes inextricably intertwined, but some protein effectors play roles in both pathways. For example, mitofusin (Mfn) 2 is one of two outer mitochondrial membrane proteins (Mfn1 and Mfn2) whose function to promote mitochondrial tethering and outer membrane fusion is largely redundant. However, Mfn2 has a seemingly unique role in mitophagy regulated by its PINK1 kinase phosphorylation status: when not phosphorylated by PINK1, Mfn2 (and Mfn1) adopts an open conformation that permits mitofusins on one mitochondria to interact in trans with mitofusins on neighboring mitochondria, physically tethering the two organelles and preparing them for actual fusion of their outer membranes (76, 141). However, PINK1-mediated phosphorylation of Mfn2 on serine 378 favors a closed protein conformation that is nonpermissive for tethering (239). PINK1 also phosphorylates Mfn2 on Thr111 and Ser442, enabling Mfn2 binding of cytosolic Parkin, which as discussed below is likely to be an important mechanism for initial Parkin recruitment to damaged mitochondria (33). The interested reader is referred to Reference 59 for a detailed examination of recently published data on the structure and function of mitofusins and exploration of new questions raised by recent structural and biochemical studies.
The role in mitophagy of mitochondrial fission mediated by dynamin-related protein 1 (Drp1) seems straightforward, as described above: asymmetrical fission sequesters defective or damaged mitochondrial components in one of two daughter mitochondria, which is therefore depolarized and recognized as such by the PINK1-Parkin mechanism, removed by autophagosomal engulfment, and transferred to lysosomes for degradation (299). How damaged proteins are preferentially incorporated into the ill-fated daughter organelle during fission remains a mystery and may simply be the inevitable consequence of failure of damaged/unfolded proteins to be properly integrated into or maintained within the highly structured, almost crystalline organization of respiratory complexes that occupy most of mitochondrial volume (266). It is also notable that fission-promoting Drp1, which is normally cytosolic and recruited to mitochondria in preparation for fission (219), can physically interact with fusion-promoting Mfn2, which like Mfn1 is constitutively mitochondrial on the outer membrane (106). The functional consequences of Drp1-Mfn2 complex formation remain unknown but suggest another level of crosstalk between mitochondrial fission, fusion, and mitophagy.
B. Mitophagy as a Mechanism for Mitochondrial Quality Control
As introduced above, the conventional concept of mitochondrial quality control derives from the view that these endosymbionts-turned-organelles are first and foremost the engines of oxidative phosphorylation, but that they can transform into potentially lethal generators of cytotoxic ROS (294). This functional duality mandates cell processes that identify, sequester, and ultimately remove damaged ROS-producing mitochondria, thereby maintaining the fitness of the mitochondrial collective. Moreover, because ROS have been implicated in senescence and aging (47) and damaging mutations of mitochondrial DNA can cause experimental (148, 297) and clinical (307) disease, mitochondrial quality control is widely assumed to be a necessary homeostatic function as well as a response induced by cellular or mitochondrial stress.
A frequent underlying anthropomorphic assumption when considering mitochondrial population homeostasis is that the mitochondrial life cycle is linear, like ours. Mitochondria are “born” or assembled through biogenesis (231), they live for some defined period, and (like us) they undergo age-related declines in function requiring repair (as through fusion-mediated complementation with a younger/healthier mitochondrion; like an organ transplant) (253, 337) or they are removed via mitophagy to mitochondrial hospice. These misconceptions are patterned after the wrong domain of life; mitochondria are not derived from eukaryotes, but from primordial bacteria (91, 330). Like their ancestors, mitochondria reproduce using symmetric fission: one parent mitochondrion divides into two daughter organelles that grow by adding new components provided by the cell through the process of mitochondrial biogenesis and/or by fusing with other mitochondria. Thus mitochondrial damage too great to be repaired represents a dead-end that interrupts the replicative cycle. Indeed, damaged or dysfunctional mitochondria must be eliminated so that they cannot contaminate healthy members of the cell’s mitochondrial population. This is the purpose of mitochondrial quality control.
The purpose of mitophagy is to both functionally sequester and physically eliminate dysfunctional, potentially cytotoxic mitochondria before they can directly or indirectly injure the host cell. The mitochondrion needs to signal, or the host cell needs to detect, mitochondrial dysfunction at the level of individual organelles. Thus the mitophagy quality control “on switch” must originate at the damaged mitochondrion. Known mitophagy triggers include mitochondrial depolarization, ROS production, and protein misfolding (119, 181, 203), and the common mitophagy effector is PINK1 kinase, which senses these inputs. The elegant molecular mechanism underlying damage sensing and mitophagy activation by PINK1 kinase were elucidated by Youle and colleagues (203, 226). Briefly, PINK1 is encoded by the host cell genome, translated in the cytosolic ribosomal apparatus, and must be actively imported into mitochondria through the standard outer and inner membrane transport mechanisms, TOM and TIM (100) (FIGURE 3A). The seminal observation that revealed the key role of PINK1 in mitophagy signaling was that PINK1 levels increase in damaged or depolarized mitochondria because the normal import-and-immediately-degrade PINK1 pathway is interrupted at the degradation step. Unprocessed 63-kDa PINK1 is transported across the outer mitochondrial membrane by the TOM complex, delivered to the inner mitochondrial membrane translocase TIM, and under normal conditions is rapidly cleaved there by PARL (118, 187) (FIGURE 3A). Proteolytic processing generates a 52-kDa PINK1 fragment that moves back to the cytosol and is eliminated by N-Degron directed proteasomal degradation (329). Consequently, PINK1 protein levels in healthy mitochondria are vanishingly low, and there is no measurable PINK1 kinase activity (203). However, in damaged mitochondria, TIM-mediated translocation of PINK1 across the inner mitochondrial membrane is impaired, protecting PINK1 from degradation by PARL (FIGURE 3B). As a consequence, PINK1 maintains its association with TOM on the outer mitochondrial membrane (155) and phosphorylates available substrates (215), including proteins like Mfn2 and ubiquitinated outer membrane proteins that attract Parkin (33, 144) (FIGURE 4). We previously likened this mechanism to a “dead-man switch” (61), in which healthy mitochondria avoid mitophagy by actively degrading PINK1, but damaged mitochondria lacking the vigor to constantly degrade PINK1 become its substrate, triggering Parkin-mediated and other events that target the organelle for mitophagic elimination.
FIGURE 3.

Regulation of PINK1. A: newly synthesized PINK1 is immediately imported into healthy mitochondria with an intact membrane potential (ΔΨ) by the TOM/TIM import machinery. PINK1 undergoes proteolytic cleavage by the mitochondrial intramembrane protease PARL. The cleaved PINK1 retrotranslocates to the cytosol where it is subjected to ubiquitination by E3 ubiquitin ligases UBR1, UBR2, and UBR4 and proteasomal degradation. B: upon mitochondrial damage and loss of ΔΨ, import of PINK1 is abrogated, and it accumulates on the outer mitochondrial membrane which leads to recruitment of Parkin.
FIGURE 4.

Activation of PINK1/Parkin-mediated mitophagy. PINK1 accumulates on the outer mitochondrial membrane (OMM) in the absence of ΔΨ. PINK1 recruits and activates Parkin in a process involving PINK1-mediated phosphorylation of Mfn2, ubiquitin (Ub), and Parkin. Activated Parkin conjugates ubiquitin to various proteins in the OMM. The ubiquitin chains on proteins in the OMM are recognized by autophagy adaptors which in turn tether the ubiquitinated cargo to the autophagosome via binding to lipidated LC3.
The recent observations that PINK1 phosphorylates free ubiquitin (122, 143) as well as ubiquitin complexes on outer mitochondrial membrane (OMM) proteins (216) suggested that Parkin binding to phospho-ubiquitylated OMM proteins either triggers or amplifies mitochondrial Parkin recruitment (216, 226, 282). Because Parkin is necessary to create ubiquitylated OMM proteins for PINK1 to act upon, we favor the idea that binding of Parkin to PINK1-phosphorylated ubiquitin chains on OMM proteins serves to amplify, rather than trigger, the PINK1-Parkin mitophagy pathway. PINK1-mediated phosphorylation of Mfn2 on Thr111 and Ser442 simultaneously recruits Parkin to mitochondria through Parkin-Mfn2 binding (33) and suppresses Mfn2-mediated fusion (90), while concurrent PINK1-mediated phosphorylation of free ubiquitin activates Parkin’s E3 ubiquitin ligase activity (122, 143); the accumulated effect is to localize and activate Parkin on the target mitochondrion, thus initiating mitophagy signaling (FIGURE 4). In contrast, PINK1-mediated phosphorylation of ubiquitin on OMM proteins previously acted upon by Parkin would amplify mitophagy signaling (216).
The physical and mechanistic links between PINK1, Mfn2, and Parkin provide a mechanism for simultaneously triggering mitophagy and interdicting mitochondrial fusion, which is necessary to prevent damaged mitochondria from spreading their dysfunction, a situation we have referred to as “mitochondrial contagion” (16). PINK1-mediated phosphorylation transforms Mfn2 from a fusion protein into a Parkin receptor (90) in a manner that is more rapid and direct than the proposed mechanism of Parkin-mediated Mfn2 ubiquitination, selective extraction, and proteasomal degradation (84, 228, 232, 290, 347). In the latter scenario, Parkin-mediated ubiquitination of Mfn1 and Mfn2 selectively targets these fusion-promoting proteins for proteasomal degradation, functionally quarantining the organelle until it is physically removed by autophagosomes and lysosomes (124, 348). However, there is no evidence that Parkin selectively ubiquitinates mitochondrial fusion proteins. Indeed, the pro-fission protein Drp1 is also a Parkin substrate (308), and its proteosomal degradation would shift the fission/fusion equilibrium in the wrong direction (i.e., favoring more fusion). Recent work shows that Parkin promiscuously ubiquitinates a hundred or more OMM proteins (29, 246) and that this overall process is central to attracting autophagosomes (144).
C. Mitophagy as a Mechanism for Mitochondrial Quantity Control
As is true for any population, mitochondrial number is a combined function of the rate at which new members are introduced and their longevity within the population. When mitochondrial insertion [typically through replicative fission (191)], combined with biogenesis (62) and removal (by mitophagy or autophagy) are balanced, the population remains stable. Regulation of mitochondrial biogenesis and mitophagy has been described in many contexts, implying that mitochondrial homeostasis requires careful orchestration of input and output. Recent studies have elucidated an unexpected role for mitochondrial dynamism as a third process, acting in concert with biogenesis and mitophagy, for controlling mitochondrial quantity.
Compared with other organs, mitochondria of in vivo cardiac myocytes are hypodynamic and long-lived. Fusion and fission occur infrequently (274), mitochondrial interactions such as “kiss and run” are detectable but not routine (66, 108, 170), and mitochondrial turnover measured directly or indirectly takes longer in hearts than in other mitochondrial-rich tissues such as liver (35). It was somewhat surprising therefore that genetic interruption of mitochondrial fusion (combined ablation of outer mitochondrial membrane fusion proteins Mfn1 and Mfn2) or fission (ablation of fission protein Drp1) in adult mouse hearts provoked rapidly progressing and ultimately lethal cardiomyopathies (112, 277). Moreover, while the outcome was the same after interrupting mitochondrial fusion or fission (death in 6–7 wk), the processes by which this was achieved were completely different. Fusion-defective mouse hearts developed eccentric hypertrophy and accumulated dysfunctional mitochondria; interrupting fusion increased mitochondrial number but decreased mitochondrial quality, suggesting a defect in mitochondrial quality control (275). The functionally reciprocal genetic intervention evoked opposing effects: fission-defective hearts developed dilated cardiomyopathy with cardiomyocyte drop-out, and suffered from markedly reduced numbers of (seemingly healthy) mitochondria. Again, this suggested a defect in mitochondrial quality control, but with hyperfunctioning so that the mitophagic set point was altered in a manner such that normal mitochondria were mitophagically eliminated. Because mitochondrial biogenesis was suppressed in both the fusion- and fission-defective hearts, it cannot underlie changes in mitochondrial quantity. Instead, reciprocal changes in mitophagic markers (277) supported a central role for mitochondrial dynamism as a modulator of mitophagy that, in turn, regulates mitochondrial quantity.
The same group recently extended this work, comparing the consequences on adult mouse hearts of interrupting mitochondrial fusion (Mfn1/Mfn2 double cardiac knockout), suppressing mitochondrial fission (Drp1 cardiac knockout), and completely abrogating mitochondrial dynamics (fusion and fission-defective mouse hearts; Mfn1/Mfn2/Drp1 triple cardiac knockout) (275). Remarkably, combining one rapidly lethal cardiomyopathy with the other moderated both phenotypes, delaying cardiac failure and death. Thus, in mammalian hearts as in yeast and Caenorhabditis elegans (13, 221), absence of mitochondrial dynamism appears to be less harmful to the organism than an imbalance between fission and fusion, in either direction. Moreover, the major abnormality in fusion and fission defective hearts was of mitochondrial quantity, not quality. Combined ablation of Mfn1, Mfn2, and Drp1 in young adult mice provoked progressive accumulation of mitochondria to such an extent that the myofibrillar structure within cardiac myocytes was disrupted, with mitochondria occupying the central ~80% of the cell and sarcomeres displaced to the cardiomyocyte periphery. Increased mitochondrial numbers and mass were paralleled in this model by increased heart mass and cardiac myocyte volume, suggesting that observed cardiac “hypertrophy” reflected an infiltration of mitochondria rather than typical generation of additional sarcomeres. Other than the approximate doubling in content within cardiac myocytes, the adynamic mitochondria themselves were only slightly abnormal: substrate-stimulated ATP production was intact and there was no indication of increased ER stress. Rather, it was concluded that the mitochondria were exhibiting signs of senescence including heterogeneity in size, a tendency for increased maximal uncoupling of the electron transport chain from ATP synthesis resulting in slightly increased ROS production, and a markedly increased unfolded protein response. The greatly expanded population of “aged” mitochondria was mechanistically linked to mitochondrial adynamism through impaired mitophagy, as in the parent Mfn1/Mfn2-deficient heart mice (275). Thus mitophagy dependent on mitochondrial dynamism is an important mechanism for controlling mitochondrial quantity, as well as quality.
D. Mitophagy for Mitochondrial Replacement During Metabolic Transitions
Mitochondria may exhibit no dysfunction as organelles, but may still be bioenergetically mismatched to a given pathophysiological context. For example, during mammalian development and with the formation of a functioning circulatory system, the early embryo transitions from being a largely anaerobic organism to a partially aerobic organism in a hypoxic environment (269). This transition is accompanied by a proliferation of mitochondria and the beginning of a dependence on mitochondrial metabolism. With the use of the heart as a well-studied example, in adults, cardiac contractions can be sustained only seconds without mitochondrial-generated ATP. In the midterm fetus however, the priority is myocardial growth, and circulatory flow can vary greatly without endangering the fetus (which explains why all but the most severe congenital heart defects become life threatening only after birth). Cardiomyocyte mitochondria in fetal hearts differ from those in adult hearts in several ways. 1) Fetal heart mitochondria prefer carbohydrate as substrate for ATP production, whereas adult heart mitochondria prefer fatty acids and branched chain amino acids (269). This is likely because fetal hearts use fatty acids to synthesize membrane lipids and amino acids to synthesize proteins; growth is the priority and the molecular building blocks for growth are therefore reserved for that purpose. 2) Fetal heart mitochondria are less abundant than in adult hearts, and 3) mitochondria of fetal cardiac myocytes are morphologically distinct, being longer and thinner than their ovoid adult counterparts. Each of these factors transitions from the fetal to an adult phenotype shortly after birth.
Mammalian birth provokes massive changes in metabolism. Arterial oxygen content increases dramatically as a result of the newly functioning pulmonary circulation and gas exchange (272). Thus, not only does the type of hemoglobin in the blood have to change to accommodate more oxygen (168), but mitochondria must be able to manage the oxygen physiologically as the terminal electron acceptor for ATP synthase, rather than as an incidental electron acceptor for production of ROS. Moreover, mitochondrial substrate preference in the heart transitions to fatty acids, which are abundant in mother’s milk (289). Finally, cardiomyocyte mitochondria assume their adult form and proper subcellular distribution between myofilaments running the long axis of the cell (90). Over the past decade it has been popular to refer to these changes as metabolic “reprogramming,” invoking transcriptional mechanisms driven by master metabolic regulators such as peroxisome proliferator-activated receptor gamma coactivator 1 (PGC-1) and implying that the mitochondrial metabolic hardware simply needs a software update (in the form of newly expressed genes) to adjust to neonatal life in an oxygen abundant, fatty acid-abundant, pump function-critical environment.
Given the parallel changes in morphology and metabolism, Dorn and colleagues (90) were skeptical that mitochondria have readily adjustable metabolic substrate and oxygen utilization. Instead, they hypothesized that mitochondria are generated by cells for particular pathophysiological contexts and must be “switched out” (through accelerated turnover) when this context changes. In the context of the newborn heart, this would produce mature adult mitochondria. They further posited that the Parkin mitophagy apparatus might play a role in generalized mitochondrial turnover by scaling up the same cellular apparatus used for quality and quantity control. For the reasons detailed above, they tested this notion in perinatal hearts by genetically deleting Parkin in cardiomyocytes using a floxed Parkin allele mouse in combination with a cardiomyocyte-specific Cre that is activated at birth (90). Whereas cardiomyocyte-directed genetic ablation of Parkin had no measurable effects in adult mice, in neonatal mice it prevented normal cardiomyocyte mitochondrial maturation and was rapidly lethal. Although this result suggested that Parkin has an important role in the perinatal heart that might involve mitochondrial maturation, gene deletion is a molecular sledge hammer that abrogates all Parkin functionality, i.e., mitophagy and everything else. Therefore, they also selectively interrupted Parkin-mediated mitophagy in the neonatal heart using the more nuanced approach of conditionally expressing a Mfn2 mutant (Mfn2 Thr111Ala/Ser442Ala, or Mfn2 AA) that functions normally for mitochondrial fusion, but does not bind Parkin. As with Parkin ablation, Mfn2 AA expression had no adverse effects when expressed in adult hearts, or from weaning. When expressed in the heart from birth however, lethal cardiomyopathies developed characterized by retention of fetal mitochondrial morphometry and the fetal metabolic profile; both mitochondria and cardiac metabolism failed to mature when Parkin-Mfn2 mitophagic signaling was interrupted. They concluded that “persistence of fetal carbohydrate-metabolizing mitochondria in adult Mfn2 AA hearts revealed a requirement for organelle removal through the PINK1-Mfn2-Parkin mitophagy mechanism before mitochondrial transitioning to normal adult fatty acid metabolism” and that “the Parkin-Mfn2 interaction drives general mitophagic turnover of fetal mitochondria in the perinatal heart, enabling their replacement with mitochondria incorporating biogenically derived metabolic systems optimized for the high energetic demands of contracting adult hearts” (90).
E. Mitophagy in the Removal of Paternal Mitochondria
Paternal mitochondria enter the oocyte cytoplasm upon fertilization, but mature mammalian cells contain only maternal mitochondria. Thus the paternal mitochondria are selectively eliminated during embryogenesis (3, 227, 240, 248, 314). Accumulating evidence indicates that mitophagy is the main mechanism responsible for the selective elimination of paternal mitochondria. Two groups reported independently that autophagy was rapidly induced after fertilization in C. elegans where it selectively eliminated sperm components, including mitochondria, in the embryos (3, 248). More importantly, abrogation of autophagy prevented the maternal inheritance of mitochondria. An early study by Sutovsky et al. (287) reported that elimination of paternal mitochondria in mammalian cells involved labeling of mitochondria by ubiquitination. Although most sperm membranous organelles were found to be ubiquitinated before elimination by autophagosomes, paternal mitochondria were surprisingly not ubiquitinated in C. elegans (3). This suggests that the elimination of paternal mitochondria in C. elegans is not mediated via an E3 ubiquitin ligase such as Parkin. Recently, Prohibitin 2 (PHB2), a protein that is localized to the inner mitochondrial membrane, was identified to function as a mitophagy receptor that can directly bind to LC3II on the autophagosome (314). PHB2 is required for paternal mitochondrial elimination in C. elegans and selective paternal inactivation of phb-2 impaired elimination of paternal mitochondrial in embryos (314). However, exactly how PHB2 promotes clearance of sperm mitochondria still needs to be determined. Because PHB2 is localized in the inner mitochondrial membrane, it is not readily accessible to bind to LC3II on the autophagosome. The outer mitochondrial membrane must presumably be broken down to expose PHB2 before mitophagy of paternal mitochondria. Parkin-mediated ubiquitination and subsequent proteasomal degradation can promote breakdown of the outer membrane (29, 336), but since the paternal mitochondria are not ubqiuitinated, it is unclear how the OMM is broken down to expose PHB2. It is also unknown if PHB2 is required for elimination of paternal mitochondria in mammalian embryos.
Whereas ubiquitination does not appear to be involved in paternal clearance of mitochondria in C. elegans, it seems to be required in Drosophila melanogaster and mammalian cells. In Drosophila, clearance of paternal mitochondria requires ubiquitination and the adaptor protein p62/SQSTM1. Because Parkin is not required (227), other E3 ubiquitin ligases appear to perform this particular mitophagy function. Recently, Rojansky et al. (240) demonstrated that two different E3 ubiquitin ligases, MUL1 and Parkin, coordinate to ensure elimination of paternal mitochondria in mouse embryos. They found that MUL1 (also known as Mulan and Mapl), a mitochondria-localized E3 ligase, works in parallel with Parkin in mitophagy. Knockdown of either Parkin or Mul1 had modest effects on the elimination of paternal mitochondria in embryo; only simultaneous knockdown of Parkin and MUL1 resulted in a substantial retention of paternal mitochondria, suggesting that they can compensate for each other in eliminating mitochondria (240). How paternal mitochondria, but not maternal mitochondria, are selectively targeted by MUL1 and Parkin for degradation and whether PHB2 also participates in the downstream clearance remains to be investigated. Overall, these studies confirm that mitophagy plays an important role in eliminating paternal mitochondria in embryos but that the mode of mitophagy (PINK1/Parkin vs. mitophagy receptors) might not be conserved between species.
F. Mitophagy in Stem Cell Maintenance and Differentiation
Because mitochondria are key factors in dictating stem cell function and fate (2, 81, 153, 197, 217), mitophagy indirectly regulates stem cell function. In adult tissues, stem cells exist in hypoxic niches in a quiescent state with low metabolic activity. They contain a reduced number of immature mitochondria and rely primarily on glycolysis to meet energetic needs (129). To maintain quiescence and stemness, stem cells continuously repress oxidative metabolism and eliminate actively respiring mitochondria via mitophagy (103). It was also determined that mitophagy plays a critical role in ensuring that hematopoietic stem cells (HSCs) stay “stem cells” and do not differentiate: autophagy-deficient Atg12−/− HSCs contained increased numbers of elongated, fused mitochondria and were more metabolically active than control HSC. By comparing mitochondria in Parkin−/− and Atg12−/− HSCs, it was determined that Parkin-deficient HSCs contained mitochondria with reduced membrane potential, suggesting a role for Parkin-mediated mitophagy in the clearance of damaged mitochondria. In contrast, mitochondria in Atg12-deficient HSCs had increased mitochondrial membrane potential, indicating accumulation of more active mitochondria. Transplantation experiments with Atg12−/− HSCs into irradiated mice confirmed both impaired self-renewal and lineage commitment in the absence of functional autophagy. In contrast, Parkin−/− HSCs were indistinguishable from wild-type HSCs in mouse transplantation experiments (103), suggesting that Parkin plays a minimal role in clearing metabolically active mitochondria in HSCs. A key role for Parkin-independent mitophagy in differentiation was recently confirmed in a study using stem cells isolated from adult hearts (153). This study found that Parkin was undetectable in various resident stem cells isolated from adult human and mouse cardiac tissues. Instead, the different adult stem cells expressed high levels of the mitophagy receptors. Mitophagy receptor-mediated mitochondrial elimination was required for formation of a functional mitochondrial network during differentiation (153), confirming the critical role of mitophagy in metabolic remodeling. Overall, these studies demonstrate the critical role for mitophagy in regulating both stem cell homeostasis and differentiation.
G. Mitophagy in Cellular Reprogramming
Somatic cells reprogramming into induced pluripotent stem cells (iPSCs) is accompanied by changes in mitochondrial number, composition, structure, and function. While differentiation entails activation of mitochondrial biogenesis to increase mitochondrial content and a switch from glycolysis to oxidative phosphorylation, reprogramming requires a substantial reduction of mitochondrial content and a switch from oxidative phosphorylation to glycolysis (322). Mitophagy plays a pivotal role in reducing mitochondrial mass during cellular reprogramming as described by Vazquez-Martin et al. (304) who discovered that loss of PINK1-dependent mitophagy reduced the rate and efficiency of iPSC reprogramming. In this study, reprogramming of PINK1-deficient mouse embryonic fibroblasts (MEFs) resulted in iPSCs containing a mixture of mature and immature mitochondria. These iPSCs were also unstable and displayed a strong propensity to undergo spontaneous differentiation (304).
A role for BNIP3L/Nix-mediated mitophagy in differentiation was originally reported in erythrocyte maturation (244). More recently, Nix-mediated mitophagy was identified as the major mechanism for mitochondrial elimination during reprogramming of MEFs (325). Here, reprogramming was associated with Nix upregulation, and Nix knockdown reduced mitochondrial clearance during reprogramming. This group also found mitochondrial sequestration inside LC3II/Rab5-positive autophagosomal/endosomal vesicles (325), indicating cooperation between the autophagy and endosomal pathways in removing mitochondria during reprogramming.
IV. MITOPHAGY AND DISEASE
A. Mitophagy and Neurodegenerative Diseases
Due to their high metabolic requirements, a healthy pool of mitochondria is critical for neuronal function and survival. Neurons are long-lived cells, and continuous mitochondrial quality control ensures cell function and survival throughout the person’s lifespan. Moreover, neurons such as the motor and sensory neurons serving arms and legs can be over a meter long from cell body to terminal synapse, requiring mitochondria to be distributed over long physical distances, which complicates mitochondrial turnover. Therefore, it is not surprising that mitochondrial dysfunction and defects in mitophagy have been linked to several neurodegenerative diseases.
1. Parkinson’s disease
Parkinson’s disease (PD) is a neurodegenerative disorder characterized by the loss of dopaminergic (DA) neurons in the substantia nigra, provoking progressive movement disorders including resting tremor, bradykinesia, and loss of facial expression (48). PD is most frequently a sporadic disease, but ~10% of PD cases exhibit familial inheritance, revealing an underlying genetic cause (249). Although aging and several environmental and genetic factors have been linked to the etiology of PD, the exact underlying pathobiology is not well understood. Studies of sporadic-, familial- and pharmacologically induced forms of PD have each implicated mitochondrial dysfunction in the progression of the disease, suggesting a central role for mitochondrial fitness (21, 82). Moreover, the identification of loss-of-function mutations in Parkin and PINK1 in hereditary PD, and the subsequent discovery that genetic ablation of these two genes in fruit flies can evoke some of the characteristics of PD, have led to a widespread acceptance that mitophagy defects underlie PD. However, Parkin and PINK1 ablation in mice does not phenocopy clinical PD, and whether a mitophagy defect is directly responsible for development of PD in humans requires confirmation.
As discussed earlier, because PINK1 and Parkin act in a common pathway involving mitophagy and defects in the PINK1/Parkin pathway cause mitochondrial dysfunction in vitro, it has been assumed that impaired mitophagy is the underlying cause in familial PD. Currently, there is little evidence that impaired Parkin-dependent mitophagy is responsible for the loss of DA neurons in vivo, and studies have yet to demonstrate exactly how impaired PINK1-Parkin signaling contributes to PD pathogenesis and loss of dopaminergic neurons. In addition, both brain and heart are mitochondria-rich tissues, but Parkin-deficient mice fail to recapitulate the neurological symptoms observed in humans diagnosed with PD (158), and Parkin null adult hearts have normal mitochondria and no negative effect on cardiac function (147, 276). Thus this suggests that either alternative mitochondrial quality control mechanisms are compensating for the lack of Parkin or mitochondrial quality control under baseline conditions is not the main function of Parkin. Moreover, POLG mutator mice have a proofreading deficiency in the DNA polymerase γ and accumulate dysfunctional mitochondria due to the accelerated generation of mtDNA mutations (148, 297). Parkin-deficient mice do not display signs of neurodegeneration (88), but POLG mutator mice on a Parkin-deficient background have increased loss of DA neurons (225). Although this supports the idea that Parkin protects the DA neurons from mitochondrial dysfunction in vivo, it is still uncertain that mitophagy is the primary underlying mechanism.
