

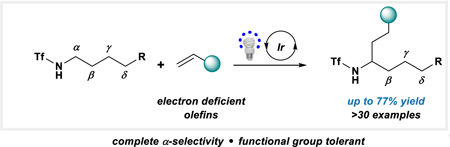
Abstract
The synthetic utility of tertiary amines to oxidatively generate α-amino radicals is well established; however, primary amines remain challenging due to competitive side reactions. This report describes the site-selective α-functionalization of primary amine derivatives through the generation of α-amino radical intermediates. Employing visible light photoredox catalysis, primary sulfonamides are coupled with electron deficient alkenes to efficiently and mildly construct C-C bonds. Interestingly, a divergence between intermolecular HAT catalysis and intramolecular [1,5]-HAT was observed through precise manipulation of the protecting group. This dichotomy was leveraged to achieve excellent α/δ site-selectivity.
Keywords: α-functionalization, hydrogen-atom-transfer, photoredox catalysis, α-amino radical
Graphical abstract
Layout 2:
COMMUNICATION
The judicious choice of nitrogen protecting group allows the site-selective functionalization of primary amines to proceed alpha. Under photoredox catalysis conditions, a variety of alkene acceptors participate in good yields and excellent selectivities.
The formation of α-amino radicals using visible-light photoredox catalysis has garnered significant attention as a mild method to construct C-C bonds.[1] Electron-rich tertiary amines can be oxidized to generate nitrogen radical cations, allowing facile access to α-amino radicals after deprotonation of the α-C-H bond.[ 2 ] Competitive N-alkylation events for primary and secondary amines can hinder the formation of α-amino radicals (Figure 1a).[3] A cleavable functionality at the α-position may be pre-installed to circumvent undesired reactivity by accessing α-amino radicals (Figure 1b).[4] Hydrogen atom transfer (HAT) catalysts have been successfully implemented to achieve α-C-H functionalization of acylated secondary amine derivatives devoid of α-pre-functionalization.[5] Our group has recently reported the selective α-functionalization of primary aliphatic amines to afford γ-lactams under dual photoredox and HAT catalysis utilizing CO2 as an activating group, wherein we identified an electrostatic acceleration of the HAT event between the quinuclidinium cation and carbamate anion. [6] We recognized, however, that ring closure is not always desired and manipulation of primary amines themselves can be challenging in multi-step synthetic planning. For this reason, we further explored the impact of different common activating or protecting groups searching for a broadly applicable α-alkylation of primary amine derivatives.
Figure 1.
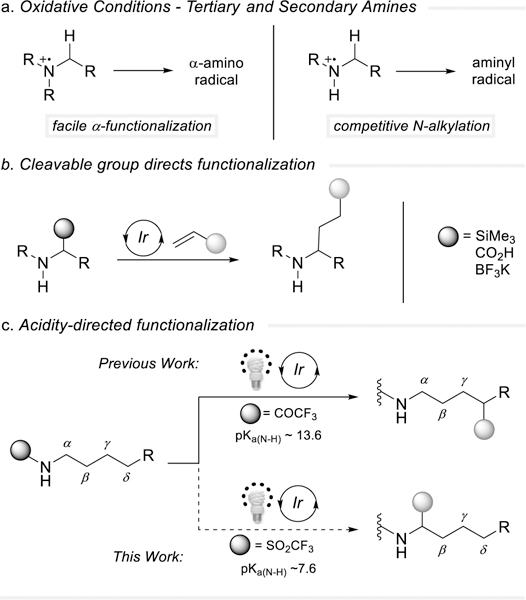
Photoredox functionalization: (a) tertiary vs secondary amines (b) installation of cleavable functionality to direct α-amino radical formation (c) acidity-controlled site-selectivity.