Additional common substrates for PINK1 and Parkin have been identified, such as MIRO (312) and PARIS (262), linking these proteins to nonmitophagic pathways critical to proper mitochondrial functioning. MIRO interacts with Mfn2 to regulate mitochondrial motility (8, 192), while PARIS functions as a transcriptional repressor of PGC-1α, itself a master transcriptional co-regulator of mitochondrial biogenesis (262, 281). PARIS is increased in both sporadic and familial PD brains which leads to PGC-1α repression and reduced mitochondrial biogenesis. Recent studies have revealed that Parkin preferentially ubiquitinates PARIS that has been phosphorylated by PINK1 (159), uncovering additional biological complexity within PINK1-Parkin signaling pathways. Defects in PINK1 lead to accumulation of nonphosphorylated PARIS, which is not efficiently recognized by Parkin. Parkin also has non-mitochondrial substrates and functions that may be indirectly related to mitochondrial health and function. The identification and validation of currently unrecognized substrates is likely to clarify the pathways that underlie neurodegeneration in PD and ascertain whether compromised mitophagy plays a role in sporadic or age-associated PD; autophagic activity declines with age (180, 263), leading to impaired elimination of mitochondria by Parkin-mediated and Parkin-independent mitophagy.
It is important to keep in mind that most of our knowledge of the molecular mechanism underlying PINK1/Parkin-mediated mitophagy stems from in vitro experiments in immortalized cell lines using mitochondrial toxins combined with overexpression of proteins. Although mitochondrial poisons are good tools to study mitophagy, they do not mimic physiological conditions in vivo. Also, such experiments on PINK1 and Parkin are biased toward mitophagy and have impeded our understanding of other pathways that are regulated by Parkin. Many cell lines, such as HeLa cells, have undetectable levels of Parkin and still have a perfectly healthy mitochondria, suggesting that Parkin is not crucial for survival in these cells. Because it has been challenging to validate these findings in vivo, the physiological relevance of these findings has been questioned recently. Therefore, it will be important for future studies to translate the in vitro findings to more relevant in vivo models.
2. Alzheimer’s disease
Alzheimer’s disease (AD) is the most common neurodegenerative disorder and is characterized by severe memory loss and cognitive dysfunction due to loss of neurons and synapses. The pathological characteristics of AD are the accumulation of β-amyloid (Aβ) plaques derived from proteolytic processing of the amyloid precursor protein and intracellular neurofibrillary tangles of hyperphosphorylated Tau protein (pTau) (101, 116). As with PD, mitochondrial dysfunction is a hallmark of AD and is posited to be an underlying cause of the Aβ and pTau pathologies in AD. Thus mitochondrial dysfunction precedes the accumulation of Aβ deposits in the brains of AD mouse models (68, 177, 333), and administration of toxins that compromise mitochondrial function accelerates the Aβ pathology (32). It has been speculated that reduced cellular ATP levels and excessive ROS production that result from mitochondrial impairment contribute to aberrant processing of the amyloid precursor protein and pTau, leading to formation of Aβ plaques and neurofibrillary tangles (184). The underlying cause of impaired mitochondrial function in AD is unclear, but emerging evidence suggests that impaired mitophagy contributes to accumulation of dysfunctional neuronal mitochondria. Over a decade ago, Nixon et al. (210) observed that autophagosomes accumulated in AD brains. Likewise, autophagosomes accumulated in neuronal dendrites and soma before the appearance of Aβ plaques in a mouse model of AD disease (338). Subsequent studies have verified that autophagosomes accumulate in dystrophic neurites of AD brains, animal AD model brains, and AD cellular models, suggesting that autophagic flux is impaired (128, 162, 166, 334). Ye et al. (334) observed that Parkin-mediated mitophagy was robust in neurons derived from amyloid precursor protein transgenic mice and AD patient brains, but mitochondria were accumulating in autophagosomes as a consequence of impaired lysosomal function. This suggests both that autophagic flux is impaired in AD neurons and that intact autophagic flux is a requirement for effective mitophagy.
Overall, these results are consistent with the notion that compromised mitophagy in AD results from impaired fusion between autophagosomes and lysosomes. It is not known, however, whether dysfunctional mitochondria accumulate because there is a defect in mitophagy, or whether mitochondrial dysfunction precedes the impairment of autophagy. It is even possible that the dysfunctional mitochondria contribute to the defect in autophagy.
3. Huntington disease
Huntington disease (HD) is a hereditary progressive and disabling neurodegenerative disorder characterized by loss of motor coordination, cognitive decline, and progressive behavioral disturbances (241). HD is caused by mutations in the gene encoding the Huntingtin (Htt) protein leading to production of an abnormal protein having expanded (i.e., >36) polyglutamine (polyQ) repeats at the NH2 terminus (255). The mutant Htt protein is misfolded and forms cytotoxic aggregates that disrupt many cellular functions. The rate of protein aggregation, and therefore the extent of cytotoxicity, is directly proportional to the length of polyQ expansion (251). Htt is widely expressed during development and interacts with a number of different proteins impacting a variety of physiological processes such as axonal trafficking, regulation of gene transcription, and cell survival (255). Recent studies have demonstrated that Htt also plays a role in selective autophagy and mitochondrial quality control. It was initially observed that the majority of autophagosomes in neurons in HD lacked cargo, despite exhibiting normal autophagic flux (178), suggesting either a defect in autophagosomal cargo recognition or sequestration. Subsequently, it was discovered that Htt functions as a scaffold protein to regulate selective autophagy. Specifically, Htt was found to modulate both cargo recognition efficiency and autophagosome initiation by interacting simultaneously with the autophagy adaptor p62/SQSTM1 and the autophagy initiating kinase Ulk1 (242). This dual interaction allows for selective formation of an autophagosome around the cargo to ensure its efficient sequestration. A limitation of this study is that they did not test if mutant Htt still interacted with p62/SQSTM1 and Ulk1 and whether their functions were altered. It is possible that, depending of the length of the polyQ repeats, some mutant Htt proteins still interact with these proteins, thereby sequestering them away from binding to their cargo.
As discussed in section VIIIC, dysfunctional mitochondria can be directly sequestered in lysosomes via microautophagy mediated by glyceraldehyde-3-phosphate dehydrogenase (GAPDH) (111, 335). Remarkably, the presence of expanded polyglutamine repeats in the mutant Htt inhibits this GAPDH-mediated form of mitophagy (111): mutant Htt selectively associates with GAPDH at the damaged mitochondria, thereby inhibiting their engulfment into lysosomes and leading to accumulation of damaged mitochondria and increased cell death. Taken together, these findings suggest that impaired mitophagy might contribute as one of the underlying mechanism of HD as a consequence of direct effects of mutant Htt on mitophagy pathway proteins.
4. Amyotrophic lateral sclerosis
Amyotrophic lateral sclerosis (ALS) is characterized by degeneration of motor neurons, leading to progressive muscle weakness, paralysis, and respiratory failure (43). Based on evidence from experimental mouse models and patients, mitochondrial abnormalities are implicated as a major contributor to ALS (174, 247, 321). ALS motor neurons progressively accumulate damaged mitochondria, suggesting that impaired mitochondrial quality control could also be one of the underlying pathogenic factors. While most cases of ALS appear to be sporadic, ~10% are familial. Mitochondrial defects have been described in ALS-linked mutations of mitochondrial superoxide dismutase (SOD1) that degrades ROS and in ALS-linked mutations of autophagy/mitophagy genes encoding OPTN (179), TBK1 (77), and VCP/p97 (10). For instance, mutations in the autophagy adaptor OPTN are associated with impaired Parkin-mediated mitophagy in motor neurons (321). In addition, TBK1 plays a key role in selective autophagy and is responsible for phosphorylating a number of autophagy adaptors including p62/SQSTM1, OPTN, and NDP52, thus enhancing their ability to bind LC3II and ubiquitinated cargo. Loss-of-function mutations in TBK1 decrease activation of these autophagy adaptors, evoking decreased mitophagy in the motor neurons (194). Finally, VCP/p97 is recruited to mitochondria whose proteins have been ubiquitinated by Parkin and therefore plays an important role in Parkin-mediated mitophagy (290); missense mutations of VCP in familial ALS compromise mitochondrial clearance (132).
Clearly, ALS-linked mutations in OPTN, TBK1, and VCP/p97 can interfere with mitophagy, suggesting that inefficient turnover of damaged mitochondria could represent a key pathophysiological mechanism contributing to ALS. Although the above studies implicate defective mitophagy as the underlying cause of neurodegeneration, it is still unclear exactly how much mitophagy contributes to the disease development. Most of the mutant proteins that impair mitophagy also play key roles in other cellular processes.
B. Mitophagy and Cardiovascular Diseases
Mitophagy plays a fundamental role in the homeostasis of the cardiovascular system, and impaired mitophagy has been implicated in various cardiovascular pathologies. As discussed in detail in section IIID, Parkin-mediated mitophagy is activated in response to metabolic changes and is critical for removal of fetal mitochondria during cardiac metabolic remodeling in the perinatal period (90). Thus cardiac specific deletion of Parkin during the perinatal period is lethal due to the disruption of normal mitochondrial maturation and retention of fetal mitochondria in myocytes.
Moreover, to limit the damage induced by dysfunctional mitochondria following myocardial ischemia, myocytes activate mitophagy to clear damaged mitochondria before they can do harm to the cell. Not surprisingly, studies have reported that impaired mitophagy in the heart leads to increased susceptibility to stress and heart failure development (112, 147). In addition, Parkin is rapidly upregulated in the heart in response to challenge and mitochondrial damage (98, 147, 276, 326), revealing that Parkin-mediated mitophagy is most important in the adaptation to cellular stress in the adult heart. In addition, Parkin-deficient mice have increased accumulation of dysfunctional mitochondria in the border zone after a myocardial infarction which leads to increased ventricular remodeling and development of heart failure (147). Mortality in the Parkin-deficient mice is also significantly increased compared with wild-type mice, confirming the importance of Parkin in adapting to stress associated with a myocardial infarction.
Sadoshima and colleagues (264) have observed that mitophagy is transiently activated in mouse hearts between 3 and 7 days after pressure overload induced by microsurgical transaortic constriction (TAC) and determined that reduced mitophagy correlated with mitochondrial dysfunction. Parkin knockout mice exposed to chronic cardiac pressure-overload induced by TAC experience adverse cardiac remodeling and pathological hypertrophy compared with wild-type mice (98). Interestingly, although the authors observed reduced mitophagy in Parkin-deficient hearts, they found that the primary function of Parkin in this context was to promote degradation of C/EBP homologous protein (CHOP), a known regulator of endoplasmic reticulum (ER) stress-initiated apoptosis. Thus Parkin’s function was to prevent maladaptive hypertrophy by inhibiting apoptosis in myocytes rather than promoting mitophagy. In vivo studies on PINK1 confirm the importance of this pathway in protecting against stress. It was initially reported that PINK1 deficiency leads to development of left ventricular dysfunction and cardiac hypertrophy at 2 mo of age (18). PINK1-deficient mice are also more susceptible to myocardial ischemia/reperfusion (I/R) injury (267), and transgenic overexpression of PINK1 in the heart leads to reduced cell death and decreased infarct size after I/R (309). Taken together, available data point to the importance of PINK1/Parkin signaling in the adaptation to metabolic demands and stress in the heart and suggest that homeostatic mitochondrial quality control is largely the domain of other pathways.
Doxorubicin (Dox) is a potent chemotherapeutic agent that is used for the treatment of many different cancers (271). However, its use is limited due to its adverse effects on the heart. The mechanisms underlying Dox-mediated cardiotoxicity have been extensively investigated, but it is still unclear how Dox exposure can cause damage to the heart which is not manifest until sometimes many years later. Dox can directly target mitochondria, and mitochondrial damage is central to Dox-mediated cardiotoxicity (14). Based on the fact that Dox directly damages mitochondria, it is very likely that mitophagy is activated in myocytes to counteract the effect of Dox. To date, only a few studies have investigated the effect of Dox on the mitophagy pathway. Hoshino et al. (105) reported that elimination of mitochondria via mitophagy was reduced after Dox exposure, suggesting that Dox exposure impaired the mitophagy pathway. In contrast, Hull et al. (109) reported that Dox exposure initially reduced mitophagy, but at 2 wk post-injection, mitophagy was enhanced as evident from reduced mitochondrial content. The opposing findings in these two studies are most likely due to differences in cumulative Dox dose and using mice with different genetic backgrounds. It is possible that insufficient mitophagy in myocytes is one of the contributors to the late-onset cardiotoxicity that can develop years after completion of the treatment in some patients. The reduced mitochondrial quality control will lead to accumulation of dysfunctional mitochondria over time until a certain threshold has been reached and the cell can no longer function and will undergo cell death.
C. Mitophagy and Cancer
There is evidence that defects in the mitophagy machinery may have a role in the progression of cancer. However, its role is still controversial and seems to depend on the type of cancer. A growing number of studies have observed a correlation between impaired Parkin activity and enhanced cancer development. Many tumors including glioblastoma (305), breast (259) and ovarian (50) cancers have deletion or loss-of-function mutations in PARK2. Also, Parkin-deficient mice develop hepatocellular carcinoma (78) and are more susceptible to gamma-irradiation-induced tumorigenesis (340). This supports the hypothesis that Parkin can function as a tumor suppressor, but whether this is due to its role in mitophagy is currently unclear. It has been proposed that mitophagy suppresses initiation of cancer by eliminating cytotoxic mitochondria to limit oxidative stress. However, as a cytosolic E3 ubiquitin ligase, Parkin’s function is not limited to mitophagy, but it can also target cytosolic proteins for proteasomal-mediated degradation. For instance, Parkin can regulate levels of cell cycle regulators, such as cyclin-dependent kinases and cyclins, and Parkin deficiency leads to accelerated cell cycle progression (157). Hence, it is possible that Parkin has different functions in dividing cells and postmitotic cells such as neurons and myocytes.
BNIP3-mediated mitophagy has also been reported to function as a tumor suppressor mechanism. Chourasia et al. (39) discovered that BNIP3-dependent mitophagy functioned to limit mitochondrial mass and ROS production in growing tumors. Also, loss of BNIP3 led to accumulation of dysfunctional mitochondria and a switch to aerobic glycolysis which correlated with increased tumor growth and progression to metastasis in a mouse model of mammary tumorigenesis (39). This group found that triple negative breast cancer commonly lacks BNIP3 and proposed that BNIP3 could be used to predict progression to metastasis. In contrast, another study recently reported that BNIP3-mediated mitophagy functioned to promote survival of colorectal cancer cells (163). In this study, the authors found that suppression of BNIP3-mediated mitophagy in colorectal cancer cells led to accumulation of dysfunctional mitochondria and initiation of apoptosis (71). While loss of BNIP3 in both types of cancers above led to accumulation of dysfunctional mitochondria, the end results differed drastically with survival and a metabolic shift to aerobic glycolysis in mammary tumor cells while activating cell death in colorectal cancer cells. Thus the function of BNIP3-mediated mitophagy in cancer cells might be dependent on the type of cell.
D. Mitophagy in Skeletal Muscle
1. Mitophagy in satellite cells and muscle regeneration
In contrast to brain and heart, skeletal muscle has the ability to regenerate after damage. Skeletal muscle contains a unique population of resident stem cell, also known as satellite cells, that reside between the basal lamina and the sarcolemma (20). Satellite cells exist in a quiescence state under normal conditions, but are rapidly activated in response to tissue damage. The activated cells proliferate and differentiate to form new myofibers. Recent studies have discovered that the number and function of the satellite cells in skeletal muscles decline with age or in muscular disorders (28, 72, 279), and this is due, at least in part, to reduced autophagic activity (72, 83). Similar to HSCs (103), satellite cells maintain their quiescent state through autophagy (83). A study compared the transcriptomes of quiescent satellite cells to activated cells and identified autophagy to be the most predominant pathway in the quiescent state. The authors also found that autophagic activity and mitophagy were reduced in aged satellite cells which led to accumulation of dysfunctional mitochondria and senescence of cells. Restoring autophagic activity in the old satellite cells pharmacologically or genetically restored mitophagy and rescued their regenerative capacity, while disrupting autophagy led to loss of the satellite cell and reduced muscle regeneration (83). This suggests that basal autophagy and mitophagy are required for maintenance of the adult quiescent stem-cell population.
2. Mitophagy in exercise
High-intensity exercise increases the demand for ATP, and to sustain muscle contraction the cell must generate more ATP, in part by enhancing mitochondrial respiration. ROS generation is a byproduct of oxidative phosphorylation, and intensive exercise is associated with mitochondrial oxidative stress and damage (52). During the recovery from exercise, there is activation of mitophagy to selectively degrade damaged mitochondria in the muscle cells. Studies in mice with muscle-specific disruption of autophagy have confirmed the importance of autophagy in removing mitochondria that are damaged during exercise (171). Mitophagy prevents the accumulation of dysfunctional mitochondria during damaging muscle contraction. Surprisingly, selective disruption of autophagy in skeletal muscle did not affect physical performance where no significant differences in exercise capacity between muscle specific Atg7f/f and systemic Atg7−/− mice were observed (171). Instead, lack of autophagy in the skeletal muscle led to accumulation of dysfunctional mitochondria in response to eccentric contraction, confirming an essential role for autophagy during muscle repair post-exercise. Intriguingly, this suggests that exercise allows for selective elimination of weak mitochondria that could potentially become harmful to the cell. One of the beneficial effects of exercise is to ensure a population of young functional mitochondria in muscle cells.
The mechanisms underlying exercise-induced mitophagy have yet to be fully elucidated. The AMP-activated protein kinase (AMPK)-Ulk1 signaling pathway has been implicated in activating mitophagy in skeletal muscle during the recovery after acute exercise (25, 152). Whether the AMPK-Ulk1 pathway promotes clearance of mitochondria via the PINK1/Parkin or the mitophagy receptor pathways is still unclear. Studies have reported that PINK1/Parkin transcripts and protein levels are increased (250) or unchanged (117, 152) in response to exercise. However, this variability is likely due to type and duration of exercise as well as species examined. Alternatively, studies have reported that exercise is associated with upregulation of BNIP3 in skeletal muscle (120, 167, 341).
E. Mitophagy in Lung Injury
The lung is a highly complex organ and composed of over 40 different cell types. Several of the cell types have high energy demand and are therefore highly enriched in mitochondria. These include bronchial epithelial cells that are involved in moving particles out of the lungs through movements of their cilia, alveolar macrophages that are in charge of removing particles from the respiratory surface, and the bronchial and vascular smooth muscle cells that contract and relax the lungs (64). Lung cells are constantly exposed to insults such as particles and pollutants in the air, as well as bacteria and viruses. Thus both autophagy and mitophagy function as important defense systems to ensure cell survival in this stressful environment. A defect in these pathways contributes to increased susceptibility to lung injury and respiratory diseases. Mitochondrial dysfunction is a common pathological feature in the development of both acute and chronic lung disease (44). Changes in mitophagy have been observed in many different lung pathologies, including acute lung injury, chronic obstructive pulmonary disease, and pulmonary fibrosis. However, whether mitophagy is increased or decreased and whether the altered activity is protective or detrimental appear to depend on the cell type and the insult or disease.
1. Acute lung injury
Acute lung injury can be caused by exposure to environmental toxins, infection, or sepsis. It is characterized by severe inflammation and loss of epithelial cells which leads to alveolar-capillary barrier breakdown and respiratory failure. Mitophagy has been reported to be protective in acute lung injury (175, 286, 343). For instance, patients with acute respiratory failure are administered high levels of oxygen. However, oxygen treatment has a narrow therapeutic range and hyperoxia can induce lung injury and increase the mortality in these patients (74). When investigating signaling pathways that are altered during hyperoxia in the lungs, it was discovered that activation of mitophagy functions as an important protective mechanism in this setting. PINK1 expression was increased in lungs in mice exposed to hyperoxia, and PINK1-deficient mice were more susceptible to hyperoxia than wild-type mice (343). Lung endothelial-targeted PINK1 knockdown increased lung permeability and reduced survival, while lung-targeted PINK1 overexpression decreased susceptibility to hyperoxia in vivo. These findings suggest a protective role for enhanced PINK1-mediated mitophagy in hyperoxia-induced lung injury. In another study using a model of acute lung injury involving pulmonary Staphylococcus aureus infection, the infection led to simultaneous activation of PINK1/Parkin-mediated mitophagy and mitochondrial biogenesis in the alveolar region of the lung (286). Specifically, this study noted that although the infection led to substantial mitochondrial damage in the alveolar region, the surviving epithelial cells had increased mitophagy and mitochondrial biogenesis. Furthermore, sepsis is a systemic inflammatory response that causes mitochondrial damage and acute lung injury and increased mitophagy functions to reduce the susceptibility to sepsis-induced lung injury (175). Overall, these studies confirm the protective role of mitophagy in acute lung injury.
2. Chronic obstructive pulmonary disease
The role of mitophagy in chronic lung disorders, such as chronic obstructive pulmonary disease (COPD), is less clear. Cigarette smoke is known to be a major underlying cause of COPD, and acute exposure to cigarette smoke extracts leads to mitochondrial dysfunction and induction of mitophagy in cells (115, 193). However, how the activation of mitophagy affects lung function and its role in the development of COPD is currently controversial. One study compared protein levels of mitophagy activators in lung tissue from nonsmokers, smokers, and patients diagnosed with COPD. They found that Parkin was increased in lung homogenates from smokers but significantly decreased in lungs of COPD patients compared with nonsmokers. This suggests that mitophagy is activated in lungs of smokers but reduced with the development of COPD (115). In addition, this study found that a key function of PINK1/Parkin-mediated mitophagy in epithelial cells was to prevent epithelial cells from becoming senescent. Senescence of alveolar and airway epithelial cells has been implicated in COPD because it prevents these cells from replacing injured/dead cells through cell division (150). Thus insufficient mitophagy in lungs of chronic smokers might be a contributor of COPD development. In contrast, Mizumura et al. (193) reported that activation of mitophagy by cigarette smoke directly contributed to the pathogenesis of COPD by activating necroptosis in epithelial cells. Necroptosis is a regulated form of necrotic cell death (303), but how mitophagy functions as an upstream regulator of necroptosis remains to be elucidated. Both of these studies used human bronchial epithelial cells (BEAS-2B) and cigarette smoke extracts (CSE) in their studies. The main difference between these studies is the concentration of CSE used in the experiments. Ito et al. (115) used 1% CSE in their senescence study, while Mizumura et al. (193) used 20% CSE in their studies on necroptosis. Thus it is clear that the differences in the experimental design accounts for the different outcomes and that further studies are needed to understand the dose-dependent effects on CSE on various cellular processes.
3. Idiopathic pulmonary fibrosis
Idiopathic pulmonary fibrosis (IPF) is a progressive chronic lung disease, and it has been proposed that repeated small injuries to the alveolar epithelial cells lead to inflammation and recruitment of fibroblasts to the site of injury (320). The fibroblasts differentiate into myofibroblasts that are responsible for generating the excessive extracellular matrix leading to pulmonary fibrosis. Altered mitophagy in the lung in IPF has been implicated in the development of lung fibrosis in IPF, but whether it is protective or detrimental appears to be cell dependent. For instance, alveolar macrophages play a key role in the pathogenesis of pulmonary fibrosis by initiating the immune response. Larson-Casey et al. (154) found that alveolar macrophages from IPF patients had increased mitophagy which correlated with their resistance to apoptosis during fibrosis. They also found that mitophagy was required for macrophage expression of transforming growth factor (TGF)-β1 expression which is responsible for fibroblast differentiation. These findings were confirmed in vivo where bleomycin injury in wild-type mice led to increased PINK1/Parkin-mediated mitophagy in alveolar macrophages. Parkin−/− mice were resistant to the bleomycin, and Parkin−/−macrophages had increased apoptosis (154). These findings suggest that macrophage mitophagy is critical for the pathogenesis of fibrosis.
In contrast, Kobayashi et al. (137) observed reduced mitophagy in fibroblasts during the development of lung fibrosis in IPF. ROS play an important role in regulating the differentiation of fibroblasts into myofibroblasts and PINK1/Parkin-mediated mitophagy eliminated ROS producing mitochondria and prevented the differentiation of the fibroblasts. This study also found reduced levels of Parkin in fibroblasts isolated from IPF patients’ lungs which correlated with accumulation of p62/SQSTM1 and ubiquitinated proteins (137), an indication of insufficient mitophagy. To further elucidate the functional consequence of impaired mitophagy in lungs, the authors also utilized the bleomycin-induced lung fibrosis models in Parkin-deficient mice. In contrast to the study by Larson-Casey et al. (154), this group found that bleomycin-treated Parkin knockout mice had enhanced lung fibrosis development compared with wild-type mice (137). The underlying reasons for these two conflicting findings are currently unclear. The two groups used the same Parkin-deficient mouse model (220), used same dose and mode of administration of bleomycin (2 U/kg), and evaluated lung fibrosis at similar time points (20 vs. 21 days). This also demonstrates the need to develop cell specific Parkin null mice to study its true role in specific cell types in various diseases.
Finally, Patel et al. (224) reported that PINK1 was increased in lung tissue from IPF patients. TGF-β stimulation increases expression of a number of genes involved in mitophagy including BECN1 and Atg5 (288), and studies on the cellular levels confirmed that TGF-β1 treatment led to increased expression of PINK1 in alveolar epithelial cells. They also found that PINK1 deficiency exacerbates bleomycin-induced lung fibrosis. Type II alveolar epithelial cells were also more susceptible to TGF-β1-induced apoptosis. However, although PINK1 levels are increased, insufficient formation of autophagosomes observed in IPF will lead to reduced degradation of mitochondria (6). It is also important to note that this study found that PINK1−/− mice were more susceptible to bleomycin-induced fibrosis in lungs, suggesting a protective role for mitophagy. In contrast to the other two conflicting studies, this study used a higher dose of bleomycin (3 U/kg).
F. Mitophagy in Liver Homeostasis and Injury
Since its initial discovery in hepatocytes, mitophagy has been confirmed to be important for maintaining mitochondrial homeostasis in liver cells under baseline conditions. For instance, mice deficient in the mitophagy protein BNIP3 have increased mitochondrial mass in the liver, suggesting that there is an imbalance in mitochondrial turnover in the absence of BNIP3 (87). Mitophagy also protects against various liver pathologies such as alcoholic liver disease and acetaminophen overdose.
Alcoholic liver disease and acetaminophen overdose are two common causes of severe liver disease and liver failure. Both binge and chronic alcohol consumption can cause damage to liver. Mitochondrial damage, ROS generation, and steatosis are characteristics of both acute and chronic alcohol exposure (204). Parkin is highly expressed in liver, and Parkin null mice have greater alcohol-induced mitochondrial damage, steatosis, and liver injury compared with wild type (318). Interestingly, mitophagy was still induced in livers of Parkin-null mice after alcohol exposure, but to a significantly reduced extent (318). This confirms the presence of other mitophagy mechanisms that can compensate for the lack of Parkin, but the lower level of mitophagy is insufficient to completely protect the liver against stressors such as alcohol.
The liver is responsible for metabolizing and detoxifying many drugs, which also makes the liver cells susceptible to damage by drugs. Acetaminophen overdose is a major cause of acute liver failure and the hepatotoxicity is caused by reactive acetaminophen metabolites that can target mitochondrial components. This leads to mitochondrial dysfunction, production of ROS, and opening of the mitochondrial permeability transition pore. Pharmacological activation of autophagy with rapamycin protected against acetaminophen-induced liver injury while suppression of autophagy using chloroquine exacerbated the liver injury (208). Similarly, acute knockdown of Parkin in vivo enhanced liver injury (318), suggesting that autophagy and mitophagy are protecting against hepatotoxicity in this setting. Intriguingly, acetaminophen exposure produced the opposite result in global Parkin−/− mice and hepatocyte-specific Atg5 null mice, which were resistant to acetaminophen hepatotoxicity (208, 318). It was discovered that the resistance of the knockout mice was due to activation compensatory pathways. For instance, Parkin has numerous substrates and some of them are not regulators of mitophagy, and it was found that increased levels of the pro-survival protein MCL-1 in the mitochondria provided protection in hepatocytes in Parkin−/− mice. MCL-1 is a Parkin substrate and subject to Parkin-mediated degradation (27). These findings also suggest that caution must be used when studying susceptibility to injury in chronic knockout mice.
G. Mitophagy in Innate Immunity
Mitochondria play a key role in innate immune cell function and fate (188). In response to injury or infection, damaged cells release factors that are detected by immune cells which will then initiate the inflammatory response. At baseline conditions, cells of the immune system are relatively quiescent, but upon initiation of inflammation, they switch to an activated state which involves increased mitochondrial content and enhanced respiration. Therefore, a healthy mitochondrial population is essential for immune system homeostasis, and disrupting autophagy in these cells leads to accumulation of damaged mitochondria and reduced survival (229, 319).
Moreover, given the bacterial origin of mitochondria, it is not surprising that xenophagy, a selective form of autophagy used to eliminate invading bacteria, shares a common regulatory mechanism with mitophagy. It has been reported that genetic polymorphisms in Parkin is associated with increased susceptibility to bacterial infections in humans (4, 190), and Parkin-deficient mice are more susceptible to Mycobacterium tuberculosis infection (176). Also, Manzanillo et al. (176) found that Parkin played an important role in the innate defense against M. tuberculosis by promoting xenophagy. Specifically, Parkin was recruited to M. tuberculosis-containing phagosomes where it facilitated linkage of ubiquitin chains to label them for autophagy. Consequently, lack of Parkin led to impaired bacterial elimination.