We sought to develop a protocol for amine C-H functionalization in which the site-selectivity can be achieved through the judicious choice of the directing group on nitrogen. Independently, our group and the Knowles group demonstrated the robustness of δ-C-(sp3)-H alkylation through [1,5]-hydrogen atom transfer. [7] Utilizing trifluoroacetamides, amidyl radicals are formed under oxidative conditions to remotely activate the δ-C-H bond (Figure 1c), an undesirable pathway for α-derivatization. We reasoned that using a more acidifying functionality could pivot reactivity towards the activation of α-C-(sp3)-H bonds by leveraging the following consequences: a change in the nature of the protecting group results in a change in the bond strength and the pKa of the N-H. [8]
After surveying a variety of amine protecting groups, we gratifyingly observed promising reactivity and selectivity using tri-fluoromethanesulfonamides. We were delighted to observe α-functionalization of 1a in 50% yield with benzyl acrylate in the presence of a carbonate base, photocatalyst A, and blue light (Table 1, entry 1). A superior yield was obtained when using [Ir(dF-CH3-ppy)2(dtbbpy)PF6] (B) [9] as the photocatalyst, and subjecting quinuclidine to the reaction conditions (77%, Table 1, entry 6). Control experiments confirmed photocatalyst, base, and light were all essential for successful α-alkylation of triflamide 1a (see SI for details). Desired product formation was not obtained with weaker electron-withdrawing groups on nitrogen (Table 1, entries 7–9).
Table 1.
Reaction Optimization
 | |||||
|---|---|---|---|---|---|
| entry | PG | Photocatalyst | R | Base | Yield(%)b |
| 1 | Tf | A | Bn | CS2CO3 | 50 |
| 2 | Tf | A | Bn | K2CO3 | 54 |
| 3 | Tf | A | Bn | K3PO4 | 58 |
| 4 | Tf | B | Bn | K3PO4 | 65 |
| 5 | Tf | A | tBu | quinuclidine | 71 |
| 6 | Tf | B | tBu | quinuclidine | 77 c |
| 7 | COCF3 | B | tBu | quinuclidine | 0 |
| g | Ts | B | tBu | quinuclidine | 0 |
| 9 | Ac | B | tBu | quinuclidine | 0 |
 | |||||
[a]1a (0.1 mmol), alkene (0.15 or 0.3 mmol), base (0.2 mmol), photocatalyst (2.0 mol%), DMF (0.2M), 34 W blue LED, ~ 40 °C, 16h.
[b]Yields determined by 1H NMR using trimethoxybenzene as an internal standard.
[c]Yield of isolated product.
[d]PG = protecting group; Tf = triflyl; Ts = tosyl; Ac = acetyl; DMF = dimethylformamide; Bn = benzyl; tBu = tert-butyl; Et = ethyl; Me = methyl.
With optimized conditions in hand, we sought to investigate the alkene scope with n-propyl triflamide 1a as the substrate (Scheme 1). Both acrylates devoid of a-substituents (3aa-3ad) as well as methyl-methacrylate (3ae) give product in acceptable yields. Acrylates containing β-substituents (3af) lead to a moderate drop in reactivity (33%) except in the case of highly activated dimethyl fumarate (3f), which proceeds in moderate yield (53%). In addition to acrylates, enones provide the desired α-functionalization (3ah-3ai). Additional Michael-acceptors including vinyl sulfones (3aj), vinyl phosphonates (3ak) and acrylonitrile (3al-3am) are well tolerated. Dimethylacrylamide (3an) is incorporated with a slightly compromised yield (38%). Of note is the successful use of electron-deficient styrene derivatives as coupling partners (3ao-3aq) as well as heteroarene-substituted olefins (3ar). [10]
Scheme 1.