Several studies have reported a link between autophagy, mitophagy, and inflammasome activation. The NLRP3 inflammasome is the most well-known intracellular pattern recognition receptor, and it is activated by dysfunctional mitochondria (198, 345). NLRP3 senses excessive mitochondrial ROS and mtDNA damage and localizes to these mitochondria where it promotes the release of mtDNA into the cytosol. The activated inflammasome then proceeds to initiate an inflammatory response via caspase-1 activation, resulting in the maturation and secretion of pro-inflammatory cytokine interleukin (IL)-1β and IL-18 (188). When mitophagy is diminished, the accumulation of dysfunctional mitochondria can lead to excessive NLRP3 inflammasome activation and chronic inflammation (198, 345). For instance, autophagy-deficient macrophages accumulate dysfunctional mitochondria which leads to activation of the NLRP3 inflammasome and release of mtDNA into the cytosol in response to lipopolysaccharide and ATP (345). Moreover, Kim et al. (131) demonstrated that induction of mitophagy in macrophages suppressed prolonged NLRP3 inflammasome activation, while impaired mitophagy led to excessive inflammasome activation and increased mortality in sepsis. Finally, the mitophagy adaptor p62/SQSTM1 functions to prevent excessive inflammasome activation by promoting mitophagy of damaged mitochondria that can cause NLRP3-inflammasome activation (344). Overall, these studies support the notion that mitophagy functions to limit NLRP3 inflammasome activation and the inflammatory response and that a defect in mitophagy can lead to chronic inflammation.
V. MITOPHAGY AND THE AUTOPHAGY MACHINERY
Our knowledge of mitophagy and its regulators has increased tremendously in the past decade, and mitophagy has clearly emerged to be a more complex process than previously thought. It is clear that mitophagy plays a role in regulating multiple physiological processes ranging from metabolic remodeling and mitochondrial mass. Here, we provide an overview of the key proteins involved in regulating mitophagy (TABLE 1).
Table 1.
Key mitophagy regulators
| Gene | Protein | Molecular Function | Physiological Functions and Disease Links | Reference Nos. |
|---|---|---|---|---|
| Park6 | PINK1 | Mitochondrial serine/threonine protein kinase. Phosphorylates ubiquitin, Mfn2, and Parkin to initiate mitophagy. | Recruits Parkin to mitochondria. Loss-of-function mutations contribute to familial Parkinson's disease. | 33, 42, 118, 122, 143, 155, 181, 187, 203, 215, 223, 300, 306 |
| Park2 | Parkin | Cytosolic E3 ubiquitin ligase. Ubiquitinates proteins to label them for proteasomal or autophagic degradation. | Labels mitochondria for mitophagic degradation. Induces mitophagy during metabolic remodeling and in response to stress in myocardium. Involved in mitophagy of paternal mitochondria in mouse embryos. Loss-of-function mutations contribute to familial Parkinson's disease. | 33, 84, 90, 92, 134, 147, 181, 202, 240, 261, 276 |
| Mul1 | Mul1 (Mulan, Mapl) | Mitochondrial E3 ubiquitin ligase. | Participates in the elimination of paternal mitochondria in mouse embryos. | 240 |
| Atg32 | Atg32 | Mitophagy receptor in yeast. Interacts with Atg8 and Atg11. | Involved in selective removal of mitochondria in yeast. | 5, 123, 140, 213 |
| Bnip3 | BNIP3 | Mitophagy receptor in the OMM that interacts directly with LC3B. | Activates mitophagy during hypoxia or increased oxidative stress. Regulates mitochondrial content in tissues. Loss of Bnip3 can promote tumor metastasis. | 9, 39, 60, 87, 99, 235, 237 |
| Bnip3L | BNIP3L/Nix | Bnip3 homologue and mitophagy receptor. Interacts preferentially with GABARAP. | Programmed mitophagy and regulation of mitochondrial mass in tissues. Involved in clearance of mitochondria in erythrocyte differentiation and in reprogramming of somatic cells. Can compensate for Parkin deficiency. | 55, 60, 69, 139, 211, 244, 257 |
| Fundc1 | FUNDC1 | Mitophagy receptor that interacts with LC3. Regulated by MARCH5, PGAM5, and Ulk1. | Involved in hypoxia-mediated mitophagy. | 31, 37, 145, 169, 323 |
| Phb2 | Prohibitin2 | Mitophagy receptor in the inner mitochondrial membrane. | Involved in removal of paternal mitochondria. | 314 |
| Becn1 | Beclin1 | Scaffolding protein involved in regulating autophagy and mitophagy. | Facilitates translocation of Parkin from cytosol to mitochondria and ensures formation of autophagosomes at MAMs via its interaction with PINK1. | 38, 86 |
| Gapdh | GAPDH | Regulates microautophagy of mitochondria. Catalytically inactive GAPDH associates with damaged mitochondria and promotes their engulfment directly into lysosomes for degradation. | Involved in eliminating damaged mitochondria after exposure to I/R. Impaired GAPDH-induced micromitophagy contributes to the pathology of Huntington disease. | 111, 335 |
| Ambra1 | Ambra1 | Positive regulator of autophagy. Can function as a mitophagy inducer at the mitochondria. Localizes to mitochondria by interacting with Bcl-2. Contains a LIR and interacts with LC3. | Promote Parkin independent mitophagy but can also augment Parkin-mediated mitophagy. | 73, 283, 301 |
| Fkbp8 | FKBP8 | Mitophagy receptor localized to the OMM. Preferentially interacts with LC3A via its LIR motif. | Promotes mitophagy of damaged mitochondria in a Parkin-independent manner. Escapes from the mitochondria to the endoplasmic reticulum after recruitment of LC3A to avoid degradation. | 17 |
| Bcl2l13 | BCL2L13 | Mammalian homologue of Atg32. Contains a LIR and interacts with LC3. | Promotes mitophagy of damaged mitochondria in a Parkin-independent manner. | 195 |
| N/A | Cardiolipin | Lipid unique to mitochondria that is localized in the inner membrane. Interacts with LC3. | Redistributes to the outer membrane of damaged mitochondria where it interacts with LC3 to promote mitophagy. | 40 |
| Sqstm1 | p62/SQSTM1 | Autophagy adaptor. Binds polyubiquitinated proteins to facilitate their removal by autophagy. | Recruited to damaged ubiquitinated mitochondria. Promotes perinuclear aggregation of mitochondria which may allow for more efficient mitophagy. | 85, 156, 201, 214 |
| Optn | OPTN/Optineurin | Autophagy adaptor for Parkin-mediated mitophagy. Binds both ubiquitinated targets on the mitochondria via its UBD and to LC3 on the phagophore via its LIR. | Recruited to damaged ubiquitinated mitochondria. Facilitates their removal by anchoring them to the autophagosome. Mutations have been linked to ALS. | 102, 156, 179, 236, 317 |
| Calcoco2 | NDP52 (nuclear dot protein 52) | Autophagy adaptor for Parkin-mediated mitophagy. Binds both ubiquitinated targets on the mitochondria via its UBD and to LC3 on the phagophore via its LIR. | Recruited to damaged ubiquitinated mitochondria. Facilitates their removal by anchoring them to the autophagosome. | 102, 156 |
| Tax1bp1 | TAX1BP1 | Autophagy adaptor | Recruited to damaged ubiquitinated mitochondria. Facilitates their removal by anchoring them to the autophagosome. | 156 |
I/R, ischemia/reperfusion; GAPDH, glyceraldehyde-3-phosphate dehydrogenase; ALS, amyotrophic lateral sclerosis.
A. Induction of Autophagy
The different stages of autophagy can be divided into five events: induction, nucleation, expansion, fusion, and degradation, orchestrated by the autophagy-related gene (Atg) proteins (FIGURE 5). To date, more than 40 Atg proteins have been identified in yeast, and many of these are conserved in mammalian cells. The Atg proteins form numerous different complexes that are sequentially engaged to regulate those events: 1) the Atg1/Unc-51 like autophagy activating kinase 1 (Ulk1) complex (consisting of Ulk1/2, Atg13, and Fip200); 2) the class III phosphatidylinositol 3-kinase (PI3K) complex composed of VPS34 (Vps34 in yeast), p150 (Vps15 in yeast), and Beclin 1 (Atg6 in yeast); 3) the Atg12-Atg5 conjugation system; 4) the Atg8/LC3 conjugation system; and 5) the Atg9 cycling system (135, 218).
FIGURE 5.

Formation of autophagosomes. Activation of the Ulk1/2-Atg13-Fip200 complex by AMP-activated protein kinase (AMPK) initiates the formation of the phagophore through activation of the class III phosphatidylinositol 3-kinase (PI3K) complex composed of Vps34-Vps15-Beclin1-Atg14L. Atg9-positive vesicles contribute membrane to the growing phagophore. The subsequent elongation and closure of the membrane are mediated by two ubiquitin-like conjugation pathways: the Atg5–Atg12 and the microtubule-associated protein 1 light chain 3 (LC3) pathways. 1) The Atg12-Atg5 conjugating system consists of Atg12 (ubiquitin-like module), Atg5 (substrate), Atg7 (E1-like enzyme), and Atg10 (E2-like enzyme). The Atg12–Atg5 conjugate is formed by Atg7 and Atg10. Atg12–Atg5 forms a final complex with Atg16 to form the E3 enzyme for the LC3/Atg8 conjugating system. 2) The LC3 (Atg8) conjugating system consists of LC3 (ubiquitin-like module), Atg4 (a cysteine protease), phosphatidylethanolamine (PE, substrate), Atg7 (E1-like enzyme), Atg3 (E2-like enzyme), and Atg5–Atg12–Atg16 (E3-like enzyme).
The initiating event for autophagy is induction or nucleation of the membrane that will become an autophagosome, followed by membrane expansion to form the double-membrane phagophore. Initiation begins with the activation of the Atg1/Ulk1 kinase complex which subsequently recruits the PI3K complex (Vps34-Vps15-Beclin 1) to the phagophore assembly site (PAS). The PI3K complex begins a nucleation process that will recruit additional Atg proteins involved in the expansion of the phagophore. Specifically, the complex is responsible for the production of phosphatidylinositol-3-phosphate (114, 130), which is important for localization of some Atg proteins, including Atg18 and Atg2, enabling recruitment of LC3/Atg8, Atg9, and Atg12 to the PAS. Elongation of the phagophore membrane requires two ubiquitin-like conjugation pathways: 1) the Atg5-Atg12-Atg16 complex and 2) conjugation of phosphatidylethanolamine (PE) to LC3/Atg8. Atg12 is conjugated to Atg5 via the action of the E1 and E2 enzymes Atg7 and Atg10, and then Atg5-Atg12 conjugate binds Atg16 to form the Atg12-Atg5-Atg16 complex (7). The Atg12-Atg5-Atg16 complex forms an E3-like ligase complex that conjugates Atg8/LC3 to PE. PE-conjugated Atg8/LC3 is required both for expansion of autophagic membranes and for recognition of cargo for the autophagosomes (7). In mammalian cells, there are six Atg8 orthologs categorized as members of either the LC3 (LC3A, LC3B, LC3C) or GABARAP (GABARAP, GABARAPL1, GABARAPL2) subfamilies (315). As the only transmembrane protein of the autophagy core machinery, Atg9 is localized to various membrane structures and is required for expansion of the phagophore. Atg9 cycles between the PAS and various sites that provide donor membrane for the growing phagophore (218, 234).
B. Beclin 1
Beclin 1 is a well-known autophagy protein and regulates autophagosome nucleation and maturation (161). Beclin 1 functions as a scaffolding protein and forms distinct complexes to regulate these steps in the autophagy pathway. Because of its role in regulating autophagy, defects in Beclin 1 also impact mitophagy. However, recent studies have identified a more direct role for Beclin 1 in regulating PINK1/Parkin-mediated mitophagy. Choubey et al. (38) reported that Beclin 1 interacts with Parkin in the cytosol and regulates its translocation to depolarized mitochondria. Interestingly, the interaction between Beclin 1 and Parkin is increased upon activation of mitophagy, suggesting that Beclin 1 might facilitate the translocation of Parkin to mitochondria. More recently, it was reported that Beclin 1 is responsible for initiating formation of the autophagosome at the mitochondria during mitophagy (86). Mitochondria are known to form contact sites with the ER via mitochondria-associated membranes (MAM). MAM are part of the ER that can be reversibly tethered to mitochondria (15). The autophagic machinery assembles at these sites to initiate formation of autophagosomes (95). Specifically, Gelmetti et al. (86) demonstrated that Beclin 1 accumulates at MAMs upon mitochondrial depolarization where it interacts with PINK1 to initiate formation of the autophagosome. The presence of PINK1, but not Parkin, was required for both Beclin 1 localization to MAMs and autophagosome formation during mitophagy. Overall, these studies suggest that Beclin 1 can regulate mitophagy at two distinct steps: 1) facilitates translocation of Parkin from cytosol to mitochondria and 2) ensures formation of autophagosomes at MAMs via its interaction with PINK1.
C. AMPK: Upstream Activator of Mitophagy
Autophagy activation in the face of bioenergetic stress is directed by the AMPK, a “master metabolic regulator” that orchestrates cellular energy sensing and activation of general autophagy under conditions of compromised cellular energy status (280). It is increasingly evident that, in addition to autophagy initiation, AMPK also has a central role in sensing mitochondrial damage and promoting mitophagy (65, 165).
Two distinct mechanisms by which AMPK directly regulates mitophagy have been identified to date. First, AMPK can activate mitophagy via Ulk1 (65, 292). The importance of Ulk1 in mitophagy was uncovered after it was observed that Ulk1-deficient reticulocytes exhibit delayed mitochondrial clearance during red blood cell development (104, 151), suggesting a specific role in mitochondrial clearance. The central mitophagic function of Ulk1 was determined to phosphorylate mitochondrial Fundc1, a mitophagy receptor, leading to enhanced interaction between Fundc1 and autophagosomal LC3 (323). The direct connection between AMPK and Ulk1 in the selective activation of mitophagy was first demonstrated by Egan et al. (65), who identified Ulk1 as an AMPK substrate and revealed that AMPK-mediated phosphorylation of Ulk1 was required for mitophagy during energy stress. The functional importance of AMPK-mediated phosphorylation of Ulk1 was further shown because overexpression of wild-type Ulk1, but not a phosphorylation-resistant Ulk1 mutant, restored mitochondrial content in Ulk1−/− cells. Subsequent studies have linked the phosphorylation of Ulk1 at Ser-555 by AMPK with the translocation of Ulk1 to mitochondria: mutating Ser-555 in Ulk1 to alanine (S555A) abolished the translocation of ULK1 to mitochondria under hypoxia (292). In addition, deletion of Ulk1 hampered induction of mitophagy induction during hypoxia, while overexpression of the phosphomimetic Ulk1 S555D rescued mitophagy induction in Ulk1−/− MEFs.
A second mechanism by which AMPK can regulate mitophagy is to promote assembly of the autophagy machinery directly at the damaged mitochondria. AMPK specifically associates with damaged mitochondria where it recruits the Atg16 complex, in an elegant mechanism that evokes directed expansion of the autophagosome membrane by the particular cargo to be degraded (165). The association of AMPK with mitochondria and autophagic machinery depends upon N-myristoylation of AMPKβ, which mediates membrane association (212). In contrast, nonselective autophagy can occur independently of AMPK N-myristoylation (165). It is obvious that assembly of the autophagosome directly at the dysfunctional mitochondrion via this AMPK/Atg16 mechanism is a key factor that maintains an efficient mitochondrial quality control apparatus.
D. ROS: Positive Regulators of Mitophagy
While ROS have long been known for their damage-promoting effects, it is now clear ROS also function as important signaling molecules and regulate key processes such as mitochondrial biogenesis (258). Interestingly, recent studies have shown that mitochondrial ROS can also activate mitophagy. This was first reported by Song et al. (273) who discovered a differential effect of low versus high overexpression of a mitochondrial-targeted catalase (mito-CAT) on mitochondrial dysfunction induced by loss of Mfn2 in mouse hearts. This study discovered that transgenic mice with low overexpression of mito-CAT were protected against Mfn2 deletion in myocytes by neutralizing excess mitochondrial ROS production. Unexpectedly, transgenic mice with high overexpression of mito-CAT produced the opposite results and further exacerbated the cardiomyopathy in Mfn2-deficient hearts. The authors found that the complete suppression of mitochondrial ROS in hearts with high overexpression of mito-CAT led to abrogation of mitophagy. Moreover, it is known that the thyroid hormone triiodothyronine (T3) increases oxidative phosphorylation in cells, which also leads to enhanced production of ROS that could potentially damage mitochondria. However, T3-mediated increases in mitochondrial ROS also led to activation of mitophagy via the AMPK/Ulk1 pathway (270). Disruption of Ulk1-dependent mitophagy led to accumulation of dysfunctional mitochondria in response to T3 treatment, confirming that the concurrent increase in mitochondrial respiration and mitophagy by T3 maintains a healthy population of actively respiring mitochondria.
VI. PROMOTERS OF MITOPHAGY
A. Ubiquitin and Mitophagy Adaptor Proteins
Ubiquitin is central to PINK1/Parkin-mediated mitophagy because specific linkages on mitochondrial proteins direct mitochondria for mitophagic elimination via adaptor proteins. Several adaptor proteins have been identified as participating in mitophagy, including p62/SQSTM1 (85, 201, 214), NDP52 (nuclear dot protein 52 kDa) (102, 156), Optineurin (OPTN) (102, 156, 321), and TAX1BP1 (156, 321). Multiple autophagy adaptor proteins are recruited to the ubiquitinated mitochondria which ensures their rapid removal.
Early studies suggested that p62/SQSTM1 was responsible for directing dysfunctional mitochondria to the autophagosome (85), but subsequent investigations revealed that the main function of p62/SQSTM1 is in the aggregation or clustering of damaged mitochondria, thus allowing for more efficient mitophagy (201, 214). Okatsu et al. (214) demonstrated that deletion of p62/SQSTM1 in mouse embryonic fibroblasts did not prevent degradation of mitochondria, but instead resulted in a loss of mitochondrial perinuclear clustering. Similarly, Narendra et al. (201) reported that genetic deletion or siRNA depletion of p62/SQSTM1 abrogated aggregation without inhibiting Parkin-induced mitophagy. Instead, recent studies suggest that OPTN and NDP52 are the primary autophagy adaptors for Parkin-mediated mitophagy (156, 321).
Autophagy adaptor proteins can undergo Tank-binding kinase 1 (TBK1)-mediated phosphorylation which increases their affinity for ubiquitin and LC3 (183, 236, 317). In addition, the GTPase Rab35 reportedly can act downstream of TBK1 in recruiting the autophagy adaptor NDP52 to damaged mitochondria (189). In this study, activated (GTP-bound) Rab35 localized specifically to damaged mitochondria where it recruited NDP52 in a manner dependent on TBK1 activity; GTP-bound Rab35 both directly interacted with NDP52 and facilitated interactions between NDP52 and ubiquitin. The observation that NDP52 failed to translocate to impaired mitochondria in Rab35 knockout cells confirmed a critical role of Rab35 in recruiting NDP52, but it remains unknown if Rab35 is also involved in recruiting other mitophagy adaptors to damaged mitochondria.
B. Atg32
Atg32 is a 60-kDa protein anchored in the OMM via a COOH-terminal transmembrane domain (5, 140) and functions as a mitophagy receptor in yeast. The NH2-terminal cytosolic region of Atg32 contains a WXXL-like motif required both for binding to Atg8 and for mitophagy to proceed. Interestingly, when this NH2-terminal region with the WXXL motif is artificially anchored on peroxisomes, the peroxisomes are degraded by autophagy (140), suggesting that the cytosolic portion of Atg32 is sufficient to function as an autophagy/mitophagy receptor. Further investigation into molecular mechanisms underlying the regulation of mitophagy uncovered that phosphorylation serves as an activator of mitophagy. Specifically, phosphorylation of Ser-114 on Atg32 acts as an initial molecular switch to activate selective mitochondrial degradation (5).
There are still some important questions in yeast cells that remain to be answered. Although it is clear that dysfunctional mitochondria are selectively degraded by mitophagy, the physiological importance of mitophagy in yeast is still unclear. Mitophagy selectively degrades mitochondria in yeast, but there is no experimental evidence that mitophagy selects damaged mitochondria for degradation. Also, no defects in mitochondria have been identified in Atg32-null yeast. Thus the precise role of Atg32 in regulating mitochondrial quality and/or quantity remains unknown. Indeed, one of the most vexing unanswered questions about mitophagy is whether mitophagy can select damaged mitochondria as a cargo in yeast. There is no doubt that mitophagy selectively degrades mitochondria in these cells, but identification of how mitophagy distinguishes damaged from healthy mitochondria in yeast has yet to be established.
C. BNIP3 and BNIP3L/Nix
BNIP3 (BCL2 and adenovirus E1B 19-kDa interacting protein 3) and its homologue BNIP3-like (BNIP3L, also known as Nix) were initially identified as mitochondria-localized pro-death proteins and classified as atypical members of the BH3-only family (30). Early studies on BNIP3 reported that cell death was mediated via opening of the mitochondrial permeability transition pore (235, 302) and via activation of Bax/Bak (146). Similarly, Nix was reported to activate both apoptotic and necrotic cell death (34, 55, 57). Additional in vivo studies demonstrated that BNIP3 promoted cell death in the heart in response to ischemic injury (56, 235) and doxorubicin exposure (51). Nix activated cell death in myocytes during hemodynamic pressure overload which led to development of heart failure (58, 339). Interestingly, it was also observed that BNIP3 and Nix overexpression in cells was always associated with formation of autophagosomes (12, 94, 302). It is now well established that these proteins function as mitophagy receptors (94, 211, 237, 256). They are both anchored in the outer mitochondrial membrane via a transmembrane domain in the COOH terminus (207). Their cytosolic NH2 terminus contains a LIR motif which facilitates the interaction with LC3/GAPBARAP on the phagophore membrane, in a manner similar to Atg32 in yeast. There seems to be specificity in the Atg8 family protein that they preferentially interact with. Thus Nix preferentially binds to GABARAP and LC3A (211, 256) while BNIP3 binds to LC3B (99), although the mechanistic underpinnings for why these mitophagy receptors interact preferentially with different Atg8-family proteins is still unknown.
Many in vitro and in vivo studies have now confirmed that, in addition to promoting programmed cell death, BNIP3 and Nix also function to promote mitophagy. For instance, genetic deletion of BNIP3 leads to increased mitochondrial mass in mouse livers (87), and lack of both Nix and BNIP3 leads to accelerated accumulation of abnormal mitochondria in mouse hearts (60). This suggests that lack of BNIP3/Nix-mediated mitophagy leads to impaired mitochondrial turnover in these tissues. In addition, BNIP3 has been observed to activate mitophagy during hypoxia (9, 39, 113), while Nix also functions in programmed mitophagy (69, 153, 244, 325). Further evidence that they can function as dual regulators of mitophagy and programmed cell death in the same cell was provided by studies in erythrocyte development. During early stages of erythropoiesis, Nix regulates the number of differentiating erythrocytes by inducing apoptosis (55). However, in the terminal stages of erythrocyte maturation, Nix-mediated mitophagy becomes critical in eliminating mitochondria (211, 244). As dual regulators of two distinct processes, BNIP3 and Nix coordinate the mitochondrial quality control and cell death pathways depending on the context. Thus it is likely that the main objective of BNIP3/Nix-mediated mitophagy is to maintain a healthy population of mitochondria in cells by rapidly eliminating any defective mitochondria. However, chronic or excessive stress can lead to extensive mitochondrial damage which exceeds the capacity of mitophagic elimination. Under these conditions, BNIP3 and Nix will activate programmed cell death to eliminate the cell.
In addition, there is little evidence that, as is the case with Parkin, BNIP3 and Nix play a role in eliminating depolarized mitochondria in cells, allthough there is some evidence of crosstalk where they can enhance Parkin-mediated mitophagy as well as compensate for the lack of functional Parkin. It was reported that BNIP3 did not require PINK1 or Parkin to induce mitophagy, but it enhanced PINK1/Parkin mitophagy in response to carbonyl cyanide m-chlorophenyl hydrazone (CCCP) treatment (342). Interestingly, BNIP3 was found to directly interact with PINK1 which led to accumulation of PINK1 on the outer membrane of mitochondria, increased Parkin recruitment, and enhanced mitophagy (342). Similarly, Nix was found to stimulate Parkin-mediated mitophagy in response to mitochondrial depolarization with CCCP (89). More recently, it was reported that Nix can compensate for a defect in the PINK1/Parkin pathway. Koentjoro et al. (138) identified a carrier with homozygous PARK2 mutation who had not developed PD in her seventies despite the lack of functional Parkin. This suggested the existence of a compensatory mechanism against Parkin deficiency. In a followup study, this group found that fibroblasts derived from this patient had normal mitochondria as demonstrated by normal mitochondrial membrane potential and ATP production, preserved mitochondrial respiration, and intact mitophagy, indicating the presence of a Parkin-independent mitophagy (139). The authors identified Nix to be responsible for maintaining mitochondrial health by mediating mitophagy. Also, Nix overexpression restored mitophagy and improved mitochondrial function in cell lines generated from PD patients with PINK1 or Parkin mutations (139). This suggests that there is crosstalk between the different mitophagy mechanisms.
D. Fundc1
Fundc1 is another mitochondrial resident receptor in mammalian cells, regulated by phosphorylation and mediating hypoxia-induced mitophagy (169). Under normal situations, Fundc1 is phosphorylated by Src kinase and CK2 on Tyr-18 and Ser-13, respectively, which prevents it from interacting with LC3 (31). In response to hypoxia or loss of mitochondrial membrane potential, Fundc1 is dephosphorylated by the mitochondrial protein phosphatase PGAM5 which allows it to interact with LC3 and induce mitophagy (31). The NH2-terminal region containing the LIR is exposed to the cytosol, and overexpression of a chimera consisting of Fundc1 NH2 terminus fused to the mitochondrial transmembrane domain of BCL-XL is sufficient to induce mitophagy during hypoxia (145). Mutational analysis of Ser-13 and Tyr-18 in the chimera confirmed that these residues need to be unphosphorylated for the cytosolic portion of Fundc1 to interact with LC3, indicating that dephosphorylation serves as a molecular switch to turn on Fundc1-mediated mitophagy. Although dephosphorylation of Ser-13 and Tyr-18 is required for the interaction with LC3, another group reported that Fundc1 is also phosphorylated by Ulk1 at Ser-17 upon activation of mitophagy which also enhances its interaction with LC3 (323). It is interesting to note that the Src phosphorylation site is located in Fundc1’s LIR motif (Y18-E19-V20-L21) while the Ulk1 phosphorylation site is located immediately preceding the LIR.
A recent study also reported that the resident mitochondrial E3 ligase MARCH5 is a negative regulator of Fundc1-mediated mitophagy. Fundc1 is a MARCH5 substrate, and MARCH5 mediates ubiquitination of Fundc1 to promote its proteasomal degradation during hypoxia (37). The authors suggested that MARCH5-mediated degradation of Fundc1 reduces mitophagy and prevents unnecessary degradation of mitochondria during acute hypoxia. However, prolonged hypoxia will lead to dephosphorylation of remaining Fundc1 and activation of mitophagy.
E. Others: BCL2L13, FKBP8, Ambra1, and Cardiolipin
Several additional, but less well studied, autophagy receptors have been identified in mammalian cells. These include BCL2L13 (195), FKBP8 (17), and Ambra1 (283). Based on the key characteristics of yeast mitophagy receptor Atg32, WXXL like motifs, mitochondrial localization, and single membrane-spanning topology, Murakawa et al. (195) screened the public protein databases for mammalian homologues. In this screen, they identified BCL2L13 as a putative mammalian Atg32 homologue. They found that BCL2L13 localized to OMM, interacted with LC3B through the conserved LIR motif, and induced mitophagy when overexpressed in cells (195). BCL2L13 also induced mitophagy independent of Parkin and restored mitophagy in Atg32-deficient yeast cells. Similar to BNIP3 and Nix, BCL2L13 can also function as a pro-death protein where overexpression of BCL2L13 triggers caspase-3 activation and cytochrome c release from mitochondria in HEK293T cells (125). However, the molecular mechanism underlying the activation of its pro-mitophagy versus pro-death functions are still unknown.
Moreover, a yeast two-hybrid screen identified FKBP8 as a putative mitophagy receptor (17). FKBP8 is a member of the FK506‐binding protein (FKBP) family and is anchored in the outer mitochondrial membrane (265). FKBP8 contains an NH2‐terminal LIR motif, and it interacts with lipidated LC3A both in vitro and in vivo. FKBP8 efficiently recruited LC3A to damaged mitochondria (17). Interestingly, even when acting as a mitophagy receptor, FKBP8 avoided degradation by escaping from mitochondria to the ER. This has been reported for other proteins as well (243).