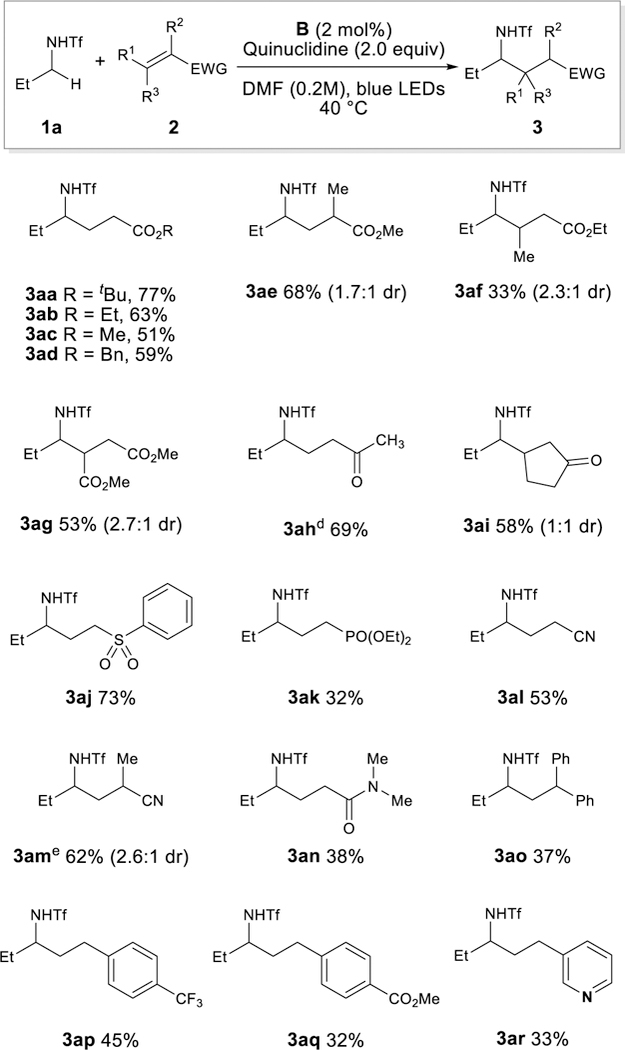
Olefin Scope. [a]1a (0.1 mmol), alkene (0.15 or 0.3 mmol), quinuclidine (0.2 mmol), [Ir(dF-CH3-ppy)2(dtbbpy)PF6] (2.0 mol%), DMF (0.2M), 34 W blue LED, ~ 40 °C, 16h. [b]Yield of isolated products. [c]dr was determined by 1H NMR of isolated product. [d]1.2 equiv. of 3-buten-2-one was used. [e]0.3 mmol scale. fEWG = electron withdrawing group; Ph = phenyl.
We examined the amine scope using either tert-butyl acrylate (2a) or ethyl acrylate (2b) as the coupling partner (Scheme 2). Substrates containing secondary α-C(sp3)-H bonds (3ba-3da) provide product in good yields, whereas tertiary (3ea-fa) α-positions show lower levels of reactivity. Interestingly, methyl-triflamide (3ga) as substrate leads to a 34% yield of the dialkylated product as nearly the sole product. Altering the ratio of triflamide and olefin coupling partner did not suppress dialkylation. We reasoned the second alkylation event occurs at a faster rate due to the resulting radical stability from the first (primary carbon radical) versus second hydrogen atom abstraction event (secondary carbon radical). Nearby electron-withdrawing groups create a more difficult alkylation event (3hb, 34%) presumably due to decreased hydridicity of the triflamide α-C-H bond. Absence of over-alkylation in the formation of products 3ba and 3ca can be attributed to this mode of deactivation in conjunction with a sterically demanding environment. Furthermore, the formation of fully branched 3ea and 3fa proceeds in lower yield. This methodology also proved tolerant of heterocyclic derivatives (3ia-3ja).
Scheme 2.
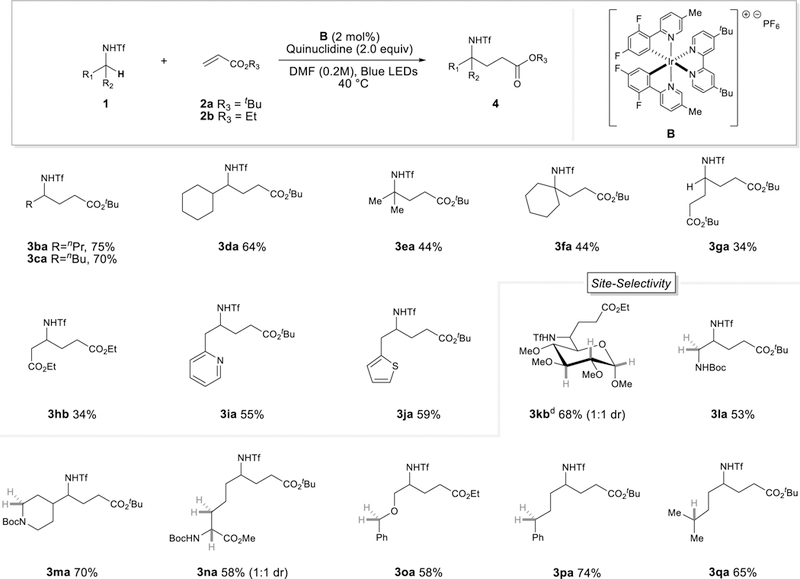
Triflamide Scope. [a]1a (0.1 mmol), alkene (0.15 or 0.3 mmol), quinuclidine (0.2 mmol), [Ir(dF-CF3-ppy)2(dtbbpy)PF6] (2.0 mol%), DMF (0.2M), 34 W blue LED, ~ 40 °C, 16h. [b]Yield of isolated products are reported. [c]dr was determined by 1H NMR of isolated product. [d]0.3 mmol scale. [f]Boc = tert-butyloxycarbonyl.
To demonstrate the robustness of our site-selective α-alkylation, we sought to functionalize sulfonamides bearing additional potential sites of activation. A glucose derivative containing multiple abstractable tertiary C-(sp3)-H bonds affords the desired α-functionalization selectively to form 3kb in 68% yield. The inclusion of primary (3la) and secondary Boc-protected amines (3ma) provide a competitive site for α-functionalization. Previous work from MacMillan demonstrates a quinuclidine radical cation can abstract a hydrogen atom from these sites. [5] Gratifyingly, alkylation occurs site-selectively at the α-C(sp3)-H triflamide site. A lysine-derived triflamide also participates delivering 3na as a single constitutional isomer in 58% yield. Preference for the selective formation of α-amino radicals over [1,5]-HAT path-ways is further demonstrated with products 3oa-3qa. This selectivity is quite remarkable considering our previous work with trifluoroacetamide directing groups which activate the δ-C-H bond via [1,5]-HAT.
Several mechanistic experiments proved enlightening. Stern-Volmer quenching studies (Figure 3b) reveal a strong kinetic preference for the single electron oxidation of quinuclidine (E1/2red = 1.1 V vs SCE in DMF) over the triflamide anion (E1/2red = 1.2 V vs SCE in DMF) by the excited state of B. A deuterium labelling experiment reveals a lack of appreciable deuteration at the position alpha to the ester suggesting that a chain transfer mechanism by direct HAT from another molecule of substrate to the enoyl radical is a minor pathway at best. Taken together, we propose the following mechanism to explain these observations. Under quinuclidine conditions, a dual HAT-photoredox catalytic cycle is proposed (Scheme 3a).[ 11 ] The highly electrophilic quinuclidinium radical cation (VI) abstracts an activated hydridic α-hydrogen of the triflamide anion (I) to deliver α-amino radical anion (II). [12] Subsequent radical trapping by an electron-deficient olefin coupling partner will furnish a carbon-centered radical (III). Single electron reduction of III by the reduced iridium photocatalyst E1/2red = IrIII/IrII = −1.42 V vs SCE) and a final protonation event affords the desired product (VIII) closing the catalytic cycle.
Scheme 3.