Ambra1 (activating molecule in Beclin 1-regulated autophagy) is known to interact with Beclin 1 to positively regulate autophagy (73). Although Ambra1 lacks a mitochondrial targeting motif, it can localize to mitochondria by interacting with proteins in the outer mitochondrial membrane such as Bcl-2 (284). It was initially observed that Ambra1 could augment Parkin-mediated mitophagy in cells (301). Subsequently, Ambra1 was found to both increase Parkin-mediated mitochondrial clearance and promote mitophagy in the absence of Parkin (283). This study also discovered that Ambra1 contains a LIR and directly interacts with LC3 to induce mitophagy independent of Parkin (283). Similarly to overexpression of other mitophagy receptors such as Nix and BNIP3, targeting Ambra1 to mitochondria alone was sufficient to induce mitochondrial depolarization and massive mitophagy in both wild-type and Parkin−/− cells. Interestingly, the authors found that the mitochondria were still ubiquitinated in the absence of Parkin, suggesting that Ambra1 can coordinate with other E3 ubiquitin ligases to label the mitochondria, although the identities of those E3 ubiquitin ligases are unknown. As a mitophagy receptor, Ambra1 is unique because it does not integrate in the OMM like the other receptors.
Finally, cardiolipin is a unique mitochondrial phospholipid in the inner mitochondrial membrane that is critical for mitochondrial respiration and energy metabolism (252). Cardiolipin interacts with various proteins involved in mitochondrial bioenergetic processes, such as subunits in the respiratory chain complexes and mitochondrial substrate carriers. Studies suggest that this interaction with cardiolipin is required for their stabilization and activity (245). More recent studies have uncovered that cardiolipin can also function as a positive regulator of mitophagy (40). Upon mitochondrial injury, cardiolipin is redistributed to the outer mitochondrial membrane (40, 121). Similar to mitophagy receptors, cardiolipin binds directly to both Beclin 1 (107) and to LC3 (40) to facilitate mitophagy.
VII. DEUBIQUITINASES AS NEGATIVE REGULATORS OF MITOPHAGY
Ubiquitin is a signal for mitophagy in the PINK1/Parkin pathway; the balance between ubiquitination and deubiquitination governs the mitophagic activity. Ubiquitination is a reversible posttranslational modification reciprocally regulated by ubiquitin ligases that add ubiquitin to proteins and deubiquitylase enzymes (DUBs) that remove ubiquitin (41). Several DUBs, including USP8, USP15, USP30, and USP35, have been identified as important regulators of Parkin-mediated mitophagy (19, 45, 46, 63). While we have developed a detailed understanding of central roles played by the E3 ubiquitin ligase Parkin and ubiquitin itself in mitophagy, the contributions of DUBs as mitophagy regulators are less well understood.
USP30 is a resident mitochondrial deubiquitinase anchored in the OMM (200). USP30 opposes mitophagy by removing ubiquitin from Parkin substrates such as MIRO1 and TOM20 (19, 313). Overexpression of USP30 reduces ubiquitination of mitochondrial proteins and prevents Parkin-mediated degradation of mitochondria (FIGURE 6A). USP30 can also delay Parkin translocation to depolarized mitochondria, although the responsible mechanisms are unclear. Interestingly, USP30 is also a Parkin substrate, and Parkin-mediated ubiquitination leads to its proteosomal degradation (19), thereby removing this brake on mitophagy.
FIGURE 6.

Deubiquitinating enzymes regulate mitophagy. A: USP30 is anchored in the mitochondrial outer membrane and removes the ubiquitin from Parkin substrates to inhibit mitophagy. B: USP35 counteracts Parkin-mediated mitophagy on healthy mitochondria. USP35 dissociates from depolarized mitochondria to allow mitophagy to proceed. C: Parkin activity is inhibited at baseline via auto-ubiquitination. USP8 preferentially removes ubiquitin conjugates from Parkin. This is required for efficient recruitment of Parkin to depolarized mitochondria.
UPS35 is another DUB localized at healthy polarized mitochondria. In contrast to USP30, USP35 rapidly dissociates from depolarized mitochondria upon induction of mitophagy (FIGURE 6B). Knockdown of USP35 leads to reduced levels of the mitochondrial fusion protein, Mfn2, supporting opposing roles for mitochondrial USP35 and Parkin for maintaining mitochondrial Mfn2 levels (313). Studies in cells have demonstrated that mitochondria undergo fusion during starvation which protects them against mitophagy (89, 233). Thus it is possible that the presence of USP35 can prevent unnecessary degradation of mitochondria, for example, during energy-limiting conditions such as starvation.
Finally, DUBs can function as positive regulators of Parkin-mediated mitophagy. Many E3 ubiquitin ligases, including Parkin, are inhibited at baseline via auto-ubiquitination (296). In contrast to the other DUBs involved in counteracting mitophagy, USP8 has little effect on the ubiquitination of known mitochondrial Parkin substrates (63). Instead, USP8 is responsible for removing the K6-linked ubiquitin chains from Parkin to promote its activation (FIGURE 6C). Interestingly, the deubiquitination of Parkin by USP8 is a prerequisite for recruitment of Parkin to depolarized mitochondria and subsequent mitophagy (63). Overall, these studies suggest that the regulation of Parkin-mediated mitophagy involves the complex coordination of several DUBs.
VIII. ALTERNATIVE MECHANISM OF MITOPHAGY
For a long time, it was assumed that traditional Atg5/7-dependent autophagy was the only available mechanism by which organelles could be eliminated. However, it is now clear that cells have evolved a number of mechanisms to remove either the entire mitochondrion or just the damaged components. The existence of these pathways first became evident when cells and mouse models with global or tissue specific defects in autophagy developed normally and cells deficient in critical autophagy genes were, unexpectedly, still capable of eliminating damaged or redundant organelles (149, 182, 209, 291). To date, four noncanonical mitophagy/mitochondrial quality control mechanisms have been identified: alternative autophagy, the endosomal pathway, microautophagy, and mitochondrial-derived vesicles (FIGURES 7 and 8).
FIGURE 7.

Noncanonical mitophagy pathways. A: alternative Atg5/7-independent autophagy. The autophagosomes are generated from Rab9+ vesicles derived from the trans-Golgi. Double membrane vesicles containing cargo fuse with lysosomes. B: endosomal-mediated elimination of mitochondria. Mitochondria are engulfed into Rab5+ early endosomes that mature into late endosomes before fusing with the lysosomes. C: in microautophagy, mitochondria are directly engulfed by lysosomes.
FIGURE 8.

Generation of mitochondrial-derived vesicles (MDVs) with distinct cargo. Formation of single-membrane vesicles occurs upon mitochondrial exposure to external reactive oxygen species (ROS). In contrast, double-membrane vesicles result from the production of excess ROS inside mitochondria and involve PINK1/Parkin. The MDVs are targeted to the lysosome for degradation of their contents.
A. Alternative Autophagy
Because Atg5 was known to play a critical role in autophagy, the observation that Atg5-deficient mouse embryos develop normally until the perinatal period was unexpected (149). Other surprising phenotypes of Atg5-deficient mice were delineated that suggested a parallel and redundant mitophagy pathway: despite a requirement for mitophagy to achieve maturational elimination of mitochondria in erythrocytes (142), degraded mitochondria were detected in vacuoles of Atg5−/− erythrocytes (209); during development, epithelial cells covering the anterior surface of the lens eliminate their organelles to become transparent fibers (11), and autophagy deficient mice have normal lens fiber cells without organelles (182). This constellation of findings implicated an alternative pathway that could compensate for the lack of traditional Atg5-mediated autophagy during development.
Further support for an alternative mitophagy pathway came from the discovery that double membrane autophagic vesicles could form in cells independently of traditional Atg5/Atg7-dependent autophagy. This form of autophagy was first documented in autophagy-deficient MEFs in which treatment with the topoisomerase inhibitor etoposide induced formation of double membrane autophagosomes in both Atg5- and Atg7-deficient MEFs (209). Notably, the vesicles were morphologically indistinguishable from conventional autophagic structures, and similar to traditional autophagy, their formation was dependent on Ulk1 (Atg1) and Beclin 1 (Atg6). Thus traditional and alternative autophagy have common initiation steps at the level of Ulk1 and Beclin 1, but the two pathways diverge at the expansion and elongation steps. It is currently unknown how the extension and closure of autophagic membranes are regulated during alternative autophagy.
Although relatively little is still known about the components and regulation of the alternative autophagy pathway, a few characteristics have been identified that distinguish it from traditional autophagy. First, the small GTPase Rab9 that helps transport proteins between late endosomes and the trans-Golgi network (172) is required for alternative autophagy (209) (FIGURE 7A). Also, while most traditional autophagosomes are derived mainly from the ER, the generation of alternative autophagosomes appears to originate exclusively from the trans-Golgi (209). Another unique feature of alternative autophagy is the inhibition of this pathway by Brefeldin A (BFA), that disrupts cis-Golgi. In contrast, traditional autophagy is not inhibited by BFA treatment (230). This distinction emphasizes the unique importance of the Golgi to autophagosome formation via the alternative pathway.
Recent studies using systemic/germline and tissue-specific Atg-gene knockout mice have generated fresh insights into the physiological roles of alternative autophagy. Early on, the presence of mitochondria inside the Rab9-positive vesicles was noted (209), and subsequent studies determined the importance of Rab9 in the selective conditional clearance of mitochondria. For instance, alternative autophagy pathways are central to mitochondrial clearance during erythrocyte maturation (104). During terminal differentiation, erythrocytes use autophagy to eliminate their organelles, including mitochondria. However, erythrocyte maturation appears normal in Atg5−/− embryos (182), suggesting that alternative autophagy is responsible for clearing the mitochondria. In contrast, Ulk1 is required to initiate formation of autophagosomes in both traditional and alternative autophagy and Ulk1−/− erythrocytes failed to clear mitochondria during maturation (104). Of note, the mitophagy receptor Nix is also required for clearance of mitochondria in erythrocyte differentiation (244, 257), suggesting that the alternative autophagy pathway could be utilized by the mitophagy receptors. However, determining the nature of any functional relationships between Ulk1, Nix, and the alternative mitophagy pathway requires detailed investigation.
Alternative autophagy has also been reported to participate in the iPSC reprogramming process. Compared with most differentiated cells, stem cells have reduced mitochondrial mass and primarily rely on glycolysis for ATP generation (75). Thus iPSC reprogramming leading to cellular differentiation involves not only a metabolic switch from mitochondrial oxidation to glycolysis, but also substantial mitophagic mitochondrial culling. Ma et al. (173) recently reported that the alternative autophagy pathway regulates mitochondrial clearance during iPSC reprogramming. They found that knockdown or deletion of Atg5 in fibroblasts had little effect on fibroblast reprogramming. In contrast, disrupting alternative autophagy by knockdown of Ulk1 or Rab9, or by BFA treatment, significantly inhibited both mitochondrial clearance and reprogramming. These results collectively suggest that iPSC reprogramming induces Atg5-independent and Rab9/Ulk1-dependent autophagy. Based on current information, it seems reasonable to conclude that the function of alternative mitophagy under these settings is to facilitate metabolic reprogramming by suppressing mitochondria-mediated oxidation.
B. Early Endosomal-Mediated Degradation of Mitochondria
The endosomal-lysosomal degradation pathway is primarily known for its role in the degradation of cell-surface receptors and other proteins that reside in the plasma membrane (268). In this process, proteins are internalized from the plasma membrane and delivered to an early endosome compartment. From there, they are either recycled back to the plasma membrane or sorted to late endosomes (also called multivesicular bodies or MVBs) for degradation. In this manner, endosomes play a crucial role in the regulation of cell signaling. Similar to autophagy, ubiquitin modification of proteins can target them for degradation via the endosomal pathway. Recent studies have found that this pathway can also degrade cytosolic cargo such as α-synuclein (295). α-Synuclein aggregation has been linked to both familial and sporadic PD.
More recently, Gustafsson’s group (96) discovered how the endosomal-lysosomal pathway can facilitate the clearance of depolarized mitochondria (FIGURE 7B). In this study mitochondrial clearance required the presence of functional Parkin and involved internalization of mitochondria into Rab5-positive early endosomes via the sorting complex required for transport (ESCRT) complexes (96). The ESCRT machinery performs its action at the endosomal membrane and is responsible for sequestering ubiquitinated cargo into intraluminal vesicles (ILVs) by inward budding into the endosomal lumen (26). After fusion of the late endosomes/MVB with the lysosome, ILVs are broken down within the lysosomal lumen. Interestingly, the early endosomal mitochondrial clearance pathway could also be utilized by the mitophagy receptor BNIP3, and this still occurred in the absence of Parkin (97).
C. Microautophagy
An understudied and perhaps underappreciated mechanism of mitochondrial elimination occurs via a process called microautophagy. Here, the cytosolic cargo is directly delivered into the lysosome by invagination of the membrane (FIGURE 7C). Microautophagy has mainly been described in yeast cells and is recognized for being induced by nitrogen starvation (164). A variety of organelles have been identified to be cellular targets of microautophagy in yeast, including nucleus (238), ER (254), and mitochondria (133). Recent studies have described a physiological function for microautophagy in multicellular organisms, including in mouse embryonic development (127), and in the selective clearance of mitochondria in cardiac myocytes and neurons (111, 335): GAPDH is a well-characterized glycolytic enzyme (298), but emerging evidence indicates that GAPDH can also regulate processes independent of its role in energy metabolism (23, 293). Relevant to the focus of this review, GAPDH has been implicated in microautophagy where it functions to target mitochondria directly to the lysosomes. Yogalingam et al. (335) reported that GAPDH can selectively associate with damaged mitochondria in myocytes exposed to I/R to promote their engulfment directly into lysosomes. GAPDH can undergo oxidative modifications leading to catalytic inactivation (110). Thus oxidative stress was found to inactivate GAPDH and induce mitophagy via microautophagy (111), and oxidative modification of GAPDH during I/R might function as a signaling switch to initiating mitophagy of damaged mitochondria during conditions of increased oxidative stress. More recently, impaired GAPDH-induced micromitophagy was implicated as a contributor to the pathology of HD (111): the expanded polyglutamine repeats of mutant Htt selectively associated with mitochondrial GAPDH. This led to accumulation of damaged mitochondria due to inhibition of GAPDH‐mediated delivery of damaged mitochondria into lysosomes. On the basis of these examples, it is can be safely concluded that microautophagy plays an important role in clearing mitochondria in cells, but much is unknown about how this pathway is activated and how in mammalian cells cargo is selected for degradation by this pathway.
D. Mitochondrial Derived Vesicles
When there is limited mitochondrial damage, it is not necessary to eliminate the entire mitochondrion. Instead, the mitochondrion can undergo local repair by exporting the damaged components via mitochondrial derived vesicles (MDVs). These small vesicles, ranging between 70 and 150 nm in diameter, bud from the outer mitochondrial membrane via a process that is independent of the mitochondrial fission protein Drp1 (205). The MDVs can be either single- or double-membraned vesicles and contain distinct mitochondrial cargo depending on the cellular stress (285) (FIGURE 8). MDVs were initially identified as transporting cargo from mitochondria to the peroxisomes (205), but subsequent studies identified MDVs containing damaged, oxidized cargo targeted for lysosomal degradation (186, 278), implicating the MDV pathway as a noncanonical mechanism for mitochondrial quality control.
MDVs are continuously generated under basal conditions, but their formation is rapidly increased by mitochondrial/cellular exposure to oxidative stress. For instance, MDVs are formed at steady state at baseline in heart tissue, but are rapidly increased in response to doxorubicin exposure (24). Importantly, the MDV cargo differs depending on whether the oxidant stress originates from within or outside the mitochondria. Thus external ROS production leads to the generation of single-membraned MDVs enriched in OMM proteins such as TOM20 and VDAC, and lacking matrix or inner mitochondrial membrane markers (278). In contrast, production of ROS inside mitochondria leads to formation of double-membraned MDVs containing matrix proteins such as pyruvate dehydrogenase and complex III subunit core2, but lacking outer membrane VDAC and TOM20 (186). Notably, the generation of double-membraned MDVs to deliver oxidized mitochondrial proteins to lysosomes requires Parkin and PINK1 at the budding site (186), and point mutations in Parkin (C431F, K211N) linked to PD lead to impairment in the MDV pathway, mechanistically linking the MDV pathway to familial PD and suggesting that the PINK1/Parkin pathway plays a role in mitochondrial quality control in both canonical mitophagy and the MDV pathway.
At the present time, less is known than unknown about the molecular machinery regulating membrane dynamics driving formation of MDVs. In one recent advance, Vps35 was implicated as a regulator of this pathway. Vps35 is recognized to be a key component of the membrane protein-recycling retromer complex and is involved in mediating protein trafficking from the endosomes to the trans-Golgi network or through direct recycling to the plasma membrane (310). Recent studies have extended the function of Vps35 to the formation of MDVs (22, 311): Wang et al. (311) reported that the mitochondrial fission protein Drp1 is a cargo of MDVs and that Vps35 is responsible for directing the Drp1 complexes to the MDVs for trafficking to degradative lysosomes. Mutations in the Vps35 gene are associated with autosomal-dominant PD (260, 346), and the PD-linked mutations in Vps35 increase Drp1 at the mitochondria, increase turnover in MDVs, and provoke excessive mitochondrial fission and cell death (311). This constellation of observations suggests a previously unsuspected pathophysiological link between impaired MDV-mediated mitochondrial quality control and PD.
Taken together, these studies and accumulating data strongly suggest that MDVs function in mitochondrial quality control by forming a first line of defense against mitochondrial damage. This concept is supported by the finding that MDVs are formed before autophagosomes in response to stress (278). Cells such as neurons and cardiac myocytes that are highly dependent on mitochondrial respiration to meet metabolic demands cannot tolerate the removal of large portions of their mitochondrial collective via conventional mitophagy. Thus the MDV pathway allows the mitochondrion to undergo local repair to ensure that its function is maintained.
E. Chaperone-Mediated Autophagy
Chaperone-mediated autophagy (CMA) is another important cellular quality control mechanism that involves the delivery and selective degradation of proteins in lysosomes (126). Proteins that are CMA substrates contain a KFERQ-like motif that is recognized by the cytosolic chaperone heat shock-cognate protein of 70 kDa (Hsc70). Hsc70 and its co-chaperones, including COOH terminus of HSC70-interacting protein (CHIP), heat shock protein 40 (HSP40), and HSP70-HSP90 organizing protein (HOP), are responsible for unfolding the substrate and delivering it to the surface of the lysosome where it is then transported into the lumen of the lysosome by lysosome-associated membrane protein type 2A (LAMP2A) (126). Interestingly, CMA is activated by the same stressors that activate autophagy and mitophagy, including oxidative stress, starvation, and hypoxia. Also, there is crosstalk between the various autophagy pathways, and when CMA is inhibited, macroautophagy can compensate and degrade CMA substrates. However, the reverse scenario is less effective since CMA cannot degrade bulky cardo such a protein aggregates and organelles. By degrading key regulators, CMA can influence various cellular processes. Although there are no published reports on the direct regulation of mitophagy by CMA, some of the validated CMA substrates have been reported to regulate mitophagy. For instance, GAPDH is a bona fide substrate of CMA, and GAPDH can function as a regulator of micromitophagy (111), i.e., the direct engulfment of damaged mitochondria into lysosomes (see sect. VIIIC). Thus degradation of GAPDH by CMA will inhibit this process. Hexokinase II is another CMA substrate (324), and hexokinase II has been reported to be required for efficient translocation of Parkin to mitochondria (185). Although these studies have not directly assessed the link between these substrates and mitophagy, future studies will serve to further validate the important role of CMA in the regulation of mitophagy.
IX. CONCLUSIONS AND CLINICAL APPLICATIONS
Here, we have tried to provide a comprehensive overview of recent progress elucidating the cellular mechanisms for, and organ/systemic consequences of, mitochondrial regulation by mitophagy and related processes. Given the rapid advances and accumulating information described, it is reasonable to posit about possible clinical applications (TABLE 2). Conceptually, there seems to be a powerful rationale for enhancing mitophagic mitochondrial quality control in diseases where mitochondrial dysfunction is a causal or contributory factor, for modulating mitophagic mitochondrial quantity control when abnormally increased or decreased mitochondrial mass is a pathological factor, or for intervening in mitophagic mitochondrial turnover during metabolic remodeling in conditions where this is either an inadequate or maladaptive response. Here we propose three conditions that will need to be met before recently acquired knowledge can be translated into new and useful tools:
Table 2.
Targeting Mitophagy in Disease
| Tissue | Disease | Characteristics | Approach | Reference Nos. |
|---|---|---|---|---|
| Brain | Parkinson’s disease | Defects in mitophagy, reduced mitochondrial biogenesis and mitochondrial dysfunction. Loss of dopaminergic neurons. | Increase or restore mitophagy | 21, 82, 134, 159, 225, 300 |
| Alzheimer’s disease | Accumulation of dysfunctional mitochondria, formation of Aβ plaques and neurofibrillary tangles. Loss of neurons and synapses. | Increase autophagy/mitophagy | 128, 162, 166, 177, 210, 333, 334 | |
| Huntington disease | Misfolding of Htt leading to formation of cytotoxic aggregates. Defects in autophagy cargo recognition and inhibition of micromitophagy. Accumulation of damaged mitochondria and increased cell death. | Increase autophagy and mitophagy | 111, 178, 242, 255 | |
| Amyotrophic lateral sclerosis | Mutations in genes linked to regulation of autophagy/mitophagy. Decreased mitophagy and loss of motor neurons. | Increase autophagy or restore mitophagy | 132, 194, 247, 321 | |
| Heart | Myocardial infarction | Mitophagy is activated in response to ischemic/energetic stress. Impaired mitophagy leads to accumulation of damaged mitochondria, loss of myocytes, and heart failure development. | Increase mitophagy | 112, 147, 267, 309 |
| Pressure overload | Autophagy and mitophagy are increased during the acute phase of pressure overload but decreased in the chronic phase. Decreased mitophagy correlates with accumulation of dysfunctional mitochondria and cardiac dysfunction. | Increase mitophagy | 18, 98, 264 | |
| Doxorubicin-induced cardiotoxicity | Controversial studies have reported both recued and increased mitophagy after exposure to the drug. | Unclear | 105, 109 | |
| Various tissues | Cancer | Mitophagy functions as a tumor suppressor mechanism in certain cancers. Impaired mitophagy correlates with enhanced cancer development. | Unclear | 39, 50, 71, 78, 163, 259, 305, 340 |
| Lungs | Acute lung injury | Increased mitophagy protects lung cells against hyperoxia, S. aureus infection and sepsis. | Increase autophagy/mitophagy | 175, 286, 343 |
| Chronic obstructive pulmonary disease | Controversial: insufficient mitophagy leads to senescence of lung epithelial cells, while enhanced mitophagy leads to activation of necroptosis in lung epithelial cells. Both conditions contribute to COPD. | Unclear | 115, 193 | |
| Idiopathic pulmonary fibrosis | Alteration in mitophagy is cell dependent. | Unclear | 6, 137, 154, 224 | |
| Increased mitophagy in alveolar macrophages leads to apoptosis resistance and increased inflammation. Reduced mitophagy in fibroblasts enhances the fibrotic response. | ||||
| Liver | Alcoholic liver disease and acetaminophen overdose | Activation of autophagy and mitophagy protect against mitochondrial damage and liver injury. | Increase autophagy/mitophagy | 204, 208, 318 |
| Immune system | Inflammation | Impaired autophagy/mitophagy leads to the reduced elimination of pathogens. Impaired mitophagy also leads to excess NLRP3 inflammasome activation and chronic inflammation. | Increase autophagy/mitophagy | 4, 131, 176, 190, 198, 344, 345 |
COPD, chronic obstructive pulmonary disease.
First, there must be unambiguous evidence that inadequate, overzealous, or dysregulated mitophagy causes or contributes to a specific clinical disease. Given the wealth of experimental data from cell and animal models demonstrating that abnormalities in individual mitophagy effectors or dysregulation of integrated mitophagic processes can provoke diseaselike phenotypes, this does not seem like such a heavy lift. But, upon careful consideration, it is difficult to identify even a single human condition known to be caused by abnormal mitophagy. The counterargument nominates PD as the prototypical disease of inadequate mitophagy, and this is almost certainly correct in flies. But as we noted above, a clear causal relationship between the specific genetic mutations in mitophagy effectors PINK1 and Parkin that underlie hereditary or early-onset PD and impaired mitophagy has been difficult to establish in the human condition. It is indisputable that neuronal mitochondria are abnormal in clinical PD. However, evidence that these abnormalities accrue from defects in the PINK1-Parkin mitophagy pathway is lacking, and other chronic neurodegenerative conditions without primary abnormalities in mitophagy (e.g., ALS and HD) exhibit similar or more dramatic mitochondrial fragmentation, depolarization, and increased ROS production, undercutting the thesis that mitochondrial abnormalities characteristic of PD are necessarily primary or causal. Of course, as with all monogenic diseases, gene editing remains an option, when the technology becomes sufficiently advanced to do so. Correcting underlying mutations is a therapeutic approach that should be agnostic as to underlying mechanism.
The quandary is greater when altered or impaired mitophagy is invoked as a contributory factor in diseases lacking a primary mitochondrial genesis, such as tissue ischemia. There are two general paradigms. Most commonly, mitophagic impairment is implicated in the context of observed mitochondrial degeneration, and dysfunctional mitochondria are then designated as factors that adversely impact cell survival by actively producing cytotoxic ROS or by inadequately generating ATP to fuel cellular repair. Less commonly, mitophagy is nominated, perhaps in combination with impaired biogenesis, as a mechanism for mitochondrial (and therefore bioenergetic) insufficiency. Although abnormal mitophagy is not the source of pathology in either of these scenarios, the underlying problems with mitochondrial quality or quantity seem theoretically amenable to mitophagy manipulation. We think that therapeutically fine-tuning mitophagy, which is already highly regulated at both the cellular level (where the overall mitophagic set point can be adjusted according to level of stress), and the organelle level (where individual mitochondria falling along a continuum of levels of dysfunction are subject to the binary decision of retain vs remove), will be difficult whether or not usable tools to do so exist. Not only is mitophagy exquisitely regulated under normal conditions as a function of developmental state, metabolic demand, and cellular senescence, it is potently induced at the individual cellular level as a compensatory response to stress or injury. Compared with our own cells with their complex intrinsic regulatory mechanisms, systemically or locally administered mitophagy activators and suppressors (see below) appear to be relatively crude instruments with no ability to discriminate between normal versus disease, and therefore with the potential to disrupt in unanticipated ways both homeostatic and reactive orchestration of mitochondrial repair, biogenic renewal, and mitophagic removal.
Second, to successfully modulate mitophagy, we need clinically translatable tools having the requisite selectivity and exhibiting acceptable patient tolerance. We are witnessing an ongoing effort directed toward transitioning from preclinical studies in animal models to feasibility trails in human subjects using repurposed United States Food and Drug Administration-approved drugs that can target autophagy (but not mitochondrial-specific autophagy). Candidates include rapamycin that activates autophagy and chloroquine that inhibits autophagy (80). Even to modulate cell-wide autophagy, selectivity becomes an important issue limiting the clinical utility of these compounds. For example, chloroquine interrupts autophagy by raising the pH of normally acidic lysosomes. Consequently, any lysosome-dependent process in any cell will be impacted. However, when the goal is modulation of mitochondrial-selective autophagy, i.e., mitophagy, there are not even likely candidates. Most in vivo research studies have relied on standard transgenic “gain-of-function” and germ-line or temporally defined/tissue-targeted gene ablation “loss-of-function” approaches to modulate mitophagy. More recently, transgenic introduction of engineered mutant mitophagy effectors that specifically promote or interrupt mitophagy without impacting other cell processes was described (90). Notwithstanding delineation of hundreds of genetic causes for human diseases and the promise of “gene therapy,” it has been generally difficult to translate genetic gain- or loss-of-function approaches to the clinic (we are also still anxiously awaiting much promised flying cars).
Whether successful therapeutic targeting of mitophagy employs genetic or as-yet-undiscovered pharmacological tools, redundancy of mitochondrial quality control pathways and parallel functioning of mitophagy mediators in other cell-critical pathways will almost certainly complicate clinical introduction. As examples, we noted above that BNIP3/Nix can activate either mitophagy or programmed cell death, depending on cellular context. Also, Parkin can ubiquitinate and mark for degradation nonmitochondrial proteins. On the other hand, suppressing PINK-Parkin mitophagy is almost certain to enable alternate and counteractive mitophagic processes. For each of these examples, functional overlap or biological redundancy increases the chances of undesirable off-target effects. It seems that the journey towards therapeutically targeting mitophagy has barely begun.