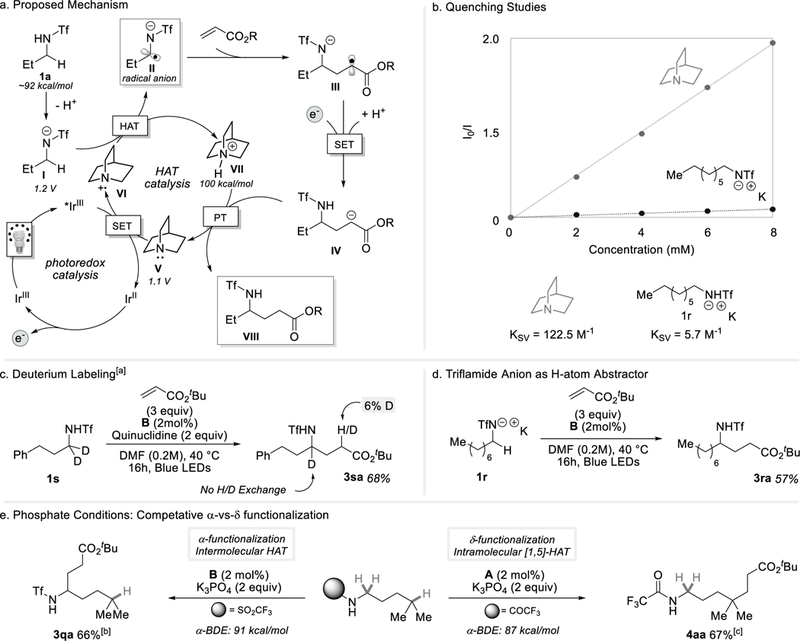
Mechanistic Interrogation. [a]No deuterium/hydrogen scrambling was observed α-to the nitrogen. Deuterium incorporation was measured in the product. Reaction was repeated under phosphate conditions in which ~1% incorporation was observed. [b]Phosphate conditions [c]trifluoroacetamide (0.1 mmol), alkene (0.15 or 0.3 mmol), K3PO4 (0.2 mmol), [Ir(dF-CF3-ppy)2(dtbbpy)PF6] (2.0 mol%), PhCF3 (0.4M), blue LED. [c]Ksv = Stervn-Volmer quenching constant.
More significant, perhaps, is the question of mechanism in the presence of phosphate as a base (entry 3 in Table 1). While phosphate has been implicated as a potential HAT catalyst, [13] we believe its dominant role is to deprotonate the acidic triflamide forming the anion in high concentration. A control experiment revealed that the potassium salt of the triflamide undergoes the α-alkylation reaction in the absence of added phosphate or quinuclidine in similar yield (Scheme 3d). As mentioned above, Stern-Volmer studies support triflamide oxidation by the excited state of the photocatalyst. Thus, we suggest the N-centered triflamidyl radical undergoes intermolecular HAT from another molecule of triflamide anion delivering the C-centered radical. Subjecting iso-hexylamine derivatives to the photoredox catalyzed C-H activation reaction (Scheme 3e) we find that trifluoroacetamides site-selectively functionalize at the δ-position (4aa) whereas triflamides conserve α-activation (3qa). This divergence in reactivity can be attributed to the resulting nitrogen radical stability and extent of deprotonation of the N-H bond. Computational studies suggest the nitrogen radical from a trifluoroacetamide is less stable than that from a triflamide.[8] Under phosphate conditions, trifluoroacetamides lie toward heavily protonated, thus, α-C-H bond activation is minimized when compared to their triflamide counter-partners. In the protonated state, the trifluoroacetamide nitrogen-center radical will experience a larger driving force for intamolecular [1,5]-HAT due to nitrogen radical instability and the absence of activated α-C-H bonds in solution. The more stable nitrogen-centered radical present on triflamide acts as an intermolecular hydrogen atom abstractor due to the activation of the α-C-H bond by the anionic charge. This pivot in reactivity allows for the selective functionalization of both α-and δ-C-H bonds depending on the installed nitrogen protecting group
In summary, we have developed a site-selective, visible-light driven photocatalyzed α-functionalization of primary amines. Key to success is the use of a trifluoromethanesulfonyl group on nitrogen which allows full deprotonation of the N-H bond and renders the α-C-H bond more hydridic and susceptible to intermolecular hydrogen atom transfer. Our reaction allows the formation of a C-C bond at the α position of primary amines derivatives through coupling α-amino radicals and electron-de ficient alkenes.
Supplementary Material
Acknowledgements
We thank NIGMS (GM125206) for support. J.C.K.C. thanks the Croucher Foundation for financial support.
Footnotes
Supporting information for this article is given via a link at the end of the document.
Contributor Information
Melissa A. Ashley, Department of Chemistry, Columbia University, New York, New York 10027 (USA) tr2504@columbia.edu
Chiaki Yamauchi, Department of Chemistry, Columbia University, New York, New York 10027 (USA) tr2504@columbia.edu.