Third, we need clinically applicable diagnostics to assay mitophagy in human subjects. Developing reliable and practical clinical diagnostic tests for mitophagy may be the preeminent immediate requisite for bench-to-bedside translation, as detecting abnormal mitophagy in disease and measuring the mitophagic response to experimental clinical therapeutics will be essential to addressing the above two needs. The battery of investigative assays to identify mitophagy and measure mitophagic flux are not readily applied to either human research or patient care. Currently, assessment of mitophagy in people is limited to genetic testing, as for PINK1 and Parkin mutations, or histological analyses that are limited to providing a snapshot picture of a small sample of diseased tissue. Thus histology can detect an increased number of autophagosomes or the presence of dysfunctional mitochondria, but is incapable of distinguishing between stimulated autophagy versus interrupted autophagosome delivery to lysosomes as the underlying cause, which would determine therapeutic approach. Some progress is being made in clinically relevant animal models identifying circulating biomarkers of mitochondrial disease (54), but these are not specific to mitophagy and if they were would not distinguish between healthy reactive mitophagy and pathological causal mitophagy.
In conclusion, mitophagy is not as straightforward as previously thought. The biochemical events mediating mitophagy exhibit parallelism, functional redundancy, alternate effectors, and crosstalk with mitochondrial dynamics, biogenesis, and programmed cell death pathways. At a more holistic level, mitophagy is orchestrated in concert with, and feeds back to, cell development, growth, and metabolic remodeling. This level of biological complexity has not evolved simply to challenge biomedical researchers, but because mitochondrial quality control is critically important to life, and likely especially so for postmitotic cells such as neurons and cardiac myocytes. In this context, the ability of mitophagic mechanisms to resist attack by genetic, environmental, or external biological factors through induction of alternate platers or pathways ensures that healthy relationships are maintained between mitochondrial endosymbionts and their completely dependent hosts, us.
GRANTS
Å. B. Gustafsson is supported by an American Heart Association Established Investigator Award and by National Institutes of Health (NIH) Grants R21AG052280, R01HL132300, R01HL138560, and P01HL085577. G. W. Dorn II is supported by NIH/National Heart, Lung, and Blood Institute Grant R35 135736.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
ACKNOWLEDGMENTS
Address for reprint requests and other correspondence: Å. Gustafsson, Univ. of California, San Diego, 9500 Gilman Dr. #0751, La Jolla, CA 92093–0751 (e-mail: asag@ucsd.edu).
REFERENCES
- 1.Abeliovich H, Zarei M, Rigbolt KT, Youle RJ, Dengjel J. Involvement of mitochondrial dynamics in the segregation of mitochondrial matrix proteins during stationary phase mitophagy. Nat Commun 4: 2789, 2013. doi: 10.1038/ncomms3789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ahlqvist KJ, Hämäläinen RH, Yatsuga S, Uutela M, Terzioglu M, Götz A, Forsström S, Salven P, Angers-Loustau A, Kopra OH, Tyynismaa H, Larsson NG, Wartiovaara K, Prolla T, Trifunovic A, Suomalainen A. Somatic progenitor cell vulnerability to mitochondrial DNA mutagenesis underlies progeroid phenotypes in Polg mutator mice. Cell Metab 15: 100–109, 2012. doi: 10.1016/j.cmet.2011.11.012. [DOI] [PubMed] [Google Scholar]
- 3.Al Rawi S, Louvet-Vallée S, Djeddi A, Sachse M, Culetto E, Hajjar C, Boyd L, Legouis R, Galy V. Postfertilization autophagy of sperm organelles prevents paternal mitochondrial DNA transmission. Science 334: 1144–1147, 2011. doi: 10.1126/science.1211878. [DOI] [PubMed] [Google Scholar]
- 4.Ali S, Vollaard AM, Widjaja S, Surjadi C, van de Vosse E, van Dissel JT. PARK2/PACRG polymorphisms and susceptibility to typhoid and paratyphoid fever. Clin Exp Immunol 144: 425–431, 2006. doi: 10.1111/j.1365-2249.2006.03087.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aoki Y, Kanki T, Hirota Y, Kurihara Y, Saigusa T, Uchiumi T, Kang D. Phosphorylation of Serine 114 on Atg32 mediates mitophagy. Mol Biol Cell 22: 3206–3217, 2011. doi: 10.1091/mbc.e11-02-0145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Araya J, Kojima J, Takasaka N, Ito S, Fujii S, Hara H, Yanagisawa H, Kobayashi K, Tsurushige C, Kawaishi M, Kamiya N, Hirano J, Odaka M, Morikawa T, Nishimura SL, Kawabata Y, Hano H, Nakayama K, Kuwano K. Insufficient autophagy in idiopathic pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol 304: L56–L69, 2013. doi: 10.1152/ajplung.00213.2012. [DOI] [PubMed] [Google Scholar]
- 7.Ariosa AR, Klionsky DJ. Autophagy core machinery: overcoming spatial barriers in neurons. J Mol Med (Berl) 94: 1217–1227, 2016. doi: 10.1007/s00109-016-1461-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Baloh RH, Schmidt RE, Pestronk A, Milbrandt J. Altered axonal mitochondrial transport in the pathogenesis of Charcot-Marie-Tooth disease from mitofusin 2 mutations. J Neurosci 27: 422–430, 2007. doi: 10.1523/JNEUROSCI.4798-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Band M, Joel A, Hernandez A, Avivi A. Hypoxia-induced BNIP3 expression and mitophagy: in vivo comparison of the rat and the hypoxia-tolerant mole rat, Spalax ehrenbergi. FASEB J 23: 2327–2335, 2009. doi: 10.1096/fj.08-122978. [DOI] [PubMed] [Google Scholar]
- 10.Barthelme D, Jauregui R, Sauer RT. An ALS disease mutation in Cdc48/p97 impairs 20S proteasome binding and proteolytic communication. Protein Sci 24: 1521–1527, 2015. doi: 10.1002/pro.2740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bassnett S. On the mechanism of organelle degradation in the vertebrate lens. Exp Eye Res 88: 133–139, 2009. doi: 10.1016/j.exer.2008.08.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bellot G, Garcia-Medina R, Gounon P, Chiche J, Roux D, Pouysségur J, Mazure NM. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol 29: 2570–2581, 2009. doi: 10.1128/MCB.00166-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Bernhardt D, Müller M, Reichert AS, Osiewacz HD. Simultaneous impairment of mitochondrial fission and fusion reduces mitophagy and shortens replicative lifespan. Sci Rep 5: 7885, 2015. doi: 10.1038/srep07885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Berthiaume JM, Wallace KB. Adriamycin-induced oxidative mitochondrial cardiotoxicity. Cell Biol Toxicol 23: 15–25, 2007. doi: 10.1007/s10565-006-0140-y. [DOI] [PubMed] [Google Scholar]
- 15.Betz C, Stracka D, Prescianotto-Baschong C, Frieden M, Demaurex N, Hall MN. mTOR complex 2-Akt signaling at mitochondria-associated endoplasmic reticulum membranes (MAM) regulates mitochondrial physiology. Proc Natl Acad Sci USA 110: 12526–12534, 2013. doi: 10.1073/pnas.1302455110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bhandari P, Song M, Chen Y, Burelle Y, Dorn GW II. Mitochondrial contagion induced by Parkin deficiency in Drosophila hearts and its containment by suppressing mitofusin. Circ Res 114: 257–265, 2014. doi: 10.1161/CIRCRESAHA.114.302734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bhujabal Z, Birgisdottir AB, Sjøttem E, Brenne HB, Øvervatn A, Habisov S, Kirkin V, Lamark T, Johansen T. FKBP8 recruits LC3A to mediate Parkin-independent mitophagy. EMBO Rep 18: 947–961, 2017. doi: 10.15252/embr.201643147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Billia F, Hauck L, Konecny F, Rao V, Shen J, Mak TW. PTEN-inducible kinase 1 (PINK1)/Park6 is indispensable for normal heart function. Proc Natl Acad Sci USA 108: 9572–9577, 2011. doi: 10.1073/pnas.1106291108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Bingol B, Tea JS, Phu L, Reichelt M, Bakalarski CE, Song Q, Foreman O, Kirkpatrick DS, Sheng M. The mitochondrial deubiquitinase USP30 opposes parkin-mediated mitophagy. Nature 510: 370–375, 2014. doi: 10.1038/nature13418. [DOI] [PubMed] [Google Scholar]
- 20.Blau HM, Cosgrove BD, Ho AT. The central role of muscle stem cells in regenerative failure with aging. Nat Med 21: 854–862, 2015. doi: 10.1038/nm.3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Bose A, Beal MF. Mitochondrial dysfunction in Parkinson’s disease. J Neurochem 139, Suppl 1: 216–231, 2016. doi: 10.1111/jnc.13731. [DOI] [PubMed] [Google Scholar]
- 22.Braschi E, Goyon V, Zunino R, Mohanty A, Xu L, McBride HM. Vps35 mediates vesicle transport between the mitochondria and peroxisomes. Curr Biol 20: 1310–1315, 2010. doi: 10.1016/j.cub.2010.05.066. [DOI] [PubMed] [Google Scholar]
- 23.Bryksin AV, Laktionov PP. Role of glyceraldehyde-3-phosphate dehydrogenase in vesicular transport from golgi apparatus to endoplasmic reticulum. Biochemistry (Mosc) 73: 619–625, 2008. doi: 10.1134/S0006297908060011. [DOI] [PubMed] [Google Scholar]
- 24.Cadete VJ, Deschênes S, Cuillerier A, Brisebois F, Sugiura A, Vincent A, Turnbull D, Picard M, McBride HM, Burelle Y. Formation of mitochondrial-derived vesicles is an active and physiologically relevant mitochondrial quality control process in the cardiac system. J Physiol 594: 5343–5362, 2016. doi: 10.1113/JP272703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Call JA, Wilson RJ, Laker RC, Zhang M, Kundu M, Yan Z. Ulk1-mediated autophagy plays an essential role in mitochondrial remodeling and functional regeneration of skeletal muscle. Am J Physiol Cell Physiol 312: C724–C732, 2017. doi: 10.1152/ajpcell.00348.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Campsteijn C, Vietri M, Stenmark H. Novel ESCRT functions in cell biology: spiraling out of control? Curr Opin Cell Biol 41: 1–8, 2016. doi: 10.1016/j.ceb.2016.03.008. [DOI] [PubMed] [Google Scholar]
- 27.Carroll RG, Hollville E, Martin SJ. Parkin sensitizes toward apoptosis induced by mitochondrial depolarization through promoting degradation of Mcl-1. Cell Reports 9: 1538–1553, 2014. doi: 10.1016/j.celrep.2014.10.046. [DOI] [PubMed] [Google Scholar]
- 28.Chakkalakal JV, Jones KM, Basson MA, Brack AS. The aged niche disrupts muscle stem cell quiescence. Nature 490: 355–360, 2012. doi: 10.1038/nature11438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chan NC, Salazar AM, Pham AH, Sweredoski MJ, Kolawa NJ, Graham RL, Hess S, Chan DC. Broad activation of the ubiquitin-proteasome system by Parkin is critical for mitophagy. Hum Mol Genet 20: 1726–1737, 2011. doi: 10.1093/hmg/ddr048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chen G, Cizeau J, Vande Velde C, Park JH, Bozek G, Bolton J, Shi L, Dubik D, Greenberg A. Nix and Nip3 form a subfamily of pro-apoptotic mitochondrial proteins. J Biol Chem 274: 7–10, 1999. doi: 10.1074/jbc.274.1.7. [DOI] [PubMed] [Google Scholar]
- 31.Chen G, Han Z, Feng D, Chen Y, Chen L, Wu H, Huang L, Zhou C, Cai X, Fu C, Duan L, Wang X, Liu L, Liu X, Shen Y, Zhu Y, Chen Q. A regulatory signaling loop comprising the PGAM5 phosphatase and CK2 controls receptor-mediated mitophagy. Mol Cell 54: 362–377, 2014. doi: 10.1016/j.molcel.2014.02.034. [DOI] [PubMed] [Google Scholar]
- 32.Chen L, Na R, Boldt E, Ran Q. NLRP3 inflammasome activation by mitochondrial reactive oxygen species plays a key role in long-term cognitive impairment induced by paraquat exposure. Neurobiol Aging 36: 2533–2543, 2015. doi: 10.1016/j.neurobiolaging.2015.05.018. [DOI] [PubMed] [Google Scholar]
- 33.Chen Y, Dorn GW II. PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science 340: 471–475, 2013. doi: 10.1126/science.1231031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Chen Y, Lewis W, Diwan A, Cheng EH, Matkovich SJ, Dorn GW II. Dual autonomous mitochondrial cell death pathways are activated by Nix/BNip3L and induce cardiomyopathy. Proc Natl Acad Sci USA 107: 9035–9042, 2010. doi: 10.1073/pnas.0914013107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen Y, Liu Y, Dorn GW II. Mitochondrial fusion is essential for organelle function and cardiac homeostasis. Circ Res 109: 1327–1331, 2011. doi: 10.1161/CIRCRESAHA.111.258723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chen YR, Zweier JL. Cardiac mitochondria and reactive oxygen species generation. Circ Res 114: 524–537, 2014. doi: 10.1161/CIRCRESAHA.114.300559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chen Z, Liu L, Cheng Q, Li Y, Wu H, Zhang W, Wang Y, Sehgal SA, Siraj S, Wang X, Wang J, Zhu Y, Chen Q. Mitochondrial E3 ligase MARCH5 regulates FUNDC1 to fine-tune hypoxic mitophagy. EMBO Rep 18: 495–509, 2017. doi: 10.15252/embr.201643309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Choubey V, Cagalinec M, Liiv J, Safiulina D, Hickey MA, Kuum M, Liiv M, Anwar T, Eskelinen EL, Kaasik A. BECN1 is involved in the initiation of mitophagy: it facilitates PARK2 translocation to mitochondria. Autophagy 10: 1105–1119, 2014. doi: 10.4161/auto.28615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chourasia AH, Tracy K, Frankenberger C, Boland ML, Sharifi MN, Drake LE, Sachleben JR, Asara JM, Locasale JW, Karczmar GS, Macleod KF. Mitophagy defects arising from BNip3 loss promote mammary tumor progression to metastasis. EMBO Rep 16: 1145–1163, 2015. doi: 10.15252/embr.201540759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Chu CT, Ji J, Dagda RK, Jiang JF, Tyurina YY, Kapralov AA, Tyurin VA, Yanamala N, Shrivastava IH, Mohammadyani D, Wang KZQ, Zhu J, Klein-Seetharaman J, Balasubramanian K, Amoscato AA, Borisenko G, Huang Z, Gusdon AM, Cheikhi A, Steer EK, Wang R, Baty C, Watkins S, Bahar I, Bayir H, Kagan VE. Cardiolipin externalization to the outer mitochondrial membrane acts as an elimination signal for mitophagy in neuronal cells. Nat Cell Biol 15: 1197–1205, 2013. doi: 10.1038/ncb2837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Clague MJ, Barsukov I, Coulson JM, Liu H, Rigden DJ, Urbé S. Deubiquitylases from genes to organism. Physiol Rev 93: 1289–1315, 2013. doi: 10.1152/physrev.00002.2013. [DOI] [PubMed] [Google Scholar]
- 42.Clark IE, Dodson MW, Jiang C, Cao JH, Huh JR, Seol JH, Yoo SJ, Hay BA, Guo M. Drosophila pink1 is required for mitochondrial function and interacts genetically with parkin. Nature 441: 1162–1166, 2006. doi: 10.1038/nature04779. [DOI] [PubMed] [Google Scholar]
- 43.Cleveland DW, Rothstein JD. From Charcot to Lou Gehrig: deciphering selective motor neuron death in ALS. Nat Rev Neurosci 2: 806–819, 2001. doi: 10.1038/35097565. [DOI] [PubMed] [Google Scholar]
- 44.Cloonan SM, Choi AM. Mitochondria in lung disease. J Clin Invest 126: 809–820, 2016. doi: 10.1172/JCI81113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Cornelissen T, Haddad D, Wauters F, Van Humbeeck C, Mandemakers W, Koentjoro B, Sue C, Gevaert K, De Strooper B, Verstreken P, Vandenberghe W. The deubiquitinase USP15 antagonizes Parkin-mediated mitochondrial ubiquitination and mitophagy. Hum Mol Genet 23: 5227–5242, 2014. doi: 10.1093/hmg/ddu244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Cunningham CN, Baughman JM, Phu L, Tea JS, Yu C, Coons M, Kirkpatrick DS, Bingol B, Corn JE. USP30 and parkin homeostatically regulate atypical ubiquitin chains on mitochondria. Nat Cell Biol 17: 160–169, 2015. doi: 10.1038/ncb3097. [DOI] [PubMed] [Google Scholar]
- 47.Dai DF, Rabinovitch PS. Cardiac aging in mice and humans: the role of mitochondrial oxidative stress. Trends Cardiovasc Med 19: 213–220, 2009. doi: 10.1016/j.tcm.2009.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dauer W, Przedborski S. Parkinson’s disease: mechanisms and models. Neuron 39: 889–909, 2003. doi: 10.1016/S0896-6273(03)00568-3. [DOI] [PubMed] [Google Scholar]
- 49.De Duve C, Wattiaux R. Functions of lysosomes. Annu Rev Physiol 28: 435–492, 1966. doi: 10.1146/annurev.ph.28.030166.002251. [DOI] [PubMed] [Google Scholar]
- 50.Denison SR, Wang F, Becker NA, Schüle B, Kock N, Phillips LA, Klein C, Smith DI. Alterations in the common fragile site gene Parkin in ovarian and other cancers. Oncogene 22: 8370–8378, 2003. doi: 10.1038/sj.onc.1207072. [DOI] [PubMed] [Google Scholar]
- 51.Dhingra R, Margulets V, Chowdhury SR, Thliveris J, Jassal D, Fernyhough P, Dorn GW II, Kirshenbaum LA. Bnip3 mediates doxorubicin-induced cardiac myocyte necrosis and mortality through changes in mitochondrial signaling. Proc Natl Acad Sci USA 111: E5537–E5544, 2014. doi: 10.1073/pnas.1414665111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Di Meo S, Venditti P. Mitochondria in exercise-induced oxidative stress. Biol Signals Recept 10: 125–140, 2001. doi: 10.1159/000046880. [DOI] [PubMed] [Google Scholar]
- 53.Ding WX, Ni HM, Li M, Liao Y, Chen X, Stolz DB, Dorn GW II, Yin XM. Nix is critical to two distinct phases of mitophagy, reactive oxygen species-mediated autophagy induction and Parkin-ubiquitin-p62-mediated mitochondrial priming. J Biol Chem 285: 27879–27890, 2010. doi: 10.1074/jbc.M110.119537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Disatnik MH, Joshi AU, Saw NL, Shamloo M, Leavitt BR, Qi X, Mochly-Rosen D. Potential biomarkers to follow the progression and treatment response of Huntington’s disease. J Exp Med 213: 2655–2669, 2016. doi: 10.1084/jem.20160776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Diwan A, Koesters AG, Odley AM, Pushkaran S, Baines CP, Spike BT, Daria D, Jegga AG, Geiger H, Aronow BJ, Molkentin JD, Macleod KF, Kalfa TA, Dorn GW II. Unrestrained erythroblast development in Nix−/− mice reveals a mechanism for apoptotic modulation of erythropoiesis. Proc Natl Acad Sci USA 104: 6794–6799, 2007. doi: 10.1073/pnas.0610666104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Diwan A, Krenz M, Syed FM, Wansapura J, Ren X, Koesters AG, Li H, Kirshenbaum LA, Hahn HS, Robbins J, Jones WK, Dorn GW II. Inhibition of ischemic cardiomyocyte apoptosis through targeted ablation of Bnip3 restrains postinfarction remodeling in mice. J Clin Invest 117: 2825–2833, 2007. doi: 10.1172/JCI32490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Diwan A, Matkovich SJ, Yuan Q, Zhao W, Yatani A, Brown JH, Molkentin JD, Kranias EG, Dorn GW II. Endoplasmic reticulum-mitochondria crosstalk in NIX-mediated murine cell death. J Clin Invest 119: 203–212, 2009. doi: 10.1172/JCI36445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Diwan A, Wansapura J, Syed FM, Matkovich SJ, Lorenz JN, Dorn GW II. Nix-mediated apoptosis links myocardial fibrosis, cardiac remodeling, and hypertrophy decompensation. Circulation 117: 396–404, 2008. doi: 10.1161/CIRCULATIONAHA.107.727073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dorn GW., II Evolving Concepts of Mitochondrial Dynamics. Annu Rev Physiol 81: 2018. doi: 10.1146/annurev-physiol-020518-114358. [DOI] [PubMed] [Google Scholar]
- 60.Dorn GW., II Mitochondrial pruning by Nix and BNip3: an essential function for cardiac-expressed death factors. J Cardiovasc Transl Res 3: 374–383, 2010. doi: 10.1007/s12265-010-9174-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Dorn GW II, Kitsis RN. The mitochondrial dynamism-mitophagy-cell death interactome: multiple roles performed by members of a mitochondrial molecular ensemble. Circ Res 116: 167–182, 2015. doi: 10.1161/CIRCRESAHA.116.303554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Dorn GW II, Vega RB, Kelly DP. Mitochondrial biogenesis and dynamics in the developing and diseased heart. Genes Dev 29: 1981–1991, 2015. doi: 10.1101/gad.269894.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Durcan TM, Tang MY, Pérusse JR, Dashti EA, Aguileta MA, McLelland GL, Gros P, Shaler TA, Faubert D, Coulombe B, Fon EA. USP8 regulates mitophagy by removing K6-linked ubiquitin conjugates from parkin. EMBO J 33: 2473–2491, 2014. doi: 10.15252/embj.201489729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Eapen MS, Myers S, Walters EH, Sohal SS. Airway inflammation in chronic obstructive pulmonary disease (COPD): a true paradox. Expert Rev Respir Med 11: 827–839, 2017. doi: 10.1080/17476348.2017.1360769. [DOI] [PubMed] [Google Scholar]
- 65.Egan DF, Shackelford DB, Mihaylova MM, Gelino S, Kohnz RA, Mair W, Vasquez DS, Joshi A, Gwinn DM, Taylor R, Asara JM, Fitzpatrick J, Dillin A, Viollet B, Kundu M, Hansen M, Shaw RJ. Phosphorylation of ULK1 (hATG1) by AMP-activated protein kinase connects energy sensing to mitophagy. Science 331: 456–461, 2011. doi: 10.1126/science.1196371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Eisner V, Cupo RR, Gao E, Csordás G, Slovinsky WS, Paillard M, Cheng L, Ibetti J, Chen SR, Chuprun JK, Hoek JB, Koch WJ, Hajnóczky G. Mitochondrial fusion dynamics is robust in the heart and depends on calcium oscillations and contractile activity. Proc Natl Acad Sci USA 114: E859–E868, 2017. doi: 10.1073/pnas.1617288114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Elmore SP, Qian T, Grissom SF, Lemasters JJ. The mitochondrial permeability transition initiates autophagy in rat hepatocytes. FASEB J 15: 2286–2287, 2001. doi: 10.1096/fj.01-0206fje. [DOI] [PubMed] [Google Scholar]
- 68.Esposito L, Raber J, Kekonius L, Yan F, Yu GQ, Bien-Ly N, Puoliväli J, Scearce-Levie K, Masliah E, Mucke L. Reduction in mitochondrial superoxide dismutase modulates Alzheimer’s disease-like pathology and accelerates the onset of behavioral changes in human amyloid precursor protein transgenic mice. J Neurosci 26: 5167–5179, 2006. doi: 10.1523/JNEUROSCI.0482-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Esteban-Martínez L, Sierra-Filardi E, McGreal RS, Salazar-Roa M, Mariño G, Seco E, Durand S, Enot D, Graña O, Malumbres M, Cvekl A, Cuervo AM, Kroemer G, Boya P. Programmed mitophagy is essential for the glycolytic switch during cell differentiation. EMBO J 36: 1688–1706, 2017. doi: 10.15252/embj.201695916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Exner N, Treske B, Paquet D, Holmström K, Schiesling C, Gispert S, Carballo-Carbajal I, Berg D, Hoepken HH, Gasser T, Krüger R, Winklhofer KF, Vogel F, Reichert AS, Auburger G, Kahle PJ, Schmid B, Haass C. Loss-of-function of human PINK1 results in mitochondrial pathology and can be rescued by parkin. J Neurosci 27: 12413–12418, 2007. doi: 10.1523/JNEUROSCI.0719-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Feng X, Liu X, Zhang W, Xiao W. p53 directly suppresses BNIP3 expression to protect against hypoxia-induced cell death. EMBO J 30: 3397–3415, 2011. doi: 10.1038/emboj.2011.248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fiacco E, Castagnetti F, Bianconi V, Madaro L, De Bardi M, Nazio F, D’Amico A, Bertini E, Cecconi F, Puri PL, Latella L. Autophagy regulates satellite cell ability to regenerate normal and dystrophic muscles. Cell Death Differ 23: 1839–1849, 2016. doi: 10.1038/cdd.2016.70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fimia GM, Stoykova A, Romagnoli A, Giunta L, Di Bartolomeo S, Nardacci R, Corazzari M, Fuoco C, Ucar A, Schwartz P, Gruss P, Piacentini M, Chowdhury K, Cecconi F. Ambra1 regulates autophagy and development of the nervous system. Nature 447: 1121–1125, 2007. doi: 10.1038/nature05925. [DOI] [PubMed] [Google Scholar]
- 74.Fisher AB. Oxygen therapy. Side effects and toxicity. Am Rev Respir Dis 122, 5P2: 61–69, 1980. doi: 10.1164/arrd.1980.122.5P2.61. [DOI] [PubMed] [Google Scholar]
- 75.Folmes CD, Nelson TJ, Martinez-Fernandez A, Arrell DK, Lindor JZ, Dzeja PP, Ikeda Y, Perez-Terzic C, Terzic A. Somatic oxidative bioenergetics transitions into pluripotency-dependent glycolysis to facilitate nuclear reprogramming. Cell Metab 14: 264–271, 2011. doi: 10.1016/j.cmet.2011.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Franco A, Kitsis RN, Fleischer JA, Gavathiotis E, Kornfeld OS, Gong G, Biris N, Benz A, Qvit N, Donnelly SK, Chen Y, Mennerick S, Hodgson L, Mochly-Rosen D, Dorn GW II. Correcting mitochondrial fusion by manipulating mitofusin conformations. Nature 540: 74–79, 2016. doi: 10.1038/nature20156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Freischmidt A, Wieland T, Richter B, Ruf W, Schaeffer V, Müller K, Marroquin N, Nordin F, Hübers A, Weydt P, Pinto S, Press R, Millecamps S, Molko N, Bernard E, Desnuelle C, Soriani MH, Dorst J, Graf E, Nordström U, Feiler MS, Putz S, Boeckers TM, Meyer T, Winkler AS, Winkelman J, de Carvalho M, Thal DR, Otto M, Brännström T, Volk AE, Kursula P, Danzer KM, Lichtner P, Dikic I, Meitinger T, Ludolph AC, Strom TM, Andersen PM, Weishaupt JH. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat Neurosci 18: 631–636, 2015. doi: 10.1038/nn.4000. [DOI] [PubMed] [Google Scholar]
- 78.Fujiwara M, Marusawa H, Wang HQ, Iwai A, Ikeuchi K, Imai Y, Kataoka A, Nukina N, Takahashi R, Chiba T. Parkin as a tumor suppressor gene for hepatocellular carcinoma. Oncogene 27: 6002–6011, 2008. doi: 10.1038/onc.2008.199. [DOI] [PubMed] [Google Scholar]
- 79.Galan JM, Haguenauer-Tsapis R. Ubiquitin lys63 is involved in ubiquitination of a yeast plasma membrane protein. EMBO J 16: 5847–5854, 1997. doi: 10.1093/emboj/16.19.5847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Galluzzi L, Bravo-San Pedro JM, Levine B, Green DR, Kroemer G. Pharmacological modulation of autophagy: therapeutic potential and persisting obstacles. Nat Rev Drug Discov 16: 487–511, 2017. doi: 10.1038/nrd.2017.22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gan B, Hu J, Jiang S, Liu Y, Sahin E, Zhuang L, Fletcher-Sananikone E, Colla S, Wang YA, Chin L, Depinho RA. Lkb1 regulates quiescence and metabolic homeostasis of haematopoietic stem cells. Nature 468: 701–704, 2010. doi: 10.1038/nature09595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Gao F, Yang J, Wang D, Li C, Fu Y, Wang H, He W, Zhang J. Mitophagy in Parkinson’s Disease: Pathogenic and Therapeutic Implications. Front Neurol 8: 527, 2017. doi: 10.3389/fneur.2017.00527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.García-Prat L, Martínez-Vicente M, Perdiguero E, Ortet L, Rodríguez-Ubreva J, Rebollo E, Ruiz-Bonilla V, Gutarra S, Ballestar E, Serrano AL, Sandri M, Muñoz-Cánoves P. Autophagy maintains stemness by preventing senescence. Nature 529: 37–42, 2016. doi: 10.1038/nature16187. [DOI] [PubMed] [Google Scholar]
- 84.Gegg ME, Cooper JM, Chau KY, Rojo M, Schapira AH, Taanman JW. Mitofusin 1 and mitofusin 2 are ubiquitinated in a PINK1/parkin-dependent manner upon induction of mitophagy. Hum Mol Genet 19: 4861–4870, 2010. doi: 10.1093/hmg/ddq419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Geisler S, Holmström KM, Skujat D, Fiesel FC, Rothfuss OC, Kahle PJ, Springer W. PINK1/Parkin-mediated mitophagy is dependent on VDAC1 and p62/SQSTM1. Nat Cell Biol 12: 119–131, 2010. doi: 10.1038/ncb2012. [DOI] [PubMed] [Google Scholar]
- 86.Gelmetti V, De Rosa P, Torosantucci L, Marini ES, Romagnoli A, Di Rienzo M, Arena G, Vignone D, Fimia GM, Valente EM. PINK1 and BECN1 relocalize at mitochondria-associated membranes during mitophagy and promote ER-mitochondria tethering and autophagosome formation. Autophagy 13: 654–669, 2017. doi: 10.1080/15548627.2016.1277309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Glick D, Zhang W, Beaton M, Marsboom G, Gruber M, Simon MC, Hart J, Dorn GW II, Brady MJ, Macleod KF. BNip3 regulates mitochondrial function and lipid metabolism in the liver. Mol Cell Biol 32: 2570–2584, 2012. doi: 10.1128/MCB.00167-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Goldberg MS, Fleming SM, Palacino JJ, Cepeda C, Lam HA, Bhatnagar A, Meloni EG, Wu N, Ackerson LC, Klapstein GJ, Gajendiran M, Roth BL, Chesselet MF, Maidment NT, Levine MS, Shen J. Parkin-deficient mice exhibit nigrostriatal deficits but not loss of dopaminergic neurons. J Biol Chem 278: 43628–43635, 2003. doi: 10.1074/jbc.M308947200. [DOI] [PubMed] [Google Scholar]
- 89.Gomes LC, Di Benedetto G, Scorrano L. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat Cell Biol 13: 589–598, 2011. doi: 10.1038/ncb2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Gong G, Song M, Csordas G, Kelly DP, Matkovich SJ, Dorn GW II. Parkin-mediated mitophagy directs perinatal cardiac metabolic maturation in mice. Science 350: aad2459, 2015. doi: 10.1126/science.aad2459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Gray MW, Burger G, Lang BF. Mitochondrial evolution. Science 283: 1476–1481, 1999. doi: 10.1126/science.283.5407.1476. [DOI] [PubMed] [Google Scholar]
- 92.Greene JC, Whitworth AJ, Kuo I, Andrews LA, Feany MB, Pallanck LJ. Mitochondrial pathology and apoptotic muscle degeneration in Drosophila parkin mutants. Proc Natl Acad Sci USA 100: 4078–4083, 2003. doi: 10.1073/pnas.0737556100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Guo M. Drosophila as a model to study mitochondrial dysfunction in Parkinson’s disease. Cold Spring Harb Perspect Med 2: a009944, 2012. doi: 10.1101/cshperspect.a009944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Hamacher-Brady A, Brady NR, Logue SE, Sayen MR, Jinno M, Kirshenbaum LA, Gottlieb RA, Gustafsson AB. Response to myocardial ischemia/reperfusion injury involves Bnip3 and autophagy. Cell Death Differ 14: 146–157, 2007. doi: 10.1038/sj.cdd.4401936. [DOI] [PubMed] [Google Scholar]
- 95.Hamasaki M, Furuta N, Matsuda A, Nezu A, Yamamoto A, Fujita N, Oomori H, Noda T, Haraguchi T, Hiraoka Y, Amano A, Yoshimori T. Autophagosomes form at ER-mitochondria contact sites. Nature 495: 389–393, 2013. doi: 10.1038/nature11910. [DOI] [PubMed] [Google Scholar]
- 96.Hammerling BC, Najor RH, Cortez MQ, Shires SE, Leon LJ, Gonzalez ER, Boassa D, Phan S, Thor A, Jimenez RE, Li H, Kitsis RN, Dorn GW II, Sadoshima J, Ellisman MH, Gustafsson AB. A Rab5 endosomal pathway mediates Parkin-dependent mitochondrial clearance. Nat Commun 8: 14050, 2017. doi: 10.1038/ncomms14050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hammerling BC, Shires SE, Leon LJ, Cortez MQ, Gustafsson AB. Isolation of Rab5-positive endosomes reveals a new mitochondrial degradation pathway utilized by BNIP3 and Parkin. Small GTPases 11: 1–8, 2017. doi: 10.1080/21541248.2017.1342749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Han K, Hassanzadeh S, Singh K, Menazza S, Nguyen TT, Stevens MV, Nguyen A, San H, Anderson SA, Lin Y, Zou J, Murphy E, Sack MN. Parkin regulation of CHOP modulates susceptibility to cardiac endoplasmic reticulum stress. Sci Rep 7: 2093, 2017. doi: 10.1038/s41598-017-02339-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hanna RA, Quinsay MN, Orogo AM, Giang K, Rikka S, Gustafsson AB. Microtubule-associated protein 1 light chain 3 (LC3) interacts with Bnip3 protein to selectively remove endoplasmic reticulum and mitochondria via autophagy. J Biol Chem 287: 19094–19104, 2012. doi: 10.1074/jbc.M111.322933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Harbauer AB, Zahedi RP, Sickmann A, Pfanner N, Meisinger C. The protein import machinery of mitochondria-a regulatory hub in metabolism, stress, and disease. Cell Metab 19: 357–372, 2014. doi: 10.1016/j.cmet.2014.01.010. [DOI] [PubMed] [Google Scholar]
- 101.Harrington CR. The molecular pathology of Alzheimer’s disease. Neuroimaging Clin N Am 22: 11–22, 2012. doi: 10.1016/j.nic.2011.11.003. [DOI] [PubMed] [Google Scholar]
- 102.Heo JM, Ordureau A, Paulo JA, Rinehart J, Harper JW. The PINK1-PARKIN Mitochondrial Ubiquitylation Pathway Drives a Program of OPTN/NDP52 Recruitment and TBK1 Activation to Promote Mitophagy. Mol Cell 60: 7–20, 2015. doi: 10.1016/j.molcel.2015.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ho TT, Warr MR, Adelman ER, Lansinger OM, Flach J, Verovskaya EV, Figueroa ME, Passegué E. Autophagy maintains the metabolism and function of young and old stem cells. Nature 543: 205–210, 2017. doi: 10.1038/nature21388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Honda S, Arakawa S, Nishida Y, Yamaguchi H, Ishii E, Shimizu S. Ulk1-mediated Atg5-independent macroautophagy mediates elimination of mitochondria from embryonic reticulocytes. Nat Commun 5: 4004, 2014. doi: 10.1038/ncomms5004. [DOI] [PubMed] [Google Scholar]
- 105.Hoshino A, Mita Y, Okawa Y, Ariyoshi M, Iwai-Kanai E, Ueyama T, Ikeda K, Ogata T, Matoba S. Cytosolic p53 inhibits Parkin-mediated mitophagy and promotes mitochondrial dysfunction in the mouse heart. Nat Commun 4: 2308, 2013. doi: 10.1038/ncomms3308. [DOI] [PubMed] [Google Scholar]
- 106.Huang P, Galloway CA, Yoon Y. Control of mitochondrial morphology through differential interactions of mitochondrial fusion and fission proteins. PLoS One 6: e20655, 2011. doi: 10.1371/journal.pone.0020655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Huang W, Choi W, Hu W, Mi N, Guo Q, Ma M, Liu M, Tian Y, Lu P, Wang FL, Deng H, Liu L, Gao N, Yu L, Shi Y. Crystal structure and biochemical analyses reveal Beclin 1 as a novel membrane binding protein. Cell Res 22: 473–489, 2012. doi: 10.1038/cr.2012.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Huang X, Sun L, Ji S, Zhao T, Zhang W, Xu J, Zhang J, Wang Y, Wang X, Franzini-Armstrong C, Zheng M, Cheng H. Kissing and nanotunneling mediate intermitochondrial communication in the heart. Proc Natl Acad Sci USA 110: 2846–2851, 2013. doi: 10.1073/pnas.1300741110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Hull TD, Boddu R, Guo L, Tisher CC, Traylor AM, Patel B, Joseph R, Prabhu SD, Suliman HB, Piantadosi CA, Agarwal A, George JF. Heme oxygenase-1 regulates mitochondrial quality control in the heart. JCI Insight 1: e85817, 2016. doi: 10.1172/jci.insight.85817. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Hwang NR, Yim SH, Kim YM, Jeong J, Song EJ, Lee Y, Lee JH, Choi S, Lee KJ. Oxidative modifications of glyceraldehyde-3-phosphate dehydrogenase play a key role in its multiple cellular functions. Biochem J 423: 253–264, 2009. doi: 10.1042/BJ20090854. [DOI] [PubMed] [Google Scholar]
- 111.Hwang S, Disatnik MH, Mochly-Rosen D. Impaired GAPDH-induced mitophagy contributes to the pathology of Huntington’s disease. EMBO Mol Med 7: 1307–1326, 2015. doi: 10.15252/emmm.201505256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Ikeda Y, Shirakabe A, Maejima Y, Zhai P, Sciarretta S, Toli J, Nomura M, Mihara K, Egashira K, Ohishi M, Abdellatif M, Sadoshima J. Endogenous Drp1 mediates mitochondrial autophagy and protects the heart against energy stress. Circ Res 116: 264–278, 2015. doi: 10.1161/CIRCRESAHA.116.303356. [DOI] [PubMed] [Google Scholar]
- 113.Ishihara M, Urushido M, Hamada K, Matsumoto T, Shimamura Y, Ogata K, Inoue K, Taniguchi Y, Horino T, Fujieda M, Fujimoto S, Terada Y. Sestrin-2 and BNIP3 regulate autophagy and mitophagy in renal tubular cells in acute kidney injury. Am J Physiol Renal Physiol 305: F495–F509, 2013. doi: 10.1152/ajprenal.00642.2012. [DOI] [PubMed] [Google Scholar]
- 114.Itakura E, Kishi C, Inoue K, Mizushima N. Beclin 1 forms two distinct phosphatidylinositol 3-kinase complexes with mammalian Atg14 and UVRAG. Mol Biol Cell 19: 5360–5372, 2008. doi: 10.1091/mbc.e08-01-0080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Ito S, Araya J, Kurita Y, Kobayashi K, Takasaka N, Yoshida M, Hara H, Minagawa S, Wakui H, Fujii S, Kojima J, Shimizu K, Numata T, Kawaishi M, Odaka M, Morikawa T, Harada T, Nishimura SL, Kaneko Y, Nakayama K, Kuwano K. PARK2-mediated mitophagy is involved in regulation of HBEC senescence in COPD pathogenesis. Autophagy 11: 547–559, 2015. doi: 10.1080/15548627.2015.1017190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Ittner LM, Götz J. Amyloid-β and tau–a toxic pas de deux in Alzheimer’s disease. Nat Rev Neurosci 12: 67–72, 2011. doi: 10.1038/nrn2967. [DOI] [PubMed] [Google Scholar]
- 117.Jamart C, Naslain D, Gilson H, Francaux M. Higher activation of autophagy in skeletal muscle of mice during endurance exercise in the fasted state. Am J Physiol Endocrinol Metab 305: E964–E974, 2013. doi: 10.1152/ajpendo.00270.2013. [DOI] [PubMed] [Google Scholar]
- 118.Jin SM, Lazarou M, Wang C, Kane LA, Narendra DP, Youle RJ. Mitochondrial membrane potential regulates PINK1 import and proteolytic destabilization by PARL. J Cell Biol 191: 933–942, 2010. doi: 10.1083/jcb.201008084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Jin SM, Youle RJ. The accumulation of misfolded proteins in the mitochondrial matrix is sensed by PINK1 to induce PARK2/Parkin-mediated mitophagy of polarized mitochondria. Autophagy 9: 1750–1757, 2013. doi: 10.4161/auto.26122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ju JS, Jeon SI, Park JY, Lee JY, Lee SC, Cho KJ, Jeong JM. Autophagy plays a role in skeletal muscle mitochondrial biogenesis in an endurance exercise-trained condition. J Physiol Sci 66: 417–430, 2016. doi: 10.1007/s12576-016-0440-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Kagan VE, Jiang J, Huang Z, Tyurina YY, Desbourdes C, Cottet-Rousselle C, Dar HH, Verma M, Tyurin VA, Kapralov AA, Cheikhi A, Mao G, Stolz D, St. Croix CM, Watkins S, Shen Z, Li Y, Greenberg ML, Tokarska-Schlattner M, Boissan M, Lacombe ML, Epand RM, Chu CT, Mallampalli RK, Bayır H, Schlattner U. NDPK-D (NM23-H4)-mediated externalization of cardiolipin enables elimination of depolarized mitochondria by mitophagy. Cell Death Differ 23: 1140–1151, 2016. doi: 10.1038/cdd.2015.160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kane LA, Lazarou M, Fogel AI, Li Y, Yamano K, Sarraf SA, Banerjee S, Youle RJ. PINK1 phosphorylates ubiquitin to activate Parkin E3 ubiquitin ligase activity. J Cell Biol 205: 143–153, 2014. doi: 10.1083/jcb.201402104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Kanki T, Wang K, Cao Y, Baba M, Klionsky DJ. Atg32 is a mitochondrial protein that confers selectivity during mitophagy. Dev Cell 17: 98–109, 2009. doi: 10.1016/j.devcel.2009.06.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Karbowski M, Youle RJ. Regulating mitochondrial outer membrane proteins by ubiquitination and proteasomal degradation. Curr Opin Cell Biol 23: 476–482, 2011. doi: 10.1016/j.ceb.2011.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Kataoka T, Holler N, Micheau O, Martinon F, Tinel A, Hofmann K, Tschopp J. Bcl-rambo, a novel Bcl-2 homologue that induces apoptosis via its unique C-terminal extension. J Biol Chem 276: 19548–19554, 2001. doi: 10.1074/jbc.M010520200. [DOI] [PubMed] [Google Scholar]
- 126.Kaushik S, Cuervo AM. The coming of age of chaperone-mediated autophagy. Nat Rev Mol Cell Biol 19: 365–381, 2018. doi: 10.1038/s41580-018-0001-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Kawamura N, Sun-Wada GH, Aoyama M, Harada A, Takasuga S, Sasaki T, Wada Y. Delivery of endosomes to lysosomes via microautophagy in the visceral endoderm of mouse embryos. Nat Commun 3: 1071, 2012. doi: 10.1038/ncomms2069. [DOI] [PubMed] [Google Scholar]
- 128.Kerr JS, Adriaanse BA, Greig NH, Mattson MP, Cader MZ, Bohr VA, Fang EF. Mitophagy and Alzheimer’s Disease: Cellular and Molecular Mechanisms. Trends Neurosci 40: 151–166, 2017. doi: 10.1016/j.tins.2017.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Khacho M, Slack RS. Mitochondrial activity in the regulation of stem cell self-renewal and differentiation. Curr Opin Cell Biol 49: 1–8, 2017. doi: 10.1016/j.ceb.2017.11.003. [DOI] [PubMed] [Google Scholar]
- 130.Kihara A, Kabeya Y, Ohsumi Y, Yoshimori T. Beclin-phosphatidylinositol 3-kinase complex functions at the trans-Golgi network. EMBO Rep 2: 330–335, 2001. doi: 10.1093/embo-reports/kve061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Kim MJ, Bae SH, Ryu JC, Kwon Y, Oh JH, Kwon J, Moon JS, Kim K, Miyawaki A, Lee MG, Shin J, Kim YS, Kim CH, Ryter SW, Choi AM, Rhee SG, Ryu JH, Yoon JH. SESN2/sestrin2 suppresses sepsis by inducing mitophagy and inhibiting NLRP3 activation in macrophages. Autophagy 12: 1272–1291, 2016. doi: 10.1080/15548627.2016.1183081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kim NC, Tresse E, Kolaitis RM, Molliex A, Thomas RE, Alami NH, Wang B, Joshi A, Smith RB, Ritson GP, Winborn BJ, Moore J, Lee JY, Yao TP, Pallanck L, Kundu M, Taylor JP. VCP is essential for mitochondrial quality control by PINK1/Parkin and this function is impaired by VCP mutations. [Erratum in Neuron 78: 403, 2013.] Neuron 78: 65–80, 2013. doi: 10.1016/j.neuron.2013.02.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Kissová I, Salin B, Schaeffer J, Bhatia S, Manon S, Camougrand N. Selective and non-selective autophagic degradation of mitochondria in yeast. Autophagy 3: 329–336, 2007. doi: 10.4161/auto.4034. [DOI] [PubMed] [Google Scholar]
- 134.Kitada T, Asakawa S, Hattori N, Matsumine H, Yamamura Y, Minoshima S, Yokochi M, Mizuno Y, Shimizu N. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature 392: 605–608, 1998. doi: 10.1038/33416. [DOI] [PubMed] [Google Scholar]
- 135.Klionsky DJ, Emr SD. Autophagy as a regulated pathway of cellular degradation. Science 290: 1717–1721, 2000. doi: 10.1126/science.290.5497.1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Klionsky DJ, Schulman BA. Dynamic regulation of macroautophagy by distinctive ubiquitin-like proteins. Nat Struct Mol Biol 21: 336–345, 2014. doi: 10.1038/nsmb.2787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Kobayashi K, Araya J, Minagawa S, Hara H, Saito N, Kadota T, Sato N, Yoshida M, Tsubouchi K, Kurita Y, Ito S, Fujita Y, Takasaka N, Utsumi H, Yanagisawa H, Hashimoto M, Wakui H, Kojima J, Shimizu K, Numata T, Kawaishi M, Kaneko Y, Asano H, Yamashita M, Odaka M, Morikawa T, Nakayama K, Kuwano K. Involvement of PARK2-Mediated Mitophagy in Idiopathic Pulmonary Fibrosis Pathogenesis. J Immunol 197: 504–516, 2016. doi: 10.4049/jimmunol.1600265. [DOI] [PubMed] [Google Scholar]
- 138.Koentjoro B, Park JS, Ha AD, Sue CM. Phenotypic variability of parkin mutations in single kindred. Mov Disord 27: 1299–1303, 2012. doi: 10.1002/mds.25041. [DOI] [PubMed] [Google Scholar]
- 139.Koentjoro B, Park JS, Sue CM. Nix restores mitophagy and mitochondrial function to protect against PINK1/Parkin-related Parkinson’s disease. Sci Rep 7: 44373, 2017. doi: 10.1038/srep44373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Kondo-Okamoto N, Noda NN, Suzuki SW, Nakatogawa H, Takahashi I, Matsunami M, Hashimoto A, Inagaki F, Ohsumi Y, Okamoto K. Autophagy-related protein 32 acts as autophagic degron and directly initiates mitophagy. J Biol Chem 287: 10631–10638, 2012. doi: 10.1074/jbc.M111.299917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Koshiba T, Detmer SA, Kaiser JT, Chen H, McCaffery JM, Chan DC. Structural basis of mitochondrial tethering by mitofusin complexes. Science 305: 858–862, 2004. doi: 10.1126/science.1099793. [DOI] [PubMed] [Google Scholar]
- 142.Koury MJ, Koury ST, Kopsombut P, Bondurant MC. In vitro maturation of nascent reticulocytes to erythrocytes. Blood 105: 2168–2174, 2005. doi: 10.1182/blood-2004-02-0616. [DOI] [PubMed] [Google Scholar]
- 143.Koyano F, Okatsu K, Kosako H, Tamura Y, Go E, Kimura M, Kimura Y, Tsuchiya H, Yoshihara H, Hirokawa T, Endo T, Fon EA, Trempe JF, Saeki Y, Tanaka K, Matsuda N. Ubiquitin is phosphorylated by PINK1 to activate parkin. Nature 510: 162–166, 2014. doi: 10.1038/nature13392. [DOI] [PubMed] [Google Scholar]
- 144.Kraft C, Peter M, Hofmann K. Selective autophagy: ubiquitin-mediated recognition and beyond. Nat Cell Biol 12: 836–841, 2010. doi: 10.1038/ncb0910-836. [DOI] [PubMed] [Google Scholar]
- 145.Kuang Y, Ma K, Zhou C, Ding P, Zhu Y, Chen Q, Xia B. Structural basis for the phosphorylation of FUNDC1 LIR as a molecular switch of mitophagy. Autophagy 12: 2363–2373, 2016. doi: 10.1080/15548627.2016.1238552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kubli DA, Ycaza JE, Gustafsson AB. Bnip3 mediates mitochondrial dysfunction and cell death through Bax and Bak. Biochem J 405: 407–415, 2007. doi: 10.1042/BJ20070319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Kubli DA, Zhang X, Lee Y, Hanna RA, Quinsay MN, Nguyen CK, Jimenez R, Petrosyan S, Murphy AN, Gustafsson AB. Parkin protein deficiency exacerbates cardiac injury and reduces survival following myocardial infarction. J Biol Chem 288: 915–926, 2013. doi: 10.1074/jbc.M112.411363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Kujoth GC, Hiona A, Pugh TD, Someya S, Panzer K, Wohlgemuth SE, Hofer T, Seo AY, Sullivan R, Jobling WA, Morrow JD, Van Remmen H, Sedivy JM, Yamasoba T, Tanokura M, Weindruch R, Leeuwenburgh C, Prolla TA. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 309: 481–484, 2005. doi: 10.1126/science.1112125. [DOI] [PubMed] [Google Scholar]
- 149.Kuma A, Hatano M, Matsui M, Yamamoto A, Nakaya H, Yoshimori T, Ohsumi Y, Tokuhisa T, Mizushima N. The role of autophagy during the early neonatal starvation period. Nature 432: 1032–1036, 2004. doi: 10.1038/nature03029. [DOI] [PubMed] [Google Scholar]
- 150.Kumar M, Seeger W, Voswinckel R. Senescence-associated secretory phenotype and its possible role in chronic obstructive pulmonary disease. Am J Respir Cell Mol Biol 51: 323–333, 2014. doi: 10.1165/rcmb.2013-0382PS. [DOI] [PubMed] [Google Scholar]
- 151.Kundu M, Lindsten T, Yang CY, Wu J, Zhao F, Zhang J, Selak MA, Ney PA, Thompson CB. Ulk1 plays a critical role in the autophagic clearance of mitochondria and ribosomes during reticulocyte maturation. Blood 112: 1493–1502, 2008. doi: 10.1182/blood-2008-02-137398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Laker RC, Drake JC, Wilson RJ, Lira VA, Lewellen BM, Ryall KA, Fisher CC, Zhang M, Saucerman JJ, Goodyear LJ, Kundu M, Yan Z. Ampk phosphorylation of Ulk1 is required for targeting of mitochondria to lysosomes in exercise-induced mitophagy. Nat Commun 8: 548, 2017. doi: 10.1038/s41467-017-00520-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Lampert MA, Orogo AM, Najor RH, Hammerling BC, Leon LJ, Wang BJ, Kim T, Sussman MA, Gustafsson ÅB. Nix and Fundc1-Mediated Programmed Mitophagy is Required for Mitochondrial Network Remodeling during Cardiac Progenitor Cell Differentiation. Autophagy. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Larson-Casey JL, Deshane JS, Ryan AJ, Thannickal VJ, Carter AB. Macrophage Akt1 Kinase-Mediated Mitophagy Modulates Apoptosis Resistance and Pulmonary Fibrosis. Immunity 44: 582–596, 2016. doi: 10.1016/j.immuni.2016.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Lazarou M, Jin SM, Kane LA, Youle RJ. Role of PINK1 binding to the TOM complex and alternate intracellular membranes in recruitment and activation of the E3 ligase Parkin. Dev Cell 22: 320–333, 2012. doi: 10.1016/j.devcel.2011.12.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Lazarou M, Sliter DA, Kane LA, Sarraf SA, Wang C, Burman JL, Sideris DP, Fogel AI, Youle RJ. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature 524: 309–314, 2015. doi: 10.1038/nature14893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Lee SB, Kim JJ, Nam HJ, Gao B, Yin P, Qin B, Yi SY, Ham H, Evans D, Kim SH, Zhang J, Deng M, Liu T, Zhang H, Billadeau DD, Wang L, Giaime E, Shen J, Pang YP, Jen J, van Deursen JM, Lou Z. Parkin Regulates Mitosis and Genomic Stability through Cdc20/Cdh1. Mol Cell 60: 21–34, 2015. doi: 10.1016/j.molcel.2015.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Lee Y, Dawson VL, Dawson TM. Animal models of Parkinson’s disease: vertebrate genetics. Cold Spring Harb Perspect Med 2: a009324, 2012. doi: 10.1101/cshperspect.a009324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Lee Y, Stevens DA, Kang SU, Jiang H, Lee YI, Ko HS, Scarffe LA, Umanah GE, Kang H, Ham S, Kam TI, Allen K, Brahmachari S, Kim JW, Neifert S, Yun SP, Fiesel FC, Springer W, Dawson VL, Shin JH, Dawson TM. PINK1 Primes Parkin-Mediated Ubiquitination of PARIS in Dopaminergic Neuronal Survival. Cell Reports 18: 918–932, 2017. doi: 10.1016/j.celrep.2016.12.090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev Cell 6: 463–477, 2004. doi: 10.1016/S1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- 161.Levine B, Liu R, Dong X, Zhong Q. Beclin orthologs: integrative hubs of cell signaling, membrane trafficking, and physiology. Trends Cell Biol 25: 533–544, 2015. doi: 10.1016/j.tcb.2015.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Li Q, Liu Y, Sun M. Autophagy and Alzheimer’s Disease. Cell Mol Neurobiol 37: 377–388, 2017. doi: 10.1007/s10571-016-0386-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Li Q, Qi F, Meng X, Zhu C, Gao Y. Mst1 regulates colorectal cancer stress response via inhibiting Bnip3-related mitophagy by activation of JNK/p53 pathway. Cell Biol Toxicol 34: 263–277, 2018. doi: 10.1007/s10565-017-9417-6. [DOI] [PubMed] [Google Scholar]
- 164.Li WW, Li J, Bao JK. Microautophagy: lesser-known self-eating. Cell Mol Life Sci 69: 1125–1136, 2012. doi: 10.1007/s00018-011-0865-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Liang J, Xu ZX, Ding Z, Lu Y, Yu Q, Werle KD, Zhou G, Park YY, Peng G, Gambello MJ, Mills GB. Myristoylation confers noncanonical AMPK functions in autophagy selectivity and mitochondrial surveillance. Nat Commun 6: 7926, 2015. doi: 10.1038/ncomms8926. [DOI] [PubMed] [Google Scholar]
- 166.Lipinski MM, Zheng B, Lu T, Yan Z, Py BF, Ng A, Xavier RJ, Li C, Yankner BA, Scherzer CR, Yuan J. Genome-wide analysis reveals mechanisms modulating autophagy in normal brain aging and in Alzheimer’s disease. Proc Natl Acad Sci USA 107: 14164–14169, 2010. doi: 10.1073/pnas.1009485107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Lira VA, Okutsu M, Zhang M, Greene NP, Laker RC, Breen DS, Hoehn KL, Yan Z. Autophagy is required for exercise training-induced skeletal muscle adaptation and improvement of physical performance. FASEB J 27: 4184–4193, 2013. doi: 10.1096/fj.13-228486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Lister G, Walter TK, Versmold HT, Dallman PR, Rudolph AM. Oxygen delivery in lambs: cardiovascular and hematologic development. Am J Physiol Heart Circ Physiol 237: H668–H675, 1979. [DOI] [PubMed] [Google Scholar]
- 169.Liu L, Feng D, Chen G, Chen M, Zheng Q, Song P, Ma Q, Zhu C, Wang R, Qi W, Huang L, Xue P, Li B, Wang X, Jin H, Wang J, Yang F, Liu P, Zhu Y, Sui S, Chen Q. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol 14: 177–185, 2012. doi: 10.1038/ncb2422. [DOI] [PubMed] [Google Scholar]
- 170.Liu X, Weaver D, Shirihai O, Hajnóczky G. Mitochondrial ‘kiss-and-run’: interplay between mitochondrial motility and fusion-fission dynamics. EMBO J 28: 3074–3089, 2009. doi: 10.1038/emboj.2009.255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Lo Verso F, Carnio S, Vainshtein A, Sandri M. Autophagy is not required to sustain exercise and PRKAA1/AMPK activity but is important to prevent mitochondrial damage during physical activity. Autophagy 10: 1883–1894, 2014. doi: 10.4161/auto.32154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Lombardi D, Soldati T, Riederer MA, Goda Y, Zerial M, Pfeffer SR. Rab9 functions in transport between late endosomes and the trans Golgi network. EMBO J 12: 677–682, 1993. doi: 10.1002/j.1460-2075.1993.tb05701.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Ma T, Li J, Xu Y, Yu C, Xu T, Wang H, Liu K, Cao N, Nie BM, Zhu SY, Xu S, Li K, Wei WG, Wu Y, Guan KL, Ding S. Atg5-independent autophagy regulates mitochondrial clearance and is essential for iPSC reprogramming. Nat Cell Biol 17: 1379–1387, 2015. doi: 10.1038/ncb3256. [DOI] [PubMed] [Google Scholar]