John C. K. Chu, Department of Chemistry, Colorado State University, Fort Collins, CO 80523 (USA)
Shinya Otsuka, Department of Chemistry, Columbia University, New York, New York 10027 (USA) tr2504@columbia.edu, Department of Chemistry, Graduate School of Science, Kyoto University, Sakyo-ku, Kyoto 606-8502 (Japan).
Hideki Yorimitsu, Department of Chemistry, Graduate School of Science, Kyoto University, Sakyo-ku, Kyoto 606-8502 (Japan).
Tomislav Rovis, Department of Chemistry, Columbia University, New York, New York 10027 (USA) tr2504@columbia.edu, Department of Chemistry, Colorado State University, Fort Collins, CO 80523 (USA).
References
- [1] (a).McNally A, Prier K, MacMillan DWC, Science 2011, 334, 1114–1117 [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kohls P, Jadhav D, Pandey G, Rieser O, Org. Lett. 2012, 14, 672–675. [DOI] [PubMed] [Google Scholar]; (c) Miyake Y, Nakajima K, Nishibayashi Y, J. Am. Chem. Soc. 2012, 134, 3338–3341. [DOI] [PubMed] [Google Scholar]; (d) Nakajima K, Miyake Y, Nishibayashi Y, Acc. Chem. Res. 2016, 49, 1946–1956. [DOI] [PubMed] [Google Scholar]; (e) Prier CK, Rankic DA, MacMillan DWC, Chem. Rev. 2013, 113, 5322–5363. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Beatty JW, Stephenson CRJ, Acc. Chem. Res. 2015, 48, 1474–1484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2] (a).Dinnocenzo JP, Banach TE, J. Am. Chem. Soc. 1989, 111, 8646–8653. [Google Scholar]; (b) Zhang X, Yeh S-R, Hong S, Freccero M, Albini A, Falvey DE, Mariano PS, J. Am. Chem. Soc. 1994, 116, 4211–4220. [Google Scholar]
- [3] (a).Lewis FD, Zebrowski BE, Correa PE, J. Am. Chem. Soc. 1984, 106, 187–193. [Google Scholar]; (b) Lewis FD, Correa PE, J. Am. Chem. Soc. 1984, 106, 194–198. [Google Scholar]; (c) Lewis FD, Reddy GD, Schneider S, Gahr M, J. Am. Chem. Soc. 1989, 111, 6465–6466. [Google Scholar]; (d) Das S, Dileep Kumar JS, Shivaramayya K, George MV, J. Chem. Soc, Perkin Trans. 1, 1995, 1797–1799. [Google Scholar]; (e) Cookson RC, Costa S.M. De B., Hudec J, J. Chem. Soc. D: Chem. Commun., 1969, 13, 753–754. [Google Scholar]
- [4] (a).Yoon UC, Mariano PS, Acc. Chem. Res. 1992, 25, 233–240. [Google Scholar]; (b) Cho DW, Yoon UC, Acc. Chem. Res. 2011, 44, 204–215. [DOI] [PubMed] [Google Scholar]; (c) Miyake Y, Ashida Y, Nakajima K, Nishibayashi Y, Chem. Commun. 2012, 48, 6966–6968. [DOI] [PubMed] [Google Scholar]; (d) Nakajima K, Kitagawa M, Ashida Y, Miyake Y, Y. Nishibayashi Y. Chem. Commun. 2014, 50, 8900–8903. [DOI] [PubMed] [Google Scholar]; (e) Ruiz Espelt L, McPherson IS, Wiensch EM, Yoon TP, J. Am. Chem. Soc. 2015, 137, 2452–2455. [DOI] [PMC free article] [PubMed] [Google Scholar]; (f) Lenhart D, Bauer A, Pothig A, Bach T, Chem. -Eur. J. 2016, 22, 6519–6523. [DOI] [PubMed] [Google Scholar]; (g) Chu L, Ohta C, Zuo Z, MacMillan DWC, J. Am. Chem. Soc. 2014, 136, 10886–10889. [DOI] [PMC free article] [PubMed] [Google Scholar]; (h) Yasu Y, Koike T, Akita M, Adv. Synth. Catal. 2012, 354, 3414–3420. [Google Scholar]; (i) Miyazawa K, Koike T, Akita M, Adv. Synth. Catal. 2014, 356, 2749–2755. [Google Scholar]; (j) Fan L, Jia J, Hou H, Lefebvre Q, Rueping M, Chem. Eur. J. 2016, 22, 16437–16440. [DOI] [PubMed] [Google Scholar]; (k) Zuo Z, Cong H, Li W, Choi J, Fu GC, MacMillan DWC, J. Am. Chem. Soc. 2016, 138, 1832–1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5] (a).Le C, Liang Y, Evans RW, Li X, MacMillan DWC, Nature. 2017, 547, 79–83. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Shaw MH, Shurtleff VW, Terrett JA, Cuthbertson JD, MacMillan DWC, Science 2016, 352, 1304–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Alternately, Nicewicz and coworkers recently described the formation of α-amido alkyl radicals from photoredox catalyzed direct oxidation of the tertiary amides; see: McManus JB, Pnushka NPR, Nicewicz DA, J. Am. Chem. Soc. 2018, 140,9056–9060. [DOI] [PubMed] [Google Scholar]
- [6].Ye J, Kalvet I, Schoenebeck F, Rovis T, Nat. Chem. 2018, 10, 1037–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7] (a).Chu JCK, T. Rovis T. Nature 2016, 539, 272–275. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Choi GJ, Zhu Q, Miller DC, Gu CJ, Knowles RR, Nature 2016, 539, 268–271. [DOI] [PMC free article] [PubMed] [Google Scholar]; (c) Chen D-F, Chu JCK, Rovis T, J. Am. Chem. Soc. 2017, 139, 14897–14900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].For nitrogen-radical stability, see: D. Šakić, H. Zipse, Adv. Synth. Catal.2016, 358, 3983–3991. For discussions pertaining to the activation of C-H bonds toward abstraction please see: (a) Bietti M, Agnew. Chem. Int. Ed. 2018, 57, 16618–16637. [DOI] [PubMed] [Google Scholar]; (b) Salamone M, Carboni G, Bietti M, J. Org. Chem. 2016, 81, 9269–9278. [DOI] [PubMed] [Google Scholar]; (c) Roberts BP, Chem. Soc. Rev. 1999, 28, 25–35. [Google Scholar]; (d) Morris M, Chan B, Radom L, J. Phys. Chem. A, 2014, 118, 2810–2819. [DOI] [PubMed] [Google Scholar]; (e) O’Reilly RJ, Chan B, Taylor MS, Ivanic S, Bacskay GB, Easton CJ, Radom L, J. Am. Chem. Soc. 2011, 133, 16553–16559. [DOI] [PubMed] [Google Scholar]; (f) Salamone M, DiLabio GA, Bietti M, J. Am. Chem. Soc. 2011, 133, 16625–16634. [DOI] [PubMed] [Google Scholar]
- [9].Ladouceur S, Fortin D, Zysman-Colman E, Inorg. Chem. 2011, 50, 11514–11526. Photocatalyst B has a slightly higher reduction potential (−1.43 V vs SCE) than photocatalyst A (−1.37 V vs SCE). A common by-product observed during optimization was a 1:2 adduct resulting from oligomerization of acrylate off the enoyl radical formed from the initial addition. Photocatalyst B may allow for a more facile reduction of this resulting carbon-centered radical and minimize undesired reactivity. [DOI] [PubMed] [Google Scholar]
- [10].Background styrene polymerization is somewhat competitive to desired product formation.
- [11].The reaction proceeds with catalytic amounts of quinuclidine; however, byproducts that are observed include those arising from acrylate oligomerization (from III). Increasing the equivalents of quinuclidine ensures high reaction efficiency. As seen in Table 1, K3PO4 gives comparable yields.
- [12].We cannot discount an electrostatic attraction between VI and the triflamide anion, analogous to what we observe with CO2-derived carbamates (see ref. 6).
- [13].Margrey KA, Czaplyski WL, Nicewicz DA, Alexanian EJ, J. Am. Chem. Soc. 2018, 140, 4213–4217 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.