- 174.Magrané J, Cortez C, Gan WB, Manfredi G. Abnormal mitochondrial transport and morphology are common pathological denominators in SOD1 and TDP43 ALS mouse models. Hum Mol Genet 23: 1413–1424, 2014. doi: 10.1093/hmg/ddt528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Mannam P, Shinn AS, Srivastava A, Neamu RF, Walker WE, Bohanon M, Merkel J, Kang MJ, Dela Cruz CS, Ahasic AM, Pisani MA, Trentalange M, West AP, Shadel GS, Elias JA, Lee PJ. MKK3 regulates mitochondrial biogenesis and mitophagy in sepsis-induced lung injury. Am J Physiol Lung Cell Mol Physiol 306: L604–L619, 2014. doi: 10.1152/ajplung.00272.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Manzanillo PS, Ayres JS, Watson RO, Collins AC, Souza G, Rae CS, Schneider DS, Nakamura K, Shiloh MU, Cox JS. The ubiquitin ligase parkin mediates resistance to intracellular pathogens. Nature 501: 512–516, 2013. doi: 10.1038/nature12566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Mao P, Manczak M, Calkins MJ, Truong Q, Reddy TP, Reddy AP, Shirendeb U, Lo HH, Rabinovitch PS, Reddy PH. Mitochondria-targeted catalase reduces abnormal APP processing, amyloid β production and BACE1 in a mouse model of Alzheimer’s disease: implications for neuroprotection and lifespan extension. Hum Mol Genet 21: 2973–2990, 2012. doi: 10.1093/hmg/dds128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Martinez-Vicente M, Talloczy Z, Wong E, Tang G, Koga H, Kaushik S, de Vries R, Arias E, Harris S, Sulzer D, Cuervo AM. Cargo recognition failure is responsible for inefficient autophagy in Huntington’s disease. Nat Neurosci 13: 567–576, 2010. doi: 10.1038/nn.2528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Maruyama H, Morino H, Ito H, Izumi Y, Kato H, Watanabe Y, Kinoshita Y, Kamada M, Nodera H, Suzuki H, Komure O, Matsuura S, Kobatake K, Morimoto N, Abe K, Suzuki N, Aoki M, Kawata A, Hirai T, Kato T, Ogasawara K, Hirano A, Takumi T, Kusaka H, Hagiwara K, Kaji R, Kawakami H. Mutations of optineurin in amyotrophic lateral sclerosis. Nature 465: 223–226, 2010. doi: 10.1038/nature08971. [DOI] [PubMed] [Google Scholar]
- 180.Marzetti E, Csiszar A, Dutta D, Balagopal G, Calvani R, Leeuwenburgh C. Role of mitochondrial dysfunction and altered autophagy in cardiovascular aging and disease: from mechanisms to therapeutics. Am J Physiol Heart Circ Physiol 305: H459–H476, 2013. doi: 10.1152/ajpheart.00936.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Matsuda N, Sato S, Shiba K, Okatsu K, Saisho K, Gautier CA, Sou YS, Saiki S, Kawajiri S, Sato F, Kimura M, Komatsu M, Hattori N, Tanaka K. PINK1 stabilized by mitochondrial depolarization recruits Parkin to damaged mitochondria and activates latent Parkin for mitophagy. J Cell Biol 189: 211–221, 2010. doi: 10.1083/jcb.200910140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Matsui M, Yamamoto A, Kuma A, Ohsumi Y, Mizushima N. Organelle degradation during the lens and erythroid differentiation is independent of autophagy. Biochem Biophys Res Commun 339: 485–489, 2006. doi: 10.1016/j.bbrc.2005.11.044. [DOI] [PubMed] [Google Scholar]
- 183.Matsumoto G, Shimogori T, Hattori N, Nukina N. TBK1 controls autophagosomal engulfment of polyubiquitinated mitochondria through p62/SQSTM1 phosphorylation. Hum Mol Genet 24: 4429–4442, 2015. doi: 10.1093/hmg/ddv179. [DOI] [PubMed] [Google Scholar]
- 184.Mattson MP, Gleichmann M, Cheng A. Mitochondria in neuroplasticity and neurological disorders. Neuron 60: 748–766, 2008. doi: 10.1016/j.neuron.2008.10.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.McCoy MK, Kaganovich A, Rudenko IN, Ding J, Cookson MR. Hexokinase activity is required for recruitment of parkin to depolarized mitochondria. Hum Mol Genet 23: 145–156, 2014. doi: 10.1093/hmg/ddt407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.McLelland GL, Soubannier V, Chen CX, McBride HM, Fon EA. Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J 33: 282–295, 2014. doi: 10.1002/embj.201385902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Meissner C, Lorenz H, Weihofen A, Selkoe DJ, Lemberg MK. The mitochondrial intramembrane protease PARL cleaves human Pink1 to regulate Pink1 trafficking. J Neurochem 117: 856–867, 2011. doi: 10.1111/j.1471-4159.2011.07253.x. [DOI] [PubMed] [Google Scholar]
- 188.Mills EL, Kelly B, O’Neill LAJ. Mitochondria are the powerhouses of immunity. Nat Immunol 18: 488–498, 2017. doi: 10.1038/ni.3704. [DOI] [PubMed] [Google Scholar]
- 189.Minowa-Nozawa A, Nozawa T, Okamoto-Furuta K, Kohda H, Nakagawa I. Rab35 GTPase recruits NDP52 to autophagy targets. EMBO J 36: 2790–2807, 2017. doi: 10.15252/embj.201796463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Mira MT, Alcaïs A, Van Thuc N, Moraes MO, Di Flumeri C, Hong Thai V, Chi Phuong M, Thu Huong N, Ngoc Ba N, Xuan Khoa P, Sarno EN, Alter A, Montpetit A, Moraes ME, Moraes JR, Doré C, Gallant CJ, Lepage P, Verner A, van de Vosse E, Hudson TJ, Abel L, Schurr E. Susceptibility to leprosy is associated with PARK2 and PACRG. Nature 427: 636–640, 2004. doi: 10.1038/nature02326. [DOI] [PubMed] [Google Scholar]
- 191.Mishra P, Chan DC. Mitochondrial dynamics and inheritance during cell division, development and disease. Nat Rev Mol Cell Biol 15: 634–646, 2014. doi: 10.1038/nrm3877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Misko A, Jiang S, Wegorzewska I, Milbrandt J, Baloh RH. Mitofusin 2 is necessary for transport of axonal mitochondria and interacts with the Miro/Milton complex. J Neurosci 30: 4232–4240, 2010. doi: 10.1523/JNEUROSCI.6248-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Mizumura K, Cloonan SM, Nakahira K, Bhashyam AR, Cervo M, Kitada T, Glass K, Owen CA, Mahmood A, Washko GR, Hashimoto S, Ryter SW, Choi AM. Mitophagy-dependent necroptosis contributes to the pathogenesis of COPD. J Clin Invest 124: 3987–4003, 2014. doi: 10.1172/JCI74985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Moore AS, Holzbaur EL. Dynamic recruitment and activation of ALS-associated TBK1 with its target optineurin are required for efficient mitophagy. Proc Natl Acad Sci USA 113: E3349–E3358, 2016. doi: 10.1073/pnas.1523810113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Murakawa T, Yamaguchi O, Hashimoto A, Hikoso S, Takeda T, Oka T, Yasui H, Ueda H, Akazawa Y, Nakayama H, Taneike M, Misaka T, Omiya S, Shah AM, Yamamoto A, Nishida K, Ohsumi Y, Okamoto K, Sakata Y, Otsu K. Bcl-2-like protein 13 is a mammalian Atg32 homologue that mediates mitophagy and mitochondrial fragmentation. Nat Commun 6: 7527, 2015. doi: 10.1038/ncomms8527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Murphy MP. How mitochondria produce reactive oxygen species. Biochem J 417: 1–13, 2009. doi: 10.1042/BJ20081386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Nakada D, Saunders TL, Morrison SJ. Lkb1 regulates cell cycle and energy metabolism in haematopoietic stem cells. Nature 468: 653–658, 2010. doi: 10.1038/nature09571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Nakahira K, Haspel JA, Rathinam VA, Lee SJ, Dolinay T, Lam HC, Englert JA, Rabinovitch M, Cernadas M, Kim HP, Fitzgerald KA, Ryter SW, Choi AM. Autophagy proteins regulate innate immune responses by inhibiting the release of mitochondrial DNA mediated by the NALP3 inflammasome. Nat Immunol 12: 222–230, 2011. doi: 10.1038/ni.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Nakamoto RK, Baylis Scanlon JA, Al-Shawi MK. The rotary mechanism of the ATP synthase. Arch Biochem Biophys 476: 43–50, 2008. doi: 10.1016/j.abb.2008.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Nakamura N, Hirose S. Regulation of mitochondrial morphology by USP30, a deubiquitinating enzyme present in the mitochondrial outer membrane. Mol Biol Cell 19: 1903–1911, 2008. doi: 10.1091/mbc.e07-11-1103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Narendra D, Kane LA, Hauser DN, Fearnley IM, Youle RJ. p62/SQSTM1 is required for Parkin-induced mitochondrial clustering but not mitophagy; VDAC1 is dispensable for both. Autophagy 6: 1090–1106, 2010. doi: 10.4161/auto.6.8.13426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Narendra D, Tanaka A, Suen DF, Youle RJ. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J Cell Biol 183: 795–803, 2008. doi: 10.1083/jcb.200809125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Narendra DP, Jin SM, Tanaka A, Suen DF, Gautier CA, Shen J, Cookson MR, Youle RJ. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol 8: e1000298, 2010. doi: 10.1371/journal.pbio.1000298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Nassir F, Ibdah JA. Role of mitochondria in alcoholic liver disease. World J Gastroenterol 20: 2136–2142, 2014. doi: 10.3748/wjg.v20.i9.2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Neuspiel M, Schauss AC, Braschi E, Zunino R, Rippstein P, Rachubinski RA, Andrade-Navarro MA, McBride HM. Cargo-selected transport from the mitochondria to peroxisomes is mediated by vesicular carriers. Curr Biol 18: 102–108, 2008. doi: 10.1016/j.cub.2007.12.038. [DOI] [PubMed] [Google Scholar]
- 206.Newton K, Matsumoto ML, Wertz IE, Kirkpatrick DS, Lill JR, Tan J, Dugger D, Gordon N, Sidhu SS, Fellouse FA, Komuves L, French DM, Ferrando RE, Lam C, Compaan D, Yu C, Bosanac I, Hymowitz SG, Kelley RF, Dixit VM. Ubiquitin chain editing revealed by polyubiquitin linkage-specific antibodies. Cell 134: 668–678, 2008. doi: 10.1016/j.cell.2008.07.039. [DOI] [PubMed] [Google Scholar]
- 207.Ney PA. Mitochondrial autophagy: origins, significance, and role of BNIP3 and NIX. Biochim Biophys Acta 1853, Pt B: 2775–2783, 2015. doi: 10.1016/j.bbamcr.2015.02.022. [DOI] [PubMed] [Google Scholar]
- 208.Ni HM, Bockus A, Boggess N, Jaeschke H, Ding WX. Activation of autophagy protects against acetaminophen-induced hepatotoxicity. Hepatology 55: 222–232, 2012. doi: 10.1002/hep.24690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Nishida Y, Arakawa S, Fujitani K, Yamaguchi H, Mizuta T, Kanaseki T, Komatsu M, Otsu K, Tsujimoto Y, Shimizu S. Discovery of Atg5/Atg7-independent alternative macroautophagy. Nature 461: 654–658, 2009. doi: 10.1038/nature08455. [DOI] [PubMed] [Google Scholar]
- 210.Nixon RA, Wegiel J, Kumar A, Yu WH, Peterhoff C, Cataldo A, Cuervo AM. Extensive involvement of autophagy in Alzheimer disease: an immuno-electron microscopy study. J Neuropathol Exp Neurol 64: 113–122, 2005. doi: 10.1093/jnen/64.2.113. [DOI] [PubMed] [Google Scholar]
- 211.Novak I, Kirkin V, McEwan DG, Zhang J, Wild P, Rozenknop A, Rogov V, Löhr F, Popovic D, Occhipinti A, Reichert AS, Terzic J, Dötsch V, Ney PA, Dikic I. Nix is a selective autophagy receptor for mitochondrial clearance. EMBO Rep 11: 45–51, 2010. doi: 10.1038/embor.2009.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Oakhill JS, Chen ZP, Scott JW, Steel R, Castelli LA, Ling N, Macaulay SL, Kemp BE. β-Subunit myristoylation is the gatekeeper for initiating metabolic stress sensing by AMP-activated protein kinase (AMPK). Proc Natl Acad Sci USA 107: 19237–19241, 2010. doi: 10.1073/pnas.1009705107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Okamoto K, Kondo-Okamoto N, Ohsumi Y. Mitochondria-anchored receptor Atg32 mediates degradation of mitochondria via selective autophagy. Dev Cell 17: 87–97, 2009. doi: 10.1016/j.devcel.2009.06.013. [DOI] [PubMed] [Google Scholar]
- 214.Okatsu K, Saisho K, Shimanuki M, Nakada K, Shitara H, Sou YS, Kimura M, Sato S, Hattori N, Komatsu M, Tanaka K, Matsuda N. p62/SQSTM1 cooperates with Parkin for perinuclear clustering of depolarized mitochondria. Genes Cells 15: 887–900, 2010. doi: 10.1111/j.1365-2443.2010.01426.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 215.Okatsu K, Uno M, Koyano F, Go E, Kimura M, Oka T, Tanaka K, Matsuda N. A dimeric PINK1-containing complex on depolarized mitochondria stimulates Parkin recruitment. J Biol Chem 288: 36372–36384, 2013. doi: 10.1074/jbc.M113.509653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 216.Ordureau A, Sarraf SA, Duda DM, Heo JM, Jedrychowski MP, Sviderskiy VO, Olszewski JL, Koerber JT, Xie T, Beausoleil SA, Wells JA, Gygi SP, Schulman BA, Harper JW. Quantitative proteomics reveal a feedforward mechanism for mitochondrial PARKIN translocation and ubiquitin chain synthesis. [Erratum in Mol Cell 56: 462, 2014.] Mol Cell 56: 360–375, 2014. doi: 10.1016/j.molcel.2014.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Orogo AM, Gonzalez ER, Kubli DA, Baptista IL, Ong SB, Prolla TA, Sussman MA, Murphy AN, Gustafsson AB. Accumulation of Mitochondrial DNA Mutations Disrupts Cardiac Progenitor Cell Function and Reduces Survival. [Correction in J Biol Chem 282: 11348, 2017.] J Biol Chem 290: 22061–22075, 2015. doi: 10.1074/jbc.M115.649657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 218.Orsi A, Razi M, Dooley HC, Robinson D, Weston AE, Collinson LM, Tooze SA. Dynamic and transient interactions of Atg9 with autophagosomes, but not membrane integration, are required for autophagy. Mol Biol Cell 23: 1860–1873, 2012. doi: 10.1091/mbc.e11-09-0746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Otera H, Ishihara N, Mihara K. New insights into the function and regulation of mitochondrial fission. Biochim Biophys Acta 1833: 1256–1268, 2013. doi: 10.1016/j.bbamcr.2013.02.002. [DOI] [PubMed] [Google Scholar]
- 220.Palacino JJ, Sagi D, Goldberg MS, Krauss S, Motz C, Wacker M, Klose J, Shen J. Mitochondrial dysfunction and oxidative damage in parkin-deficient mice. J Biol Chem 279: 18614–18622, 2004. doi: 10.1074/jbc.M401135200. [DOI] [PubMed] [Google Scholar]
- 221.Palikaras K, Lionaki E, Tavernarakis N. Coordination of mitophagy and mitochondrial biogenesis during ageing in C. elegans. Nature 521: 525–528, 2015. doi: 10.1038/nature14300. [DOI] [PubMed] [Google Scholar]
- 222.Pankiv S, Clausen TH, Lamark T, Brech A, Bruun JA, Outzen H, Øvervatn A, Bjørkøy G, Johansen T. p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J Biol Chem 282: 24131–24145, 2007. doi: 10.1074/jbc.M702824200. [DOI] [PubMed] [Google Scholar]
- 223.Park J, Lee SB, Lee S, Kim Y, Song S, Kim S, Bae E, Kim J, Shong M, Kim JM, Chung J. Mitochondrial dysfunction in Drosophila PINK1 mutants is complemented by parkin. Nature 441: 1157–1161, 2006. doi: 10.1038/nature04788. [DOI] [PubMed] [Google Scholar]
- 224.Patel AS, Song JW, Chu SG, Mizumura K, Osorio JC, Shi Y, El-Chemaly S, Lee CG, Rosas IO, Elias JA, Choi AM, Morse D. Epithelial cell mitochondrial dysfunction and PINK1 are induced by transforming growth factor-beta1 in pulmonary fibrosis. PLoS One 10: e0121246, 2015. doi: 10.1371/journal.pone.0121246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Pickrell AM, Huang CH, Kennedy SR, Ordureau A, Sideris DP, Hoekstra JG, Harper JW, Youle RJ. Endogenous Parkin Preserves Dopaminergic Substantia Nigral Neurons following Mitochondrial DNA Mutagenic Stress. Neuron 87: 371–381, 2015. doi: 10.1016/j.neuron.2015.06.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 226.Pickrell AM, Youle RJ. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 85: 257–273, 2015. doi: 10.1016/j.neuron.2014.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Politi Y, Gal L, Kalifa Y, Ravid L, Elazar Z, Arama E. Paternal mitochondrial destruction after fertilization is mediated by a common endocytic and autophagic pathway in Drosophila. Dev Cell 29: 305–320, 2014. doi: 10.1016/j.devcel.2014.04.005. [DOI] [PubMed] [Google Scholar]
- 228.Poole AC, Thomas RE, Yu S, Vincow ES, Pallanck L. The mitochondrial fusion-promoting factor mitofusin is a substrate of the PINK1/parkin pathway. PLoS One 5: e10054, 2010. doi: 10.1371/journal.pone.0010054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Pua HH, Guo J, Komatsu M, He YW. Autophagy is essential for mitochondrial clearance in mature T lymphocytes. J Immunol 182: 4046–4055, 2009. doi: 10.4049/jimmunol.0801143. [DOI] [PubMed] [Google Scholar]
- 230.Purhonen P, Pursiainen K, Reunanen H. Effects of brefeldin A on autophagy in cultured rat fibroblasts. Eur J Cell Biol 74: 63–67, 1997. [PubMed] [Google Scholar]
- 231.Rabinowitz M, Swift H. Mitochondrial nucleic acids and their relation to the biogenesis of mitochondria. Physiol Rev 50: 376–427, 1970. doi: 10.1152/physrev.1970.50.3.376. [DOI] [PubMed] [Google Scholar]
- 232.Rakovic A, Grünewald A, Kottwitz J, Brüggemann N, Pramstaller PP, Lohmann K, Klein C. Mutations in PINK1 and Parkin impair ubiquitination of Mitofusins in human fibroblasts. PLoS One 6: e16746, 2011. doi: 10.1371/journal.pone.0016746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Rambold AS, Kostelecky B, Elia N, Lippincott-Schwartz J. Tubular network formation protects mitochondria from autophagosomal degradation during nutrient starvation. Proc Natl Acad Sci USA 108: 10190–10195, 2011. doi: 10.1073/pnas.1107402108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 234.Reggiori F, Shintani T, Chong H, Nair U, Klionsky DJ. Atg9 cycles between mitochondria and the pre-autophagosomal structure in yeasts. Autophagy 1: 101–109, 2005. doi: 10.4161/auto.1.2.1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 235.Regula KM, Ens K, Kirshenbaum LA. Inducible expression of BNIP3 provokes mitochondrial defects and hypoxia-mediated cell death of ventricular myocytes. Circ Res 91: 226–231, 2002. doi: 10.1161/01.RES.0000029232.42227.16. [DOI] [PubMed] [Google Scholar]
- 236.Richter B, Sliter DA, Herhaus L, Stolz A, Wang C, Beli P, Zaffagnini G, Wild P, Martens S, Wagner SA, Youle RJ, Dikic I. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc Natl Acad Sci USA 113: 4039–4044, 2016. doi: 10.1073/pnas.1523926113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 237.Rikka S, Quinsay MN, Thomas RL, Kubli DA, Zhang X, Murphy AN, Gustafsson AB. Bnip3 impairs mitochondrial bioenergetics and stimulates mitochondrial turnover. Cell Death Differ 18: 721–731, 2011. doi: 10.1038/cdd.2010.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 238.Roberts P, Moshitch-Moshkovitz S, Kvam E, O’Toole E, Winey M, Goldfarb DS. Piecemeal microautophagy of nucleus in Saccharomyces cerevisiae. Mol Biol Cell 14: 129–141, 2003. doi: 10.1091/mbc.e02-08-0483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 239.Rocha AG, Franco A, Krezel AM, Rumsey JM, Alberti JM, Knight WC, Biris N, Zacharioudakis E, Janetka JW, Baloh RH, Kitsis RN, Mochly-Rosen D, Townsend RR, Gavathiotis E, Dorn GW II. MFN2 agonists reverse mitochondrial defects in preclinical models of Charcot-Marie-Tooth disease type 2A. Science 360: 336–341, 2018. doi: 10.1126/science.aao1785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 240.Rojansky R, Cha MY, Chan DC. Elimination of paternal mitochondria in mouse embryos occurs through autophagic degradation dependent on PARKIN and MUL1. eLife 5: e17896, 2016. doi: 10.7554/eLife.17896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 241.Ross CA, Tabrizi SJ. Huntington’s disease: from molecular pathogenesis to clinical treatment. Lancet Neurol 10: 83–98, 2011. doi: 10.1016/S1474-4422(10)70245-3. [DOI] [PubMed] [Google Scholar]
- 242.Rui YN, Xu Z, Patel B, Chen Z, Chen D, Tito A, David G, Sun Y, Stimming EF, Bellen HJ, Cuervo AM, Zhang S. Huntingtin functions as a scaffold for selective macroautophagy. Nat Cell Biol 17: 262–275, 2015. doi: 10.1038/ncb3101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 243.Saita S, Shirane M, Nakayama KI. Selective escape of proteins from the mitochondria during mitophagy. Nat Commun 4: 1410, 2013. doi: 10.1038/ncomms2400. [DOI] [PubMed] [Google Scholar]
- 244.Sandoval H, Thiagarajan P, Dasgupta SK, Schumacher A, Prchal JT, Chen M, Wang J. Essential role for Nix in autophagic maturation of erythroid cells. Nature 454: 232–235, 2008. doi: 10.1038/nature07006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 245.Santucci R, Sinibaldi F, Polticelli F, Fiorucci L. Role of cardiolipin in mitochondrial diseases and apoptosis. Curr Med Chem 21: 2702–2714, 2014. doi: 10.2174/0929867321666140414112156. [DOI] [PubMed] [Google Scholar]
- 246.Sarraf SA, Raman M, Guarani-Pereira V, Sowa ME, Huttlin EL, Gygi SP, Harper JW. Landscape of the PARKIN-dependent ubiquitylome in response to mitochondrial depolarization. Nature 496: 372–376, 2013. doi: 10.1038/nature12043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 247.Sasaki S, Iwata M. Mitochondrial alterations in the spinal cord of patients with sporadic amyotrophic lateral sclerosis. J Neuropathol Exp Neurol 66: 10–16, 2007. doi: 10.1097/nen.0b013e31802c396b. [DOI] [PubMed] [Google Scholar]
- 248.Sato M, Sato K. Degradation of paternal mitochondria by fertilization-triggered autophagy in C. elegans embryos. Science 334: 1141–1144, 2011. doi: 10.1126/science.1210333. [DOI] [PubMed] [Google Scholar]
- 249.Schapira AH. Mitochondria in the aetiology and pathogenesis of Parkinson’s disease. Lancet Neurol 7: 97–109, 2008. doi: 10.1016/S1474-4422(07)70327-7. [DOI] [PubMed] [Google Scholar]
- 250.Scheele C, Petrovic N, Faghihi MA, Lassmann T, Fredriksson K, Rooyackers O, Wahlestedt C, Good L, Timmons JA. The human PINK1 locus is regulated in vivo by a non-coding natural antisense RNA during modulation of mitochondrial function. BMC Genomics 8: 74, 2007. doi: 10.1186/1471-2164-8-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 251.Scherzinger E, Sittler A, Schweiger K, Heiser V, Lurz R, Hasenbank R, Bates GP, Lehrach H, Wanker EE. Self-assembly of polyglutamine-containing huntingtin fragments into amyloid-like fibrils: implications for Huntington’s disease pathology. Proc Natl Acad Sci USA 96: 4604–4609, 1999. doi: 10.1073/pnas.96.8.4604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 252.Schlame M, Ren M, Xu Y, Greenberg ML, Haller I. Molecular symmetry in mitochondrial cardiolipins. Chem Phys Lipids 138: 38–49, 2005. doi: 10.1016/j.chemphyslip.2005.08.002. [DOI] [PubMed] [Google Scholar]
- 253.Schon EA, Gilkerson RW. Functional complementation of mitochondrial DNAs: mobilizing mitochondrial genetics against dysfunction. Biochim Biophys Acta 1800: 245–249, 2010. doi: 10.1016/j.bbagen.2009.07.007. [DOI] [PubMed] [Google Scholar]
- 254.Schuck S, Gallagher CM, Walter P. ER-phagy mediates selective degradation of endoplasmic reticulum independently of the core autophagy machinery. J Cell Sci 127: 4078–4088, 2014. doi: 10.1242/jcs.154716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 255.Schulte J, Littleton JT. The biological function of the Huntingtin protein and its relevance to Huntington’s Disease pathology. Curr Trends Neurol 5: 65–78, 2011. [PMC free article] [PubMed] [Google Scholar]
- 256.Schwarten M, Mohrlüder J, Ma P, Stoldt M, Thielmann Y, Stangler T, Hersch N, Hoffmann B, Merkel R, Willbold D. Nix directly binds to GABARAP: a possible crosstalk between apoptosis and autophagy. Autophagy 5: 690–698, 2009. doi: 10.4161/auto.5.5.8494. [DOI] [PubMed] [Google Scholar]
- 257.Schweers RL, Zhang J, Randall MS, Loyd MR, Li W, Dorsey FC, Kundu M, Opferman JT, Cleveland JL, Miller JL, Ney PA. NIX is required for programmed mitochondrial clearance during reticulocyte maturation. Proc Natl Acad Sci USA 104: 19500–19505, 2007. doi: 10.1073/pnas.0708818104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 258.Shadel GS, Horvath TL. Mitochondrial ROS signaling in organismal homeostasis. Cell 163: 560–569, 2015. doi: 10.1016/j.cell.2015.10.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 259.Shah SP, Roth A, Goya R, Oloumi A, Ha G, Zhao Y, Turashvili G, Ding J, Tse K, Haffari G, Bashashati A, Prentice LM, Khattra J, Burleigh A, Yap D, Bernard V, McPherson A, Shumansky K, Crisan A, Giuliany R, Heravi-Moussavi A, Rosner J, Lai D, Birol I, Varhol R, Tam A, Dhalla N, Zeng T, Ma K, Chan SK, Griffith M, Moradian A, Cheng SW, Morin GB, Watson P, Gelmon K, Chia S, Chin SF, Curtis C, Rueda OM, Pharoah PD, Damaraju S, Mackey J, Hoon K, Harkins T, Tadigotla V, Sigaroudinia M, Gascard P, Tlsty T, Costello JF, Meyer IM, Eaves CJ, Wasserman WW, Jones S, Huntsman D, Hirst M, Caldas C, Marra MA, Aparicio S. The clonal and mutational evolution spectrum of primary triple-negative breast cancers. Nature 486: 395–399, 2012. doi: 10.1038/nature10933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 260.Sharma M, Ioannidis JP, Aasly JO, Annesi G, Brice A, Bertram L, Bozi M, Barcikowska M, Crosiers D, Clarke CE, Facheris MF, Farrer M, Garraux G, Gispert S, Auburger G, Vilariño-Güell C, Hadjigeorgiou GM, Hicks AA, Hattori N, Jeon BS, Jamrozik Z, Krygowska-Wajs A, Lesage S, Lill CM, Lin JJ, Lynch T, Lichtner P, Lang AE, Libioulle C, Murata M, Mok V, Jasinska-Myga B, Mellick GD, Morrison KE, Meitnger T, Zimprich A, Opala G, Pramstaller PP, Pichler I, Park SS, Quattrone A, Rogaeva E, Ross OA, Stefanis L, Stockton JD, Satake W, Silburn PA, Strom TM, Theuns J, Tan EK, Toda T, Tomiyama H, Uitti RJ, Van Broeckhoven C, Wirdefeldt K, Wszolek Z, Xiromerisiou G, Yomono HS, Yueh KC, Zhao Y, Gasser T, Maraganore D, Krüger R; GEOPD consortium . A multi-centre clinico-genetic analysis of the VPS35 gene in Parkinson disease indicates reduced penetrance for disease-associated variants. J Med Genet 49: 721–726, 2012. doi: 10.1136/jmedgenet-2012-101155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 261.Shimura H, Hattori N, Kubo S, Mizuno Y, Asakawa S, Minoshima S, Shimizu N, Iwai K, Chiba T, Tanaka K, Suzuki T. Familial Parkinson disease gene product, parkin, is a ubiquitin-protein ligase. Nat Genet 25: 302–305, 2000. doi: 10.1038/77060. [DOI] [PubMed] [Google Scholar]
- 262.Shin JH, Ko HS, Kang H, Lee Y, Lee YI, Pletinkova O, Troconso JC, Dawson VL, Dawson TM. PARIS (ZNF746) repression of PGC-1α contributes to neurodegeneration in Parkinson’s disease. Cell 144: 689–702, 2011. doi: 10.1016/j.cell.2011.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 263.Shirakabe A, Ikeda Y, Sciarretta S, Zablocki DK, Sadoshima J. Aging and Autophagy in the Heart. Circ Res 118: 1563–1576, 2016. doi: 10.1161/CIRCRESAHA.116.307474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 264.Shirakabe A, Zhai P, Ikeda Y, Saito T, Maejima Y, Hsu CP, Nomura M, Egashira K, Levine B, Sadoshima J. Drp1-Dependent Mitochondrial Autophagy Plays a Protective Role Against Pressure Overload-Induced Mitochondrial Dysfunction and Heart Failure. Circulation 133: 1249–1263, 2016. doi: 10.1161/CIRCULATIONAHA.115.020502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 265.Shirane M, Nakayama KI. Inherent calcineurin inhibitor FKBP38 targets Bcl-2 to mitochondria and inhibits apoptosis. Nat Cell Biol 5: 28–37, 2003. doi: 10.1038/ncb894. [DOI] [PubMed] [Google Scholar]
- 266.Shirihai OS, Song M, Dorn GW II. How mitochondrial dynamism orchestrates mitophagy. Circ Res 116: 1835–1849, 2015. doi: 10.1161/CIRCRESAHA.116.306374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 267.Siddall HK, Yellon DM, Ong SB, Mukherjee UA, Burke N, Hall AR, Angelova PR, Ludtmann MH, Deas E, Davidson SM, Mocanu MM, Hausenloy DJ. Loss of PINK1 increases the heart’s vulnerability to ischemia-reperfusion injury. PLoS One 8: e62400, 2013. doi: 10.1371/journal.pone.0062400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 268.Sigismund S, Confalonieri S, Ciliberto A, Polo S, Scita G, Di Fiore PP. Endocytosis and signaling: cell logistics shape the eukaryotic cell plan. Physiol Rev 92: 273–366, 2012. doi: 10.1152/physrev.00005.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 269.Simon MC, Keith B. The role of oxygen availability in embryonic development and stem cell function. Nat Rev Mol Cell Biol 9: 285–296, 2008. doi: 10.1038/nrm2354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 270.Sinha RA, Singh BK, Zhou J, Wu Y, Farah BL, Ohba K, Lesmana R, Gooding J, Bay BH, Yen PM. Thyroid hormone induction of mitochondrial activity is coupled to mitophagy via ROS-AMPK-ULK1 signaling. Autophagy 11: 1341–1357, 2015. doi: 10.1080/15548627.2015.1061849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 271.Smith LA, Cornelius VR, Plummer CJ, Levitt G, Verrill M, Canney P, Jones A. Cardiotoxicity of anthracycline agents for the treatment of cancer: systematic review and meta-analysis of randomised controlled trials. BMC Cancer 10: 337, 2010. doi: 10.1186/1471-2407-10-337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 272.Smolich JJ, Berger PJ, Walker AM. Interrelation between ventricular function, myocardial blood flow, and O2 consumption changes at birth in lambs. Am J Physiol Heart Circ Physiol 270: H741–H749, 1996. doi: 10.1152/ajpheart.1996.270.2.H741. [DOI] [PubMed] [Google Scholar]
- 273.Song M, Chen Y, Gong G, Murphy E, Rabinovitch PS, Dorn GW II. Super-suppression of mitochondrial reactive oxygen species signaling impairs compensatory autophagy in primary mitophagic cardiomyopathy. Circ Res 115: 348–353, 2014. doi: 10.1161/CIRCRESAHA.115.304384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 274.Song M, Dorn GW II. Mitoconfusion: noncanonical functioning of dynamism factors in static mitochondria of the heart. Cell Metab 21: 195–205, 2015. doi: 10.1016/j.cmet.2014.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 275.Song M, Franco A, Fleischer JA, Zhang L, Dorn GW II. Abrogating Mitochondrial Dynamics in Mouse Hearts Accelerates Mitochondrial Senescence. Cell Metab 26: 872–883.e5, 2017. doi: 10.1016/j.cmet.2017.09.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 276.Song M, Gong G, Burelle Y, Gustafsson AB, Kitsis RN, Matkovich SJ, Dorn GW II. Interdependence of Parkin-Mediated Mitophagy and Mitochondrial Fission in Adult Mouse Hearts. Circ Res 117: 346–351, 2015. doi: 10.1161/CIRCRESAHA.117.306859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 277.Song M, Mihara K, Chen Y, Scorrano L, Dorn GW II. Mitochondrial fission and fusion factors reciprocally orchestrate mitophagic culling in mouse hearts and cultured fibroblasts. Cell Metab 21: 273–286, 2015. doi: 10.1016/j.cmet.2014.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 278.Soubannier V, McLelland GL, Zunino R, Braschi E, Rippstein P, Fon EA, McBride HM. A vesicular transport pathway shuttles cargo from mitochondria to lysosomes. Curr Biol 22: 135–141, 2012. doi: 10.1016/j.cub.2011.11.057. [DOI] [PubMed] [Google Scholar]
- 279.Sousa-Victor P, Gutarra S, García-Prat L, Rodriguez-Ubreva J, Ortet L, Ruiz-Bonilla V, Jardí M, Ballestar E, González S, Serrano AL, Perdiguero E, Muñoz-Cánoves P. Geriatric muscle stem cells switch reversible quiescence into senescence. Nature 506: 316–321, 2014. doi: 10.1038/nature13013. [DOI] [PubMed] [Google Scholar]
- 280.Steinberg GR, Kemp BE. AMPK in Health and Disease. Physiol Rev 89: 1025–1078, 2009. doi: 10.1152/physrev.00011.2008. [DOI] [PubMed] [Google Scholar]
- 281.Stevens DA, Lee Y, Kang HC, Lee BD, Lee YI, Bower A, Jiang H, Kang SU, Andrabi SA, Dawson VL, Shin JH, Dawson TM. Parkin loss leads to PARIS-dependent declines in mitochondrial mass and respiration. Proc Natl Acad Sci USA 112: 11696–11701, 2015. doi: 10.1073/pnas.1500624112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 282.Stolz A, Dikic I. PINK1-PARKIN interplay: down to ubiquitin phosphorylation. Mol Cell 56: 341–342, 2014. doi: 10.1016/j.molcel.2014.10.022. [DOI] [PubMed] [Google Scholar]
- 283.Strappazzon F, Nazio F, Corrado M, Cianfanelli V, Romagnoli A, Fimia GM, Campello S, Nardacci R, Piacentini M, Campanella M, Cecconi F. AMBRA1 is able to induce mitophagy via LC3 binding, regardless of PARKIN and p62/SQSTM1 [Erratum in Cell Death Differ 22: 517, 2015.]. Cell Death Differ 22: 419–432, 2015. doi: 10.1038/cdd.2014.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 284.Strappazzon F, Vietri-Rudan M, Campello S, Nazio F, Florenzano F, Fimia GM, Piacentini M, Levine B, Cecconi F. Mitochondrial BCL-2 inhibits AMBRA1-induced autophagy. EMBO J 30: 1195–1208, 2011. doi: 10.1038/emboj.2011.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 285.Sugiura A, McLelland GL, Fon EA, McBride HM. A new pathway for mitochondrial quality control: mitochondrial-derived vesicles. EMBO J 33: 2142–2156, 2014. doi: 10.15252/embj.201488104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 286.Suliman HB, Kraft B, Bartz R, Chen L, Welty-Wolf KE, Piantadosi CA. Mitochondrial quality control in alveolar epithelial cells damaged by S. aureus pneumonia in mice. Am J Physiol Lung Cell Mol Physiol 313: L699–L709, 2017. doi: 10.1152/ajplung.00197.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 287.Sutovsky P, Moreno RD, Ramalho-Santos J, Dominko T, Simerly C, Schatten G. Ubiquitin tag for sperm mitochondria. Nature 402: 371–372, 1999. doi: 10.1038/46466. [DOI] [PubMed] [Google Scholar]
- 288.Suzuki HI, Kiyono K, Miyazono K. Regulation of autophagy by transforming growth factor-β (TGF-β) signaling. Autophagy 6: 645–647, 2010. doi: 10.4161/auto.6.5.12046. [DOI] [PubMed] [Google Scholar]
- 289.Taegtmeyer H, Sen S, Vela D. Return to the fetal gene program: a suggested metabolic link to gene expression in the heart. Ann N Y Acad Sci 1188: 191–198, 2010. doi: 10.1111/j.1749-6632.2009.05100.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 290.Tanaka A, Cleland MM, Xu S, Narendra DP, Suen DF, Karbowski M, Youle RJ. Proteasome and p97 mediate mitophagy and degradation of mitofusins induced by Parkin. J Cell Biol 191: 1367–1380, 2010. doi: 10.1083/jcb.201007013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 291.Taneike M, Yamaguchi O, Nakai A, Hikoso S, Takeda T, Mizote I, Oka T, Tamai T, Oyabu J, Murakawa T, Nishida K, Shimizu T, Hori M, Komuro I, Takuji Shirasawa TS, Mizushima N, Otsu K. Inhibition of autophagy in the heart induces age-related cardiomyopathy. Autophagy 6: 600–606, 2010. doi: 10.4161/auto.6.5.11947. [DOI] [PubMed] [Google Scholar]
- 292.Tian W, Li W, Chen Y, Yan Z, Huang X, Zhuang H, Zhong W, Chen Y, Wu W, Lin C, Chen H, Hou X, Zhang L, Sui S, Zhao B, Hu Z, Li L, Feng D. Phosphorylation of ULK1 by AMPK regulates translocation of ULK1 to mitochondria and mitophagy. FEBS Lett 589: 1847–1854, 2015. doi: 10.1016/j.febslet.2015.05.020. [DOI] [PubMed] [Google Scholar]
- 293.Tisdale EJ. Glyceraldehyde-3-phosphate dehydrogenase is phosphorylated by protein kinase Ciota/lambda and plays a role in microtubule dynamics in the early secretory pathway. J Biol Chem 277: 3334–3341, 2002. doi: 10.1074/jbc.M109744200. [DOI] [PubMed] [Google Scholar]
- 294.Tocchi A, Quarles EK, Basisty N, Gitari L, Rabinovitch PS. Mitochondrial dysfunction in cardiac aging. Biochim Biophys Acta 1847: 1424–1433, 2015. doi: 10.1016/j.bbabio.2015.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 295.Tofaris GK, Kim HT, Hourez R, Jung JW, Kim KP, Goldberg AL. Ubiquitin ligase Nedd4 promotes alpha-synuclein degradation by the endosomal-lysosomal pathway. Proc Natl Acad Sci USA 108: 17004–17009, 2011. doi: 10.1073/pnas.1109356108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 296.Trempe JF, Sauvé V, Grenier K, Seirafi M, Tang MY, Ménade M, Al-Abdul-Wahid S, Krett J, Wong K, Kozlov G, Nagar B, Fon EA, Gehring K. Structure of parkin reveals mechanisms for ubiquitin ligase activation. Science 340: 1451–1455, 2013. doi: 10.1126/science.1237908. [DOI] [PubMed] [Google Scholar]
- 297.Trifunovic A, Wredenberg A, Falkenberg M, Spelbrink JN, Rovio AT, Bruder CE, Bohlooly-Y M, Gidlöf S, Oldfors A, Wibom R, Törnell J, Jacobs HT, Larsson NG. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 429: 417–423, 2004. doi: 10.1038/nature02517. [DOI] [PubMed] [Google Scholar]
- 298.Tristan C, Shahani N, Sedlak TW, Sawa A. The diverse functions of GAPDH: views from different subcellular compartments. Cell Signal 23: 317–323, 2011. doi: 10.1016/j.cellsig.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 299.Twig G, Elorza A, Molina AJ, Mohamed H, Wikstrom JD, Walzer G, Stiles L, Haigh SE, Katz S, Las G, Alroy J, Wu M, Py BF, Yuan J, Deeney JT, Corkey BE, Shirihai OS. Fission and selective fusion govern mitochondrial segregation and elimination by autophagy. EMBO J 27: 433–446, 2008. doi: 10.1038/sj.emboj.7601963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 300.Valente EM, Abou-Sleiman PM, Caputo V, Muqit MM, Harvey K, Gispert S, Ali Z, Del Turco D, Bentivoglio AR, Healy DG, Albanese A, Nussbaum R, González-Maldonado R, Deller T, Salvi S, Cortelli P, Gilks WP, Latchman DS, Harvey RJ, Dallapiccola B, Auburger G, Wood NW. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science 304: 1158–1160, 2004. doi: 10.1126/science.1096284. [DOI] [PubMed] [Google Scholar]
- 301.Van Humbeeck C, Cornelissen T, Hofkens H, Mandemakers W, Gevaert K, De Strooper B, Vandenberghe W. Parkin interacts with Ambra1 to induce mitophagy. J Neurosci 31: 10249–10261, 2011. doi: 10.1523/JNEUROSCI.1917-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 302.Vande Velde C, Cizeau J, Dubik D, Alimonti J, Brown T, Israels S, Hakem R, Greenberg AH. BNIP3 and genetic control of necrosis-like cell death through the mitochondrial permeability transition pore. Mol Cell Biol 20: 5454–5468, 2000. doi: 10.1128/MCB.20.15.5454-5468.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 303.Vandenabeele P, Galluzzi L, Vanden Berghe T, Kroemer G. Molecular mechanisms of necroptosis: an ordered cellular explosion. Nat Rev Mol Cell Biol 11: 700–714, 2010. doi: 10.1038/nrm2970. [DOI] [PubMed] [Google Scholar]
- 304.Vazquez-Martin A, Van den Haute C, Cufí S, Corominas-Faja B, Cuyàs E, Lopez-Bonet E, Rodriguez-Gallego E, Fernández-Arroyo S, Joven J, Baekelandt V, Menendez JA. Mitophagy-driven mitochondrial rejuvenation regulates stem cell fate. Aging (Albany NY) 8: 1330–1352, 2016. doi: 10.18632/aging.100976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 305.Veeriah S, Taylor BS, Meng S, Fang F, Yilmaz E, Vivanco I, Janakiraman M, Schultz N, Hanrahan AJ, Pao W, Ladanyi M, Sander C, Heguy A, Holland EC, Paty PB, Mischel PS, Liau L, Cloughesy TF, Mellinghoff IK, Solit DB, Chan TA. Somatic mutations of the Parkinson’s disease-associated gene PARK2 in glioblastoma and other human malignancies. Nat Genet 42: 77–82, 2010. doi: 10.1038/ng.491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 306.Vives-Bauza C, Zhou C, Huang Y, Cui M, de Vries RL, Kim J, May J, Tocilescu MA, Liu W, Ko HS, Magrané J, Moore DJ, Dawson VL, Grailhe R, Dawson TM, Li C, Tieu K, Przedborski S. PINK1-dependent recruitment of Parkin to mitochondria in mitophagy. Proc Natl Acad Sci USA 107: 378–383, 2010. doi: 10.1073/pnas.0911187107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 307.Wallace DC. Mitochondrial DNA variation in human radiation and disease. Cell 163: 33–38, 2015. doi: 10.1016/j.cell.2015.08.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 308.Wang H, Song P, Du L, Tian W, Yue W, Liu M, Li D, Wang B, Zhu Y, Cao C, Zhou J, Chen Q. Parkin ubiquitinates Drp1 for proteasome-dependent degradation: implication of dysregulated mitochondrial dynamics in Parkinson disease. J Biol Chem 286: 11649–11658, 2011. doi: 10.1074/jbc.M110.144238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 309.Wang K, Zhou LY, Wang JX, Wang Y, Sun T, Zhao B, Yang YJ, An T, Long B, Li N, Liu CY, Gong Y, Gao JN, Dong YH, Zhang J, Li PF. E2F1-dependent miR-421 regulates mitochondrial fragmentation and myocardial infarction by targeting Pink1. Nat Commun 6: 7619, 2015. doi: 10.1038/ncomms8619. [DOI] [PubMed] [Google Scholar]
- 310.Wang S, Bellen HJ. The retromer complex in development and disease. Development 142: 2392–2396, 2015. doi: 10.1242/dev.123737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 311.Wang W, Wang X, Fujioka H, Hoppel C, Whone AL, Caldwell MA, Cullen PJ, Liu J, Zhu X. Parkinson’s disease-associated mutant VPS35 causes mitochondrial dysfunction by recycling DLP1 complexes. Nat Med 22: 54–63, 2016. doi: 10.1038/nm.3983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 312.Wang X, Winter D, Ashrafi G, Schlehe J, Wong YL, Selkoe D, Rice S, Steen J, LaVoie MJ, Schwarz TL. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 147: 893–906, 2011. doi: 10.1016/j.cell.2011.10.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 313.Wang Y, Serricchio M, Jauregui M, Shanbhag R, Stoltz T, Di Paolo CT, Kim PK, McQuibban GA. Deubiquitinating enzymes regulate PARK2-mediated mitophagy. Autophagy 11: 595–606, 2015. doi: 10.1080/15548627.2015.1034408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 314.Wei Y, Chiang WC, Sumpter R Jr, Mishra P, Levine B. Prohibitin 2 Is an Inner Mitochondrial Membrane Mitophagy Receptor. Cell 168: 224–238.e10, 2017. doi: 10.1016/j.cell.2016.11.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 315.Weidberg H, Shvets E, Shpilka T, Shimron F, Shinder V, Elazar Z. LC3 and GATE-16/GABARAP subfamilies are both essential yet act differently in autophagosome biogenesis. EMBO J 29: 1792–1802, 2010. doi: 10.1038/emboj.2010.74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 316.Wikstrom JD, Twig G, Shirihai OS. What can mitochondrial heterogeneity tell us about mitochondrial dynamics and autophagy? Int J Biochem Cell Biol 41: 1914–1927, 2009. doi: 10.1016/j.biocel.2009.06.006. [DOI] [PubMed] [Google Scholar]
- 317.Wild P, Farhan H, McEwan DG, Wagner S, Rogov VV, Brady NR, Richter B, Korac J, Waidmann O, Choudhary C, Dötsch V, Bumann D, Dikic I. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science 333: 228–233, 2011. doi: 10.1126/science.1205405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 318.Williams JA, Ni HM, Ding Y, Ding WX. Parkin regulates mitophagy and mitochondrial function to protect against alcohol-induced liver injury and steatosis in mice. Am J Physiol Gastrointest Liver Physiol 309: G324–G340, 2015. doi: 10.1152/ajpgi.00108.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 319.Willinger T, Flavell RA. Canonical autophagy dependent on the class III phosphoinositide-3 kinase Vps34 is required for naive T-cell homeostasis. Proc Natl Acad Sci USA 109: 8670–8675, 2012. doi: 10.1073/pnas.1205305109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 320.Wolters PJ, Collard HR, Jones KD. Pathogenesis of idiopathic pulmonary fibrosis. Annu Rev Pathol 9: 157–179, 2014. doi: 10.1146/annurev-pathol-012513-104706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 321.Wong YC, Holzbaur EL. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc Natl Acad Sci USA 111: E4439–E4448, 2014. doi: 10.1073/pnas.1405752111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 322.Wu J, Ocampo A, Belmonte JCI. Cellular Metabolism and Induced Pluripotency. Cell 166: 1371–1385, 2016. doi: 10.1016/j.cell.2016.08.008. [DOI] [PubMed] [Google Scholar]
- 323.Wu W, Tian W, Hu Z, Chen G, Huang L, Li W, Zhang X, Xue P, Zhou C, Liu L, Zhu Y, Zhang X, Li L, Zhang L, Sui S, Zhao B, Feng D. ULK1 translocates to mitochondria and phosphorylates FUNDC1 to regulate mitophagy. EMBO Rep 15: 566–575, 2014. doi: 10.1002/embr.201438501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 324.Xia HG, Najafov A, Geng J, Galan-Acosta L, Han X, Guo Y, Shan B, Zhang Y, Norberg E, Zhang T, Pan L, Liu J, Coloff JL, Ofengeim D, Zhu H, Wu K, Cai Y, Yates JR, Zhu Z, Yuan J, Vakifahmetoglu-Norberg H. Degradation of HK2 by chaperone-mediated autophagy promotes metabolic catastrophe and cell death. [Correction in J Cell Biol 212: 881, 2016.] J Cell Biol 210: 705–716, 2015. doi: 10.1083/jcb.201503044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 325.Xiang G, Yang L, Long Q, Chen K, Tang H, Wu Y, Liu Z, Zhou Y, Qi J, Zheng L, Liu W, Ying Z, Fan W, Shi H, Li H, Lin X, Gao M, Liu J, Bao F, Li L, Duan L, Li M, Liu X. BNIP3L-dependent mitophagy accounts for mitochondrial clearance during 3 factors-induced somatic cell reprogramming. Autophagy 13: 1543–1555, 2017. doi: 10.1080/15548627.2017.1338545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 326.Xu X, Hua Y, Nair S, Bucala R, Ren J. Macrophage migration inhibitory factor deletion exacerbates pressure overload-induced cardiac hypertrophy through mitigating autophagy. Hypertension 63: 490–499, 2014. doi: 10.1161/HYPERTENSIONAHA.113.02219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 327.Xue L, Fletcher GC, Tolkovsky AM. Mitochondria are selectively eliminated from eukaryotic cells after blockade of caspases during apoptosis. Curr Biol 11: 361–365, 2001. doi: 10.1016/S0960-9822(01)00100-2. [DOI] [PubMed] [Google Scholar]
- 328.Yamaguchi O, Murakawa T, Nishida K, Otsu K. Receptor-mediated mitophagy. J Mol Cell Cardiol 95: 50–56, 2016. doi: 10.1016/j.yjmcc.2016.03.010. [DOI] [PubMed] [Google Scholar]
- 329.Yamano K, Youle RJ. PINK1 is degraded through the N-end rule pathway. Autophagy 9: 1758–1769, 2013. doi: 10.4161/auto.24633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 330.Yang D, Oyaizu Y, Oyaizu H, Olsen GJ, Woese CR. Mitochondrial origins. Proc Natl Acad Sci USA 82: 4443–4447, 1985. doi: 10.1073/pnas.82.13.4443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 331.Yang JY, Yang WY. Bit-by-bit autophagic removal of parkin-labelled mitochondria. Nat Commun 4: 2428, 2013. doi: 10.1038/ncomms3428. [DOI] [PubMed] [Google Scholar]
- 332.Yang Z, Klionsky DJ. Eaten alive: a history of macroautophagy. Nat Cell Biol 12: 814–822, 2010. doi: 10.1038/ncb0910-814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 333.Yao J, Irwin RW, Zhao L, Nilsen J, Hamilton RT, Brinton RD. Mitochondrial bioenergetic deficit precedes Alzheimer’s pathology in female mouse model of Alzheimer’s disease. Proc Natl Acad Sci USA 106: 14670–14675, 2009. doi: 10.1073/pnas.0903563106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 334.Ye X, Sun X, Starovoytov V, Cai Q. Parkin-mediated mitophagy in mutant hAPP neurons and Alzheimer’s disease patient brains. Hum Mol Genet 24: 2938–2951, 2015. doi: 10.1093/hmg/ddv056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 335.Yogalingam G, Hwang S, Ferreira JC, Mochly-Rosen D. Glyceraldehyde-3-phosphate dehydrogenase (GAPDH) phosphorylation by protein kinase Cδ (PKCδ) inhibits mitochondria elimination by lysosomal-like structures following ischemia and reoxygenation-induced injury. J Biol Chem 288: 18947–18960, 2013. doi: 10.1074/jbc.M113.466870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 336.Yoshii SR, Kishi C, Ishihara N, Mizushima N. Parkin mediates proteasome-dependent protein degradation and rupture of the outer mitochondrial membrane. J Biol Chem 286: 19630–19640, 2011. doi: 10.1074/jbc.M110.209338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 337.Youle RJ, van der Bliek AM. Mitochondrial fission, fusion, and stress. Science 337: 1062–1065, 2012. doi: 10.1126/science.1219855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 338.Yu WH, Cuervo AM, Kumar A, Peterhoff CM, Schmidt SD, Lee JH, Mohan PS, Mercken M, Farmery MR, Tjernberg LO, Jiang Y, Duff K, Uchiyama Y, Näslund J, Mathews PM, Cataldo AM, Nixon RA. Macroautophagy–a novel Beta-amyloid peptide-generating pathway activated in Alzheimer’s disease. J Cell Biol 171: 87–98, 2005. doi: 10.1083/jcb.200505082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 339.Yussman MG, Toyokawa T, Odley A, Lynch RA, Wu G, Colbert MC, Aronow BJ, Lorenz JN, Dorn GW II. Mitochondrial death protein Nix is induced in cardiac hypertrophy and triggers apoptotic cardiomyopathy. Nat Med 8: 725–730, 2002. doi: 10.1038/nm719. [DOI] [PubMed] [Google Scholar]
- 340.Zhang C, Lin M, Wu R, Wang X, Yang B, Levine AJ, Hu W, Feng Z. Parkin, a p53 target gene, mediates the role of p53 in glucose metabolism and the Warburg effect. Proc Natl Acad Sci USA 108: 16259–16264, 2011. doi: 10.1073/pnas.1113884108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 341.Zhang Q, Zheng J, Qiu J, Wu X, Xu Y, Shen W, Sun M. ALDH2 restores exhaustive exercise-induced mitochondrial dysfunction in skeletal muscle. Biochem Biophys Res Commun 485: 753–760, 2017. doi: 10.1016/j.bbrc.2017.02.124. [DOI] [PubMed] [Google Scholar]
- 342.Zhang T, Xue L, Li L, Tang C, Wan Z, Wang R, Tan J, Tan Y, Han H, Tian R, Billiar TR, Tao WA, Zhang Z. BNIP3 Protein Suppresses PINK1 Kinase Proteolytic Cleavage to Promote Mitophagy. J Biol Chem 291: 21616–21629, 2016. doi: 10.1074/jbc.M116.733410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 343.Zhang Y, Sauler M, Shinn AS, Gong H, Haslip M, Shan P, Mannam P, Lee PJ. Endothelial PINK1 mediates the protective effects of NLRP3 deficiency during lethal oxidant injury. J Immunol 192: 5296–5304, 2014. doi: 10.4049/jimmunol.1400653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 344.Zhong Z, Umemura A, Sanchez-Lopez E, Liang S, Shalapour S, Wong J, He F, Boassa D, Perkins G, Ali SR, McGeough MD, Ellisman MH, Seki E, Gustafsson AB, Hoffman HM, Diaz-Meco MT, Moscat J, Karin M. NF-κB Restricts Inflammasome Activation via Elimination of Damaged Mitochondria. Cell 164: 896–910, 2016. doi: 10.1016/j.cell.2015.12.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 345.Zhou R, Yazdi AS, Menu P, Tschopp J. A role for mitochondria in NLRP3 inflammasome activation. [Corrigendum in Nature 475: 122, 2011.] Nature 469: 221–225, 2011. doi: 10.1038/nature09663. [DOI] [PubMed] [Google Scholar]
- 346.Zimprich A, Benet-Pagès A, Struhal W, Graf E, Eck SH, Offman MN, Haubenberger D, Spielberger S, Schulte EC, Lichtner P, Rossle SC, Klopp N, Wolf E, Seppi K, Pirker W, Presslauer S, Mollenhauer B, Katzenschlager R, Foki T, Hotzy C, Reinthaler E, Harutyunyan A, Kralovics R, Peters A, Zimprich F, Brücke T, Poewe W, Auff E, Trenkwalder C, Rost B, Ransmayr G, Winkelmann J, Meitinger T, Strom TM. A mutation in VPS35, encoding a subunit of the retromer complex, causes late-onset Parkinson disease. Am J Hum Genet 89: 168–175, 2011. doi: 10.1016/j.ajhg.2011.06.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 347.Ziviani E, Tao RN, Whitworth AJ. Drosophila parkin requires PINK1 for mitochondrial translocation and ubiquitinates mitofusin. Proc Natl Acad Sci USA 107: 5018–5023, 2010. doi: 10.1073/pnas.0913485107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 348.Ziviani E, Whitworth AJ. How could Parkin-mediated ubiquitination of mitofusin promote mitophagy? Autophagy 6: 660–662, 2010. doi: 10.4161/auto.6.5.12242. [DOI] [PMC free article] [PubMed] [Google Scholar]